

Abstract
Neuroinflammation is a common feature of many neurodegenerative diseases. It fosters a dysfunctional neuron–microglia–astrocyte crosstalk that, in turn, maintains microglial cells in a perniciously reactive state that often enhances neuronal damage. The molecular components that mediate this critical communication are not fully explored. Here, we show that secreted frizzled‐related protein 1 (SFRP1), a multifunctional regulator of cell‐to‐cell communication, is part of the cellular crosstalk underlying neuroinflammation. In mouse models of acute and chronic neuroinflammation, SFRP1, largely astrocyte‐derived, promotes and sustains microglial activation, and thus a chronic inflammatory state. SFRP1 promotes the upregulation of components of the hypoxia‐induced factor‐dependent inflammatory pathway and, to a lower extent, of those downstream of the nuclear factor‐kappa B. We thus propose that SFRP1 acts as an astrocyte‐to‐microglia amplifier of neuroinflammation, representing a potential valuable therapeutic target for counteracting the harmful effect of chronic inflammation in several neurodegenerative diseases.
Keywords: activated microglia, Alzheimer's disease, HIF pathway, multiple sclerosis, reactive astrocytes
Subject Categories: Immunology, Molecular Biology of Disease, Neuroscience
In response to neuroinflammation, astrocytes secrete more SFRP1 protein. SFRP1 enhances glial activation and microglia responses by sustaining robust expression of the down‐stream targets of the HIF pathway.
Introduction
Degeneration of neurons occurs in a variety of rare and common pathological conditions of the central nervous system (CNS) including Alzheimer’s disease (AD) or multiple sclerosis (MS). CNS function is supported by a robust neuron–glia crosstalk (Jha et al, 2019; Marinelli et al, 2019) so that neuronal damage is almost invariably associated with the activation of two types of glial cells: microglia and astrocytes. The prompt glial response to insults brings about an inflammatory reaction that favours tissue healing and helps restoring CNS homeostasis (Ransohoff, 2016; Escartin et al, 2019). However, excessive glial activation, as often occurs in neurodegeneration, is itself a cause of neuronal damage, which, in turn, establishes a state of pernicious chronic neuroinflammation (Frank‐Cannon et al, 2009; Glass et al, 2010; Perry & Holmes, 2014; Escartin et al, 2019). Consistent with an important cellular crosstalk, glial cell dysfunction, either as a consequence of hyper‐ or hypofunctionality, can also be the cause of neuronal damage, rather than its consequence, strongly contributing to the progression of neurodegenerative diseases (Ransohoff, 2016; Sarlus & Heneka, 2017; Hickman et al, 2018; Escartin et al, 2019). Although this is nowadays a widely accepted idea, there is still only partial information on the molecular components that sustain a dys‐ or hyperfunctional state of glial cells and an abnormal glia–neuron crosstalk. Here, we have investigated whether SFRP1 may represent one of such components.
SFRP1 is a small, secreted and dispersible protein with functions that have been related to Wnt signalling (Esteve & Bovolenta, 2010) and ADAM10 activity (Esteve et al, 2011a), although its interaction with other factors such as RANKL (receptor activator of nuclear factor‐kappa B ligand), integrins (Bovolenta et al, 2008) and thrombospondin‐1 (Martin‐Manso et al, 2011) has also been reported. In particular, Wnt signalling not only has been mostly implicated in moulding neurodegenerative synaptic changes (Palomer et al, 2019) but also contributes to neuroinflammation, although this role is still ill‐defined (Aghaizu et al, 2020). ADAM10, a member of the A Disintegrin and Metalloprotease family of plasma membrane proteins (Saftig & Lichtenthaler, 2015), acts as an α‐secretase (or sheddase; Lichtenthaler et al, 2018) for many neuronal or glial expressed substrates, which participate in the control of microglial activation (Marinelli et al, 2019). These include TREM2 (triggering receptor expressed on myeloid) (Kleinberger et al, 2014) and CD200 and CX3CL1 (Hundhausen et al, 2007; Wong et al, 2016). ADAM10 also sheds proteins involved in synaptic plasticity, such as N‐cadherin or the amyloid precursor protein (APP) (Musardo et al, 2014; Saftig & Lichtenthaler, 2015). Consistently, genetic inactivation of Adam10 in mice causes neuroinflammation and loss of synaptic plasticity (Prox et al, 2013), whereas genetic studies in humans have demonstrated a link between impaired ADAM10 activity and AD (Suh et al, 2013; Kunkle et al, 2019), in part linked to its non‐amyloidogenic processing of APP (Kuhn et al, 2010). Supporting this possibility, we have recently shown that elevated levels of SFRP1 contribute to AD pathogenesis, acting as an endogenous negative modulator of ADAM10 (Esteve et al, 2019b). SFRP1 upregulation correlates with poor ADAM10‐mediated processing of APP and N‐cadherin, whereas neutralization of its activity prevents the appearance of AD pathological traits, including glial cell activation (Esteve et al, 2019b).
Sfrp1 is abundantly expressed in mammalian radial glial progenitors of the developing CNS (Campanelli et al, 2008; Esteve et al, 2011a, 2019a) but is largely downregulated in the adult brain with the exception of restricted neurogenic areas (Augustine et al, 2001; Zhang et al, 2016; Esteve et al, 2019a). Besides in human neurodegenerative diseases (Blalock et al, 2004; Esteve et al, 2019b; Folke et al, 2019; Bai et al, 2020; preprint: Johnson et al, 2021), SFRP1‐upregulated expression has been reported in several other inflammatory conditions such as periodontitis, rheumatoid arthritis, uropathies or pulmonary emphysema (Esteve & Bovolenta, 2010; Claudel et al, 2019). An upregulation has been also observed in aged brains (Folke et al, 2019), in which a low‐grade chronic inflammation is present (Youm et al, 2013). Notwithstanding, whether SFRP1 is directly involved in the modulation of neuroinflammation remains unexplored.
By addressing this issue, here we show that SFRP1 is a novel mediator of the astrocyte‐to‐microglia crosstalk that underlies mammalian CNS inflammation. In mice, SFRP1, largely astrocyte‐derived, is sufficient to activate microglial cells and to amplify their response to distinct acute and chronic neuroinflammatory challenges, sustaining their chronic activation. From a molecular point of view, SFRP1 allows for the full expression of downstream targets of the transcription factors hypoxia‐induced factors (HIFs) and, to a lesser extent, nuclear factor‐kappa B (NF‐κB), which are mediators of neuroinflammatory responses (Helton et al, 2005; Kaltschmidt & Kaltschmidt, 2009). Thus, neutralizing SFRP1 function may represent a strategy to counteract pernicious chronic neuroinflammation that contributes to many human neurodegenerative conditions.
Results
Acute brain neuroinflammation elevates astrocyte‐specific levels of SFRP1 expression
To evaluate a possible link between SFRP1 and neuroinflammation, we induced acute brain inflammation by injecting bacterial lipopolysaccharides (LPS; Wright, 1999) or control saline into the somatosensory cortex of 3‐month‐old Sfrp1 +/ βgal;CX3CR1 +/ GFP mice (n = 4), which allow the simultaneous identification of microglial (GFP; Jung et al, 2000) and Sfrp1‐producing (nuclear βgal‐positive; Satoh et al, 2006) cells. In a separate set of experiments, we performed similar injections in wt and Sfrp1 +/ βgal mice (n = 5). Immunofluorescence of the brains three days after injection, when inflammation is at its peak (Rivest, 2009), revealed a broader βgal immunoreactivity (reporter of Sfrp1 expression) in LPS vs saline‐treated animals (Fig 1A), largely localized in GFAP+ astrocytes (Fig 1A), as also reported in demyelinating or kainic acid‐induced CNS lesions (Huang et al, 2020; García‐Velázquez & Arias, 2021). Using RNAscope, we had previously detected Sfrp1 mRNA in some Iba+ microglial cells surrounding amyloid plaques present in an AD‐like mouse model (Esteve et al, 2019b). However, we failed to detect βgal+ signal in these cells after LPS injection in neither Sfrp1 +/ βgal;CX3CR1 +/ GFP (Fig 1A) or Sfrp1 +/ βgal (Fig 1C). This observation was confirmed by RNA‐seq analysis of isolated microglial cells (see the last section of the results) and likely reflects the reported heterogeneous and disease‐dependent activation of microglial cell (Bachiller et al, 2018).
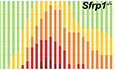
Figure 1. Astrocytes upregulate SFRP1 expression upon LPS stimulation.
-
A–DConfocal image analysis of cryostat sections from adult CX3CR1 +/ GFP ; Sfrp1 +/ βgal (A, B); Sfrp1 +/ βgal (C) and CX3CR1 +/ GFP ;Sfrp1 −/− (D) and mouse brains three days after intracortical infusion of saline or LPS. Sections were immunostained for βgal (magenta, green in C) and Iba1 (green in A; red in C) or Sfrp1 (magenta) and GFP (green, B and D), and GFAP (cyan in A, B, D, red in C). Arrowheads indicate βgal/GFAP (A) and Sfrp1/GFP co‐localization (B). No Sfrp1 protein is detected in the null mice independently of the treatment (two bottom lines). Scale bar: 25 μm.
-
EELISA determination of Sfrp1 levels in brain extracts from 3‐month‐old wt and Sfrp1 −/− mice. WT mice were injected either with saline or with LPS. Three days after injection, the region around the injected side (10 mm3 cortical cube) was isolated and SFRP1 content compared with that present in similar region of non‐injected or Sfrp1 −/− mice used as negative control (n = 5 mice for each group). Error bars represent standard error. Statistical significance: ns P > 0.5 ****P < 0.0001; one‐way ANOVA followed by the Bonferroni multiple comparisons test.
Immunodetection of SFRP1 with specific antibodies (Esteve et al, 2019b) confirmed an increased SFRP1 production after LPS compared with saline injections (Fig 1B). Notably, SFRP1 protein distribution in saline‐injected animals was comparable to that of non‐injected mouse brains (Esteve et al, 2019b). Consistent with the secreted and dispersible nature of SFRP1 (Mii & Taira, 2009; Esteve et al, 2011b), immunosignal was widely distributed in the brain parenchyma (arrows, Fig 1B) and localized to the choroid plexus (not shown) but completely absent in Sfrp1 −/− brains (Fig 1D), as previously reported (Esteve et al, 2019b). The use of a highly specific ELISA (Esteve et al, 2019b) further confirmed a significant increase of SFRP1 levels in extracts of small cortical tissue samples—dissected in the proximity of the injection site—from LPS‐treated animals (Fig 1E) as compared to that present in equivalent brain regions from non‐injected or saline‐injected animals (Fig 1E).
These data indicate that the level of brain SFRP1 increases in response to a bacterial lipopolysaccharide and that astrocytes are likely the most abundant source of SFRP1.
In vivo Sfrp1 gene addition is sufficient to trigger and sustain glial cell activation
We next reasoned that if SFRP1 is indeed associated with neuroinflammation, its forced expression should be sufficient to activate glial cells. To test this possibility, we next infused lentiviral particles (LV) containing Sfrp1‐IRES‐Gfp or control IRES‐Gfp into the lateral ventricle of 8‐ to 10‐week‐old wt mice (n = 13 per group; Figs 2 and EV1A). As expected by the injection site, GFP+ (used to determine infection efficiency) LV‐transduced cells included cells lining along the wall of the lateral ventricle, the choroid plexus, GFAP+ astrocytes and, to a lesser extent, cells of the rostral migratory stream (Fig EV1A and B). Immunohistochemistry of cortical sections 1 month after injection showed a significantly higher presence of SFRP1 protein at the infected site (Fig 2A) associated with Sox9+, GFAP+ and S100 β+ reactive astrocytes (Escartin et al, 2021) and CD45+, Clec7a+ and Iba1+ microglial cells as compared to LV‐IRES‐Gfp control animals (Figs 2A and B, and EV1C). Iba1 immunoreactivity was accumulated around the injection site (Fig 2A and B), and many cells had a round amoeboid morphology and a significantly higher CD45 expression (Figs 2B and EV1D), characteristics of activated microglia (Heneka et al, 2014). CD45hi round cells could also represent blood‐borne infiltrating macrophages (Fig EV1D), although the molecular and functional difference between the two cell types is currently a matter of debate (Grassivaro et al, 2020; Honarpisheh et al, 2020). A wider distribution of hyperphosphorylated tau, which often appears as a brain response to inflammation and degeneration (Ising et al, 2019), was also detected in LV‐Sfrp1‐IRES‐Gfp‐transduced vs control brains with a significantly wider distribution (Fig 2A and B). Analysis of a different set of LV‐transduced animals (n = 4) 5 months after LV delivery showed a persistent microglial activation in SFRP1‐ vs GFP‐treated animals, with more CD45+ cells than those observed at 1‐month post‐injection (Fig EV1D). Furthermore, several CD45hi round‐shaped microglia/macrophages were detected in the parenchyma, especially in the proximity of transduced cells (white arrowheads in Fig EV1D).
