

Abstract
Collagens are the most abundant proteins in the body and comprise the basement membranes and stroma through which cancerous invasion occurs; however, a pro-neoplastic function for mutant collagens is undefined. Here we identify COL11A1 mutations in 66 of 100 cutaneous squamous cell carcinomas (cSCCs), the second most common U.S. cancer, concentrated in a triple helical region known to produce trans-dominant collagens. Analysis of COL11A1 and other collagen genes found that they are mutated across common epithelial malignancies. Knockout of mutant COL11A1 impairs cSCC tumorigenesis in vivo. Compared to otherwise genetically identical COL11A1 wild-type tissue, gene-edited mutant COL11A1 skin is characterized by induction of β1 integrin targets and accelerated neoplastic invasion. In mosaic tissue, mutant COL11A1 cells enhanced invasion by neighboring wild-type cells. These results suggest that specific collagens are commonly mutated in cancer and that mutant collagens may accelerate this process.
Subject terms: Cancer, Genetics
Introduction
Cancers of the epithelial tissues that line body surfaces account for more than 90% of human malignancies and the vast majority of cancer mortality [1]. Arising in tissues, such as the skin, lung, prostate, colon, and breast, biologic malignancy of epithelial neoplasms requires neoplastic cells to invade through their underlying epithelial basement membranes into surrounding stromal tissue [2]. Understanding the mechanisms that enable local invasion in epithelial cancer progression may identify targets to interrupt the process before it leads to the negative cancer health impacts of local tissue destruction and distant metastases. The extracellular environment provides cues to enable local cancer invasion, however, the pro-invasion factors secreted from cancer cells themselves are still being characterized.
The collagen gene family [3, 4] encodes fibrillar and non-fibrillar collagen components of both basement membranes and of the extracellular stromal matrix through which cancer cells must penetrate during local neoplastic invasion. Collagens have thus been primarily appreciated for their roles in sustaining normal tissue structure as well as their functions as passive barriers to cancer cell invasion, although recent data support a role in regulating tumor immunity [5]. A number of enzymes can modify collagen proteins, including matrix metalloproteinases (MMPs) [6], which can cleave collagens to disrupt collagen matrix networks. MMP cleavage of collagens has been extensively studied in neoplasia, with a focus on efforts to develop therapeutics that inhibit this process [7]. Mutations in collagen genes themselves, however, have not been widely noted in cancer. An exception to this is COL2A1, which was observed in one study to be mutated in 37% of spontaneous human chondrosarcomas [8]. The biologic significance of COL2A1 mutations in this setting, however, is unknown. The collagen mutations evident in databases, such as those of the cancer genome atlas (TCGA), have been largely overlooked, perhaps because collagen proteins are encoded by large genes that may be susceptible to accumulating background mutations during carcinogenesis.
Among collagen genes, COL11A1 is of notable interest in cancer [9]. COL11A1 encodes the type XI collagen α1 chain, a minor fibrillar collagen whose major known action is to regulate the diameter of other major collagen protein fibrils [10]. COL11A1 overexpression has been associated with poorer progression-free survival in multiple studies [11]. The association of COL11A1 overexpression with decreased cancer survival spans a diverse array of neoplasms, including head and neck, breast, ovarian, colon, and lung, suggesting a potentially broad role for COL11A1 in a wide spectrum of cancers [9, 12–14]. Further supporting a possible role for COL11A1 in cancer are observational data linking it to angiogenesis, drug resistance and metastasis [9]. Finally, COL11A1 is a protein binding partner of COL2A1, noted above, suggesting that disruption of the COL11A1–COL2A1-containing heterotrimer could play potential a role in cancer. Mutant COL11A1, however, has not been characterized in cancer and a functional role for the COL11A1 protein in promoting neoplastic progression to cancerous invasion in tissue has not been studied.
Cutaneous squamous cell carcinoma (cSCC) has a U.S. annual incidence estimated as high as 2.7 million [15] and may cause comparable numbers of deaths to malignant melanoma [16]. cSCC commonly arises in sun-exposed skin and progresses from premalignant actinic keratoses and cSCC in situ to locally invasive cancer by invasion through the epidermal basement membrane. cSCC is thus a prototype of epithelial malignancy, which progresses through discrete local stages prior to metastasis. The latter occurs in ~4% of cSCC patients, with roughly a third of metastatic cSCC leading to death [17]. Because of the accessibility and tractability of skin to organoid formation and grafting, human skin tissue has been amenable to production of a variety of cancer models that accurately reflect the tissue architecture and gene expression observed in spontaneous cSCC arising in humans [18–20]. A number of mutations have been reported in cSCC studies, with TP53 the most commonly mutated gene noted [21]. A number of other genes implicated in cancer pathogenesis have also been observed to be recurrently mutated in human cSCC, such as NOTCH1, CDKN2A, KNSTRN, and HRAS [22–25]. cSCC, however, was not a focus of TCGA efforts and its mutational spectrum has not been fully characterized.
To begin to address this, we analyzed the mutational spectrum of 100 cSCC exomes as well as 100 patient- and site-matched normal control skin samples, including 67 previously published tissue pairs and 33 new for this study. Surprisingly, COL11A1 was identified among the top recurrently mutated genes in cSCC. COL11A1 point mutations were found to both substantially boost local neoplastic invasion in vivo as well as to be required for subcutaneous tumorigenic growth in cSCC models. Mutant COL11A1-activated β1 integrin targets and accelerated neoplastic invasion non-cell autonomously. In TCGA data, collagen gene mutations were found widely across other common epithelial cancers. Rather than predominantly functioning as passive structural conduits for cancer invasion, therefore, collagens are also frequently mutated in epithelial cancers where they can accelerate neoplastic progression.
