

Key Points
Question
What is the clinical spectrum of calcium-release deficiency syndrome (CRDS), a recently described arrhythmogenic syndrome caused by a ryanodine receptor 2 (RyR2) loss-of-function variant?
Findings
In this cohort study of patients undergoing testing for CRDS and family-based screening, 6 RyR2 loss-of-function variants were found in 6 probands and 13 family members with life-threatening arrhythmias without evident catecholaminergic polymorphic ventricular tachycardia during exercise testing; variants were associated with normal or near-normal exercise testing results, features consistent with CRDS. Ventricular fibrillation was preceded by a spontaneous ectopy pattern similar to the long-burst, long-pause, short-coupled ventricular extra-stimulus CRDS protocol.
Meaning
These findings suggest that CRDS is an emerging syndrome that differs from RyR2 gain-of-function catecholaminergic polymorphic ventricular tachycardia.
This cohort study uses in vitro functional characterization of candidate RyR2 variants to assess the diagnosis of calcium-release deficiency syndrome in probands and their relatives.
Abstract
Importance
Calcium-release deficiency syndrome (CRDS), which is caused by loss-of-function variants in cardiac ryanodine receptor 2 (RyR2), is an emerging cause of ventricular fibrillation. However, the lack of complex polymorphic/bidirectional ventricular tachyarrhythmias during exercise stress testing (EST) may distinguish it from catecholaminergic polymorphic ventricular tachycardia (CPVT). Recently, in the first clinical series describing the condition, mouse and human studies showed that the long-burst, long-pause, short-coupled ventricular extra stimulus (LBLPS) electrophysiology protocol reliably induced CRDS ventricular arrhythmias. Data from larger populations with CRDS and its associated spectrum of disease are lacking.
Objective
To further insight into CRDS through international collaboration.
Design, Setting, and Participants
In this multicenter observational cohort study, probands with unexplained life-threatening arrhythmic events and an ultrarare RyR2 variant were identified. Variants were expressed in HEK293 cells and subjected to caffeine stimulation to determine their functional impact. Data were collected from September 1, 2012, to March 6, 2021, and analyzed from August 9, 2015, to March 6, 2021.
Main Outcomes and Measures
The functional association of RyR2 variants found in putative cases of CRDS and the associated clinical phenotype(s).
Results
Of 10 RyR2 variants found in 10 probands, 6 were loss-of-function, consistent with CRDS (p.E4451del, p.F4499C, p.V4606E, p.R4608Q, p.R4608W, and p.Q2275H) (in 4 [67%] male and 2 [33%] female probands; median age at presentation, 22 [IQR, 8-34] years). In 5 probands with a documented trigger, 3 were catecholamine driven. During EST, 3 probands with CRDS had no arrhythmias, 1 had a monomorphic couplet, and 2 could not undergo EST (deceased). Relatives of the decedents carrying the RyR2 variant did not have EST results consistent with CPVT. After screening 3 families, 13 relatives were diagnosed with CRDS, including 3 with previous arrhythmic events (23%). None had complex ventricular tachyarrhythmias during EST. Among the 19 confirmed cases with CRDS, 10 had at least 1 life-threatening event at presentation and/or during a median follow-up of 7 (IQR, 6-18) years. Two of the 3 device-detected ventricular fibrillation episodes were induced by a spontaneous LBLPS-like sequence. β-Blockers were used in 16 of 17 surviving patients (94%). Three of 16 individuals who were reportedly adherent to β-blocker therapy (19%) had breakthrough events.
Conclusions and Relevance
The results of this study suggest that calcium-release deficiency syndrome due to RyR2 loss-of-function variants mechanistically and phenotypically differs from CPVT. Ventricular fibrillation may be precipitated by a spontaneous LBLPS-like sequence of ectopy; however, CRDS remains difficult to recognize clinically. These data highlight the need for better diagnostic tools and treatments for this emerging condition.
Introduction
A distinct inherited arrhythmia syndrome caused by loss-of-function (LOF) variants in RYR2 (OMIM 180902)–coded cardiac ryanodine receptor 2 (RyR2) has recently been described, termed calcium-release deficiency syndrome (CRDS).1 Patients with CRDS are at risk for ventricular fibrillation but do not appear to have provocable ventricular tachyarrhythmias during exercise stress testing (EST). This contrasts with gain-of-function (GOF) variants in RyR2 that lead to spontaneous calcium ion (Ca2+) release and catecholaminergic polymorphic ventricular tachycardia (CPVT),2 which can be diagnosed when bidirectional/polymorphic ventricular tachyarrhythmias are induced by EST.3 Patients with CRDS are vulnerable to ventricular fibrillation through a distinct pathophysiology that can be potentially diagnosed using a novel electrophysiology protocol, consisting of a long-burst, long-pause, and short-coupled (LBLPS) ventricular extra stimulus.1 Whether this pattern spontaneously causes ventricular fibrillation in patients with CRDS is unknown.