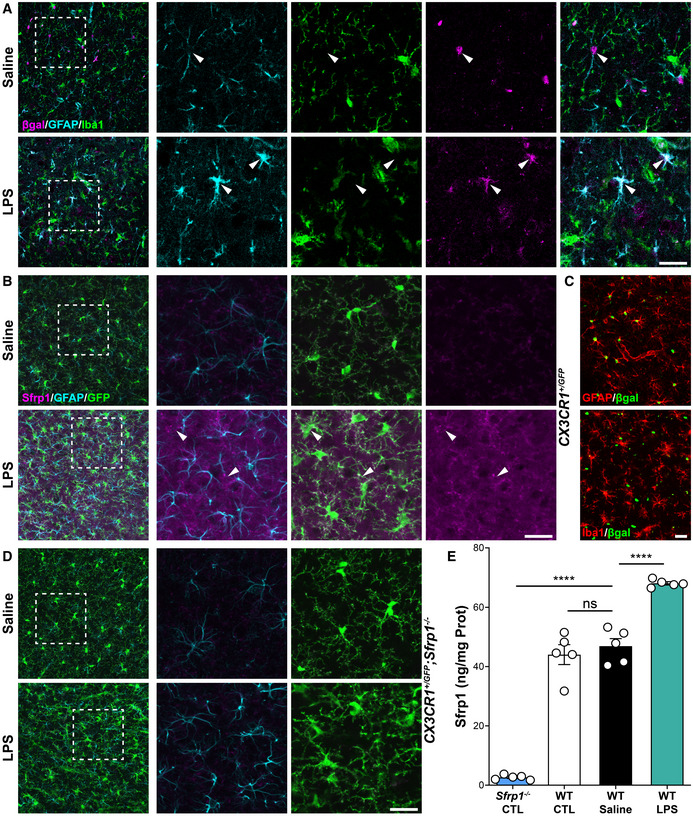
Figure 2. SFRP1 is sufficient and required to enhance glial cell activation upon LPS treatment.
-
ACoronal cryostat sections of LV‐IRES‐Gfp‐ or LV‐Sfrp1‐IRES‐Gfp‐infected brains 1 month post‐infusion immunostained for SFRP1 (magenta), Iba1 (green), GFAP (cyan) or TauP (red). Lv, lateral ventricle. Scale bar: 100 μm.
-
BThe graph shows the normalized level of GFAP, Iba1 and CD45 immunoreactivity (IR) and the area occupied by TauP signal (n = 24 acquisitions, white dots; N = 3 mice, black dots, for each group), normalized to LV‐IRES‐Gfp‐infected brains. Error bars represent standard error. Statistical significance: *P < 0.05; **P < 0.01; ****P < 0.0001 by two‐sided Student’s t‐test.
-
C, DCoronal sections from wt and Sfrp1 −/− mouse brains three days after infusion of saline or LPS immunostained for GFAP (cyan, C) or CD45 (magenta, D). The images are high‐power views of the somatosensory cortex (lower power view in Fig EV1D). Scale bar: 60 μm.
-
E, FThe graphs show the normalized levels of immunoreactivity (IR) for GFAP (E) and CD45 (F, P = 0.006) present in cortical sections (n = 24 acquisitions white dots; from N = 3 animals, black dots, per group) from wt and Sfrp1 −/− animals infused with saline or LPS. Bars represent standard error. Statistical significance calculated per biological replicas is indicated in green and that based on number of acquisitions in black. ** or ## P < 0.01; *** or ### P < 0.001; and **** or #### P < 0.0001 by two‐way ANOVA followed by Bonferroni's multiple comparisons test. * and # indicate significance between genotypes and treatments, respectively.
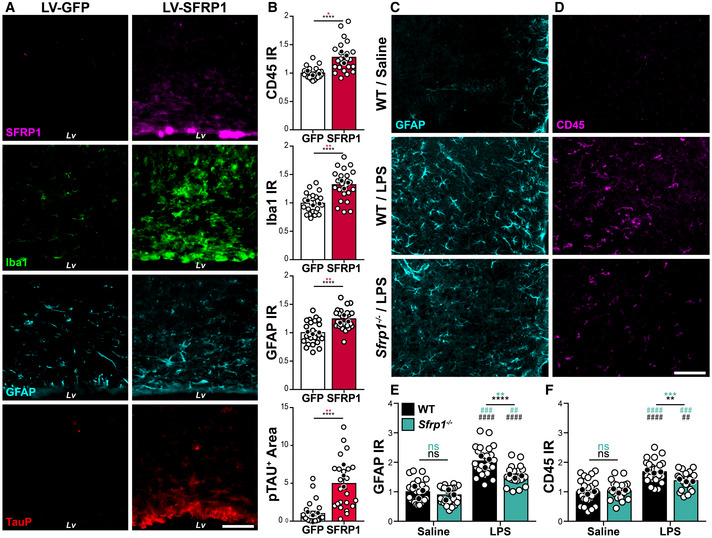
Figure EV1. SFRP1 enhances glial cell activation.
- On the left, diagram of the observed GFP distribution (green dots) after lentiviral (LV) particles' delivery into the lateral ventricle (Lv). The dashed boxes represent areas shown in (B) and (D), 1 points to the wall of the lateral ventricle, and 2 to the Rostral Migratory Stream. Images on the right show examples of immunostaining against GFP protein (GFP signal was no longer visible, unless detected with immunohistochemistry) in the Lv of brains transduced with LV‐IRES‐Gfp or LV‐Sfrp1‐IRES‐Gfp 5 months (m) post‐infusion. Scale bar: 100 μm.
- Double immunostaining against GFP (green) and GFAP (magenta) of brains transduced with LV‐Sfrp1‐IRES‐Gfp, indicating that transduced cells include astrocytes. Arrowheads indicate GFP+ astrocytes. Scale bar: 50 μm.
- The graphs show the number of Sox9 immunopositive cells, S100β and Cleac7a immune intensity level and CD45 immunopositive area in LV‐Sfrp1‐IRES‐Gfp‐infected brains normalized against the mean value obtained in LV‐IRES‐Gfp‐infected control brains. Dots represent the mean value obtained by the analysis of 6 sections for each biological replica (n = 4–6, mice per group). Error bars represent standard error. Statistical significance: *P < 0.05; **P < 0.01 by two‐sided Student’s t‐test.
- Coronal sections from LV‐IRES‐Gfp‐ or LV‐Sfrp1‐IRES‐Gfp‐infected brains 1 or 5 months (m) post‐infusion (PI). Sections were immunostained for CD45. Arrowheads indicate CD45hi‐positive macrophages or lymphocytes infiltrated in the parenchyma after prolonged LV‐Sfrp1‐IRES‐Gfp infection. High‐power images 1 and 2 were taken from the regions indicated with grey dotted lines in A. Scale bar: 50 μm.
- Coronal sections from wt and Sfrp1 −/− mouse brains three days after infusion of saline or LPS, immunostained for GFAP. The white arrows indicate the injection site; the area boxed with white lines is represented at high power in Fig 2. Scale bar: 100 μm.
Together, these results indicate that SFRP1 is sufficient to trigger an inflammatory response in glial cells, which persists for prolonged periods of time.
Sfrp1 is required for amplifying CNS inflammatory response
Given that SFRP1 was sufficient to induce glial cell activation, we next asked whether it was also a necessary component of the CNS inflammatory response. To this end, we took advantage of Sfrp1 −/− mice, in which SFRP1 protein is completely undetectable (Esteve et al, 2019b) (Fig 1B and C). These mice have a slightly shorter and thicker cortex that however does not affect their life span, reproduction rate or cognitive and motor behaviour and present no evident neuronal defects (Esteve et al, 2019a, 2019b). Furthermore, their content of astrocytes and microglial cells was undistinguishable from that of wt mice (Fig 2C–F). We thus compared the effect of intracortical LPS infusion into the brains of 3‐month‐old wt and Sfrp1 −/− mice (Fig 2C and D). In the somatosensory cortex of wt brains (Fig EV1E), LPS but not saline treatment caused the appearance of GFAP+ reactive astrocytes (Fig 2C) and CD45+ reactive microglia (Fig 2D). In contrast, Sfrp1 −/− littermates presented fewer and less immunopositive astrocytes and a significant reduction in CD45+ reactive microglia/macrophages (Fig 2C and D), further supporting that SFRP1 is relevant for the activation of both astrocytes and microglial cells. Quantitation of immunoreactivity among different genotypes and treatments confirmed no significant differences between the two saline‐treated genotypes but showed a significantly lower response of Sfrp1 −/− mice to LPS as compared to wt (Fig 2E and F). Nevertheless, Sfrp1 −/− mice do not completely lose their response to LPS, given that this is significantly different from that observed in saline‐injected mice (Fig 2E and F). This suggests that SFRP1 is involved in boosting inflammation.
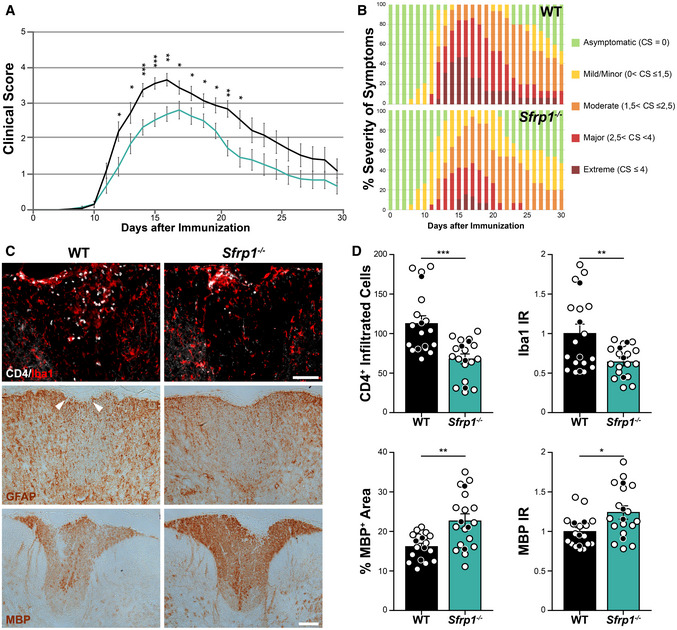
To evaluate a possible specificity of this response, we induced experimental autoimmune encephalomyelitis (EAE) in wt and Sfrp1 −/− mice. This is a widely used experimental model for MS, a human inflammatory‐demyelinating disease. In EAE, CNS inflammation and gliosis occur as a consequence of a strong autoimmune response against the peripheral exposure to myelin components (Constantinescu et al, 2011), thus representing a neuroinflammatory paradigm quite different from the direct LPS intracerebral infusion. Female littermates of the two genotypes were immunized following a standard protocol, and animals were scored for the development/remission of their clinical symptoms over the course of a month (Borroto et al, 2016). Animals were classified with a standard 0 to 5 rank based on their paralysis degree, with 0 corresponding to the absence of symptoms and 5 to a moribund condition (Borroto et al, 2016). In wt mice (n = 19), tail limping—the first symptom of the disease—became apparent around 8 days after immunization with a subsequent rapid progression so that, by 16 days, most of the wt animals presented hindlimb paralysis followed by a slow recovery (Fig 3A). Notably, 47% of the immunized wt mice developed extreme and protracted symptoms (Fig 3A and B). The response of Sfrp1 −/− mice (n = 19) to immunization was instead slower and milder: only 16% of them developed extreme symptoms, and their recovery was significantly faster (Fig 3A and B). Immunostaining of spinal cord sections before immunization showed no difference between wt and Sfrp1 −/− in astrocytes, microglia or myelin distribution (not shown). In contrast, in animals (n = 4) sacrificed 16 days after immunization, there was a significant reduction in pathological signs in Sfrp1 −/− vs wt mice, including infiltration of CD4+/Iba‐ lymphocytes (no CD4+/Iba+ monocytes were instead detected), presence of Iba1+ macrophages/activated microglial cells and loss of MBP+ myelin (Fig 3C and D). There was no significant difference in the amount of GFAP labelling between wt and Sfrp1 −/−; however, in all null mice analysed there was basically no disruption of the astrocytic pial surface otherwise observed in all wt mice (Fig 3C).
