

Abstract
We developed a resource, the Arabidopsis PeptideAtlas (www.peptideatlas.org/builds/arabidopsis/), to solve central questions about the Arabidopsis thaliana proteome, such as the significance of protein splice forms and post-translational modifications (PTMs), or simply to obtain reliable information about specific proteins. PeptideAtlas is based on published mass spectrometry (MS) data collected through ProteomeXchange and reanalyzed through a uniform processing and metadata annotation pipeline. All matched MS-derived peptide data are linked to spectral, technical, and biological metadata. Nearly 40 million out of ∼143 million MS/MS (tandem MS) spectra were matched to the reference genome Araport11, identifying ∼0.5 million unique peptides and 17,858 uniquely identified proteins (only isoform per gene) at the highest confidence level (false discovery rate 0.0004; 2 non-nested peptides ≥9 amino acid each), assigned canonical proteins, and 3,543 lower-confidence proteins. Physicochemical protein properties were evaluated for targeted identification of unobserved proteins. Additional proteins and isoforms currently not in Araport11 were identified that were generated from pseudogenes, alternative start, stops, and/or splice variants, and small Open Reading Frames; these features should be considered when updating the Arabidopsis genome. Phosphorylation can be inspected through a sophisticated PTM viewer. PeptideAtlas is integrated with community resources including TAIR, tracks in JBrowse, PPDB, and UniProtKB. Subsequent PeptideAtlas builds will incorporate millions more MS/MS data.
A web resource providing the global community with mass spectrometry-based Arabidopsis proteome information and its spectral, technical, and biological metadata integrated with TAIR and JBrowse.
Introduction
Arabidopsis (Arabidopsis thaliana) was the first plant species whose nuclear genome was sequenced and has served as a model species for plant biology research for the last ∼25 years (Provart et al., 2016). The current Arabidopsis genome release version 11 (Araport11) contains 27,655 protein-coding gene loci represented by 48,359 transcripts (Cheng et al., 2017). The collective set of proteins in Arabidopsis, referred to as the proteome, carries out essential functions in metabolism, gene expression, signal transduction, transport, and more. The proteome not only varies with time, development, and (a)biotic conditions, but also undergoes a wide range of dynamic reversible and irreversible post-translational modifications (PTMs; e.g. phosphorylation, ubiquitination, acetylation). Furthermore, proteins are distributed across subcellular locations, such as the various organelles, and many proteins often stably or dynamically interact with other proteins. Whereas genome sequencing technologies combined with large-scale RNA-seq data and computation can predict the theoretical set of protein-coding genes in an organism, cell-type specific and subcellular protein abundance, protein PTMs, and protein interactions cannot be predicted but must be experimentally determined at the protein level. Furthermore, even the best annotated genomes (such as the Arabidopsis and human genomes) cannot easily predict which mRNA splice forms result in proteins; indeed, the impact of alternative splicing on the human and other proteomes is still under debate (Blencowe, 2017; Tress et al., 2017). The use of proteomics data for plant genome annotation has only very recently begun to make a more systematic impact under the term “proteogenomics” (Castellana et al., 2014; Walley and Briggs, 2015; Chapman and Bellgard, 2017; Zhu et al., 2017; Ren et al., 2019). This has included the genomes of Arabidopsis (Zhu et al., 2017; Zhang et al., 2019), rice (Oryza sativa; Ren et al., 2019; Chen et al., 2020), maize (Zea mays; Castellana et al., 2014), grape (Vitis vinifera; Chapman and Bellgard, 2017), and sweet potato (Ipomoea batatas; Al-Mohanna et al., 2019).
Initial mass spectrometry (MS)-based plant proteomics studies appeared in the year 2000 with investigations of the proteomes of maize and pea (Chang et al., 2000; Peltier et al., 2000; van Wijk, 2000) at a time when there were no sequenced plant genomes, instead relying on expressed sequence tag assemblies. With the release of the first partial Arabidopsis (ecotype Columbia-0) genome sequence (Initiative, 2000), Arabidopsis rapidly became the organism of choice for plant proteomics studies. Initially, MS was used for the study of subcellular organelles such as chloroplasts and mitochondria (Millar et al., 2001; Peltier et al., 2002; Schubert et al., 2002; Ytterberg, 2002), plant structures such as pollen (Mayfield et al., 2001), and protein complexes (Peltier et al., 2001). MS-based proteomics has since become increasingly successful for studying proteomes of different plant organs, cell types, and (subcellular) compartments as well as the many plant protein PTMs, such as phosphorylation (Stecker et al., 2014; Balmant et al., 2016), lysine acetylation (Hosp et al., 2017), ubiquitination (Vierstra, 2012) and SUMOylation (Miura and Hasegawa, 2010), redox modifications (Akter et al., 2015; Waszczak et al., 2015), N-terminal acetylation (Rowland et al., 2015; Willems et al., 2017), and lysine acetylation (Hartl et al., 2017). For recent reviews on PTMs in plants, see Friso and van Wijk (2015) and Millar et al. (2019). Proteomics has also been extensively used to study plant responses to (a)biotic conditions, and plant developmental processes in e.g. roots, seeds, and leaves, reviewed in Vanderschuren et al. (2013) and Ruiz-May et al. (2019). The progress of proteomics research of plants has been regularly reviewed, mostly in an attempt to consolidate plant proteome information, including protein detection, various PTMs, abundance measurements, and to provide updates of plant proteomics and MS technologies and plant proteome databases (Tan et al., 2017; Misra, 2018). A range of plant proteome databases have been developed by individual laboratories, mostly for Arabidopsis proteins. These databases are typically focused quite narrowly toward a particular aspect of plant proteomics, such as subcellular compartments (San Clemente and Jamet, 2015; Salvi et al., 2018), protein localization (SUBA and PPDB; Sun et al., 2009; Tanz et al., 2013), or PTMs (Schulze et al., 2015; Willems et al., 2019). Most recently, a comprehensive Arabidopsis proteome database (ATHENA) has released to allow mining of a large-scale experimental proteome dataset involving multiple tissue types, as published in Mergner et al. (2020). Many of the MS data for Arabidopsis were collected through MASCP GATOR (Joshi et al., 2011; Mann et al., 2013), which was an aggregation portal for proteomic data produced by the community that united a large collection of specialized resources. However, GATOR has been discontinued, thus leaving a void for the Arabidopsis community.
The global scientific community has developed a wide range of initiatives to capture and store highly data-rich MS-based proteomics information using standardized bioinformatics workflows and file formats (Orchard et al., 2003; Deutsch et al., 2017a). The ProteomeXchange consortium (http://www.proteomexchange.org/) coordinates standard data submission and dissemination pipelines across the main proteomics repositories and promotes submission of all published datasets and open data policies in the field (Vizcaino et al., 2014; Deutsch et al., 2017b, 2020). The consortium has made tremendous progress in getting the community to deposit its datasets in conjunction with publication of an article. Currently, there are well over 15,000 released ProteomeXchange datasets (PXDs). Many plant journals such as The Plant Cell, Plant Physiology, Plant Journal, Molecular Plant, and others strongly encourage MS data deposition for publications that rely on MS-based proteomics. Currently (at the time of submission), ProteomeXchange has over 1,200 released PXDs for proteome datasets from many plant species (and a few from algae), of which approximately 425 PXDs are from Arabidopsis.
PeptideAtlas (http://www.peptideatlas.org/) reprocesses MS datasets available through ProteomeXchange with the trans-proteomic pipeline (TPP; Keller et al., 2005; Deutsch et al., 2015; Slagel et al., 2015) and makes an integrated view of the results available to the community. So far, PeptideAtlas has focused heavily on the human proteome, beginning with the first publication in 2005 (Desiere et al., 2005) and continuing with ongoing contributions to the Human Proteome Project (HPP) including yearly advances in coverage of the human proteome (Omenn et al., 2019; Omenn et al., 2020). However, PeptideAtlas has also created builds for several other species, including pig (Sus scrofa; Hesselager et al., 2016), chicken (Gallus gallus; McCord et al., 2017), cow (Bos taurus; Bislev et al., 2012), the pathogens Pseudomonas aeruginosa (33757883) and Candida albicans (Vialas et al., 2014), and the yeast Saccharomyces cerevisiae (King et al., 2006). Yet, PeptideAtlas builds have not been created for any plant species. Given the significant amount of PXD submissions for plants, particularly Arabidopsis, this provides a unique opportunity to take full advantage of the rapidly growing amounts of MS-based proteomics data for Arabidopsis in order to build a thorough understanding of the observed Arabidopsis proteome.
The current report describes a project that will take advantage of the current and anticipated submissions to ProteomeXchange by reanalyzing these data through the TPP to generate PeptideAtlas builds for Arabidopsis and in later stages additional plant species. This freely available Arabidopsis PeptideAtlas provides the global community with high quality, fully reprocessed MS-based proteome information together with its metadata. This resource can be used to solve central questions about the Arabidopsis proteome, such as the significance of protein splice forms, PTMs, or simply to obtain reliable information about specific protein sets of interest without the need to be an expert in MS. The Arabidopsis PeptideAtlas provides immediate insight into: (1) which Arabidopsis proteins have been identified and with how much protein sequence coverage; (2) relative protein abundance based on the frequency of observations across datasets and sampling across plant organs, cell types, organelles, (a)biotic treatments, development, and complexes; (3) enrichment for specific post-translational modifications; (4) which proteins have not yet been observed (the “dark” proteome); and (5) specific information to improve genome annotation, including the discovery of protein-coding small Open Reading Frames (sORFs). PeptideAtlas differs from other databases such as ATHENA and Plant PTM Viewer in that the raw MS data from laboratories around the world and available in ProteomeXchange are reprocessed. All identified peptides, PTMs, and MS/MS (tandem MS) spectra in PeptideAtlas are linked to the metaData collected from the PXDs, publications, and frequently from additional information from the submitting labs. We envision that these Arabidopsis PeptideAtlas builds will stimulate laboratories around the world to submit their proteomics and MS data to ProteomeXchange, further accelerating our knowledge about the expression and PTMs of plant proteins.
Materials and methods
Selection and downloads of ProteomeXchange submissions
PXDs were selected based on several criteria, including mass spectrometer type, with preference for Orbitrap-type instruments from Thermo (Q Exactive models, LTQ-Orbitrap Velos/Elite, Orbitrap Fusion Lumos), submissions from 2018 and 2019, and samples including subcellular fractions or specific PTMs. The rationale is provided in the “Results and Discussion”. Raw files for the selected PXDs were downloaded from ProteomeXchange. Supplemental Data Set S1 provides the final 52 selected PXDs and information about instrument, sample (e.g. subcellular proteome, plant organ), number of raw files and MS/MS spectra (searched and matched), identified proteins and peptides, submitting lab and associated publication, as well as several informative key words.
Extraction and annotation of metadata
For each selected dataset, we obtained information associated with the submission, as well as the publication if available. This information was used to determine search parameters and to provide meaningful tags that describe the samples in some detail. These tags are visible for the relevant proteins in PeptideAtlas. If needed, we contacted the submitters for more information about the raw files. To facilitate the metadata assignments and association to specific raw files, we developed a metadata annotation system that aims to provide detailed information about each matched spectrum for the users of PeptideAtlas. Where possible, we incorporated controlled vocabularies for plant parts and developmental stages, growth conditions, sample purification methods, as well as protein/peptide labeling and processing steps (e.g. type of enzyme used for generation of peptides). These controlled vocabularies are from the Planteome (PO, PECO; https://github.com/Planteome), Gene Ontology (http://geneontology.org/), as well as PSI-MS (http://www.psidev.info/groups/mass-spectrometry;Mayer et al., 2013), Unimod (https://www.unimod.org;Creasy and Cottrell, 2004), PSI-MOD (https://www.ebi.ac.uk/ols/ontologies/mod;Montecchi-Palazzi et al., 2008), and the Experimental Factor Ontology (https://www.ebi.ac.uk/ols/ontologies/efo). These metadata can be viewed for each identified protein in PeptideAtlas.
Assembly of the protein search space
We assembled a comprehensive protein search space comprising the predicted Arabidopsis protein sequences from: (1) Araport11 (Cheng et al., 2017); (2) TAIR10 (Lamesch et al., 2012); (3) UniProtKB (UniProt, 2020); (4) RefSeq (https://www.ncbi.nlm.nih.gov/refseq;Li et al., 2020); (5) the repository ARA-PEPs (http://www.biw.kuleuven.be/CSB/ARA-PEPs;Hazarika et al., 2017), with 7,901 sORFs, 16,809 low molecular weight peptides and proteins (LWs; between 26 and 250 amino acids (aa); median 37 aa), and 607 novel stress-induced peptides (SIPs), most of which are currently not annotated in TAIR10 or Araport11; (6) data from Dr Eve Wurtele (Iowa State University) assembled based on RNA-seq data; (7) GFP, RFP, and YFP protein sequences commonly used as reporters and affinity enrichments; and (8) 116 contaminant protein sequences frequently observed in proteome samples (e.g. keratins, trypsin, BSA; https://www.thegpm.org/crap/). Table 1 shows the number of sequences for each set, their overlap, and unique protein sequences.
Table 1.
The assembly of protein sequences from different sources used as the protein search space, and the respective number of total, distinct, and unique sequences in each source, as well as the sequence-identical intersection among sources
| Source | Sequences | Distinct | Unique | TAIR10 | UniProtKB | RefSeq | LW | SIPs | sORFs | IowaORFs |
|---|---|---|---|---|---|---|---|---|---|---|
| Araport11 | 48,359 | 40,784 | 26 | 31,134 | 38,703 | 40,653 | 0 | 0 | 0 | 1,147 |
| TAIR10 | 35,386 | 32,785 | 1,501 | 29,405 | 31,032 | 0 | 0 | 0 | 1,057 | |
| UniProtKB | 39,346 | 39,277 | 383 | 38,673 | 0 | 0 | 0 | 1,116 | ||
| RefSeq | 48,265 | 40,709 | 6 | 0 | 0 | 0 | 1,116 | |||
| ARA-PEP: LW | 16,809 | 16,628 | 16,478 | 21 | 129 | 0 | ||||
| ARA-PEP: SIPs | 607 | 606 | 565 | 20 | 0 | |||||
| ARA-PEP: sORFs | 7,901 | 7,764 | 7,615 | 0 | ||||||
| IowaORFs | 7,481 | 7,270 | 6,116 | |||||||
| Total Nonredundant | 73,816 |
The TPP data processing pipeline
For all selected datasets, the vendor-format raw files were downloaded from the hosting ProteomeXchange repository, converted to mzML files (Martens et al., 2011) using ThermoRawFileParser (Hulstaert et al., 2020) for Thermo Fisher Scientific instruments or the msconvert tool for SCIEX wiff files from the ProteoWizard toolkit (Chambers et al., 2012), and analyzed with the TPP. The TPP analysis consisted of sequence database searching with Comet (Eng and Deutsch, 2020) and post-search validation with several additional TPP tools as follows: PeptideProphet (Keller et al., 2002) was run to assign probabilities of being correct for each peptide-spectrum match (PSM) using semi-parametric modeling of the Comet expect scores with z-score accurate mass modeling of precursor m/z deltas. These probabilities were further refined via corroboration with other PSMs, such as multiple PSMs to the same peptide sequence but different peptidoforms or charge states, using the iProphet tool (Shteynberg et al., 2011).
For datasets in which trypsin was used as the protease to cleave proteins into peptides, two parallel searches were performed, one with full tryptic specificity and one with semi-tryptic specificity. The semi-tryptic searches were carried out with the following possible variable modifications (maximum of five per peptide): oxidation of Met or Trp (+15.9949), acetylation of Lys (+42.0106), peptide N-terminal Gln to pyro-Glu (−17.0265), peptide N-terminal Glu to pyro-Glu (−18.0106), deamidation of Asn or Gln (+0.9840), peptide N-term acetylation (+42.0106), and if peptides were specifically affinity enriched for phosphopeptides, also phosphorylation of Ser, Thr, or Tyr (+79.9663). For the full tryptic searches, we also added oxidation of Pro or His (+15.99) and formylation of peptide N-termini, Ser, or Thr (+27.9949). We deliberately restricted these PTMs to only full tryptic (rather than also allowing semi-tryptic) to reduce the search space and computational needs. Formylation is a very common chemical modification that occurs in extracted proteins/peptides during sample processing, whereas Pro and His oxidation is observed less frequently, but nevertheless at significant levels (Verrastro et al., 2015; Hawkins and Davies, 2019). In both semi-tryptic and full tryptic searches, fixed modifications for carbamidomethylation of Cys (+57.0215) if treated with reductant and iodoacetamide and isobaric tag modifications (tandem mass tag [TMT], isobaric tags for relative and absolute quantitation [iTRAQ]) were applied as appropriate. Both variable and fixed modifications were applied to dimethyl-labeled datasets as appropriate. Four missed cleavages were allowed (Arg-Pro and Lys-Pro do not count as missed cleavages). Several datasets were generated using other proteases (GluC, ArgC, Chymotrypsin); these datasets were processed similarly to those generated by trypsin except that the relevant enzyme was chosen. Some of the datasets contain the analysis of extracted peptidomes in which no protease treatment was used, and these datasets were searched using the term “no enzyme”.
PeptideAtlas assembly
In order to create the combined PeptideAtlas build of all experiments, all datasets were thresholded at a probability that yields an iProphet model-based false discovery rate (FDR) of 0.001 at the peptide level. The exact probability varies from experiment to experiment depending on how well the modeling can separate correct from incorrect information. This probability threshold is typically greater than 0.99. As more and more experiments are combined, the total FDR increases unless the threshold is made more stringent (Deutsch et al., 2016b). The final iProphet model-based peptide sequence level FDR across all experiments is 0.001, corresponding to a PSM-level FDR of 0.0001. Throughout the procedure, decoy identifications are retained and then used to compute final decoy-based FDRs. The decoy-based PSM-level FDR is 0.0001 (4,843 decoy PSMs out of 40 million), the peptide sequence-level FDR is 0.001 (746 decoy sequences out of 535,000), and the final protein-level FDR is 0.03 (683 decoy proteins out of 21,297). Because of the tiered system, quality MS/MS spectra that are matched to a peptide are never lost, even if a single matched peptide by itself cannot confidently identify a protein.
Protein identification confidence levels and classification
Proteins are identified at different confidence levels using standardized assignments to different confidence levels based on various attributes and relationships to other proteins using a relatively complex but precise 10-tier system developed over many years for the human proteome PeptideAtlas (Farrah et al., 2011; Table 2, panel A). We simplified this 10-tier system to a simpler four category system in (Table 2, panel B), which is more accessible to nonexperts, and used this to summarize most of our findings. For all protein identifications and categorizations, all peptides must first meet the stringent PSM threshold already described above. For both systems, the highest confidence level category is the “canonical” category (tier 1), which requires at least two uniquely mapping nonnested (one not inside the other) peptides at least 9 aa long with a total coverage of at least 18 aa, as required by the HPP guidelines (Deutsch et al., 2019; Table 2, panel A and B). The decoy-based canonical protein FDR is 0.0005 (only eight decoys remain out of 18,045 canonical sequences including contaminants and contributed sequences).
Table 2.
