

Abstract
Background and Purpose
(R)‐Ketamine (arketamine) may have utility as a rapidly acting antidepressant. While (R)‐ketamine has lower potency than (R,S)‐ketamine to inhibit NMDA receptors in vitro, the extent to which (R)‐ketamine shares the NMDA receptor‐mediated adverse effects of (R,S)‐ketamine in vivo has not been fully characterised. Furthermore, (R)‐ketamine is metabolised to (2R,6R)‐hydroxynorketamine (HNK), which may contribute to its antidepressant‐relevant actions.
Experimental Approach
Using mice, we compared (R)‐ketamine with a deuterated form of the drug (6,6‐dideutero‐(R)‐ketamine, (R)‐d2‐ketamine), which hinders its metabolism to (2R,6R)‐HNK, in behavioural tests predicting antidepressant responses. We also examined the actions of intracerebroventricularly infused (2R,6R)‐HNK. Further, we quantified putative NMDA receptor inhibition‐mediated adverse effects of (R)‐ketamine.
Key Results
(R)‐d2‐Ketamine was identical to (R)‐ketamine in binding to and functionally inhibiting NMDA receptors but hindered (R)‐ketamine's metabolism to (2R,6R)‐HNK. (R)‐Ketamine exerted greater potency than (R)‐d2‐ketamine in several antidepressant‐sensitive behavioural measures, consistent with a role of (2R,6R)‐HNK in the actions of (R)‐ketamine. There were dose‐dependent sustained antidepressant‐relevant actions of (2R,6R)‐HNK following intracerebroventricular administration. (R)‐Ketamine exerted NMDA receptor inhibition‐mediated behaviours similar to (R,S)‐ketamine, including locomotor stimulation, conditioned‐place preference, prepulse inhibition deficits, and motor incoordination, with approximately half the potency of the racemic drug.
Conclusions and Implications
Metabolism of (R)‐ketamine to (2R,6R)‐HNK increases the potency of (R)‐ketamine to exert antidepressant‐relevant actions in mice. Adverse effects of (R)‐ketamine require higher doses than those necessary for antidepressant‐sensitive behavioural changes in mice. However, our data revealing that (R)‐ketamine's adverse effects are elicited at sub‐anaesthetic doses indicate a potential risk for sensory dissociation and abuse liability.
Abbreviations
- CA1
CA1 region of the hippocampus
- fEPSPs
field EPSPs
- HNK
hydroxynorketamine
- MK‐801
dizocilpine
- OFT
open‐field test
- PPI
prepulse inhibition
What is already known
(R)‐Ketamine is a putative rapid‐acting antidepressant drug candidate.
(R)‐Ketamine induces more potent antidepressant‐relevant actions in rodent tests compared with (S)‐ketamine.
What this study adds
(R)‐Ketamine exerts rapid‐acting antidepressant‐relevant actions in part via its conversion to the (2R,6R)‐hydroxynorketamine metabolite.
(R)‐Ketamine induces adverse effects including ataxia, sensory dissociation, hyperactivity, and conditioned‐place preference.
What is the clinical significance
Adverse effects of (R)‐ketamine at sub‐anaesthetic doses suggest a risk for unsupervised use.
1. INTRODUCTION
Current pharmacological treatments for depression, which rely upon direct activation of monoamine systems, have a delayed therapeutic onset of several months, and ~30% of patients remain treatment resistant (Rush et al., 2006). During the last decade, evidence has accumulated supporting the feasibility of pharmacologically delivering rapidly acting antidepressant effects in depressed patients. Specifically, (R,S)‐ketamine exerts rapid (within hours) and sustained antidepressant responses following administration of a single sub‐anaesthetic dose in treatment‐resistant patients (e.g., Murrough et al., 2013; Singh et al., 2016; Zarate et al., 2006). Despite these promising findings, a widespread use of (R,S)‐ketamine for the routine treatment of depression is restricted due to its abuse liability and dissociative properties, even at low, sub‐anaesthetic doses (Krystal et al., 1994; Sassano‐Higgins, Baron, Juarez, Esmaili, & Gold, 2016; Zanos, Moaddel, et al., 2018).