Figure 3. Sfrp1 −/− mice develop a milder form of EAE.
-
A, BTime course analysis and severity of the symptoms in wt and Sfrp1 −/− mice after EAE induction. In A, means are depicted with black (wt, n = 19) and cyan (Sfrp1 −/−, n = 19) lines. In B, data are expressed as % of the total number of analysed animals (n = 19 per genotype). In (A), error bars represent standard error.
-
CWt and Sfrp1 −/− mice immunized with MOG and sacrificed 16 days post‐immunization. Cryostat sections of the thoracic spinal cords were stained with antibodies against CD4, Iba1, GFAP and MBP. Images show the region dorsal fasciculus. White arrowheads point to the disruption of the pia surface in wt. Scale bar: 100μm.
-
DQuantification of CD4+ infiltrated lymphocytes, Iba1‐normalized immunoreactivity, MBP‐normalized immunoreactivity and MBP immune‐reactive area in the dorsal fasciculus of spinal cord sections from wt and Sfrp1 −/− mice 16 days after immunization (n = 16 acquisitions, white dots; from N = 4 animals, black dots, per genotype). Error bars represent standard error. Statistic refers to number of acquisitions.
Data information: *P < 0.05, **P < 0.01 and ***P < 0.001 by the Kolmogorov–Smirnov test followed by the Mann–Whitney U nonparametric test comparing mice of the same day after immunization (A) or two‐sided Student’s t‐test (D).
All in all, these data indicate that SFRP1 is commonly required for a robust neuroinflammatory response, likely contributing to astrocyte‐to‐microglia crosstalk.
Astrocyte‐derived Sfrp1 is required for a robust response of microglial cells to damage
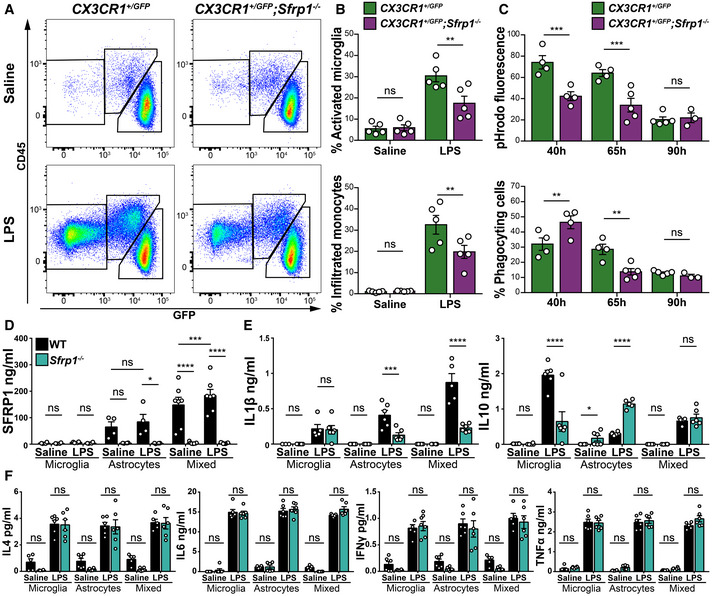
The latter possibility found support in the observation that astrocytes but not microglial cells are the main SFRP1 source in the brain, although the protein has a widespread distribution including around microglial cells (Fig 1), as well as in the remarkably poor microglial activation (Figs 2 and 3) and myeloid cell recruitment (Fig 3C) observed in the absence of Sfrp1. To further explore this possibility, we used flow cytometric analysis to determine the proportion of the different CD11b+ myeloid populations (Greter et al, 2015) in the cortex of CX3CR1 +/ GFP and CX3CR1 +/ GFP ; Sfrp1 −/− mice 3 days after intracerebroventricular LPS or saline administration (Figs 4A and EV2A). In the absence of Sfrp1, there was a significant decrease in LPS‐induced infiltration of CD11b+/CD45+/GFP− monocytes and in the proportion of microglial cells that passed from a CD11b+/CD45lo/GFP+ surveying to a CD11b+/CD45hi/ GFP+ activated state (Figs 4A and B, and EV2A). Furthermore, FACS analysis of isolated GFP‐positive microglial cells (Fig EV2B–D) revealed that, upon infusion of pHrodo E.coli BioParticles (Kapellos et al, 2016) into the third ventricle, CD11b+/CD45hi/ GFP+ activated microglial cells derived from CX3CR1 +/ GFP ; Sfrp1 −/− mouse brains showed less cumulative phagocytized fluorescent signal than those isolated from CX3CR1 +/ GFP brains at both 40 and 65 h post‐infusion (Figs 4C and EV2B–D), whereas CD11b+/CD45lo/GFP+ surveying microglial cells showed a very limited phagocytic activity in both genotypes (Fig EV2B). Notably, at 40 h the percentage of phagocyting microglial cells in CX3CR1 +/ GFP; Sfrp1 −/− mice was significantly higher than in controls (Fig 4C), but this proportion underwent a drastic reduction at 65 h (Fig 4C). These data suggest that in the absence of SFRP1, microglial cells present a less efficient phagocyting activity that, at earlier time points, might be compensated by increasing cell recruitment. A differential rate of phagocytosis vs degradation or the acidity of the lysosomal compartment (Sole‐Domenech et al, 2016) may also explain the time course of phagocytosis and the lower phagocytic signal observed in mutant microglia.
Figure 4. SFRP1 amplifies LPS‐induced microglial activation and modifies their cytokine secretion.
-
ARepresentative cytometric plots of CD11b+ populations present in the cortex of CX3CR1GFP /+ or CX3CR1GFP /+ ;Sfrp1 −/− mice 3 days after saline or LPS intraventricular infusion. Gating was set for isolating the following myeloid cell populations: CD11b+; CD45lo; GFP+ surveying microglia, CD11b+; CD45+; GFP+ activated microglia and CD11b+;CD45+;GFP‐ infiltrated monocytes.
-
BQuantification of the percentage of activated microglia and infiltrated monocyte present in Sfrp1 −/− and controls brains 3 days after infusion of saline or LPS (n = 5 animals per genotype and condition). Error bars represent standard error. Statistical significance: **P < 0.01 by two‐way ANOVA followed by Bonferroni's multiple comparisons test.
-
CQuantification of the total fluorescence from phagocytized pHrodo‐labelled E. coli BioParticles and percentage of phagocyting cells in the CD11b+;GFP+ microglial population (n = 3–5 animals per genotype and condition). Error bars represent standard error. Statistical significance: **P < 0.01 and ***P < 0.001 by two‐way ANOVA followed by Bonferroni' s multiple comparisons test.
-
DELISA determination of SFRP1 levels present in the media of microglia (n = 4 cultures per genotype), astrocytes (n = 4 cultures per genotype) or mixed astrocytes and microglial (2:1, n = 7 cultures per genotype) cultures derived from wt or Sfrp1 −/− pups exposed for 24 h to saline or LPS (1 μg/ml). Error bars represent standard error. Statistical significance: *P < 0.05, ***P < 0.001 and ****P < 0.0001 by one‐way ANOVA followed by the Bonferroni test.
-
E, FSecretory profile of cytokines present in the medium of microglia, astrocytes or mixed astrocytes and microglial (2:1) culture as above (n = 5–6 cultures type—microglia, astrocytes or mixed cultures—and genotype). The content of IFN‐γ, TNF‐α, IL‐1β, IL‐4, IL‐6 and IL‐10 was determined by multiplex ELISA. Values are represented in absolute concentration. Error bars represent standard error. Statistical significance: ns, P > 0.05, *P < 0.05, ***P < 0.001 and ****P < 0.0001 by two‐way ANOVA followed by Bonferroni's multiple comparisons test.
Figure EV2. SFRP1 enhances microglial cell activation and phagocytosis.
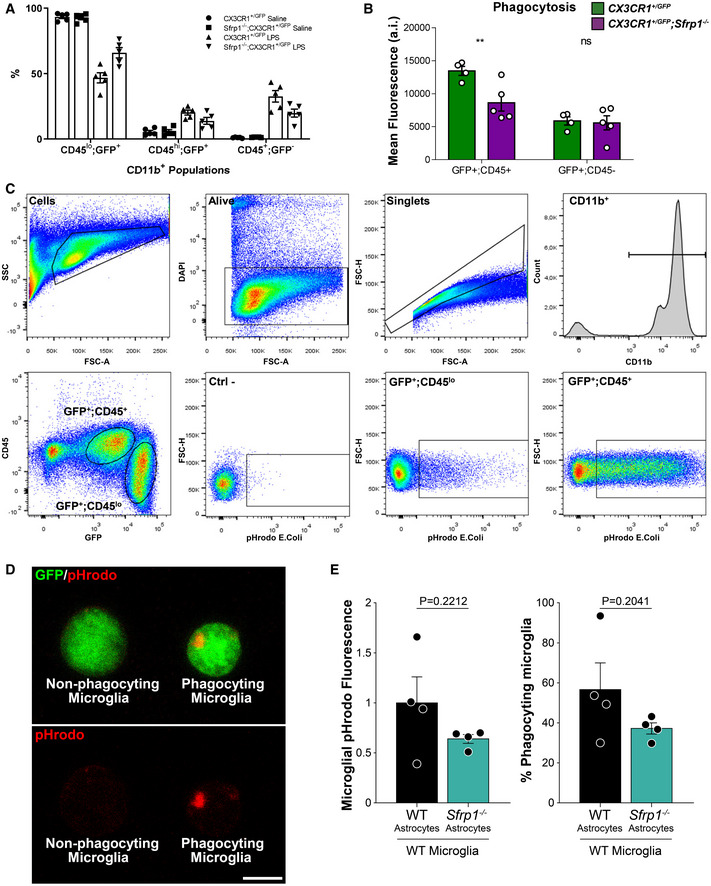
- The graph shows the % of CD11b+ populations present in the cortex of CX3CR1GFP /+ or CX3CR1GFP /+ ;Sfrp1 −/− mice 3 days after saline or LPS intraventricular infusion (n = 5 per genotype and condition). Gating was set for isolating the following myeloid cell populations: CD11b+; CD45lo; GFP+ surveying microglia, CD11b+; CD45+; GFP+ activated microglia and CD11b+; CD45+; GFP‐ infiltrated monocytes. Error bars represent standard error.
- The graph shows the pHrodo mean fluorescent signal present in GFP/CD45hi and GFP/CD45low cell populations isolated from cortex of CX3CR1GFP /+ or CX3CR1GFP /+ ;Sfrp1 −/− mice 3 days after pHrodo intraventricular infusion (n = 4/5 per genotype respectively). Error bars represent standard error. Statistical significance: **P < 0.01 by two‐way ANOVA followed by Bonferroni's multiple comparisons test.
- Representative cytometric plots of brain cells suspension showing gating of individualized live cells CD11b+; CX3CR1+ recognized as microglial cells for pHrodo‐labelled E. coli BioParticles quantitation. Comparison of microglial cells exposed (GFP+; CD45lo and GFP+; CD45+ or not exposed (Ctrl‐) to pHrodo‐labelled E. coli shows specific pHrodo recognition.
- Representative confocal image of FACS‐sorted GFP+ microglia. Note specific pHrodo+ lysosomal inclusion of E.coli particles in phagocyting vs non‐phagocyting microglia. Scale bar: 5μm.