Protein identification confidence tiers and categories in the Arabidopsis PeptideAtlas build
| Categorya | Definition |
|---|---|
| Tier 1: Canonical | Protein has at least two uniquely mapping non-nested peptides of at least 9 residues with at least 18 residues of total coverage. |
| Tier 2: Indistinguishable representative | Protein is selected as the representative of a set of proteins that are different in sequence but cannot be disambiguated based on the detected peptides. All peptides are shared with all group members. Others are “Indistinguishable”. |
| Tier 3: Representative | Protein is selected a representative in a situation more complex than a set of indistinguishable, where several proteins have shared peptides and at least some of the proteins must have been detected but it is not possible to determine which ones. |
| Tier 4: Marginally distinguished | Protein that shares several peptides with a canonical protein, but also has one uniquely mapping peptide that appears to distinguish it from the canonical. |
| Tier 5: Weak | Protein has at least one uniquely mapping peptide of 9 residues in length but does not meet the criteria for canonical. |
| Tier 6: Insufficient evidence | Protein has one or more uniquely mapping peptides but none reach 9 residues in length. |
| Tier 7: Indistinguishable | Protein is part of a set of proteins that cannot be disambiguated and it not selected as a leader of the group. |
| Tier 8: Subsumed | Protein has only shared peptides and is not needed to explain the peptide evidence. |
| Tier 9: Identical | Protein has an identical protein sequence to another one, and this one is effectively removed from category competition. Its partner may be canonical. |
| Tier 10: Not observed | Protein has no peptides above our PSM significance threshold. It may have low significant PSMs, but these are not considered. |
|
| |
| Categoryb | Definition |
|
| |
| Canonical (as in tier 1 in Table 2a) | Protein has at least two uniquely mapping non-nested peptides of at least 9 residues with at least 18 residues of total coverage |
| Uncertain (tiers 2–7 in Table 2a) | Protein has too few uniquely mapping peptides of ≥ 9 aa to qualify for canonical status and may also have one or more shared peptides with other proteins. |
| Redundant (tiers 8 and 9 in Table 2a) | Protein has only peptides that are can be assigned to other entries and thus these proteins are not needed to explain the observed peptide evidence. |
| Not Observed (tier 10 in Table 2a) | Protein has no peptides above our PSM significance threshold. It may have low significance PSMs, but these are not considered. |
Panel A: List of protein identification confidence tiers in the Arabidopsis PeptideAtlas build. Note that for each gene locus, only one gene model was counted using model .1 as default, unless there were specific matched peptides that could specifically distinguish more than one model, thereby receiving classification as tier 1 or 2.
Panel B: List of protein identification confidence tiers in the Arabidopsis PeptideAtlas build. Note that for each gene locus, only one gene model was counted using model .1 as default, unless there were specific matched peptides that could specifically distinguish more than one model, thereby receiving classification as canonical or uncertain.
The 10-tier system: When a group of proteins cannot be disambiguated because they contain shared peptides, one or more “leaders” of the group are categorized as “indistinguishable representative” (tier 2) or “representative” (tier 3; Table 2, panel A). This means that the protein or one of its close siblings is detected, but it is not possible to disambiguate them. The “marginally distinguished” category (tier 4) means that the protein shares peptides with a canonical entry but has some additional uniquely mapping peptide evidence that is however not sufficient to raise it to the canonical level. The “weak” category (tier 5; Table 2, panel A) means that there is at least one uniquely mapping peptide that is nine or more residues long, but the evidence does not meet the criteria for being canonical. The “insufficient evidence” category (tier 6) means that all the uniquely mapping peptides are less than nine residues long. While even one uniquely mapping peptide in theory uniquely identifies a protein, these guidelines guard against false positives due to our imperfect understanding of the reference proteome and incomplete b and y ion series identifications, which can lead to amino acid order transpositions and protein misassignment. Tiers 7, 8, and 9 describe proteins that share all their peptides with one or more proteins in an earlier tier, and thus are not needed to explain the available peptide evidence. Finally, all other proteins that lack any matched peptides observed above our minimum PSM significance threshold are categorized as “not observed” proteins (tier 10; Table 2, panel A).
The four-category system: In the simpler four-category system, proteins that have no uniquely mapping peptides but do not qualify as canonical (same as tier 1) are categorized as “uncertain” (Table 2, panel B), corresponding to the sum of tiers 2–6 in Table 2, panel A). Proteins are categorized as “redundant” if they have only shared peptides that can be assigned to other entries, and thus these proteins are not needed to explain the observed peptide evidence (tiers 7–9). Finally, all other proteins that completely lack any peptides observed at our minimum PSM significance threshold are categorized as “not observed” (tier 10).
Handling of gene models and splice forms
The 27,655 protein-coding genes in Araport11 are represented by 48,359 gene models (transcript isoforms), which are identified by the digit after the AT identifier (e.g. AT1G10000.1). We refer to the translations of these gene models as protein isoforms. Most protein isoforms are very similar (differing in only a few amino acid residues, often at the N- or C-terminus) or even identical at the protein level. It is often hard to distinguish between different protein isoforms due to the incomplete sequence coverage inherent to most MS proteomics workflows. For the assignment of canonical proteins (at least two uniquely mapping peptides identified; Table 2, panel A and B), we selected by default only one of the protein isoforms as the canonical protein; this was labeled as the model “.1” isoform unless one of the other isoforms had a higher number of matched peptides. However, if other protein isoforms did have detected peptides that are unique compared to the canonical protein isoform (e.g. perhaps due to the presence of a different exon), they can be given tier 1 or less confident tier status depending on the nature of the additional uniquely mapping peptides (length and numbers; Table 2, panel A and B). If the other protein isoforms do not have any uniquely mapping peptides amongst all protein isoforms (for that gene), they are classified as redundant (tiers 7–9 in the more complex system).
Physicochemical properties and functions of proteins
To characterize the canonical and unobserved proteomes, physicochemical properties were calculated or predicted using various web-based tools. These include: protein length, mass, GRAVY index, isoelectric point (pI), number of transmembrane domains (http://www.cbs.dtu.dk/services/TMHMM), and sorting sequences for the ER, plastids, and mitochondria (http://www.cbs.dtu.dk/services/TargetP-1.0/).
Integration of PeptideAtlas results in other web-based resources
PeptideAtlas is accessible through its web interface at http://peptideatlas.org. Furthermore, direct links are provided between PeptideAtlas and PPDB (http://ppdb.tc.cornell.edu/), UniProtKB (https://www.uniprot.org/), TAIR (https://www.arabidopsis.org/), Plant PTM Viewer (https://www.psb.ugent.be/webtools/ptm-viewer/), PhosPhAt (http://phosphat.uni-hohenheim.de/), SUBA4 (https://suba.live/), and several more, and soon also ATHENA (http://athena.proteomics.wzw.tum.de:5002/master_arabidopsisshiny/) at the level of protein entries. Links to matched peptide entries in PeptideAtlas are available in the Arabidopsis annotated genome through a specific track in JBrowse at https://jbrowse.arabidopsis.org.
Results and discussion
Overview of the generation and output of the Arabidopsis PeptideAtlas
Figure 1 provides an overview of the generation of the first build of the Arabidopsis PeptideAtlas. The project started by collecting all available MS datasets for Arabidopsis from ProteomeXchange; we refer to these datasets as PXDs. A subset of PXDs was selected (see ‘Selection of PXDs for the first build’), and detailed information about the samples and MS acquisition within each PXD was collected and annotated using a newly built in-house metadata annotation system. Selected PXDs were processed through the TPP to match MS data to peptides and proteins (including selected PTMs) in Araport11, TAIR10, a collection of small peptides, as well as other predicted proteins (Table 1). The genome annotation of Araport11 was used as the default (see “Materials and methods”). For each analyzed PXD, we calculated the MS/MS spectral match rate to peptides as a measure of MS/MS data quality as well as data processing. In case of a very low match rate (<10%), we reevaluated the search parameters and, if needed, reran the search with adjusted parameters. Following rigorous evaluation using sophisticated dedicated algorithms to control FDRs and PTM site verification (Shteynberg et al., 2019), as well redundancy removal (avoiding identical predicted proteins listed under different protein identifiers), identified proteins were classified into a 10-tier system, ranging from very high-confidence identifications to low-confidence identifications (Table 2, panel A). This tiered system allowed us to capture confidently matched peptides even if by themselves these peptides do not confidently identify a protein. Thus, the tiered system prevents the loss of any valuable MS/MS spectra. We also provide a simpler four-category system in which tiers 2–7 are folded into a single category (Table 2, panel B). The identified proteome was then evaluated for physicochemical properties, predicted subcellular localization, and function. Protein entries in PeptideAtlas are directly linked to TAIR, PPDB, UniProtKB, the Plant PTM Viewer, PhosPhAt, SUBA4, and ATHENA (Figure 1). Peptides are mapped to the Arabidopsis genome on specific tracks through the genome browser JBrowse. After in-depth evaluation of the identified proteome coverage from this first build and feedback from the international research community, we will select additional PXDs for subsequent PeptideAtlas builds, as discussed further below in the section “The next Arabidopsis PeptideAtlas build”. We aim to widely advertise and inform the community through seminars, workshops, and tutorials. In the remainder of this article, we will provide more detail about and insights into this PeptideAtlas build and the observed Arabidopsis proteome.
Figure 1.

Graphical overview of the Arabidopsis PeptideAtlas project and generation of the first build presented here. Specific steps and components are numbered.
Features of publicly available Arabidopsis PXDs
At the start of building the first Arabidopsis PeptideAtlas in the fall of 2019, we first reviewed all PXDs available through the ProteomeXchange interface for Arabidopsis, and we continued to do so as the project progressed into 2020. We verified if indeed the plant material was A. thaliana (and also checked the ecotype), scored each submission for the type of MS instrument(s) with which the data were collected, and collected information about nature of the samples (e.g. organ, subcellular fraction, enrichment for specific PTMs). Figure 2 summarizes some of this information for all 356 Arabidopsis PXDs until July 15, 2020. The first Arabidopsis PXD available in ProteomeXchange was from 2012 (we note that earlier submissions to PRIDE (Perez-Riverol et al., 2018) were not transferred to ProteomeXchange), and the number of datasets exponentially increased in subsequent years, resulting in 357 available PXDs from some 200 different laboratories by July 2020 (Figure 2A). A wide range of MS instruments was used to acquire these data (Figure 2B). There were just four submissions that used MALDI-TOF-TOF instruments, and the majority (82%) used different generations of Orbitrap-based instruments from Thermo Fisher Scientific (Eliuk and Makarov, 2015; Makarov, 2019). The sensitivity and throughput of MS have dramatically increased over subsequent generations of MS instruments, and this should also be reflected in increasing proteome coverage with newer PXDs.
Figure 2.

Features of PXDs for Arabidopsis available via ProteomeXchange through July 15, 2020. Information about these PXDs was obtained from the submitted metadata and/or accompanying publications. A, Accumulative PXDs with verified Arabidopsis content by year (2010-7/2020). B, Type of MS Instrument (LTQ-Orbitrap–Velos/Elite/XT [Thermo], Orbitrap-LumosFusion [Thermo], QExactive [Thermo], Synapt [Waters], TripleTOF5600/6600 [Sciex], ImpactII/MaXis [Bruker], other). C, Arabidopsis subcellular fractions (plastid, mitochondria, peroxisomes, vacuole, nucleus, apoplast/extracellular, cytosol, ER/Golgi/PM). D, Post-translational modifications that were specifically enriched prior to MS analysis (phosphorylation, acetylation [N-term or Lys], ubiquitination, cysteine oxidation, glycosylation, sumoylation, lipidation, other).
Based on keywords and information associated with each PXD, Figure 2C gives an impression of the types of subcellular fractions analyzed across these 356 PXDs. For simplicity, we grouped various keywords into 10 sample types, which showed a strong interest in proteomes from chloroplasts (often specific sub-organellar fractions such as thylakoids, stroma, or envelope). It should be noted that many of the PXDs did not involve a specific subcellular fraction, but rather analyzed proteome extracts (either just soluble or total detergent-extracted proteomes) from whole seedlings, plant parts (e.g. roots, flowers, rosettes) without further subfractionation. Whereas all PXDs allowed for one or more common PTMs that are either (often) induced after protein extraction (e.g. oxidation of Met or Trp, cyclization of Gln and Glu, deamidation of Asn or Gln, and carbamidomethylation of Cys), a subset of PXDs specifically focused on selected PTMs that often require affinity enrichment or labeling (Figure 2D). A significant portion of PXDs focused on protein phosphorylation, N-terminal or lysine acetylation, ubiquitination, and various cysteine modifications. Finally, these proteomics analyses were motivated by a wide range of biological questions, including the effects of abiotic stress (e.g. cold, heat, light, oxidation, metals, touch), biotic stress/plant immunity (e.g. Pseudomonas syringae, flagellin), developmental questions (e.g. seed development and germination), and circadian rhythms.
Selection of PXDs for the first build
Because it was not feasible to process all available PXDs for the first PeptideAtlas build (due to time and computing constraints), we focused mostly on those PXDs that were generated by the high mass accuracy Orbitrap-type instrument types since they were by far the most frequently used (∼82% of all PXDs; Figure 2B) and to simplify the data analysis and better control FDRs. Table 3 provides key information about the final 52 PXDs used in this first build; additional details are provided in Supplemental Data Set S1. One of the PXDs (PXD012710) containing a very large dataset was acquired on a TripleTOF5600 instrument (Table 3). The majority of selected PXDs were from 2019 (∼40%), with additional PXDs from 2015 to 2018. We also added the recent 2020 PXD (PXD013868) associated with Mergner et al. (2020) because it included a very large amount of MS data, including phosphorylated proteins, sampled across 30 different Arabidopsis tissues. Finally, most PXDs were from ecotype Colombia 0 (Col-0) since this is the reference Arabidopsis ecotype that was originally sequenced and on which the Arabidopsis Araport11 and previous TAIR genome annotations are based. However, one PXD used Wassilewskij, and several PXDs used ecotype Landsberg erecta mostly for cell cultures (ordered from the Arabidopsis Biological Resource Center; PSB-D (CCL84840) and PSB-L (CCL84841)).
Table 3.
Summarizing information about the 52 selected PXD datasets for this first PeptideAtlas build. This includes PXD number, publication, number of matched MS/MS spectra and % match rate, the number of identified proteins (canonical and groups of proteins), the number of matched distinct MS/MS peptides, the MS instrument, information about the sample (plant part, subcellular fraction, enrichment for PTMs). An extended table with additional information is provided as Supplemental Data Set S1
| Data Set identifier | Publication | Matched No. of MS/MS Spectra | Matched MS/MS Spectra (%) | No. of Distinct Peptides | Instrument | Plant Parts | Subcellular Fraction | N-terminome and Specific PTM Analysis |
|---|---|---|---|---|---|---|---|---|
| PXD000136 | Hesse et al. (2016) | 22,419 | 0.16 | 4,569 | LTQ FT | RL | Chloroplast | |
| PXD000546 | Tomizioli et al. (2014) | 120,945 | 0.43 | 8,858 | LTQ Orbitrap Velos | RL | Chloroplast | |
| PXD002069 | Linster et al. (2015) | 213,875 | 0.08 | 7,230 | LTQ Orbitrap Velos | RL | Acetylation of N-term and lysine | |
| PXD006651 | Hartl et al. (2017) | 159,852 | 0.61 | 26,626 | Q Exactive | RL | Chloroplast | lysine acetylation |
| PXD006652 | Hartl et al. (2017) | 114,752 | 0.25 | 15,734 | Q Exactive | RL | Chloroplast | lysine acetylation |
| PXD008663 | Castrec et al. (2018) | 172,302 | 0.05a | 6,906 | LTQ Orbitrap Velos | RL | N-term & lysine acetylation | |
| PXD007630 | Koskela et al. (2018) | 166,166 | 0.35 | 15,184 | Q Exactive | RL | Chloroplast | N-terminal/lysine acetylation |
| PXD001855 | Venne et al. (2015) | 36,078 | 0.12 | 12,721 | Q Exactive | Slg | N-terminome (ChaFRADIC) | |
| PXD004896 | Willems et al. (2017) | 87,053 | 0.13 | 31,022 | LTQ Orbitrap | CC (Ler) | N-terminome (COFRADIC) | |
| PXD000660 | Köhler et al. (2015) | 11,795 | 0.12 | 3,280 | LTQ Orbitrap Velos | RL | Chloroplast | N-terminome (TAILS) |
| PXD001719 | Zhang et al. (2015) | 39,966 | 0.20 | 13,154 | LTQ Orbitrap Velos | Rt | N-terminome (TAILS) | |
| PXD001473 | Lin et al. (2015) | 13,313 | 0.08 | 655 | LTQ Orbitrap XL | CC (Ler) | Phosphorylation | |
| PXD004276 | Choudhary et al. (2016) | 59,548 | 0.19 | 12,591 | LTQ Orbitrap | Sdl | Phosphorylation | |
| PXD004599 | Mattei et al. (2016) | 11,582 | 0.13 | 2,371 | LTQ Orbitrap | Sdl | Phosphorylation | |
| PXD005600 | Schonberg et al. (2017) | 45,886 | 0.13 | 2,462 | LTQ Orbitrap Velos | RL | Chloroplast | Phosphorylation |
| PXD008355 | Van Leene et al. (2019) | 374,427 | 0.28 | 21,350 | Q Exactive | CC (Ler) | Phosphorylation | |
| PXD009016 | Zhang et al. (2019b) | 94,355 | 0.15 | 13,443 | Q Exactive | RL | Phosphorylation | |
| PXD013646 | Furtauer et al. (2019) | 2,374,645 | 0.21 | 35,719 | Q Exactive; LTQ Orbitrap Elite | RL (Ler) | Phosphorylation | |
| PXD013868 | Mergner et al. (2020) | 15,180,331 | 0.27 | 388,665 | Q Exactive HF | 30 tissue types | Phosphorylation | |
| PXD000869 | Zhang et al. (2018) | 62,763 | 0.40 | 4,616 | LTQ Orbitrap Velos | RL | Chloroplast | |
| PXD000908 | Baerenfaller et al. (2015) | 466,846 | 0.22 | 19,973 | LTQ Orbitrap XL | RL | ||
| PXD001207 | Köhler et al. (2015) | 26,393 | 0.35 | 7,554 | LTQ Orbitrap Velos | RL | Chloroplast | |
| PXD002160 | Correa-Galvis et al (2016) | 77,605 | 0.14 | 3,638 | LTQ Orbitrap Elite | RL | Chloroplast | |
| PXD002186 | Nishimura et al. (2015) | 257,800 | 0.45 | 9,575 | LTQ Orbitrap | RL | Chloroplast | |
| PXD003162 | Lundquist et al. (2017) | 247,024 | 0.22 | 13,092 | LTQ Orbitrap Elite | RL | Chloroplast | |
| PXD003516 | Wang et al. (2016) | 25,046 | 0.17 | 12,277 | Q Exactive | RL | Chloroplast | |
| PXD003684 | Bhuiyan et al. (2016) | 130,083 | 0.32 | 8,337 | LTQ Orbitrap | RL | Chloroplast | |
| PXD004025 | Al Shweiki et al. (2017) | 559,931 | 0.39 | 22,301 | LTQ Orbitrap Velos | RL | ||
| PXD004742 | Subramanian et al (2016) | 161,009 | 0.45 | 8,788 | LTQ Orbitrap Velos | RL | ||
| PXD005740 | Hander et al (2019) | 1,388 | 0.02b | 860 | Q exactive | SDL – Rt and RL | ||
| PXD006113 | Brocard et al. (2017) | 153,376 | 0.23 | 13,578 | LTQ Orbitrap | RL | Lipid droplet | |
| PXD006328 | Strehmel et al. (2017) | 32,278 | 0.10 | 6,268 | Q Exactive | Rt | Exudate | |
| PXD006347 | Nee et al. (2017) | 9,155 | 0.03c | 1,838 | Q Exactive | S | ||
| PXD006800 | Brault et al. (2019) | 238,014 | 0.47 | 28,668 | Q Exactive | CC (Ler) | Total cell extract, plasmodesmata, plasma membrane, microsome, and cell wall | |
| PXD006806 | Brault et al. (2019) | 634,158 | 0.73 | 40,233 | Q Exactive | CC (Ler) | Plasmodesmata, plasma membrane, microsome, and cell wall | |
| PXD006848 | Seaton et al. (2018) | 1,609,008 | 0.54 | 51,874 | LTQ Orbitrap Velos | RL | ||
| PXD010324 | Waltz et al (2019) | 402,558 | 0.31 | 17,491 | Q Exactive | F; CC (dark) | Mitochondria | |
| PXD010545 | Bouchnak et al. (2019) | 66,782 | 0.19 | 16,619 | Q Exactive | RL (WS) | Chloroplast | |
| PXD010730 | Wu et al. (2019b) | 629,552 | 0.44 | 27,754 | Q Exactive | RL | ||
| PXD011088 | Rugen et al. (2019) | 589,487 | 0.23 | 22,692 | Q Exactive | RL; CC (Col-0) | Mitochondria | |
| PXD011483 | McLoughlin et al. (2019) | 2,897,014 | 0.38 | 44,923 | Q Exactive | RL, Sdl | Protein aggregates | |
| PXD011716 | Kosmacz et al. (2019) | 121,240 | 0.17 | 24,030 | Q Exactive | Sdl | Stress granule | |
| PXD011759 | Wu et al. (2019a) | 840,034 | 0.45 | 40,953 | Q Exactive | Sdl | ||
| PXD012708 | Zhang et al. (2019b) | 6,316,858 | 0.47 | 239,706 | Orbitrap Fusion Lumos | 10 plant partsd | ||
| PXD012710 | Zhang et al. (2019b) | 2,077,659 | 0.15 | 123,441 | TripleTOF 5600 | 11 plant partsd | ||
| PXD013005 | Wu et al. (2019b) | 731,387 | 0.35 | 41,004 | Q Exactive | Sdl | ||
| PXD013325 | Jiang et al. (2019) | 15,139 | 0.23 | 4,558 | LTQ Orbitrap Elite | RL | ||
| PXD013494 | Montandon et al. (2019) | 29,810 | 0.19 | 3,782 | LTQ Orbitrap | RL | Chloroplast | |
| PXD013637 | Hu et al. (2019) | 77,866 | 0.14 | 15,379 | Q Exactive | RL | ||
| PXD017189 | Bhuiyan et al. (2020) | 59,880 | 0.31 | 5,203 | LTQ Orbitrap | RL | ||
| PXD017380 | Not published | 267,019 | 0.12 | 24,271 | Q Exactive | RL | Chloroplast | |
| PXD017400 | Not published | 367,359 | 0.17 | 19,279 | Q Exactive | RL | Chloroplast | |
| Total | 39,480,811 |
Rosette leaves – RL; seedling – Sdl; root – Rt; flowers – F; seed – S; cell culture – CC.
