

Abstract
Halophilic and osmotolerant yeast Debaryomyces hansenii has a high potential for cell factory applications due to its resistance to harsh environmental factors and compatibility with a wide substrate range. However, currently available genetic techniques do not allow the full potential of D. hansenii as a cell factory to be harnessed. Moreover, most of the currently available tools rely on the use of auxotrophic markers that are not suitable in wild-type prototrophic strains. In addition, the preferred non-homologous end-joining (NHEJ) DNA damage repair mechanism poses further challenges when precise gene targeting is required. In this study, we present a novel plasmid-based CRISPRCUG/Cas9 method for easy and efficient gene editing of the prototrophic strains of D. hansenii. Our toolset design is based on a dominant marker and facilitates quick assembly of the vectors expressing Cas9 and single or multiple single-guide RNAs (sgRNAs) that provide the possibility for multiplex gene engineering even in prototrophic strains. Moreover, we have constructed NHEJ-deficient D. hansenii that enable our CRISPRCUG/Cas9 tools to support the highly efficient introduction of point mutations and single/double gene deletions. Importantly, we also demonstrate that 90-nt single-stranded DNA oligonucleotides are sufficient for direct repair of DNA breaks induced by sgRNA-Cas9, resulting in precise edits reaching 100% efficiencies. In conclusion, tools developed in this study will greatly advance basic and applied research in D. hansenii. In addition, we envision that our tools can be rapidly adapted for gene editing of other non-conventional yeast species including the ones belonging to the CUG clade.
Keywords: CRISPR-Cas9; , CUG clade; , panARS; , homologous recombination; , KU70
1. Introduction
Yeasts are widely used for basic research and as hosts for the production of various scientifically and industrially important products (1–3). Yet, most applied research depends on the conventional platforms, i.e. Saccharomyces cerevisiae. One reason for this choice is that significant genomic engineering of yeast hosts is often required for heterologous bio-production, and a well-established genetic toolbox is already in place for S. cerevisiae. Unfortunately, laboratory strains of S. cerevisiae often fail to perform when exposed to real industrial conditions. On the other hand, it is often possible to find non-conventional yeasts (NCYs) that may cope with these conditions; and these NCYs may therefore serve as attractive production alternatives as they can directly be applied for various bio-processes (4–6). However, a production strategy based on NCYs is not straightforward, especially in non-model organisms, due to the lack of genetic engineering tools and because they are genetically less characterized.
Debaryomyces hansenii is a hemiascomycetous NCY, which for a long time has been of high interest for basic and applied research fields. This yeast species has drawn attention due to its ability to withstand high osmotic pressure, high salinity (up to 25% NaCl) and low water activity (7–9). Because of these characteristics, D. hansenii is widely applied in dairy and fermented/cured food industries. In the context of biotechnological applications, D. hansenii can consume a wide range of carbon sources and accumulate high levels of lipids or biosynthesize commercially feasible quantities of several valuable products, i.e. xylitol and flavonoids (8, 10, 11). In addition, it can produce killer toxins that inhibit the growth of competing microorganisms (12). Microbial production in cell factories with such properties has great potential in applications such as bio-production of third-generation biofuels (13–15). Despite the obvious potential of D. hansenii, the field of basic research in this organism is underdeveloped, and heterologous production possibilities of this yeast remain unexploited due to the lack of robust genetic engineering techniques.
The slow development of genetic tools for D. hansenii has been affected by several factors. For example, efficient transformation protocols were not available until recent years (16, 17). There are only few known circular plasmids that can propagate in D. hansenii; however, they rely on auxotrophic markers and are only applicable for specific laboratory strains (18–20). In addition, this yeast belongs to the CTG-clade that uses the ambiguous CUG codon that is mainly translated into serine instead of leucine (21). This fact further complicates the heterologous expression in this yeast as all genes of interest (GOIs) need to be codon-optimized to exclude CUG codons. Finally, but very importantly, genetic engineering of this yeast has been very limited as the non-homologous end-joining (NHEJ) pathway is preferred for integrating DNA into chromosomes (16). This fact makes it very difficult to introduce precise genome alterations via homology-directed gene targeting and gene editing in this species (16, 18). Discovery of CRISPR/Cas9 and its successful application have revolutionized the genetic engineering and synthetic biology fields in many species (22). This technology has been successfully adapted for genome engineering of several NCYs (23). Indeed, a recent study has recently demonstrated the application of CRISPR/Cas9 in D. hansenii; however, it was mainly used for gene inactivation via flawed NHEJ repair, while attempts to perform homologous recombination (HR)-directed edits had low success (16). Thus, there is a high need for an improved genetic toolbox for D. hansenii that allows for swift and robust gene targeting and gene editing. In fact, to realize the full potential of D. hansenii in bio-production and to obtain a better understanding of the peculiar physiology of this yeast, it is mandatory to develop techniques that make it possible to perform accurate gene deletions and to introduce specific point mutations with high efficiency and in a short time frame.
In this work, we have developed a highly efficient CRISPRCUG/Cas9 gene engineering toolbox for D. hansenii, which is applicable to even prototrophic strains. Importantly, we have disrupted the NHEJ pathway to enable precise Cas9-mediated introduction of point mutations and marker-free gene deletions via HR. Lastly, we have demonstrated that short single-stranded oligonucleotides are sufficient to mediate repair of Cas9-induced double-strand brakes (DSBs), thus significantly reducing time and cost needed for the construction of complex gene-targeting substrates (GTSs).
2. Materials and methods
2.1. Strains and cultivation media
Escherichia coli DH5α strain was used for maintenance of plasmids and was cultivated in Lysogeny Broth (24) medium supplemented with 100 mg/l ampicillin (Sigma) or 50 mg/l kanamycin (Sigma) depending on the selection marker.
Two prototrophic D. hansenii strains were used in this work: a wild-type sDIV115 (Source: CBS 767) and an NHEJ-deficient sDIV165 (ΔKU70, source: this study). For maintenance and transformation of D. hansenii, a yeast extract peptone dextrose (YPD) medium containing 1% yeast extract, 2% peptone and 2% glucose (and 2% of agar for solid media) was used. For the selection of transformed strains, YPD medium was supplemented (after autoclavation) by addition of 100 mg/l nourseothricin (NTC; Werner BioAgents). For replica-plating experiments, synthetic complete (SC) medium was prepared as described previously (25), except the leucine concentration that was increased to 60 mg/l. In addition, three drop-out media were used to screen mutant strains for auxotrophy: SC-ade (lacking adenine), SC-his (lacking histidine) and SC-ura (lacking uracil).
