

ABSTRACT
A growing body of evidence supports the important role of molecular charge on antibody pharmacokinetics (PK), yet a quantitative description of the effect of charge on systemic and tissue disposition of antibodies is still lacking. Consequently, we have systematically engineered complementarity-determining regions (CDRs) of trastuzumab to create a series of variants with an isoelectric point (pI) range of 6.3–8.9 and a variable region (Fv) charge range of −8.9 to +10.9 (at pH 5.5), and have investigated in vitro and in vivo disposition of these molecules. These monoclonal antibodies (mAbs) exhibited incrementally enhanced binding to cell surfaces and cellular uptake with increased positive charge in antigen-negative cells. After single intravenous dosing in mice, a bell-shaped relationship between systemic exposure and Fv charge was observed, with both extended negative and positive charge patches leading to more rapid nonspecific clearance. Whole-body PK experiments revealed that, although overall exposures of most variants in the tissues were very similar, positive charge of mAbs led to significantly enhanced tissue:plasma concentration ratios for most tissues. In well-perfused organs such as liver, spleen, and kidney, the positive charge variants show superior accumulation. In tissues with continuous capillaries such as fat, muscle, skin, and bone, plasma concentrations governed tissue exposures. The in vitro and in vivo disposition data presented here facilitate better understanding of the impact of charge modifications on antibody PK, and suggest that alteration in the charge may help to improve tissue:plasma concentration ratios for mAbs in certain tissues. The data presented here also paves the way for the development of physiologically based pharmacokinetic models of mAbs that incorporate charge variations.
KEYWORDS: Antibody pharmacokinetics, charge modification, tissue distribution
Introduction
Monoclonal antibodies (mAbs) have emerged as vital therapeutics, owing to their high affinity, specificity, capacity to elicit immune responses, and relatively long half-life. Their favorable pharmacokinetic (PK) characteristics can be greatly attributed to pH-dependent interactions with the neonatal Fc receptor (FcRn). Antibodies in the vascular space are taken up by endothelial cells via fluid-phase pinocytosis, and within the endosomes they bind to FcRn at an acidic pH. FcRn-bound mAbs are cycled back on the cell surface, where they dissociate from FcRn due to negligible binding at physiological pH. As such, the tight binding to FcRn at pH 6 and efficient release at pH 7.4 are critical for an efficient recycling process and thus for maintaining a long plasma residence time of mAbs. Antibodies that are not bound to FcRn in the endosomes are subjected to lysosomal degradation via a process classified as nonspecific elimination.1 Another clearance mechanism for mAbs is target-mediated drug disposition (TMDD), which is most prominent at relatively lower doses. So far, a plethora of mAbs has been tested in the clinic, and huge inter-antibody variability in the PK profiles have been observed. Molecular size,2,3 glycosylation,4 immunogenicity,5 TMDD,6 and FcRn binding have all been shown to impact the PK of protein therapeutics.7,8 However, the majority of therapeutic and investigational mAbs have similar molecular weights and glycosylation patterns, and even in the absence of TMDD and immunogenicity, mAbs with similar FcRn binding kinetics have still exhibited disparate plasma PK profiles. As such, there is a need to better understand other molecular properties that can affect the PK of mAbs.
Among many physicochemical properties that may account for the variability in the PK of mAbs, charge is one of the most studied properties. In 2010, Igawa et al. demonstrated that IgG1 and IgG4 antibodies with lower isoelectric point (pI) had lower clearance and longer half-life.9 However, when a large panel of antibodies with a wide range of pI/charge values were analyzed, no correlation between clearance and pI/charge was found.10 In another retrospective analysis, Sharma et al. found that both variable regions with extremely negative and extremely positive charges exhibit fast clearance.11,12 In addition to net charge and pI values, charge distribution also appears to play a role in determining antibody PK. Disrupting positive charge patches on complementarity-determining regions (CDRs) of an antibody without altering pI could improve its plasma exposure.13,14 Generally speaking, the relationships between clearance and antibody charge have been reported to be monotonic, bell-shaped, or uncorrelated, using various metrics for charge and charge distribution. One caveat of these studies is that only one or two variants derived from each parental antibody were used for PK comparison, making it challenging to demonstrate a continuous quantitative structure-pharmacokinetic relationship (QSPKR) over a wide range of antibody charge. As such, it is desirable to accurately recapitulate the relationship between antibody charge and clearance using a series of antibody variants with incremental changes in charge and minimal differences in other molecular properties.
Furthermore, mechanisms underlying potential effects of charge on mAb PK remain arguable. It has been widely accepted that positive charges on the surface of antibodies may interact with negatively charged cell membrane or extracellular matrix (ECM), ultimately leading to faster nonspecific cellular uptake and degradation.15 Another hypothesis is that positive charges in Fv can prolong the association of an antibody with FcRn at pH 7.4, thus preventing its recycling and increasing its degradation.16 What makes the later mechanism less generalizable is the fact that the effect of charge on PK could be observed in both wild-type (WT) and FcRn knockout mice.9,17 Therefore, it is important to clarify if the lower systemic exposure of positively charged antibodies mainly originates from electrostatic interactions with cell membrane/ECM, or is commonly dependent on altered FcRn interactions.
So far, several full physiologically based pharmacokinetic (PBPK) and minimal PBPK models have revealed correlations between antibody charge and fitted parameters such as pinocytosis rate and pre-systemic clearance following subcutaneous dosing.18,19 While such findings are intriguing, only plasma PK data were incorporated in model fitting, compromising the validness of the concluded correlations. Indeed, most of the investigations of the effect of charge variations on antibody PK focus on systemic PK only. Although plasma exposures can reflect overall clearance, it is tissue concentrations that are oftentimes more relevant in terms of efficacy and toxicity. Among the few biodistribution studies in this matter, radiolabeled drugs, such as 111In and 125I labeled antibodies, were frequently used. While these experiments are powerful in identifying the “past” and the “present” of mAbs, it can be difficult to discern if the observed accumulation of antibodies in certain tissues is a result of enhanced extravasation or an artifact of antibody accumulation in the blood. Accordingly, tissue PK data from complementary approaches are warranted to further understand the impact of antibody charge on distribution into various tissues, and to validate PBPK models that incorporate antibody charge as a covariate.
In this study, we have systematically engineered an existing mAb, trastuzumab, to create a series of charge variants that cover a wide range of pI values, total charge, and charge patches, without considering target binding affinity. In vitro cellular PK and in vivo systemic PK experiments were conducted to seek an in vitro-in vivo correlation. Whole-body tissue PK of these charge variants was determined using both enzyme-linked immunosorbent assay (ELISA) and imaging-based approaches. Different proposed mechanisms underlying the effect of charge on mAb PK were examined experimentally, and various physicochemical properties of the charge variants were characterized to understand if the observed PK differences were exclusively due to charge variation.
Results
Before any protein engineering efforts were made, we digitized PK profiles of various marketed mAbs in mouse and human, to examine if the PK variabilities can be explained by charge, and if any representative mAbs are appropriate for further investigation. These mAbs are reported to have pI values from 6.1 to 9.4, all measured by imaged capillary isoelectric focusing (icIEF) in one study.20 Saturating doses were selected to minimize interference of TMDD, and only the PK data without indications of immunogenicity were extracted. No trend was observed in dose-normalized serum PK profiles, clearance, or terminal half-life for mAbs with diverse pI values (Fig. S1). Of note, these antibodies differ in immunoglobulin (IgG) subclasses, Fv framework sequences, and FcRn binding properties, which can all confound the analysis, necessitating the generation of a series of nested (i.e., more closely related) mAbs that have minimal differences in other properties than charge.