N-acetoxy-[2H3] succinimide (C6D3H4NO4) treatment labeling resulting N-terminal and Lys acetylation as well as O-acetylation of Ser, Thr, and Tyr side-chains but then reversed through hydrolysis.
Affinity pulldown GFP tagged protein.
Native peptides (peptidome); no digests.
Ten plant parts: rosette leaves, cauline leaf, stems, flower, pollen, siliques, seeds, cotyledons, root, root cell culture.
We aimed to have representation across as many plant parts as possible to maximize proteome coverage, including those proteins that are specifically expressed in specific parts of the plant (see Table 3 and Figure 3). Figure 3A shows the number of MS runs for the different types of plant samples. The vast majority of MS runs (61%) were done on the major green tissues, including whole rosettes, specific leaf stages, cauline leaves, stems, and petioles. Fourteen percent of the MS runs were done on whole siliques, seeds at different developmental stages, or embryos isolated from seeds. Root samples (tips, whole roots, or even root exudates) were analyzed in 7.4% of the MS runs, whereas whole flowers or specific flower parts were used in 5% of the MS runs. Cell cultures were used in 6.6% of the MS runs. Finally, a smaller number of MS runs (0.5%–1.4%) were from hypocotyls, callus, pollen, cotyledons, or young seedlings (including roots, cotyledons, and a few leaves). For most of these MS runs, there was no further subcellular fractionation, and the proteome was either extracted in the presence of the strong ionic detergent SDS or in the absence of detergent, resulting in the extracted total cellular proteome including membrane proteins or just the soluble proteome, respectively. However, for nearly 20% of the MS runs, subcellular fractions were isolated from the plant parts, in particular isolated chloroplasts or sub-chloroplast compartments (thylakoids, stroma, envelopes, nucleoids, or plastoglobules), but also mitochondrial fractions (mostly ribosomes; Figure 3B). Other subcellular fractions included cytosolic lipid droplets, cytosolic stress granules, root exudate, and enriched plasmodesmata fractions (Figure 3B). There was a relatively high number of chloroplast samples because the proteomes of chloroplasts have been the subject of many of the PXDs over the last 10 years (Figure 2C), and also due to our own expertise and interest in chloroplasts.
Figure 3.

Key features of the samples used for the raw files (MS runs) for the 52 selected PXDs for the first Arabidopsis PeptideAtlas build. The count is based on the number of MS runs (raw files) for each part. A, Arabidopsis plant parts—hypocotyl, callus, pollen, cotyledon, flower parts (sepal/petal/carpel/stamen/pedicle, seed/septum/embryo), seedling (all parts of a young plant—root-hypocotyl/cotyledons/few young leaves, mostly collected from plates or liquid culture), root (tip/exudate/zone), cell culture, rosette parts (rosette/leaf/petiole/cauline leaf/senescing leaf/stem/internode). B, Arabidopsis subcellular fractions specifically analyzed are stress granule, mitochondrion, plasmodesmata, root exudate, cytosolic lipid droplet, chloroplast (black), and the specific fractions thylakoid (orange), plastoglobuli (blue). C, MS runs of samples that were specifically prepared to analyze PTMs (phosphorylation, acetylation of the N-terminus, and/or lysine) or to determine the physiological N-terminus using N-terminome enrichment techniques (TAILS, COFRADIC, or ChaFRADIC).
To support recognition and annotation of the N-termini of mature proteins (including after maturation processes such as cleavage of signal Peptides [sPs]), we selected several PXDs in which specific N-terminal labeling and enrichment techniques (TAILS [Marino et al., 2015]; COFRADIC [Staes et al., 2011]) were used to identify the N-termini of accumulated proteins, protein-derived signaling peptides, or protein degradation products (Figure 3C). Finally, the set of PXDs also included the most widely studied PTMs, i.e. phosphorylation and N-terminal or lysine acetylation (Figure 3C). Future Arabidopsis PeptideAtlas builds will aim to complement the current set of PXDs (see “The next Arabidopsis PeptideAtlas build”).
The identified proteome in the first PeptideAtlas build and MS support
Unless one uses “de novo” annotation, MS data can only lead to the identification of peptides and proteins by searching these MS data against an assembly of predicted, putative proteins. Proteins or peptides not represented in this protein search space cannot be identified. “De novo” annotation is in principle possible, and various software programs have been published (reviewed in Vitorino et al., 2020). However, it is hard to judge the quality of such searches; searching the different Arabidopsis genome annotations complemented with other speculative sequences is more efficient. Therefore, we assembled a comprehensive set of sequences from a variety of key sources (Table 1). These include Araport11 (the most recent [2017] annotation of the Arabidopsis genome), TAIR10 as the precursor of Araport11, RefSeq, and UniProtKB, a large collection of putative and speculative peptides encoded by sORFs (assembled in ARA-PEP; Hazarika et al., 2017), as well as a collection of highly expressed putative orphan ORFs from E. Wurtele (Iowa State). This set included a total of 204,154 sequence identifiers with significant redundancy and smaller numbers of unique proteins for each source (see Table 1); overall, these represented 73,816 unique amino acid sequences. After downloading PXD raw MS files, file conversions, and sample annotations, the MS data were searched against this total protein space (see “Materials and methods”). We searched in several iterations to optimize the search parameters (mostly variable and fixed PTMs) and search time. In particular, PXDs involving stable isotope dimethylation for N-terminomics and lysine acetylation (see Table 3) required particular attention, because these can lead to different mass shifts depending on the isotopes employed (+28 [2xC12H3] for light; +32 [2xC12HD2] or +34 [2xC12D3 or C13HD2] for heavy). Also, the use of TMT or iTRAQ labeling used for multiplexing and comparative proteomics required careful attention and verification of metadata.
The finalized searches and post-search processing for control of FDRs resulted in the matching of nearly 40 million out of approximately 143 million submitted MS/MS spectra, leading to the identification of 535,340 distinct peptides matching to 17,858 canonical proteins, as well as 1,942 uncertain and 1,600 redundant proteins for which identification is ambiguous due to shared peptides or lower evidence levels (http://www.peptideatlas.org/builds/arabidopsis/). For the remaining 6,255 proteins in Araport11, there were no observed matching peptides (Table 3; Supplemental Data Set S2). The overall match rate of MS/MS spectra to peptides was 28%, but this match rate varied dramatically across PXDs, from 2% to 74% (Table 3), with an average and median match rate of 26% and 22%, respectively. For those PXDs where we obtained a low match rate, we re-evaluated the search parameters to ensure that we did not overlook specific sample treatments that could affect the optimal search parameters (e.g. labeling techniques). The low match rate (<10%) was in most cases observed for N-terminomics and acetylation (N-terminal and lysine) studies involving dimethyl-labeling possibly combined with TAILS or COFRADIC/ChaFRADIC and in other cases involving affinity purification with a specific bait or analysis of the secreted peptidome from roots (Table 3). Other explanations for variations in match rate are often related to the acquisition parameters, in particular low thresholds for MS/MS acquisition and/or the lack of repeat MS/MS scans, resulting in low-quality MS/MS spectra. We did not detect an obvious relationship between MS/MS match rate and instrument type across the PXDs.
Figure 4 shows the number of distinct (nonredundant) peptides (irrespective of PTMs; Figure 4A) and distinct identified canonical proteins (Figure 4B) as a function of the cumulative number of matched MS/MS spectra ordered by PXD identifier (from low to high or old to new) for the first Arabidopsis PeptideAtlas. To better understand the underlying data for this PeptideAtlas build, we calculated the frequency distributions of peptide charge state, missed cleavages, and peptide length for the ∼40 million matched MS/MS spectra (Figure 5). The vast majority of matched spectra had a charge state of +2 (60%), +3 (34%), or 4+ (5.6%) and minor amounts of 1+ (0.09%), 5+ (0.71%), or 6+ (0.08%; Figure 5A). The majority of matched tryptic PSMs (77%) did not have a missed cleavage, whereas 20%, 3%, and 0.1% had 1, 2, or 3 missed cleavages, respectively (Figure 5B). Allowing for missed cleavages can potentially increase the false peptide discovery rate because it increases the peptide search space, but it is not uncommon that missed cleavages occur, and it does allow for increased sequence coverage and detection of N- and C-termini and splice junctions. We observed a wide range of matched peptide lengths, with 7 aa being the shortest sequence allowed (Figure 5C). 99% of all matched peptides were between 7 and 35 aa long, with the most frequent peptide length of 12 aa.
Figure 4.

Number of distinct (non-redundant) peptides (left panel) and identified canonical proteins (right panel) as a function of the cumulative number of PSMs (peptide-spectrum matches) for the first Arabidopsis PeptideAtlas. The cumulative count is ordered by PXD identifier (from low to high or old to new). The build is based on 266 experiments across the 52 selected PXDs, where each PXD may be decomposed into several experiments/samples (when such information can be determined). The PSM FDR is 0.0001. A, Number of distinct (non-redundant) peptides as a function of the cumulative number of MS/MS spectra matched. 535,000 distinct peptides are identified at a peptide-level FDR of 0.001. Areas in blue indicate the total number of distinct peptides in each experiment, whereas areas in red indicate the cumulative number of identified peptides from the current and previous experiments. B, Number of distinct (non-redundant) canonical proteins as a function of the cumulative number of MS/MS spectra matched at a canonical protein-level FDR of 0.0005. Areas in blue indicate the total number of distinct canonical proteins in each experiment, whereas area in red indicates the cumulative number of identified canonical proteins from the current and previous experiments.
Figure 5.

Key statistics of matched MS/MS data for this PeptideAtlas build. A, Frequency distribution of peptide charge state (z). B, Frequency distribution of missed cleavages for tryptic peptides. Note that when R or K is followed by P, trypsin does not cleave and hence these are not counted towards missed cleavages. Values for 3 and 4 missed cleavages are 0.11% and 0.02%, respectively. C, Frequency distribution of peptide length (aa).
Mapping the Araport11 proteome and splice forms
Because the Araport11 annotation is the most common reference used by the Arabidopsis community compared to TAIR10, RefSeq, and UniProtKB, the default protein identifier for sets of identical protein sequences (across all sources) was always from Araport11. Araport11 has 27,655 protein-coding genes with 48,359 gene model or transcript isoforms (Cheng et al., 2017), representing 40,784 unique protein amino acid sequences; it should be noted that the difference between transcript isoforms for a gene are often very minor at the amino acid level. For comparison, TAIR10 has 27,416 genes and 35,386 transcript isoforms, representing 32,785 unique proteins; 1651 protein sequences are found TAIR10 but not in Araport11 (at 100% sequence identity; Table 1). The vast majority of peptide sequences (>99%) in this first build matched to proteins in Araport11 (Table 4) with the remainder matching to sequences in one or more of the other sources (Table 5). We assigned multiple confidence levels of protein identification using a sophisticated tiered system (with 10 tiers) similar to that developed for the human PeptideAtlas (Deutsch et al., 2016a; Table 2, panel A). These 10 tiers allowed us to precisely distinguish different evidence levels of protein identification, including the use of peptides that are matched to multiple proteins (see “Materials and methods”). Figure 6A shows a schematic explanation for the tier system, and Figure 6, B–D provides specific examples from this PeptideAtlas build. These 10-tier assignments were then also condensed in a simplified classification of proteins identified using just four categories (Table 2, panel B) to provide a simpler overview of the identified proteome. The overall number of identified proteins for both classification systems is displayed in the PeptideAtlas browser. (http://www.peptideatlas.org/builds/arabidopsis/). The tiered system allowed us to capture matched peptides even if by themselves these peptides did not confidently identify a protein; thus the tier systems prevents the loss of any valuable MS/MS spectra.
Table 4.
Proteins identified in Araport11 for each of the four confidence categories by nuclear chromosome (1–5), mitochondrial (M), and plastid chromosome (C).
| Chromosome | Entries | Canonical, n (%) | Uncertain, n (%) | Redundant, n (%) | Not observed, n (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| M | 122 | 15 | 12.3 | 10 | 8.2 | 17 | 13.9 | 80 | 65.6 |
| C | 88 | 59 | 67.0 | 15 | 17.0 | 7 | 8.0 | 7 | 8.0 |
| 1 | 7,156 | 4,622 | 64.6 | 545 | 7.6 | 397 | 5.5 | 1,592 | 22.2 |
| 2 | 4,317 | 2,695 | 62.4 | 291 | 6.8 | 247 | 5.7 | 1,084 | 25.1 |
| 3 | 5,460 | 3,561 | 65.2 | 365 | 6.7 | 308 | 5.6 | 1,226 | 22.5 |
| 4 | 4,180 | 2,723 | 65.1 | 306 | 7.3 | 230 | 5.5 | 921 | 22.0 |
| 5 | 6,332 | 4,183 | 66.1 | 410 | 6.5 | 394 | 6.2 | 1,345 | 21.2 |
| Total | 27,655 | 17,858 | 64.6 | 1,942 | 7.0 | 1,600 | 5.8 | 6,255 | 22.6 |
Table 5.
Peptides and proteins not identified in Araport11 but identified in one of the other Arabidopsis sources.
| Hierarchy | Primary Protein Match | No. of peptides | Total Peptide Frequency | No. of Primary Proteins | No. of Peptides (≥3 Observations) | Total Peptide Frequency | No. of Primary Proteins | No. of Primary Proteins (≥ 2 Distinct Peptides; Each ≥3x) |
|---|---|---|---|---|---|---|---|---|
| 1 | Araport11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | TAIR10 | 409 | 23,003 | 61 | 258 | 22,801 | 43 | 29 |
| 3 | UniProt | 526 | 53,928 | 78 | 343 | 43,682 | 60 | 49 |
| 4 | RefSeq | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | LW | 73 | 285 | 54 | 19 | 222 | 10 | 2 |
| 6 | SIPs | 4 | 11 | 4 | 1 | 8 | 1 | 0 |
| 7 | sORFs | 46 | 206 | 33 | 19 | 175 | 13 | 4 |
| 8 | Iowa | 351 | 7,390 | 109 | 188 | 7,179 | 50 | 22 |
| Total | 1409 | 84,823 | 339 | 828 | 74,067 | 177 | 106 |
Figure 6.

Explanation and examples of the tiered identification system. A, Schematic depiction of the tiered protein identification system. Protein sequences are represented by a simple line, and identified peptides (PSMs) are shown as filled rectangles of different colors. Peptides contributing to identification at the highest confidence level (tier 1— canonical) are shown in green (must be at least 9 aa). Peptides of seven or more amino acids are shown in blue. Peptides of 7 or 8 aa are shown in amber. Peptides of fewer than 7 aa are never considered for protein identification and are shown in black. Also, any PSMs below a minimum build threshold of 0.001 PSM-level FDR are shown in black. This panel shows eight scenarios where either a single protein or a group of proteins is identified. B, This panel shows a case where three proteins were identified in a group. Two identical proteins, AT2G32180 (PTAC8) and AT2G32650 (PTAC18-like), were identified as having nine distinct peptides. Because these proteins are identical in sequence, one cannot distinguish them; one was designated as the indistinguishable representative (tier 2) and the other as indistinguishable (tier 7). A third protein with partial sequence identity, AT2G27402, was identified by a subset of these distinct peptides and was therefore assigned to tier 8 (subsumed) because this protein is not needed to explain these PSMs. An amino acid alignment between PTAC8/PTAC8-Like and AT2G27402 shows the residues that were part of the identified peptides (boxed in red). C, This panel shows an example of a tier 5 identification (weak), i.e. AT1G27330.1. This is a small RAMP4 (68 aa) with one predicted transmembrane domain in the C-terminal portion, a positive GRAVY index (0.034), and three nested or overlapping peptides, each identified multiple times across several independent PXD datasets and publications. Moreover, the N-terminal region contains eight closely spaced lysine and arginine residues, which would generate very short (3–5 aa) peptides that are too small to be considered as supported evidence by MS/MS. D, Figure 6D shows an example of a tier 6 identification, i.e. a steroid dehydrogenase (ATDET2/DWARF6; AT2G38050.1) involved in the brassinolide biosynthetic pathway. It has five or six predicted transmembrane domains and a positive GRAVY index of 0.132. This protein was identified in two publications across some 40 different sample types with a 9 aa N-terminal peptide (just downstream of a hydrophobic region) and an 8 aa C-terminal peptide.
As described in detail in the “Materials and methods” section, we included several fixed and variable PTMs for all datasets, in addition to several enriched PTMs (e.g. phosphorylation, isotope labels) that were only applicable to specific PXDs. There are hundreds of possible PTMs (see www.unimod.org), both physiological (i.e. introduced in the cell) and chemically induced during protein sample preparation and analysis. Indeed, tolerant database searches (i.e. allowing for many mass modifications) showed that extracted proteomes contain many peptides that are typically unaccounted for because they contain PTMs that are not searched (Zybailov et al., 2009; Chick et al., 2015; Kong et al., 2017). Strikingly, mass modifications that are observed vary widely among different datasets. Adding more PTMs adds extra search space, allowing more MS/MS spectra to be matched but also affecting the FDR, whereas reducing the number of PTMs will result in lower sequence coverage and lower MS/MS match rates. Increasing the number of variable PTMs does also increase computational needs; we therefore empirically determined a reasonable balance between searching the most frequent PTMs and keeping computational needs practical. We therefore selected a subset of mass modifications that fit within the computational resources that we had available. Importantly, we analyzed all datasets consistently using those parameters, and the FDR was controlled by including decoy sequences.
At the highest level of confidence are the canonical proteins (Table 2, panel A and B): we identified 17,857 canonical proteins in Araport11 (Table 4; Supplemental Data Set S2). These canonical proteins have at least two uniquely mapped non-nested peptides of at least nine residues (Figure 6A). This is a very high standard of identification and follows the HPP guidelines (Deutsch et al., 2019). The empirically determined FDR was 0.0004 for this highest confidence tier (corresponding to only seven false positives across these 17,857 proteins). We note that if gene loci were represented by different protein isoforms (gene models), we assigned one isoform as the canonical protein and did not further count the other isoforms, unless there was a uniquely mapped peptide to the alternate protein model. Unless a higher isoform number (gene model) received stronger MS support, isoform #1 was selected. In 878 cases, the canonical protein was an isoform with a higher model number (653 for .2; 99 for .3; 25 for .4; 10 for .5; no identification of isoform .6 or higher was observed even for genes that have up to 27 isoforms!). Inspection of these genes for which a higher isoform number was the canonical form showed a range of scenarios that explain the specific identification of the alternative isoform instead of the default .1. These included an extra N-terminal or C-terminal protein sequence or additional internal exon due to different splicing. Most isoforms are very similar or even identical at the protein level, and in many cases it was very hard or even impossible to distinguish between protein models based on MS/MS data.