2.2. Molecular cloning techniques
All DNA fragments that constituted genetic elements used for plasmid or GTS assembly were amplified by polymerase chain reaction (PCR) using Phusion U Master Mix (ThermoFisher) according to the manufacturer’s recommendations. All PCR fragments used for cloning purposes were gel-purified using NucleoSpin Gel and PCR Clean-up Kit (MACHEREY-NAGEL) by following the manufacturer’s protocol. All primers used in this study are denoted in Table_S1 and were purchased from Integrated DNA Technologies (IDT). GTSs with ∼400 bp upstream and ∼400 bp downstream targeting sequences for deletion of GOIs were constructed using the overlay PCR method ((26), see Supplementary Figure S1 for details). All plasmids were assembled by the uracil-specific excision reagent (USER™) method by cloning PCR fragments into AsiSI/Nb.bsmI cassette by following well-established protocols ((27), also see Supplementary Figures S2–S4 for details). Plasmid minipreps were prepared using GeneElute Plasmid MiniPrep Kit (Sigma-Aldrich) according to the manufacturer’s protocol. All newly constructed DNA constructs were validated by Sanger sequencing Mix2Seq ON (Eurofins). The full list of all plasmids used and constructed in this work is listed in Table_S2. All gene sequences were optimized using IDTs’ Codon Optimization Tool (https://eu.idtdna.com).
2.3. Plasmid construction
Five Cas9-expressing plasmids were constructed as follows. A single PCR fragment containing the Cl_TDH3 promoter, CTG codon-optimized Cas9 equipped with NLS (SV40) at C-terminus and the Sc_CYC1t terminator (Cl_TDH3p-Cas9-(SNV40)-Sc_CYC1t) was amplified from pRB732 (28) and was USER cloned into a linearized pDIV116 backbone, resulting in the pDIV488 plasmid. Two promoters Dh_RNR2p and Dh_RHR2p were PCR amplified from D. hansenii genomic DNA, and single PCR fragment with Cas9-(SNV40)-Sc_CYC1t was amplified from pRB732 (28). Next, Dh_RNR2p and Dh_RHR2p fragments were individually paired with Cas9-(SNV40)-Sc_CYC1t and USER cloned into linearized pDIV066, resulting in pDIV489 and pDIV490, respectively. Likewise, same fragments were USER-cloned into the pDIV116 backbone, producing plasmids pDIV491 and pDIV492.
Plasmids expressing single-guide RNAs (sgRNAs) and Cas9 were assembled as follows. For ADE2 gene targeting constructs, three types of sgRNA cassettes were cloned individually into five Cas9-expressing vectors resulting in a total of 15 different plasmids. For version (1) sgRNA, two PCR fragments Cl_SNR52p and gRNA_ADE2-Ca_ENO1t were amplified from pRB733 (28) and assembled by USER fusion into linearized Cas9-expressing vectors, resulting in pDIV494, pDIV497, pDIV509, pDIV512 and pDIV513 plasmids. Version (2) sgRNA cassettes were assembled by fusing two PCR fragments—Dh_SCR1p amplified from D. hansenii genomic DNA and sgRNA_ ADE2-Ca_ENO1t from pRB733 (28)—into five Cas9-expressing vectors, to obtain pDIV495, pDIV498, pDIV500, pDIV502 and pDIV510 plasmids. Likewise, for the sgRNA cassette version (v3—CRISPRCUG-tRNA), two PCR fragments (Dh_TEF1-tRNAGly and sgRNA_ADE2-tRNAGly-Dh_TEF1t) were amplified from a gBlock (IDT) and cloned into the five Cas9 vectors to obtain plasmids pDIV514–518. Construction of plasmids targeting other GOIs was done only using the version (v3—CRISPRCUG-tRNA) sgRNA cassette setup (Supplementary Figure S4). Plasmids expressing Cas9 and double sgRNAs were assembled by USER cloning using the method described before ((29), see Supplementary Figures S3 and S4 for details).
2.4. Transformation of D. hansenii
D. hansenii was transformed using the previously described electroporation method (17) with slight modifications. In brief, D. hansenii cultures were incubated overnight in liquid YPD (10 ml of culture per one transformation) at 28°C and with orbital shaking at 200 rpm. Next day, when OD600 reached ∼ 2.6, cultures were centrifuged at 4500g for 10 min and the supernatant was discarded. The cell pellet was resuspended in 50 mM sodium phosphate buffer containing 25 mM dithiothreitol (DTT) and incubated at 30°C for 15 min at 150 rpm. Next, cells were washed twice with sterile Milli-Q water and resuspended in 1 ml of ice-cold 1 M sucrose. The cell suspension was centrifuged at 4500g for 10 min at 4°C and the supernatant was discarded. The cell pellet was resuspended in 175 µl of ice-cold 1 M sucrose and transferred to a pre-cooled 2-mm electroporation cuvette (BioRad). DNA was added to the cells (per transformation: 1 μg of plasmid DNA, and, when necessary, 1 μg of GTSs with long targeting sequences or 4 μg of single-stranded oligonucleotides was added individually to a given cuvette and gently mixed. Electroporation was carried out with a MicroPulser electroporator (BioRad) on manual settings at 2.3 kV. After single electroporation, pulse cells were resuspended in YPD media containing 1 M sucrose and incubated at 28°C for ∼2 h. Lastly, cells were plated on solid YPD + NTC plates and incubated at 28°C for 3 days or until visible colonies were formed.
2.5. Validation of mutant strains
The obtained D. hansenii transformants were assessed by checking for expended phenotypes. In case of ade2, plates were visually checked by counting the number of red colonies with regard to the total number of colonies formed. Moreover, plates with sgRNA_ADE2-Cas9 transformants were also replica-plated using sterile velvet cloths on SC-ade medium to confirm adenine auxotrophy. The phenotypes of his4 or ura3 mutants were assessed by replica plating on SC-his and SC-ura media, respectively. Mutant strains showing the correct phenotype were tested by colony PCR to confirm introduction of the desired mutation(s). Routinely, 10 random colonies displaying the correct phenotype were selected for PCR analysis using primers designed to bind upstream and downstream of the genomic region containing the intended mutation(s) (see Supplementary Figure S5). All colony PCRs were done using Quick-Load® Taq 2X Master Mix according to the manufacturer’s recommendation. Eventually, PCR fragments of the expected size were column-purified using NucleoSpin Gel and PCR Clean-up Kit (MACHEREY-NAGEL) and sent for Sanger sequencing using Mix2Seq ON (Eurofins) service.