Results
Somatic COL11A1 mutations in cSCC and other human cancers
We analyzed whole exome data of 100 cSCCs with patient-matched skin as control, including 33 newly generated for this study (SI Appendix, Table S1) [22–25]. These 100 cSCCs displayed a transition-rich mutational profile consistent with ultraviolet light exposure (SI Appendix, Fig. S1a) and featured mutations in genes that have previously been well-studied in cSCC, including TP53 (78%), NOTCH1 (59%), CDKN2A (41%), KNSTRN (17%), and HRAS (16%), at frequencies comparable to those seen in prior work (Fig. 1a and SI Appendix, Table S2) [21–26]. COL11A1 was the third most commonly mutated gene (66%) after TP53 and CDKN2A upon normalizing for coding sequence transcript length. Analysis of mutation type confirmed that while canonical tumor suppressors such as TP53, CDKN2A, and NOTCH1 frequently harbor premature stop codons, cSCC-associated somatic variants in COL11A1 were primarily missense mutations, suggesting functional consequences at the protein level that might be more nuanced than simple loss-of-function (Fig. 1b). The nature of COL11A1 mutations was similar across cSCC datasets examined. In total, 61% (110 of 180) of these mutations affected glycine and proline residues, notably within Gly-X-Y triple helical repeats. We confirmed COL11A1 mutations by Sanger sequencing (SI Appendix, Fig. S1b, c) and detected COL11A1 protein in every cSCC from an independent series (n = 76) of human tumors (SI Appendix, Fig. S1d, e), verifying its presence in this cancer. The predominance of missense glycine and proline substitutions (83%, 55 of 66 cSCC) concentrated in the triple helical region (Fig. 1c) indicated that these mutations might alter Gly-X-Y (with X and Y frequently proline) collagenous tripeptide [27] repeats, a phenomenon well-characterized for COL7A1 [28–30] that disrupts the collagen triple helix and produces a dominant-negative collagen protein. In total, 42% (59 of 139) mutations detected in the COL11A1 triple helical region were glycine substitutions, and 27% (38 of 139) altered a proline. Although their cancer incidence is unreported, the very rare autosomal dominant connective tissue disorders, Stickler and Marshall syndromes, can be caused by similar mutant COL11A1 glycine substitutions in the Gly-X-Y tripeptide [31, 32], indicating that such mutations can act dominantly. COL11A1 is thus the third most commonly mutated gene in cSCC, with a mutation pattern suggesting potential trans-dominant function.
Fig. 1. COL11A1 is frequently mutated in cSCC and other human cancers.

a Top mutated genes in 100 cSCC. Ranking is limited to genes with expression in keratinocytes and mutation frequency is shown in parentheses next to each gene name. b Analysis of the genes in a by mutation type. c Mutation frequency in COL11A1 coding sequence. The number of mutations per amino acid (AA) is shown. d Mutation frequency calculated from The Cancer Genome Atlas (TCGA) of collagen genes with selected frequently mutated cancer genes from the Catalog of Somatic Mutations in Cancer (COSMIC) shown for reference.
To determine if COL11A1 mutation is unique to cSCC, additional tumor types were examined. After assessment for background mutation rate differences in each tumor type, analysis of cancer sequencing data from TCGA found COL11A1 commonly mutated across multiple epithelial malignancies, including cancers of the lung, esophagus, stomach, cervix, and colon (Fig. 1d). Mutations in other collagens, such as COL6A6, COL22A1, COL6A3, COL12A1, and COL14A1, were also noted in a variety of tumor types. Cancers displaying the highest frequency of collagen gene family mutations independent of COL11A1 included cutaneous, gastrointestinal (esophageal, colorectal), urogenital (cervical, bladder), and lung neoplasms. Biologic malignancy in each of these tissues requires epithelial cells to invade through the epithelial basement membrane to penetrate into the underlying stroma. Collagen mutations are therefore a common feature of epithelial cancers.