We also identified 1,943 Araport11 proteins in the “uncertain” category encompassing tiers 2–7 (Supplemental Data Set S2). These proteins have too few uniquely mapping peptides of ≥9 aa to qualify for canonical status and may also have one or more shared peptides with other proteins. We identified 1,600 Araport11 proteins assigned to the “redundant” category encompassing tiers 8 and 9 (Supplemental Data Set S2). These proteins have only peptides that can also be assigned to other entries and thus these proteins are not needed to explain the observed peptide evidence. The overall protein FDR across all identified proteins in all 10 tiers is 0.03. This strategy allows the user to select their tolerance for error and use different subsets of proteins based on that, anywhere from 0.0004 to 0.03. This strategy is a great strength of PeptideAtlas. There is a tradeoff between the sensitivity and specificity of detection. As the FDR decreases, the overall sensitivity decreases as well; the effort to keep the false positives down comes at the expense of discarding correct identifiers that are mixed in with false identifiers. We do note that confidence thresholds in general are somewhat arbitrary, and their preference varies among different laboratories; it also depends on the purpose of the proteome analysis. For example, the HPP has opted for 1% FDR at the protein level (Deutsch et al., 2019).
Finally, there were 6,255 (6,255/27,655 = 22.6%) predicted proteins in Araport11, quite evenly distributed across the five nuclear chromosomes, for which we did not observe any peptides above our minimum PSM significance threshold (“not observed” or tier 10; Table 4;Supplemental Data Set S2). Some of these “not observed” proteins may have low significance PSMs but these are not considered as evidence for identification for PeptideAtlas. To better understand the nature of these unobserved proteins, we will compare the physicochemical properties and functions of these unobserved proteins and compare them with the canonical proteins below. In the remainder of the current section, we will show examples of identification of Araport11 proteins in the “uncertain” category (tiers 2–8; Figure 6, B–D).
Within tier 2 (indistinguishable representative), we identified at a high level of confidence 27 groups of different proteins (each with unique primary sequences within the group) but for which all group members were identified based on the same set of shared peptides (Figure 6A). At least some of the group members must have been detected, but it is not possible to determine which ones based on the detected peptides. In most cases, members of these groups share significant sequence identity/similarity, and they often have similar types of functions. One protein was selected as the representative for each group and was placed into tier 2 and the others in tier 7. An example of this scenario is the plastid-localized family of nucleoid-interacting proteins PTAC18 (AT2G32180) (selected as the indistinguishable representative in tier 2), PTAC18-like (AT2G32650; selected as an entry in tier 7), and AT2G27402 (tier 8—subsumed), as shown in Figure 6B. PTAC18 and PTAC18-like differ by only 2 aa in their protein sequences (16 kDa, 139 aa), whereas AT2G27402 is a much smaller protein (6 kDa) with high sequence identity to PTAC18. A set of overlapping and/or nested peptides matched to an N-terminal region in all three proteins, whereas several other peptides matched to PTAC8/PTAC8-like only but they did not cover their slight differences.
Tier 3, with 309 groups of different proteins identified (each protein with unique primary sequences) is similar to tier 2, but here the situation was more complex, with group members sharing one or more matched peptides and none has uniquely mapping peptides (Figure 6A). Again, one representative member of each group was selected and assigned to tier 3 and the other group members were assigned to tier 7. A total of 576 groups belonging to the tier 4 “Marginally distinguished” were identified. Proteins in this category share several peptides with a canonical protein, but also have one uniquely mapping peptide of ≥9 residues (Figure 6A). Exploring tier 4, we noticed that in many cases, the uniquely mapping peptide differed by a single amino acid change to a mapped peptide of the canonical protein in the same group. Consequently, this required careful inspection of the underlaying MS/MS spectra, paying particular attention of the coverage by b and y ions of the key peptides.
A total of 978 proteins (tier 5 “Weak”) were identified that had at least one uniquely mapping peptide of ≥9 residues but that did not meet the criteria for canonical (Figure 6A). Figure 6C shows the example of AT1G27330.1 such as a tier 5 identification. This is a small Ribosome-Associated Membrane Protein (RAMP4; 68 aa) with one predicted transmembrane domain in the C-terminal portion, a positive GRAVY index (0.034), and three nested or overlapping peptides, each identified multiple times across several independent PXD datasets and publications. Moreover, the N-terminal region contains eight closely spaced lysine and arginine residues, which would generate very short (3–5 aa) peptides that are too small to be considered as supporting evidence by MS/MS. Therefore, whereas this protein was not considered to be a canonical identification (tier 1), instead representing only a tier 5 identification, this constitutes a rather solid identification. We do note that most other identified proteins in this category only have a single distinct peptide (sometimes called “one hit wonders”, see Cottingham, 2009) and are therefore typically less reliable (even if the peptide was identified multiple times).
Fifty-three Araport11 proteins were identified in tier 6 (Insufficient evidence). These proteins have one or more uniquely mapping peptides, but none reach nine residues in length (Figure 6A). We note that most MS-based studies allow peptides as short as 7 aa to be used for protein identification, but shorter peptides are generally not considered. Hence, the 9 aa criterium applied here is relatively stringent. Figure 6D shows a tier 6 example of a steroid dehydrogenase (ATDET2/DWARF6; AT2G38050.1) involved in the brassinolide biosynthetic pathway. It has five or six predicted transmembrane domains and a positive GRAVY index of 0.132. This protein was identified in two publications across some 40 different sample types with a 9 aa N-terminal peptide (just downstream of a short hydrophobic region, perhaps comprising part of the sP) and one 8 aa C-terminal peptide. Whereas this was not a canonical identification (since both of the peptides were only 8 aa long), this appears to be a fairy robust identification, particularly considering that most of the protein does not yield suitable tryptic peptides for MS/MS analysis. Nearly, all other identifications in this tier 6 are based on a single distinct peptide are therefore potentially less reliable (“one-hit wonders” as in tier 5). However, several recent large-scale papers aiming to obtain a deep coverage of cellular proteomes provide experimental support (e.g. by MRMs or PRMs) that these so-called ‘one-hit-wonders’ can represent true identifications (Chen et al., 2014; Vandenbrouck et al., 2016). Therefore, Arabidopsis proteins identified in tiers 5 and 6 are valuable for expanding proteome coverage but require close manual scrutiny before being used as experimental support.
Sixty-nine proteins (tier 7—indistinguishable) and 1,388 proteins (tier 8—subsumed) were identified based on one or more matched peptides. However, none of these peptides were uniquely mapped, and none of these proteins were selected to be the representative of a group of identified proteins (see Figure 6A). Finally, 143 proteins were assigned to tier 9; a protein in this tier has an identical protein sequence to another one (Figure 6A), and this one is effectively removed from category competition, meaning that its partner can achieve a higher status (such as canonical) since it is not competing with identical sequences for uniqueness mapping.
Like all other plants, Arabidopsis has a small plastid genome and mitochondrial genome. Most sources recognize 88 protein-coding genes in the Arabidopsis plastid genome, with the initial sequence reported in Sato et al. (1999), and typically 33 protein-coding genes on the mitochondrial genome (Sloan et al., 2018). To our surprise (realized at the last stage of completing this first build), Araport11 (and also TAIR10) includes 122 predicted mitochondrial-encoded proteins (with identifiers starting with ATMG). Comparison of these 122 protein sequences with the recently updated sequences (33 in total) from Sloan et al. (2018) shows that only a subset does match. Several plastid- and many mitochondrial-encoded mRNAs undergo mRNA editing and/or trans-splicing, which can affect the resulting protein sequence, thus increasing the protein search space (Takenaka et al., 2013; Germain et al., 2015; Fuchs et al., 2020; Small et al., 2020). We have reached out to members of the plant community for input and advice on how to obtain the most complete set of possible organelle-encoded proteins, including their unedited and edited variants. We will revisit protein accumulation, including partial editing and possible tissue specificity, of these organelle-encoded proteins in a follow-up study. In the current build, a total 59 and 15 Araport11 plastid- and mitochondrial proteins, respectively, were identified at the highest confidence level (“canonical”; Table 4). For seven plastid and 80 mitochondrial predicted proteins, we did not observe any matched MS/MS spectra.
Identification and discovery of proteins not represented in Araport11
We identified 1,408 peptide sequences (length at least 7 aa; irrespective of PTM or charge state) that did not match to Araport11 protein sequences but instead matched to predicted amino acid sequences in one or more of the other Arabidopsis protein sources listed in Table 1 (Supplemental Data Set S3A). The number of observations of these peptides ranged from 1 (408 peptides) to 8,854. Figure 7 shows a frequency distribution for the number of peptide observations (PSMs; Table 5; Supplemental Data Set S3, B–E). When we removed peptides only observed once or twice and requiring at least two unique peptide sequences to further reduce false discovery, the number of observed protein identifiers was reduced to 106 (Table 5). It should be noted that that we applied a strict hierarchy to assign peptides to protein sequences from these additional sources. That is, even if a peptide was matched to a protein sequence in more than one source, the peptide was assigned to the sequence in the most highly ranked source (for ranking see Table 5). For instance, a peptide matched to a sequence in TAIR10 would not be used again to report a sequence in UniprotKB. In the next sections, we explore the significance for some of these 106 protein sequences.
Figure 7.
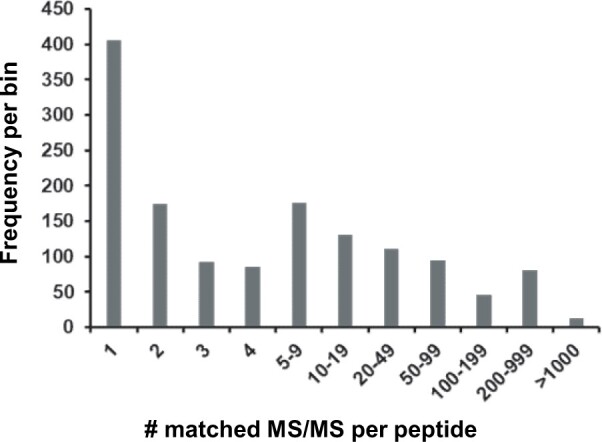
Frequency of observation for peptides not matching to Araport11 entries but matching to other Arabidopsis protein sources, including TAIR10, UniProt, sORFs, and other sources.
Proteins identified in TAIR10 and absent in Araport11
There are 32,785 distinct predicted protein sequences in TAIR10 (represented by 35,386 gene models), 1,651 of which do not have 100% identical protein sequences in Araport11. As indicated in Table 5, we identified uniquely mapping peptides for 61 proteins in TAIR10 that could not map to Araport11, and the number of unique matched peptide sequences per protein ranged from 1 to 172 (Supplemental Data Set S3B). Forty-three proteins contained at least one distinct peptide that was identified all least three times by MS/MS, and 29 proteins contained at least two unique peptide sequences that were identified at least three times (Table 5). We compared these 29 TAIR10 genes to Araport11 genes and observed 5 different scenarios that explain the peptides that were uniquely identified for TAIR10 proteins: (1) the gene was removed from Araport11 and there was no protein-coding gene in this chromosomal region (five genes, see example in Figure 8A); (2) the gene contained an alternative START sites; in all cases, the Araport11 protein was shorter (five genes); (3) the gene contained an alternative STOP site; in all cases, the Araport11 protein was shorter (three genes, see example in Figure 8B); (4) there was a mismatch within an exon (three genes), (4) different splicing events occurred due to either a change in the length of the exon or the addition or removal of an exon; in all cases, there was also a change in the START and/or STOP codon (11 genes, see example Figure 8C); and (6) finally, in two cases, the TAIR10 protein was mitochondria-encoded. Table 6 summarizes the findings for these 27 nucleus-encoded TAIR10 proteins, which should be considered for future Arabidopsis genome annotations. Figure 8, A–C shows examples by comparing the TAIR10 and Araport11 chromosomal regions, assigned gene models, and uniquely matched peptides for the TAIR10 entry.
Figure 8.

Examples of identified proteins not captured well in Araport11 but detected in TAIR10. For a complete list, see Supplemental Data Set S3, A and B and Table 6. A, AT3G12012.1 was identified in TAIR10 but is not annotated as a protein-coding gene in Araport11. The predicted protein sequence is shown with the identified residues marked in orange (left side). The specific peptides identified by MS/MS and their frequency of observation are shown (middle). The right-hand panel shows the predicted gene structure with three exons in the TAIR10 annotation. This short gene is positioned within the 5′UTR of the protein-coding gene AT3G12010.1 and is likely an expressed uORF with unknown function. AT3G12010.1 (annotated as C18orf8; 782 aa) is identical in TAIR10 and Araport11 and was identified at the canonical level (59% sequence coverage). B, Alternative protein model AT2G38255.1 (unknown protein with DUF239) with an extended C-terminus in TAIR10 exhibiting multiple detected peptides not found in the shorter Araport11 entry. This was due to an alternative STOP codon combined with a change in splicing. Consequently, AT2G38255 has seven exons in TAIR10 but six exons in Araport11; exon 7 is missing in Araport11 and exon 6 partially differs between TAIR10 and Araport11. The protein sequence alignment shows that the C-terminal region of the TAIR10 (333 aa) and Araport11 (218 aa) proteins has 218 residues; the two sequences are identical until residue number 208. Five distinct peptides match to shared regions of the TAIR10 and Araport11 entries—these are SQIWLENGPR, TGCYNTNCPGFVIISR, LTIYWTADGYK, GELNSIQFGWAVHPR, LYGDTLTR (see PeptideAtlas for details). C, Detection of TAIR10 version of AT3G52130.1 (non-Type III lipid transfer protein), with no detection of the completely different sequence for AAT3G52130.1 in Araport11. This was due to alternative START and STOP codons combined with a change in splicing; the coding frames between the two genes are different. Consequently, in the case of Araport11, the N-terminal residue is a lysine and not a methionine. mRNA accumulation is limited to young flower buds, as displayed in BAR ePlant (yellow → red scale reflects low to high expression values). The primary sequences for both proteins are: TAIR10_AT3G52130.1 (125 aa): MMMKAMRVGLAMTLLMTITVLTIVAAQQEGLQQPPPPPMLPEEEVGGCSRTFFSALVQLIPCRAAVAPFSPIPPTEICCSAVVTLGRPCLCLLANGPPLSGIDRSMALQLPQRCSANFPPCDIIN Araport11_AT3G52130.1 (123 aa): RSKRACNNHLHHQCCPRRKWEDAAGHFSPRWYSSYHVEQQLLLLARSHRPRYVALPLSHLVVLVFASLPMDLHSLALTAPWLFSSLRDALLISLPAISSTRKDISSFFSFLFSFTFLFNNLAA.
Table 6.
Twenty-seven nucleus-encoded TAIR10 proteins not identified in Araport11 based on at least two distinct peptides supported by at least three MS/MS spectra
| TAIR10 | No. of Unique Matched Peptide Sequences (Includes also Peptides Observed Only 1x or 2x) | No. of Total Frequency of Observation of Matched Peptides | Protein Name | Change in Araport11 as Compared to TAIR10 | Comments | No. of Gene Models in TAIR10 | No. of Gene Models in Araport11 |
|---|---|---|---|---|---|---|---|
| AT1G14070.1 | 12 | 28 | Xyloglucan fucosyltransferase | Removed | 2 exons; very strong support 2nd exon | 1 | N/A |
| AT2G33430.1 | 15 | 1,339 | MORF2 (also DAG protein) – editing factor | Removed | Very strong peptide support for 3 of the 3 exons | 1 | N/A |
| AT3G12012.1 (Figure 8A) | 5 | 39 | Conserved peptide upstream ORF20 | Removed | 3 exons; strong support for 2 exons | 1 | 0 |
| AT4G18120.1 | 22 | 202 | MEI2-like 2 | Removed | Very strong peptide support for 7 of the 10 exons. How are model .1 and .2 different (TAIR10)? | 2 | N/A |
| AT5G25752.1 | 7 | 235 | Rhomboid protease 11/9 (RBL11/9) | Removed | 7 exons (TAIR10). 5 peptides for first exon. One peptide for last exon | 1 | N/A |
| AT1G16780.1 | 3 | 23 | Inorganic H pyrophosphatase | ATG shifted. Araport11 is shorter | 15 (TAIR10) or 14 exons (Ara11). All Araport models start later | 1 | 3 |
| AT5G18280.2 | 3 | 478 | Apyrase 1 | ATG shifted. Araport11 is shorter | 9 exons in model 1 and 11 in model 2 for TAIR10. 9 exons in Araport11. One extra exon in model .2 supported with one short peptide; 2 peptides for first exon | 2 | 1 |
| AT5G52640.1 | 4 | 51 | Heat shock protein (HSP81-1/HSP83) | ATG shifted. Araport11 is shorter | 4 exons in both TAIR10 and araport11; strong support for 1st exon | 1 | 1 |
| AT5G63980.1 | 4 | 54 | SAL1 (FIERY1), 3(2), 5-bisphosphate nucleotidase | ATG shifted. Araport11 is shorter | 7 exons in both TAIR10 and Araport11; 4 peptides for the 1st exon | 1 | 1 |
| AT5G18280.1 | 3 | 478 | Apyrase 2 | ATG. Araport11 is shorter | 9 exons in model 1 and 11 in model 2 for TAIR10. 9 exons in Araport11. One extra exon in model .2 supported with one short peptide; 2 peptides for first exon | 2 | 1 |
| AT1G03495.1 | 4 | 26 | HXXXD-type acyl-transferase | STOP. Araport11 is shorter | One big exon – clear case | 1 | 1 |
| AT1G79920.1 | 26 | 2,006 | Heat shock protein 70 (Hsp 70) | STOP. Araport11 is shorter | 9 exons in TAIR10; 8 exons in Arport11 | 2 | 4 |
| AT4G23000.1 | 5 | 16 | Calcineurin-like metallo-phosphoesterase | STOP. Araport11 is shorter | 15 exons in TAIR10, 14 exons in Araport11. several peptides for both extra exons | 1 | 2 |
| AT5G40780.2 | 3 | 21 | Lysine histidine transporter 1 | Splicing | 8 exons in TAIR10; 8 exons for model 1, but 7 for models 2 and 3 in Araport11 | 2 | 3 |
| AT3G57180.1 | 3 | 28 | BPG2-homolog of YqeH–GTPase | Mismatch–in one exon | 3 overlapping long peptides; sequencing difference? | 1 | 1 |
| AT4G16150.1 | 3 | 18 | Calmodulin-binding transcription activator 5 | Mismatch–in one exon; likely related to a gene duplication At3G16940 and AT4G16150 | 13 exons in both TAIR10 and Araport11. sequencing mismatch in 11th exon | 1 | 1 |
| AT1G52827.1 | 3 | 191 | Cadmium tolerance 1 | ATG+STOP. Big change | 3 nested peptides for exon 1; no peptides for exon 2 | 1 | 1 |
| AT3G52130.1 (Figure 8C) | 7 | 70 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin | ATG+STOP+splicing | One exon in TAIR10; 2 exons in Araport–but first new one very short | 1 | 1 |
| AT4G16770.1 | 3 | 66 | 2-Oxoglutarate (2OG) and Fe(II)-dependent oxygenase | ATG+STOP+splicing. Araport11 is shorter | 11 exons in TAIR10 and 8 in Araport11. 2 peptides for extra C-terminal exon; splice difference for exon 7 | 1 | 1 |
| AT5G39570.1 | 172 | 14,860 | Transmembrane protein | ATG+STOP+splicing. Araport11 is shorter | 2 exons in TAIR10; 2 or 3 exons in Araport11 | 1 | 2 |
| AT4G21326.1 | 3 | 12 | Subtilase SBT12 | ATG+splicing. Araport 11 is shorter | 8 exons in TAIR10, 7 exons in Araport11 | 1 | 1 |
| AT1G23580.1 | 10 | 49 | Transmembrane protein with DUF220 | ATG+splicing. Araport11 is shorter | 4 exons in TAIR10; 2 exons in Araport11 | 1 | 1 |
| AT2G38255.1 (Figure 8B) | 5 | 14 | Hypothetical protein (DUF239) | STOP+splicing | 7 exons (TAIR10); 6 exons in (Araport11). Strong support for extra C-term exon | 1 | 1 |
| AT1G17060.1 | 7 | 33 | Cytochrome p450 72c1 | STOP+splicing | 6 exons (TAIR10) or 4 exons (Araport11); Araport 11–antisense gene–overlapping protein coding–AT1G7065) | 1 | 1 |
| AT4G16144.1 | 5 | 46 | JAB1/Mov34/MPN/PAD-1 domain protein | STOP+splicing. Araport11 is shorter | 13 exons in TAIR10 and 13 in Araport11. 4 peptides in extra C-term exon, and 1 for splice junction | 1 | 1 |
| AT4G18260.1 | 11 | 103 | Cytochrome b561/ferric reductase | STOP+splicing. Araport11 is shorter | 7 exons in TAIR10, 4 exons in Araport11. All 3 extra C-terminal exons are supported with peptides | 1 | 1 |
| AT5G02370.1 | 2 | 11 | Kinesin motor protein-related | STOP+splicing. Araport11 is shorter | 1 exons in TAIR10 but 9 in Araport11; 2 nested peptides for the last exon | 1 | 1 |
TAIR10 AT3G12012 is represented by one gene model and has three exons (Figure 8A). The MS/MS data provide very strong support for exons 1 and 2, by two and three peptides respectively, but not for the very short exon 3, which encodes just three residues (GKA) immediately downstream of a lysine residue. This third and C-terminal exon can only be observed if there were two missed cleavage in the C-terminal region resulting in the peptide SCMQHTIRGKA. This annotated gene is not present in the Araport11 genome annotation, and the description in the TAIR database states that the gene is obsolete but previously it was assigned as a conserved upstream opening reading frame (uORF) named CPuORF20 in the 5′-untranslated region (UTR) of the protein-coding gene AT3G12010 (annotated as C18orf8 in TAIR – Araport11; Figure 8A). We identified AT3G12010.1 as a canonical protein with very high sequence coverage, and the TAIR10 and Araport11 protein sequences are identical. It appears that AT3G12010 is indeed a short uORF that is expressed as a stable small protein (6.7 kDa) with unknown function.