In silico design and production of trastuzumab charge variants
Trastuzumab (TS-WT), a humanized anti-human epidermal growth factor receptor 2 (HER2) antibody, was chosen as the starting template, as it has average PK profiles in human and mouse (Fig. S1). Bumbaca et al. have successfully designed and purified 2 charge variants of trastuzumab with charge changes of +5 and −4 relative to TS-WT, respectively.12 These mutants were included in the present study as TS+5 and TS-4. Previous work aimed to introduce mutations that retain antigen-binding affinity while altering the charge; however, our objective was not to create therapeutically effective charge variants, and hence we did not intend to maintain HER2 binding during the engineering that follows. Surface-exposed residues in CDRs of TS-WT were identified in Swiss PDB Viewer, and two different mutation strategies were applied. First, all these residues were mutated to R or D for positive and negative charge variants, respectively. In the second strategy, the same WT residues were allowed to mutate to either R or K for positively charged variants, and D or E for negatively charged variants, depending on the similarity in hydrophobicity and size between WT residues and R/K or D/E. This manual approach resulted in a total of 4 mutants, and Molecular Operating Environment (MOE) predicted 2 variants to have the best stability, which were termed TS+11 and TS-8, respectively. To obtain even more extreme charge variants, a more comprehensive engineering method was required. We generated 2000 random variants in MOE, whereby up to15 CDR residues were allowed to change to either R/K or D/E, followed by in-silico assessment of their properties and hence feasibility for antibody production. Since difficulty in expression of charge variants at the extreme ends was anticipated, 3 positive variants (TS+15, TS+16, TS+17) and 3 negative variants (TS-11, TS-12, TS-14) were prioritized for downstream experiments. The mutation details are shown in Table S1.
The 10 selected mutants displayed incremental changes in positive/negative charge patch size, as well as net charge (Table 1). A stability filter of delta stability <5.5 kcal/mol was applied to identify variants with high probability to be expressed as stable proteins. Although all the negative charge variants were successfully purified from Chinese hamster ovary (CHO) cells, only one positive charge variant, TS+5, was purified with good quality. TS+11, TS+16, and TS+17 showed around 40% aggregation, whereas TS+15 was not detected at all (data not shown). TS-14, −11, −8, −4, WT, and +5 were used for further experiments, and their electrostatic surfaces are presented in Figure 1.
Table 1.
Electrostatic properties and stability calculated in MOE and purification results for the charge variants
| Code name | Source | Delta stability (kcal/mol) | Delta net charge | Positive patch size change (Ang2) | Negative patch size change (Ang2) | Positive CDR patch size change (Ang2) | Negative CDR patch size change (Ang2) | Fv charge (at pH 5.5) |
Purification result |
|---|---|---|---|---|---|---|---|---|---|
| −14 | MOE | 5.38 | −13.6 | −10 | 480 | 0 | 480 | −7.9 | Successful |
| −12 | MOE | 5.25 | −12.4 | −40 | 380 | 0 | 380 | −8.9 | Successful |
| −11 | MOE | 4.95 | −11.4 | −10 | 460 | −10 | 460 | −6.1 | Successful |
| −8 | Manual | 1.72 | −8.4 | 0 | 270 | −10 | 270 | −2.4 | Successful |
| −4 | Ref.12 | 1.15 | −3.4 | 50 | 150 | 0 | 150 | +2.3 | Successful |
| +5 | Ref.12 | 2.27 | +4.6 | 140 | −40 | 140 | −40 | +10.9 | Successful |
| +11 | Manual | −0.17 | +10.8 | 430 | −40 | 430 | −40 | +16.8 | Aggregation |
| +15 | MOE | 1.50 | +15.87 | 750 | −30 | 710 | −30 | +20.9 | Failed |
| +16 | MOE | 1.58 | +15.98 | 630 | −40 | 640 | −40 | +21.1 | Aggregation |
| +17 | MOE | 0.82 | +16.79 | 720 | −60 | 800 | −60 | +22.0 | Aggregation |
Figure 1.
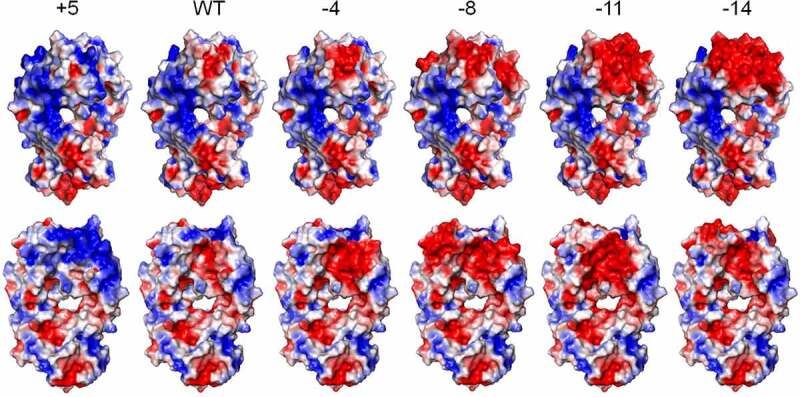
Electrostatic surface presentation of the engineered charge variants of trastuzumab. These images were made in PyMOL, and both frontal and back views are displayed. Positive charge, negative charge, and neutral patches are colored by blue, red, and white, respectively
Impact of antibody charge on cellular disposition
In an effort to understand the mechanisms underlying the potential effect of charge on antibody disposition at the cellular level, we conducted in vitro disposition experiments using Madin-Darby canine kidney (MDCK) cells. In the experimental setup, HER2 and FcRn were both absent,21–23 which makes the system appropriate for examining the interaction between charge variants and cell membrane. Cells were incubated with 100 nM mAbs for predetermined time periods, and media and cell lysates were harvested for quantification. Intracellular exposures of mAbs correlated positively with their net charges, with TS+5 showing the highest accumulation over time, and TS-11 and TS-14 were barely detectable within cells at all times (Figure 2a). Lower measured intracellular concentrations may originate from poorer stability in endosomes or compromised cellular uptake. The former assumption is less likely, since no concentration loss was detected for any charge variants in acidic buffer at 37°C for at least one week (Fig. S2B), although it cannot be precluded that these mAbs had different proteolytic degradation rates. On the other hand, nonspecific binding to negatively charged cell membranes was observed using flow cytometry. MDCK cells were incubated with charge variants on ice, followed by labeling with anti-human Fc conjugated to R-phycoerythrin (PE). Again, antibodies with more positive charge resulted in stronger cell surface binding (Figure 2b). Interestingly, intracellular area under the concentration curve (AUC) calculated from the cellular PK study demonstrated a positive correlation with mean fluorescence intensity (MFI) values obtained from the flow cytometry (Figure 2c). These observations altogether imply that antibodies with more positive charges can interact more tightly to cell membranes, which facilitates their pinocytosis and hence cellular accumulation.
Figure 2.
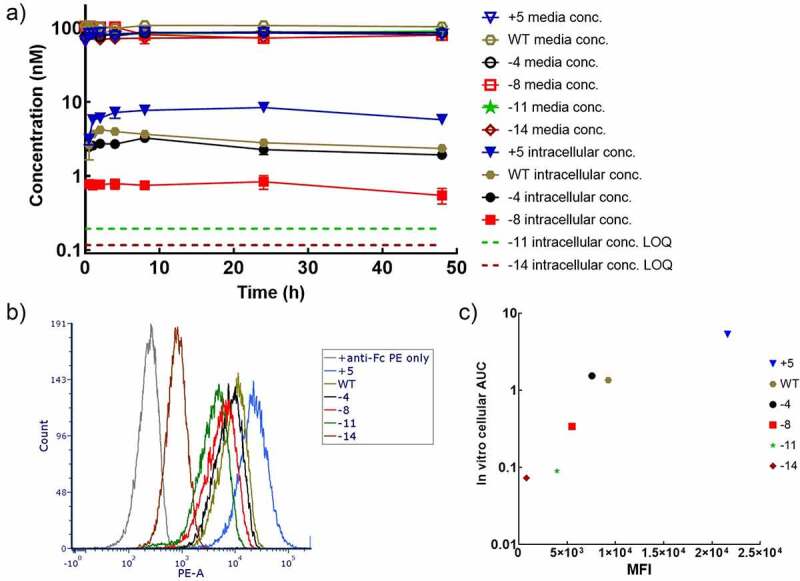
Influence of charge on cell disposition of mAbs. A) Concentration-time profiles of charge variants in the cell media and MDCK cells. B) MDCK cell surface binding of charge variants characterized in flow cytometry. C) The correlation between intracellular AUC obtained in cellular PK experiments and MFI binding signals in flow cytometry
Influence of antibody charge on plasma PK
For the plasma PK study, all the charge variants were administered at a 10 mg/kg single intravenous dose to Swiss Webster mice. This high dose was chosen to minimize known and unknown TMDD, even though trastuzumab does not cross-react with mouse HER2. These mAbs displayed biphasic or triphasic profiles with moderate variability (Figure 3a). As a typical antibody, TS-WT had a nonspecific clearance of 12.4 mL/day/kg, and a half-life of 14.2 days. TS+5 showed a 3.6-fold increase in clearance, whereas TS-4 and TS-8 decreased clearance by around 30% compared to TS-WT. Surprisingly, when antibody charge became even more negative, systemic elimination started to rise, with clearance values being 35.3 and 36.5 mL/day/kg for TS-11 and TS-14, respectively. Therefore, the relationship between clearance and antibody charge can be depicted as “U” shaped (Figure 3b). Since enhanced clearance was associated with shorter half-life in the experiment, the relationship between terminal half-life and antibody charge is bell-shaped (Figure 3c). These plasma concentration–time profiles indeed reflected whole-body elimination rates rather than varied stability, because all the charge variants were stable in isolated mouse plasma at 37°C for at least 1 week (Fig. S2A).