Mutant COL11A1 promotes tumorigenesis
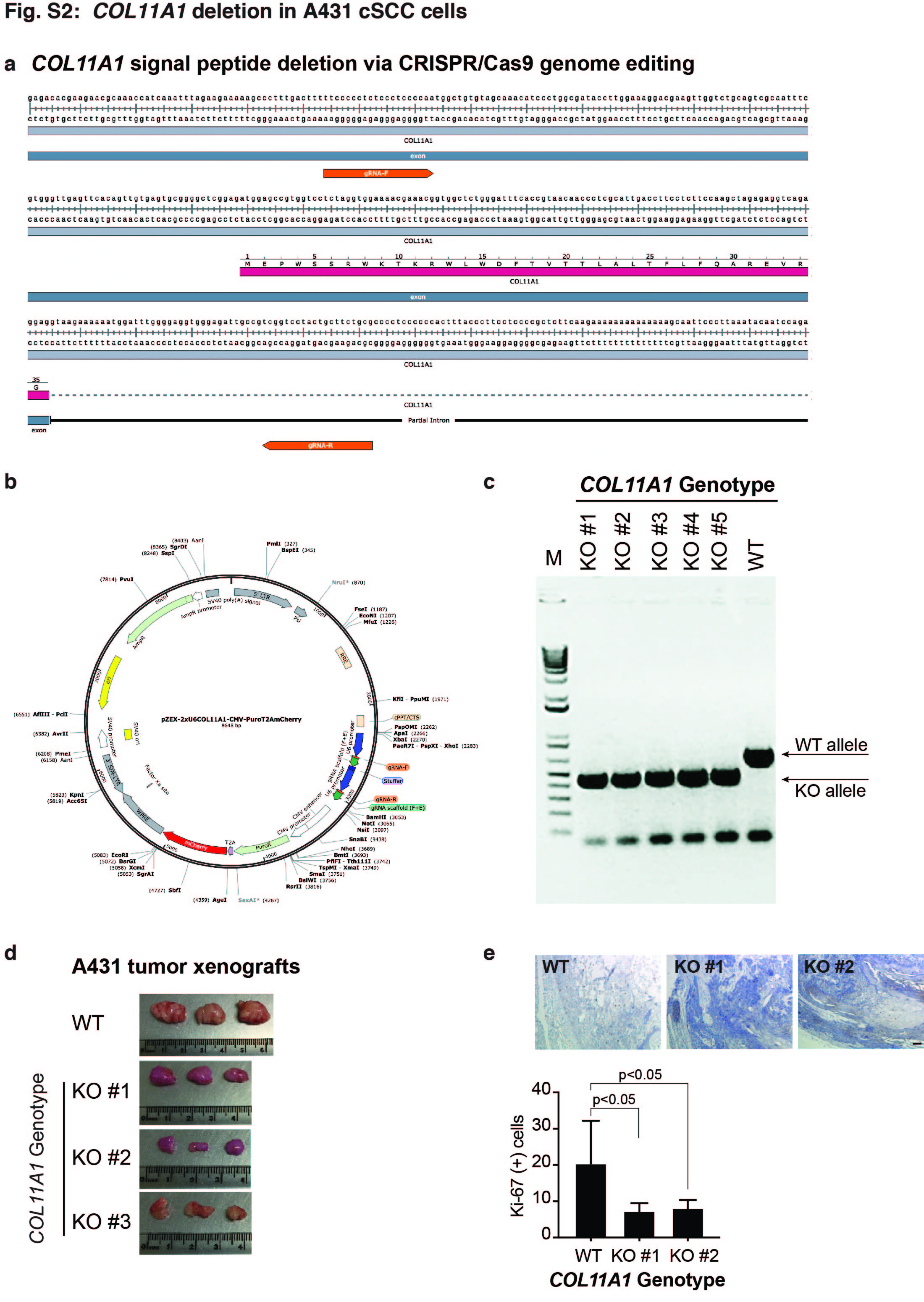
To investigate the functional consequences of mutant COL11A1 in cSCC, we established subcutaneous tumors in immune deficient mice using human A431 cSCC cells, which contain a G598A mutation in COL11A1 representative of the glycine substitutions observed in cSCCs; A431 cells produce rapidly growing intradermal and subcutaneous tumors in this setting [33, 34]. CRISPR-mediated ablation of COL11A1 in three independently derived A431 clones was associated with markedly impaired tumorigenic growth in vivo (Fig. 2a, b and SI Appendix, Fig. S2a–d). COL11A1 knockout A431 tumors displayed evidence of modestly decreased proliferation in vivo (SI Appendix, Fig. S2e). These mutant COL11A1 knockout data support the possibility that mutated COL11A1 may contribute to tumorigenesis.
Fig. 2. Mutant COL11A1 promotes tumorigenesis.

a Tumor volume of A431 cSCC subcutaneous mouse xenografts. Parental A431 cells (WT) are compared to three independently derived COL11A1 knockout clones (KO). b Tumor weight 35 days post-injection. c Isogenic, otherwise genetically identical human skin organoids programmed for invasive neoplasia by HRASG12V and Cdk4 with endogenous COL11A1 edited to either cSCC-associated G598A or synonymous G598G wild-type control. Scale bar, 50 µm; arrowheads denote invasive cells, collagen VII staining defines the epidermal basement membrane. d Invasion index (number of keratin-positive cells/100 µm of basement membrane) of c; G/A, COL11A1G598A and G/G, COL11A1G598G. e Parallel experiment from c xenografted onto scid/scid mice. Scale bar, 50 µm. f Invasion index of d.
Epithelial cancers, including cSCC, arise via neoplastic invasion of epithelial cells through the underlying basement membrane. This process of tumorigenic progression is bypassed by tissue injection of cancer cell lines. Therefore, the impact of mutant COL11A1 was next assessed in a more veridical model free of cell line-specific idiosyncrasies. A human skin tissue model of Ras-driven epidermal neoplasia, in which normal epidermis undergoes in situ neoplasia followed by progression to full invasion through the epidermal basement membrane, was therefore used to address the role of mutant COL11A1 in early malignant progression. In both organoids and skin xenografts, this model accurately recapitulates cSCC at the levels of histology, protein markers, and global gene expression [18–20]. In this model, expression of HRASG12V and Cdk4, which reflect Ras-MAP kinase activation [23–25, 35] as well as inactivation of the CDKN2A-mediated G1 restraints characteristic of cSCC [18–20, 24, 36, 37] (Fig. 1a and SI Appendix, Table S2), are sufficient to drive cancer progression by normal diploid epidermal keratinocytes.
