

Abstract
Bumped Kinase Inhibitors (BKIs), targeting Calcium-dependent Protein Kinase 1 (CDPK1) in apicomplexan parasites with a glycine gatekeeper, are promising new therapeutics for apicomplexan diseases. Here we will review advances, as well as challenges and lessons learned regarding efficacy, safety, and pharmacology, that have shaped our selection of pre-clinical candidates.
Keywords: Apicomplexa, Bumped Kinase Inhibitor, Calcium-dependent Protein Kinases, Gatekeeper, Cryptosporidium, Toxoplasma, Neospora, Sarcocystis
1. Introduction
Bumped Kinase Inhibitors (BKIs) that target apicomplexan Calcium-dependent Protein Kinases (CDPKs) have great promise for therapeutic use. Substantial improvements in safety and efficacy have been achieved using medicinal chemistry to advance a BKI towards commercial use in veterinary and human treatment of infectious apicomplexan diarrheal diseases. As the primary target for BKIs, apicomplexan CDPKs are structurally similar in a number of parasites, with BKI activity confirmed against CDPKs in Toxoplasma, Neospora, Plasmodium, Cryptosporidium, Sarcocystis, Babesia, Thelieria (Van Voorhis et al., 2017), and Cystoisospora (Shrestha et al., 2019). BKIs are effective and selective against this group of parasite kinases relative to mammalian kinases due to differences in the topology of a hydrophobic pocket within the ATP binding site (Van Voorhis et al., 2017) that is, in part, determined by the size of their respective gatekeeper residues (Larson et al., 2012). This broad range of activity with no corresponding mammalian kinase target makes BKIs ideal candidates for development as drugs for treating the diseases caused by these parasites in both humans and livestock due to the reduced risk of toxicity due to off-target inhibition of host kinases. It is noteworthy that although apicomplexan CDPKs share highly conserved ATP binding domains, sensitivity to BKIs varies based on the size of the gatekeeper residue side chain (the most important driver of compound potency) as well as characteristics and orientations of other amino acids within the ATP binding pocket (Keyloun et al, 2014). Generally, BKI potency is reduced as the size of the gatekeeper side chain increases, approaching similar levels of activity as mammalian kinases with a threonine gatekeeper. Therefore, obtaining selectivity for apicomplexan enzymes such as PfCDPK1 (Plasmodium falciparum), BbCDPK4 (Babesia bovis), EtCDPK1 (Eimeria tenella), and Theileria CDPKs (which all possess threonine gatekeeper residues similar to the human enzyme SRC) become more challenging. In contrast, Toxoplasma, Neospora, Cryptosporidium, Sarcocystis (Van Voorhis et al., 2017), Cystoisospora (Shrestha et al., 2019) and Hammondia hammondi are most sensitive to BKIs and have enzymes that share a glycine gatekeeper residue (Keyloun et al, 2014). For this reason, the focus of BKI development has mainly been prioritized around these small gatekeeper CDPKs.
A CDPK1-focused library of BKIs has been developed over the years based on a limited number of core scaffolds with diverse chemical functional groups. This focused library of compounds exhibits a wide range of pharmacokinetic/pharmacodynamic (PK/PD) properties that show superior potency in vitro and demonstrates efficacy in multiple in vivo models of disease. Despite the lack of an analogous mammalian kinase target to the apicomplexan CDPK1, some BKIs have a predilection for off-target kinases, which may be associated with undesirable side effects, as is the case for many small molecules that target kinases. In addition, BKIs are bioactive compounds beyond kinases, and non-kinase effects may also be found in this class. This unfortunately has led to a variety of liabilities such as cardiovascular and gastrointestinal (GI) side effects and teratogenicity that require modification in order to home in on a lead candidate. Furthermore, differences in the localization of target parasites need to be considered to optimize compounds for precise biopharmaceutical properties that maximize effects at the infection site. Hence, some enteric pathogens such as Cryptosporidium and Cystoisospora require large GI drug exposures but not necessarily systemic distribution, while others such as Toxoplasma and Neospora will need high systemic exposure in plasma, multiple organ tissues, and the CNS.
In this review, we describe the challenges and lessons learned in the development and testing of a library of BKIs around three separate central scaffolds: pyrrolopyrimidines (PrP), pyrazolopyrimidines (PP), and 5-aminopyrazole-4-carboxamide (AC) (Fig. 1). These include chirality, human ether-à-go-go-related gene (hERG) inhibition, in vivo cardiovascular and GI side effects, teratogenicity, general toxicity and lack of efficacy due to PK/PD properties.
Fig. 1.
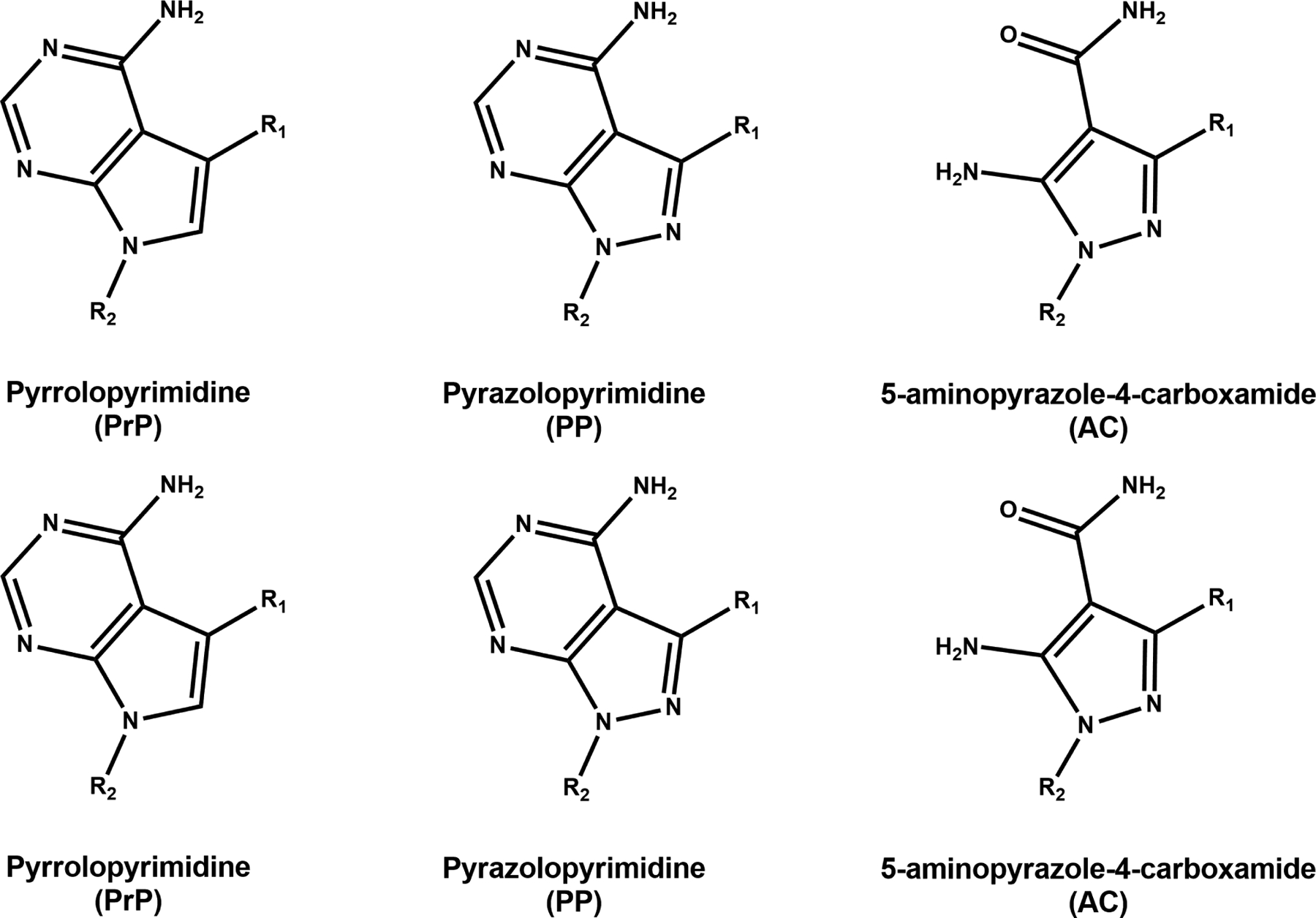
Central scaffolds of lead bumped kinase inhibitors (BKIs). BKI development and testing was centered around three main scaffolds: pyrrolopyrimidines (PrP), pyrazolopyrimidines (PP), and 5-aminopyrazole-4-carboxamides (AC).