Figure 3.
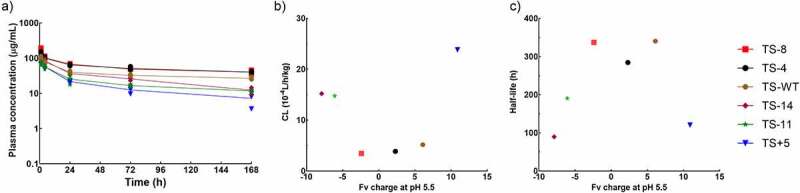
Systemic PK of charge variants. A) Plasma concentrations of charge variants over 7 days. The relationship between B) systemic clearance calculated by non-compartmental analysis and C) terminal half-life and Fv charge at pH 5.5
Physicochemical and FcRn-binding properties of charge variants
In search of explanations for the bell-shaped relationship between systemic exposure and antibody charge, we further examined whether charge engineering modified other important molecular properties, such as pI, FcRn binding, hydrophobicity, shape, stability, and nonspecific binding, which may play a role in antibody PK discrepancies.
IEF data confirm the expected positive correlation of mAb pI with positive Fv charge, and the charge variants studied cover a pI range of 6.3–8.9 (Table 2). However, adding positive charges did not always lead to a higher pI. TS-14 has a higher pI than TS-11 (6.7 vs. 6.3), and the pI of TS+5 is lower than that of TS-WT (8.7 vs. 8.9). Binding kinetics of the mAbs to mouse FcRn at pH 6 and dissociation kinetics at pH 7.4 were characterized by surface plasmon resonance (SPR). The mFcRn was immobilized on a CM5 chip, and mAbs were flowed over at pH 6, followed by dissociation phases at either pH 6 or pH 7.4. All the charge variants showed similarly good affinity to mFcRn at acidic pH (Table 2). TS+5 showed the lowest KD (2.48 nM), whereas all the negative charge variants had slightly higher KD values, which were still within twofold of the KD of TS-WT. These mAbs were all released from mFcRn rapidly at physiological pH, with dissociation half-life less than 4 s. As such, different PK profiles could not be explained by altered FcRn binding.
Table 2.
Biophysical properties and mouse FcRn binding of the charge variants. SPR was used to characterize association and dissociation kinetics of the mAbs binding to mFcRn at acidic pH, and dissociation rate constant at pH 7.4. Higher hydrophobicity was indicated by greater retention time in HIC. Melting temperatures were calculated in CD, whereas pI values were determined in IEF
| Charge variant | mFcRn binding at pH 6.0 |
mFcRn binding at pH 7.4 |
Retention time (min) | Tm (°C) | Measured pI | |||
|---|---|---|---|---|---|---|---|---|
| Kon (1/M/s) | Koff (1/s) | KD (nM) | Koff (1/s) | Dissociation half-life (s) | ||||
| TS+5 | 5.31e5 | 0.00132 | 2.48 | 0.180 | 3.85 | 4.006 | 68 | 8.7 |
| TS-WT | 3.27e5 | 0.00182 | 5.58 | 0.207 | 3.35 | 4.260 | 72 | 8.9 |
| TS-4 | 1.72e5 | 0.00137 | 7.96 | 0.176 | 3.95 | 4.386 | 70 | 7.9 |
| TS-8 | 2.03e5 | 0.00160 | 7.88 | 0.204 | 3.40 | 3.996 | 72 | 6.6 |
| TS-11 | 2.35e5 | 0.00188 | 8.00 | 0.193 | 3.59 | 4.482; 5.037 | 65 | 6.3 |
| TS-14 | 1.82e5 | 0.00170 | 9.34 | 0.194 | 3.57 | 4.572 | 62 | 6.7 |
We also evaluated hydrophobicity of the mAbs using hydrophobicity interaction chromatography (HIC). TS+5, TS-4, and TS-8 showed similar hydrophobicity to TS-WT (Table 2). Surprisingly, the two variants at the negative charge extreme, TS-11 and TS-14, exhibited greater hydrophobicity (Fig. S3). Because higher hydrophobicity is often associated with worse stability and strong nonspecific binding,11,24,25 we further measured additional biophysical properties. Circular dichroism (CD) spectra obtained at room temperature demonstrated that TS-11 and TS-14 deviated the most from other mAbs, perhaps because excessive negative charge distribution significantly altered the structure, yet all the mAbs still exhibited beta-sheet dominated secondary structures (Fig. S4A). The temperature-ramping study suggested that the melting temperatures (Tm) of TS-11 and TS-14 were lower than that of TS-WT (65°C and 62°C vs. 72°C, Table 2), but melting profiles implied good stability of all mAbs at physiological temperatures (<45°C, Fig. S4B). To detect nonspecific binding, 96-well plates were coated with bovine serum albumin (BSA), and interactions of mAbs with BSA were quantified in ELISA. TS-WT and TS-4 did not show any binding even at a concentration as high as 50 μg/mL, reflecting their favorable physicochemical properties. TS+5 displayed strong and concentration-dependent binding to BSA, whereas TS-8, TS-11, and TS-14 all showed moderate binding only when concentrations were higher than 3 μg/mL (Fig. S5A), suggesting low-affinity and high-capacity type of binding. In order to test the hypothesis of enhanced binding of positive charge variants to ECM, we also characterized mAb binding to heparin, which contributes to the negative charge of ECM, using pre-coated heparin plates in ELISA. However, significant binding to the blocking agent, BSA, would bias interpretation of the result and thus needed to be accounted for. After correction for the binding to BSA, only TS+5 was found to bind to negatively charged heparin significantly (Fig. S5C).
Whole-body PK studies of charge variants
Unexpected high systemic clearance of extreme negative charge variants did not seem to be sufficiently explained by impaired FcRn binding, hydrophobicity, stability, or nonspecific binding, as evidenced by antibody characterization mentioned above, necessitating exploration of other mechanisms. We speculated that specific tissue cells could recognize and consume TS-11 and TS-14, causing augmented drug loss in the plasma. In order to quickly identify possible organs for boosted catabolism, an in vivo fluorescence imaging approach was used. We confirmed that N-hydroxysuccinimide (NHS) chemistry was superior to maleimide reaction when conjugating Alexa Fluor 680 (AF680) to mAbs, as it had no influence on antibody PK (Fig. S6). Consequently, TS+5, WT, −8, −11, and −14 were conjugated with AF680 via lysines (dye-antibody-ratios < 2) and administered intravenously to nude mice at a single 10 mg/kg dose. Mice were imaged at 4, 24, 72, and 168 h in FMT2000 (Perkin Elmer, Fig. S7). In agreement with the plasma PK data, TS-8 displayed the longest residence in systemic circulation, whereas TS+5, −11, and −14 were cleared rapidly, as indicated by low heart concentrations. Compared to TS-WT, TS-8 exhibited much higher liver distribution, possibly due to strong signals of molecules in the blood. On the other hand, TS+5 also exhibited unusually high liver concentrations, despite its fast disappearance in the blood. Intriguingly, TS-11 and TS-14 had comparable plasma exposures, but their biodistribution profiles were distinct. TS-14 showed excessive accumulation in liver and spleen only at 4 h, and was almost invisible all over the body soon. TS-11 displayed strong and retained signals in spleen at all times.