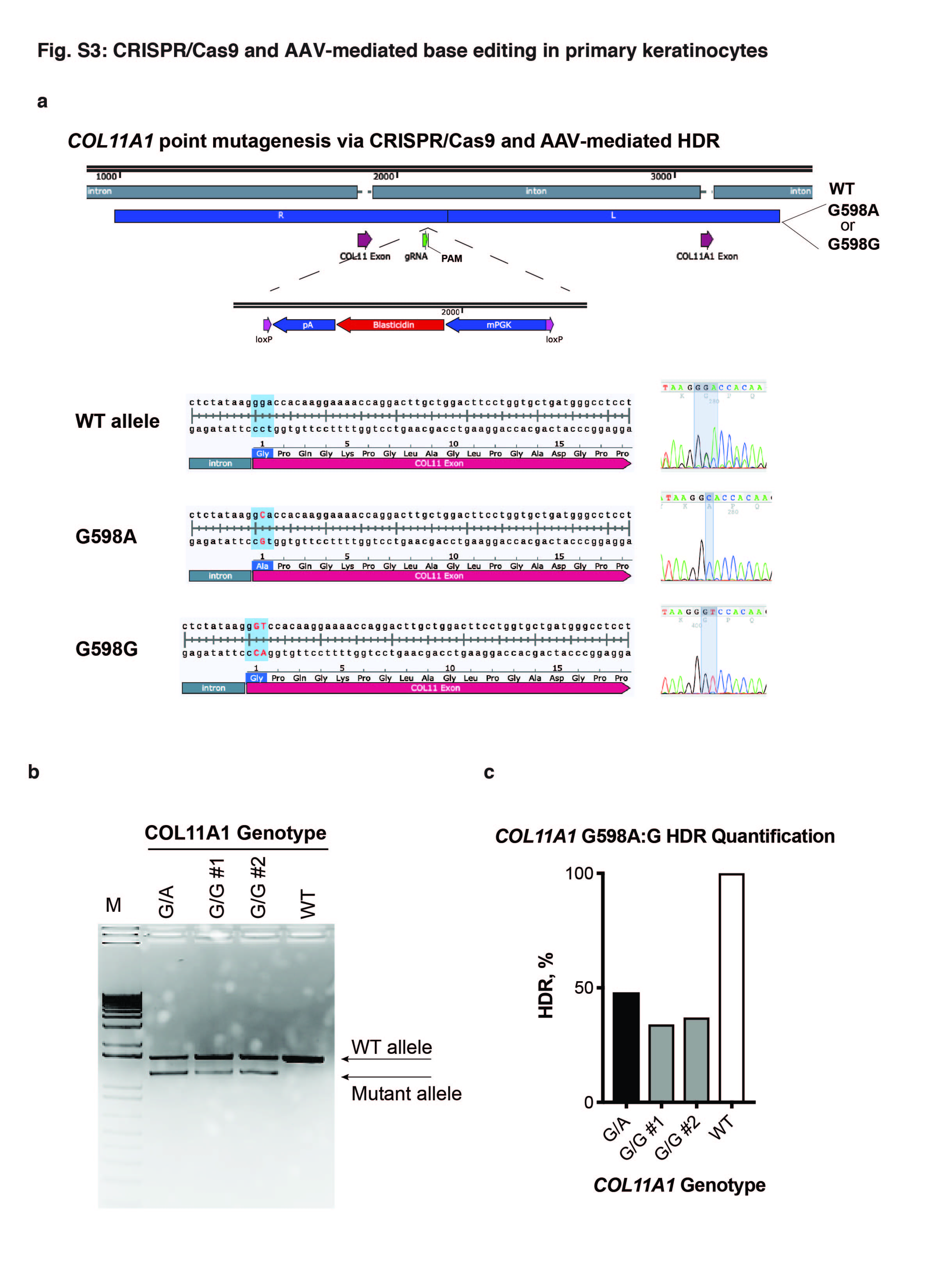
To define the impact of mutant COL11A1 on neoplastic progression to local invasion in the context of three-dimensional human skin tissue, wild-type human keratinocytes were edited to introduce the cSCC-associated COL11A1G598A mutation using a combined CRISPR-Cas9 and adeno-associated virus-driven homology directed recombination approach (SI Appendix, Fig. S3) [38–40]. In parallel, keratinocytes from the same parental cell pool were edited with an A to T substitution that preserves the native glycine residue while controlling for any non-specific effects of editing the COL11A1 locus (COL11A1G598G). These cells were used to regenerate epidermis on normal human dermis, producing two sets of human tissues for skin organoids and xenografts that differ by only a single nucleotide at the endogenous COL11A1 locus. In human skin organoids, COL11A1 mutation was associated with quantitatively increased neoplastic invasion (Fig. 2c, d) through the epidermal basement membrane into underlying dermal stroma, compared to otherwise genetically identical, gene-edited wild-type COL11A1 control tissue. This result was replicated in xenografted human skin tissue in vivo (Fig. 2e, f), further confirming that cancer-associated mutations in the endogenous COL11A1 locus are associated with enhanced neoplastic invasion in organoid tissues and in vivo.