TAIR10 AT2G38255 (unknown protein with DUF239) has seven exons in TAIR10 but six exons in Araport11; exon 7 is missing in Araport11 and exon 6 partially differs between TAIR10 and Araport11 due to a splicing difference and the presence of a STOP codon in Araport11 (Figure 8B). As illustrated in the alignment, the TAIR10 protein has 333 residues and the Araport11 protein has 218 residues; the two sequences are identical until residue number 208. The MS/MS data strongly support exon 7 with four distinct peptide sequences and the alternate exon 6 from TAIR10 with one MS/MS spectrum. Five distinct peptides match to the N-terminal portion, which is identical across TAIR10 and Araport11 (see the legend of Figure 8B). mRNA expression levels appear to be very low (no values are reported in e.g. BAR ePlant (http://bar.utoronto.ca/) or the ATTED co-expression database (https://atted.jp/), which perhaps explains the incomplete annotation of its predicted protein sequence.
AT3G52130.1 was detected in TAIR10 with seven distinct peptides and a total of 70 MS/MS spectra, but not at all in Araport11 (Figure 8C). The predicted protein sequences in TAIR10 and Araport11 are completely different due alternative START and STOP codons combined with a change in splicing; the coding frames between the two genes are different. Consequently, in the case of Araport11, the N-terminal residue is a lysine and not a methionine. Both primary protein sequences are listed in the legend (Figure 8C). A study in 2013 demonstrated that AT3G52130 is a non-Type III lipid transfer protein with transcripts nearly exclusively present in the inflorescence (flower bud stage 9; Huang et al., 2013), as also shown in BAR ePlant (Figure 8C). The TAIR10 gene assignment appears correct.
Peptides matching to UniProtKB sequences
A total of 526 peptides did not match to Araport11 or TAIR10 protein-coding sequences but matched to 78 UniProtKB identifiers (Supplemental Data Set S3C; Table 5). Considering only peptides identified at least three times reduced this to 343 peptides matching to 60 UniprotKB identifiers. Increasing the stringency further by requiring at least two distinct peptides (each observed at least three times) reduced the number of identified UniProtKB sequences to 49. To investigate the nature and significance of these UniProtKB identifications, we performed BLAST analysis of all UniprotKB sequences against Araport11 protein-coding genes (pBlast), pseudogenes (tBlastn), and the genomic sequence (tBlastn) (Supplemental Data Set S3D). We then evaluated the 49 UniprotKB identifiers that passed the stringent criteria.
Four UniprotKB ids mapped each to a different plastid-encoded protein. In three cases (ACCD, cytb559-beta, and ClpP1), the unique peptides matched only to UniprotKB and not to Araport11 and always included an RNA edited site because the Araport11 sequences did not consider the resulting amino acid change, whereas the UniprotKB sequences were corrected for the edited site. The fourth case was for YCF3 and was due to a miss-assigned N-terminus in Araport11; instead of the correct 168 aa protein, the Araport11 sequence was only 126 aa long (the first 42 aa N-terminal residues were missing). Twenty-five UniProtKB identifications mapped to 19 Araport11 mitochondrial-encoded proteins, with 5 Araport11 proteins matching to two different UniProtKB entries. In most cases, this is due to the presence of unedited forms in Araport11 and the edited form in UniProtKB. As mentioned earlier, we will further investigate the coverage of the plastid- and mitochondrial-encoded proteome in a follow-up study. Twenty UniProtKB identifications mapped each to a nucleus-encoded protein in Araport11. In a handful of cases, these best mapped to a pseudogene in Araport11—but these are likely to be actual protein-coding genes (e.g. AT3G0875, AT4G18120, AT4G13900, AT4G204033, AT4G14610). In other cases, there was one more mismatch between the UniProtKB entry and the Araport11 protein, likely related to sequence annotation.
Figure 9 shows three examples of UnipProtKB identifications that were selected because the UniProtKB protein sequence matched with relatively high significance to a predicted pseudogene in Araport11 (based on TBlastN). The first example is Q9LHK4, which encodes a large 137 kDa protein (1,241 aa; Figure 9A). TBlastN identified Araport11 AT3G30875 as a strong match. AT3G30875 is annotated as a pseudogene, but the TAIR website notes that this is probably not a pseudogene based on evidence for transcription (RNA-seq) and translation (Ribo-seq) described in (Hsu et al., 2016). We identified six peptides that uniquely mapped to Q9LHK4, two of which were observed three times, and the others only once or twice. An additional three matched peptides were shared with other proteins in protein group AT4G17140, which is a protein with repeating coiled regions of VPS13. The second example is Q9SVV9, encoding an 85 kDa protein (759 aa) that is identical to TAIR10 AT2G18120, with the exception of two gaps in the amino acid sequence alignment. This protein (AML3) is a member of the MEI2-like gene family and is annotated as a pseudogene in Araport11. In situ hybridization detected expression during early embryo development but not in vegetative or floral apices (Kaur et al., 2006). The third example is P0CC32, which encodes a 57 kDa protein (512 aa) and maps to the pseudogene AT2G04033 with similarity to the defensin-like (DEFL) family. However, careful inspection of the results for P0CC32 in PeptideAtlas showed that the UniProtKB sequence is nearly identical to a much smaller (42 kDa) chloroplast-encoded NDHB/NDH1 protein (ATCG00980) with the exception of an N-terminal region of 123 aa. P0CC32 was identified as having two unique peptides that did not match to AT2G04033 because this protein is RNA edited, and these two peptides contain one or two editing sites resulting in amino acid changes. As mentioned earlier, the Araport11 sequences are the unedited form, whereas UniProtKB does incorporate these edits. Three additional peptides were identified for P0CC32, but these were all shared with AT2G0433.
Figure 9.

Examples of UniProtKB identifications selected from the 49 identifications based on at least two distinct peptides (not matched to predicted Araport11 proteins) that were observed at least three times. For a complete listing, see Supplemental Data Set S3D. The examples were selected because the UniProtKB protein sequence matched with relative high significance to a predicted pseudogene in Araport11 (based on TBlastN). A, Q9LHK4 encodes a large 137-kDa protein (1,241 aa). TBlastN identified Araport11 AT3G30875 as a strong match (1,215 aa alignment length and E = 0). AT3G30875 is annotated as a pseudogene (nine exons are shown in Jbrowse), but the TAIR website notes that this is probably not a pseudogene based on evidence for transcription (RNA-seq) and translation (Ribo-seq) described in Hsu et al. (2016). We identified six peptides (marked with red arrows) uniquely mapping to Q9LHK4 (sequences are shown), two of which were observed three times, and the others only once or twice. An additional three matched peptides were shared with other proteins in the protein group represented by AT4G17140.3, which is a protein with repeating coiled regions of VPS13. B, Q9SVV9 encodes an 85 kDa protein (759 aa) and is identical to TAIR10 AT4G18120.2, with the exception of two gaps in the amino acid sequence alignment. This protein (AML3) is encoded by a member of the MEI2-like gene family and is annotated as a pseudogene in Araporti1. In situ hybridization detected expression during early embryo development but not in vegetative or floral apices (Kaur et al., 2006). Two peptides uniquely map to Q9SVV9 as indicated by the red arrows. The other matched peptides are shared with the protein group represented by AT4G18120.2 as indicated. C, P0CC32 encodes a 57-kDa protein (512 aa) and maps to the pseudogene AT2G04033.1 with similarity to the defensin-like (DEFL) family. However, careful inspection of the results for P0CC32 in PeptideAtlas shows that the Uniprot sequences is nearly identical to a much smaller (42 kDa) chloroplast-encoded NDHB/NDH1 protein (ATCG00980.1), with the exception of an N-terminal region of 123 aa. P0CC32 was identified as having two unique peptides that did not match to AT2G04033.1 because this protein is RNA edited, and these two peptides contain one or two editing sites resulting into amino acid changes. As mentioned in the “Results and discussion”, the Araport11 sequences are in the unedited form, whereas UniProtKB does incorporate these edits. Three additional peptides were identified for P0CC32, but these were all shared with AT2G0433.1, as indicated.
Discovery of sORFs
Transcriptomics, including using Ribo-seq, combined with a range of in silico prediction and analysis tools have predicted large numbers of sORFs in the Arabidopsis genome that could result in the accumulation of small proteins or peptides (Hanada et al., 2007; Hsu et al., 2016; Hazarika et al., 2017; Hsu and Benfey, 2018; Takahashi et al., 2019; Kage et al., 2020). These sORFs have been found in intergenic regions, introns, embedded within non-coding RNAs (ncRNAs), directly upstream of coding sequences in the 5′-UTRs (uORFs), C-terminally encoded peptides (Roberts et al., 2013), or on the anti-sense strand, and some are induced by (a)biotic stresses, sometimes assigned as SIPs (Hazarika et al., 2017; Qi et al., 2020; Takahashi et al., 2020). Recent MS studies searched these ORF collections for Arabidopsis and identified matching peptides for a relatively low percentage of predicted proteins or peptides (Zhang et al., 2019; Mergner et al., 2020; Wang et al., 2020). The assignments within this collection are low weight (LWs) proteins, SIPs, and sORFs. As indicated in Table 5 and Supplemental Data Set S3D, we identified 54, 4, and 33 LWs, SIPs and sORFs based on 74, 4, and 46 peptides, respectively. When considering only peptides observed at least three times, this was reduced to 10, 1, and 13, LWs, SIPs, and sORFs, respectively. Upon increasing the stringency by requiring two distinct peptides, each observed at least three times, only four sORFs and two LWs remained. We investigated these six most stringent hits. Figure 10 shows the identification of these elements by displaying screenshots for their identifications in PeptideAtlas showing how the peptides map to the predicted protein sequence, and how they map to the Arabidopsis genome sequence.
Figure 10.

Examples of the identification of sORFs and one LW in the PeptideAtlas build. These six examples represent the identifications that pass the stringent criterium of having at least two matched distinct peptides that are each identified three times (for more information, see Supplemental Data Set S3D). A, CONTRIB_sORFs_sORF2808 encodes a 4.9 kDa peptide (42 aa) and was identified as having three distinct and nested peptides that map to a small portion of the pseudogene AT2G20724 in Araport11. The identified peptides were all identified from dimethyl labeling (modifying both N-terminal free amines and the lysine side-chain) and enrichment studies (using TAILS or COFRADIC) as indicated. The 4.9-kDa predicted protein has a very high number of lysine and arginine residues (total 13) and upon tryptic digestion would result in only a single peptide of 7 (N-terminal methionine removed) or 8 aa. Trypsin cannot cleave the peptidyl bond of dimethylated lysine residues, which enhances the chance to observe peptides GAAFEDQVKMR and GAAFEDQVKMRALE. All four PSMs of GAFEDQVK and one of the four PSMs of GAFEDQVKMR are dimethylated and the other three are iTRAQ8plex labeled. The single PSM of GAFEDQVKMRALE is iTRAQ8plex labeled. The results suggest that AT2G20724 is not a pseudogene but rather a protein-coding gene. B, Both CONTRIB_sORFs_sORF1912 and CONTRIB_sORFs_sORF1913 mapped to the same transposon (AT2TE05755/AT2G04000) in Araport11. AT2G0400 has two exons, and the two ORFs each represent one exon. The transposon belongs to the VANDAL21 family and the DNA/MuDR superfamily and its preferred substrate for the integration of VANDAL21 is euchromatin. VANDAL21 mainly targets promotors and 5′UTR of broadly active genes that are enriched in histone marks H3K4me3 and H3K36me3 (Quesneville, 2020). C, CONTRIB_sORFs_sORF7763 encodes a 11 kDa protein (96 aa) and was identified as having five peptides that all map to exon 2 of AT5G00690 in Araport11. However, Araport11 has not assigned this as a protein-coding gene but as a “novel transcribed region”. The results suggest that AT5G00690 should be annotated as a protein-coding gene. D, CONTRIB_LW_ath_mu_ch3_8568top encodes a 16-kDa protein (136 aa) and was identified as having six peptides, three of which mapped to AT3G02345, which is annotated as a long-non-coding RNA in Araport11. Most PSMs were identified in seeds and a few others in embryos or siliques (see PeptideAtlas). BlastP with the 136 aa sequence against Araport11 found that the closest match was AT2G23148 but with a very poor E-value (0.003). BlastP against all nr proteins identified ARALYDRAFT_897225 in the lyrate ecotype as the closest match (98/117 identities for the region 20–80 aa; 1E-72). The significance of the small protein remains to be determined. E, CONTRIB_LW_ath_mu_ch5_3086top encodes a 15.6-kDa protein (131 aa) and was identified as having nine peptides, none of which map to an annotated genome element in Araport11. However, BlastP against all nr proteins identified AT5G03740 in ecotype Landsberg as the perfect match. The peptides were identified in samples from flowers and flower parts (petals, pollen, sepals, stamen) as well as siliques in two studies (Zhang et al., 2019; Mergner et al., 2020), even though these studies used samples from ecotype Col-0.
CONTRIB_sORFs_sORF2808 encodes a 4.9 kDa protein (42 aa) and was identified as having three distinct and nested peptides that map to a small portion of the pseudogene AT2G20724 in Araport11 (Figure 10A). The observed peptides were all identified from dimethyl labeling (modifying both N-terminal free amines and the lysine side chain) and enrichment studies using TAILS or COFRADIC as indicated. The 4.9 kDa predicted protein has a very high number of lysine and arginine residues (total 13) and upon tryptic digestion would result in only a single peptide of 7 aa (N-terminal methionine removed) or 8 aa. Trypsin cannot cleave the peptidyl bond of dimethylated lysine residues, which enhances the chance to observe peptide GAAFEDQVKMR and GAAFEDQVKMRALE. The results suggest that AT2G20724 is not a pseudogene but rather a protein-coding gene.
Both CONTRIB_sORFs_sORF1912 and CONTRIB_sORFs_sORF1913 mapped to the same transposon in Araport11: AT2TE05755/AT2G04000 (Figure 10B). AT2G0400 has two exons, and the two ORFs each represent one exon. The transposon belongs to the VANDAL21 family and the DNA/MuDR superfamily and its preferred substrate for the integration of VANDAL21 is euchromatin. VANDAL21 mainly targets promotors and 5′-UTRs of broadly active genes, which are enriched in the histone marks H3K4me3 and H3K36me3 (Quesneville, 2020).
CONTRIB_sORFs_sORF7763 encodes an 11-kDa protein (96 aa) and was identified as having five peptides that all map to exon 2 of AT5G00690 in Araport11 (Figure 10C). However, Araport11 has not assigned this as a protein-coding gene but as a ‘novel transcribed region’. The results suggest that AT5G00690 should be annotated as a protein-coding gene.
CONTRIB_LW_ath_mu_ch3_8568top encodes a 16 kDa protein (136 aa) and was identified as having six peptides, three of which mapped to AT3G02345, which is annotated as a long-non-coding RNA in Araport11 (Figure 10D) Most PSMs were identified in seeds and a few others in embryos or siliques (see PeptideAtlas). BlastP with the 136 aa sequence against Araport11 found that the closest match was AT2G23148 but with very poor E-value (0.003). BlastP against all nonredundant proteins identified ARALYDRAFT_897225 in the A. lyrata species as the closest match (98/117 identities for the region 20–80 aa; 1E-72). The significance of this small protein remains to be determined.
CONTRIB_LW_ath_mu_ch5_3086top encodes a 15.6-kDa protein (131 aa), was identified as having nine peptides, and maps to a region of chromosome 5 without annotated features in Araport11 (Figure 10E). In fact, they map downstream of transposon AT5TE04420. BlastP against all nr proteins identified AT5G03740 in ecotype Landsberg as the perfect match. The peptides were identified in samples from flowers and flower parts (petals, pollen, sepals, stamens) as well as siliques in two studies (Zhang et al., 2019; Mergner et al., 2020), even if these studies used samples from ecotype Col-0. AT5G03740 in Araport11 or TAIR10 is a predicted protein-coding gene but with a predicted protein sequence that is unrelated to AT5G03740 in Landsberg.
Relative abundance, physicochemical properties, and subcellular locations of the canonical and unobserved Araport11 proteome
As described above and in Table 5, ∼65% of all predicted Araport11 proteins (counting one isoform per gene) were identified as canonical proteins. To better understand this canonical proteome, we analyzed the physicochemical properties and subcellular localizations of these canonical proteins and compared this information to the complete predicted Araport11 proteome (selecting one protein isoform per gene) as well as the unobserved (“dark”) proteome.
Physicochemical properties.