2. Challenges that have shaped the selection of pre-clinical candidates
2.1. Chirality
It is estimated that more than half of all drugs currently in use are compounds that possess chirality, or ‘handedness’ (Rentsch, 2002; Nguyen et al., 2006). A compound and its chiral partner are enantiomers, mirror images sharing the same molecular formula and atom-to-atom bonding, yet lacking an internal plane of symmetry, and thus not superimposable. Chirality arises when a central atom, typically carbon, possesses four distinct substituents. These substituents can occupy different orientations in three-dimensional space, creating two versions of the same molecule, a right-handed and a left-handed form (Brooks et al., 2011). Many naturally occurring molecules such as nucleic acids, amino acids, and sugars are chiral.
From the perspective of structure-based drug development, chirality is an important factor to consider. A compound designed with a particular three-dimensional configuration in mind to allow for specific binding interactions with a target protein would not be afforded the same interactions in its chiral partner. Due to this difference in stereochemistry, most enantiomers exhibit differential efficacy, pharmacokinetics (PK), metabolism, and/or toxicity profiles (Nguyen et al., 2006; Brooks et al., 2011). As such, they should be treated as chemically distinct compounds. Most chiral drugs on the market exist as a racemic mixture of both enantiomers, typically with only one form being bioactive. In some cases, the inactive form may be toxic or possess undesired pharmacology, as in the example of thalidomide, a widely used racemate drug to treat morning sickness that was later found to have severe teratogenic effects attributed to the “inactive enantiomer”(Nguyen et al., 2006). In 1992, the United States Food and Drug Administration published guidelines that recommended stereochemical enantiomers be fully characterized with regard to their activity and safety profiles early in the drug development process and added requirements for the manufacturing process including identification and purification (U.S. Food and Drug Administration https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs First published: May, 1992, Accessed 8 March, 2020). With the goal of reducing the cost of drug development, it is advisable to avoid chirality if possible, since all testing efforts would need to be doubled to account for both chiral forms. Further costs may be associated with difficulties in chemical synthesis, purification, and scale-up. Low cost of development and cost of goods are important to both anticipated usages of BKIs in the veterinary industry and for apicomplexan infections in resource-limited regions.
Several BKIs in development were identified to possess chirality. BKI-1634, which demonstrated excellent anti-Cryptosporidium activity in vitro and in the mouse efficacy model (Hulverson et al., 2017b), contains a chiral center owing to the five-membered asymmetrical oxolane ring at the R2 position. In order to circumvent chirality, the oxolane moiety was replaced with a six-membered symmetrical oxane ring (Fig. 2). These cyclic ether derivatives, BKIs-1841 and -1842, retained in vitro potency and thus are currently being investigated for in vivo models of cryptosporidiosis and toxoplasmosis (Table 1). Another series of BKIs harboring a chiral 4-hydroxybutan-2-yl moiety at the R2 position was active against both Cryptosporidium parvum and Toxoplasma gondii in vitro, with BKIs-1712, -1713, -1768, and -1769 all exhibiting submicromolar effective concentrations that lead to a 50% reduction in growth (EC50s) (Table 1). BKIs-1713 and -1768 showed further promise in the cryptosporidiosis mouse model. To avoid chirality, an additional methyl group was introduced to form the 4-hydroxy-2-methylbutan-2-yl moiety (Fig. 2). BKIs-1745 and -1770 are examples of this modification, and both compounds retain good in vitro activities. In vivo, both compounds exhibited potent anti-Cryptosporidium efficacies in the mouse model, which prompted both to be prioritized for lead development.
Fig. 2.
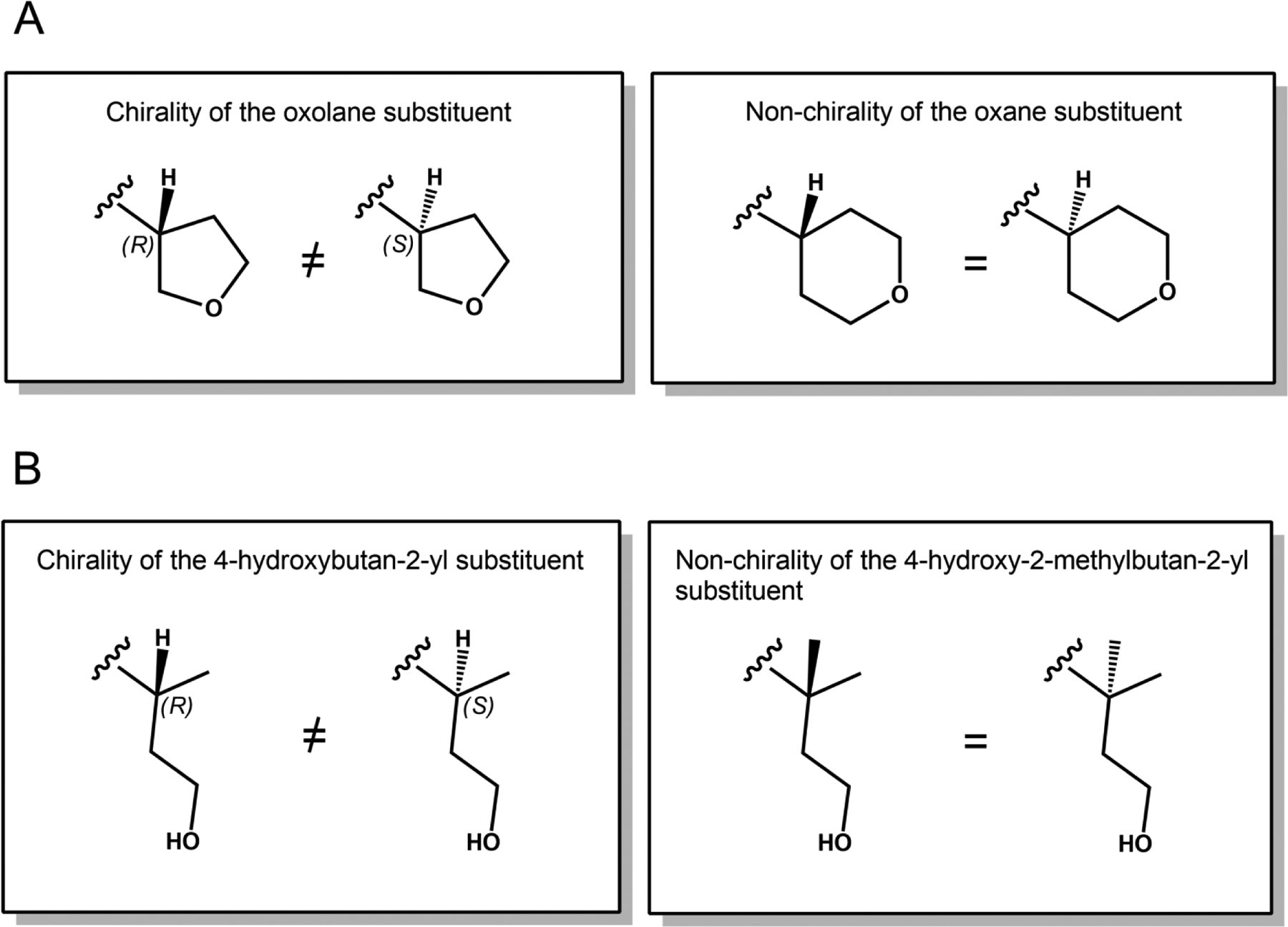
Substituents of chiral and non-chiral Bumped Kinase Inhibitors (BKIs). (A) BKI-1634’s R2 substituent contains a chiral oxolane ring. Modifying this to a six-membered oxane ring results in a loss of chirality. (B) BKIs-1712, -1713, -1768, and -1769 possess a chiral 4-hydroxybutan-2-yl R2 substituent. Addition of a methyl group to form the 4-hydroxy-2-methylbutan-2-yl substituent resolves chirality.
Table 1.