To expand our understanding of this newly recognized arrhythmic syndrome, we aimed to identify additional patients with CRDS through a multicenter collaboration. Based on the hypothesis that some atypical cases of CPVT could be CRDS and that survivors of idiopathic ventricular fibrillation are increasingly genotyped in the modern era, we searched for these phenotypes occurring in the context of an ultrarare RyR2 variant. To inform the diagnosis of CRDS, we performed in vitro functional characterization of all candidate RyR2 variants. These studies yielded 6 LOF variants, affirming that CRDS is a distinct arrhythmic entity.
Methods
Multicenter Case Collection
Through a multicenter collaboration involving the retrospective and prospective components of the Pediatric CPVT Registry (Vancouver, British Columbia, Canada), International CPVT Registry (Amsterdam, the Netherlands), and 2 additional large inherited arrhythmia referral centers (Aarhus University, Aarhus, Denmark; Western University, London, Ontario, Canada), we identified RyR2-variant–positive patients with normal left ventricular ejection fractions at presentation who had life-threatening arrhythmic events in the presence of testing results that were inconsistent with CPVT. Details of the registries are described elsewhere.4,5,6 Patients with consecutive bidirectional/polymorphic premature ventricular complexes (PVCs), consistent with a CPVT diagnosis by guidelines, were excluded.3 Patients were required to have a single ultrarare RyR2 variant (minor allele frequency, <0.00001),7 defined as pathogenic, likely pathogenic, or a variant of unknown significance, that occurred in a residue conserved across isoforms and species8,9 (further details on variant selection are provided in the eMethods in the Supplement). Broad arrhythmia and cardiomyopathy panels were usually used, although genetic testing decisions were clinically driven. Patients with other arrhythmia- or cardiomyopathy-associated variants, except benign ones, were excluded. Probands were defined as the index case with malignant ventricular arrhythmia in the family. Relatives with the familial RyR2 variant were included. Demographic details, information on the initial event, cardiac and genetic testing results, antiarrhythmic treatments, and follow-up events were collected. A life-threatening arrhythmic event was defined as a sudden unexpected death, sudden cardiac arrest, arrhythmic syncope with or without seizure, or appropriate shock from an implantable cardioverter-defibrillator (ICD). Follow-up events were determined from age at symptom onset or index cardiac assessment, whichever occurred first. Deaths at presentation were excluded from event analysis during follow-up. Ethical approval was obtained by each center in accordance with local review board policies, including informed consent for retrospective data collection if required.
RyR2 Point Sequence Variations and Caffeine-Induced Ca2+ Release Assays
All RyR2 variants identified were functionally characterized in vitro (additional details are provided in the eMethods in the Supplement). Briefly, point sequence variations in the mouse RyR2-complementary DNA were generated by the overlap extension method using polymerase chain reaction analysis.10,11 All variants were confirmed by DNA sequencing. The free cytosolic Ca2+ concentration in transfected HEK293 cells before and after repeated additions of various concentrations of caffeine was measured using fluorescence Ca2+ indicator dye (Fluo-3 am; Molecular Probes).12
Statistical Analysis
Data were collected from September 1, 2012, to March 6, 2021, and analyzed from August 9, 2015, to March 6, 2021. Data are presented as median values with IQRs or mean (SEM) as appropriate. Clinical data were tabulated in Microsoft Excel (Microsoft Office 365; Microsoft Corporation). Multiple comparisons were performed with a 2-tailed t test or 1-way analysis of variance with Bonferroni post hoc test using GraphPad Prism, version 8 (GraphPad Software). A 2-sided P < .05 was deemed statistically significant.
Results
Multicenter Collection of Candidate RyR2-CRDS Cases
Population at Baseline
This cohort included 10 symptomatic probands with ultrarare RyR2 variants without consecutive polymorphic and/or bidirectional PVCs during EST and their 14 genotypically affected relatives (Figure 1). The median age of symptom onset in probands was 12 (IQR, 9-27) years. Four probands (40%) had at least 1 first-degree relative with the familial variant available for analysis. Case-specific details are provided in eTable 1 in the Supplement.
Figure 1. Summary of Ryanodine Receptor 2 (RyR2)–Calcium-Release Deficiency Syndrome (CRDS) Case Ascertainment and Outcomes in the Multicenter Cohort.