Mutant COL11A1 gene expression and survival
To define the changes in gene expression induced by COL11A1 point mutation in epidermis undergoing Ras-driven neoplasia, RNA expression was next compared in triplicate isogenic COL11A1 mutant and triplicate wild-type tissues generated from identical donor cells that differ only in at the single-edited COL11A1 nucleotide. Compared to control-edited COL11A1 wild-type tissue, mRNA expression for 506 genes was significantly changed (increased or decreased) by COL11A1 point mutation. The 264 upregulated genes were enriched for links to β1 integrin signaling, focal adhesion, and features of extracellular matrix-receptor interactions (Fig. 3a, b). We next evaluated the association between this 264-gene signature upregulated by mutant COL11A1 and survival in head and neck SCC, which like cSCC is also a malignancy of stratified epithelium but which differs from cSCC in that it presently has long-term survival data available. Increased expression of the mutant COL11A1-activated gene signature was associated with decreased survival and demonstrated a 34% increase in hazard ratio (HR = 1.34, 95% CI = 1.14–1.59, p = 5.56e–4) and added 40% more information to a predictive model based solely on age, gender, clinical stage, and radiation therapy (Likelihood ratio test, p = 3.04e–4) (Fig. 3c and SI Appendix, Fig. S4a, b). High expression of the mutant COL11A1-activated gene signature was similarly associated with decreased survival in cervical cancer (HR = 1.41, 95% CI = 1.10–1.81, p = 6.29e–3) as well as lung SCC (HR = 1.16, 95% CI = 1.01–1.34, p = 3.79e−2), adding 27% and 26% more information, respectively, to a predictive model based on age, gender, clinical stage, and radiation therapy (Likelihood ratio test, p = 4.61e−3 and 3.66e−2) (Fig. 3d and SI Appendix, Fig. S4c–f). The gene signature upregulated by mutant COL11A1 therefore correlates with decreased survival across multiple tumor types. Consistent with a potential link to integrin-driven gene expression, mutant COL11A1 tissue displayed phosphorylated focal adhesion kinase, a downstream target of β1 integrin activation (SI Appendix, Fig. S4g). COL11A1 point mutation therefore induces a gene set associated with overall worse cancer survival that is enriched for links to integrin signaling.
Fig. 3. Genes induced by mutant COL11A1 are associated with integrin signaling and worse survival.

a Mean-centered, hierarchical clustering of 506 genes altered (adjusted p value <0.05) in isogenic human skin organoids with their endogenous COL11A1 alleles edited to cSCC-associated G598A or synonymous G598G as control and programmed to transform into invasive neoplasia by oncogenic Ras and Cdk4. FC fold change. b Enriched GO terms of genes upregulated by COL11A1G598A in RNA-Seq. c, d Kaplan–Meier estimate of overall survival using the mutant COL11A1-activated 264-gene signature in TCGA patients from the Head and Neck Squamous Cell Carcinoma (HNSC) and Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (CESC) cohorts, tumors chosen because, like cSCC, they comprise stratified epithelial cancers but which, unlike cSCC, have substantial sized cohorts of patient survival data.
Tissue mosaic for mutant COL11A1
The fact that collagens are secreted proteins, the resemblance of COL11A1 tripeptide sequence mutants in cSCC to the dominant-negative secreted mutant COL7A1 protein seen in dominant dystrophic epidermolysis bullosa [41, 42], and the evidence above supporting engagement of integrin activation in tissue by mutant COL11A1 all raised the possibility that mutant COL11A1 secreted from tumor cells might act on adjacent COL11A1 wild-type tumor cells. If so, cells expressing mutant COL11A1 protein should be able to enhance neoplastic invasion by adjacent COL11A1 wild-type tumor cells when both contain oncogenic drivers.
To test this, mosaic human epidermis was generated in which all keratinocytes express HRASG12V as well as Cdk4 and were also edited to achieve either endogenous expression of mutant COL11A1G598A or wild-type COL11A1G598G as control (Fig. 4a). A subset of epidermal cells edited to wild-type COL11A1G598G were marked with a hemagglutinin (HA)-tagged Keratin 14 expression vector and mixed at identical known percentages with unmarked COL11A1G598A or COL11A1G598G cells. Mosaic tissues with mutant COL11A1 demonstrated quantitatively enhanced Ras-Cdk4-driven invasion over tissues mosaic for wild-type COL11A1 (Fig. 4b, c) and the proportion of HA-tagged COL11A1G598G cells crossing the epidermal basement membrane was significantly higher in mosaic tissues with mutant COL11A1 compared to wild-type COL11A1 mosaic tissues (Fig. 4d). The enhanced invasion by wild-type COL11A1 cells adjacent to mutant COL11A1 cells indicates that COL11A1 mutations observed in cSCC can increase neoplastic invasion by adjacent COL11A1 wild-type tumor cells.
Fig. 4. Tissue mosaic for mutant COL11A1.

a Schematic illustrating generation of COL11A1 mosaic human skin neoplasia. b HRASG12V and Cdk4 human skin tissues generated from keratinocytes with either endogenous COL11A1 edited to cSCC-associated G598A or synonymous G598G as control; a subset of the latter group is further labeled with HA-epitope-tagged keratin 14 (G/G:K14-HA). Arrows point to the BMZ. Scale bar, 50 µm and technical replicates are shown. c Invasion index of COL11A1G598A (G/A) and COL11A1G598G (G/G) keratin 5-positive cells in b. d Invasion index of HA-tagged COL11A1G598G (G/G: K14-HA) keratinocytes in COL11A1G598A (G/A) and COL11A1G598G (G/G) tissues in b.