Activity of chiral and non-chiral Bumped Kinase Inhibitors (BKIs)
| BKI | Scaffold | Chiral | R2 substituent | TgCDPK1 IC50 (μM) | Toxoplasma gondii EC50 (μM) | CpCDPK1 IC50 (μM) | Cryptosporidium parvum EC50 (μM) | C. parvum efficacious dose in mousea |
|---|---|---|---|---|---|---|---|---|
| 1634 | AC | X | oxolane | 0.0060 | 0.0026 | 1.183 | 60 mg/kg QD | |
| 1841 | AC | oxane | 0.0301 | 0.13 | 0.0028 | 1.21 | ||
| 1842 | AC | oxane | 0.0376 | 0.17 | 0.0045 | 0.26 | ||
| 1712 | AC | X | 4-hydroxybutan-2-yl | 0.0159 | 0.367 | 0.0059 | 0.599 | |
| 1713 | AC | X | 4-hydroxybutan-2-yl | 0.0169 | 0.472 | 0.0169 | 0.472 | 60 mg/kg QD |
| 1768 | AC | X | 4-hydroxybutan-2-yl | 0.0002 | 0.128 | 0.0020 | 0.374 | 60 mg/kg QD |
| 1769 | AC | X | 4-hydroxybutan-2-yl | 0.0005 | 0.343 | 0.0020 | 0.43 | |
| 1745 | AC | 4-hydroxy-2-methylbutan-2-yl | 0.0030 | 0.366 | 0.0040 | 0.25 | 15 mg/kg QD | |
| 1770 | AC | 4-hydroxy-2-methylbutan-2-yl | 0.0002 | 0.147 | 0.0025 | 0.344 | 30 mg/kg BID |
Efficacious dose results in >2 log unit reduction in oocyst shedding after 5 days of treatment.
IC50, inhibitory concentration that leads to a 50% reduction; EC50, effective concentration that leads to a 50% reduction in growth; QD, once daily; BID, twice daily; AC, 5-aminopyrazole-4-carboxamide.
2.2. hERG liability
A commonly encountered challenge in drug discovery relates to interference of lead compounds with the hERG product, a voltage-gated potassium channel that plays an important role in regulating action potentials in a wide range of tissues including brain and smooth muscle cells and, notably, in myocardial cells (Warmke and Ganetzky, 1994; Farrelly et al., 2003). Compromised functionality of hERG channels can deplete the cardiac repolarization reserve and cause excessive lengthening of the QT interval of the cardiac action potential on the electrocardiogram (ECG), a measure of the time taken for ventricular depolarization and repolarization (Curran et al., 1995; Sanguinetti et al., 1995; Priori et al., 1996). Long QT syndrome (LQTS) is a disorder characterized by this prolongation and is associated with an increased risk of Torsades de Pointes (TdP), a heart rhythm disorder that can lead to chaotic arrhythmias and symptoms such as syncope, seizures, cardiac arrest and, in some cases, sudden death (Keating and Sanguinetti, 2001). Drugs that interfere with normal hERG channel activity can lead to acquired LQTS (Redfern et al., 2003). Several drugs from a variety of therapeutic classes have been removed from the market due to unexpected reports of sudden cardiac death associated with QT interval prolongation and TdP (Fermini and Fossa, 2003). This resulted in increased regulatory scrutiny of preclinical pharmaceuticals over the years, with mandated hERG screening and stringent guidelines on cardiac safety profiles for drugs discovered to produce QT prolongation. It is estimated that approximately 60% of preclinical drugs currently in development interfere with hERG channel activity (Redfern et al., 2003). This high promiscuity of hERG to a diversity of drugs suggests an unusual binding site that can readily accommodate small drug-like molecules. Recently, a high resolution cryogenic electron microscopy (Cryo-EM) structure of the open hERG channel revealed extended hydrophobic pockets within the central channel cavity, providing a strong structural basis for indiscriminate drug binding in hERG channels (Wang and MacKinnon, 2017). Despite these advances, consensus on the actual binding modes of drugs remains lacking. Additional structures, ideally complexed with bound drug, are necessary to further resolve this picture and help direct drug development programs away from hERG liability.
Our first encounter with hERG involved BKI-1294, an early lead progenitor from the PP series that exhibited promising anti-Toxoplasma and anti-Cryptosporidium activity in vitro and in several in vivo models. BKI-1294 was later discovered to inhibit hERG at submicromolar levels, effectively eliminating it from further development for human use. This prompted us to incorporate a high-throughput hERG screen into our pipeline to quickly evaluate the off-target propensity of BKIs and identify structural activity relationships (SAR) to inform subsequent drug design. For these higher-throughput safety screens, a functional assay that measures thallium ion flux was employed to obtain direct measurements of channel activity. Thallium ions are known to permeate through open potassium channels. Cells pre-loaded with an indicator dye that fluoresces upon binding to thallium allow for a functional indication of channel activity, since intensity of the fluorescent signal is proportional to the number of potassium channels in the open state. Using this assay system, the landscape of hERG interference was progressively defined for the BKI library. Subsequent SAR work identified strategies to steer BKI development away from potential interactions with the hERG channel. These included avoidance of the 4-piperidinemethyl moiety at the R2 position, as exemplified by BKIs -1294, and -1369, and directing lead optimization efforts around the AC scaffold, which was observed to generally exhibit lower affinity for hERG compared with PP BKIs (Ojo et al., 2014; Hulverson et al., 2019).
While the thallium flux assay is adequate for screening purposes and rank ordering of compounds according to channel activity, an important limitation is the lack of voltage control and temporal resolution afforded by traditional voltage clamp methods. Furthermore, inhibitory concentrations that lead to a 50% reduction (IC50s) as determined by ion flux assays tend to be less potent compared with electrophysiological methods, potentially leading to inadequately defined safety indices. BKI-1369, a potent anti-Cryptosporidium candidate which is structurally identical to BKI-1294, except for a quinoline moiety at the R1 position in place of naphthalene, was initially reported to have a thallium flux IC50 of 10 μM, which was >10-fold higher than that of BKI-1294 (Table 2). This moderate improvement in the hERG signal facilitated lead prioritization of BKI-1369 for subsequent large animal efficacy models and safety profiling. However, when investigated in an alternate hERG assay system that provides voltage control and physiologically relevant conditions in cardiac myocytes (the automated patch clamp system, QPatch, Sophion Biosciences, Woburn, Massachusetts, USA), the IC50 value of BKI-1369 was revealed to be 0.97 μM. This unfortunate discrepancy between fluorescence and electrophysiological determinants of hERG inhibition was also observed in a second anti-Cryptosporidium candidate, BKI 1774, as well as in a promising anti-Toxoplasma candidate, BKI-1660 (Table 2). With a maturing BKI medicinal chemistry campaign, synthesis of new compounds became more focused, and routine hERG safety profiling shifted away from the thallium flux assay to the QPatch system, as the high-throughput needs diminished and accurate resolution of hERG liability became more important in the lead evaluation process. It is prudent for drug discovery and development groups to screen compounds for hERG inhibition early on in the lead discovery and optimization process. However, it is even more important to recognize the limitations of the selected assay platform to mitigate the enormous costs associated with compound failures further down the pipeline.
Table 2.
In vitro human ether-à-go-go-related gene (hERG) inhibition and in vivo cardiovascular (CV) safety of Bumped Kinase Inhibitors (BKIs)
| BKI | Scaffold | TgCDPK1 IC50 (μM) | Toxoplasma gondii EC50 (μM) | T. gondii efficacious dose in mice | CpCDPK1 IC50 (μM) | Crypto sporidium parvum EC50 (μM) | C. parvum efficacious dose in micea | hERG Thallium Flux IC50 (μM) | hERG Qpatch IC50 (μM) | Rat CV safety (plasma conc. μM) | Dog CV safety (plasma conc. μM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1294 | PP | 0.0029 | 0.137 | 0.0010 | 2.22 | 100 mg/kg QD | 0.79 | ||||
| 1369 | PP | 0.0061 | 0.275 | 0.0009 | 2.5 | 30 mg/kg QD | ~10 | 0.97 | ↓dP/dt & ↓CO (9.3 μM); ↓MAP & ↓HR (50 μM); ↑SVR (129 μM); ↑QTcV (1.7 μM) | ||
| 1517 | AC | 0.0021 | 0.25715 | 60 mg/kg QD | 0.0014 | 0.649 | 30 mg/kg BID | >30 | >16.7 | dose dependent ↓MAP (13 μM) | |
| 1660 | PrP | 0.0091 | 0.2713 | 10 mg/kg QD | 0.0164 | 4.58 | 30 | 4.97 | |||
| 1673 | AC | 0.0033 | 0.1166 | 10 mg/kg QD | 0.0043 | 0.351 | 30 mg/kg QD | 27 | > 17.0 | ↓MAP & ↓HR (>290 μM); ↑dP/dt (27 μM) | |
| 1708 | AC | 0.0026 | 0.0695 | 20 mg/kg QD | 0.0007 | 0.412 | 15 mg/kg QD | 13 | 12.3 | no changes (60 μM) | no changes (95 μM) |
| 1748 | AC | 0.0068 | 0.0629 | 20 mg/kg QD | 0.0031 | 0.214 | 60 mg/kg QD | >30 | >21.5 | mild ↓MAP; mild ↑dP/dt (48 μM) | ↓ SVR (18 μM); ↑HR & ↑CO (18 μM), ↑dP/dt (57 μM) |
| 1750 | AC | 0.0101 | 0.112 | 0.0069 | 0.464 | 60 mg/kg QD | 30 | >30 | dose dependent ↑dP/dt (6.4 μM) | ||
| 1768 | AC | 0.0002 | 0.128 | 0.0020 | 0.374 | 60 mg/kg QD | >30 | >23.2 | |||
| 1770 | AC | 0.0002 | 0.147 | 0.0025 | 0.344 | 30 mg/kg BID | >30 | >17.3 | no changes (20 μM) | ||
| 1774 | AC | <0.0001 | 0.072 | 0.4131 | 0.73 | 60 mg/kg QD | >30 | 3.2 |
Efficacious dose results in >2 log unit reduction in infection.