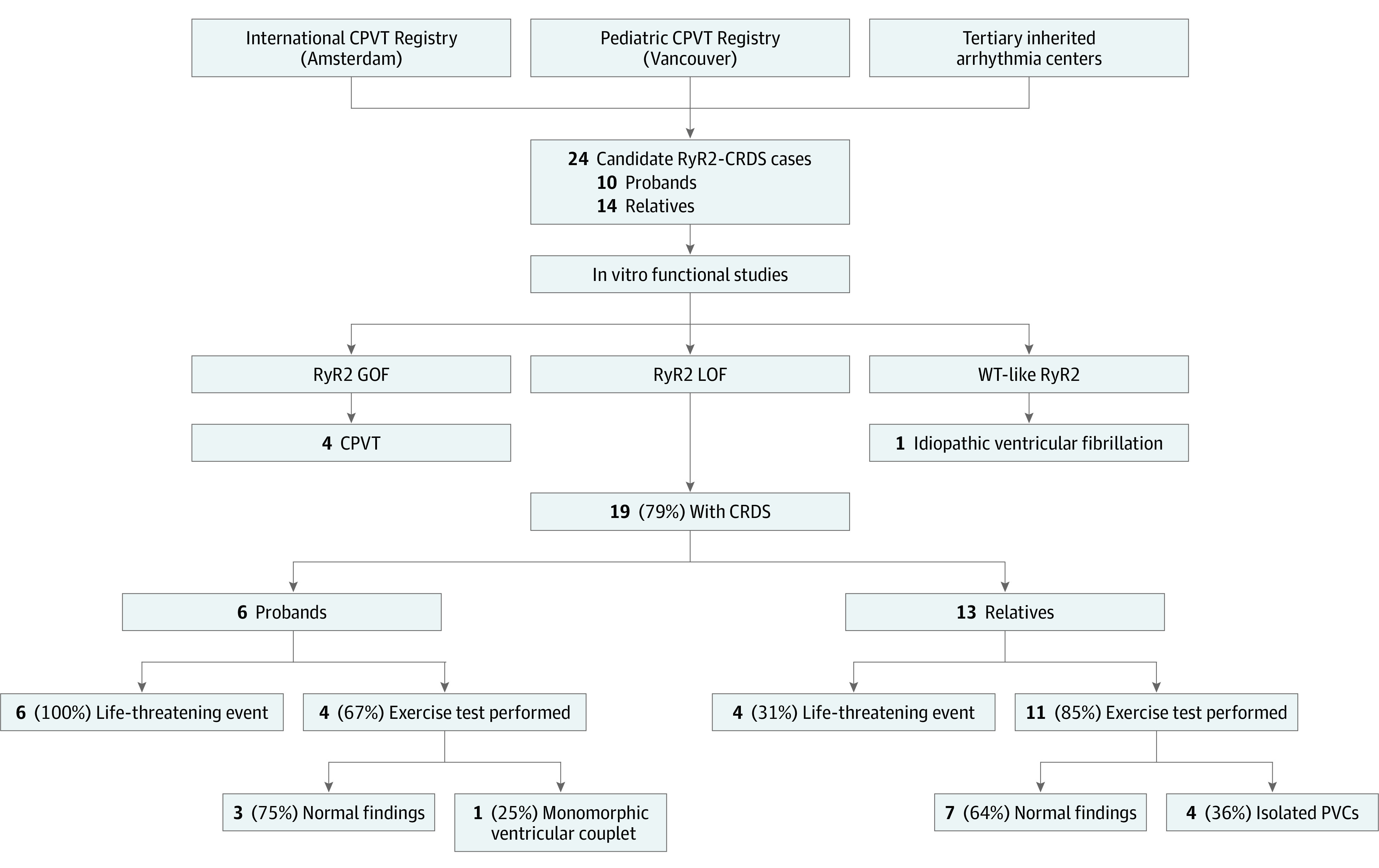
CPVT indicates catecholaminergic polymorphic ventricular tachycardia; GOF, gain of function; LOF, loss of function; PVC, premature ventricular complex; and WT, wild-type.
Functional Characterization of Candidate RyR2-CRDS Variants
To assess the functional consequence of the candidate variants, we generated all 10 RyR2 variants (Table and Figure 2) using site-directed mutagenesis and determined their responses to caffeine activation in HEK293 cells (Figure 3 and eFigure 1 in the Supplement). Consecutive additions of caffeine (0.05-1.0mM) progressively increased the level of Ca2+ release in wild-type RyR2–expressing cells, but further additions of caffeine (2.5-5.0mM) decreased it owing to store Ca2+ depletion by prior caffeine additions. RyR2 variants p.G3988D, p.G4772S, and p.D4956E markedly enhanced the response of RyR2 to activation by caffeine, especially at low concentrations, consistent with GOF (Figure 3 and eFigure 2 in the Supplement). In contrast, variants p.Q2275H, p.E4451del, p.F4499C, p.V4606E, p.R4608Q, and p.R4608W markedly inhibited or completely abolished caffeine response, consistent with RyR2 LOF (Figure 3 and eFigure 2 in the Supplement). The RyR2 p.I4867T variant displayed caffeine responses similar to the wild-type–RyR2 channel (eFigure 2 in the Supplement). eFigure 2 in the Supplement shows the association between caffeine-induced Ca2+ release and the cumulative caffeine concentration in HEK293 cells transfected with wild-type RyR2 and mutants.