Owing to relatively low organ resolution, poor sensitivity, limited quantification power, and interferences from blood drug concentrations in the imaging study, we decided to further conduct biodistribution experiments with whole-body perfusion using unlabeled molecules and ELISA, to unravel extravasation behaviors and tissue cell accumulation of charge variants. Charge variants were intravenously dosed to Swiss Webster mice at 10 mg/kg, and perfusion was performed prior to tissue collection at various time points. TS+5, TS-WT, and TS-8 were quantified in liver, spleen, lung, kidney, fat, heart, brain, pancreas, muscle, skin, and bone using ELISA, whereas TS-11 and TS-14 were only quantified in 4 well-perfused organs highlighted in the imaging study – liver, spleen, lung, and kidney (Figure 4). Astoundingly, most of the mAbs showed very similar concentration–time profiles in all tissues, except TS+5, which had extraordinarily high distribution into liver, spleen, and kidney at early time points, and TS-14 showed steep slopes as seen in its plasma PK profile. TS-8 exhibited only slightly higher exposures in fat, pancreas, muscle, skin, and bone. However, when tissue to plasma concentration ratios (T/P) were plotted over time, the discrepancies between mAb variants became substantial (Figure 5). Similar trends were observed in their biodistribution coefficients (BC), calculated as AUC6-168h, tissue/AUC6-168h, plasma (Table 3). TS+5 demonstrated the highest BC values in all tissues, and the degrees of divergence with other 2 mAbs were the greatest in liver, spleen, lung, kidney, and heart. In liver, spleen, lung, kidney, heart, and brain, TS-WT exhibited higher BC values than TS-8, whereas in the other tissues both variants yielded similar T/P values. These results suggest that although charge variants resulted in comparable levels of tissue accumulation, considering their significantly different plasma PK, their extravasation extents (and maybe even extravasation process) must have varied considerably.
Figure 4.
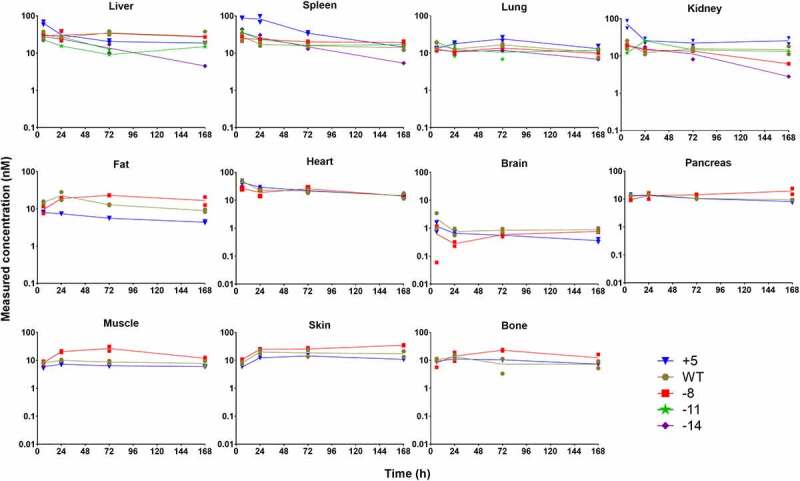
Whole-body PK of charge variants in mouse following 10 mg/kg intravenous dosing characterized using ELISA
Figure 5.
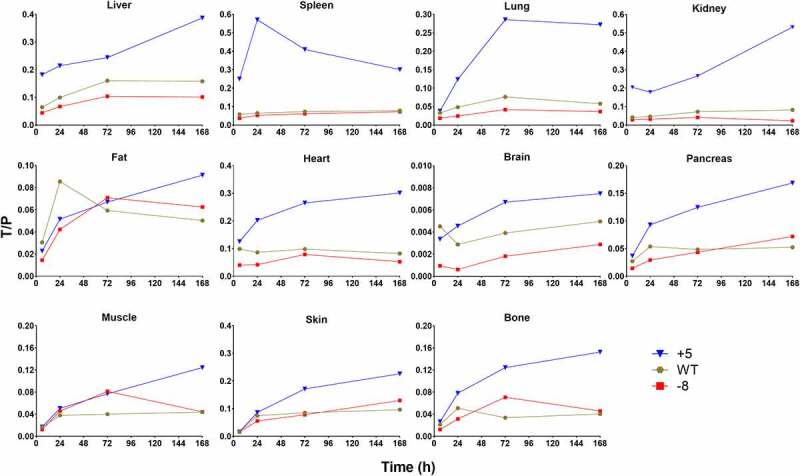
Tissue/plasma concentration ratios over time in mouse tissues
Table 3.
Calculated biodistribution coefficients (BC, %) for charge variants in each tissue
| Variant | Heart | Liver | Lung | Kidney | Spleen | Pancreas | Fat | Skin | muscle | Bone | Brain |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TS-8 | 5.92 | 8.74 | 3.37 | 3.34 | 5.89 | 4.32 | 5.57 | 7.73 | 5.66 | 4.89 | 0.165 |
| TS-WT | 9.17 | 13.4 | 6.04 | 6.47 | 7.03 | 4.80 | 6.00 | 7.72 | 3.77 | 3.79 | 0.398 |
| TS+5 | 22.5 | 24.4 | 18.8 | 26.6 | 41.0 | 10.5 | 5.74 | 12.4 | 6.45 | 9.59 | 0.553 |
Discussion
Charge has been recognized as an important molecular property that contributes toward antibody PK and PK variability. To create antibody variants with different charge properties for experimental investigation, chemical conjugation has been used traditionally.26,27 This approach allows fine tuning of charge modification, but it also introduces great charge heterogeneity, making the derived QSPKR more ambiguous. Switching of antibody framework sequences has also been applied,28 but the attainable pI or charge range is very narrow with this approach. Recently, more researchers have endeavored to introduce CDR mutations that modify antibody charge without compromising antigen binding affinity.12 This is the ideal situation, but such mutations are very difficult to discover, and can limit the charge/pI range as well. Therefore, here we disregarded target binding, and managed to purify TS-14, TS-11, TS-8, TS-4, and TS+5 variants designed by CDR mutagenesis on trastuzumab. These variants together with TS-WT were used to investigate the effect of charge on in vitro and in vivo disposition of antibodies.
It is hypothesized that positive charge patches on antibodies can interact with negatively charged ECM or cell membrane components via electrostatic interactions,15 and retention in the interstitial space or on cell surface makes antibodies more likely to be internalized into endosomes via pinocytosis, leading to higher nonspecific clearance.29 We determined binding of charge variants to heparin, a type of glycosaminoglycan (GAG) present both in ECM and cell membrane,30 and only the positive charge variant, TS+5, exhibited noticeable heparin binding. On the other hand, we observed stronger binding to MDCK cell membrane and augmented intracellular accumulation as antibody charge became more positive, which confirms the hypothesis that positive charge patches interact more with cell membrane, leading to faster uptake and clearance.
One would expect higher in vitro cellular accumulation to translate into faster nonspecific clearance in vivo, i.e., TS-14 and TS+5 should theoretically have the slowest and fastest elimination, respectively. Following a 10 mg/kg bolus dose of TS+5, TS-WT, TS-4, and TS-8, we indeed observed incrementally improved plasma exposures. Strikingly, as the antibody charge got even more negative than TS-8, the systemic clearance became much more rapid. Such a “U” shaped relationship between clearance and antibody net charge has rarely been described and never been explained before,11 perhaps because previously reported charge variants covered a broad range of charge/pI, but were not engineered in such a way that they contained incremental changes in the charge.
The inconsistency between in vitro disposition and in vivo PK data sparked our interest in studying other important properties that were not captured in the in vitro MDCK system. FcRn binding, the main mechanism that controls long half-life of antibodies and is absent in MDCK cells, was evaluated first. In SPR, these variants exhibited within 4-fold differences in their KD values for FcRn binding at pH 6, as well as equally rapid release from mFcRn with dissociation half-lives of 3.40–3.95 s, making FcRn binding unlikely to account for faster elimination of TS-11 and TS-14. Moreover, it has been suggested that positive charge areas of Fab regions can come in proximity to the negative charge patch of FcRn that reinforces the interaction at pH 7.4, ultimately causing shorter antibody half-life.16,31,32 But our finding precludes FcRn-mediated enhanced retention at neutral pH as a contributor to poor PK of more positively charged mAbs. It is still possible that our SPR assay at pH 7.4 could not resolve small differences in FcRn dissociation occurring throughout recycling.16,33 Of note, due to the poor translatability of rodent FcRn binding to human FcRn binding, it remains unknown if the impact of positive charge on human FcRn binding will also remain negligible, and if the magnitude of the influence of charge on PK will be similar between mouse and humans.