QD, once daily; BID, twice daily; CV, cardiovascular; MAP, mean arterial pressure; HR, heart rate; CO, cardiac output; SVR, systemic vascular resistance; dP/dt, cardiac contractility; QTcV, Van der Water corrected QT interval (time taken for ventricular depolarization and repolarization); IC50, inhibitory concentration that leads to a 50% reduction;; EC50, effective concentration that leads to a 50% reduction in growth; AC, 5-aminopyrazole-4-carboxamide; PrP, pyrrolopyrimidine; PP, pyrazolopyrimidine.
While hERG liability is an important assessment of cardiovascular (CV) safety for human-relevant pharmacology, the underlying mechanisms responsible for ventricular action potential repolarization are similarly tied to hERG channel function in many other species such as guinea pigs, rabbits, dogs, and horses (Wymore et al., 1997; Chen et al., 2005; Pedersen et al., 2015), and as such, liabilities relating to hERG activity would extend to these veterinary species as well. For some species such as rats and mice, hERG does not appear to contribute significantly to repolarization (Wymore et al., 1997; Priest et al., 2008; Rosati et al., 2008). In addition, variations in hERG protein sequence may also translate to topological differences in the pore structure that limits promiscuous binding of drugs, such as in bovine species (Ficker et al., 1998). Although the hERG IC50 at ~1 μM precluded the use of BKI-1369 in humans, the compound demonstrated a generous safety window in calf models to warrant continued development as a bovine cryptosporidiosis therapeutic. Clinical relevance of CV safety models should be considered to avoid unnecessary compound attrition, particularly in the case of veterinary usage.
2.3. In vivo cardiovascular safety
Despite the utility of hERG assays as a screen for CV liability, no in vitro platform can fully resolve the cardiac safety profile of a test compound. In vivo investigations are required to account for PK/PD properties and ancillary liabilities of the drug (and its associated metabolites) that may act synergistically to compound CV risks, or antagonistically to counteract negative effects associated with hERG interference. Beyond QT interval prolongation, drug-induced effects on CV function can include changes to other electrophysiological parameters (e.g. PR interval and QRS complex) as well as to hemodynamic parameters such as mean arterial pressure (MAP), cardiac output (CO), systemic vascular resistance (SVR), myocardial contractility (dP/dt), and heart rate (HR). Furthermore, the inverse relationship of HR and QT interval suggests that drug-induced effects on the QT interval can be masked by changes in HR. For this reason, ECG-recorded QT intervals are typically normalized for the impact of HR using various correction factors such as the Van der Water formula (Van de Water et al., 1989; Spence et al., 1998).
Lead BKIs are screened for CV safety with three escalating 30 min i.v. infusions targeting therapeutic-to-supratherapeutic exposures in anesthetized rats to allow monitoring of drug-induced changes in HR, MAP, and cardiac contractility (dP/dt@50) (Segreti et al., 2008). Rats are a frequently used model for preclinical CV safety testing (Guth, 2007; Jacob, 2010) since they share similar cardiac morphologies with humans and comparable expression profiles of cardiac ion channel proteins. However, as noted earlier, hERG does not play a significant role in repolarization of the cardiac action potential in rats. Thus, hERG-related QT interval prolongation may not be capitulated in a rat CV assay.
In vivo ECG monitoring is one of the most direct measures of cardiac safety (Picard and Lacroix, 2003). Dogs are the most commonly used non-rodent species for preclinical CV testing (Greaves, 1998; Leishman et al., 2012) because they share remarkably similar ion channel expression profiles and activity with humans, and thus are a strong predictor for CV safety (Wymore et al., 1997; Gralinski, 2003; Hanson et al., 2006). Therefore, for investigation of QT interval changes, subsequent testing is performed in an analogous anesthetized dog CV model, where addition of ECG recording allows for monitoring of the Van de Water-corrected QT interval (QTcV). CO is also measured, and SVR is calculated using the MAP and CO values.
As expected, BKI-1369 exhibited significant dose-dependent increases in QTcV starting around a plasma concentration of 1.7 μM in the dog CV assay, signifying risk for drug-induced LQTS (Table 2). In addition, decreases in CO and negative inotropic effects were biologically significant at 9.3 μM, and a decrease in MAP and complete CV collapse occurred at higher exposures (>50 μM). Since cardiovascular effects have not been detected at 3X maximal therapeutic doses in calves, development continues for this compound for calf cryptosporidiosis (unpublished data).
Common cardioactive trends shared by BKIs tested in the rat CV model include dose-dependent decreases in MAP and positive inotropy (Table 2). For BKI-1748, a lead candidate for toxoplasmosis and neosporosis therapy, the positive inotropy observed at the highest exposures (48 μM) in the rat assay model was recapitulated in the dog CV assay. Moreover, the dog CV study detected additional CV effects such as elevated HR and CO and decreased SVR at 18 μM, which may limit the overall CV safety window for this compound. Work is ongoing to define the minimal therapeutic exposure of BKI-1748 needed for successful therapy for toxoplasmosis and neosporosis and to fully characterize its safety index.
BKI-1708, a highly efficacious lead candidate for cryptosporidiosis, produced a modest hERG signal in both thallium flux and QPatch assays (~13 μM). Curiously, subsequent in vivo experiments in dogs did not show a change in QTcV through the highest exposure (95 μM) nor exhibit any other CV issues. This could be due to plasma protein binding of BKI-1708, which is greater than 99% for dogs and human plasma. It is likely that the free fraction of 1708 available to influence QTcV was less than 1 μM in these experiments. This suggests that compounds with moderate hERG issues should not be quickly abandoned and, instead, may warrant further investigation if their efficacy profiles demonstrate promise. As exemplified in this case, both in vitro and in vivo studies are important for an accurate assessment of CV safety.
2.4. Teratogenicity
A successful drug candidate for the treatment of toxoplasmosis and neosporosis requires both systemic exposures that extend to the CNS for clearance of tissue cyst bradyzoites, and transplacental exposure for clearance of fetal infection. As such, any drug in development for these diseases must be deemed safe for use in pregnancy and lack teratogenic effects at therapeutic levels. In humans, teratogens have the biggest impact on the fetus during the first trimester of development (van Gelder et al., 2010). Because the developing fetus is undergoing rapid cell proliferation, it is extremely vulnerable to agents that interfere with this process. Therefore, teratogenesis can be thought of as the threshold at which the fetus is unable to restore itself due to proliferation-related damage inflicted by an agent (Finnell, 1999). Drugs that target essential genes are more likely to injure embryonic tissue (Yao and Rzhetsky, 2008). As kinases play crucial regulatory roles in the signaling pathways of cell cycle progression, differentiation, metabolism, and death, unintended inhibition may result in perturbations in pathways that regulate these processes. Despite the specificity afforded by the small gatekeeper residue of CDPK, as ATP-competitive inhibitors, BKIs still carry the risk of off-target binding in the highly-conserved ATP binding pockets of host kinases. As part of our standard drug testing pipeline, BKIs are screened against a representative panel of approximately 100 human kinases for potential off-target activities. Although BKI kinome profiles are generally clean (>10 μM IC50s), they appear to share a disposition for the same off-target host kinases, notably 3-phosphoinositide dependent protein kinase 3 (PKD3) and mitogen-activated protein kinase 1 (MEK1) (Table 3). This is likely due to a flexible gatekeeper residue within the ATP-binding pockets of these kinases that makes them more accommodating to BKIs. This also suggests that BKI drug development will need to factor in the collateral impact of inhibiting these off-target host kinases, as medicinal chemistry efforts to avoid them may be difficult or not feasible.