- The graphs show the quantification of the normalized total fluorescence from phagocytized pHrodo‐labelled E. coli BioParticles and percentage of CD11b+ phagocyting cells in the presence of wt or Sfrp1 −/− astrocytes (n = 4 cultures per condition). Error bars represent standard error. Statistical significance: Student’s t‐test.
This effect could be direct or mediated by a more complex mechanism associated with SFRP1‐producing astrocytes. To test these possibilities, we exposed wt microglial cell suspensions to pHrodo E. coli BioParticles in the presence or absence of wt or Sfrp1 −/− astrocytes. After 1‐h incubation, the presence of phagocytized fluorescent signal of CD11b+ microglial cells was evaluated by FACS analysis. In the presence of Sfrp1 −/− astrocytes, both the phagocytized fluorescent signal and the proportion of phagocyting microglial cells were lower than those observed in the presence of wt astrocytes (Fig EV2E). Although in both cases the difference was not significant, this observation points to a cell non‐autonomous effect.
Collectively, these data support the contention that astrocyte‐derived SFRP1 modulates microglial activation in response to an inflammatory challenge and favours their efficient function. To assess that there is indeed an astrocyte‐to‐microglial flux of information, we next established microglia, astrocyte or astrocyte–microglia mixed cultures (2:1, close to the reported ratio existent in the mouse cortex; Keller et al, 2018) from wt and Sfrp1 −/− neonatal mouse cortices, using the same density in each case. ELISA determination confirmed the presence of SFRP1 only in culture media from wt astrocytes or mixed cultures, which was increased after LPS treatment (Fig 4D), further supporting the astrocytic origin of SFRP1 and the notion that SFRP1 levels increase upon an inflammatory stimulus. Notably, in the mixed culture, although the total amount of astrocytes was one third less than that present in the pure astrocytic cultures, SFRP1 levels were significantly increased in both the presence and absence of LPS (Fig 4D), suggesting that microglia somehow influences SFRP1 secretion. We next used a multiplex ELISA to determine cytokines' release (IFN‐γ, TNF‐α, IL‐1β, IL‐4, IL‐6 and IL‐10) in the media of the three culture types (Fig 4E and F), as a measure of their LPS‐induced activation (Heneka et al, 2014). At 24 h, LPS but not saline treatment enhanced the accumulation of all cytokines tested in the culture media of both purified microglia and astrocyte cultures, with no significant differences between genotypes in the case of IFN‐γ, IL‐4, IL‐6 and TNF‐α (Fig 4F). Sfrp1 −/− astrocytes but not microglial cells produced instead significantly lower levels of IL‐1β upon LPS treatment, and this difference was more pronounced in the mixed cultures (Fig 4E). Notably, in the presence of LPS, IL‐10 production was significantly increased in the media of Sfrp1 −/− astrocytic cultures but decreased in the corresponding microglia media as compared to wt (Fig 4E). These opposite changes likely explain why there was no difference in IL‐10 production between wt and mutant mixed cultures.
Taken together, these results indicate that SFRP1 may be both a target and an effector of the astrocyte–microglia crosstalk during inflammation. In particular, astrocyte‐derived SFRP1 influences the response of both astrocytes and microglial cells to an inflammatory stimulus, modifying their ability to secrete at least two specific cytokines: IL‐1β and IL‐10.
Sfrp1 allows for the full expression of downstream targets of HIF transcription factors
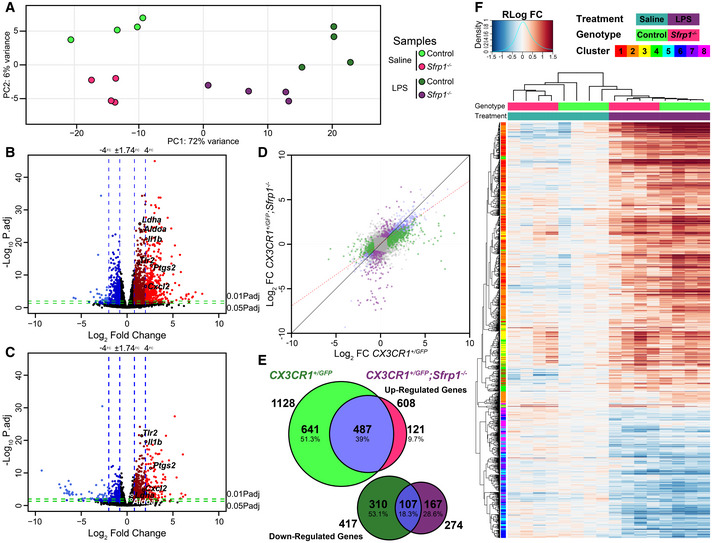
We next reasoned that if SFRP1 influences microglial activation, its absence should modify their transcriptomic profiling towards a less activated or surveying state. To test this possibility, we used fluorescence‐associated cell sorting to purify GFP+ microglial cells from the brain of CX3CR1 +/ GFP and CX3CR1 +/ GFP ; Sfrp1 −/− mice (n = 4). Cells were purified three days after the injection of LPS or saline and used to obtain the corresponding transcriptomic profiles. Confirming previous results, Sfrp1 mRNA was not detected at any significant levels in the microglial transcriptome independently of the genotype or treatment (Fig EV3A and data deposited in ERP119668/PRJEB3647). We next compared the gene expression signature of CX3CR1 +/ GFP ‐ and CX3CR1 +/ GFP ; Sfrp1 −/−‐derived microglial cells in response to LPS (Fig 5). Principal component analysis (Fig 5A, Dataset EV1A–D) and volcano plot comparisons (Fig EV3B) demonstrated LPS as the main source of variation (72% variance), whereas only 6% of the total variations could be attributed to the genotype after secondary component analysis, well in line with the observation that SFRP1 is poorly expressed in the adult brain under homeostatic conditions (Fig 1; Esteve et al, 2019b). The relatively central position of LPS‐treated Sfrp1 −/− microglia supported their milder inflammatory response (Figs 2 and 3), which was further confirmed when overall gene expression variations of LPS‐treated control and Sfrp1 −/− microglia were plotted and compared (Fig 5B and C). Linear regression analysis of this comparison demonstrated a 30% reduction in the global transcriptomic response induced by LPS in the absence of Sfrp1 (slope of red dotted line, Fig 5D). LPS treatment in control microglia induced the upregulation of 1,128 genes, 487 of which were shared with Sfrp1 −/− microglia (Dataset EV1A–B). LPS treatment also induced the upregulation of 121 and the downregulation of 167 microglial genes in the absence of SFRP1 (Fig 5E; Dataset EV1A–D). Genes associated with metabolic pathways (i.e. AldoA, LdhA or Pygl), the cell cycle (i.e. Gas6) or immune regulators (i.e. Mif, Mefv) were highly upregulated in controls but not in Sfrp1 −/− microglia (Dataset EV1A–D), whereas genes downstream of the Toll‐like 4 receptor (TLR4), such as Tlr2, were upregulated at similar levels in both genotypes (Dataset EV1A–D). TLR4 is fundamental for LPS recognition and the activation of the immediate inflammatory response (Ransohoff & Perry, 2009), strongly suggesting that, in the absence of Sfrp1, microglial cells retain their prompt response to damage.
Figure EV3. Non‐cell‐autonomous effect of Sfrp1 on microglial response to LPS.
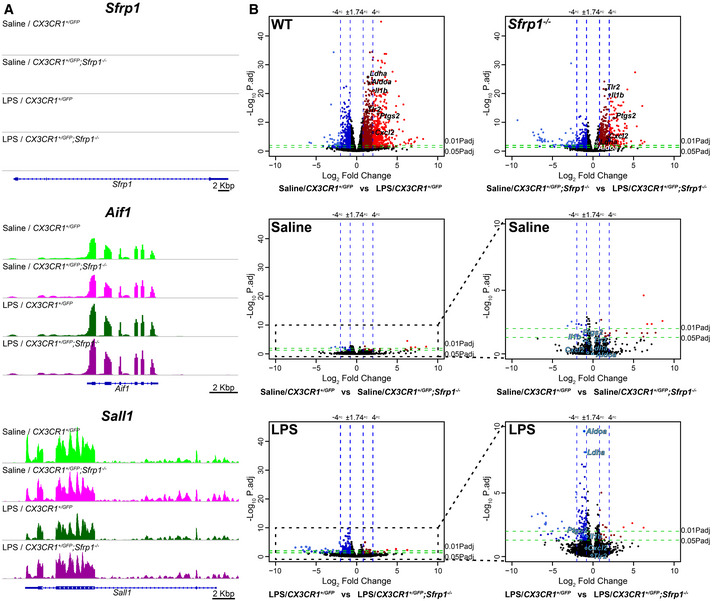
- Integrative Genomics Viewer transcription profile of Sfrp1 and the microglial specific genes Aif1 and Sall1 in microglial cells isolated from saline and LPS treatment of CX3CR1GFP /+ and CX3CR1GFP /+ ;Sfrp1 −/− mice. Scale bar: 2 Kbp.
- Volcano plots of differential gene expression from CX3CR1GFP /+ or CX3CR1GFP /+ ;Sfrp1 −/− microglial cell in response to saline or LPS. Data are represented as log2 fold change vs ‐Log10 adjusted P‐value by the Wald test corrected for multiple comparisons by the Benjamini and Hochberg method. Note that expression variations associated with genotype in saline‐treated animals are minimal. The effect of the genotype is evident only after LPS treatment. Genes with an expression change higher than 75% and an adjusted P‐value < 0.05 are highlighted in dark red (+FC) and dark blue (−FC). Genes with an expression change higher than 400% and an adjusted P‐value < 0.01 are highlighted in light red (+FC) and light blue (−FC).
Figure 5. SFRP1 enhances microglial cells’ transcriptional response to LPS treatment.
-
APrincipal component analysis with the 1,000 most variable genes from microglial cells isolated from CX3CR1GFP /+ and CX3CR1GFP /+ ;Sfrp1 −/− mouse brains three days after saline or LPS intracerebroventricular infusion. The analysis depicts sample clusterization by genotype and treatment.
-
B, CVolcano plots of differential gene expression from CX3CR1GFP /+ (B) or CX3CR1GFP /+ ;Sfrp1 −/− (C) microglial cell in response to LPS. Data are represented as log2 fold change vs ‐Log10 adjusted P‐value by the Wald test corrected for multiple comparisons by the Benjamini and Hochberg method. Blue vertical lines represent an increase of 75 and 400% in the expression levels, respectively. Green horizontal lines represent a 0.05 or 0.01 adjusted P‐value of statistical significance, respectively. Genes with an expression change higher than 75% and an adjusted P‐value lower than 0.05 are highlighted in dark red (+FC) and dark blue (−FC). Genes with an expression change higher than 400% and an adjusted P‐value lower than 0.01 are coloured in light red (+FC) and light blue (−FC).
-
DScatterplot of differentially expressed genes in CX3CR1GFP /+ and CX3CR1GFP /+; Sfrp1 −/− microglia; the red line represents linear regression (slope 0.704014 P‐value: < 2.2e‐16 by the F‐test followed by Bonferroni's correction) of the differential expression, indicating response attenuation. Coloured dots represent response, which are exclusive for each genotype as represented in (E).
-
EVenn diagram showing the extent of differential gene expression in response to LPS in CX3CR1GFP /+ or CX3CR1GFP /+; Sfrp1 −/− microglial cells. Fold change (75%) and adjusted P‐value < 0.05 cut‐off by the Wald test corrected for multiple comparisons by the Benjamini and Hochberg method.
-
FHeatmap showing fold changes of regularized log‐transformed gene‐level RNA‐seq counts with hierarchical clustering of samples and differentially expressed genes in response to LPS in CX3CR1GFP /+ or CX3CR1GFP /+; Sfrp1 −/− microglial cells.