Rapidly acting antidepressant drugs exert acute neurobiological actions (when these compounds are still in the brain), which induce long‐lasting neuroadaptations and synaptic plasticity changes that are hypothesised to be responsible for their sustained effects long after the drugs are fully eliminated (see Zanos, Thompson, et al., 2018). One hypothesis for (R,S)‐ketamine's antidepressant mechanism of action is via inhibition of NMDA receptors (Autry et al., 2011; Li et al., 2010; Maeng et al., 2008; also see Zanos & Gould, 2018). However, other NMDA receptor antagonists lack the full complement of fast‐acting, robust, and/or sustained antidepressant effects of (R,S)‐ketamine in humans (Newport et al., 2015), thus challenging the NMDA receptor inhibition hypothesis of (R,S)‐ketamine's antidepressant actions. Preclinical studies have revealed that the (R)‐ketamine enantiomer, which has ~4‐fold lower affinity/potency to inhibit the NMDA receptor compared with (S)‐ketamine (Ebert, Mikkelsen, Thorkildsen, & Borgbjerg, 1997; Zeilhofer, Swandulla, Geisslinger, & Brune, 1992), exerts more potent and longer lasting actions in behavioural and biochemical assays considered relevant to (R,S)‐ketamine's antidepressant effects (Fukumoto et al., 2017; Yang, Qu, Fujita, et al., 2017; Yang et al., 2018; Yang et al., 2015; Zanos et al., 2016; Zhang, Li, & Hashimoto, 2014). This greater potency of (R)‐ketamine compared to (S)‐ketamine is in agreement with NMDA receptor inhibition not being the primary mechanism involved in (R,S)‐ketamine's antidepressant efficacy. However, there is less controversy regarding NMDA receptor inhibition being a mediator for many of the acute adverse effects of (R,S)‐ketamine including motor incoordination, perception and sensory changes, and psychotomimetic properties, as well as rewarding/abuse potential (Krystal et al., 1994; Sassano‐Higgins et al., 2016; Wan et al., 2015; Zanos, Moaddel, et al., 2018).
Both (R)‐ketamine and (S)‐ketamine are rapidly and stereoselectively metabolised to norketamines, dehydronorketamines, hydroxyketamines, and hydroxynorketamines (HNKs), via the action of liver P450 enzymes (see Portmann et al., 2010). The (2S,6S;2R,6R)‐HNK metabolite is the major HNK metabolite found in the plasma of humans, as well as in the plasma and brain of rodents, following antidepressant‐relevant dosing of (R,S)‐ketamine (Zanos et al., 2016; Zarate et al., 2012). Metabolism of peripherally administered (R,S)‐ketamine to its (2S,6S;2R,6R)‐HNK metabolite was found necessary for its full antidepressant‐relevant actions in mice (Zanos et al., 2016). Notably, the (2R,6R)‐HNK metabolite, which is solely derived from the (R)‐ketamine enantiomer, has been identified to rapidly cross the blood brain barrier and exert rapid antidepressant‐like effects in many animal tests (Chou et al., 2018; Fukumoto et al., 2017; Highland et al., 2018; Pham et al., 2018; Zanos et al., 2016). The finding that (2R,6R)‐HNK is more potent in inducing antidepressant‐relevant behavioural actions compared to (2S,6S)‐HNK (Chou et al., 2018; Zanos et al., 2016) is in line with the greater antidepressant‐relevant potency of (R)‐ketamine compared with the (S)‐ketamine enantiomer in rodents (Fukumoto et al., 2017; Yang, Qu, Fujita, et al., 2017; Yang et al., 2018; Yang et al., 2015; Zanos et al., 2016). Although there is evidence that (R)‐ketamine exerts antidepressant‐relevant actions independent of its conversion to (2R,6R)‐HNK in some animal tests (Shirayama & Hashimoto, 2017; Shirayama & Hashimoto, 2018; Yamaguchi et al., 2018; Zhang, Fujita, & Hashimoto, 2018; Zhang, Toki, et al., 2018), whether metabolism of (R)‐ketamine contributes to the actions of the drug is unclear.