Table 3.
Off-target kinases and mouse pregnancy safety of Bumped Kinase Inhibitors (BKIs)
| BKI | Scaffold | TgCDPK1 IC50 (μM) | Toxoplasma gondii EC50 (μM) | T. gondii efficacious dose in micea | Human Src IC50 (μM) | Human PKD3 IC50 (μM) | Human MEK1 IC50 (μM) | Mouse pregnancy safety dose | Fertility (pregnant dams) | Total pups born | Pups survived (3 days post birth) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1517 | AC | 0.0021 | 0.25715 | 60 mg/kg QD | >30 | 1.11 | 10 | 50 mg/kg | 2 of 7 | 0 | |
| 1649 | PrP | 0.0032 | 0.0838 | 6 mg/kg QD | >10 | 0.045 | 1.55 | 6 mg/kg | 4 of 6 | 4 | 0 (0%) |
| 1673 | AC | 0.0033 | 0.1166 | 10 mg/kg QD | >10 | 0.181 | 0.0243 | 60 mg/kg | 0 of 6 | 0 | |
| 1713 | AC | 0.0169 | 0.472 | >10 | 60 mg/kg | 4 of 6 | 20 | 16 (80%) | |||
| 1748 | AC | 0.0068 | 0.0629 | 20 mg/kg QD | >10 | 0.302 | 1.12 | 60 mg/kg | 3 of 6 | 5 | 4 (80%) |
| 1748 | 20 mg/kg | 4 of 6 | 20 | 20 (100%) | |||||||
| 1748 | 5 mg/kg | 3 of 6 | 14 | 14 (100%) | |||||||
| 1751 | AC | 0.0060 | 0.0415 | >10 | 0.2 | 0.9 | 60 mg/kg | 1 of 6 | 0 | ||
| 1757 | AC | 0.0017 | 0.0725 | 20 mg/kg QD | >10 | 0.995 | 0.082 | 60 mg/kg | 4 of 6 | 15 | 12 (80%) |
| 1774 | AC | 0.0000 | 0.072 | >10 | 60 mg/kg | 4 of 6 | 22 | 19 (86%) | |||
| 1796 | PP | 0.0033 | 0.025 | >10 | 0.213 | 2.44 | 25 mg/kg | 0 of 6 | 0 |
Efficacious dose results in ≥2 log reduction in infection.
QD, once daily; IC50, inhibitory concentration that leads to a 50% reduction; EC50, effective concentration that leads to a 50% reduction in growth; Src, proto-oncogene human tyrosine-protein kinase; TgCDPK1, Toxoplasma gondii calcium-dependent protein kinase 1; PKD, 3-phosphoinositide dependent protein kinase 3; MEK1, mitogen-activated protein kinase 1; AC, 5-aminopyrazole-4-carboxamide; PrP, pyrrolopyrimidine; PP, pyrazolopyrimidine
PKD3 is a serine/threonine kinase expressed in a wide variety of tissues and is linked to several intercellular signaling pathways (Hayashi et al., 1999). PKD3 is also reported to promote cell proliferation in prostate cancer cell lines (Chen et al., 2008) and is associated with the mitotic apparatus including spindle, centrosome, and midbody formation (Papazyan et al., 2008), which suggests a role in the regulation of cell cycle events. Further studies of PKD3 expression in mouse embryo development revealed strong differential expression during organogenesis, especially in cardiac and skeletal muscles, suggesting its relevance in the development of these tissues (Ellwanger et al., 2008). MEK1 is a dual-specificity kinase that plays a role in activating the ERK/MAPK cascade, a signal transduction pathway involved in cell fate determination and regulation of cell proliferation and differentiation. The role of MEK1 in mouse development was elucidated with gene disruption studies in embryonic stem cells where a loss of MEK1 function led to embryos dying after 10.5 days of gestation (Giroux et al., 1999). Embryo defects were limited to pale yolk sacs that lacked blood circulation, with no other obvious deformities. However, placental tissues from MEK1 null embryos were abnormal, with reduced vascularization and a compacted labyrinthine region. This suggests that MEK1 plays a crucial role in blood vessel formation and circulation during placentogenesis. These results suggest that both kinases are important for the developing fetus, and compounds that inhibit their normal function may result in teratogenesis.
Although the mechanisms by which drugs cause developmental defects are not fully understood, teratogenic effects are generally avoidable if the dose-response relationship and the threshold for toxicological effects can be fully defined (Finnell, 1999) and the drug can still be dosed high enough to retain its therapeutic activity. We experienced this with BKI-1748, a lead toxoplasmosis candidate that demonstrated mild teratogenic effects in a mouse pregnancy safety model when administered at 60 mg/kg for 5 days, where one dam aborted before term, one dam died after delivery, and 20% of pups failed to survive. However, when administered at 20 mg/kg and 5 mg/kg, all pregnant dams produced viable pups that survived beyond 3 days after delivery (Table 3). One limitation of the mouse pregnancy safety model is that it is difficult to separate maternal health issues from fetal effects such as teratogenesis. As discussed above, BKI-1748 has cardiovascular effects in rats and dogs at plasma levels over 40 μM. These high plasma levels are expected with BKI-1748 given at 60 mg/kg daily, and we speculate that the negative effects on maternal cardiovascular performance may have caused the fetal loss seen at this dose.
The mouse model of pregnancy safety remains part of our drug development campaign for toxoplasmosis and neosporosis. However, as a routine screening platform, these experiments can be expensive due to low throughput, labor intensive breeding programs, and the amount of test compound required for 5 days of dosing. In addition, differences between human placenta and mouse placenta lead to questions about the applicability of the pregnant mouse screen (Georgiades et al., 2002). Thus, it is desirable to have an intermediate screen for teratogens with higher throughput, lower cost, and quicker turnaround of results. For these reasons, we are currently investigating the utility of a zebrafish developmental safety model as a pre-screen to allow for more efficient testing of BKIs (Weigt et al., 2011).
2.5. Gastrointestinal side effects
Oral treatment is the ideal administration route for most of the apicomplexan diseases targeted by BKIs. This leads to a direct exposure of drug to gastrointestinal (GI) tissues as it is absorbed or, in the case of BKIs that are not highly absorbed, passing through the GI tract before being excreted. Previously examined BKI-1369 (Hulverson et al., 2017a, 2017b) was shown to be highly efficacious against Cryptosporidium in mice. This efficacy can be attributed to the high concentrations and long exposure times of BKI-1369 within the various tissues of the murine GI tract after oral dosing. Following a 30 mg/kg oral dose, plasma concentration peaked at 3–4 μM, while GI tissue concentrations peaked at much higher levels: >76 μM in the duodenum, 39 μM in the jejunum/ileum, 20 μM in the cecum, and 23 μM in the ascending colon (Table 4). Furthermore, these concentrations remained at 8–11 μM throughout a 12 h period in all GI sections except the duodenum, even as plasma levels were eliminated to below 1 μM (Hulverson et al., 2017a). This demonstrates that with a stable BKI dosed orally, the GI tract will be exposed to significantly higher levels of compound than plasma. It should be noted that these increased concentrations in the GI tract make it more difficult to obtain an adequate therapeutic window between side effects and efficacy, if the major side effects are localized to the GI tract.
Table 4.