Consistent with this observation, hierarchical clustering of the samples demonstrated treatment‐dependent similarities, but with Sfrp1 −/− microglia showing an attenuated response to LPS treatment as compared to wt (Fig 5F). Hierarchical clustering of the differentially expressed genes presented a pattern of covariance that was further analysed by Z‐score covariance unsupervised clustering (Fig 5F). This analysis generated 4 different clusters of upregulated genes, which were analysed for enrichment of regulatory elements and gene ontology annotations (Fig EV4). Genes regulated by E2F transcription factors and implicated in cell cycle regulation composed the less abundant 3 and 4 clusters. Genes belonging to cluster 3, involved in the regulation of chromosomal segregation and spindle organization, were slightly more upregulated in control microglia. On the contrary, genes belonging to cluster 4 and involved in chromatin assembly and DNA packing were upregulated in Sfrp1 −/− microglia, suggesting possible differences in the length of the cell cycle (Fig EV4).
Figure EV4. SFRP1 microglial inflammatory response depends on HIF factors.
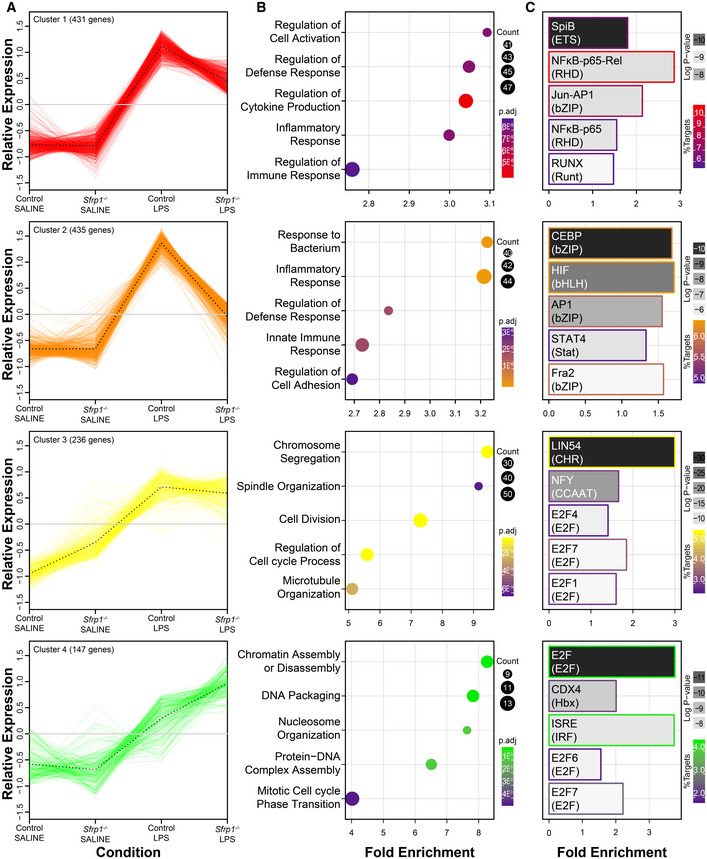
- Representation of the four upregulated k‐means unsupervised clusters of upregulated genes in response to LPS in CX3CR1GFP /+ or CX3CR1GFP /+; Sfrp1 −/− microglial cells. Eight different clusters were generated by Z‐score covariation. The clusters are indicated in the heatmap of Fig 5F. Dotted lines indicate the mean variation of the genes included in each cluster.
- Gene ontology enrichment analysis of different clusters and GO biological process annotated is represented by fold enrichment and colour‐coded by adjusted P‐value of enrichment by the hypergeometric test corrected for multiple comparisons by the Benjamini and Hochberg method, and number of genes of each cluster in that term.
- Regulatory element analysis of different clusters. Specifically enriched transcription factors are represented by fold enrichment relative to the overall transcripts present in the microglia transcriptome. Coloured by log odds detection threshold by the hypergeometric test corrected for multiple comparisons by the Benjamini and Hochberg method, and the percentage of total targets for each transcription factor present in the cluster.
Clusters 1 and 2 were the largest clusters and included genes that mediate the inflammatory response and regulators of the defence response (Fig EV4A and B). Both clusters were strongly upregulated in control microglia in response to LPS (Fig EV4A): genes in cluster 1 showed significant enrichment in NF‐kB TF binding sites at the promoter regions, whereas those putatively controlled by HIF transcription factors were enriched in cluster 2 (Fig EV4C; Table EV1E–F). In microglia derived from Sfrp1 −/− mice, the expression of a few genes belonging to cluster 1 was decreased (Fig EV4A; Dataset EV1E–F), whereas that of genes belonging to cluster 2 was basically abrogated (Fig EV4A; Dataset EV1E–F). Ingenuity Pathway Analysis network representation of the NF‐κB and HIF downstream targets showed that the absence of SFRP1 has a greater effect on downstream targets of HIF (Fig 6A). Genes downstream NF‐κB, such as Tlr2 and ApoE, showed LPS‐dependent upregulation with levels almost similar in the two genotypes, whereas the expression of the homeostatic Cx3cr1 and Trem2 genes was unchanged (Fig 6B; Dataset EV1E). Of note, a number of genes shared between the NF‐κB and HIF pathways, such as Cxcl2, Cxcl3 and Ptgs2, were strongly upregulated in LPS‐treated control microglia but not in those derived from Sfrp1 −/− brains (Fig 6A). This difference, albeit at a slightly lower level, was also observed for IL‐1β and IL‐10 (Fig 6A) supporting the differences observed in cell culture media (Fig 4E). Similarly, other LPS‐induced HIF targets failed to be upregulated in the absence of Sfrp1 and maintained levels of expression comparable to those of saline‐treated control microglia (Fig 6A and B; Dataset EV1F). These included Vegfa, Sod2, Mif, Aldoa and Ldha. Notably, Hif1α expression was similar to that observed in LPS‐treated controls (Dataset EV1F), possibly in line with the notion that Hif1 levels and activity are largely modulated by post‐translational modifications and proteolysis (Choudhry & Harris, 2018).
Figure 6. SFRP1 induces HIF target genes' upregulation during neuroinflammation.
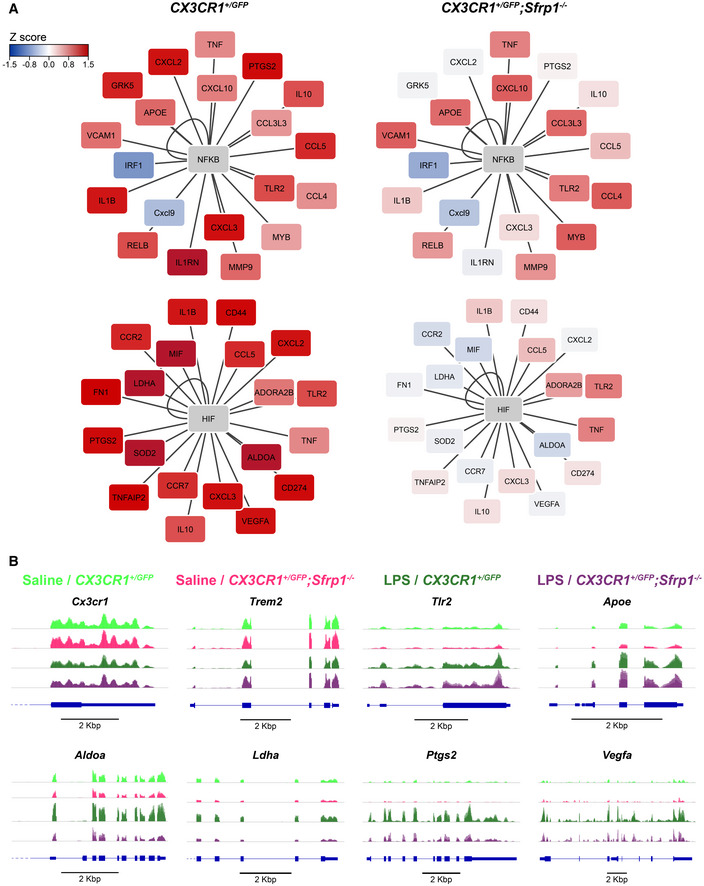
- Ingenuity Pathway Analysis network representation of NF‐κB and HIF downstream targets for CX3CR1GFP /+ and CX3CR1GFP /+ ;Sfrp1 −/− microglial cells. Coloured by Z‐score alteration of their expression after LPS stimulus.
- Integrative Genomics Viewer transcription profile of represented genes in microglial cells isolated for saline‐ and LPS‐treated CX3CR1GFP /+ and CX3CR1GFP /+ ;Sfrp1 −/− mouse brains. Scale bar: 2 Kbp.
All in all, these results support the idea that in the absence of Sfrp1, microglial cells can sense and respond to an inflammatory insult undertaking an initial response. SFRP1 is, however, required to enhance this response allowing for the full activation of microglia‐mediated inflammation in the brain.
Discussion
Microglial cells transit from a surveying to an activated state in response to adverse signals derived from the surrounding environment (Kierdorf & Prinz, 2013). However, both insufficient or prolonged microglial activation is harmful to the brain so that an elaborated neuron–microglia–astrocyte crosstalk is in charge of shaping the brain immune response to damage (Colonna & Butovsky, 2017). The molecular components that mediate the flux of information from neurons to microglia (i.e. the CD200/CD200R signalling system) (Kierdorf & Prinz, 2013) or from microglia to astrocytes (i.e. cytokines and NO) (Jha et al, 2019) have been in part identified. In contrast, there is perhaps less information on how astrocytes communicate with microglial cells (Jha et al, 2019). Our study unveils that SFRP1 is part of the molecular signals that astrocytes provide to microglial cells to enhance their inflammatory response. In response to damage, reactive astrocytes produce and secrete SFRP1, which, in turn, increases the number of activated microglial cells and fosters the expression of HIF and, to a much lower extent, NF‐kB downstream targets in microglial cells. SFRP1 persistency is, however, pernicious as it sustains a chronic inflammatory state. Indeed, its inactivation significantly reduces the prolonged neuroinflammation associated with EAE. These observations indicate that SFRP1 is a potential valuable target to counteract the harmful effect of prolonged inflammatory conditions, such as those present in MS or AD.
SFRP1 function has been studied in many developmental contexts largely linked to the control of cell specification, proliferation and differentiation (Bovolenta et al, 2008), but its homeostatic role in adult tissues is less clear. Its poor or absent expression (or that of other SFRP family members) due to promoter hypermethylation is considered a sign of bad prognosis in different types of tumour (Dahl et al, 2007; Esteve & Bovolenta, 2010; Baharudin et al, 2020), although Sfrp1 −/− mice are viable, fertile and do not form spontaneous tumour (Trevant et al, 2008), indicating that SFRP1 absence per se is not tumorigenic. On the other hand, our finding unveils that elevation of SFRP1 levels has also pathological consequences but related to neuroinflammation (this study) and neurodegeneration (Esteve et al, 2019b). As an advantage, SFRP1 neutralization in AD‐like mice counteracts disease pathology without any apparent side effect (Esteve et al, 2019b). In the specific case of neuroinflammation, neutralization of SFRP1 function should have less effect on the initial acute inflammatory phase, which is a necessary step towards pathogen elimination, tissue repair and homeostasis restoration (Colonna & Butovsky, 2017). Abrogation of SFRP1 function does not prevent this acute response given that, in its absence, both LPS infusion and MOG immunization induce an initial inflammatory reaction, albeit at somewhat lower levels. Furthermore, activated microglial cells and infiltrated monocytes can still be isolated from the brain of Sfrp1 −/− mice, although at significantly decreased numbers. These microglial cells retain phagocytic competence, and no significant difference was observed for most of the LPS‐induced cytokines released from purified microglial cells obtained from Sfrp1 −/− and wt brains. Nevertheless, the significant less phagocytic efficiency observed in vivo and the reduced ability of isolated mutant microglia to secrete IL‐10 in response to LPS may be linked to the small (6%) transcriptomic variations observed between control and mutant microglia (Fig 5). This small variation may reflect that astrocyte‐derived SFRP1 could influence the development of microglial cells and/or change their behaviour in homeostatic conditions. Although we cannot fully exclude these possibilities, we did not find any significant variations in the number and shape of microglial cells in Sfrp1 −/− early postnatal brains and SFRP1 is very poorly expressed in the adult mammalian brain under homeostatic conditions (Esteve et al, 2019b, and this study). Our transcriptomic analysis further shows that Sfrp1 −/− microglia cells retain their capacity of sensing LPS, providing strong support to the implication of SFRP1 in the amplification of the inflammatory response. Indeed, Tlr2 expression, a read‐out of LPS‐mediated TLR4 activation (Rivest, 2009), is increased (and to a comparable extent) in microglial cells from both genotypes. A similar consideration applies to a large fraction of the genes related to the NFκB‐dependent pathway, which is largely related to the acute inflammatory response (Kaltschmidt & Kaltschmidt, 2009).