In rodent tests, using a limited range of doses, (R)‐ketamine has been reported to be devoid of the NMDA receptor inhibition‐mediated adverse effects that are observed following administration of (R,S)‐ketamine, including hyperlocomotion, disruption of prepulse inhibition (PPI), and rewarding properties (Hashimoto, Kakiuchi, Ohba, Nishiyama, & Tsukada, 2017; Tian, Dong, Fujita, Fujita, & Hashimoto, 2018; Yang, Han, Zhang, Ren, & Hashimoto, 2016; Yang et al., 2015). However, at least one human clinical study revealed some adverse events following (R)‐ketamine administration, similar to those reported with (R,S)‐ketamine and (S)‐ketamine (Mathisen, Skjelbred, Skoglund, & Oye, 1995), including motor incoordination, proprioceptive disturbances, and hallucinations (Mathisen et al., 1995). Indeed, a complete lack of NMDA receptor‐mediated adverse effects is inconsistent with its ~2‐fold lower potency, compared to (R,S)‐ketamine, to inhibit the NMDA receptor in vitro (Ebert et al., 1997; Zeilhofer et al., 1992).
Here, we tested whether metabolism of (R)‐ketamine to (2R,6R)‐HNK contributes towards (R)‐ketamine's behavioural effects in (R,S)‐ketamine‐sensitive mouse tests predicting antidepressant efficacy. We used an (R)‐ketamine analogue that cannot readily be metabolised to (2R,6R)‐HNK, as well as direct administration of (2R,6R)‐HNK into the brain to assess for the dose dependency of brain‐specific effects. We also looked for potential (R)‐ketamine adverse effects by testing its potency, compared to (R,S)‐ketamine, to induce psychomotor stimulation, motor incoordination, sensory deficits, and reward‐related conditioning in mice.
2. METHODS
2.1. Animal welfare and ethical statement
All animal care and experimental procedures were conducted in full accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Maryland, Baltimore Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
Male and female CD‐1 mice (Charles River Laboratories, Raleigh, NC, USA; 9–11 weeks of age at the start of the experiment; RRID:IMSR_CRL:22) were kept in a temperature‐ and humidity‐controlled environment and maintained in a 12:12‐hr light/dark cycle (lights on at 7:00 a.m.). Mice were housed in groups of four or five per cage (Super Mouse 750 cage with Allerzone Filter Top; 7.718 in. W × 13.338 in. L × 6.462 in. H; Lab Products, Inc., DE, USA). Cages contained corn cob bedding processed to ¼ in. in nominal dimension (product # 7097; Envigo, Dublin, VA, USA) with a single piece of nestlet (50 mm × 50 mm × 5 mm; Animal Specialties and Provisions, PA, USA). Animals had ad libitum access to food (normal chow diet pellets; product # 2018; Envigo, Dublin, VA, USA) and water. Mice were acclimatised to the new environment for at least 7 days prior to the start of the experiments.
For the electrophysiological experiments, male CD‐1 mice were anaesthetised by a 3‐min exposure to 3% isoflurane, then decapitated, and the brains immediately excised. For the [3H]dizocilpine (MK‐801) binding assays, Sprague Dawley male and female rat brains were used (product code: RAT00BRAINUZN; BioIVT, Westbury, NY, USA) and combined for the experiments. Rats were 7–14 weeks old, housed in standard rat cages (23 in. deep × 13 in. wide × 8 in. tall), equipped with aspen bedding and sani‐chip, and no extra environmental enrichment was provided. Water and food (Teklad diet 2018) were available ad libitum. Rats were killed via CO2 asphyxiation.
Rodents were used in this study, as intact circuits are required to assess the in vivo neurobiological mechanisms of antidepressant drug pharmacokinetics and efficacy. All procedures were carried out to minimise the number of animals used and their suffering.