3. Results
3.1. Design and validation of CRISPRCUG/Cas9 vector set
With the aim of introducing efficient CRISPR technology in D. hansenii and possible other CUG clade yeasts, we first set out to develop CRISPR shuttle vectors with the ability to encode Cas9 and one or more sgRNAs. Such a plasmid should be reasonably stable and produce Cas9 and sgRNA at levels that promote efficient CRISPR-based genetic engineering. Moreover, coding sequences for sgRNA production should be easy to insert. At the start of this project, only few functional plasmid elements were available for D. hansenii, and we therefore decided to construct a set of CRISPR shuttle vectors to increase the chance of constructing at least one functional plasmid.
All plasmids in the set contain an E. coli vector backbone and four yeast specific elements: a changeable sgRNA expression cassette, a CUG codon-free and codon-optimized Cas9CUG gene from Streptococcus pyogenes, a CUG codon-free and codon-optimized selection marker and, lastly, for vector maintenance, an autonomously replicating sequence (ARS) or a centromere ARS fusion (CEN/ARS, see Figure 1A). Using different variants of the individual four elements and a combinatorial assembly, we constructed 15 different CRISPRCUG vectors (see below and Figure 1A and Supplementary Figure S2).
Figure 1.
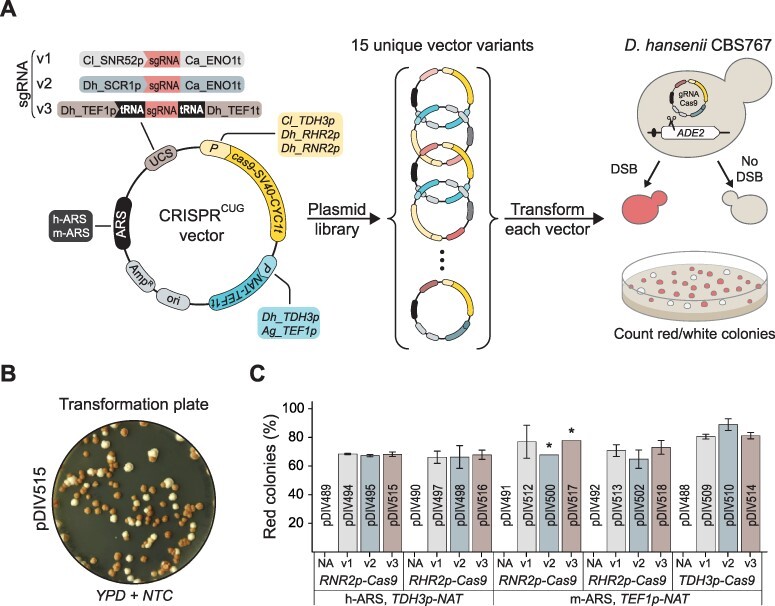
CRISPRCUG vector design and validation. (A) An overview of the experimental strategy. To the left, sgRNA-Cas9 vectors are assembled by combining variable parts: autonomously replicating sequences (h-ARS and m-ARS), sgRNA expression cassettes (v1, v2 and v3), promoters driving expression of CAS9 (Cl_TDH3p, Dh_RHR2p and Dh_RNR2p) and promoters driving the NAT marker (Dh_TDH3p and Ag_TEF1p). Middle, a library of vectors expressing an sgRNA targeting ADE2 is generated by combining different parts. To the right, sgRNA-Cas9 vectors expressing the sgRNA_ADE2 are individually transformed into a wild-type D. hansenii strain for inducing NHEJ-mediated mutagenesis of the ADE2 locus. Wild-type strains are white, and strains harboring mutated and dysfunctional ade2 genes accumulate red pigment. (B) An image of a typical transformation plate, D. hansenii CBS767 transformed with pDIV515 plasmid. (C) Results of CRISPRCUG/Cas9 vector validation represented as an average fraction of red colonies versus all colonies per each sgRNA-Cas9 variant. Error bars represent standard deviation based on biological duplicates. UCS—USER cloning site, DSB—double-strand break, Ca—Candida albicans, Cl—Candida lusitaniae, Dh—Debaryomyces hansenii, Ag—Ashbya gossypii. *—Data based only on one out of two experiments.
For selection, we used two different elements based on the NAT marker that provides resistance to the drug NTC and therefore allows for selection in prototrophic strains. To allow for expression in D. hansenii, the NATCUG gene was equipped with either Cl_TDH3p or Ag_TEF1p promoter in combination with the Ag_TEF1t terminator (see the ‘Materials and methods’ section). For plasmid maintenance, we constructed two elements. The first, h-ARS, contains a fusion of two ARS sequences CfARS16 from Candida famata (30) and panARS from Kluyveromyces lactis (31), and the second, m-ARS, contains a centromere and three ARS sequences and is a fusion of CEN6/ARSH4 from Saccharomyces cerevisiae (32) and h-ARS (see Supplementary Figures S2 and S6). We next tested the functionality of the four elements by constructing two basic vectors pDIV066, containing h-ARS and NAT controlled by the Cl_TDH3p promoter, and pDIV116, containing m-ARS and NAT controlled by the Ag_TEF1p promoter, which we transformed into D. hansenii. In both cases, we obtained high transformation efficiencies (>3000 CFU/μg of plasmid DNA, see Supplementary Figure S6), indicating that all four elements are functional.
For the production of Cas9 in D. hansenii, we constructed three cas9CUG expression cassettes based on a cas9CUG gene constructed by Norton et al. (28). Specifically, we used the part of the gene containing the cas9CUG open reading frame (ORF) fused in frame with a sequence encoding SV40 NLS and the Sc_CYC1t terminator from S. cerevisiae. This fragment was individually fused to three different promoters: one from Candida lusitaniae, Cl_TDH3p, and two from D. hansenii, Dh_RHR2p and Dh_RNR2p (see the ‘Materials and methods’ section). For the production of sgRNA, we constructed three different expression cassettes (see Figure 1A and Supplementary Figure S2). Two cassettes directly produce sgRNA by using different RNA pol III promoters, Cl_SNR52p (v1) or Dh_SRC1p (v2) in combination with the Candida albicans Ca_ENO1t terminator. The third expression cassette (v3) contains the Pol II promoter Dh_TEF1p and the Dh_TEF1t terminator. In this design, the sgRNA is flanked by native tRNAGly sequences and is liberated from the transcript by the transfer RNA (tRNA) processing system (33).