Brain response not only to LPS but also to other harmful signals depends on the fast reaction of functional microglial cells (Holm et al, 2012), whereas astrocytes and neurons have a minor role in this response despite their reported expression of the TLR4 receptor (Bowman et al, 2003; Rolls et al, 2007). Reactive microglial cells thereafter induce astrocyte activation that, in turn, feeds back on microglial function (Jha et al, 2019). Fitting our data into this loop, we propose that upregulation and release of SFRP1 from astrocytes occur as part of their microglia‐mediated early activation, as supported by the enhanced secreted levels observed when astrocytes and microglial cells were cultured together. Secreted SFRP1 then acts on microglial cells amplifying a HIF‐dependent inflammatory response and thus a significant increase in specific cytokine secretion, further impinging upon inflammation. Supporting SFRP1 implication in this astrocyte–microglia loop, astrocytic Sfrp1 −/− mice show decreased GFAP expression and thus are also less reactive, likely producing lower levels of other microglia‐activating molecules. This implies that, as long as SFRP1 upregulation persists, neuroinflammation persists, contributing to its chronicization. This idea is well in agreement with our observation that genetic inactivation of Sfrp1 significantly limits the severity and progression of EAE, a condition that, as MS, is characterized by persistent microglial activation. Both EAE and MS are driven by the infiltration of peripheral macrophages and lymphocytes (Ajami et al, 2011). We cannot exclude that the lack of peripherally expressed Sfrp1 may limit this infiltration and thus the disease, especially because SFRP1 has been shown to influence lymphocytes’ differentiation (Lee et al, 2012). Notwithstanding, there is strong evidence that endogenous microglial activation is critical for EAE onset and maintenance (Becher et al, 2001; Heppner et al, 2005) and the SFRP1 gene is hypomethylated in brain samples from MS patients (Chomyk et al, 2017), likely promoting an abnormal protein increase. Thus, reduced microglial activation is a plausible cause of the milder EAE symptoms observed in Sfrp1 −/− mice.
SFRP1 upregulation in other neurodegenerative diseases characterized by the presence of chronic inflammation further supports a role of SFRP1 microglial activation. An example is Glaucoma (Wang et al, 2008) or AD (Esteve et al, 2019b; Bai et al, 2020; preprint: Johnson et al, 2021), in which antibody‐mediated neutralization of SFRP1 strongly decreases different AD pathological features, including neuroinflammation (Esteve et al, 2019b). Notably, SFRP1‐mediated inhibition of ADAM10 contributes to the generation of toxic amyloid peptides in AD patients, whereas SFRP1 deficiency decreases amyloid burden (Esteve et al, 2019b). Low neuroinflammation upon SFRP1 neutralization could therefore be secondary to this reduction (Esteve et al, 2019b). The present study shows that this is not necessarily the case as SFRP1 acts directly on microglial cells, suggesting that SFRP1 simultaneously impinges upon multiple pathological events in AD. A possible SFRP1‐mediated activation of microglial cells in AD finds further support in our transcriptomic analysis that links SFRP1 with HIF signalling. Indeed, many LPS‐induced genes regulated or coregulated by HIF were not activated in the absence of Sfrp1, although Hif1α expression was not modified, possibly because its levels are largely post‐translationally regulated (Choudhry & Harris, 2018). Consistent with our observations, a number of recent studies have shown that microglial cells isolated from AD‐like mouse models undergo important metabolic changes with the activation of the HIF pathway (Ulland et al, 2017; Wendeln et al, 2018; Baik et al, 2019). Furthermore, Hif1α is involved in astrocytic proinflammatory activity (Wheeler et al, 2019) and its activity seems to be dysregulated in association with other genes genetically linked to AD risk, suggesting that HIF‐1α may be detrimental in AD pathology (Wendeln et al, 2018).
The precise molecular interactions underlying SFRP1‐mediated HIF pathway activation are at the moment an open question. Microglial cells express members of the ADAM family of metalloproteases (Kleinberger et al, 2014), including ADAM10, which has been shown to be upregulated in both neurons and microglial cells during microglia‐mediated synaptic pruning (Gunner et al, 2019). Furthermore, SFRP1 effectively inhibits ADAM enzymatic activity in cultured microglial cells (our own observations). Given that TREM2 is a proven substrate of ADAM10/17 (Kleinberger et al, 2014), it seems plausible to postulate that SFRP1 may modulate the shedding of TREM2 on the surface of microglial cells. This shedding occurs, for example, in response to LPS‐induced activation of TLR4 (Turnbull et al, 2006; Piccio et al, 2007), thereby attenuating microglial activation (Turnbull et al, 2006). Notably, microglial cells deficient in TREM2 undergo only a partial and abortive activation and remain locked in an almost homeostatic state (Keren‐Shaul et al, 2017; Mazaheri et al, 2017). It is thus tempting to speculate that in SFRP1 absence, enhanced shedding of TREM2 may reduce microglial activation as we have observed in Sfrp1 −/− mice, whereas high SFRP1 levels, by interfering with ADAM function, may enhance TREM2 signalling. In addition (or alternatively), SFRP1 may interfere with ADAM10‐mediated shedding of the neuronal ligands CX3CL1 and CD200 preventing the generation of their soluble forms (Hundhausen et al, 2007; Wong et al, 2016) and thus the activation of their respective microglial receptors, CX3CR1 and CD200R (Jung et al, 2000; Wright et al, 2000). Unfortunately, lack of appropriate biochemical tools has prevented us from verifying these possibilities in our mouse models, leaving this question open for future studies.
An alternative and non‐mutually exclusive possibility involves SFRP1 function as a Wnt signalling modulator. The role of this pathway in microglial activation is, however, somewhat controversial, perhaps in line with the notion that Wnt pathway activity is highly context‐dependent. For example, studies in vitro have shown that Wnt3a and Wnt5a can counteract LPS‐induced microglial activation (Halleskog & Schulte, 2013). In contrast, in preterm‐born infants and in different postnatal animal models of injured‐induced neuroinflammation, downregulation of the expression of Wnt signalling components seems a prerequisite for proinflammatory activation of microglia (Van Steenwinckel et al, 2019). SFRP1 upregulation could lead to the same net effect, inducing at least the downregulation of the expression of Axin2 or Lef1, genes that targets and read‐out of Wnt/βcatenin pathway activation. Notably, however, we found no evidence of changes in the expression of these genes in our RNA‐seq analysis.
In conclusion, we have shown that astrocyte‐derived SFRP1 plays an important role in shaping microglial response to CNS damage, sustaining chronic neuroinflammation. This effect might not be limited to the brain, as SFRP1 upregulation has been reported in different pathological conditions associated with inflammation or fibrosis such as periodontitis, rheumatoid arthritis, uropathies or pulmonary emphysema (Esteve & Bovolenta, 2010; Claudel et al, 2019). Therefore, once analysed the possible side effect, SFRP1 neutralization (Esteve et al, 2019b) may represent a promising therapeutic avenue to treat a wide variety of chronic pathological conditions.
Materials and Methods
Animals
Newborn and adult mice of both sexes were used, unless otherwise indicated. Mice were maintained in pathogen‐free conditions at the CBMSO animal facilities, following current national and European guidelines (Directive 2010/63/EU). Experimental procedures were approved by the CBMSO and Comunidad Autónoma de Madrid ethical committees. Sfrp1 −/− mice were generated by inter‐cross of the Sfrp1 −/− ; Sfrp2 +/− mice in a mixed 129/C57BL/6 background as described (Esteve et al, 2011a) and then back‐crossed at least 4X with C57BL/6J to unify the background. Wild‐type (wt) animals were littermates selected from heterozygous crosses. Breeding pairs of CX3CR1::GFP mice (Jung et al, 2000) were kindly provided by Prof. J Avila, CBMSO. Mice were further crossed with the Sfrp1 −/− to obtain CX3CR1::GFP; Sfrp1 −/−.
Stereotactic LPS infusion
LPS or saline was infused into the brain parenchyma of 10‐ to 12‐week‐old littermates from wt and Sfrp1 −/− or CX3CR1 +/ GFP and CX3CR1 +/ GFP; Sfrp1 −/− mice. Animals were anaesthetized with 4% Isoflurane (Forane, AbbVie Farmacéutica) vaporized into a sealed anaesthetic induction chamber (SurgiVet, Smiths Medical) and placed in a stereotaxic apparatus (Stoelting). Anaesthesia was maintained at 2.5% in 250 ml/min oxygen flow. Saline (2.5 µl) or LPS (5 µg; Escherichia coli 0111:B4; Sigma‐Aldrich) was delivered through a small skull window using a Quintessential Stereotaxic Injector (Stoelting) coupled to a 10 µl syringe with a 34G needle (Hamilton) at the rate of 0.5 µl/min using the following bregma coordinates: 0.0 mm A‐P; −1.0 mm lateral, and −1.5 mm D/V. Mice were let survive for three days and then sacrificed and processed for further analysis. Delivery of pHrodo Red E. coli BioParticles Conjugate (Molecular Probes) was performed with a similar procedure at −2.5 mm A‐P; 0.0 mm lateral, and −2.3 mm D‐V from bregma. Lentiviral vectors (2.5 μl), generated as described below, were delivered −0.5 mm A‐P; 1.0 mm lateral and −2.3 mm D‐V from bregma. Mice were let survive 1–5 months and then analysed.
Lentiviral (LV) particle generation
LV particles carrying IRES‐Gfp or Sfrp1‐IRES‐Gfp were obtained by transient transfection of mycoplasma‐free HEK‐293T cells (Marin et al, 2016; Merten et al, 2016). Cells were transfected employing a three‐plasmid HIV‐derived and VSV‐pseudotyped LV system kindly provided by M.K. Collins, University College London, UK; A. Thrasher, Institute of Child Health, UK; and D. Trono, Ecole Polytechnique Fédérale de Lausanne, Switzerland. Culture supernatants were collected two and three days after transfection and ultracentrifuged. Pellets containing the concentrated particles were resuspended in PBS. Functional titre of the viral preparations was determined by transfection of HEK‐293T cells (1 × 108 TU/ml, in both cases).
Experimental Autoimmune Encephalomyelitis (EAE)
Chronic EAE was induced as described (Borroto et al, 2016). Briefly, 8‐ to 10‐week‐old female C57BL/6J and Sfrp1 −/− littermates were injected bilaterally in subcutaneous femoral region with 150 mg of MOG35–55 (Espikem) emulsified with Freund's complete adjuvant (Sigma‐Aldrich), supplemented with Mycobacterium tuberculosis (1mg/ml; H37Ra strain from Difco), followed by two intraperitoneal injections of pertussis toxin (200 ng; Sigma‐Aldrich) separated by 48h. An observer blind to the animals’ genotype weighed and inspected the animals daily to detect the appearance of clinical signs according to the following classification: (0) no overt signs of disease; (1) weakness at the tail distal portion; (1.5) complete tail flaccidity; (2) moderate hindlimb weakness; (2.5) severe hindlimb weakness; (3) ataxia; (3.5) partial hindlimb paralysis; (4) complete hindlimb paralysis; (4.5) complete hindlimb paralysis with muscle stiffness; and (5) moribund state and hence sacrificed according to the ethical procedures. A representative pool of mice was anaesthetized and perfused intracardially sixteen days after immunization, when symptoms picked, for histological analysis. Other animals were let recover. Spinal cords were dissected and processed.