Discussion
Here we show that COL11A1 is recurrently mutated in cSCC and that its cancer-associated mutation boosts neoplastic invasion in tissue. Mutant COL11A1 enhanced invasion in both organoids and in vivo xenografts of human skin tissue and its targeted disruption blocked subcutaneous tumor growth as well. When compared to otherwise identical isogenic skin tissue, COL11A1 point mutation engaged a gene expression program associated with β1 integrin activation. COL11A1 was found to be mutated in additional epithelial cancers along with additional collagens, including COL6A6, COL22A1, COL6A3, COL12A1, and COL14A1. We did not observe a strong correlation between COL11A1 mutations and mutations in COL2A1, COL11A2, COL5A1, or COL5A2, which encode distinct alpha chains that can form heterotrimers with collagen XI (SI Appendix, Fig. S5a, b). Similarly, examination of helix-disrupting somatic variants in the most frequently mutated collagen genes did not reveal strong mutational co-occurrence with COL11A1 (SI Appendix, Fig. S5b). Mutant collagens include not just fibrillar collagens but also non-fibrillar basement membrane collagens, such as COL4A4, which was highly mutated across its multiple domains in cSCC (SI Appendix, Fig. S5c). These data suggest that mutations in a variety of collagen genes may play potential roles in cancer pathogenesis across diverse cancer types.
Acceleration of neoplastic invasion by mutant collagens may occur via a number of mechanisms. The trimeric structure of collagens make them susceptible to disruption by dominant-negative mutants, much like another structural family of multimers, the keratins [43]. Such mutants may disrupt structural integrity of fibrillar and non-fibrillar collagenous components of the very barriers they normally maintain. Additionally, and possibly as a consequence of such disruption of collagen multimers, possibly in a trans-dominant fashion for mutants such as G598A in COL11A1, mutant collagens may trigger receptors on cancer cells, such as integrins, which normally sense alterations in the extracellular environment in normal wound repair [44]. The precise factors promoting the increased invasion observed in mutant COL11A1 tissue, however, are not defined and may include synergy with well-characterized secreted factors, such as TGFB. The high incidence of COL11A1 mutations in the primary cSCC tumors studied here suggest that COL11A1 could act primarily in the process of local tumorigenesis as opposed to functioning as a strong driver of metastatic spread in this setting.
Based on similarities in gene expression programs, cancer has been compared to wounds that do not heal [45], suggesting that aberrant activation of wounding programs, such as those that drive cellular migration, may contribute to cancer progression. In the case of COL11A1, because it helps control fibril diameter of major stromal constituent collagens, such as type I collagens, its mutation in cSCC may disrupt the normal dermal collagenous matrix in such a fashion as to trigger such an aberrant wounding response. Assessment of collagen fibrils in COL11A1 wild-type and mutant tissues stained with Masson’s trichrome to highlight collagens did not show quantitatively significant differences in our model cSCCs (Fig. S6A), however, more detailed ultrastructural studies are needed to definitively assess such potential impacts. Interestingly, COL11A1 is not widely overexpressed in cSCCs, at either the mRNA [46] or protein levels, as shown here, raising the possibility that COL11A1 mutations may in fact lead to a less stable protein, as is seen with other cancer mutant proteins. More generally, the common occurrence of collagen mutations in human cancer raises the possibility that these and other potential mechanisms operate via a variety of collagens in diverse epithelial malignancies.
Tumor cells interact with extracellular matrix proteins as they traverse both the basement membrane as well as its underlying stroma. Collagens comprise central proteinaceous components of both structures and are normally secreted into these locations by overlying epithelial cells as well as by stromal cells, most notably fibroblasts [47–53]. Interestingly, laser capture microdissection and sequencing of fibroblast regions of COL11A1 mutant cSCC found only wild-type COL11A1 sequence (Fig. S6B), consistent with a model in which epithelial cells serve as the source of mutant COL11A1 in cSCC. The present data thus support a model in which tumor cell-expressed mutant collagens boost a central process in cancer progression, namely invasion through the underlying epithelial basement membrane into the underlying stroma.
Materials and methods
Please see additional methods in Supplementary Information.
Tumor tissues
Cutaneous cSCC and patient-matched normal adjacent skin were collected following informed consent under a protocol approved by the Institutional Review Board at Stanford University. All tissues were analyzed by frozen section histology and samples with heavy neutrophilic infiltrate or widespread necrosis were excluded. Genomic DNA was isolated from all specimens using the DNeasy Blood and Tissue kit (Qiagen). Whole exome capture (Agilent SureSelect Human All Exon V5), library preparation, and sequencing were performed by Centrillion Technologies (Palo Alto, CA).
Immunohistochemistry
COL11A1 immunohistochemistry (1:300, Biorbyt) was performed on a skin cancer and normal tissue microarray (Biomax) by the Stanford University Human Pathology/Histology Service Center. Ki-67 immunohistochemistry (1:200, Dako) was similarly performed on A431 xenograft tumors.
RNA isolation and sequencing
Total RNA was then isolated from epidermis of the organoid skin samples using the QIAshredder (Qiagen) and Trizol (Thermo Fisher) followed by DNA removal with the TURBO DNA-free kit (Ambion) according to the manufacturer’s instructions. RNA integrity was verified using an Agilent 2100 Bioanalyzer. RNA-Seq libraries were prepared with the mRNA Seq Sample Prep Kit (Illumina, Inc.) as recommended by the manufacturer. Seventy-five bp paired-end sequencing reads were obtained using the Illumina HiSeq platform. DESeq was used to call differential gene expression with an adjusted p value cutoff of p < 0.05 [54].