2.2. Tissue distribution and clearance measurements of (R)‐ and (R)‐d2‐ketamine and metabolites
At 10, 30, 60, or 120 min following an intraperitoneal injection of 10 mg·kg−1 of (R)‐ketamine or (R)‐d2‐ketamine, male CD‐1 mice (n = 4 per treatment or time point) were exposed to 3% isoflurane, and after achievement of complete anaesthesia (2‐min exposure), mice were decapitated. Trunk blood was collected in EDTA (30 μl)‐containing Eppendorf tubes and kept on ice for ~20 min and then centrifuged at 8,000 g for 6 min (4°C). Following trunk blood collection, whole brains were immediately collected, frozen in dry ice, and stored at −80°C until analysis. The 10‐mg·kg−1 dose was chosen based on our previous findings that (R)‐ketamine exerts potent antidepressant‐relevant behaviours at this dose in CD‐1 mice (Zanos et al., 2016). For pharmacokinetic analyses, we used four mice per group. This number of mice is sufficient to conclusively indicate relative plasma and brain levels of compounds (Ette, Kelman, Howie, & Whiting, 1995).
The concentrations of (R)‐ketamine and 6,6‐dideuteroketamine ((R)‐d2‐ketamine) and their respective metabolites in brain tissue were determined by achiral LC–MS following a previously described method (Zanos et al., 2016), with slight modifications. The analysis was accomplished using an Eclipse XDB‐C18 guard column (4.6 mm × 12.5 mm) and a Varian Pursuit XRs 5 C18 analytical column (250 mm × 4.0 mm ID, 5 μm; Varian, Palo Alto, CA, USA). The mobile phase consisted of ammonium acetate [5 mM, pH 7.6] as Component A and acetonitrile as Component B. A linear gradient was run as follows: 0 min 20% B; 5 min 20% B; 15 min 80% B; and 20 min 20% B at a flow rate of 0.4 ml·min−1. The total run time was 30 min per sample. For plasma and brain samples, the calibration standards ranged from 2,000 to 31.25 ng·ml−1 for (R)‐ketamine, (R)‐norketamine, (2R,6R)‐HNK, (R)‐d2‐ketamine, (R)‐d2‐norketamine, and d‐(2R,6R)‐HNK. The quantitation of (R)‐ketamine, (R)‐d2‐ketamine, and their respective metabolites was accomplished by calculating area ratios using d4‐ketamine (10 μl of 10‐μg·ml−1 solution) as the internal standard. The MS/MS analysis was performed using a triple quadrupole mass spectrometer model API 4000 system from Applied Biosystems/MDS Sciex equipped with Turbo Ion Spray® (Applied Biosystems, Foster City, CA, USA). The data were acquired and analysed using Analyst version 1.4.2 (Applied Biosystems). Positive electrospray ionisation data were acquired using optimised multiple reaction monitoring for each metabolite using the following transitions for (R)‐ketamine: 238 → 125 (ketamine), 224 → 125 (norketamine), and 240 → 125 (HNK) and for (R)‐d2‐ketamine: 240 → 125 (d2‐ketamine), 226 → 125 (d2‐norketamine), 241 → 125 (d‐(2,6)‐HNK), 242–125 (d2‐(2,5)‐HNK), and (d2‐(2,4)‐HNK). Lower quantification limits for ketamine and norketamine were 0.00391 μg·ml−1 for plasma and 0.01775 μg·g−1 for brain. For the HNK levels, lower quantification limits were 0.00781 μg·ml−1 for plasma and 0.0355 μg·g−1 for the brain.
2.2.1. Pharmacokinetic analysis
The pharmacokinetic parameters for ketamine, norketamine, and HNK (2–5 time points, depending on the last measurable concentration of each analyte in each matrix) were calculated using non‐compartmental analysis (Model 200) in the pharmacokinetic software Phoenix WinNonlin (version 8.0, Certara, St. Louis, MO). The area under the plasma and tissue concentration versus time curve (AUC) was calculated using the linear trapezoidal method. The slope of the apparent terminal phase was estimated by log linear regression using at least 3 data points, and the terminal rate constant (λ) was derived from the slope. AUC0 → ∞ was calculated as the sum of the AUC0 → t (where t is the time of the last measurable concentration) and Ct/λ. The apparent terminal t 1/2 was calculated as 0.693/λ.