Table. Demographic and Clinical Characteristics of Patients Confirmed to Have LOF and GOF Variants In Vitro.
| Characteristic | Patient groupa | |||
|---|---|---|---|---|
| LOF variant (n = 19) | GOF variant (n = 4) | |||
| Probands (n = 6) | Relatives (n = 13) | Probands (n = 3) | Relatives (n = 1) | |
| Age, median (IQR), y | 22 (8-34) | 20 (10-53) | 11 | 14 |
| Sex | ||||
| Male | 1 (17) | 6 (46) | 3 (100) | 1 (100) |
| Female | 5 (83) | 7 (54) | 0 | 0 |
| Arrhythmic event(s) at diagnosis | 6 (100) | 3 (23) | 3 (100) | 1 (100) |
| Exertion/emotion trigger | 3 (50) | 0 | 3 (100) | 1 (100) |
| No trigger | 2 (33) | 3 (100) | 0 | 0 |
| Unknown trigger | 1 (17) | 0 | 0 | 0 |
| Exercise stress testing performed | 4 (67) | 11 (84) | 3 (100) | 1 (100) |
| Normal | 3 (75) | 7 (64) | 0 | 1 (100) |
| PVCs only | 0 | 4 (36) | 3 (100) | 0 |
| Monomorphic couplet | 1 (25) | 0 | 0 | 0 |
| β-Blocker treatmentb | 4 (100) | 12 (92) | 3 (100) | 1 (100) |
| ICD implantb | 3 (50) | 1 (8) | 3 (100) | 1 (100) |
| Atrial arrhythmias | 3 (50) | 0 | 1 (33) | 0 |
| Deceased | 3 (50) | 0 | 0 | 0 |
Abbreviations: GOF, gain-of-function; ICD, implantable cardioverter-defibrillator; LOF, loss-of-function; PVC, premature ventricular complex.
Unless otherwise indicated, data are expressed as number (%) of patients. Note that 1 of the 24 patients (4%) is not included because the variant was wild-type–like during caffeine stimulation (proband 10, with the RyR2-p.I4867T variant). No patient with an LOF variant had abnormal cardiac imaging at presentation, but 2 had minimally abnormal electrocardiograms (nonspecific ST/T wave changes and incomplete right bundle branch block with poor R wave progression).
Excludes patients with sudden unexpected death at presentation.
Figure 2. Location of Ryanodine Receptor 2 (RyR2) Sequence Variants in the 3-Dimensional (3-D) Structure of RyR2.

A, Schematic diagram of the linear sequence of RyR2 showing major structural domains of RyR2 (blue boxes). Orange boxes indicate 4 disease-associated variant clusters (variant hotspots). RyR2 variants identified and characterized in this study are indicated beneath their corresponding domains. B, Locations of RyR2 variants in the 3-D structure of RyR2 (Protein Data Bank identification code 6JI0). Locations of the cytosolic calcium ion (Ca2+) binding sites, U-motif, C-terminal domain (CTD), S2 to S3 loop, and S1 to S6 helixes are also depicted. The 3-D locations of RyR2 residues, Q2275 and E4451, have not been resolved; therefore, the p.Q2275H and p.E4451del variants are not shown in the 3-D structure.
Figure 3. Responses of Wild-Type (WT) Ryanodine Receptor (RyR2) and Mutants to Caffeine Activation.

The association between caffeine-induced calcium ion (Ca2+) release and the cumulative caffeine concentration in HEK293 cells transfected with WT RyR2 and mutants. The amplitude of caffeine-induced Ca2+ release at each caffeine concentration was normalized to that of the maximum caffeine peak for each experiment. Data are shown as the mean (SEM) for 5-12 patients (P < .05 vs WT [1-way analysis of variance with a Bonferroni post hoc test]). The caffeine response curves of loss-of-function RyR2 variants (p.Q2275H, p.E4451del, p.F4499C, p.V4606E, p.R4608Q, and p.R4608W) are depicted in blue; gain-of-function RyR2 variants (p.G3988D, p.G4772S, and p.D4956E), in orange; and WT-like variant (p.I4867T), in gray.
Presentation and Follow-up of RyR2-CRDS Cases
Among the 10 RyR2 variants subjected to caffeine stimulation in HEK293 cells, 6 had an LOF mechanism consistent with CRDS, 3 were GOF, and 1 was similar to wild-type RyR2. The eResults in the Supplement describes the patients who did not have LOF mechanisms consistent with CRDS based on in vitro testing. In the CRDS cohort, 10 of 19 patients with RyR2-CRDS (53%) had at least 1 life-threatening event at presentation and/or during follow-up. The baseline findings of all the patients with CRDS appear in the Table and eTable 1 in the Supplement.