We calculated molecular weight (kDa), overall hydrophobicity based on the GRAVY index (positive and negative values are hydrophobic and hydrophilic, respectively), and pI for all full-length predicted, canonical, and unobserved in Araport11. The distribution of calculated properties for each group are displayed as histograms and violin plots, and mean, median, and mode values for molecular weight and GRAVY are also shown (Figure 11A). The canonical proteome had a higher mean and median molecular weight than the total proteome shifted by 7–9 kDa, whereas the mode dramatically increased by 22.5 kDa. In contrast, the unobserved proteome was strongly skewed toward proteins of lower molecular weight, as reflected by much lower values for mean, median, and mode (Figure 11A). However, only a few had one or no predicted full tryptic peptides, and most are theoretically amenable to detection by MS/MS following tryptic digestions. The canonical proteome showed a narrower distribution for GRAVY index values, lacking proteins with GRAVY >1 (very hydrophobic proteins) but also lacking the most hydrophilic proteins, such as a family of very basic and small ribosomal L41 homologs (AT1G56045, AT2G40205, AT3G08520, AT3G11120, AT3G56020) as well as a very small and acidic predicted replication factor (AT5G03710). The unobserved proteins with high GRAVY values are mostly low molecular weight proteins (<10 kDa) with one or two predicted transmembrane domains. Many of these low mass unobserved proteins have no known function, but a subset is well-known small thylakoid integral membrane proteins of photosystem II (e.g. psbZ, psbM, psbK, psbJ). The pI plots show a bimodal distribution, with relatively few proteins with a pI around 7.5, as is generally observed for many other cellular proteomes (Schwartz et al., 2001; Kiraga et al., 2007). The general explanation for this modality is that proteins are generally least soluble near their pIs and that the physiological pH within cells is typically around ∼7 to ∼7.5; hence proteins tend to be more soluble at acidic or basic pH values. The canonical proteome has a similar pI distribution to the total predicted proteome, but is somewhat enriched for low pI proteins, whereas the unobserved proteome has a broader pI distribution (Figure 11A). We conclude that pI per se is not a strong predictor for protein discovery by MS/MS.
Figure 11.

Distribution of the physicochemical properties and subcellular locations of the predicted (27,655 proteins), canonical (17,857 proteins), and unobserved (6,255 proteins) in the Araport11 proteome. A, Frequency distributions for size, GRAVY, and pI for the three proteomes shown as histograms and violin plots. The table shows mean, median, and mode (min–max bin value) of molecular weight and GRAVY for the three proteomes. B, Distribution of subcellular localizations of nucleus-encoded proteins in the three proteomes based on predicted sP (secreted–gray), cTP (plastid–green), and mTP (mitochondria–yellow).
Subcellular localization.
The PXDs analyzed for this first build include samples from a wide variety of plant parts and several subcellular locations (Figure 3). To get an impression of proteome coverage across subcellular localizations for the total, canonical and unobserved proteomes, we compared the predicted subcellular localizations for secreted proteins (ER, Golgi, PM, cell wall, and vacuole) based on sP, plastids based on the chloroplast Transit Peptide (cTP), and mitochondria based on the mitochondrial Transit Peptide (mTP) using the well-documented localization predictor TargetP. 20%, 15%, or 11% of all predicted proteins have a predicted sP, cTP, or mTP, respectively. The canonical proteome was somewhat enriched for chloroplast proteins (18% cTP), whereas the unobserved proteome was strongly underrepresented in proteins with predicted cTP (9% cTP; Figure 10C). This is a rough estimate (given the uncertainties of predictions and alternative sorting mechanisms) but nevertheless suggests that the plastid proteome is relatively well covered at the canonical level. In future builds, we will include additional organellar datasets and other specific subcellular localized proteomes and we will then compare protein coverage against the various protein localization databases, such as SUBA (Hooper et al., 2017) and PPDB (Sun et al., 2004).
Protein abundance.
Determining protein abundance by MS is challenging because this greatly depends on the physicochemical properties of the peptides and the number of suitable peptides for a given protein (Ankney et al., 2018; Calderon-Celis et al., 2018). Accurate and absolute protein quantification is however possible, particularly when including “spike-in” proteotypic peptides generated by chemical synthesis (AQUA) or through expression in Escherichia coli (QConcat; Ankney et al., 2018; Calderon-Celis et al., 2018). However, these spike-in experiments are costly and are typically done at a small scale, targeting just dozens of proteins. In the context of this PeptideAtlas, we determined relative abundance for the canonical proteins based on the number of PSMs normalized to the length of the protein (as number of amino acids). Furthermore, we refined that abundance by calculating the apportioned PSMs, which is the summation of the uniquely mapping PSMs and a portion of the shared PSMs based on the ratio between uniquely mapping peptides of the canonical protein and protein(s) with which the peptides were shared. The apportioned PSMs ranged from 2 to 785,178, and when normalized to protein length (aa), ranged from 0.0018 to 1,639, which is a dynamic range of nearly six orders of magnitude (Figure 12A). The five proteins with the highest relative abundance were large and small subunits of RUBISCO and CF1β of the thylakoid ATP synthase (ATCG00490, AT1G67090, AT5G38410, AT2G39730, and ATCG00480). As a complementary measure of relative abundance, we calculated the relative protein sequence coverage for canonical proteins, i.e. the percentage of residues of the primary sequence that are part of the matched peptides (%; Figure 12B). Sequence coverage ranged from 2% to 100%. The correlation between relative abundance and sequence coverage is positive but poor (Figure 12C), which is expected given that sequence coverage depends on the availability of suitable tryptic peptides for MS/MS analysis and because many proteins accumulate without their cleavable sP. Because this PeptideAtlas is built on a wide range of samples, and some tissues, subcellular fractions, or proteins are likely over- or undersampled, these relative abundances only provide a rough abundance estimate. However, the abundance estimate is nevertheless a useful attribute when investigating protein function.
Figure 12.

Relative abundance of the canonical proteins in Araport11 across the peptide atlas build. A, Relative abundance for the canonical proteins based on the number of apportioned PSMs normalized to the length of the protein (based on number of amino acids). B, Relative protein sequence coverage for canonical proteins based on sequence coverage, i.e. the % of residues of the primary sequence that are part of the matched peptides (%). C, Correlation between relative protein abundance (log10 [PSMs/length]) and sequence coverage (%).
PTMs
Plant proteins undergo various physiological (in vivo) post-translational modifications that can often best be detected using specific enrichment methods, e.g. phosphorylation, acetylation, ubiquitination, SUMOylation, and cysteine (redox) modifications (Friso and van Wijk, 2015; Augustine and Vierstra, 2018; Vu et al., 2018; Sandalio et al., 2019; Moller et al., 2020). As discussed earlier, we selected PXDs that included specific peptide enrichment for phosphorylation and lysine acetylation, as well as processing events at the N-termini of proteins using various N-terminomics techniques (TAILS, COFRADIC, and ChaFRADIC), in most cases combined with protein or peptide dimethylation (with/without stable isotope) to label N-terminal α-amines as well as ε-amines on the side chain of lysine (Figure 3C and Table 3; Supplemental Data Set S1). N-terminal acetylation is a very common PTM in the cytosol that mostly occurs co-translationally by ribosome-associated N-terminal acetyltransferases (Linster and Wirtz, 2018). In addition, a large portion of chloroplast-localized nucleus- as well as plastid-encoded proteins also undergo N-terminal acetylation (Zybailov et al., 2008; Rowland et al., 2015; Giglione and Meinnel, 2021). N-terminal acetylation can affect protein stability, localization, and protein interactions. Lysine acetylation plays critical roles in regulating gene expression by modifying histones in the nucleus as well as other proteins involved in a wide range of activities, located across different subcellular compartments, including the cytosol, mitochondria, and plastids (Hartl et al., 2017; Hosp et al., 2017; Fussl et al., 2018; Bolter et al., 2020). Phosphorylation is the most well-studied PTM in Arabidopsis and other plants (Silva-Sanchez et al., 2015; Millar et al., 2019; and in other eukaryotes). Phosphorylation occurs at serine, threonine, and tyrosine residues, and the distribution of phosphorylated serine (pS), phosphorylated threonine (pT), and phosphorylated tyrosine (pY) is ∼80%–85%, ∼10%–15%, and 0%–5%, respectively, in large-scale plant (meta) studies (van Wijk et al., 2014; Mergner et al., 2020).
There are several specialized Plant PTM databases, in particular Plant PTM viewer and PhosPhat, each containing assembled experimental PTM data for Arabidopsis (or other species) identified by MS in mostly large-scale experiments by different research groups. These data were obtained by direct submission, extracted from publications, or generated in-house. Plant PTM viewer includes data from five plant species, and it currently reports 24 different PTM types, with 165.193 PTMs in 55,920 proteins for Arabidopsis (we note that Araport11 has only 40,784 unique protein sequences; Table 1). PhosPhat specifically concerns phosphorylation and currently reports 9,159 phosphoproteins based on 19,100 unique tryptic phosphopeptides with an overall pS:pT:pY ratio of 72:22:6. Each of these databases has its own strengths, and PeptideAtlas is therefore linked to these PTM databases to provide easy access and comparisons at the individual protein level. The strength of PeptideAtlas is that all raw data are processed using the same search algorithms and carefully controlled FDR. Moreover, each psite and each MS/MS spectrum is linked back to the original PXD and its metadata. Furthermore, PeptideAtlas searches all data against the most recent Arabidopsis genome annotation (currently Araport11) as well as additional sequences (see Table 1).
For PTM analysis in this first PeptideAtlas build, we focused our efforts and resources on building a new PeptideAtlas tool to provide detailed and comprehensive information about protein phosphorylation and phospho-site (p-site) determination. The PeptideAtlas build provides an in-depth view of observed p-sites including statistical significance and associated spectra. All localization p-site probabilities are computed with the TPP tool PTMProphet (Shteynberg et al., 2019) after running iProphet for each dataset. PTMProphet considers all possible permutations of positions of the phosphates reported by Comet and computes Bayesian probabilities that a phosphate is located at each potential STY site based on the subtle differences in spectrum peaks expected for the different permutations. A high probability (e.g. P > 0.95) indicates a high likelihood that a phosphate was present at a site based on the spectral evidence; a low probability near 0 (e.g. P < 0.05) indicates high confidence that a phosphate was not at a site; a probability near 0.5 indicates the inability to localize the position of the phosphate with confidence based on the available mass spectrum peaks. Considering only p-sites with a score of P > 0.95 or P > 0.99 and considering only canonical proteins, the current PeptideAtlas build identified 31,988 p-sites for 7,778 canonical proteins (44% of the canonical proteins; Supplemental Data Set S4). The site distribution of S, T, and Y was 85% pS, 14% pT, and 0.9% pY, which is consistent with the results of prior metadata analysis (van Wijk et al., 2014). When considering only p-sites with three or more identifications at P > 0.95 or P > 0.99, the number of p-sites was 18,789, corresponding to 5,984 (34%) canonical p-proteins. When considering only p-sites with three or more identifications at P > 0.99, the number of p-sites was 16,028, corresponding to 5,565 (31%) canonical proteins. This increased stringency decreased pY to 0.003%–0.006% but did not significantly affect the pS and pT ratio. Finally, PeptideAtlas also provides information about the number of phosphorylations per peptide (Supplemental Data Set S4).
We compared the data sources currently used for phosphorylation analysis in Plant PTM Viewer and PhosPhat to the PXDs that were so far included for phosphoproteome analysis in this first PeptideAtlas build. PTM viewer currently used data from 45 publications for Arabidopsis that involved phosphorylation, which were published between 2004 and 2020. The majority (37) were from publications prior to 2015. PhosPhat is based on 45 publications (published between 2004 and 2019) that are listed p-proteome data sources, with the majority (27) published prior to 2015. PeptideAtlas Build #1 includes eight publications that include phosphorylation experiments, all published since 2015 (Table 3). There are three publications for each PhosPhat and PTM viewer that overlap with PeptideAtlas. We then did a direct comparison with a comprehensive study (Van Leene et al., 2019) that used for all three databases. Using MaxQuant, Van Leene report a total of 4988 psites (minimum localization probability P > 0.75), 4,427 of which were considered high confidence (localization probability P > 0.90). PSM, protein, and site FDR were set at 0.01 in MaxQuant. In PeptideAtlas, we identified 4,880 high-confidence p-sites (localization P > 0.95 based on PTMprophet). We found 2,771 high-confidence p-sites in common, with an additional 2,109 unique to PeptideAtlas and 1,656 unique to (Van Leene et al., 2019), thus showing an excellent overlap in reported high-quality p-sites between these very different data search workflows.
Figure 13 demonstrates the functionality of PeptideAtlas for p-site analysis by showing examples for the chloroplast Calcium Sensor Protein (CAS; Figure 13). This is one of many examples in which a protein was identified as a canonical protein and for which phosphorylation was previously demonstrated to have important functional and physiological significance. CAS (AT5G23060.1) was identified with 81% protein sequence coverage, and PeptideAtlas shows only one identified p-peptide, i.e. the C-terminal peptide SGTKFLPSSD identified 114 times (Figure 13, A and B). In 84 cases, the p-site at position T380 was identified with the highest significance (0.99 ≤ P ≤ 1.0; in green) and additional observations for this site at lower probabilities (Figure 13, C and D). There are three serines (S378, S385, S386) in this peptide (no tyrosines), but p-sites were assigned only very low probabilities (see Figures 13C (inset) and 13D), indicating high confidence that the detected phosphate was not positioned at those sites. Panel 13D provides a numerical summary of the p-site observations and also provides information about specific sample enrichment for p-peptides based on metadata information collected from the individual PXD submissions. This shows that the peptide SGTKFLPSSD was observed 13 times in samples that were not enriched for p-peptides and 114 times as a single phosphorylated peptide from enriched samples. Finally, Figure 13E shows a detailed peptide view of the phosphorylated peptide SGTKLPSSD and p-site identification scores. The lower panel shows information at the individual spectral level with hyperlinked access to the annotated spectra. Together, this strongly suggests that CAS is phosphorylated at T380 and very unlikely at S378 (although some spectra are ambiguous). A recent paper suggested that T376, S378, and T380 are the major p-sites based on phosphoproteomics and in vitro phosphorylation assays of CAS variants (Cutolo et al., 2019), with an earlier study suggesting T380 as the main p-site (Vainonen et al., 2008). PeptideAtlas showed no sequence coverage for T376 despite the very high sequence coverage of the protein (possible peptides with one missed cleavage are SFGT376RSGTK or IIPAASRSFGT376R or SFGT376RSGTK, but these were not observed; Figure 13, B and C). All p-peptides that covered S378 indicated either high confidence not at that site, or ambiguous evidence.
Figure 13.

Illustration of the functionality of PeptideAtlas for the determination of phospho-sites based on the example of the thylakoid CAS AT5G23060.1. A, Coverage by MS/MS for the primary sequence (81.3%) with identified residues in red. The predicted cleavable chloroplast sP is indicated in blue, but the spectral evidence suggests that the real sP is shorter. B, Matched peptides projected on the primary sequence. Darker red rectangles indicate higher numbers of PSMs for each peptide. The predicted thylakoid transmembrane domain is indicated as a blue rectangle (we show the N- and C-termini as facing the chloroplast stromal site based on Cutolo et al. (2019)). C, Probabilities of localization of phosphates on potential sites (as indicated by the colored bars) along the complete protein sequence. The inset provides a close-up view of the C-terminal peptide SGTKLPSSD and the frequency of specific p-sites, color-coded by localization probability. This shows e.g. that the phosphorylated peptide was observed 114 times and that pT380 was observed 84 times at the highest significance level. D, This small table provides a numerical summary of the p-site observations and information about specific sample enrichment for phospho-peptides based on metadata information collected from the individual PXD submissions. This shows that the peptide SGTKFLPSSD was observed 13 times in samples that were not enriched for p-peptides and 114 times as a single p-peptide from enriched samples. Explanation for columns: Offset – residue number from start; Residue – amino acid; nObs–Total observed PTM spectra for the site; One_mod–the number of PSMs with a single phosphate covering the site. Two_mods–the number of PSMs that have two observed phosphates (i.e. doubly phosphorylated). Over_two_mods–the number of PSMs covering the site that have more than two phosphates. nP < 0.01 − PTMProphet probability < 0.01; nP < 0.05 − PTMProphet probability ≥ 0.01 and < 0.05; nP < 0.20 − PTMProphet probability ≥ 0.05 and ≤ 0.20; nP 0.2–0.8 − PTMProphet probability > 0.20 and < 0.80; nP > 0.80 − PTMProphet probability ≥ 0.80 and < 0.95; nP>0.95 − PTMProphet probability ≥ 0.95 and < 0.99; nP>0.99 − PTMProphet probability ≥ 0.99; no-choice − Number of PSMs covering this site for which there was no choice in the localization of the PTM. Only one residue was an S, T, or Y; enriched-with-mod − Number of PSMs covering this site with phospho modification on this site, and originating from a phospho-enriched sample; enriched-but-non-mod − Number of PSMs covering this site with no phospho modification anywhere on the peptide, but yet originating from a phospho-enriched sample; nonenriched − Number of PSMs covering this site from a nonenriched sample (phospho not considered in the search). E, Detailed view of the phosphorylated peptide SGTKLPSSD and p-site localization probability distributions. The lower panel shows information at an individual spectral level with hyperlinked access to the annotated spectra.
At a later stage, we will build similar tools for other PTMs, particularly for N-terminal and lysine acetylation. Both are important physiological PTMs that affect protein localization, protein stability, and protein–protein interactions that have functional connections to the metabolic state of the cell through intracellular concentrations of acetyl-CoA. In addition to these in vivo PTMs, several PTMs are often generated during sample preparation due to exposure to organic solvents (e.g. formic acid leading to the formylation of Ser, Thr, and N-termini), (thio)urea (N-terminal or Lys carbamylation), reducing agents and oxygen, unpolymerized acrylamide (Cys propionamide), and low or high pH (cyclization of N-terminal Gln or Glu into pyro-Glu), as reviewed in Friso and van Wijk (2015). A large-scale proteomics study of Arabidopsis leaf extracts addressed the frequency of PTMs that do not require specific affinity enrichment based on a dataset of 1.5 million MS/MS spectra acquired at a mass resolution of 100,000 on an LTQ-Orbitrap instrument followed by error-tolerant searches and systematic validation based on LC retention time (Zybailov et al., 2009; Majsec et al., 2017). This showed, for example, that modification of Met and N-terminal Gln into Met-ox and pyro-Glu, respectively, showed by far the highest modification frequencies in seedlings (80% of all M observed and 46% of all N-terminal Q), followed by N-terminal formylation (1.5% of all N-termini) most likely induced during sample analysis, as well as deamidation of Asn/Gln (∼1.2% of all observed Asn/Gln). Several of these nonenzymatic PTMs (in particular deamidation, oxidation, and formylation) can also occur in vivo and therefore cannot be simply dismissed as artifacts but need to be considered as potential regulators. As mentioned earlier, to improve protein coverage and to match more MS/MS spectra to modified peptides, several of these PTMs were included in all our MS searches (see “Materials and methods”).
Integration with community resources and use of the Arabidopsis PeptideAtlas
This first build is freely available in an interactive manner at the PeptideAtlas website (http://peptideatlas.org/builds/arabidopsis). The results are also made available via web services, allowing easy access to formatted data via external software. We also provide download access to the entire build, which allows anyone to integrate large amounts of the data into their analyses or resource. The build is available as a set of text files, a fully structured XML file, and a MySQL dump that enables easy ingestion into a local MySQL relational database for querying by expert users. Data are already being pulled via web services by UniProtKB, and links are established at the protein identifier level (for Araport11 sequences) with TAIR https://www.arabidopsis.org/ and the PPDB http://ppdb.tc.cornell.edu/. The matched peptide data for Araport11 genes in PeptideAtlas are also integrated on a specific track in the Arabidopsis JB browser at https://jbrowse.arabidopsis.org/. We will work with TAIR to further validate and incorporate peptides matched to non-Araport11 sequences, such as those for the sORFs (Tanya Berardini, personal communication).
The next Arabidopsis PeptideAtlas build
The objectives for the next build will be to discover proteins that have so far not been confidently identified in the current build. As illustrated in Figure 11, reasons for the lack of protein identification can include unfavorable physicochemical properties (small, hydrophobic, acidic pI), generally very low copy numbers (e.g. for ion channels and some transcription factors), or if expression is limited to specialized cell types or subcellular locations only present in smaller numbers or under very specific developmental or environmental conditions. We envision three strategies to increase the detection of such proteins, namely (1) include PXDs of very specific cell types or specialized subcellular fractions, (2) include PXDs that concern specific protein complexes or protein affinity enrichments, and (3) include PXDs that are enriched for specific post-translational modifications. We will also include PXDs that appear to have very high dynamic resolution and sensitivity, e.g. by using the latest technologies in MS and/or sample fractionation.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Data Set S1. Detailed information about the 52 selected PXD files for this first PeptideAtlas build.