In vivo subcutaneous tumor formation assays
All experiments were performed with the approval of the Stanford University Administrative Panel on Laboratory Animal Care. In total, 1 × 106 A431 clones engineered to contain deletion in COL11A1 were suspended in a volume of 150 μl containing 50% Matrigel (BD biosciences) and injected with a 31 g needle into the subcutaneous space of immunodeficient 6–8 week old SHO female mice (Charles River). Tumor growth kinetics were monitored by caliper measurements weekly for 5 weeks. Tumor volume was calculated using formula V = 1/2(L × W × H), where L—length, W—width, and H—height of the tumor in mm. Tumors were explanted and weighed at the end of the experiment.
Each A431 clone was injected subcutaneously into a single mouse flank (n = 5 mice per clone). Parental A431 cells were injected subcutaneously into the contralateral flank (n = 15 mice). The sample size for these experiments was selected based on results from pilot studies and prior experience with tumor xenografts in mice. Sample exclusion criteria were preestablished and comprised of technical failures such as mis-injection of cells. Physical randomization was performed using animal tag number. Animal studies were not performed in a blinded fashion.
In vivo skin xenograft model
For the composite skin graft production, neoplastic organoids consist of G/G or G/A keratinocytes were prepared as described above and at day 6 grafted on NOD SCID Gamma mice (Jackson) as previously described [55]. Briefly, 6–8 week old mice were anesthetized using isofluorane and after shaving the hair from the mouse flank, a rectangular region of mouse skin (~1.6 × 1.4 cm) was removed using a scalpel. A human skin organoid was then sutured to the mouse skin and dressed with the non-adherent dressing TELFA (Tyco Healthcare/Kendall) secured by Tegaderm (3M Health Care) and Coverlet adhesive dressing (BSN-JOBST). Finally, a double layer of Co-Flex (Andover) was wrapped around the mouse. The dressing was removed 9–12 days post-grafting and grafts were then further characterized. Five mice received COL11A1G598A xenografts and five received COL11A1G598G xenografts.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the Lee and Khavari Labs for helpful discussions, the Department of Veterans Affairs, Office of Research and Development (CSL, PAK), VA Merit Review BX001409 and NIH CA142635 and AR43799 (PAK). CSL, ZS, and PAK conceived the project, analyzed data, and wrote the manuscript. CSL, AM, RMS, and ZS performed experiments; SZA performed tissue acquisition; TB and YC analyzed data. Exome and RNA sequencing data may be accessed in GEO under accession #GSE179297. Instructions and code to reproduce results from this manuscript are available at https://github.com/tjbencomo/col11a1-manuscript.
Author contributions
CSL and PAK conceived the project; CSL, ZS, AM, TB, LEE, YC, SZA, and RMS provided key reagents and performed experiments; CSL, ZS, TB, YC, and PAK analyzed data; and CSL and PAK wrote the manuscript with input from all authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Carolyn S. Lee, Email: carilee@stanford.edu
Paul A. Khavari, Email: khavari@stanford.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s41388-021-02013-y.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Stuelten CH, Parent CA, Montell DJ. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer. 2018;18:296–312. doi: 10.1038/nrc.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorushanova A, Delgado LM, Wu Z, Shologu N, Kshirsagar A, Raghunath R, et al. The collagen suprafamily: from biosynthesis to advanced biomaterial development. Adv Mater. 2019;31:e1801651. doi: 10.1002/adma.201801651. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–65. doi: 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2015;2:26–34. doi: 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923–6. doi: 10.1038/ng.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raglow Z, Thomas SM. Tumor matrix protein collagen XIα1 in cancer. Cancer Lett. 2015;357:448–53. doi: 10.1016/j.canlet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hida M, Hamanaka R, Okamoto O, Yamashita K, Sasaki T, Yoshioka H, et al. Nuclear factor Y (NF-Y) regulates the proximal promoter activity of the mouse collagen α1(XI) gene (Col11a1) in chondrocytes. Vitr Cell Dev Biol Anim. 2014;50:358–66. doi: 10.1007/s11626-013-9692-3. [DOI] [PubMed] [Google Scholar]
- 11.Jia D, Liu Z, Deng N, Tan TZ, Huang RY-J, Taylor-Harding B, et al. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. 2016;382:203–14. doi: 10.1016/j.canlet.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y-H, Chang T-H, Huang Y-F, Huang H-D, Chou C-Y. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene. 2014;33:3432–40. doi: 10.1038/onc.2013.307. [DOI] [PubMed] [Google Scholar]
- 13.Shen L, Yang M, Lin Q, Zhang Z, Zhu B, Miao C. COL11A1 is overexpressed in recurrent non-small cell lung cancer and promotes cell proliferation, migration, invasion and drug resistance. Oncol Rep. 2016;36:877–85. doi: 10.3892/or.2016.4869. [DOI] [PubMed] [Google Scholar]
- 14.Chong I-W, Chang M-Y, Chang H-C, Yu Y-P, Sheu C-C, Tsai J-R, et al. Great potential of a panel of multiple hMTH1, SPD, ITGA11 and COL11A1 markers for diagnosis of patients with non-small cell lung cancer. Oncol Rep. 2006;16:981–8. [PubMed] [Google Scholar]
- 15.Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence estimate of nonmelanoma skin cancer (Keratinocyte Carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015;151:1081–6. doi: 10.1001/jamadermatol.2015.1187. [DOI] [PubMed] [Google Scholar]
- 16.Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018;78:237–47. doi: 10.1016/j.jaad.2017.08.059. [DOI] [PubMed] [Google Scholar]
- 17.Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. 2013;68:957–66. doi: 10.1016/j.jaad.2012.11.037. [DOI] [PubMed] [Google Scholar]
- 18.Reuter JA, Ortiz-Urda S, Kretz M, Garcia J, Scholl FA, Pasmooij AMG, et al. Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell. 2009;15:477–88. doi: 10.1016/j.ccr.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridky TW, Chow JM, Wong DJ, Khavari PA. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med. 2010;16:1450–5. doi: 10.1038/nm.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, et al. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat Med. 2013;19:626–30. doi: 10.1038/nm.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–8. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, Dimon M, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014;134:2630–8. doi: 10.1038/jid.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durinck S, Ho C, Wang NJ, Liao W, Jakkula LR, Collisson EA, et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Disco. 2011;1:137–43. doi: 10.1158/2159-8290.CD-11-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CS, Bhaduri A, Mah A, Johnson WL, Ungewickell A, Aros CJ, et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat Genet. 2014;46:1060–2. doi: 10.1038/ng.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582–92. doi: 10.1158/1078-0432.CCR-14-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA. 2011;108:17761–6. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heidemann E, Roth W. Synthesis and investigation of collagen model peptides. In: Polymerizations and polymer properties. Berlin, Heidelberg: Springer; 1982. p. 143–203.