Presentations of Patients With RyR2-CRDS
Six probands were identified to have CRDS (4 [67%] male and 2 [33%] female). The median age at presentation was 22 (IQR, 8-34) years. Presentations included sudden unexpected death in 2 probands (33%), sudden cardiac arrest in 1 proband (17%), syncope in 2 probands (33%), and syncope and seizures in 1 proband (17%). All 4 probands who survived their index event underwent EST and echocardiography (proband 1 with the p.Q2275H variant; proband 3 with the p.F4499C variant; proband 4 with the p.V4606E variant; and proband 6 with the p.R4608W variant). Three probands (75%) had no inducible arrhythmias during exercise and no structural heart disease. These included proband 1, with the p.Q2275H variant, who had an unprovoked syncope, supraventricular tachycardia, and later sudden cardiac arrest and was the only patient with a central domain variant. Proband 3, with the p.F4499C variant, had childhood-onset emotional syncope and later underwent genetic testing for channelopathy after an exertional sudden cardiac arrest and development of atrial fibrillation years later. Proband 6, with the p.R4608W variant, had resuscitated exertion–related sudden cardiac arrest. Proband 4, who survived an initial arrhythmic presentation, had an abnormal EST and echocardiographic findings; this proband first experienced supraventricular tachycardia in infancy and later experienced syncope and seizures. Electrocardiographic monitoring showed atrial tachycardia and isolated PVCs, and echocardiography demonstrated mild left ventricular dilation with normal ejection fraction. EST results during follow-up showed a single monomorphic ventricular couplet inconsistent with a CPVT phenotype. The proband was hospitalized later in childhood for new progressive systolic dysfunction that culminated in a fatal bradyarrhythmia. Postmortem evaluation demonstrated severe cardiomegaly, focal areas of myocardial injury of unclear significance, and pulmonary congestion. Electron microscopy of the cardiac tissue showed myocyte hypertrophy with focal interstitial fibrosis. The RyR2-p.V4606E variant entirely abolished response to caffeine stimulation in vitro, consistent with severe LOF (Figure 3 and Figure 4). No other variants were identified on a 165-gene commercial panel for cardiomyopathy and channelopathy. The proband was adopted without a known family history of cardiac disease.
Figure 4. Device Recordings of Captured Calcium-Release Deficiency Syndrome (CRDS) Arrhythmic Events.

A, Electrogram leading up to ventricular fibrillation (VF) in the genetically affected first-degree relative of proband 3 showing sinus tachycardia with stable cycle length, 2 premature ventricular complexes (PVCs), a long pause, a sinus beat, and a shorter coupled PVC, precipitating polymorphic ventricular tachycardia/VF. B, Dot plot of appropriate implantable cardioverter-defibrillator high-rate detection in second VF episode in proband 1 with RyR2-p.Q2275H variant. A indicates atrial; V, ventricular.
Regarding the probands who died of cardiac arrest at presentation, proband 5, with the p.R4608Q variant, had an adrenergic trigger preceding death, and proband 2, with the p.E4451del variant, did not have a known inciting trigger. They both had postmortem evaluations demonstrating structurally normal hearts and genetic sequencing revealing the RyR2-p.E4451del and -p.R4608Q variants, respectively. Their genotypically affected relatives had EST results not diagnostic of CPVT (all normal or isolated PVCs only) (eTable 1 in the Supplement).
Of 13 relatives of probands with CRDS with a median age of 20 (IQR, 10-53) years at first evaluation and/or symptom, 3 had a previous life-threatening event (23%). These included sudden cardiac arrest in 1 relative and syncope in 2 relatives, all from a single family with the RyR2-p.R4608Q variant in 10 relatives. No arrhythmic events were provoked by known adrenergic stimuli. Among the 13 relatives of probands with CRDS, 11 had EST screening (85%); of these, 7 had no inducible arrhythmias (64%) and 4 had isolated PVCs (36%).
RyR2-CRDS Arrhythmic Events During Follow-up
During a median follow-up of 7 (IQR, 6-18) years after initial symptoms and/or evaluation among 17 patients who survived their index event, 4 patients with CRDS (24%), including 2 probands and 2 affected relatives, experienced at least 1 life-threatening arrhythmic event. Proband 3, with the RyR2-p.F4499C variant, had an exertional sudden cardiac arrest and an ICD shock when not receiving β-blockers during follow-up. This proband’s affected relative also had exertion-related ventricular fibrillation requiring multiple ICD therapies after recurrently missing nadolol therapy (eTable 1 in the Supplement). The relative’s ventricular fibrillation was induced by a long burst of sinus tachycardia, followed by 2 PVCs, a long pause, and then a short-coupled PVC-triggering polymorphic ventricular tachycardia/fibrillation (Figure 4A). Proband 1 with the RyR2-p.Q2275H variant had an unprovoked sudden cardiac arrest 2 years after presentation, followed by an appropriate ICD shock, all while taking metoprolol. The device recording showed sinus tachycardia, followed by a pause, and then polymorphic ventricular tachycardia/fibrillation (Figure 4B). The spontaneous onset of these events was similar to the LBLPS protocol shown to induce ventricular fibrillation in mice and humans with CRDS.1 Finally, 1 relative in the large p.R4608Q kindred experienced a sudden unexpected death during follow-up (eTable 1 in the Supplement). This individual had an aborted sudden cardiac arrest attributed to atherosclerosis of the mid–left anterior descending artery requiring percutaneous coronary intervention in his fourth decade of life. Unfortunately, the details regarding this remote event were unavailable. Later, after the relative presented with sudden unexpected death and a postmortem evaluation identified the RyR2-p.R4608Q variant, the patient who had coronary stenting died suddenly during exertion more than 10 years after index sudden cardiac arrest without ever having an EST. Results of postmortem evaluation were consistent with a primary arrhythmic event. The patient carried the familial RyR2 variant, which cosegregated in 3 generations. The unconfirmed cause of this patient’s arrhythmias is now suspected to be CRDS.