To test the functionality of all six elements, they were introduced into pDIV066 and pDIV116 in a combinatorial fashion, except that insertion of Cl_TDH3p-Cas9CUG into pDIV066 was omitted as the NATCUG marker in this plasmid is already equipped with this promoter. Importantly, an sgRNA gene producing an sgRNA matching a target sequence positioned right next to an NGG PAM sequence in ADE2 (DEHA2G13772g) was introduced in all plasmids. The resulting 15 different plasmids (pDIV494–pDIV518, see Supplementary Figure S2) as well as five corresponding control plasmids that do not contain an sgRNA expression cassette were transformed into D. hansenii to test for their ability to induce sgRNA_ADE2-Cas9-directed mutations into ADE2 via erroneous NHEJ DNA repair. To this end, we exploited that mutation of ADE2 can be easily assessed by visual screening as ade2 cells accumulate red pigment (34). As expected, transformants were readily achieved in all cases (see Figure 1B and Supplementary Figure S7), and we then determined the ratio of red and white colonies as a measure of the efficiency of mutagenesis for all experiments. As expected, no red colonies were obtained with the five corresponding control vectors that only encode Cas9 (see Supplementary Figure S7). In contrast, with all sgRNA_ADE2-Cas9 vectors, significant amounts of red colonies were obtained. To our surprise, we observed almost identical ratios of red to white colonies (∼70–90%) on all transformation plates (see Figure 1C). Lastly, seven random red colonies were selected and examined by Sanger sequencing, and these analyses confirmed that mutations were indeed induced by Cas9 as they were introduced at the site matching sgRNA_ADE2. Moreover, all events were small 1–5-bp indels (see Supplementary Figure S8), which is typical for flawed NHEJ repair of Cas9-induced DNA DBSs (35). In conclusion, all combinations of the vector parts led to plasmids producing sgRNA_ADE2-Cas9 complexes at levels that appear to produce desirable mutations with more or less the same efficiency. The highest efficiency (∼85%) was achieved with pDIV510 that features a heterologous Cl_TDH3p promoter driving Cas9CUG expression (Figure 1C). However, transformants obtained with this plasmid displayed an increased heterogeneity in colony size, which potentially could be due to high toxic levels of the Cas9 nuclease (16). Together, the results above strongly indicate that our CRISPRCUG technology works robustly in D. hansenii.
Since all plasmid designs appeared to perform equally well for Cas9-induced mutagenesis in ADE2, we selected pDIV515 for all subsequent experiments. This design features the pDIV066 backbone (h-ARS for plasmid replication), the Dh_RNR2p-driven Cas9 and a tRNA-based sgRNA expression cassette (v3—CRISPRCUG-tRNA), and it produces transformants, which appear homogenous in size. Moreover, it has the following advantageous features: (i) a small size, (ii) an RNA pol II promoter driving sgRNA production, which does not terminate at homopolymers of > 4 bp (e.g. TTTT) expanding the possible targeting sequence space, and (iii) the tRNAGly-sgRNA-tRNAGly arrangement that enables simple assembly of single and multiple sgRNA expression cassettes ((29); Supplementary Figure S3), and the latter sets the stage for multiplex applications. Below, we refer to plasmids based on this design as CRISPRCUG-tRNA vectors.
3.2. Disruption of the NHEJ pathway in D. hansenii
A key feature of CRISPR technology is its ability to induce specific DNA DSBs that can be used to stimulate template-directed genetic engineering allowing specific sequences to be deleted, integrated or accurately mutated. In these cases, the desired modifications are introduced via template-directed HR rather than by error-prone NHEJ repair. In species where NHEJ is the dominant pathway for integrating DNA into the genome, it is often advantageous to eliminate NHEJ by deleting a crucial gene in this pathway. Indeed, in the absence of NHEJ, exact CRISPR-based genome modifications can be performed with a very high efficiency even without using a selectable marker. Encouragingly in this context, multiple studies in different non-conventional yeasts have reported to display significantly improved HR efficiencies when the NHEJ pathway is disrupted (for reviews, see (23, 36)). To explore whether deficient NHEJ also benefits CRISPR experiments in D. hansenii, we therefore inactivated the NHEJ pathway in this species by deleting KU70 (DEHA2F10208g), a protein that is essential for NHEJ repair. Specifically, this was achieved by co-transforming D. hansenii with a linear GTS designed to remove the KU70 coding sequences (Supplementary Figure S9) and a CRISPRCUG-tRNA vector encoding Cas9 and an sgRNA targeting KU70. Specifically, the GTS was synthesized as a fusion of two PCR fragments containing 400 bp of upstream and downstream sequences. The fusion was mediated via a small common 34-bp synthetic sequence, which we can use to identify the strain (Supplementary Figures S1 and S9).
In this experiment, transformants were readily obtained, and 20 colonies were tested by diagnostic PCR. Only one of these colonies appeared to contain the desired ku70 deletion, indicating that CRISPR-mediated gene targeting may be difficult in the presence of an active NHEJ pathway. We therefore used Sanger sequencing analysis to confirm that KU70 was indeed deleted via HR using the designed GTS sequence as the template for repair (Supplementary Figure S9). The validated NHEJ-deficient D. hansenii strain was termed ‘sDIV165’ and was applied in all subsequent CRISPR experiments.
3.3. Highly efficient gene targeting is possible in NHEJ-deficient D. hansenii
Three target genes ADE2, HIS4 (DEHA2F09086g) and URA3 (DEHA2A11968g) were selected to investigate whether CRISPR-mediated genetic engineering can be performed efficiently in the sDIV165 (Δku70) strain. Successful deletion of these target genes by CRISPR-mediated gene targeting is easy to score as the resulting mutant strains produce auxotrophic phenotypes. Moreover, the mutant strains can serve as useful markers in future genetic engineering experiments. For each gene, we constructed a marker-free GTS composed as a fusion of ∼400 bp of upstream and downstream sequences of the target gene (see Figure 2A and Methods). To design CRISPRCUG-tRNA vectors, we identified a PAM sequence in each gene ∼15–150 bp downstream of the start codon of each GOI, which served to define target sequences of sgRNAs (see Figure 2A and Table_S 3). Next, three CRISPRCUG-tRNA plasmids (pDIV515, pDIV558 and pDIV568) expressing sgRNAs matching the desired target sequences were constructed and tested for their ability to induce breaks in the desired gene. To evaluate this activity, we exploit that in the absence of NHEJ and a homologous repair template, DNA DSBs induced by CRISPR nucleases are expected to be lethal. We have previously used this fact to develop a simple test, TAPE (technology to assess protospacer efficiency), to measure whether a given sgRNA is able to efficiently induce a DNA DSB at the desired locus in yeast S. cerevisiae and in a string of filamentous fungi (29, 37). Specifically, in this test, a CRISPRCUG-tRNA plasmid expressing the relevant sgRNA and the corresponding empty CRISPRCUG plasmid (which does not produce any sgRNA) are transformed in parallel into the target strain using the same stoichiometric amounts of each plasmid and in the presence or absence of a repair template, e.g. a GTS (see Supplementary Figure S10). Reduced amounts of transformants obtained with the CRISPRCUG-tRNA plasmid alone relative to the numbers obtained with the empty CRISPRCUG plasmid as well as to the numbers obtained in the co-transformation experiment that includes the CRISPRCUG-tRNA vector and a repair template indicate the formation of DNA DSBs.