Primary cultures
Glial primary cultures were established from cerebral cortices of C57BL/6J or Sfrp1 −/− 1‐ to 3‐day‐old pups dissected in Ca2+/Mg2+‐free Hank’s Balanced Salt Solution (HBSS, Invitrogen) and processed following the standard procedures (Bovolenta et al, 1993). Cells were plated in Dulbecco’s modified Eagle’s medium and F12 nutrient mixture (DMEM/F12, Invitrogen) containing 10% FCS (Invitrogen) and gentamycin (Sigma‐Aldrich). Cortices from two pups were platted in a 75‐cm2 flask pretreated with Poly‐D‐Lys (P7280, Sigma‐Aldrich) and cultured at 37°C in a humidified 5% CO2 incubator. After 24h, the medium was refreshed and supplemented with m‐CSF1 to improve microglial survival. Cultures were let reach confluency (about 2 weeks) without further changes. Microglial cells were purified by mechanical detachment of mixed glial cultures (3 h at 37°C and 150 rpm), recovered by medium centrifugation (5 min; 1,000 rpm) (Bovolenta et al, 1993) and plated replacing only 50% of the medium to promote microglial proliferation. Astrocytes were instead recovered after two consecutive trypsin–EDTA treatments (0.25% trypsin (Invitrogen), 1 mM EDTA in PBS at 37°C for 3 and 15 min) in which cells recovered after first treatment were discarded. After adding DMEM/F12 with 10% FCS, cells were centrifuged for 5 min at 1,000 rpm and resuspended in the same medium. Mixed cultures were established from the isolated astrocyte–microglia population using a ratio of 2:1 to reflect the reported proportion present in the mouse cortex (Keller et al, 2018). In each case, cells were seeded on multiwell culture plates (Falcon) at the final density of 105 cells/cm2. Cells were let settle for 48h and thereafter treated with either LPS (1 µg/ml) or saline in DMEM/F12 without serum. After 24 h of treatment, the cell media were then collected and analysed.
Immunohistochemistry
Adult mice were perfused transcardially with 4% PFA in PB 0.1 M (wt/vol). Brains were removed, post‐fixed by immersion overnight and then washed for 24 h in PBS, on a rocking platform at 4°C. Spinal cords were extracted after body post‐fixation and washing. Tissues were incubated in a 30% sucrose‐PB solution (wt/vol) for 24 h, embedded in a 7.5% gelatine and 15% sucrose solution (wt/vol), frozen on dry ice and, if necessary, stored at −80°C until serially sectioned coronally at 15 µm of thickness using a cryostat (Leica). Histological and cytological immunostaining was performed following standard protocols, after antigen retrieval (10 mM citrate buffer, pH6, for 5 min at 110°C in a boiling chamber, Biocare Medical). Primary antibodies (Table EV1) were incubated ON at RT. The following secondary antibodies were incubated for 1h at RT: Alexa 488‐ or Alexa 594‐conjugated donkey anti‐rabbit or anti‐mouse (1:1,000); goat anti‐rat (1:3,000; Molecular Probes, Invitrogen) or anti‐chicken (1:2,000, Abcam); biotin‐conjugated goat anti‐mouse or anti‐rabbit (1:500, Jackson Lab); Alexa 488 and Alexa 594 (1:500; Molecular Probes, Invitrogen); or POD‐conjugated (Jackson Lab) streptavidin followed by reaction with 3,3‐diaminobenzidine (0.05%; Sigma) and 0.03% H2O2. For immunofluorescence, sections were counterstained with Hoechst (Sigma‐Aldrich). Tissue was analysed with a DMCTR5000 microscope equipped with a DFC350Fx monochrome camera or a DFC500 colour camera (Leica Microsystems) or with a LSM710 confocal imaging system coupled to an AxioImager.M2 vertical microscope (Zeiss) or a LSM800 coupled to an Axio Observer inverted microscope (Zeiss). Fluorescence was quantified with ImageJ software (National Institute of Health) using 12–14 sections per analysed brain.
ELISA
The levels of specific cytokines present in the glial culture media were determined by electrochemiluminescence using 96‐well V‐PLEX plates for proinflammatory mouse panel 1 (K15048D) or custom mouse cytokine V‐PLEX plates for IFN‐γ, IL‐1β, IL‐4, IL‐6, IL‐10 and TNF‐α, following the manufacturer’s indications and using a QuickPlex SQ 120 reader (Meso Scale Discovery). SFRP1 presents in glial culture medium or in the RIPA fraction of brain lysates was determined with a capture ELISA (Esteve et al, 2019b). Culture media were diluted five‐fold, and brain lysates used at 0.1 µg/µl protein concentration were determined with BCA protein assay (Thermo Scientific). Values were determined at 450‐nm wavelength using a FLUOstar OPTIMA microtitre plate reader (BMG LABTECH).
Fluorescence‐Activated Cell Sorting (FACS) and flow cytometric analysis
CX3CR1 +/ GFP and CX3CR1 +/ GFP; Sfrp1 −/− 10‐ to 12 week‐‐old male littermates were treated with LPS or saline as described. After 3 days, animals were perfused with ice‐cold saline and brains collected on ice‐cold, HBSS Ca2+, Mg2+‐free (Invitrogen). After meninges' removal, cortices were isolated, finely chopped and digested for 20 min in DMEM with GlutaMAX (Invitrogen) containing papain 20 U/ml (Worthington Biochemical Corporation), DNase (50 U/ml, Sigma‐Aldrich) and L‐cysteine (1 mM, MERCK). After addition of 20% FCS (Invitrogen), the tissue was mechanically dissociated and filtered through a 35‐µm nylon strainer (Falcon). Microglial cells were separated from myelin/debris by isotonic Percoll gradient centrifugation (35% Fisher Scientific) at 1,200 g for 45 min at 4°C. The pellet was recovered and sequentially incubated with anti‐mouse CD16/CD32 (1:250, BD Pharmingen; for 15 min at 4°C) followed by rat anti‐CD11b and PerCP‐Cy5.5 rat anti‐mouse CD45 (1:200, BD Pharmingen; for 30 min at 4°C), all in 2% BSA, 5mM EDTA in PBS. After washing, cells were sorted using a BD FACSAria Fusion Flow Cytometer and their signal, size and complexity acquired with DiVA8 Software (BD Pharmingen). Analysis was performed using FlowJo v10.0.7 Software (BD Pharmingen).
Determination of phagocytic activity
The content of pHrodo Red E. coli BioParticles (Molecular Probes) in CD11b‐positive microglial cells was quantified by FACS using 561‐nm excitation/585‐nm emission maxima following the manufacturer’s recommendations. The gating strategy is described in Fig EV2. When analysis was performed in cultured cells, microglial cells and astrocytes were harvested separately as described in the primary cultured methods, mixed (1:2) and then incubated with pHrodo BioParticles (5 μg/ml) for 1 h. Afterwards, cells were washed and labelled with CD11b, counterstained with DAPI and analysed by FACS.
Genomic data processing and access
RNA from sorted microglia was extracted with the TRI reagent (MERCK) according to the manufacturer’s instructions, and purified with RNeasy Lipid Mini Kit (Qiagen) following the manufacturer’s protocol, and the resulting total RNA was treated with RNase‐Free DNase Set (Qiagen). RNA quality was assessed with a Bioanalyzer 2100 system (Agilent), obtaining RIN (RNA integrity number) values between 8.8 and 10. Each sample (microglia sorted from a single mouse brain) was processed to obtain a RiboZero Stranded Gold Library (Illumina) and sequenced in HiSeq 4000 sequencer in paired‐end configuration with 150‐bp sequence reads (Illumina). Sequence quality was determined with FastQC (Babraham Bioinformatics) revealing more than 32 million mean cluster reads of over 38 quality score and 93% mean Q30. Reads were aligned with HISAT2 v2.1.0 (Kim et al, 2019) to Ensemble Mus musculus (GRCm38.94). Aligned reads were further processed using Samtools v1.9 (Li et al, 2009) and quantified to gene level using HTseq v0.11.2 (Anders et al, 2015). Whole‐genome alignments were visualized with Integrative Genomics Viewer (IGV v2.5) (Thorvaldsdottir et al, 2013). Differential expression analysis (DGE) was performed using the Bioconductor package DESeq2 v1.10.0 (Love et al, 2014). DGE data were processed with custom R scripts (R version 3.5.1, 2018) considering genes with adjusted P‐value < 0.05 and log2 fold change > ± 0.8 as significantly up‐ or downregulated. Analysis of GO terms was performed with the Bioconductor package clusterProfiler (Yu et al, 2012) and that of promoter‐based motif enrichment with HOMER (Heinz et al, 2010). RNA‐seq data sets can be accessed at the European Nucleotide Archive public repository under the following accession numbers (PRJEB36471/ERP119668).
Statistical analysis
Statistical analysis was performed using Prism v7 software (GraphPad). Different statistical tests were used as indicated in each figure footnote and represented by *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Author contributions
JRC, PE and PB conceptualized and designed the research study. JRC, MJMB, GP and MIM conducted the experiments and acquired the data. AB, JRC and BA designed the EAE study. JRC and AB conducted and analysed the study. JRC, GP, MIM, MTH, PE and PB analysed and discussed the data. FB performed cytokine analysis with the support of MPK and SS. JPLA supervised RNA‐seq analysis. JRC and PB wrote the manuscript. All authors read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Table EV1
Expanded View Figures PDF
Dataset EV1
Acknowledgements
We wish to thank Prof. J Avila, CBM, for providing a breeding pair of the CX3CR1::GFP mice, and Dr. Juan Perea, CBM, for help and advice on glial cultures, the CBM image analysis, genomic and cytometric facilities for their support and advice during the course of this study. This work was supported by grants from the Spanish AEI (BFU2013‐43213‐P; BFU2016‐75412‐R with FEDER support and PID2019‐104186RB‐I00), Fundación Tatiana Pérez de Guzmán el Bueno and CIBERER to PB. JRC (BES‐2011‐047189), GP (BES‐2017‐080318) and MIM (BES‐2014‐068797) were supported by FPI fellowships from the AEI. We also acknowledge a CBM Institutional Grant from the Fundación Ramon Areces.
EMBO reports (2021) 22: e51696.
Data availability
RNA‐seq data sets can be accessed at the European Nucleotide Archive public repository under the following accession numbers (PRJEB36471/ERP119668) at: https://www.ebi.ac.uk/ena/browser/view/PRJEB36471.