Treatment of RyR2-CRDS During Follow-up
β-Blockers were prescribed to 16 of 17 patients with CRDS (94%) who survived their sentinel event or were identified by family screening. Most took bisoprolol (12 of 16 [75%]). Among treated patients, there were 9 life-threatening arrhythmic events in 4 patients during a median of 8 (IQR, 6-20) years. Nonadherence to β-blocker therapy was implicated in 5 events (55%), leaving 3 of 16 patients (19%) with at least 1 breakthrough event(s) while reportedly adherent to β-blocker therapy. One patient took flecainide acetate (proband 4 with p.V4606E variant), indicated for atrial arrhythmias, which persisted despite catheter ablation and β-blocker treatment (eTable 2 in the Supplement summarizes the medication data). Implantable cardioverter-defibrillators were used in 3 of 4 surviving probands and in 1 relative for primary prevention based on his genetic findings and family history of sudden unexpected death. Shocks for exertion-induced ventricular fibrillation occurred in 2 of these patients while not receiving β-blockers. The appropriate ICD shock in proband 1 occurred during metoprolol therapy.
Discussion
This multicenter cohort study reinforces the existence of an arrhythmia syndrome caused by impairment of RyR2-mediated Ca2+ release. This condition, termed CRDS, may be distinguished from CPVT related to GOF RyR2 variants by its propensity for ventricular fibrillation without polymorphic/bidirectional ventricular tachyarrhythmias during exercise testing. Despite this phenotype, most CRDS arrhythmias are preceded by adrenergic stimuli, which may contribute to CPVT misdiagnosis. At present, there are no validated risk stratification tools or therapies for CRDS, highlighting the need for further research in this area.
Twenty years ago, GOF variants in RyR2 were shown to cause CPVT.2,13 However, it was unknown why some patients with CPVT had normal EST results.14 The initial variant linked to this phenomenon was RyR2-p.A4860G,14 which was later shown to cause LOF rather than GOF.15,16 This finding led Jiang et al15 and Roston et al17 to hypothesize that RyR2 LOF resulted in a unique arrhythmia syndrome. In the ensuing years, case reports linking ventricular fibrillation to RyR2 LOF variants were published.18,19 To more definitively link RyR2 LOF variants to unexplained ventricular fibrillation, we initiated a multicenter effort dedicated to identifying candidate RyR2 LOF variants. This collaboration recently yielded 6 novel RyR2 LOF variants found in surviving patients with catecholamine-triggered ventricular fibrillation, all of whom had normal EST results.1 Humans and knock-in mice possessing LOF variants reliably developed ventricular fibrillation during a programmed electrical stimulation protocol consisting of an LBLPS electrophysiology protocol.1 Collectively, these efforts spanning nearly 2 decades confirmed that RyR2 LOF variants lead to a unique arrhythmia syndrome, now termed CRDS. To date, fewer than 10 confirmed cases have been reported. To expand on CRDS case discovery, we searched for patients meeting criteria believed to be associated with CRDS based on prior observations,1,15,18,19 which included the following: (1) CRDS EST results lack polymorphic and/or bidirectional complex ventricular tachyarrhythmias; (2) CRDS may be incorrectly diagnosed as CPVT in patients with unexplained RyR2-associated ventricular fibrillation; and (3) CRDS variants causing an LOF localize to areas of structural importance in RyR2. These criteria identified 10 putative RyR2-CRDS variants from 24 patients, 6 of which were confirmed to be LOF variants. After family screening, 19 patients had diagnoses of CRDS. This study increases the number of confirmed CRDS cases from less than 10 to nearly 30 in the literature.1,15,18,19,20
Our data highlight several challenges in CRDS. The first is that making a diagnosis is unlikely based on contemporary practice. As shown herein, despite a suspicion or misdiagnosis of CPVT, most probands screened for CRDS met criteria for idiopathic ventricular fibrillation.3,21 This is important because until recently,21 guidelines advised against genetic testing for idiopathic ventricular fibrillation,22 leading to CRDS underrecognition. A second challenge is that a subset of relatives of probands with CRDS had isolated PVCs during EST, whereas 3 probands with isolated PVCs had GOF RyR2 variants, suggestive of atypical CPVT manifesting a minimally penetrant EST. Therefore, an exercise and genetic test are too nonspecific to identify CRDS and would lead to gross overdiagnosis given that the population rate of rare benign RyR2 variation exceeds 3%.23 Because a reliance on in vitro testing to clarify mechanism is impractical, it is important to identify clinical features of CRDS. In this study, probands with CRDS typically had an isolated event in young adulthood. In contrast, probands with CPVT present with events in early adolescence and have high recurrence rates.5,24 RyR2 C-terminal localization is common in CRDS, although this region also forms a CPVT hotspot.9 The most promising diagnostic test is the LBLPS electrophysiology protocol,1 supported herein by the first spontaneously captured LBLPS-like onset patterns for ventricular fibrillation in 2 surviving patients with CRDS (Figure 4). These tracings indirectly show that a specific and improbable series of events likely needs to occur for CRDS to manifest, which may explain why penetrance is incomplete and recurrences are rare, unlike in CPVT. We did not perform the LBLPS electrophysiology protocol in our patients because no asymptomatic carriers had ICDs, which we had previously used to deliver the protocol to avoid the invasive complications of an experimental procedure. Importantly, although approximately half of patients had events under adrenergic circumstances, a burst of sinus/supraventricular tachycardia may still be an initiation requirement based on the recordings captured. Finally, it is important to note that although CRDS and CPVT often appeared distinguishable based on a combination of in vitro functional analysis and EST findings, in some situations (particularly patients with low grades of ventricular ectopy during exercise), it may be difficult to rule out the possibility of atypical GOF RyR2-CPVT. This could be explained by the severity of functional impact (either GOF or LOF) that a variant has on RyR2, whereby the arrhythmic phenotype is more likely to be mild or absent in the setting of a less-damaging but pathogenic variant. Further studies are needed on RyR2 pertaining to these questions.