Figure 2.
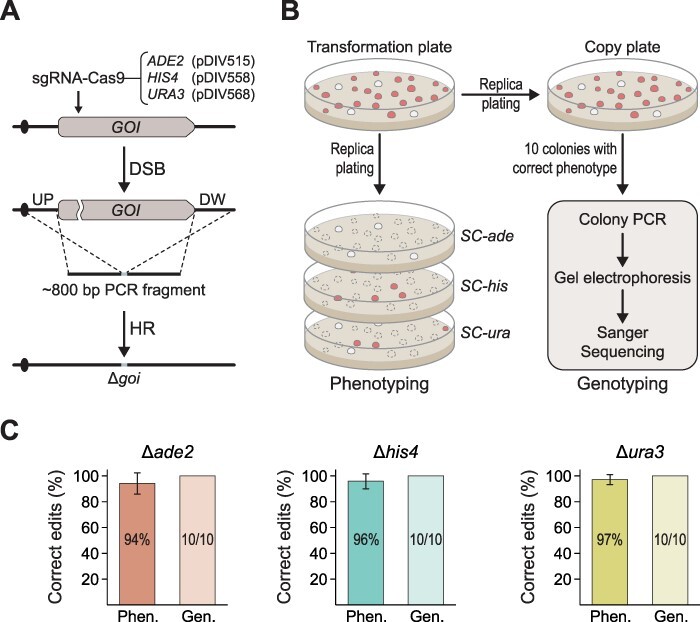
CRISPR/Cas9-mediated gene targeting via homologous-directed recombination (HDR). (A) Gene deletion strategy using locus-specific GTSs with 400-bp targeting sequences as DNA repair template. GOI—gene of interest. (B) Schematic representation for scoring and validation strategy of desired gene deletions. (C) Gene deletion efficiencies of three loci (ADE2, HIS4 and URA3). Error bars represent standard deviations based on three biological replicates. Phen. (phenotype) shows in % how many of the transformants displayed the mutant phenotype, and Gen. (genotype) shows in % how many of the mutants produce the expected band representing gene deletion in a diagnostic PCR. For the latter experiment, absolute numbers of correct versus total mutants analyzed are indicated within the individual columns. Error bars represent standard deviations calculated from three biological replicates.
With the aim of deleting the three target genes individually, we did the relevant transformations according to the TAPE strategy. Importantly, when the three CRISPRCUG-tRNA plasmids were transformed into D. hansenii, no transformants were obtained. In contrast, when the empty CRISPRCUG plasmid as well as when the CRISPRCUG-tRNA plasmids were co-transformed along with the relevant GTS, colonies were easily obtained, strongly indicating that the sgRNAs successfully stimulated gene targeting (Table_S 4). This view was strongly supported when the transformants were analyzed in more detail (see Figure 2B). Specifically, we observed that 94–97% of all colonies obtained in the three deletion experiments showed the correct phenotype. Moreover, 10 randomly picked colonies with the correct phenotype were tested by diagnostic PCR, and they all produced the band sizes expected from strains where gene deletions were accurately dictated by the GTS (Figure 2C and Supplementary Figure S11). Lastly, for each gene deletion, one transformant was validated by Sanger sequencing. Based on these results, we conclude that our method can be robustly applied for efficient CRISPR/Cas9-mediated gene targeting in D. hansenii.
3.4. CRISPRCUG/Cas9 tool is compatible with multiplex gene targeting in D. hansenii
Building cell factories for bio-production often requires multiple rounds of genetic engineering; hence, if multiplex gene editing would be possible, it would further speed up basic and applied research in D. hansenii. Inspired by our successful single gene deletion results, we proceeded to investigate whether multiple gene targeting is possible using CRISPRCUG-tRNA vectors. Exploiting that the specific arrangement of our sgRNA expression cassette, tRNAGly-sgRNA-tRNAGly, enables a simple strategy for construction expression cassettes with the ability to deliver multiple sgRNAs, we next constructed two 2x-sgRNA-Cas9 plasmids (pDIV560 and pDIV561) that allows both the ADE2 and HIS4 genes to be simultaneously targeted (see Figure 3A and Supplementary Figure S3). The two 2x-sgRNA-Cas9 plasmid variants were designed to address whether the relative positions of the two sgRNA coding sequences in the expression cassette affect gene deletion efficiencies. Hence, the positions of the two sequences are swapped relative to each other in the two plasmids (see Figure 3A).
Figure 3.
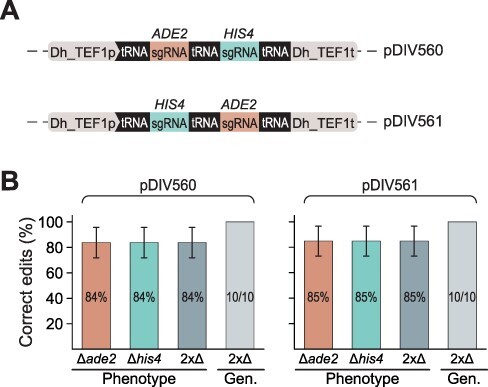
Experimental setup for multiplex gene deletions. (A) Structure of double sgRNA expressing cassettes. (B) Double gene deletion efficiencies using the pDIV560 (sgRNA_ADE2-HIS4) construct and the pDIV561 (sgRNA_HIS4-ADE2) construct. For each experiment, Phenotype shows in % how many of the transformants displayed the mutant phenotype, and Gen. (genotype) shows in % how many of the mutants produce the expected band representing gene deletion in a diagnostic PCR. For the latter experiment, absolute numbers of correct versus total mutants analyzed are indicated within the individual columns. Error bars represent standard deviations calculated from three biological replicates.
To introduce the two gene deletions simultaneously, we next co-transformed the sDIV165 strain in triplicate with the two GTSs (the same ones that were used for single gene deletions, see Figure 2A) and one of the two 2x-sgRNA-Cas9 plasmids. In this way, we tested the ability of both plasmids to support multiplexing. Like in the experiments designed to introduce single gene deletions, colonies formed only on transformation plates where both GTSs were provided (Table_S 4). Phenotypic validation of mutants (Figure 2B) demonstrated that ∼85% of all mutants displayed the phenotype expected if both gene deletions were achieved (see Figure 3B). Interestingly, we did not find any colonies displaying the phenotype that would result from cases where only one out of the two target genes was deleted. In addition, for 10 mutant strains showing the correct phenotype, subsequent PCR tests showed that all tested colonies had both genes deleted via HR using the GTSs as repair templates (see Figure 3B and Supplementary Figure S12). Lastly, we note that both 2x-sgRNA-Cas9 plasmids produced similar results, indicating that the positioning of the sgRNA coding sequences in the sgRNA expression cassette did not affect the gene deletion efficiency of the two GOIs. Altogether, we conclude that our system efficiently supports the introduction of at least two gene deletions simultaneously.