References
- Aghaizu ND, Jin H, Whiting PJ (2020) Dysregulated Wnt signalling in the Alzheimer’s brain. Brain Sc 10: 902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14: 1142–1149 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine C, Gunnersen J, Spirkoska V, Tan SS (2001) Place‐ and time‐dependent expression of mouse sFRP‐1 during development of the cerebral neocortex. Mech Dev 109: 395–397 [DOI] [PubMed] [Google Scholar]
- Bachiller S, Jiménez‐Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, Boza‐Serrano A (2018) Microglia in neurological diseases: a road map to brain‐disease dependent‐inflammatory response. Front Cell Neurosci 12: 488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharudin R, Tieng FYF, Lee LH, Ab Mutalib NS (2020) Epigenetics of SFRP1: the dual roles in human cancers. Cancers 12: 445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Wang X, Li Y, Chen P‐C, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S et al (2020) Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105: 975–991.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Lee W, Choi H, Chung S, Kim JI, Mook‐Jung I (2019) A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab 30: 493–507.e6 [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ (2001) The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med 193: 967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A 101: 2173–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto A, Reyes‐Garau D, Jimenez MA, Carrasco E, Moreno B, Martinez‐Pasamar S, Cortes JR, Perona A, Abia D, Blanco S et al (2016) First‐in‐class inhibitor of the T cell receptor for the treatment of autoimmune diseases. Sci Transl Med 8: 370ra184 [DOI] [PubMed] [Google Scholar]
- Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez‐Rios J (2008) Beyond Wnt inhibition: new functions of secreted Frizzled‐related proteins in development and disease. J Cell Sci 121: 737–746 [DOI] [PubMed] [Google Scholar]
- Bovolenta P, Wandosell F, Nieto‐Sampedro M (1993) Neurite outgrowth inhibitors associated with glial cells and glial cell lines. NeuroReport 5: 345–348 [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I (2003) Cultured astrocytes express toll‐like receptors for bacterial products. Glia 43: 281–291 [DOI] [PubMed] [Google Scholar]
- Campanelli JT, Sandrock RW, Wheatley W, Xue H, Zheng J, Liang F, Chesnut JD, Zhan M, Rao MS, Liu Y (2008) Expression profiling of human glial precursors. BMC Dev Biol 8: 102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomyk AM, Volsko C, Tripathi A, Deckard SA, Trapp BD, Fox RJ, Dutta R (2017) DNA methylation in demyelinated multiple sclerosis hippocampus. Sci Rep 7: 8696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry H, Harris AL (2018) Advances in hypoxia‐inducible factor biology. Cell Metab 27: 281–298 [DOI] [PubMed] [Google Scholar]
- Claudel M, Jouzeau JY, Cailotto F (2019) Secreted Frizzled‐related proteins (sFRPs) in osteo‐articular diseases: much more than simple antagonists of Wnt signaling? FEBS J 286: 4832–4851 [DOI] [PubMed] [Google Scholar]
- Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35: 441–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu CS, Farooqi N, O’Brien K, Gran B (2011) Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 164: 1079–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, Knüchel R, Klopocki E, Sauter G, Simon R et al (2007) Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene 26: 5680–5691 [DOI] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano‐Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A et al (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24: 312–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Guillemaud O, Carrillo‐de Sauvage MA (2019) Questions and (some) answers on reactive astrocytes. Glia 67: 2221–2247 [DOI] [PubMed] [Google Scholar]
- Esteve P, Bovolenta P (2010) The advantages and disadvantages of sfrp1 and sfrp2 expression in pathological events. Tohoku J Exp Med 221: 11–17 [DOI] [PubMed] [Google Scholar]
- Esteve P, Crespo I, Kaimakis P, Sandonis A, Bovolenta P (2019a) Sfrp1 modulates cell‐signaling events underlying telencephalic patterning, growth and differentiation. Cereb Cortex 29: 1059–1074 [DOI] [PubMed] [Google Scholar]
- Esteve P, Rueda‐Carrasco J, Inés Mateo M, Martin‐Bermejo MJ, Draffin J, Pereyra G, Sandonís Á, Crespo I, Moreno I, Aso E et al (2019b) Elevated levels of Secreted‐Frizzled‐Related‐Protein 1 contribute to Alzheimer’s disease pathogenesis. Nat Neurosci 22: 1258–1268 [DOI] [PubMed] [Google Scholar]
- Esteve P, Sandonìs A, Cardozo M, Malapeira J, Ibañez C, Crespo I, Marcos S, Gonzalez‐Garcia S, Toribio ML, Arribas J et al (2011a) SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nat Neurosci 14: 562–569 [DOI] [PubMed] [Google Scholar]
- Esteve P, Sandonis A, Ibanez C, Shimono A, Guerrero I, Bovolenta P (2011b) Secreted frizzled‐related proteins are required for Wnt/beta‐catenin signalling activation in the vertebrate optic cup. Development 138: 4179–4184 [DOI] [PubMed] [Google Scholar]
- Folke J, Pakkenberg B, Brudek T (2019) Impaired Wnt signaling in the prefrontal cortex of Alzheimer’s Disease. Mol Neurobiol 56: 873–891 [DOI] [PubMed] [Google Scholar]
- Frank‐Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener 4: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Velázquez L, Arias C (2021) Differential regulation of Wnt signaling components during hippocampal reorganization after entorhinal cortex lesion. Cell Mol Neurobiol 41: 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassivaro F, Menon R, Acquaviva M, Ottoboni L, Ruffini F, Bergamaschi A, Muzio L, Farina C, Martino G (2020) Convergence between microglia and peripheral macrophages phenotype during development and neuroinflammation. J Neurosci 40: 784–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, Croxford AL (2015) Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol 6: 249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, Nagy MA, Liu L, Bemiller SM, Kim K‐W et al (2019) Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci 22: 1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halleskog C, Schulte G (2013) WNT‐3A and WNT‐5A counteract lipopolysaccharide‐induced pro‐inflammatory changes in mouse primary microglia. J Neurochem 125: 803–808 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S et al (2005) Brain‐specific knock‐out of hypoxia‐inducible factor‐1alpha reduces rather than increases hypoxic‐ischemic damage. J Neurosci 25: 4099–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: 463–477 [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J et al (2005) Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11: 146–152 [DOI] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, El Khoury J (2018) Microglia in neurodegeneration. Nat Neurosci 21: 1359–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T (2012) Microglia are required for astroglial Toll‐like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 60: 630–638 [DOI] [PubMed] [Google Scholar]
- Honarpisheh P, Lee J, Banerjee A, Blasco‐Conesa MP, Honarpisheh P, d’Aigle J, Mamun AA, Ritzel RM, Chauhan A, Ganesh BP et al (2020) Potential caveats of putative microglia‐specific markers for assessment of age‐related cerebrovascular neuroinflammation. J Neuroinflammation 17: 366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Choi MH, Huang H, Wang X, Chang YC, Kim JY (2020) Demyelination regulates the circadian transcription factor BMAL1 to signal adult neural stem cells to initiate oligodendrogenesis. Cell Rep 33: 108394 [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Schulte A, Schulz B, Andrzejewski MG, Schwarz N, von Hundelshausen P, Winter U, Paliga K, Reiss K, Saftig P et al (2007) Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol 178: 8064–8072 [DOI] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira‐Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575: 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jo M, Kim JH, Suk K (2019) Microglia‐astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25: 227–240 [DOI] [PubMed] [Google Scholar]
- Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, Liu J, Betarbet R, Ping L, Yin L et al (2021) Large‐Scale deep multi‐layer analysis of Alzheimer’s disease brain reveals strong proteomic disease‐related changes not observed at the RNA level systems biology. bioRxiv 10.1101/2021.04.05.438450 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C (2009) NF‐kappaB in the nervous system. Cold Spring Harb Perspect Biol 1: a001271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapellos TS, Taylor L, Lee H, Cowley SA, James WS, Iqbal AJ, Greaves DR (2016) A novel real time imaging platform to quantify macrophage phagocytosis. Biochem Pharmacol 116: 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller D, Erö C, Markram H (2018) Cell densities in the mouse brain: a systematic review. Front Neuroanat 12: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, David E, Baruch K, Lara‐Astaiso D, Toth B et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169: 1276–1290.e17 [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M (2013) Factors regulating microglia activation. Front Cell Neurosci 7: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra86 [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF (2010) ADAM10 is the physiologically relevant, constitutive alpha‐secretase of the amyloid precursor protein in primary neurons. EMBO J 29: 3020–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier‐Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie‐Wolf A et al (2019) Genetic meta‐analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51: 414–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Lee KA, Yoon HB, Yoo SA, Park YW, Chung Y, Kim WU, Kang CY (2012) The Wnt inhibitor secreted Frizzled‐Related Protein 1 (sFRP1) promotes human Th17 differentiation. Eur J Immunol 42: 2564–2573 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Lemberg MK, Fluhrer R (2018) Proteolytic ectodomain shedding of membrane proteins in mammals‐hardware, concepts, and recent developments. EMBO J 37: e99456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin V, Stornaiuolo A, Piovan C, Corna S, Bossi S, Pema M, Giuliani E, Scavullo C, Zucchelli E, Bordignon C et al (2016) RD‐MolPack technology for the constitutive production of self‐inactivating lentiviral vectors pseudotyped with the nontoxic RD114‐TR envelope. Mol Ther Methods Clin Dev 3: 16033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Basilico B, Marrone MC, Ragozzino D (2019) Microglia‐neuron crosstalk: signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol 94: 138–151 [DOI] [PubMed] [Google Scholar]
- Martin‐Manso G, Calzada MJ, Chuman Y, Sipes JM, Xavier CP, Wolf V, Kuznetsova SA, Rubin JS, Roberts DD (2011) sFRP‐1 binds via its netrin‐related motif to the N‐module of thrombospondin‐1 and blocks thrombospondin‐1 stimulation of MDA‐MB‐231 breast carcinoma cell adhesion and migration. Arch Biochem Biophys 509: 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trumbach D, Wurst W et al (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18: 1186–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merten OW, Hebben M, Bovolenta C (2016) Production of lentiviral vectors. Mol Ther Methods Clin Dev 3: 16017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mii Y, Taira M (2009) Secreted Frizzled‐related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development 136: 4083–4088 [DOI] [PubMed] [Google Scholar]
- Musardo S, Marcello E, Gardoni F, Di Luca M (2014) ADAM10 in synaptic physiology and pathology. Neurodegener Dis 13: 72–74 [DOI] [PubMed] [Google Scholar]
- Palomer E, Buechler J, Salinas PC (2019) Wnt signaling deregulation in the aging and Alzheimer’s brain. Front Cell Neurosci 13: 227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10: 217–224 [DOI] [PubMed] [Google Scholar]
- Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina‐Bordignon P (2007) Blockade of TREM‐2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 37: 1290–1301 [DOI] [PubMed] [Google Scholar]
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D'Hooge R, Stroobants S, Ahmed T, Balschun D, Willem M et al (2013) Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci 33: 12915–12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016) How neuroinflammation contributes to neurodegeneration. Science 353: 777–783 [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27: 119–145 [DOI] [PubMed] [Google Scholar]
- Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9: 429–439 [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M (2007) Toll‐like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol 9: 1081–1088 [DOI] [PubMed] [Google Scholar]
- Saftig P, Lichtenthaler SF (2015) The alpha secretase ADAM10: a metalloprotease with multiple functions in the brain. Prog Neurogibol 135: 1–20 [DOI] [PubMed] [Google Scholar]
- Sarlus H, Heneka MT (2017) Microglia in Alzheimer’s disease. J Clin Invest 127: 3240–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh W, Gotoh T, Tsunematsu Y, Aizawa S, Shimono A (2006) Sfrp1 and Sfrp2 regulate anteroposterior axis elongation and somite segmentation during mouse embryogenesis. Development 133: 989–999 [DOI] [PubMed] [Google Scholar]
- Sole‐Domenech S, Cruz DL, Capetillo‐Zarate E, Maxfield FR (2016) The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res Rev 32: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, Tanzi RE (2013) ADAM10 missense mutations potentiate beta‐amyloid accumulation by impairing prodomain chaperone function. Neuron 80: 385–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevant B, Gaur T, Hussain S, Symons J, Komm BS, Bodine PV, Stein GS, Lian JB (2008) Expression of secreted frizzled related protein 1, a Wnt antagonist, in brain, kidney, and skeleton is dispensable for normal embryonic development. J Cell Physiol 217: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang S‐C, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A et al (2017) TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170: 649–663.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steenwinckel J, Schang AL, Krishnan ML, Degos V, Delahaye‐Duriez A, Bokobza C, Csaba Z, Verdonk F, Montane A, Sigaut S et al (2019) Decreased microglial Wnt/beta‐catenin signalling drives microglial pro‐inflammatory activation in the developing brain. Brain 142: 3806–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W‐H, McNatt LG, Pang I‐H, Millar JC, Hellberg PE, Hellberg MH, Steely HT, Rubin JS, Fingert JH, Sheffield VC et al (2008) Increased expression of the WNT antagonist sFRP‐1 in glaucoma elevates intraocular pressure. J Clin Invest 118: 1056–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln A‐C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A et al (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556: 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MA, Jaronen M, Covacu R, Zandee SEJ, Scalisi G, Rothhammer V, Tjon EC, Chao C‐C, Kenison JE, Blain M et al (2019) Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176: 581–596.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Zhu F, Khatri I, Huo Q, Spaner DE, Gorczynski RM (2016) Characterization of CD200 ectodomain shedding. PLoS One 11: e0152073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN (2000) Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 13: 233–242 [DOI] [PubMed] [Google Scholar]
- Wright SD (1999) Toll, a new piece in the puzzle of innate immunity. J Exp Med 189: 605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm Y‐H, Grant R, McCabe L, Albarado D, Nguyen K, Ravussin A, Pistell P, Newman S, Carter R, Laque A et al (2013) Canonical Nlrp3 inflammasome links systemic low‐grade inflammation to functional decline in aging. Cell Metab 18: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE, Sloan S, Clarke L, Caneda C, Plaza C, Blumenthal P, Vogel H, Steinberg G, Edwards M, Li G et al (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89: 37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]