There are virtually no human data on potential CRDS therapies. Most probands and affected relatives in this cohort were receiving a β-blocker, with breakthrough events occurring in approximately 1 in 5 patients. Flecainide and quinidine are proposed options for CRDS based on mouse models,1 but only 1 patient took flecainide in our study, precluding efficacy analysis. A single genetically affected relative received a prophylactic ICD in this cohort and subsequently had multiple appropriate device therapies. Whether a primary prevention ICD is indicated in select patients with CRDS is not presently known. Accordingly, the optimal risk stratification and pharmacological and invasive management among relatives carrying the familial CRDS variant is uncertain.
The findings from proband 4, with the RyR2-p.V4606E variant, warrant specific comment. This proband was the only patient to develop fatal childhood-onset cardiomyopathy after recurrent arrhythmias in infancy. Her variant abolished all measurable caffeine responsiveness in vitro, a finding not seen previously. Although RyR2 is associated with cardiomyopathy,25,26 a mechanistic connection between RyR2 LOF and cardiomyopathy has not been established. This individual is now the second known patient with CRDS and cardiomyopathy18; the first patient carried the RyR2-p.I4855M variant, causing a lesser degree of LOF perturbation.18
Limitations
This was a retrospective study, which introduces inherent limitations. We attempted to acquire key data; however, remote details were not retained in all records. Data on medication dates, adherence, and titration were often unavailable, limiting analysis on the protectiveness of β-blockers. The breadth and type of testing performed in each patient was nonuniform. Case ascertainment was based on published features of CRDS owing to the lack of diagnostic standards available. Therefore, other CRDS phenotypes may exist. We are continuing to characterize putative CRDS cases in both registries, because the searches undertaken for this study were not exhaustive. Last, there are important potential limitations of the caffeine-induced Ca2+ release assay in HEK293 cells to consider. Although this assay has been used and described extensively in the literature for analyzing RyR2 variants in patients with CPVT, some RyR2 variants likely alter channel function differently in other assays and in vivo models. These mechanisms may not be adequately assessed by the caffeine-induced Ca2+ release assay. As a result, the functional impact of RyR2 variants that exhibit a caffeine response similar to that of the wild-type RyR2 is unclear, because they have yet to be tested using different functional assays.
Conclusions
The findings of this multicenter cohort study suggest that CRDS caused by RyR2 LOF variants is an emerging inherited arrhythmia syndrome that is mechanistically and often phenotypically different from CPVT. The combination of unexplained ventricular fibrillation, an ultrarare RyR2 variant, and an EST result not suggestive of CPVT should raise suspicion for CRDS. Diagnosis before a potentially fatal event remains challenging, and β-blockers are often used with limited supporting evidence. The identification of a validated diagnostic test and therapy are needed to mitigate the risk of sudden death due to CRDS.