Bumped Kinase Inhibitor (BKI) related gastrointestinal and systemic toxicity in mice
| BKI | Dose at which toxicity was observed (mg/kg) | Observed toxicity | Observed maximum plasma exposure at toxic dose (μM) | Observed maximum gastrointestinal tissue exposure after single dose (μM) |
|---|---|---|---|---|
| 1369 | 300 (QD in mice) | Interference with digestion and peristalsis | 8.3 ± 0.2 | 30 mg/kg dose: 76 ± 41 (duodenum) 39 ± 4 (jejunum/ileum) 20 ± 6 (cecum) 23 ± 9 (ascending colon) |
| 1534 | 100 (QD in mice) 10 (BID in calves) | Interference with digestion and peristalsis | 1.8 ± 0.9 | ND |
| 1553 | 150 (QD in mice) 15 (BID in calves) | Interference with digestion and peristalsis, gastric bleeding in mice and calves | 102 ± 20 | 10 mg/kg dose: 41 ± 35 (duodenum) 56 ± 14 (jejunum/ileum) 9 ± 1.5 (cecum) 27 ± 3 (ascending colon) |
| 1608 | 100 (QD in mice) | Interference with digestion and peristalsis | 9.9 ± 3 | ND |
| 1649 | 50 (QD in mice) | Extreme signs of distress after 7 doses, including lack of grooming, weight loss, lethargy, hunched posture | 307 ± 27 | ND |
| 1673 | 250 (QD in mice) | Slight signs of distress after 7 doses, including lack of grooming, weight loss | 72 ± 5 | ND |
| 1749 | 60 (QD in mice) | GI tissue inflammation | ND | ND |
| 1751 | 60 (QD in mice) | Signs of distress after 4 doses, including lack of grooming, weight loss, lethargy | 25 ± 6 | ND |
| 1779 | 60 (QD in mice) | GI tissue inflammation | ND | ND |
ND, not determined; QD, once daily; BID, twice daily; GI, gastrointestinal
BKI-1369 has low off-target kinase activity and showed no signs of GI side effects up to 150 mg/kg dosed daily. However, at 300 and 500 mg/kg daily, upon dissection, mouse stomachs were packed full of undigested food, suggesting a disruption to peristalsis (Hulverson et al., 2017a). Given the large number of kinases, receptors, signaling pathways, and motor proteins involved in smooth muscle contraction and relaxation within the GI tract, it is reasonable to assume that very high tissue concentrations and off-target activity led to this disruption.
Fortunately, these functional side effects were only observed for BKI-1369 at doses and concentrations that were significantly higher than the efficacious dose. This has not been the case for all BKIs. BKI-1534 showed similar disruptions to digestion or peristalsis in mice after only five doses of 100 mg/kg (Hulverson et al., 2017a). Since this was still well above the efficacious dose of 20 mg/kg (Arnold et al., 2017), BKI-1534 was progressed to the C. parvum calf efficacy model with dosing at 10 mg/kg twice daily. Unlike prior BKIs that were safe and effective at similar doses (Schaefer et al., 2016), dosing with BKI-1534 led to the development of lack of stool formation, followed by bloody diarrhea in treated calves (Table 4), and the study had to be suspended prematurely. Similarly, high doses of BKI-1553 (150 mg/kg daily for 3 days) led to apparent decreased peristalsis and gastric bleeding in mice after multiple doses, and resulted in bloody diarrhea with escalated doses (15 mg/kg twice daily) in calves (data not previously published), despite this compound being safe and partially efficacious at lower doses (Schaefer et al., 2016).
This concerning pattern of GI side effects translating from mice to calves for certain compounds led to the elimination of several other BKIs from further consideration. After the observations with BKIs -1534 and -1553, mice were screened for signs of GI issues prior to any further efficacy testing in calves. BKI-1608 (PMID 30830766) also led to stomachs packed with undigested food, thus apparent disruption of peristalsis in mice when dosed at 100 mg/kg for 4 days. BKIs-1749 and -1779 (Huang et al., 2019) showed signs of GI issues that upon dissection were attributed at least partially to inflammation of the GI tissues after 4 days of dosing at 60 mg/kg in T. gondii-infected CD-1 mice.
2.6. Systemic toxicity
Many of the BKIs tested were demonstrated to be efficacious and showed no signs of toxicity when administered at relatively low doses. However, throughout their development, the different central scaffolds used for BKIs, including AC, PP and PrP (Huang et al., 2015, 2017, 2019; Hulverson et al., 2019; Vidadala et al., 2016, 2018), have demonstrated wide variabilities in bioavailability, metabolic stability, systemic exposures, and other pharmacokinetic properties. Depending upon the infectious agent to be treated, high systemic exposures may be desirable, as in the case of Toxoplasma and Neospora, or may be unnecessary, as in the case of Cryptosporidium, which is localized to the gut. For treatment of enteric pathogens, high systemic drug exposure may be a liability, as increased exposure would reduce the therapeutic index between plasma concentrations at efficacious levels and those that lead to systemic toxicity.
Several BKIs exhibited very high peak concentrations in plasma after a single dose. These include maximum concentrations (Cmax) of 69 μM for AC BKI-1712 and 80 μM for AC BKI-1751. Other BKIs have shown slow clearance after oral dosing, including PrP BKI-1649 and PP BKI-1553, resulting in accumulating plasma concentrations with multiple doses. The slow clearance of BKI-1369 allowed accumulation in plasma to concentrations of >300 μM when dosed at 50 mg/kg for 5 days, which resulted in multiple signs of toxicity (Hulverson et al., 2017a). BKI-1553 levels also similarly accumulated in calves when dosed at 15 mg/kg twice daily, leading to the aforementioned development of bloody diarrhea. The high Cmax of BKI-1751 led to observed plasma concentrations of >30 μM at 2 h after dosing and 3–5 μM at 24 h when administered at 60 mg/kg daily. Signs of toxicity were observed with this dose by the fourth administration (Table 4).
It has been difficult to sufficiently reduce the efficacious dose to obtain low enough plasma concentrations to avoid toxicity in these cases of high Cmax and/or slow clearance. This has led to the advancement of BKIs with lower bioavailability for the treatment of GI pathogens and a stringent evaluation of minimum therapeutic doses for pathogens that require higher systemic exposures, such as Toxoplasma and Neospora, in order to avoid toxicity and maximize the therapeutic safety window.
2.7. Lack of sufficient efficacy
Many compounds that were advanced to in vivo efficacy testing failed to sufficiently treat infection. This was often dependent upon many of the properties of the compounds and how they relate to the pathology of the infection being treated.
For Cryptosporidium, it has been determined that for both the PP and AC series of BKIs, systemic exposure does not directly correlate with efficacy. Many BKIs with relatively low Cmax and area-under-the-curve (AUC) values have proven very efficacious in both mice and calves, including PP BKIs-1294 and -1369 (Hulverson et al., 2017a, 2017b; Schaefer et al., 2016) and AC BKIs-1770 and -1708 (Huang et al., 2019). Others have also proven efficacious with significantly higher exposures than these, such as PrP BKI-1649 (Hulverson et al., 2017b) and AC BKI-1673 (Huang et al., 2019). Still others with either low exposure, such as BKI-1772, or high exposure, such as BKI-1757, have shown very little efficacy (Huang et al., 2019). For this reason, in the case of Cryptosporidium, other chemical, metabolic, and/or PK properties of the compounds must be determining efficacy.
For the AC compounds used for Cryptosporidium, it was determined that solubility had a strong correlation with efficacy (Huang et al., 2019). It is hypothesized that without reaching a specific solubility threshold, the compound could precipitate into the intestinal lumen as the pH along the GI tract changes, leaving it unavailable to enter the infected enterocytes. For the PP compounds, two possible explanations were explored. First, it was determined that a potent compound would fail if it did not reach sufficient exposure throughout the entire GI tract in an infected animal. This was shown when BKI-1294 reached high levels in all tissues of the GI tract and significantly reduced infection in mice, while BKI-1553 failed to reach high levels in the ascending colon and subsequently failed to prove efficacious (Arnold et al., 2017). Second, it was found that drug efflux transporters, such as P-glycoprotein (P-gp), can reduce enterocyte levels and diminish efficacy of BKIs if they have high efflux ratios (Arnold et al., 2019). As a case in point, BKI-1318 failed to be efficacious not due to potency, but because it had a much higher efflux ratio than compounds that were efficacious, such as BKI-1369. When the P-gp pumps were inhibited, the efficacy of BKI-1318 was greatly improved.
For Toxoplasma, systemic exposure is of obvious importance since the infection spreads through the blood and causes severe disease manifestations in the heart, lungs, liver, eyes, and brain. This necessity makes it more difficult to avoid possible toxicities. Studies are ongoing to determine the pharmacodynamic properties of several BKIs for treatment of toxoplasmosis in mice in order to achieve the minimum possible systemic exposures while maintaining efficacy.