3.5. Short single-stranded oligonucleotides are sufficient for Cas9-mediated gene editing
In baker’s yeast, S. cerevisiae, short double-stranded oligonucleotides are routinely used for gene editing in CRISPR/Cas9 experiments (38). Conversely, single-stranded DNA 90-mers are used for efficient gene editing in filamentous fungi (29). Inspired by the latter, we investigated whether single-stranded 90-mers can be applied as repair templates to the direct formation of gene deletions and point mutations. For this purpose, we designed oligonucleotides allowing either deletion or introduction of a premature stop codon in the ADE2 gene (Figure 4A and B). Specifically, the oligonucleotide dictating ADE2 deletion is composed by a fusion of 45-nt upstream and downstream ADE2 sequences. The oligonucleotide dictating the introduction of the stop codon contains the mutagenic sequence in the middle of this repair template. Moreover, successful introduction of the stop codon produces a novel XbaI recognition sequence, which can be used in subsequent validation of mutant strains. For both types of experiments, two complementary repair oligonucleotides were synthesized and used individually for each type of experiment. This design allowed us to address whether DNA DSB repair depends on the ability of the oligonucleotide to bind to the coding or to the non-coding DNA strand.
Figure 4.
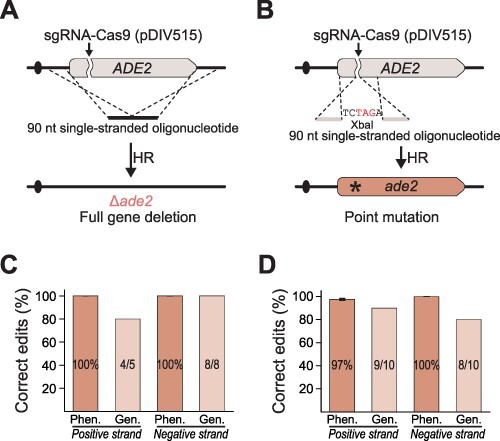
Experimental setup for oligo-mediated gene editing. 90-nt single-stranded oligonucleotides were used as the repair template for CRISPR-mediated gene editing (see text for details). Experimental schemes for introducing (A) a gene deletion and (B) a point mutation (*) into the ADE2 gene. (C) Gene deletion efficiencies and (D) point mutation efficiencies. Phen. (phenotype) shows in % how many of the transformants displayed the mutant phenotype, and Gen. (genotype) shows in % how many of the mutants contained the expected XbaI site. For the latter experiment, absolute numbers of correct versus total mutants analyzed are indicated within the individual columns. Error bars represent standard deviations calculated from three biological replicates.
Next, we individually co-transformed the sDIV165 strain with each of the four repair oligonucleotides along with pDIV515, which encodes an sgRNA targeting ADE2. In gene deletion experiments, only few (one to four) transformants were obtained in the six individual trials. However and importantly, all 13 colonies displayed the correct phenotype, and when examined by diagnostic PCR reactions, all except one generated the expected band representing the desired gene deletion (Figure 4C and Supplementary Figure S13). During these analyses, we did not detect any noteworthy difference between the results obtained with strains generated by using repair oligonucleotides binding to either the coding strand or the non-coding strand.
In the experiments designed to implement a stop-codon mutation, transformants were readily achieved using the repair templates binding to the coding strand (>80 in average) and to the non-coding strand (>65 in average). Importantly, in both cases, the vast majority of the transformants were red, indicating successful mutagenesis (Figure 4D). For each set of experiments, 10 transformants were examined by diagnostic PCR followed by XbaI restriction enzyme digestion. In 17 out of the 20 cases, the mutant strains appeared to contain the desired mutation as the PCR fragments could be cleaved by XbaI. As for the gene deletion experiment, both mutagenic repair templates appear to mutate ADE2 with the same efficiency, indicating that that the ability of the two oligonucleotides to serve as repair templates does not depend on whether it binds to the coding strand or the non-coding strand. In a final test, two of the PCR fragments, one from each experimental set, were Sanger sequenced. In both cases, the desired mutation was incorporated at the expected site in the ade2 locus, and no additional mutations were observed (see Supplementary Figure S13).
4. Discussion
Discovery of CRISPR/Cas9 technology has made it possible to work with non-model yeast species that previously were inaccessible to gene editing and engineering (23, 36). In this study, we have expanded the genetic engineering toolbox of D. hansenii and developed a NTC selectable CRISPRCUG/Cas9 vector set that can be efficiently used to perform CRISPR experiments in prototrophic strains. Prior to this report, it has not been possible to perform efficient CRISPR-mediated gene-targeting experiments in D. hansenii. Here, we show that this obstacle is due to active NHEJ repair and that highly efficient gene targeting can be achieved in an NHEJ-deficient strain similar to what has been observed in other yeasts and filamentous fungi where NHEJ is the dominant DNA integration pathway (23, 29, 36). Efficient gene targeting sets the stage for introducing defined genome alterations like specific point mutations and accurate gene deletions as well it allows for inserting genes into desirable loci or extending ORFs by sequences coding for epitope, purification or fluorescent protein tags. Importantly, we demonstrate that by using the CRISPRCUG-tRNA vector, multiplexing is possible as we were able to efficiently delete two genes simultaneously benefitting from the ability of this vector type to produce more than one sgRNA species. The presence of the ku70 mutation may in some cases influence the final phenotype of the strain. For example, deletion of YKU70 in S. cerevisiae confers a temperature-sensitive phenotype and influences telomere length as well as expression of genes close to telomeres (39). We therefore recommend to revert the mutated ku70 locus before strain characterization. This can be done in a single CRISPR-based step (29), e.g. like we propose here, by using the small synthetic sequence scar left at the ku70 locus after gene deletion as a target sequence for Cas9 in combination with a GTS containing the wild-type KU70 gene (Supplementary Figure S9).