2.3. [3H]MK‐801 displacement binding
The [3H]MK‐801 displacement binding assays were performed by the NIMH Psychoactive Drug Screening Program. Sprague Dawley (RRID:MGI:5651135) rat brains (obtained from BioIVT, Westbury, NY, USA; product code: RAT00BRAINUZN) were homogenised, and membrane fractions were collected from a mixture of male and female tissue. Aliquotted membranes were stored at −80°C until use. Membrane pellets were washed five times with an ice‐cold buffer (20‐mM HEPES, 1‐mM EDTA, pH 7.0) before use. The binding assays were performed in 96‐well plates using 5‐nM [3H]MK‐801 and rat brain membranes (100 μg per well) in a final volume of 125 μl per well in the NMDA receptor binding buffer (20‐mM HEPES, 1‐mM EDTA, 100‐μM glutamate, 100‐μM glycine, pH 7.0). Tested compounds were first distributed in 96‐well plates (25 μl per well at 5× of final concentrations ranging from 0.1 nM to 10 μM, 11 points) in triplicate. The radioligand [3H]MK‐801 was added (50 μl per well at 2.5× of final 5 nM) to all wells. Reactions were initiated with the addition of 50 μl rat brain membrane preparation and were incubated for 1 hr in the dark at room temperature. The reactions were collected via rapid filtration onto Whatman GF/B glass fibre filters presoaked with 0.3% polyethyleneimine using a 96‐well Brandel harvester, followed by three rapid washes each with 500‐μl chilled wash buffer (50‐mM Tris HCl, pH 7.4). Filters were microwave‐dried, and then the scintillation cocktail was melted onto the filter material on a hot plate. The radioactivity retained on the filters was counted in a MicroBeta scintillation counter. All assays were performed in triplicates.
2.4. Hippocampal slice electrophysiology
Measurement of NMDA receptor‐mediated field EPSPs (fEPSPs) was performed at Schaffer collateral‐CA1 synapses in mouse hippocampal slices. Male CD‐1 mice were anaesthetised using 3% isoflurane for 2 min, then decapitated, and the brains immediately excised. The hippocampal region was then dissected and embedded in a 2% agar block. Transverse slices (400 μm thick) were prepared, using a vibratome (1000 plus; Fisher Scientific, USA), with the tissue submerged in an ice‐cold ACSF storage solution composed of 124‐mM NaCl, 3‐mM KCl, 1.25‐mM NaH2PO4, 1.5‐mM MgSO4, 2.5‐mM CaCl2, 26‐mM NaHCO3, and 10‐mM glucose, oxygenated with carbogen (95% O2/5% CO2), and maintained in a recovery chamber for at least 60 min prior to recording.
NMDA receptor‐fEPSPs were recorded from mouse hippocampal slices that were placed in a submersion‐type recording chamber and superfused at a rate of 1 ml·min−1 at room temperature (20–22°C) with nominally Mg2+‐free ACSF solution (identical composition to storage ACSF, but without MgSO4) and bubbled with carbogen. Picrotoxin (final concentration: 100 μM) and CGP52432 (final concentration: 2 μM) were added to block GABA A and GABA B receptors, respectively. A concentric bipolar tungsten stimulating electrode was placed in stratum radiatum to stimulate Schaffer collateral afferents, and a recording pipette (3–5 MΩ) filled with Mg2+‐free ACSF solution was placed ~500–750 μm from the stimulating electrode into the stratum radiatum of CA1 field of the hippocampal slices.
Stimuli (0.1 to 1 mA) were delivered at 0.05 Hz to stimulate the Schaffer collaterals and evoke fEPSPs that were amplified 1,000×, filtered at 3 kHz, and digitised at 10 kHz using an NPI EXT‐02B amplifier (NPI Electronic GmbH, Tamm, Germany). Following a 10‐min period to ensure stability of the responses, slices were superfused for 15 min with DNQX (50 μM)‐containing ACSF to block AMPA receptor‐mediated fEPSPs. Subsequently, slices were superfused for an additional 30 min with ACSF containing vehicle (0.9% saline), (R)‐ketamine, or (R)‐d2‐ketamine (from a 0.9% saline solution; different concentrations included 2, 6, 20, and 60 μM) in addition to DNQX (50 μM). Any remaining NMDA receptor‐mediated fEPSPs were then blocked by superfusion of the slices with ACSF containing the NMDA receptor antagonist D‐AP5 (80 μM) for 15 min.