In addition to FcRn binding, other possible mechanisms contributing to the bell shape of plasma exposure vs. antibody charge relationship were considered individually. Differential stability of mAbs was likely to cause apparently different measured concentrations in cells or plasma, but CD data indicated that both molecules did not experience drastic conformational changes at temperatures below 45°C, and all the mAbs were very stable in either plasma or acidic buffer that mimicked endosomal environment for at least 7 d. Increased hydrophobicity may trigger nonspecific interactions and thus broad tissue sequestration and catabolism.34 TS-11 and TS-14 were the most hydrophobic among the mAb variants tested in HIC, although they had the lowest calculated hydrophobicity index based on the Eisenberg scale values. Further structural analysis is necessary to understand why introducing many negatively charged residues in CDRs caused higher hydrophobicity. Nevertheless, the aberrant PK of TS-11 and TS-14 did not originate from nonspecific binding triggered by hydrophobicity, as they only showed negligible binding to MDCK cell surface, and their binding to BSA was similar to that of TS-8, which displayed favorable plasma PK. Another plausible explanation for the unexpected fast elimination of TS-11 and TS-14 is unintended, specific off-target binding.35,36 As a surrogate, in vivo fluorescence imaging following mAb administration was performed to locate organs that may be responsible for unusually high catabolism. Despite similar plasma exposures of TS-11 and TS-14, TS-11 mainly distributed into the spleen where it was eliminated slowly, whereas TS-14 accumulated in the liver but was soon cleared. It remains to be seen whether extreme negative charge modification necessarily leads to increased systemic clearance, and whether liver or spleen tend to recognize and take up mAbs with extended negative charge patches.
We also characterized tissue PK of TS-8, TS-WT, and TS+5, since they demonstrated incremental systemic clearance for known reasons, as well as a modest charge range. Consistent with previous studies that reported that high pI variants were highly catabolized in liver, spleen, or kidney,28 we observed enhanced accumulation of TS+5 in these organs. TS+5 exhibited around double concentration compared to the other mAbs at 6 h, after which its concentrations converged with those of other variants. Liver has been recognized as an important organ for eliminating cationic macromolecules, and receptors for cationic proteins have been identified on hepatic Kupffer cells and endothelial cells.26,37,38 Additionally, TS+5 showed higher concentrations than other mAbs in spleen for at least 72 h. Correspondingly, an electron microscopic cytochemical study of the rat spleen suggested very intense negative charges on the endothelial cells of blood vessels that transport blood into and out of the red pulp,39 which may be responsible for retaining positively charged mAbs. Moreover, concentrations of TS+5 were superior to other mAbs in the kidney at all times studied, which is not unexpected, as the negatively charged endothelial cells and glomerular basement membrane is known to facilitate filtration of cationic molecules,40 and may analogously enhance cellular uptake of positively charged antibodies. It is noteworthy that TS+5 only displayed greater concentrations in tissues with either fenestrated (kidney) or discontinuous (liver, spleen) capillaries; however, in organs with continuous capillaries, such as fat, muscle, skin, and brain, plasma exposure was the overriding factor that controlled tissue concentrations (i.e., TS-8 and TS+5 had the highest and lowest tissue exposures, respectively). We have observed similar results with a different antibody, where enhanced accumulation of a positive charge variant of an anti-CD44 antibody was found in liver and spleen, but not in muscle.41 Interestingly, when tissue to plasma concentration ratios were plotted against time, strong charge-dependent relationships were found in most tissues. TS+5 showed the fastest distribution into all tissues, whereas TS-WT demonstrated faster extravasation than TS-8 in liver, lung, kidney, heart, and brain. TS+5 demonstrated the highest BC values in all tissues, and the degrees of divergence with the other 2 mAbs were the greatest in liver, spleen, lung, kidney, and heart. In liver, spleen, lung, kidney, heart, and brain, TS-WT exhibited higher BC values than TS-8, whereas in the other tissues both variants yielded similar tissue to plasma concentration ratios. These results suggest that positively charged mAbs can accomplish higher tissue to plasma concentration ratios.
Our results also facilitate discussion regarding development of more potent mAbs or drug-delivery strategies using charge variants of mAbs. There are a few scenarios where a positively charged mAb can be beneficial. First, this type of mAb may provide higher exposure at the site-of-action compared to neutral mAbs at similar doses, which can lead to attainment of better target engagement at the same dose or similar target engagement at lesser doses. Since similar/better target engagement at the site-of-action and much reduced systemic exposure can be accomplished using positively charged mAbs, they can provide better therapeutic index for mAbs whose toxicity stems from systemic exposure. Second, such mAbs can be used to develop antibody-drug conjugates (ADCs) with enhanced tissue/tumor penetration and increased intracellular delivery. However, the fact that positive charge simultaneously boosts exposures in other tissues as well (e.g., liver, spleen and kidney), the ADC would have to be made with linkers that show specific cleavage in the presence of tumor- or organ-specific chemical triggers (e.g., Fe(II), β-galactosidase, and sulfatase) to minimize undesired drug release in untargeted organs. Third, local administration routes such as intravitreal or intratumoral injections can benefit from positive charge variants, since these mAbs can demonstrate higher retention in the tissues and local administration can alleviate excessive distribution of these mAbs into highly perfused organs. However, these hypotheses require further experimental validation. On the other hand, outstanding positive charge patches on mAbs can also serve as a risk factor in drug development because both suboptimal plasma exposure and augmented tissue distribution can negatively impact drug efficacy and toxicity.
It is also important to note that our study has several limitations. First, we failed to produce extremely positively charged molecules due to challenges in antibody production. This limits the number of variants studied and the range of antibody charge covered, and makes the conclusions less generalizable. Second, we still cannot unravel the mechanisms for accelerated clearance for TS-11 and TS-14, and ascertain if extended negative charge patches necessarily result in poorer PK. Third, although the whole-body PK studies well-differentiated mAbs in terms of their systemic and tissue exposures, the homogenization-based analytical assay failed to tell the percentages of molecules bound to endothelium or tissue cell surface, inside cells, or trapped in ECM. It would be interesting to further investigate the sub-tissue-level distribution of charge variants using immunohistochemistry,42 or quantitative analysis of interstitial fluid, cell membrane-bound mAbs, and mAbs in cell lysate.43,44 Lastly, we did not evaluate the magnitude of the effect of charge on antibody PK in the presence of TMDD or when given at various dose levels. Interestingly, relative to other mAbs, TS+5 showed a drastic drop in concentrations at early time points, and a terminal slope that is not too steep, which is analogous to the TMDD profile. Indeed, the involved “target” could be the putative receptors for nonspecific binding, and the BC values can be even higher and half-life can be even shorter for TS+5 when a lower dose is delivered. Thus, the high-capacity and moderate-to-high affinity nature of binding between putative receptors and positive charge mAb variants can make this nonspecific elimination pathway still significant, even in the presence of TMDD caused by the therapeutic targets.
In summary, using a series of charge variants derived from the parental antibody, trastuzumab, we demonstrated a trend of enhanced cellular binding and uptake with increased antibody charge. The relationship between nonspecific systemic clearance in mouse and antibody charge appears “U” shaped, with Fv charge of −2.5-+2.5 leading to optimal systemic PK profiles. The unexpectedly rapid elimination of antibody variants with extended negative charge patches could not be explained by altered FcRn binding, nonspecific binding, or stability, and needs further mechanistic investigation. Biodistribution studies of 3 mAbs with incremental charges indicate that higher positive charges substantially improve extravasation into various tissues, but the overall tissue exposures of these antibodies did not differ to a great extent in most tissues. Nonetheless, our data suggest that alteration in the charge of antibodies may help in improving tissue:plasma concentration ratio for mAbs in certain tissues.