Cell factory development is likely to rely increasingly on high-throughput genetic engineering technologies. To this end, construction of GTSs represents a laborious and expensive step and may therefore constitute a significant barrier toward large-scale genetic engineering experiments. Accordingly, it would be beneficial whether gene targeting and gene editing could be achieved via simpler and less expensive GTSs. We therefore investigated whether single-stranded oligonucleotides could serve as templates for gene editing in D. hansenii. The latter strategy has been successfully applied for gene-targeting experiments in different Aspergillus species (29). In our setup, we reached gene-deletion and point-mutation efficiencies comparable to those observed for, e.g., A. nidulans when we used single-stranded 90-mer to modify the ADE2 gene (Figure 4). In this context, we note that the efficiency of oligonucleotide-directed mutagenesis does not appear to depend on whether the oligonucleotides bind to the coding or non-coding strand at the DNA DSB, and this is also what we observed in A. nidulans in a similar experiment (29).
Despite that both the point mutation and the deletion of ADE2 were achieved with very high efficiencies, we noted that the number of transformants obtained with the CRISPRCUG-tRNA plasmid in the gene-deletion experiment were ∼34-fold lower than those obtained in the point-mutation experiment. This fact probably reflects that repair of the Cas9-induced DNA DSB in ADE2 is more difficult when the deletion oligonucleotide, rather than the point-mutation oligonucleotide, is used as a template for repair and that more cells die due to unfinished DNA repair. To this end, we note that in the case of the point mutation, the oligonucleotide acts as the repair template right at the Cas9-induced DNA DSB, whereas in the case of the deletion, the repair oligonucleotide needs to bind to two regions that are both quite far (∼130–1560 bps) from the Cas9-induced DNA DSB. We speculate that the binding of the deletion-oligonucleotide constitutes a barrier for efficient repair in this experimental setup. A higher efficiency may be accomplished by using a strategy based on the formation of two Cas9-induced DNA DSBs, one at each end of the sequence to be deleted. In fact, we have used this strategy in A. nidulans to accomplish high numbers of correctly modified transformants (29), and it can easily be implemented in D. hansenii using our CRISPRCUG-tRNA plasmid, which is designed to accommodate multiple sgRNA coding sequences. The fact that oligonucleotides can be used as repair templates during HR repair to mediate the formation of gene deletions and point mutations will dramatically cut the costs of the individual experiments in large genetic engineering efforts. In comparison, construction of a classical GTS for introducing a gene deletion requires four primers, a PCR to generate the upstream and downstream sequences and a fusion PCR to generate the GTS. Hence, we envision that oligonucleotide-mediated gene targeting and gene editing will serve as leading technologies in high-throughput genetic engineering experiments.
The CRISPRCUG vectors we have developed for CRISPR experiments in D. hansenii may likely work in other yeasts belonging to the CUG clade. Indeed, the vectors presented here can be easily tested in new hosts since they are equipped with the dominant NAT marker, and it can therefore be used directly in transformable wild-type isolates. During the project, we surprisingly noted that all the different CRISPRCUG plasmids we constructed were able to induce Cas9-mediated mutagenesis with more or less the same efficiency. This may simply reflect that it is relatively easy to implement CRISPR technology in D. hansenii, and we stress that this may not be the case in other yeasts. Hence, the entire vector set may be useful in efforts aiming at introducing CRISPR technology in other yeasts. To this end, we note that many of the elements in these vectors actually originate from other species than D. hansenii, suggesting that they may likely be functional in other yeasts as well. Altogether, we believe that the tools presented here will be highly useful in academic and industrial communities that work with non-conventional yeasts.
Supplementary Material
Acknowledgments
The authors thank Richard J. Bennett for kindly providing pRB732 and pRB733 plasmids and Fabiano J. Contesini for critically reading this manuscript.
Contributor Information
Tomas Strucko, Department of Biotechnology and Biomedicine, Section for Synthetic Biology, Technical University of Denmark, Kongens Lyngby, Hovedstaden, Denmark.
Niklas L Andersen, Department of Biotechnology and Biomedicine, Section for Synthetic Biology, Technical University of Denmark, Kongens Lyngby, Hovedstaden, Denmark.
Mikkel R Mahler, Department of Biotechnology and Biomedicine, Section for Synthetic Biology, Technical University of Denmark, Kongens Lyngby, Hovedstaden, Denmark.
José L Martínez, Department of Biotechnology and Biomedicine, Section for Synthetic Biology, Technical University of Denmark, Kongens Lyngby, Hovedstaden, Denmark.
Uffe H Mortensen, Department of Biotechnology and Biomedicine, Section for Synthetic Biology, Technical University of Denmark, Kongens Lyngby, Hovedstaden, Denmark.
Supplementary data
Supplementary data are available at SYNBIO Online.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Funding
Innovation Fund Denmark [6150-00031B.9]; Novo Nordisk Fonden within the framework of the Fermentation Based Biomanufacturing Initiative ([NNF17SA0031362); AIM-Bio program [NNF19SA0057794].
Conflict of interest statement.
The authors declare that they have no conflict of interest.
Material availability
A subset of CRISPRCUG/Cas9 vectors are available at Addgene (https://www.addgene.org/Uffe_Mortensen/).