- 28.Uitto J, Pulkkinen L, Christiano AM. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: mutations in the type VII collagen and kalinin (laminin 5) genes. J Invest Dermatol. 1994;103:39S–46S. doi: 10.1038/jid.1994.8. [DOI] [PubMed] [Google Scholar]
- 29.Kuivaniemi H, Tromp G, Prockop DJ. Mutations in collagen genes: causes of rare and some common diseases in humans. FASEB J. 1991;5:2052–60. doi: 10.1096/fasebj.5.7.2010058. [DOI] [PubMed] [Google Scholar]
- 30.Kivirikko KI. Collagens and their abnormalities in a wide spectrum of diseases. Ann Med. 1993;25:113–26. doi: 10.3109/07853899309164153. [DOI] [PubMed] [Google Scholar]
- 31.Richards AJ, Yates JR, Williams R, Payne SJ, Pope FM, Scott JD, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet. 1996;5:1339–43. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
- 32.Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65:974–83. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang JY, Adams AE, Ridky TW, Tao S, Khavari PA. Tumor necrosis factor receptor 1/c-Jun-NH2-kinase signaling promotes human neoplasia. Cancer Res. 2007;67:3827–34. doi: 10.1158/0008-5472.CAN-06-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 35.van der Schroeff JG, Evers LM, Boot AJ, Bos JL. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J Invest Dermatol. 1990;94:423–5. doi: 10.1111/1523-1747.ep12874504. [DOI] [PubMed] [Google Scholar]
- 36.Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 37.Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8:1105–14. doi: 10.1038/nm779. [DOI] [PubMed] [Google Scholar]
- 38.Bak RO, Dever DP, Reinisch A, Cruz Hernandez D, Majeti R, Porteus MH. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. Elife. 2017;6. 10.7554/eLife.27873. [DOI] [PMC free article] [PubMed]
- 39.Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13:358–76. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J-S, Bonifant C, Bunz F, Lane WS, Waldman T. Epitope tagging of endogenous genes in diverse human cell lines. Nucleic Acids Res. 2008;36:e127. doi: 10.1093/nar/gkn566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammami-Hauasli N, Schumann H, Raghunath M, Kilgus O, Lüthi U, Luger T, et al. Some, but not all, glycine substitution mutations in COL7A1 result in intracellular accumulation of collagen VII, loss of anchoring fibrils, and skin blistering. J Biol Chem. 1998;273:19228–34. doi: 10.1074/jbc.273.30.19228. [DOI] [PubMed] [Google Scholar]
- 42.Fritsch A, Spassov S, Elfert S, Schlosser A, Gache Y, Meneguzzi G, et al. Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. J Biol Chem. 2009;284:30248–56. doi: 10.1074/jbc.M109.045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLean WHI, Moore CBT. Keratin disorders: from gene to therapy. Hum Mol Genet. 2011;20:R189–197. doi: 10.1093/hmg/ddr379. [DOI] [PubMed] [Google Scholar]
- 44.Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–67. doi: 10.1016/j.ccell.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dvorak HF. Tumors: wounds that do not heal. Similarities tumor Strom Gener wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 46.Lee CS, Mah A, Aros CJ, Lopez-Pajares V, Bhaduri A, Webster DE, et al. Cancer-associated long noncoding RNA SMRT-2 controls epidermal differentiation. J Invest Dermatol. 2018;138:1445–9. doi: 10.1016/j.jid.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Supp DM, Hahn JM, Combs KA, McFarland KL, Schwentker A, Boissy RE, et al. Collagen VII expression is required in both keratinocytes and fibroblasts for anchoring fibril formation in bilayer engineered skin substitutes. Cell Transpl. 2019;28:1242–56. doi: 10.1177/0963689719857657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marinkovich MP, Keene DR, Rimberg CS, Burgeson RE. Cellular origin of the dermal-epidermal basement membrane. Dev Dyn. 1993;197:255–67. doi: 10.1002/aja.1001970404. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling somatic SNVs and indels with Mutect2. bioRxiv. 2019:861054. 10.1101/861054.
- 52.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–4. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choate KA, Medalie DA, Morgan JR, Khavari PA. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat Med. 1996;2:1263–7. doi: 10.1038/nm1196-1263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.