eMethods. Selection of Cases and Procedures
eResults. Summary of the Non–RyR2-CRDS Population
eTable 1. Clinical Description of Patients Included in the Entire Cohort Evaluated for CRDS
eTable 2. Medications Used in Patients With CRDS
eFigure 1. Caffeine-Induced Ca2+ Release in HEK293 Cells Expressing Wild-Type RyR2 and Sequence Variants
eFigure 2. Effect of RyR2 Sequence Variants on Caffeine Activation
eReferences
References
- 1.Sun B, Yao J, Ni M, et al. Cardiac ryanodine receptor calcium release deficiency syndrome. Sci Transl Med. 2021;13(579):eaba7287. doi: 10.1126/scitranslmed.aba7287 [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196-200. doi: 10.1161/01.CIR.103.2.196 [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932-1963. doi: 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 4.van der Werf C, Lieve KV, Bos JM, et al. Implantable cardioverter-defibrillators in previously undiagnosed patients with catecholaminergic polymorphic ventricular tachycardia resuscitated from sudden cardiac arrest. Eur Heart J. 2019;40(35):2953-2961. doi: 10.1093/eurheartj/ehz309 [DOI] [PubMed] [Google Scholar]
- 5.Roston TM, Vinocur JM, Maginot KR, et al. Catecholaminergic polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. 2015;8(3):633-642. doi: 10.1161/CIRCEP.114.002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roston TM, Yuchi Z, Kannankeril PJ, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace. 2018;20(3):541-547. doi: 10.1093/europace/euw389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuchi Z, Van Petegem F. Ryanodine receptors under the magnifying lens: insights and limitations of cryo-electron microscopy and X-ray crystallography studies. Cell Calcium. 2016;59(5):209-227. doi: 10.1016/j.ceca.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 9.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108(7):871-883. doi: 10.1161/CIRCRESAHA.110.226845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51-59. doi: 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 11.Zhao M, Li P, Li X, Zhang L, Winkfein RJ, Chen SR. Molecular identification of the ryanodine receptor pore-forming segment. J Biol Chem. 1999;274(37):25971-25974. doi: 10.1074/jbc.274.37.25971 [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Wang R, Chen B, et al. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat Med. 2014;20(2):184-192. doi: 10.1038/nm.3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RYR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103(4):485-490. doi: 10.1161/01.CIR.103.4.485 [DOI] [PubMed] [Google Scholar]
- 14.Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69-74. doi: 10.1161/01.CIR.0000020013.73106.D8 [DOI] [PubMed] [Google Scholar]
- 15.Jiang D, Chen W, Wang R, Zhang L, Chen SR. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci U S A. 2007;104(46):18309-18314. doi: 10.1073/pnas.0706573104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao YT, Valdivia CR, Gurrola GB, et al. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc Natl Acad Sci U S A. 2015;112(13):E1669-E1677. doi: 10.1073/pnas.1419795112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roston TM, Sanatani S, Chen SR. Suppression-of-function mutations in the cardiac ryanodine receptor: emerging evidence for a novel arrhythmia syndrome? Heart Rhythm. 2017;14(1):108-109. doi: 10.1016/j.hrthm.2016.11.004 [DOI] [PubMed] [Google Scholar]
- 18.Roston TM, Guo W, Krahn AD, et al. A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular non-compaction and atypical catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol. 2017;50(2):227-233. doi: 10.1016/j.jelectrocard.2016.09.006 [DOI] [PubMed] [Google Scholar]
- 19.Fujii Y, Itoh H, Ohno S, et al. A type 2 ryanodine receptor variant associated with reduced Ca2+ release and short-coupled torsade de pointe ventricular arrhythmia. Heart Rhythm. 2017;14(1):98-107. doi: 10.1016/j.hrthm.2016.10.015 [DOI] [PubMed] [Google Scholar]
- 20.Paech C, Gebauer RA, Karstedt J, Marschall C, Bollmann A, Husser D. Ryanodine receptor mutations presenting as idiopathic ventricular fibrillation: a report on two novel familial compound mutations, c.6224T>C and c.13781A>G, with the clinical presentation of idiopathic ventricular fibrillation. Pediatr Cardiol. 2014;35(8):1437-1441. doi: 10.1007/s00246-014-0950-2 [DOI] [PubMed] [Google Scholar]
- 21.Stiles MK, Wilde AAM, Abrams DJ, et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm. 2021;18(1):e1-e50. doi: 10.1016/j.hrthm.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308-1339. doi: 10.1016/j.hrthm.2011.05.020 [DOI] [PubMed] [Google Scholar]
- 23.Kapplinger JD, Pundi KN, Larson NB, et al. Yield of the genetic test in suspected catecholaminergic polymorphic ventricular tachycardia and implications for test interpretation. Circ Genom Precis Med. 2018;11(2):e001424. doi: 10.1161/CIRCGEN.116.001424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leenhardt A, Glaser E, Burguera M, Nürnberg M, Maison-Blanche P, Coumel P. Short-coupled variant of torsade de pointes: a new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation. 1994;89(1):206-215. doi: 10.1161/01.CIR.89.1.206 [DOI] [PubMed] [Google Scholar]
- 25.Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10(3):189-194. doi: 10.1093/hmg/10.3.189 [DOI] [PubMed] [Google Scholar]
- 26.Bhuiyan ZA, van den Berg MP, van Tintelen JP, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116(14):1569-1576. doi: 10.1161/CIRCULATIONAHA.107.711606 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods. Selection of Cases and Procedures
eResults. Summary of the Non–RyR2-CRDS Population
eTable 1. Clinical Description of Patients Included in the Entire Cohort Evaluated for CRDS
eTable 2. Medications Used in Patients With CRDS
eFigure 1. Caffeine-Induced Ca2+ Release in HEK293 Cells Expressing Wild-Type RyR2 and Sequence Variants
eFigure 2. Effect of RyR2 Sequence Variants on Caffeine Activation
eReferences