3. Concluding remarks
The drug discovery and development enterprise is a complex, iterative process that requires integration of multiple disciplines for the successful characterization, optimization, and delivery of a therapeutic candidate. The efficiency of this process is of the utmost importance, as the attrition rate for drugs in development is high, and failures late in the pipeline come at a substantial economic cost and can result in the loss of many years of work invested into the campaign. The reasons for failure commonly involve toxicity and a lack of efficacy due to poor PK and bioavailability properties (Prentis et al., 1988). These challenges require a delicate balance between an emphasis on the activity of the candidate and its pharmaceutical properties for successful interpretation, and a plan of action for improvements and advancement (Kerns and Di, 2003).
Our experience with BKI drug development over the years led us to recognize commonalities in the reasons for compound attrition. These include undesirable chemical properties such as chirality, hERG liability, CV issues, teratogenicity, systemic toxicity, GI side effects, and lack of efficacy due to inadequate PK properties. By identifying and defining these challenges, our hope is to be able to anticipate and overcome potential pitfalls in the journey to successful clinical therapeutics for apicomplexan diseases.
Highlights.
BKIs that target CDPK1 in apicomplexan parasites with a glycine gatekeeper are promising new therapeutics.
Numerous challenges have shaped our selection of pre-clinical BKI candidates.
These include chirality, hERG, teratogenicity, cardiovascular, GI, and toxicity issues, and poor efficacy due to PK/PD.
Acknowledgments
This manuscript and studies reported herein were supported by the National Institute of Allergy and Infectious Diseases (USA) (award numbers R01AI089441, R01AI111341, R21AI123690); and the National Institute of Child Health and Human Development of the National Institutes of Health (USA) (award number R01HD080670). The project was also supported by the Agriculture and Food Research Initiative from the United States Department of Agriculture National Institute of Food and Agriculture (Competitive Grant no. 2014-06183) and from the U.S. Department of Veterans Affairs Biomedical Laboratory Research and Development (award number BX002440). The authors would like to thank and acknowledge the following AbbVie employees: Xiaoqin Liu and Jason Segreti for rat CV testing and Yevgeniya Koshman, Brandan Bird, and Amanda Wilsey for dog CV testing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: WCVV is an owner/officer of ParaTheraTech Inc, a company which is seeking to bring bumped kinase inhibitors to the animal health market.
References
- Arnold SLM, Choi R, Hulverson MA, Schaefer DA, Vinayak S, Vidadala RSR, McCloskey MC, Whitman GR, Huang W, Barrett LK, Ojo KK, Fan E, Maly DJ, Riggs MW, Striepen B, Van Voorhis WC, 2017. Necessity of Bumped Kinase Inhibitor Gastrointestinal Exposure in Treating Cryptosporidium Infection. J Infect Dis 216, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SLM, Choi R, Hulverson MA, Whitman GR, McCloskey MC, Dorr CS, Vidadala RSR, Khatod M, Morada M, Barrett LK, Maly DJ, Yarlett N, Van Voorhis WC, 2019. P-glycoprotein mediated efflux reduces the in vivo efficacy of a therapeutic targeting the gastrointestinal parasite Cryptosporidium. J Infect Dis 220, 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks WH, Guida WC, Daniel KG, 2011. The significance of chirality in drug design and development. Curr Top Med Chem 11, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Deng F, Singh SV, Wang QJ, 2008. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res 68, 3844–3853. [DOI] [PubMed] [Google Scholar]
- Chen X, Cass JD, Bradley JA, Dahm CM, Sun Z, Kadyszewski E, Engwall MJ, Zhou J, 2005. QT prolongation and proarrhythmia by moxifloxacin: concordance of preclinical models in relation to clinical outcome. Br J Pharmacol 146, 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT, 1995. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803. [DOI] [PubMed] [Google Scholar]
- Ellwanger K, Pfizenmaier K, Lutz S, Hausser A, 2008. Expression patterns of protein kinase D 3 during mouse development. BMC Dev Biol 8, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly AM, Ro S, Callaghan BP, Khoyi MA, Fleming N, Horowitz B, Sanders KM, Keef KD, 2003. Expression and function of KCNH2 (HERG) in the human jejunum. Am J Physiol Gastrointest Liver Physiol 284, G883–895. [DOI] [PubMed] [Google Scholar]
- Fermini B, Fossa AA, 2003. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov 2, 439–447. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM, 1998. Molecular determinants of dofetilide block of HERG K+ channels. Circulation Res 82, 386–395. [DOI] [PubMed] [Google Scholar]
- Finnell RH, 1999. Teratology: general considerations and principles. J Allergy Clin Immunol Pract 103, S337–342. [DOI] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith AC, Burton GJ, 2002. Comparative developmental anatomy of the murine and human definitive placentae. Placenta 23, 3–19. [DOI] [PubMed] [Google Scholar]
- Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J, 1999. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol : CB 9, 369–372. [DOI] [PubMed] [Google Scholar]
- Gralinski MR, 2003. The dog’s role in the preclinical assessment of QT interval prolongation. Toxicol Pathol 31 Suppl, 11–16. [DOI] [PubMed] [Google Scholar]
- Greaves P, 1998. Patterns of drug-induced cardiovascular pathology in the beagle dog: relevance for humans. Exp Toxicol Pathol 50, 283–293. [DOI] [PubMed] [Google Scholar]
- Guth BD, 2007. Preclinical cardiovascular risk assessment in modern drug development. Toxicol Sci 97, 4–20. [DOI] [PubMed] [Google Scholar]
- Hanson LA, Bass AS, Gintant G, Mittelstadt S, Rampe D, Thomas K, 2006. ILSI-HESI cardiovascular safety subcommittee initiative: evaluation of three non-clinical models of QT prolongation. J Pharmacol Toxicol Methods 54, 116–129. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Seki N, Hattori A, Kozuma S, Saito T, 1999. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta 1450, 99–106. [DOI] [PubMed] [Google Scholar]
- Huang W, Choi R, Hulverson MA, Zhang Z, McCloskey MC, Schaefer DA, Whitman GR, Barrett LK, Vidadala RSR, Riggs MW, Maly DJ, Van Voorhis WC, Ojo KK, Fan E, 2017. 5-Aminopyrazole-4-Carboxamide-Based Compounds Prevent the Growth of Cryptosporidium parvum. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Hulverson MA, Choi R, Arnold SLM, Zhang Z, McCloskey MC, Whitman GR, Hackman RC, Rivas KL, Barrett LK, Ojo KK, Van Voorhis WC, Fan E, 2019. Development of 5-Aminopyrazole-4-carboxamide-based Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J Med Chem 62, 3135–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Ojo KK, Zhang Z, Rivas K, Vidadala RS, Scheele S, DeRocher AE, Choi R, Hulverson MA, Barrett LK, Bruzual I, Siddaramaiah LK, Kerchner KM, Kurnick MD, Freiberg GM, Kempf D, Hol WG, Merritt EA, Neckermann G, de Hostos EL, Isoherranen N, Maly DJ, Parsons M, Doggett JS, Van Voorhis WC, Fan E, 2015. SAR Studies of 5-Aminopyrazole-4-carboxamide Analogues as Potent and Selective Inhibitors of Toxoplasma gondii CDPK1. ACS Med Chem Lett 6, 1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulverson MA, Bruzual I, McConnell EV, Huang W, Vidadala RSR, Choi R, Arnold SLM, Whitman GR, McCloskey MC, Barrett LK, Rivas KL, Scheele S, DeRocher AE, Parsons M, Ojo KK, Maly DJ, Fan E, Van Voorhis WC, Doggett JS, 2019. Pharmacokinetics and In Vivo Efficacy of Pyrazolopyrimidine, Pyrrolopyrimidine, and 5-Aminopyrazole-4-Carboxamide Bumped Kinase Inhibitors against Toxoplasmosis. J Infect Dis 219, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulverson MA, Choi R, Arnold SLM, Schaefer DA, Hemphill A, McCloskey MC, Betzer DP, Muller J, Vidadala RSR, Whitman GR, Rivas KL, Barrett LK, Hackman RC, Love MS, McNamara CW, Shaughnessy TK, Kondratiuk A, Kurnick M, Banfor PN, Lynch JJ, Freiberg GM, Kempf DJ, Maly DJ, Riggs MW, Ojo KK, Van Voorhis WC, 2017a. Advances in bumped kinase inhibitors for human and animal therapy for cryptosporidiosis. Int J Parasitol 47, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulverson MA, Vinayak S, Choi R, Schaefer DA, Castellanos-Gonzalez A, Vidadala RSR, Brooks CF, Herbert GT, Betzer DP, Whitman GR, Sparks HN, Arnold SLM, Rivas KL, Barrett LK, White AC Jr., Maly DJ, Riggs MW, Striepen B, Van Voorhis WC, Ojo KK, 2017b. Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J Infect Dis 215, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob HJ, 2010. The rat: a model used in biomedical research. Methods Mol Biol (Clifton, N.J.) 597, 1–11. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC, 2001. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104, 569–580. [DOI] [PubMed] [Google Scholar]