Materials and Methods
In silico design and characterization
In Swiss PDB Viewer, surface-exposed residues in CDRs of trastuzumab (PDB code: 1n8z) were identified using 30% solvent accessibility as the criterion. A total of 4 variants were manually designed by doing the following: 1) solvent-exposed CDR residues were all mutated to R for positive charge variants, and to D for negative charge variants, or 2) the same CDR residues were mutated to R or K for positive charge variants, and to D or E for negative ones, based on similarity in hydrophobicity or size between the parental residues and R/K/D/E.45 Additionally, 2000 random mutations of 15 positions in the CDRs of trastuzumab to R/K or D/E were performed in MOE (MOE2019, Chemical Computing Group). Parameters regarding stability, net charge change, and patch size changes were calculated in MOE for ranking these candidate molecules. Fv charge at different pHs were calculated using primary sequences and the PROTEIN CALCULATOR v3.4 tool (http://protcalc.sourceforge.net/). Electrostatic surfaces of charge variants were calculated and rendered in PyMOL (Schrödinger) with the APBS plugin.
Antibody production and purification
VH and VL genes for the variants were acquired from Synbio Technologies, and cloned into our in-house antibody expression vector IGK-FRT. The constructed plasmids were stably transfected into CHO cells with the LipofectamineTM 3000 Transfection Reagent (Thermo Fisher Scientific, cat# L3000015). Transfected cells were screened with 1 mg/mL hygromycin for 2 weeks, and the positive clones were further expanded in CD CHO AGT medium (Thermo Fisher Scientific, cat# 12490017) for 3 weeks. Media supernatants containing antibodies were harvested, and antibodies were purified using HiTrapTM Protein G HP column (GE Healthcare, cat# 17040501) in the NGC chromatography system (Bio-Rad) and buffer exchanged into phosphate-buffered saline (PBS) for storage at 4°C. The identity and purity of the purified antibodies were confirmed using native and reduced SDS-PAGE. Concentrations of antibodies were measured using the NanoDrop Spectrophotometer (Thermo Fisher Scientific).
Cellular uptake assay
MDCK cells were seeded in 6-well plates (Corning, cat# 353224) 1 day prior to experiments, with triplicates for each treatment. The media were replaced by 2 mL of 100 nM IgGs in media, and the cells were then incubated at 37°C for 30 min, 1, 2, 4, 8, 24, and 48 h. At each time point, the media were collected, and the cells were harvested by trypsinization followed by cold PBS wash. The cell number was counted with 0.4% trypan blue solution in the CountessTM II FL Automated Cell Counter (Thermo Fisher Scientific). For every 2 × 105 cells, 10 μL of RIPA buffer (Thermo Fisher Scientific, cat# 89901) containing 1× HaltTM protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, cat# 78441) was added to lyse the cells and to release intracellular IgGs. After lysis on ice for 90 min, the cell lysate was obtained from the supernatant following centrifugation at 13,000 rpm for 2 min. The media and cell lysate samples were kept at −20°C until ELISA quantification.
Flow cytometry
MDCK cells at a density of 5 × 105 cells/tube were incubated with 100 μL of PBS with 0.5% BSA (PBS-BSA) containing 100 μg/mL of IgGs on ice for 1 h. Following washing twice with ice-cold PBS-BSA, cells were reconstituted in 100 μL of cold PBS-BSA with 5 μg/mL of anti-human Fc antibody conjugated to R-PE (Abcam, cat# 98596) for 1 h on ice. After washing twice with cold PBS-BSA, cell pellets were reconstituted in 300 μL of PBS-BSA for flow cytometry in MACSQuant Analyzer 10 (Miltenyi Biotec). Flow cytometry results were analyzed and plotted in FCS Express 6 (De Novo Software).
Stability test
IgGs were prepared in mouse plasma at around 100 μg/mL or in 0.2 M citrate buffer (pH 5.8) at around 500 μg/mL. Three aliquots of 100 μL from each IgG preparation were transferred into individual microcentrifuge tubes, and kept in a humidified incubator with 5% CO2 at 37°C. At 0, 72, and 168 h, samples were transferred to −80°C until ELISA quantification.
Isoelectric focusing
IEF was conducted in the Pharmacia PhastSystem machine (Uppsala, Sweden) using precast PhastGel IEF 3–9 (GE Healthcare, cat# 17054301). Around 1 μL of 2–5 μg antibodies were loaded at the middle of the gel with the help of PhastGel sample applicators (GE Healthcare, cat# 18161801). IEF separation method was as follows: preconditioning at 2000 V, 2.5 mA, 3.5 W, for 75 Vh; sample application at 200 V, 2.5 mA, 3.5 W, for 15 Vh; 2000 V, 2.5 mA, 3.5 W, for 410 Vh at 15°C. Upon completion of electrophoresis, the gel was stained with the Bio-Safe Coomassie G-250 Stain (Bio-Rad, cat# 1610786) for 30 min, followed by brief wash in distilled water. The pI markers (GE Healthcare, cat# 17047101) were used to establish standard curves.
Circular dichroism
CD spectra of charge variants were obtained by scanning 100 µg/mL of antibodies in PBS over 200–260 nm in JASCO J-815 spectrometer equipped with a digital temperature controller. Unfolding of antibodies was monitored by measuring the ellipticity at 208 nm in a temperature range of 25–90°C, with a heating rate of 1°C/min and a holding time of 2 min for every 5°C increment.
Surface plasmon resonance
Binding of the charge variants to mouse FcRn was characterized on a Reichert SR7500 system (Buffalo, Ny). First, a carboxymethyl dextran hydrogel surface sensor chip (Reichert Technologies, cat# 13206066) was activated by a 400 mM of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC, Sigma-Aldrich, cat# 22980) and 100 mM of NHS (Sigma-Aldrich, cat# 24500) solution at a flow rate of 25 for 8 min. Then, 15 μg/mL of mouse FcRn non-covalently linked with mouse beta-2-microglobulin (R&D Systems) in 10 mM acetate buffer (pH 5.0) was injected at 25 μL/min for 30 s over the left channel of the chip. Unreacted chip surface was blocked by injecting 1 M ethanolamine (pH 8.5) at 25 μL/min for 10 min. After equilibrium, the immobilized mFcRn heterodimer intensity was determined to be 300 μRIU. To characterize binding kinetics at pH 6.0, PBS containing 0.005% Tween 20 (PBST) at pH 6.0 served as the running buffer and IgGs were prepared in PBST (pH 6.0) at a concentration range of 1–100 nM. The association and dissociation times were 1.5 and 5 min, respectively. To determine dissociation kinetics at pH 7.4, PBST at pH 7.4 was used as the running buffer. IgGs in PBST (pH 6.0) at 1–100 nM were injected for 1.5 min and allowed to dissociate for 2 min. For the experiments at both pHs, the temperature of measurement was 25°C, the flow rate was 25 μL/min, buffer controls were run before each IgG series, and the regeneration condition was flowing PBST (pH 8.0) for 1.5 min. SPR data were fitted using a 1:1 Langmuir model in Scrubber 2 (BioLogic Software).
Hydrophobicity interaction chromatography
HIC was conducted in Agilent 1200 HPLC system using the nonporous TSKgel Butyl-NPR column (Tosoh Bioscience, cat# 14947). IgGs were prepared as 1–2 mg/mL solutions in PBS, and 10 μL of each sample was injected for analysis. The mobile phase A was 1.5 M ammonium sulfate in 25 mM sodium phosphate at pH 7.0, and the mobile phase B consisted of 75% (v/v) aqueous solution of 25 mM sodium phosphate at pH 6.95 and 25% isopropanol. The separation condition was a linear gradient of 0–100% B over 12 minutes followed by 100% B for 6 minutes at a flow rate of 0.8 mL/min.
Biodistribution study using unlabeled IgGs
The in vivo study was approved by the institutional animal care and use committee (IACUC) of SUNY Buffalo (protocol# PHC29035Y). Male Swiss Webster mice weighing 26–30 g (Charles River) were injected with 10 mg/kg of IgGs via the penile vein. For the plasma PK study, blood samples were drawn from the retro-orbital sites of 3 mice at each predetermined time point and collected in EDTA-coated microcentrifuge tubes. Blood samples were centrifuged at 2000 g for 20 min at 4°C, and plasma was separated and stored at −80°C for further analysis. For the tissue PK study, at 6, 24, 72, and 168 h, two mice were perfused followed by cervical dislocation. Briefly, around 1.5 mL blood was first extracted via cardiac puncture, then 8 mL of PBS containing 5 U/mL heparin was injected into the apex of the left ventricle at the rate of 2 mL/min and allowed to exit via a small cut of the right ventricle. Heart, lung, liver, spleen, pancreas, kidney, fat, bone, muscle, skin and brain were harvested, blotted dry, snap frozen in liquid nitrogen, and stored at −80°C for further analysis.