The NMDA receptor‐mediated fEPSPs slopes were quantified by calculating the slope at the first linear 4 ms of the response and then normalised to the amplitude of the fibre volley, using the pCLAMP software suite (Molecular Devices, version 10.3.1.4; RRID:SCR_011323). Pre‐wash‐in (fEPSP slope average measured during the first five sweeps of the 30‐min drug wash‐in) and post‐wash‐in (fEPSP slope average measured during last five sweeps of the 30‐min drug wash‐in) values were calculated. The inhibition produced by the test substance was quantified by subtracting the final slope in the presence of drug from the baseline slope and then dividing that by the difference between the baseline slope and any remaining NMDA receptor inhibition‐insensitive slope after the addition of AP5 (calculated as the mean of the last five sweeps).
2.5. Behavioural methods
Administration of drugs and behavioural procedures were performed by male experimenters.
2.5.1. Determination of sub‐anaesthetic doses of (R)‐ketamine
To determine the minimally effective anaesthetic dose, (R)‐ketamine was administered to mice at 90‐, 120‐, 150‐, and 180‐mg·kg−1 doses (n = 7 per dose to define the dose that did not induce anaesthesia), and the duration of complete loss of righting reflex was measured in empty mouse cages by placing the mice on their backs, and the time required to completely right themselves was recorded.
2.5.2. Forced‐swim test
Male mice were tested in the forced‐swim test (FST) 24 hr post‐injection, as previously described (Zanos et al., 2016). During the FST, mice were subjected to a 6‐min swim session in clear Plexiglass cylinders (30‐cm height × 20‐cm diameter) filled with 15 cm of water (23 ± 1°C). The FST was performed in normal lighting conditions (800 lx). Sessions were recorded using a digital video camera. Immobility time, defined as passive floating with no additional activity other than that necessary to keep the animal's head above the water, was scored for the last 4 min of the 6‐min test by a trained observer.
2.5.3. Inescapable shock‐induced behaviours
Inescapable shock training (Day 1)
Inescapable shock was administered to male mice in one side of two‐chambered shuttle boxes (Coulbourn Instruments, PA, USA), with the door between the chambers closed. Following a 5‐min adaptation period, 120 inescapable foot shocks (0.45 mA, 15‐s duration, 45‐s average inter‐shock interval) were delivered. In total, we exposed 353 mice to the inescapable shock stress.
Escape deficits screening (Day 2)
During the screening session, a total of 30 escapable 3‐s foot shocks (0.45 mA, 30‐s inter‐trial interval) were delivered, and the door between the two chambers was raised simultaneously. Susceptible mice (≥5 escape failures during the last 10 screening shocks and ≥20 total escape failures) received treatment 24 hr following screening (Day 3). Resilient mice (≤20 total escape failures and ≤5 escape failures during the last 10 screening shocks) received saline injection. Out of 353 mice that were exposed to inescapable shock, 196 mice were classified as resilient and 157 as susceptible based on our criteria of susceptibility. From these mice, we treated 40 randomly selected resilient and 157 susceptible mice (see below).
Drug administration (Day 3)
For the (R)‐ketamine versus (R)‐d2‐ketamine experiment, drugs were administered to mice intraperitoneally at a volume of 7.5 ml·kg−1, using 0.9% saline solution as vehicle. For the intracerebroventricular administration experiments, (2R,6R)‐HNK was dissolved in 1× PBS buffer and was administered freehand at the volume of 10 μl at a rate of 1 μl per 2 s using Hamilton syringes (Microliter #702; Hamilton Robotics, Nevada, USA). Prior to drug administration, mice were briefly anaesthetised using 2.5% isoflurane, administered analgesia (carprofen, 5 mg·kg−1, s.c.), and moved to a stereotaxic frame to maintain anaesthesia (2.5% isoflurane) through a mouse nose cone. After achievement of complete anaesthesia, a transverse ~1‐cm midline incision was made to expose the scalp, and intracerebroventricular infusion was delivered using preselected coordinates (1.0 mm lateral and 0.3 mm anterior to the Bregma). The procedure, from anaesthesia induction to completion of the infusion and suturing, took on average 5 min per mouse. Following drug administration, mice were singly housed for the remainder of the experiment and were monitored twice daily for signs of distress or pain.