Supplemental Data Set S2. Identified proteins and unobserved proteins in the PeptideAtlas build and their assignment to the 10-tier system.
Supplemental Data Set S3. Evidence for protein identifiers not found in Araport11.
Supplemental Data Set S4. Phosphorylation observations in PeptideAtlas.
Supplementary Material
Acknowledgments
We thank members of the Scientific Advisory board Tanya Berardini, Chris Town, Nicholas Provart, Sixue Chen, and Joshua Heazlewood for advice and feedback. We thank Eve Wurtele for sending us her candidate orphan sequences for inclusion in this build.
Funding
This project was supported by a grant from the National Science Foundation #1922871 to K.J.V.W, E.W.D., and Q.S.
Conflict of interest statement. The authors have no known conflict of interest.
T.L. carried out all MS searches and created the PeptideAtlas build. Q.S. and S.S.B. carried out the physicochemical and functional property analysis of the proteome and supported analysis of sORFs and other non-protein-coding identifiers. Z.S. developed PeptideAtlas interface enhancements and assisted with the PeptideAtlas building process. L.M. developed PeptideAtlas interface enhancements and the dataset annotation tool. E.D. supervised the PeptideAtlas building process. K.J.V.W. contributed to the selection of PXDs and all aspects of specific plant biology-related issues. E.W.D. and K.J.V.W. developed this project, raised the funding, and wrote the paper.
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) are: Klaas J. van Wijk (kv35@cornell.edu) and Eric W. Deutsch (edeutsch@systemsbiology.org).
References
- Akter S, Huang J, Waszczak C, Jacques S, Gevaert K, Van Breusegem F, Messens J (2015) Cysteines under ROS attack in plants: a proteomics view. J Exp Bot 66: 2935–2944 [DOI] [PubMed] [Google Scholar]
- Al-Mohanna T, Ahsan N, Bokros NT, Dimlioglu G, Reddy KR, Shankle M, Popescu GV, Popescu SC (2019) Proteomics and proteogenomics analysis of sweetpotato (Ipomoea batatas) leaf and root. J Proteome Res 18: 2719–2734 [DOI] [PubMed] [Google Scholar]
- Al Shweiki MR, Monchgesang S, Majovsky P, Thieme D, Trutschel D, Hoehenwarter W. (2017) Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. J Proteome Res 16: 1410–1424 [DOI] [PubMed] [Google Scholar]
- Ankney JA, Muneer A, Chen X (2018) Relative and absolute quantitation in mass spectrometry-based proteomics. Annu Rev Anal Chem 11: 49–77 [DOI] [PubMed] [Google Scholar]
- Augustine RC, Vierstra RD (2018) SUMOylation: re-wiring the plant nucleus during stress and development. Curr Opin Plant Biol 45: 143–154 [DOI] [PubMed] [Google Scholar]
- Balmant KM, Zhang T, Chen S (2016) Protein phosphorylation and redox modification in stomatal guard cells. Front Physiol 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K, Massonnet C, Hennig L, Russenberger D, Sulpice R, Walsh S, Stitt M, Granier C, Gruissem W (2015) A long photoperiod relaxes energy management in Arabidopsis leaf six. Curr Plant Biol 2: 34–45 [Google Scholar]
- Bislev SL, Deutsch EW, Sun Z, Farrah T, Aebersold R, Moritz RL, Bendixen E, Codrea MC (2012) A Bovine PeptideAtlas of milk and mammary gland proteomes. Proteomics 12: 2895–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan NH, Friso G, Rowland E, Majsec K, van Wijk KJ (2016) The plastoglobule-localized metallopeptidase PGM48 is a positive regulator of senescence in Arabidopsis thaliana. Plant Cell 28: 3020–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan NH, Rowland E, Friso G, Ponnala L, Michel EJS, van Wijk KJ (2020) Autocatalytic processing and substrate specificity of Arabidopsis chloroplast glutamyl peptidase. Plant Physiol 184: 110–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe BJ (2017) The relationship between alternative splicing and proteomic complexity. Trends Biochem Sci 42: 407–408 [DOI] [PubMed] [Google Scholar]
- Bolter B, Mitterreiter MJ, Schwenkert S, Finkemeier I, Kunz HH (2020) The topology of plastid inner envelope potassium cation efflux antiporter KEA1 provides new insights into its regulatory features. Photosynth Res 145: 43–54 [DOI] [PubMed] [Google Scholar]
- Bouchnak I, Brugiere S, Moyet L, Le Gall S, Salvi D, Kuntz M, Tardif M, Rolland N (2019) Unraveling hidden components of the chloroplast envelope proteome: opportunities and limits of better MS sensitivity. Mol Cell Proteomics 18: 1285–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault ML, Petit JD, Immel F, Nicolas WJ, Glavier M, Brocard L, Gaston A, Fouche M, Hawkins TJ, Crowet JM, et al. (2019) Multiple C2 domains and transmembrane region proteins (MCTPs) tether membranes at plasmodesmata. EMBO Rep 20: e47182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocard L, Immel F, Coulon D, Esnay N, Tuphile K, Pascal S, Claverol S, Fouillen L, Bessoule JJ, Brehelin C (2017) Proteomic analysis of lipid droplets from arabidopsis aging leaves brings new insight into their biogenesis and functions. Front Plant Sci 8: 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon-Celis F, Encinar JR, Sanz-Medel A (2018) Standardization approaches in absolute quantitative proteomics with mass spectrometry. Mass Spectrom Rev 37: 715–737 [DOI] [PubMed] [Google Scholar]
- Castellana NE, Shen Z, He Y, Walley JW, Cassidy CJ, Briggs SP, Bafna V (2014) An automated proteogenomic method uses mass spectrometry to reveal novel genes in Zea mays. Mol Cell Proteomics 13: 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrec B, Dian C, Ciccone S, Ebert CL, Bienvenut WV, Le Caer JP, Steyaert JM, Giglione C, Meinnel T (2018) Structural and genomic decoding of human and plant myristoylomes reveals a definitive recognition pattern. Nat Chem Biol 14: 671–679 [DOI] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30: 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WW, Huang L, Shen M, Webster C, Burlingame AL, Roberts JK (2000) Patterns of protein synthesis and tolerance of anoxia in root tips of maize seedlings acclimated to a low-oxygen environment and identification of proteins by mass spectrometry. Plant Physiol 122: 295–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman B, Bellgard M (2017) Plant proteogenomics: improvements to the grapevine genome annotation. Proteomics 17:1700197. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu X, Zheng W, Zhang L, Yao J, Yang P (2014) Screening of missing proteins in the human liver proteome by improved MRM-approach-based targeted proteomics. J Proteome Res 13: 1969–1978 [DOI] [PubMed] [Google Scholar]
- Chen M.X, Zhu F.Y, Gao B, Ma K.L, Zhang Y, Fernie A.R, Chen X, Dai L, Ye N.H, Zhang X, et al. (2020) Full-length transcript-based proteogenomics of rice improves its genome and proteome annotation. Plant Physiol 182: 1510–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD (2017) Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J 89: 789–804 [DOI] [PubMed] [Google Scholar]
- Chick JM, Kolippakkam D, Nusinow DP, Zhai B, Rad R, Huttlin EL, Gygi SP (2015) A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol 33: 743–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary MK, Nomura Y, Shi H, Nakagami H, Somers DE (2016) Circadian profiling of the arabidopsis proteome using 2D-DIGE. Front Plant Sci 7: 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Galvis V, Poschmann G, Melzer M, Stuhler K, Jahns P (2016) PsbS interactions involved in the activation of energy dissipation in Arabidopsis. Nat Plants 2: 15225. [DOI] [PubMed] [Google Scholar]
- Cottingham K (2009) Two are not always better than one. J Proteome Res 8: 4172. [DOI] [PubMed] [Google Scholar]
- Creasy DM, Cottrell JS (2004) Unimod: protein modifications for mass spectrometry. Proteomics 4: 1534–1536 [DOI] [PubMed] [Google Scholar]
- Cutolo E, Parvin N, Ruge H, Pirayesh N, Roustan V, Weckwerth W, Teige M, Grieco M, Larosa V, Vothknecht UC (2019) The high light response in Arabidopsis requires the calcium sensor protein CAS, a target of STN7- and STN8-mediated phosphorylation. Front Plant Sci 10: 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F, Deutsch EW, Nesvizhskii AI, Mallick P, King NL, Eng JK, Aderem A, Boyle R, Brunner E, Donohoe S, et al. (2005) Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol 6: R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL (2015) Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin Appl 9: 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Sun Z, Campbell DS, Binz PA, Farrah T, Shteynberg D, Mendoza L, Omenn GS, Moritz RL (2016a) Tiered human integrated sequence search databases for shotgun proteomics. J Proteome Res 15: 4091–4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Lane L, Overall CM, Bandeira N, Baker MS, Pineau C, Moritz RL, Corrales F, Orchard S, Van Eyk JE, et al. (2019) Human proteome project mass spectrometry data interpretation guidelines 3.0. J Proteome Res 18: 4108–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Overall CM, Van Eyk JE, Baker MS, Paik YK, Weintraub ST, Lane L, Martens L, Vandenbrouck Y, Kusebauch U, et al. (2016b) Human proteome project mass spectrometry data interpretation guidelines 2.1. J Proteome Res 15: 3961–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Orchard S, Binz PA, Bittremieux W, Eisenacher M, Hermjakob H, Kawano S, Lam H, Mayer G, Menschaert G, et al. (2017a) Proteomics standards initiative: fifteen years of progress and future work. J Proteome Res 16: 4288–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, et al. (2017b) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45: D1100–D1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez-Riverol Y, Carver JJ, Kundu DJ, Garcia-Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS, et al. (2020) The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res 48: D1145–D1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliuk S, Makarov A (2015) Evolution of orbitrap mass spectrometry instrumentation. Annu Rev Anal Chem 8: 61–80 [DOI] [PubMed] [Google Scholar]
- Eng JK, Deutsch EW (2020) Extending comet for global amino acid variant and post-translational modification analysis using the PSI extended FASTA format. Proteomics 20: e1900362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, et al. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics 10: M110 006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friso G, van Wijk KJ (2015) Posttranslational protein modifications in plant metabolism. Plant Physiol 169: 1469–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs P, Rugen N, Carrie C, Elsasser M, Finkemeier I, Giese J, Hildebrandt TM, Kuhn K, Maurino VG, et al. (2020) Single organelle function and organization as estimated from Arabidopsis mitochondrial proteomics. Plant J 101: 420–441 [DOI] [PubMed] [Google Scholar]
- Furtauer L, Kustner L, Weckwerth W, Heyer AG, Nagele T (2019) Resolving subcellular plant metabolism. Plant J 100: 438–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussl M, Lassowskat I, Nee G, Koskela MM, Brunje A, Tilak P, Giese J, Leister D, Mulo P, Schwarzer D, et al. (2018) Beyond histones: new substrate proteins of lysine deacetylases in Arabidopsis nuclei. Front Plant Sci 9: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain A, Hanson M.R, Bentolila S. (2015) High-throughput quantification of chloroplast RNA editing extent using multiplex RT-PCR mass spectrometry. Plant J 83: 546–554. [DOI] [PubMed] [Google Scholar]
- Giglione C, Meinnel T (2021) Evolution-driven versatility of N terminal acetylation in photoautotrophs. Trends Plant Sci 26: 375–391 [DOI] [PubMed] [Google Scholar]
- Hanada K, Zhang X, Borevitz JO, Li WH, Shiu SH (2007) A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res 17: 632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hander T, Fernandez-Fernandez AD, Kumpf RP, Willems P, Schatowitz H, Rombaut D, Staes A, Nolf J, Pottie R, Yao P, et al. (2019) Damage on plants activates Ca(2+)-dependent metacaspases for release of immunomodulatory peptides. Science 363. [DOI] [PubMed] [Google Scholar]
- Hartl M, Fussl M, Boersema PJ, Jost JO, Kramer K, Bakirbas A, Sindlinger J, Plochinger M, Leister D, Uhrig G, et al. (2017) Lysine acetylome profiling uncovers novel histone deacetylase substrate proteins in Arabidopsis. Mol Syst Biol 13: 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins CL, Davies MJ (2019) Detection identification, and quantification of oxidative protein modifications. J Biol Chem 294: 19683–19708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazarika RR, De Coninck B, Yamamoto LR, Martin LR, Cammue BP, van Noort V (2017) ARA-PEPs: a repository of putative sORF-encoded peptides in Arabidopsis thaliana. BMC Bioinformatics 18: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse AM, Dupierris V, Adam C, Court M, Barthe D, Emadali A, Masselon C, Ferro M, Bruley C (2016) hEIDI: an intuitive application tool to organize and treat large-scale proteomics data. J Proteome Res 15: 3896–3903 [DOI] [PubMed] [Google Scholar]
- Hesselager MO, Codrea MC, Sun Z, Deutsch EW, Bennike TB, Stensballe A, Bundgaard L, Moritz RL, Bendixen E (2016) The Pig PeptideAtlas: a resource for systems biology in animal production and biomedicine. Proteomics 16: 634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper CM, Castleden IR, Tanz SK, Aryamanesh N, Millar AH (2017) SUBA4: the interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res 45: D1064–D1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp F, Lassowskat I, Santoro V, De Vleesschauwer D, Fliegner D, Redestig H, Mann M, Christian S, Hannah MA, Finkemeier I (2017) Lysine acetylation in mitochondria: from inventory to function. Mitochondrion 33: 58–71 [DOI] [PubMed] [Google Scholar]
- Hsu PY, Benfey PN (2018) Small but mighty: functional peptides encoded by small ORFs in plants. Proteomics 18: e1700038. [DOI] [PubMed] [Google Scholar]
- Hsu PY, Calviello L, Wu HL, Li FW, Rothfels CJ, Ohler U, Benfey PN (2016) Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc Natl Acad Sci USA 113: E7126–E7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Ghosh A, Stolze SC, Horvath M, Bai B, Schaefer S, Zundorf S, Liu S, Harzen A, Hajheidari M, et al. (2019) Gene modification by fast-track recombineering for cellular localization and isolation of components of plant protein complexes. Plant J 100: 411–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MD, Chen TL, Huang AH (2013) Abundant type III lipid transfer proteins in Arabidopsis tapetum are secreted to the locule and become a constituent of the pollen exine. Plant Physiol 163: 1218–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulstaert N, Shofstahl J, Sachsenberg T, Walzer M, Barsnes H, Martens L, Perez-Riverol Y (2020) ThermoRawFileParser: modular, scalable, and cross-platform RAW file conversion. J Proteome Res 19: 537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initiative TAG (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Jiang J, Chai X, Manavski N, Williams-Carrier R, He B, Brachmann A, Ji D, Ouyang M, Liu Y, Barkan A, et al. (2019) An RNA chaperone-like protein plays critical roles in chloroplast mRNA stability and translation in Arabidopsis and maize. Plant Cell 31: 1308–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi HJ, Hirsch-Hoffmann M, Baerenfaller K, Gruissem W, Baginsky S, Schmidt R, Schulze WX, Sun Q, van Wijk KJ, Egelhofer V, et al. (2011) MASCP Gator: an aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol 155: 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage U, Powell JJ, Gardiner DM, Kazan K (2020) Ribosome profiling in plants: what is NOT lost in translation? J Exp Bot 71: 5323–5332 [DOI] [PubMed] [Google Scholar]
- Kaur J, Sebastian J, Siddiqi I (2006) The Arabidopsis-mei2-like genes play a role in meiosis and vegetative growth in Arabidopsis. Plant Cell 18: 545–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Keller A, Eng J, Zhang N, Li XJ, Aebersold R (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol 1: 0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NL, Deutsch EW, Ranish JA, Nesvizhskii AI, Eddes JS, Mallick P, Eng J, Desiere F, Flory M, Martin DB, et al. (2006) Analysis of the Saccharomyces cerevisiae proteome with PeptideAtlas. Genome Biol 7: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraga J, Mackiewicz P, Mackiewicz D, Kowalczuk M, Biecek P, Polak N, Smolarczyk K, Dudek MR, Cebrat S (2007) The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics 8: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler D, Montandon C, Hause G, Majovsky P, Kessler F, Baginsky S, Agne B (2015) Characterization of chloroplast protein import without Tic56, a component of the 1-megadalton translocon at the inner envelope membrane of chloroplasts. Plant Physiol 167: 972–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI (2017) MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskela MM, Brunje A, Ivanauskaite A, Grabsztunowicz M, Lassowskat I, Neumann U, Dinh TV, Sindlinger J, Schwarzer D, Wirtz M, et al. (2018) Chloroplast acetyltransferase NSI is required for state transitions in Arabidopsis thaliana. Plant Cell 30: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmacz M, Gorka M, Schmidt S, Luzarowski M, Moreno JC, Szlachetko J, Leniak E, Sokolowska EM, Sofroni K, Schnittger A, et al. (2019) Protein and metabolite composition of Arabidopsis stress granules. New Phytol 222: 1420–1433 [DOI] [PubMed] [Google Scholar]
- Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, et al. (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40: D1202–D1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, O'Neill KR, Haft DH, DiCuccio M, Chetvernin V, Badretdin A, Coulouris G, Chitsaz F, Derbyshire MK, Durkin AS, et al. (2020) RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res 49: D1020–D1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LL, Hsu CL, Hu CW, Ko SY, Hsieh HL, Huang HC, Juan HF (2015) Integrating phosphoproteomics and bioinformatics to study brassinosteroid-regulated phosphorylation dynamics in Arabidopsis. BMC Genomics 16: 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linster E, Stephan I, Bienvenut WV, Maple-Grodem J, Myklebust LM, Huber M, Reichelt M, Sticht C, Geir Moller S, Meinnel T, et al. (2015) Downregulation of N-terminal acetylation triggers ABA-mediated drought responses in Arabidopsis. Nat Commun 6: 7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linster E, Wirtz M (2018) N-terminal acetylation: an essential protein modification emerges as an important regulator of stress responses. J Exp Bot 69: 4555–4568 [DOI] [PubMed] [Google Scholar]
- Lundquist PK, Mantegazza O, Stefanski A, Stuhler K, Weber APM (2017) Surveying the oligomeric state of Arabidopsis thaliana chloroplasts. Mol Plant 10: 197–211 [DOI] [PubMed] [Google Scholar]
- Majsec K, Bhuiyan NH, Sun Q, Kumari S, Kumar V, Ware D, van Wijk KJ (2017) The plastid and mitochondrial peptidase network in Arabidopsis thaliana: a foundation for testing genetic interactions and functions in organellar proteostasis. Plant Cell 29: 2687–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov A (2019) Orbitrap journey: taming the ion rings. Nat Commun 10: 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei B, Spinelli F, Pontiggia D, De Lorenzo G. (2016) Comprehensive analysis of the membrane phosphoproteome regulated by oligogalacturonides in Arabidopsis thaliana. Front Plant Sci 7: 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann GW, Calley PC, Joshi HJ, Heazlewood JL (2013) MASCP gator: an overview of the Arabidopsis proteomic aggregation portal. Front Plant Sci 4: 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Eckhard U, Overall CM (2015) Protein termini and their modifications revealed by positional proteomics. ACS Chem Biol 10: 1754–1764 [DOI] [PubMed] [Google Scholar]