References
- 1. Borodina I. and Nielsen J. (2014) Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol. J., 9, 609–620. [DOI] [PubMed] [Google Scholar]
- 2. Nielsen J. (2019) Yeast systems biology: model organism and cell factory. Biotechnol. J., 14, 1800421. [DOI] [PubMed] [Google Scholar]
- 3. Nandy S.K. and Srivastava R.K. (2018) A review on sustainable yeast biotechnological processes and applications. Microbiol. Res., 207, 83–90. [DOI] [PubMed] [Google Scholar]
- 4. Navarrete C. and Martínez J.L. (2020) Non-conventional yeasts as superior production platforms for sustainable fermentation based bio-manufacturing processes. AIMS Bioeng., 7, 289–305. [Google Scholar]
- 5. Navarrete C., Jacobsen I.H., Martínez J.L. and Procentese A. (2020) Cell factories for industrial production processes: current issues and emerging solutions. Processes, 8, 768. [Google Scholar]
- 6. Rebello S., Abraham A., Madhavan A., Sindhu R., Binod P., Karthika Bahuleyan A., Aneesh E.M. and Pandey A. (2018) Non-conventional yeast cell factories for sustainable bioprocesses. FEMS Microbiol. Lett., 365, fny222. [DOI] [PubMed] [Google Scholar]
- 7. Breuer U. and Harms H. (2006) Debaryomyces hansenii — an extremophilic yeast with biotechnological potential. Yeast, 23, 415–437. [DOI] [PubMed] [Google Scholar]
- 8. Prista C., Michán C., Miranda I.M. and Ramos J. (2016) The halotolerant Debaryomyces hansenii, the cinderella of non-conventional yeasts. Yeast, 33, 523–533. [DOI] [PubMed] [Google Scholar]
- 9. Navarrete C., Frost A.T., Ramos-Moreno L., Krum M.R. and Martínez J.L. (2021) A physiological characterization in controlled bioreactors reveals a novel survival strategy for Debaryomyces hansenii at high salinity. Yeast, 38, 302–315. [DOI] [PubMed] [Google Scholar]
- 10. Nobre A., Lucas C. and Leão C. (1999) Transport and utilization of hexoses and pentoses in the halotolerant yeast Debaryomyces hansenii. Appl. Environ. Microbiol., 65, 3594–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nobre A., Duarte L., Roseiro J. and Gírio F. (2002) A physiological and enzymatic study of Debaryomyces hansenii growth on xylose- and oxygen-limited chemostats. Appl. Microbiol. Biotechnol., 59, 509–516. [DOI] [PubMed] [Google Scholar]
- 12. Marquina D., Barroso J., Santos A. and Peinado J.M. (2001) Production and characteristics of Debaryomyces hansenii killer toxin. Microbiol. Res., 156, 387–391. [DOI] [PubMed] [Google Scholar]
- 13. Poblete-Castro I., Hoffmann S.L., Becker J. and Wittmann C. (2020) Cascaded valorization of seaweed using microbial cell factories. Curr. Opin. Biotechnol., 65, 102–113. [DOI] [PubMed] [Google Scholar]
- 14. Li T., Chen X., Chen J., Wu Q. and Chen G.-Q. (2014) Open and continuous fermentation: products, conditions and bioprocess economy. Biotechnol. J., 9, 1503–1511. [DOI] [PubMed] [Google Scholar]
- 15. Keasling J., Martin H.G., Lee T.S., Mukhopadhyay A., Singer S.W. and Sundstrom E. (2021) Microbial production of advanced biofuels. Nat. Rev. Microbiol., 2021, 1–15. [DOI] [PubMed] [Google Scholar]
- 16. Spasskaya D.S., Kotlov M.I., Lekanov D.S., Tutyaeva V.V., Snezhkina A.V., Kudryavtseva A.V., Karpov V.L. and Karpov D.S. (2021) CRISPR/Cas9-mediated genome engineering reveals the contribution of the 26S proteasome to the extremophilic nature of the yeast Debaryomyces hansenii. ACS Synth. Biol., 10, 297–308. [DOI] [PubMed] [Google Scholar]
- 17. Minhas A. and Biswas D. (2019) Development of an efficient transformation system for halotolerant yeast Debaryomyces hansenii CBS767. Bio-Protocol, 9, e3352.doi: 10.21769/bioprotoc.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minhas A., Biswas D. and Mondal A.K. (2009) Development of host and vector for high-efficiency transformation and gene disruption in Debaryomyces hansenii. FEMS Yeast Res., 9, 95–102. [DOI] [PubMed] [Google Scholar]
- 19. Ricaurte M.L. and Govind N.S. (1999) Construction of plasmid vectors and transformation of the marine yeast Debaryomyces hansenii. Mar. Biotechnol., 1, 15–19. [DOI] [PubMed] [Google Scholar]
- 20. Maggi R.G. and Govind N.S. (2004) Regulated expression of green fluorescent protein in Debaryomces hansenii. J. Ind. Microbiol. Biotechnol., 31, 301–310. [DOI] [PubMed] [Google Scholar]
- 21. Kawaguchi Y., Honda H., Taniguchi-Morimura J. and Iwasaki S. (1989) The codon CUG is read as serine in an asporogenic yeast Candida cylindracea. Nature, 341, 164–166. [DOI] [PubMed] [Google Scholar]
- 22. Stovicek V., Holkenbrink C. and Borodina I. (2017) CRISPR/Cas system for yeast genome engineering: advances and applications. FEMS Yeast Res., 17, fox030.doi: 10.1093/femsyr/fox030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai P., Gao J. and Zhou Y. (2019) CRISPR-mediated genome editing in non-conventional yeasts for biotechnological applications. Microb. Cell Fact., 18, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertani G. (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia Coli. J. Bacteriol., 62, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherman F. (2002) Getting started with yeast. Meth. Enzymol., 350, 3–41. [DOI] [PubMed] [Google Scholar]
- 26. Higuchi R., Krummel B., Saiki R. and General A. (1988) Method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res., 16, 7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geu-Flores F., Nour-Eldin H.H., Nielsen M.T. and Halkier B.A. (2007) USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res., 35, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Norton E.L., Sherwood R.K. and Bennett R.J. (2017) Development of a CRISPR-Cas9 system for efficient genome editing of Candida lusitaniae. mSphere, 2, e00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nødvig C.S., Hoof J.B., Kogle M.E., Jarczynska Z.D., Lehmbeck J., Klitgaard D.K. and Mortensen U.H. (2018) Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in aspergilli. Fungal Genet. Biol., 115, 78–89. [DOI] [PubMed] [Google Scholar]
- 30. Voronovsky A.A., Abbas C.A., Fayura L.R., Kshanovska B.V., Dmytruk K.V., Sybirna K.A. and Sibirny A.A. (2002) Development of a transformation system for the flavinogenic yeast Candida famata. FEMS Yeast Res., 2, 381–388. [DOI] [PubMed] [Google Scholar]
- 31. Liachko I. and Dunham M.J. (2014) An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res., 14, 364–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sikorski R.S. and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Phizicky E.M. and Hopper A.K. (2010) tRNA biology charges to the front. Genes Dev., 24, 1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roman H. (1956) Studies of gene mutation in Saccharomyces. Cold Spring Harb. Symp. Quant. Biol., 21, 175–185. [DOI] [PubMed] [Google Scholar]
- 35. Nødvig C.S., Nielsen J.B., Kogle M.E. and Mortensen U.H. (2015) A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One, 10, e0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Löbs A.K., Schwartz C. and Wheeldon I. (2017) Genome and metabolic engineering in non-conventional yeasts: current advances and applications. Synth. Syst. Biotechnol., 2, 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vanegas K.G., Lehka B.J. and Mortensen U.H. (2017) SWITCH: a dynamic CRISPR tool for genome engineering and metabolic pathway control for cell factory construction in Saccharomyces cerevisiae. Microb. Cell Fact., 16, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DiCarlo J.E., Norville J.E., Mali P., Rios X., Aach J. and Church G.M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res., 41, 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fellerhoff B., Eckardt-Schupp F. and Friedl A.A. (2000) Subtelomeric repeat amplification is associated with growth at elevated temperature in Yku70 mutants of Saccharomyces cerevisiae. Genetics, 154, 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.