Escapable shock test (Day 4)
During the test phase, the animals were placed in the shuttle boxes, and after a 5‐min adaptation period, a total of 45 shocks (0.45 mA, 30‐s inter‐trial interval) were delivered concomitantly with door opening for the first five trials, followed by a 2‐s delay for the next 40 trials. If the animal did not cross over to the other chamber, the shock was terminated after 24 s.
Sucrose preference (Days 5–6)
Mice were provided with two bottles containing either 1% sucrose solution or tap water. Twenty‐four and forty‐eight hours later, sucrose preference was measured, as sucrose solution consumed over total solution consumed, for assessing reversal of helplessness‐induced anhedonia.
Open‐field test (Day 7)
This experiment was performed at 100 lx. Mice were placed into individual open‐field arenas (50‐cm length × 50‐cm width × 38‐cm height; San Diego Instruments, CA, USA) for a 30‐min locomotor assessment. Distance travelled was automatically analysed using TopScan v2.0 (Clever Sys Inc, VA, USA; RRID:SCR_014494).
Escapable shock retest (Day 8)
In order to determine the prolonged efficacy of (2R,6R)‐HNK, mice were retested for helpless behaviour (escapable shock test; as described above) 5 days post‐drug administration.
2.5.4. Open‐field test
For assessing locomotor stimulant effects of (R)‐ketamine, we used the open‐field test (OFT), as previously published (Zanos et al., 2015). This experiment was performed at 100 lx using the same open‐field arenas described above. Male and female mice were randomly divided into six treatment groups (i.e., saline; (R,S)‐ketamine, 20 mg·kg−1; and (R)‐ketamine, 10, 20, 40, or 80 mg·kg−1). After a 60‐min habituation period, mice received treatment injection, and locomotor responses were subsequently recorded for an additional 60 min. Distance travelled was automatically analysed using TopScan v2.0 (Clever Sys Inc).
2.5.5. Prepulse inhibition
PPI was performed based on a previously published protocol (Zanos et al., 2016). Following drug administration (saline, (R,S)‐ketamine 30 mg·kg−1, or (R)‐ketamine 30, 60, and 90 mg·kg−1), mice were placed in the startle chamber for a 30‐min habituation period. The experiment started with a further 5‐min adaptation period during which the mice were exposed to a constant background noise (67 dB), followed by five initial startle stimuli (120 dB, 40‐ms duration each). Subsequently, animals were exposed to four different trial types: pulse alone trials (120 dB, 40‐ms duration), three prepulse trials of 76, 81, and 86 dB of white noise bursts (20‐ms duration) preceding a 120‐dB pulse by 100 ms, and background (67 dB) no‐stimuli trials. Each of these trials was presented five times in a pseudo‐random order. The dose of (R,S)‐ketamine (30 mg·kg−1) was selected based on a dose–response experiment we performed in a previous study (Zanos et al., 2015). Per cent PPI was calculated using the following formula: [(magnitude on pulse alone trial − magnitude on prepulse + pulse trial) / magnitude on pulse alone trial] × 100.
2.5.6. Conditioned‐place preference
The conditioned‐place preference (CPP) experiment was performed as previously published (Zanos et al., 2017). The CPP apparatus consisted of a rectangular three‐chambered box (40‐cm length × 30‐cm width × 35‐cm height; Stoelting Co., Wood Dale, IL, USA) composed of two equal sized end‐chambers (20 cm × 18 cm × 35 cm) and a central chamber (20 cm × 10 cm × 35 cm). One end‐chamber had a perforated floor and plain black walls, whereas the other end‐chamber had a smooth floor and walls with vertical black and white stripes. The CPP protocol consisted of a preconditioning phase, eight conditioning sessions, and a post‐conditioning test. On Day 1 (preconditioning phase), male and female mice were placed in the CPP apparatus and were allowed to explore all compartments for a period of 20 min. During the morning sessions of the conditioning phase (Days 2–5), saline was administered and mice were placed in their preferred end‐chamber (>50% of their time spent in that compartment compared with the other end‐chamber compartment; defined during the preconditioning phase) for 30 min. Five hours later (afternoon sessions), saline, (R,S)‐ketamine (20 mg·kg−1), or (R)‐ketamine (40 mg·kg−1) were administered, and mice were placed in their least preferred compartment for 30 min. During the post‐conditioning test session (i.e., Day 6), mice were placed in the CPP apparatus to freely explore all three compartments for 20 min. Time spent in each compartment was measured during both preconditioning and post‐conditioning sessions using TopScan v2.0 (Clever Sys Inc).