Biodistribution study characterized in FMT
This in vivo study was approved by the IACUC of SUNY Buffalo (protocol# 201900042). IgGs were conjugated to Alexa FluorTM 680 via lysines (AF680 NHS ester, Thermo Fisher Scientific, cat# A20008) or cysteines (AF680 C2 maleimide, Thermo Fisher Scientific, cat# A20344) as per the manual. Male athymic nude mice (Foxn1nu/Foxn1nu) weighing 26–28 g (The Jackson Laboratory) were injected with 10 mg/kg of NHS 680- or maleimide 680-conjugated IgGs. At 4, 24, 72, and 168 h, 2–3 mice per group were scanned under anesthesia in the FMT2000 in vivo imaging system (Perkin Elmer). For semi-quantification purpose, 2 μM of 680-IgG conjugates were used to calibrate the signal.
ELISA for quantification of IgGs
The sandwich ELISA for quantification of IgGs in in vitro and in vivo samples included the following steps: a) MaxiSorp 384-well plates (Thermo Scientific, cat# 464718) were coated with 50 μL/well of 5 μg/mL F(ab’)2 goat anti-human Fc fragment (Bethyl Laboratories, cat# A80-248A) in 20 mM Na2HPO4 overnight at 4°C, b) plates were blocked with 90 μL/well of 1% BSA at room temperature (RT) for 1 h, c) 30 μL/well of standards, quality control samples (QCs), and experimental samples were added in triplicates and incubated at RT for 2 h, d) 30 μL/well of 200 ng/μL F(ab’)2 goat anti-human F(ab’)2 conjugated to alkaline phosphatase (Bethyl Laboratories, cat# A80-249AP) in washing buffer were loaded and plates were incubated at RT for 1 h, e) 60 μL/well of 1 mg/mL p-nitrophenyl phosphate (PNPP) in 1x diethanolamine (DEA) substrate buffer was loaded, immediately followed by measurement of dA/dt at 405 nm for 40 min in Filter Max F5 microplate analyzer (Molecular Devices). All the incubation steps at RT were performed on a plate shaker. Between each step, plate wells were washed with washing buffer (PBS containing 0.05% Tween 20) for 3 times, followed by 3 washes of distilled water. Standard curves were fitted using 4-parameter logistic equations, and the typical concentration range for quantification was 1–250 ng/mL.
Separate standards and QCs were prepared for individual IgGs in corresponding matrices; in the same way as the experimental samples were processed. Sample preparation procedures varied for different applications. For the cellular PK study, media samples and cell lysates were diluted with PBS containing 0.15% BSA by 600 and 10 times, respectively. For the plasma PK and stability experiments, the samples were diluted with PBS (0.15% BSA) by 4000 times. For the whole-body PK study, on the day of ELISA quantification, tissue samples were weighted, and 400 μL RIPA buffer containing 1× HaltTM protease and phosphatase inhibitor cocktail per 100 mg tissue was added, which was considered as 5-time initial dilution. Each 5 mL centrifuge tube for homogenization contains seven stainless steel beads (Benchmark Scientific, cat# D1133-28) for skin, bone, and muscle, or 7 zirconium beads (Benchmark Scientific, cat# D1132-30TP) for all other tissues. Samples were then homogenized using Bullet Blender Homogenizer (Next Advance) at speed 8, which was repeated at least 8 times. After that, samples were kept on ice for 2 h for complete lysis, and centrifuged at 3900 rpm for 15 min at 4°C. The supernatant was collected and further diluted in RIPA buffer, incubated on ice for another 2 h for equilibrium prior to loading in ELISA plates. The overall dilution factors were 25 for brain, pancreas, and fat; 50 for kidney, lung, muscle, and bone; 100 for liver and skin; 200 for spleen and heart.
ELISA for heparin binding characterization
ELISA for detecting heparin binding consisted of the following steps: a) Heparin-coated microplates (bioWORLD, cat# 20140005) or untreated Nunc MaxiSorp 96-well plates (Thermo Fisher, cat# 442404) were blocked with 200 μL/well of 1% BSA at RT for 1 h; b) 50 μL/well of 0.2–50 μg/mL IgGs in PBS was added and incubated at RT for 1 h; c) 50 μL/well of 200 ng/μL F(ab’)2 goat anti-human F(ab’)2 conjugated to alkaline phosphatase (Bethyl Laboratories, cat# A80-249AP) in washing buffer were loaded and plates were incubated at RT for 1 h; d) 100 μL/well of 1 mg/mL PNPP in 1x DEA buffer was loaded, immediately followed by plate reading. Washing steps and plate reader setting were the same as mentioned above.
Supplementary Material
Acknowledgments
We thank Dr. Marilyn Morris for sharing the MDCK cell line; Dr. Joseph Balthasar for providing antibody standards with diverse pI values for validation of IEF method; Dr. Brandon Bordeau for his guidance and assistance in performing SPR; Vincent Chak for his help in using CD; Shengjia Wu for his instruction in FMT implementation; Hsuan-Ping Chang for her demonstration of whole-body PK studies.
Funding Statement
This research was funded by National Institute of General Medical Sciences grant [GM114179] and the Center of Protein Therapeutics at the University at Buffalo. D.K.S is also supported by National Institute of Allergy and Infectious Diseases grant [AI138195] and National Cancer Institute grants [R01CA246785 and R01CA256928].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Abbreviations
| mAbs | monoclonal antibodies |
| CDR | complementarity-determining region |
| FcRn | neonatal Fc receptor |
| SPR | surface plasmon resonance |
| pI | isoelectric point |
| PK | pharmacokinetics |
| AUC | area under the curve |
| TMDD | target-mediated drug disposition |
| ECM | extracellular matrix |
| ELISA | enzyme linked immunosorbent assay |
| KD | equilibrium dissociation constant |
References
- 1.Lencer WI, Blumberg RS.. A passionate kiss, then run: exocytosis and recycling of IgG by FcRn. Trends Cell Biol. 2005. Jan;15(1):5–13. PMID: 15653072. doi: 10.1016/j.tcb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Gill KL, Gardner I, Li L, Jamei M. A bottom-up whole-body physiologically based pharmacokinetic model to mechanistically predict tissue distribution and the rate of subcutaneous absorption of therapeutic proteins. AAPS J. 2016. Jan;18(1):156–70. PMID: 26408308; PMCID: PMCPMC6890583. doi: 10.1208/s12248-015-9819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venturoli D, Rippe B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge, and deformability. Am J Physiol Renal Physiol. 2005. Apr;288(4):F605–13. PMID: 15753324. doi: 10.1152/ajprenal.00171.2004. [DOI] [PubMed] [Google Scholar]
- 4.Falck D, Thomann M, Lechmann M, Koeleman CAM, Malik S, Jany C, Wuhrer M, Reusch D. Glycoform-resolved pharmacokinetic studies in a rat model employing glycoengineered variants of a therapeutic monoclonal antibody. MAbs. 2021. Jan-Dec;13(1):1865596. PMID: 33382957; PMCID: PMCPMC7781607. doi: 10.1080/19420862.2020.1865596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012. Jun;14(2):296–302. PMID: 22407289; PMCID: PMCPMC3326159. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mager DE, Jusko WJ.. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001. Dec;28(6):507–32. PMID: 11999290. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 7.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem PMID: 16793771. 2006. Aug 18;281(33):23514–24. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 8.Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol PMID: 19494290. 2009. Jun 15;182(12):7663–71. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]