- Kerns EH, Di L, 2003. Pharmaceutical profiling in drug discovery. Drug Discov Today 8, 316–323. [DOI] [PubMed] [Google Scholar]
- Larson ET, Ojo KK, Murphy RC, Johnson SM, Zhang Z, Kim JE, Leibly DJ, Fox AM, Reid MC, Dale EJ, Perera BG, Kim J, Hewitt SN, Hol WG, Verlinde CL, Fan E, Van Voorhis WC, Maly DJ, Merritt EA, 2012. Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. J Med Chem 55, 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leishman DJ, Beck TW, Dybdal N, Gallacher DJ, Guth BD, Holbrook M, Roche B, Wallis RM, 2012. Best practice in the conduct of key nonclinical cardiovascular assessments in drug development: current recommendations from the Safety Pharmacology Society. J Pharmacol Toxicol Methods 65, 93–101. [DOI] [PubMed] [Google Scholar]
- Nguyen LA, He H, Pham-Huy C, 2006. Chiral drugs: an overview. IntJ Biomed Sci : IJBS 2, 85–100. [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AM, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CL, Su XZ, Kappe SH, Maly DJ, Fan E, Van Voorhis WC, 2014. A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. J Infect Dis 209, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazyan R, Doche M, Waldron RT, Rozengurt E, Moyer MP, Rey O, 2008. Protein kinase D isozymes activation and localization during mitosis. Exp Cell Res 314, 3057–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen PJ, Thomsen KB, Olander ER, Hauser F, Tejada Mde L, Poulsen KL, Grubb S, Buhl R, Calloe K, Klaerke DA, 2015. Molecular Cloning and Functional Expression of the Equine K+ Channel KV11.1 (Ether a Go-Go-Related/KCNH2 Gene) and the Regulatory Subunit KCNE2 from Equine Myocardium. PloS One 10, e0138320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard S, Lacroix P, 2003. QT interval prolongation and cardiac risk assessment for novel drugs. Curr Opin Investig Drugs 4, 303–308. [PubMed] [Google Scholar]
- Prentis RA, Lis Y, Walker SR, 1988. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964–1985). Br J Clin Pharmacol 25, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT, Bell IM, Garcia ML, 2008. Role of hERG potassium channel assays in drug development. Channels (Austin, Tex.) 2, 87–93. [DOI] [PubMed] [Google Scholar]
- Priori SG, Cantu F, Schwartz PJ, 1996. The long QT syndrome: new diagnostic and therapeutic approach in the era of molecular biology. Schweiz Med Wochenschr Suppl 126, 1727–1731. [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG, 2003. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58, 32–45. [DOI] [PubMed] [Google Scholar]
- Rentsch KM, 2002. The importance of stereoselective determination of drugs in the clinical laboratory. J Biochem Biophys Methods 54, 1–9. [DOI] [PubMed] [Google Scholar]
- Rosati B, Dong M, Cheng L, Liou SR, Yan Q, Park JY, Shiang E, Sanguinetti M, Wang HS, McKinnon D, 2008. Evolution of ventricular myocyte electrophysiology. Physiological genomics 35, 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT, 1995. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307. [DOI] [PubMed] [Google Scholar]
- Schaefer DA, Betzer DP, Smith KD, Millman ZG, Michalski HC, Menchaca SE, Zambriski JA, Ojo KK, Hulverson MA, Arnold SL, Rivas KL, Vidadala RS, Huang W, Barrett LK, Maly DJ, Fan E, Van Voorhis WC, Riggs MW, 2016. Novel Bumped Kinase Inhibitors Are Safe and Effective Therapeutics in the Calf Clinical Model for Cryptosporidiosis. J Infect Dis 214, 1856–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segreti JA, Marsh KC, Polakowski JS, Fryer RM, 2008. Evoked changes in cardiovascular function in rats by infusion of levosimendan, OR-1896 [(R)-N-(4-(4-methyl-6-oxo-1,4,5,6-tetrahydropyridazin-3-yl)phenyl)acetamide], OR-1855 [(R)-6-(4-aminophenyl)-5-methyl-4,5-dihydropyridazin-3(2H)-one], dobutamine, and milrinone: comparative effects on peripheral resistance, cardiac output, dP/dt, pulse rate, and blood pressure. J Pharmacol Exp Ther 325, 331–340. [DOI] [PubMed] [Google Scholar]
- Shrestha A, Ojo KK, Koston F, Ruttkowski B, Vidadala RSR, Dorr CS, Navaluna ED, Whitman GR, Barrett KF, Barrett LK, Hulverson MA, Choi R, Michaels SA, Maly DJ, Hemphill A, Van Voorhis WC, Joachim A, 2019. Bumped kinase inhibitor 1369 is effective against Cystoisospora suis in vivo and in vitro. Int J Parasitol Drugs Drug Resist 10, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence S, Soper K, Hoe CM, Coleman J, 1998. The heart rate-corrected QT interval of conscious beagle dogs: a formula based on analysis of covariance. Toxicol Sci 45, 247–258. [DOI] [PubMed] [Google Scholar]
- Van de Water A, Verheyen J, Xhonneux R, Reneman RS, 1989. An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J Pharmacolo Method 22, 207–217. [DOI] [PubMed] [Google Scholar]
- van Gelder MM, van Rooij IA, Miller RK, Zielhuis GA, de Jong-van den Berg LT, Roeleveld N, 2010. Teratogenic mechanisms of medical drugs. Hum Reprod Update 16, 378–394. [DOI] [PubMed] [Google Scholar]
- Van Voorhis WC, Doggett JS, Parsons M, Hulverson MA, Choi R, Arnold SLM, Riggs MW, Hemphill A, Howe DK, Mealey RH, Lau AOT, Merritt EA, Maly DJ, Fan E, Ojo KK, 2017. Extended-spectrum antiprotozoal bumped kinase inhibitors: A review. Exp Parasitol 180, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidadala RS, Rivas KL, Ojo KK, Hulverson MA, Zambriski JA, Bruzual I, Schultz TL, Huang W, Zhang Z, Scheele S, DeRocher AE, Choi R, Barrett LK, Siddaramaiah LK, Hol WG, Fan E, Merritt EA, Parsons M, Freiberg G, Marsh K, Kempf DJ, Carruthers VB, Isoherranen N, Doggett JS, Van Voorhis WC, Maly DJ, 2016. Development of an Orally Available and Central Nervous System (CNS) Penetrant Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. J Med Chem 59, 6531–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidadala RSR, Golkowski M, Hulverson MA, Choi R, McCloskey MC, Whitman GR, Huang W, Arnold SLM, Barrett LK, Fan E, Merritt EA, Van Voorhis WC, Ojo KK, Maly DJ, 2018. 7 H-Pyrrolo[2,3- d]pyrimidin-4-amine-Based Inhibitors of Calcium-Dependent Protein Kinase 1 Have Distinct Inhibitory and Oral Pharmacokinetic Characteristics Compared with 1 H-Pyrazolo[3,4-d]pyrimidin-4-amine-Based Inhibitors. ACS Infect Dis 4, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, MacKinnon R, 2017. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 169, 422–430.e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B, 1994. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA 91, 3438–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt S, Huebler N, Strecker R, Braunbeck T, Broschard TH, 2011. Zebrafish (Danio rerio) embryos as a model for testing proteratogens. Toxicology 281, 25–36. [DOI] [PubMed] [Google Scholar]
- Wymore RS, Gintant GA, Wymore RT, Dixon JE, McKinnon D, Cohen IS, 1997. Tissue and species distribution of mRNA for the IKr-like K+ channel, erg. Circulation Res 80, 261–268. [DOI] [PubMed] [Google Scholar]
- Yao L, Rzhetsky A, 2008. Quantitative systems-level determinants of human genes targeted by successful drugs. Genome Res 18, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]