- Martens L, Chambers M, Sturm M, Kessner D, Levander F, Shofstahl J, Tang WH, Rompp A, Neumann S, Pizarro AD, et al. (2011) mzML–a community standard for mass spectrometry data. Mol Cell Proteomics 10: R110 000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G, Montecchi-Palazzi L, Ovelleiro D, Jones AR, Binz PA, Deutsch EW, Chambers M, Kallhardt M, Levander F, Shofstahl J, et al. (2013) The HUPO proteomics standards initiative-mass spectrometry controlled vocabulary. Database (Oxford) 2013: bat009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield JA, Fiebig A, Johnstone SE, Preuss D (2001) Gene families from the Arabidopsis thaliana pollen coat proteome. Science 292: 2482–2485 [DOI] [PubMed] [Google Scholar]
- McCord J, Sun Z, Deutsch EW, Moritz RL, Muddiman DC (2017) The PeptideAtlas of the domestic laying hen. J Proteome Res 16: 1352–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin F, Kim M, Marshall RS, Vierstra RD, Vierling E (2019) HSP101 interacts with the proteasome and promotes the clearance of ubiquitylated protein aggregates. Plant Physiol 180: 1829–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergner J, Frejno M, List M, Papacek M, Chen X, Chaudhary A, Samaras P, Richter S, Shikata H, Messerer M, et al. (2020) Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 579: 409–414 [DOI] [PubMed] [Google Scholar]
- Millar AH, Sweetlove LJ, Giege P, Leaver CJ (2001) Analysis of the Arabidopsis mitochondrial proteome. Plant Physiol 127: 1711–1727 [PMC free article] [PubMed] [Google Scholar]
- Millar AH, Heazlewood JL, Giglione C, Holdsworth MJ, Bachmair A, Schulze WX (2019) The scope, functions, and dynamics of posttranslational protein modifications. Annu Rev Plant Biol 70: 119–151 [DOI] [PubMed] [Google Scholar]
- Misra BB (2018) Updates on resources, software tools, and databases for plant proteomics in 2016-2017. Electrophoresis 39: 1543–1557 [DOI] [PubMed] [Google Scholar]
- Miura K, Hasegawa PM (2010) Sumoylation and other ubiquitin-like post-translational modifications in plants. Trends Cell Biol 20: 223–232 [DOI] [PubMed] [Google Scholar]
- Moller IM, Igamberdiev AU, Bykova NV, Finkemeier I, Rasmusson AG, Schwarzlander M (2020) Matrix redox physiology governs the regulation of plant mitochondrial metabolism through posttranslational protein modifications. Plant Cell 32: 573–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montandon C, Friso G, Liao JR, Choi J, van Wijk KJ (2019) In vivo trapping of proteins interacting with the chloroplast CLPC1 chaperone; potential substrates and adaptors. J Proteome Res 18: 2585–2600 [DOI] [PubMed] [Google Scholar]
- Montecchi-Palazzi L, Beavis R, Binz PA, Chalkley RJ, Cottrell J, Creasy D, Shofstahl J, Seymour SL, Garavelli JS (2008) The PSI-MOD community standard for representation of protein modification data. Nat Biotechnol 26: 864–866 [DOI] [PubMed] [Google Scholar]
- Nee G, Kramer K, Nakabayashi K, Yuan B, Xiang Y, Miatton E, Finkemeier I, Soppe WJJ (2017) DELAY OF GERMINATION1 requires PP2C phosphatases of the ABA signalling pathway to control seed dormancy. Nat Commun 8: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Apitz J, Friso G, Kim J, Ponnala L, Grimm B, van Wijk KJ (2015) Discovery of a unique Clp component ClpF, in chloroplasts: a proposed binary ClpF-ClpS1 adaptor complex functions in substrate recognition and delivery. Plant Cell 27: 2677–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn GS, Lane L, Overall CM, Corrales FJ, Schwenk JM, Paik YK, Van Eyk JE, Liu S, Pennington S, Snyder MP, et al. (2019) Progress on identifying and characterizing the human proteome: 2019 metrics from the HUPO Human Proteome Project. J Proteome Res 18: 4098–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn GS, Lane L, Overall CM, Cristea IM, Corrales FJ, Lindskog C, Paik YK, Van Eyk JE, Liu S, Pennington SR, et al. (2020) Research on the human proteome reaches a major milestone: >90% of predicted human proteins now credibly detected, according to the HUPO Human Proteome Project. J Proteome Res 19: 4735–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Hermjakob H, Apweiler R (2003) The proteomics standards initiative. Proteomics 3: 1374–1376 [DOI] [PubMed] [Google Scholar]
- Peltier JB, Ytterberg J, Liberles D.A, Roepstorff P, van Wijk K.J. (2001) Identification of a 350-kDa ClpP protease complex with 10 different Clp isoforms in chloroplasts of Arabidopsis thaliana. J Biol Chem 276: 16318–16327. [DOI] [PubMed] [Google Scholar]
- Peltier JB, Friso G, Kalume DE, Roepstorff P, Nilsson F, Adamska I, van Wijk KJ (2000) Proteomics of the chloroplast. Systematic identification and targeting analysis of lumenal and peripheral thylakoid proteins. Plant Cell 12: 319–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier JB, Emanuelsson O, Kalume DE, Ytterberg J, Friso G, Rudella A, Liberles DA, Soderberg L, Roepstorff P, von Heijne G, et al. (2002) Central functions of the lumenal and peripheral thylakoid proteome of Arabidopsis determined by experimentation and genome-wide prediction. Plant Cell 14: 211–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2018) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 7: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provart NJ, Alonso J, Assmann SM, Bergmann D, Brady SM, Brkljacic J, Browse J, Chapple C, Colot V, Cutler S, et al. (2016) 50 years of Arabidopsis research: highlights and future directions. New Phytol 209: 921–944 [DOI] [PubMed] [Google Scholar]
- Qi Y, Wang X, Lei P, Li H, Yan L, Zhao J, Meng J, Shao J, An L, Yu F, Liu X (2020) The chloroplast metalloproteases VAR2 and EGY1 act synergistically to regulate chloroplast development in Arabidopsis. J Biol Chem 295: 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesneville H (2020) Twenty years of transposable element analysis in the Arabidopsis thaliana genome. Mob DNA 11: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Qi D, Pugh N, Li K, Wen B, Zhou R, Xu S, Liu S, Jones AR (2019) Improvements to the rice genome annotation through large-scale analysis of RNA-Seq and proteomics data sets. Mol Cell Proteomics 18: 86–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts I, Smith S, De Rybel B, Van Den Broeke J, Smet W, De Cokere S, Mispelaere M, De Smet I, Beeckman T (2013) The CEP family in land plants: evolutionary analyses, expression studies, and role in Arabidopsis shoot development. J Exp Bot 64: 5371–5381 [DOI] [PubMed] [Google Scholar]
- Rowland E, Kim J, Bhuiyan NH, van Wijk KJ (2015) The Arabidopsis chloroplast stromal N-terminome: complexities of amino-terminal protein maturation and stability. Plant Physiol 169: 1881–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugen N, Straube H, Franken L.E, Braun HP, Eubel H (2019) Complexome profiling reveals association of PPR proteins with ribosomes in the mitochondria of plants. Mol Cell Proteomics 18: 1345–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-May E, Segura-Cabrera A, Elizalde-Contreras JM, Shannon LM, Loyola-Vargas VM (2019) A recent advance in the intracellular and extracellular redox post-translational modification of proteins in plants. J Mol Recognit 32: e2754. [DOI] [PubMed] [Google Scholar]
- Salvi D, Bournais S, Moyet L, Bouchnak I, Kuntz M, Bruley C, Rolland N (2018) AT_CHLORO: the first step when looking for information about subplastidial localization of proteins. Methods Mol Biol 1829: 395–406 [DOI] [PubMed] [Google Scholar]
- San Clemente H, Jamet E (2015) WallProtDB, a database resource for plant cell wall proteomics. Plant Methods 11: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandalio LM, Gotor C, Romero LC, Romero-Puertas MC (2019) Multilevel regulation of peroxisomal proteome by post-translational modifications. Int J Mol Sci 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Nakamura Y, Kaneko T, Asamizu E, Tabata S (1999) Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res 6: 283–290 [DOI] [PubMed] [Google Scholar]
- Schonberg A, Rodiger A, Mehwald W, Galonska J, Christ G, Helm S, Thieme D, Majovsky P, Hoehenwarter W, Baginsky S (2017) Identification of STN7/STN8 kinase targets reveals connections between electron transport, metabolism and gene expression. Plant J 90: 1176–1186 [DOI] [PubMed] [Google Scholar]
- Schubert M, Petersson UA, Haas BJ, Funk C, Schröder WP, Kieselbach T (2002) Proteome map of the chloroplast lumen of Arabidopsis thaliana. J Biol Chem 277: 8354–8365 [DOI] [PubMed] [Google Scholar]
- Schulze WX, Yao Q, Xu D (2015) Databases for plant phosphoproteomics. Methods Mol Biol 1306: 207–216 [DOI] [PubMed] [Google Scholar]
- Schwartz R, Ting CS, King J (2001) Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res 11: 703–709 [DOI] [PubMed] [Google Scholar]
- Seaton DD, Graf A, Baerenfaller K, Stitt M, Millar AJ, Gruissem W (2018) Photoperiodic control of the Arabidopsis proteome reveals a translational coincidence mechanism. Mol Syst Biol 14: e7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, Mendoza L, Moritz RL, Aebersold R, Nesvizhskii AI (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics 10: M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg DD, Deutsch EW, Campbell DS, Hoopmann MR, Kusebauch U, Lee D, Mendoza L, Midha MK, Sun Z, Whetton AD, et al. (2019) PTMProphet: fast and accurate mass modification localization for the trans-proteomic pipeline. J Proteome Res 18: 4262–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Sanchez C, Li H, Chen S (2015) Recent advances and challenges in plant phosphoproteomics. Proteomics 15: 1127–1141 [DOI] [PubMed] [Google Scholar]
- Slagel J, Mendoza L, Shteynberg D, Deutsch EW, Moritz RL (2015) Processing shotgun proteomics data on the Amazon cloud with the trans-proteomic pipeline. Mol Cell Proteomics 14: 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Wu Z, Sharbrough J (2018) Correction of persistent errors in Arabidopsis reference mitochondrial genomes. Plant Cell 30: 525–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Schallenberg-Rudinger M, Takenaka M, Mireau H, Ostersetzer-Biran O (2020) Plant organellar RNA editing: what 30 years of research has revealed. Plant J 101: 1040–1056 [DOI] [PubMed] [Google Scholar]
- Staes A, Impens F, Van Damme P, Ruttens B, Goethals M, Demol H, Timmerman E, Vandekerckhove J, Gevaert K (2011) Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat Protoc 6: 1130–1141 [DOI] [PubMed] [Google Scholar]
- Stecker KE, Minkoff BB, Sussman MR (2014) Phosphoproteomic analyses reveal early signaling events in the osmotic stress response. Plant Physiol 165: 1171–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehmel N, Hoehenwarter W, Monchgesang S, Majovsky P, Kruger S, Scheel D, Lee J (2017) Stress-related mitogen-activated protein kinases stimulate the accumulation of small molecules and proteins in Arabidopsis thaliana root exudates. Front Plant Sci 8: 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Souleimanov A, Smith DL (2016) Proteomic studies on the effects of lipo-Chitooligosaccharide and Thuricin 17 under unstressed and salt stressed conditions in Arabidopsis thaliana. Front Plant Sci 7: 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Emanuelsson O, van Wijk KJ (2004) Analysis of curated and predicted plastid subproteomes of Arabidopsis. Subcellular compartmentalization leads to distinctive proteome properties. Plant Physiol 135: 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zybailov B, Majeran W, Friso G, Olinares PD, van Wijk KJ (2009) PPDB the plant proteomics database at Cornell. Nucleic Acids Res 37: D969–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi F, Hanada K, Kondo T, Shinozaki K (2019) Hormone-like peptides and small coding genes in plant stress signaling and development. Curr Opin Plant Biol 51: 88–95 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Hayashi N, Hiragori Y, Sasaki S, Motomura T, Yamashita Y, Naito S, Takahashi A, Fuse K, Satou K, et al. (2020) Comprehensive genome-wide identification of angiosperm upstream ORFs with peptide sequences conserved in various taxonomic ranges using a novel pipeline, ESUCA. BMC Genomics 21: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka M, Zehrmann A, Verbitskiy D, Hartel B, Brennicke A (2013) RNA editing in plants and its evolution. Annu Rev Genet 47: 335–352 [DOI] [PubMed] [Google Scholar]
- Tan BC, Lim YS, Lau SE (2017) Proteomics in commercial crops: an overview. J Proteomics 169: 176–188 [DOI] [PubMed] [Google Scholar]
- Tanz SK, Castleden I, Hooper CM, Vacher M, Small I, Millar HA (2013) SUBA3: a database for integrating experimentation and prediction to define the SUBcellular location of proteins in Arabidopsis. Nucleic Acids Res 41: D1185–D1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizioli M, Lazar C, Brugiere S, Burger T, Salvi D, Gatto L, Moyet L, Breckels LM, Hesse AM, Lilley KS, et al. (2014) Deciphering thylakoid sub-compartments using a mass spectrometry-based approach. Mol Cell Proteomics 13: 2147–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Abascal F, Valencia A (2017) Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci 42: 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C (2020) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 10.1093/nar/gkaa1100 [DOI] [PMC free article] [PubMed]
- Vainonen JP, Sakuragi Y, Stael S, Tikkanen M, Allahverdiyeva Y, Paakkarinen V, Aro E, Suorsa M, Scheller HV, Vener AV, et al. (2008) Light regulation of CaS, a novel phosphoprotein in the thylakoid membrane of Arabidopsis thaliana. FEBS J 275: 1767–1777 [DOI] [PubMed] [Google Scholar]
- Van Leene J, Han C, Gadeyne A, Eeckhout D, Matthijs C, Cannoot B, De Winne N, Persiau G, Van De Slijke E, Van de Cotte B, et al. (2019) Capturing the phosphorylation and protein interaction landscape of the plant TOR kinase. Nat Plants 5: 316–327 [DOI] [PubMed] [Google Scholar]
- van Wijk KJ (2000) Proteomics of the chloroplast: experimentation and prediction. Trends Plant Sci 5: 420–425 [DOI] [PubMed] [Google Scholar]
- van Wijk KJ, Friso G, Walther D, Schulze WX (2014) Meta-analysis of Arabidopsis thaliana phospho-proteomics data reveals compartmentalization of phosphorylation motifs. Plant Cell 26: 2367–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbrouck Y, Lane L, Carapito C, Duek P, Rondel K, Bruley C, Macron C, Gonzalez de Peredo A, Coute Y, Chaoui K, et al. (2016) Looking for missing proteins in the proteome of human spermatozoa: an update. J Proteome Res 15: 3998–4019 [DOI] [PubMed] [Google Scholar]
- Vanderschuren H, Lentz E, Zainuddin I, Gruissem W (2013) Proteomics of model and crop plant species: status, current limitations and strategic advances for crop improvement. J Proteomics 93: 5–19 [DOI] [PubMed] [Google Scholar]
- Venne AS, Solari FA, Faden F, Paretti T, Dissmeyer N, Zahedi RP (2015) An improved workflow for quantitative N-terminal charge-based fractional diagonal chromatography (ChaFRADIC) to study proteolytic events in Arabidopsis thaliana. Proteomics 15: 2458–2469 [DOI] [PubMed] [Google Scholar]
- Verrastro I, Pasha S, Jensen KT, Pitt AR, Spickett CM (2015) Mass spectrometry-based methods for identifying oxidized proteins in disease: advances and challenges. Biomolecules 5: 378–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialas V, Sun Z, Loureiro y Penha CV, Carrascal M, Abian J, Monteoliva L, Deutsch EW, Aebersold R, Moritz RL, Gil C (2014) A Candida albicans PeptideAtlas. J Proteomics 97: 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra RD (2012) The expanding universe of ubiquitin and ubiquitin-like modifiers. Plant Physiol 160: 2–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitorino R, Guedes S, Trindade F, Correia I, Moura G, Carvalho P, Santos MAS, Amado F (2020) De novo sequencing of proteins by mass spectrometry. Expert Rev Proteomics 17: 595–607 [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LD, Gevaert K, De Smet I (2018) Protein language: post-translational modifications talking to each other. Trends Plant Sci 23: 1068–1080 [DOI] [PubMed] [Google Scholar]
- Walley JW, Briggs SP (2015) Dual use of peptide mass spectra: protein atlas and genome annotation. Curr Plant Biol 2: 21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yu Q, Xiong H, Wang J, Chen S, Yang Z, Dai S (2016) Proteomic insight into the response of Arabidopsis chloroplasts to darkness. PLoS One 11: e0154235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tian L, Liu H, Li X, Zhang J, Chen X, Jia X, Zheng X, Wu S, Chen Y, et al. (2020) Large-scale discovery of non-conventional peptides in maize and Arabidopsis through an integrated peptidogenomic pipeline. Mol Plant 13: 1078–1093 [DOI] [PubMed] [Google Scholar]
- Waltz F, Nguyen TT, Arrive M, Bochler A, Chicher J, Hammann P, Kuhn L, Quadrado M, Mireau H, Hashem Y (2019) Small is big in Arabidopsis mitochondrial ribosome. Nat Plants 5: 106–117 [DOI] [PubMed] [Google Scholar]
- Waszczak C, Akter S, Jacques S, Huang J, Messens J, Van Breusegem F (2015) Oxidative post-translational modifications of cysteine residues in plant signal transduction. J Exp Bot 66: 2923–2934 [DOI] [PubMed] [Google Scholar]
- Willems P, Horne A, Van Parys T, Goormachtig S, De Smet I, Botzki A, Van Breusegem F, Gevaert K (2019) The Plant PTM Viewer, a central resource for exploring plant protein modifications. Plant J 99: 752–762 [DOI] [PubMed] [Google Scholar]
- Willems P, Ndah E, Jonckheere V, Stael S, Sticker A, Martens L, Van Breusegem F, Gevaert K, Van Damme P (2017) N-terminal proteomics assisted profiling of the unexplored translation initiation landscape in Arabidopsis thaliana. Mol Cell Proteomics 16: 1064–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GZ, Meyer EH, Wu S, Bock R (2019a) Extensive posttranscriptional regulation of nuclear gene expression by plastid retrograde signals. Plant Physiol 180: 2034–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GZ, Meyer EH, Richter AS, Schuster M, Ling Q, Schottler MA, Walther D, Zoschke R, Grimm B, Jarvis RP (2019b) Control of retrograde signalling by protein import and cytosolic folding stress. Nat Plants 5: 525–538 [DOI] [PubMed] [Google Scholar]
- Ytterberg J, Peltier JB, Friso G, van Wijk KJ (2002) Identification and analysis of the thylakoid membrane proteome of Arabidopsis thaliana by sequential organic solvent extraction, gel based protein separation, RP-HPLC, MALDI-TOF MS and CapLC-Q-TOF MS. American Society for Mass Spectrometry, Orlando, FL
- Zhang H, Deery MJ, Gannon L, Powers SJ, Lilley KS, Theodoulou FL (2015) Quantitative proteomics analysis of the Arg/N-end rule pathway of targeted degradation in Arabidopsis roots. Proteomics 15: 2447–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang H, Xia Y, Xiong L (2018) The caseinolytic protease complex component CLPC1 in Arabidopsis maintains proteome and RNA homeostasis in chloroplasts. BMC Plant Biol 18: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Schneider JD, Lin C, Geng S, Ma T, Lawrence SR, Dufresne CP, Harmon AC, Chen S (2019b) MPK4 phosphorylation dynamics and interacting proteins in plant immunity. J Proteome Res 18: 826–840 [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu P, Guo T, Zhao H, Bensaddek D, Aebersold R, Xiong L (2019a) Arabidopsis proteome and the mass spectral assay library. Sci Data 6: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu FY, Chen MX, Ye NH, Shi L, Ma KL, Yang JF, Cao YY, Zhang Y, Yoshida T, Fernie AR, et al. (2017) Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J 91: 518–533 [DOI] [PubMed] [Google Scholar]
- Zybailov B, Sun Q, van Wijk KJ (2009) Workflow for large scale detection and validation of peptide modifications by RPLC-LTQ-Orbitrap: application to the Arabidopsis thaliana leaf proteome and an online modified peptide library. Anal Chem 81: 8015–8024 [DOI] [PubMed] [Google Scholar]
- Zybailov B, Rutschow H, Friso G, Rudella A, Emanuelsson O, Sun Q, van Wijk KJ (2008) Sorting signals, N-terminal modifications and abundance of the chloroplast proteome. PLoS ONE 3: e1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.