- 9.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010. May;23(5):385–92. PMID: 20159773. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 10.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012. Nov-Dec;4(6):753–60. PMID: 23778268; PMCID: PMCPMC3502242. doi: 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014. Dec 30;111(52):18601–06. PMID: 25512516; PMCID: PMCPMC4284567. doi: 10.1073/pnas.1421779112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bumbaca Yadav D, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, et al. Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics. J Biol Chem. 2015. Dec 11;290(50):29732–41. PMID: 26491012; PMCID: PMCPMC4705991. doi: 10.1074/jbc.M115.692434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. MAbs PMID: 25695748; PMCID: PMCPMC4622971. 2015;7(3):483–93. doi: 10.1080/19420862.2015.1016696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y, Cai H, Hu Z, Boswell CA, Diao J, Li C, Zhang L, Shen A, Teske CA, Zhang B, et al. Balancing the affinity and pharmacokinetics of antibodies by modulating the size of charge patches on complementarity-determining regions. J Pharm Sci. 2020. Dec;109(12):3690–96. PMID: 32910947. doi: 10.1016/j.xphs.2020.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem PMID: 21053952. 2010. Dec 15;21(12):2153–63. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- 16.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A PMID: 25918417; PMCID: PMCPMC4434771. 2015. May 12;112(19):5997–6002. doi: 10.1073/pnas.1408766112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly RL, Yu Y, Sun T, Caffry I, Lynaugh H, Brown M, Jain T, Xu Y, Wittrup KD. Target-independent variable region mediated effects on antibody clearance can be FcRn independent. MAbs PMID: 27610650; PMCID: PMCPMC5058615. 2016. Oct 8;8(7):1269–75. doi: 10.1080/19420862.2016.1208330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varkhede N, Forrest ML. Understanding the monoclonal antibody disposition after subcutaneous administration using a minimal physiologically based pharmacokinetic model. J Pharm Pharm Sci PMID: 30011390; PMCID: PMCPMC6613546. 2018;21(1s):130s–148s. doi: 10.18433/jpps30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu S, D’Argenio DZ. Predicting monoclonal antibody pharmacokinetics following subcutaneous administration via whole-body physiologically-based modeling. J Pharmacokinet Pharmacodyn. 2020. Oct;47(5):385–409. PMID: 32500362; PMCID: PMCPMC7529865. doi: 10.1007/s10928-020-09691-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goyon A, Excoffier M, Janin-Bussat MC, Bobaly B, Fekete S, Guillarme D, Beck A. Determination of isoelectric points and relative charge variants of 23 therapeutic monoclonal antibodies. J Chromatogr B Analyt Technol Biomed Life Sci. 2017. Oct 15;1065-1066:119–28. PMID: 28961486. doi: 10.1016/j.jchromb.2017.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Chung S, Nguyen V, Lin YL, Lafrance-Vanasse J, Scales SJ, Lin K, Deng R, Williams K, Sperinde G, Li JJ, et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs. 2019. Jul;11(5):942–55. PMID: 30982394; PMCID: PMCPMC6601550. doi: 10.1080/19420862.2019.1605270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Praetor A, Ellinger I, Hunziker W. Intracellular traffic of the MHC class I-like IgG Fc receptor, FcRn, expressed in epithelial MDCK cells. J Cell Sci. 1999. Jul;112(Pt 14):2291–99. PMID: 10381385. doi: 10.1242/jcs.112.14.2291. [DOI] [PubMed] [Google Scholar]
- 23.Claypool SM, Dickinson BL, Yoshida M, Lencer WI, Blumberg RS. Functional reconstitution of human FcRn in Madin-Darby canine kidney cells requires co-expressed human beta 2-microglobulin. J Biol Chem PMID: 12023961; PMCID: PMCPMC2825174. 2002. Aug 2;277(31):28038–50. doi: 10.1074/jbc.M202367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015. Jul;33(7):733–35. PMID: 26076429. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 25.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012. Jan;101(1):102–15. PMID: 21935950. doi: 10.1002/jps.22758. [DOI] [PubMed] [Google Scholar]
- 26.Hong G, Bazin-Redureau MI, Scherrmann JM. Pharmacokinetics and organ distribution of cationized colchicine-specific IgG and Fab fragments in rat. J Pharm Sci. 1999. Jan;88(1):147–53. PMID: 9874717. doi: 10.1021/js970335n. [DOI] [PubMed] [Google Scholar]
- 27.Lee HJ, Pardridge WM. Monoclonal antibody radiopharmaceuticals: cationization, pegylation, radiometal chelation, pharmacokinetics, and tumor imaging. Bioconjug Chem. 2003. May-Jun;14(3):546–53. PMID: 12757378. doi: 10.1021/bc0256648. [DOI] [PubMed] [Google Scholar]
- 28.Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. MAbs. 2014;6(5):1255–64. PMID: 25517310; PMCID: PMCPMC4623330. doi: 10.4161/mabs.29809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraft TE, Richter WF, Emrich T, Knaupp A, Schuster M, Wolfert A, Kettenberger H. Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis. MAbs. 2020. Jan-Dec;12(1):1683432. PMID: 31769731; PMCID: PMCPMC6927760. doi: 10.1080/19420862.2019.1683432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer PMID: 16069816. 2005. Jul 5;5(7):526–42. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 31.Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’Hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. MAbs. 2018. Jan;10(1):81–94. PMID: 28991504; PMCID: PMCPMC5800364. doi: 10.1080/19420862.2017.1389355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen PF, Schoch A, Larraillet V, Hilger M, Schlothauer T, Emrich T, Rand KD. A two-pronged binding mechanism of IgG to the neonatal Fc receptor controls complex stability and IgG serum half-life. Mol Cell Proteomics. 2017. Mar;16(3):451–56. PMID: 28062799; PMCID: PMCPMC5341005. doi: 10.1074/mcp.M116.064675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs. 2013. Jul-Aug;5(4):576–86. PMID: 23765230; PMCID: PMCPMC3906311. doi: 10.4161/mabs.24981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020. Jan-Dec;12(1):1743053. PMID: 32249670; PMCID: PMCPMC7153844. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bumbaca D, Wong A, Drake E, Reyes AE 2nd, Lin BC, Stephan JP, Desnoyers L, Shen BQ, Dennis MS. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs. 2011. Jul-Aug;3(4):376–86. PMID: 21540647; PMCID: PMCPMC3218534. doi: 10.4161/mabs.3.4.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vugmeyster Y, Szklut P, Wensel D, Ross J, Xu X, Awwad M, Gill D, Tchistiakov L, Warner G. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm Res. 2011. Jul;28(7):1696–706. PMID: 21424161. doi: 10.1007/s11095-011-0405-x. [DOI] [PubMed] [Google Scholar]
- 37.Takakura Y, Fujita T, Hashida M, Sezaki H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm Res PMID: 1694582. 1990. Apr 7;07(4):339–46. doi: 10.1023/a:1015807119753. [DOI] [PubMed] [Google Scholar]
- 38.Koyo N, Chiaki T, Sachi N, Yoshinobu T, Mitsuru H, Hitoshi S. Effect of electric charge on the hepatic uptake of macromolecules in the rat liver. Int J Pharm. 1990;65(1–2):7–17. doi: 10.1016/0378-5173(90)90003-M. [DOI] [Google Scholar]
- 39.Ueda H, Takehana K, Eerdunchaolu IK, Fujimori O, Shimada S, SHIMADA S. Electron microscopic cytochemical studies of anionic sites in the rat spleen. J Vet Med Sci. 2001. Mar;63(3):287–91. PMID: 11307929. doi: 10.1292/jvms.63.287. [DOI] [PubMed] [Google Scholar]
- 40.Miner JH. Glomerular filtration: the charge debate charges ahead. Kidney Int. 2008. Aug;74(3):259–61. PMID: 18626493. doi: 10.1038/ki.2008.260. [DOI] [PubMed] [Google Scholar]
- 41.Stüber JC, Rechberger KF, Miladinović SM, Pöschinger T, Zimmermann T, Villenave R, Eigenmann MJ, Kraft TE, Shah DK, Kettenberger H. Impact of charge patches on tumor disposition and biodistribution of therapeutic antibodies. bioRxiv. 2021. doi: 10.1101/2021.09.01.458024. [DOI] [Google Scholar]
- 42.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol PMID: 17362988. 2007. May 4;368(3):652–65. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 43.Wiig H, Aukland K, Tenstad O. Isolation of interstitial fluid from rat mammary tumors by a centrifugation method. Am J Physiol Heart Circ Physiol. 2003. Jan;284(1):H416–24. PMID: 12388326. doi: 10.1152/ajpheart.00327.2002. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Fagotto F. A method to separate nuclear, cytosolic, and membrane-associated signaling molecules in cultured cells. Sci Signal PMID: 22169476. 2011. Dec 13;4(203). pl2. doi: 10.1126/scisignal.2002373. [DOI] [PubMed] [Google Scholar]
- 45.Livingstone CD, Barton GJ. Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci. 1993. Dec 9;6. 745–56. PMID: 8143162. DOI: 10.1093/bioinformatics/9.6.745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.