2.5.7. Rotarod
This paradigm was performed as previously described (Zanos et al., 2016). The experiment had two phases: a training phase (5 days) and a test phase (1 day). On each of the training days, five trials (total trial duration: 3 min) were conducted with an inter‐trial interval of 2 min. Male and female mice were individually placed on the rotarod apparatus (IITC Life Science; Woodland Hills, CA, USA), and the rotor (3.75‐in. diameter) accelerated from 5 to 20 RPM over a period of 3 min. Latency to fall was recorded for each trial. Animals with an average of <100 s of latency to fall during the last training day (Day 4) were excluded from the experiment. On the test day (Day 5), mice received (intraperitoneal) injections of saline or (R)‐ketamine at the doses of 10, 20, or 40 mg·kg−1 and were tested in the rotating rod 5, 10, 15, 20, and 30 min post‐injection using the same procedure described for the training days.
2.6. Data and statistical analyses
All the data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Sample sizes were based upon our prior experience using the same paradigms. Mice were randomly assigned to each treatment group, and experimentation and data analysis were performed in a manner completely blind to experimental groups. The extra sum‐of‐squares F test was used to determine whether the one‐site or two‐site fit model is appropriate for analysing the [3H]MK‐801 binding data. The extra sum‐of‐squares F test was used for the comparison of best‐fit values between the dose response curves of (R)‐ketamine and (R)‐d2‐ketamine to displace [3H]MK‐801 binding. For the inescapable shock‐induced behaviours, the results were analysed via one‐way ANOVA. OFT distance travelled (m per 15 min) between (R)‐ketamine and (R)‐d2‐ketamine following injection was analysed using Student's unpaired t test. Sex differences in the OFT, drug‐induced sensitisation, and rotarod tests were analysed by repeated measures two‐way ANOVA. Sex differences in the rotarod training were assessed by a two‐way ANOVA. A three‐way repeated measures ANOVA was used to assess sex differences in the CPP and PPI. Since no sex difference was observed in any of these experiments, data from both male and female mice were pulled together and analysed together, as follows. Distance travelled following administration of (R,S)‐ketamine versus (R)‐ketamine was analysed via one‐way ANOVA. CPP, PPI, and rotarod data were analysed using a repeated measures two‐way ANOVA. ANOVAs were followed by Holm–Sidak's multiple comparisons post hoc test when significance was reached (i.e., P < .05). All post hoc comparison results are indicated in the figures. Statistical analyses were performed using GraphPad Prism software v6 (RRID:SCR_002798), except for the three‐way ANOVA analyses used to assess for sex differences, which were performed in Statistica v7 software. All values are expressed as the mean ± SEM. Statistical outliers (based on priori criteria) were determined and removed from the dataset using the ROUT method (Motulsky & Brown, 2006), provided by GraphPad Prism (RRID: SCR_002798); parameter used: Q = 1%. All raw data are provided in Table S1.
2.7. Nomenclature of targets and ligands
Key ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017; Alexander, Peters et al., 2017).
3. RESULTS
3.1. In vitro and in vivo characterisation of (R)‐d2‐ketamine
To test the hypothesis that production of (2R,6R)‐HNK contributes to the antidepressant‐relevant actions of (R)‐ketamine (Figure 1a), an analogue of (R)‐ketamine was synthesised by substituting the hydrogen atoms at the C6 position with deuterium ((R)‐d2‐ketamine; Figure 1b). This substitution was expected to hinder its metabolism to (2R,6R)‐HNK, without changing its pharmacodynamic properties. Indeed, in vitro characterisation using [3H]MK‐801 displacement binding revealed no difference in Ki affinity of (R)‐d2‐ketamine compared with (R)‐ketamine (Figure 1c). (R)‐Ketamine inhibited electrically evoked NMDA receptor‐mediated fEPSPs in the CA1 hippocampal region of mouse brains with an IC50 of 9.96 μM (95% confidence intervals: 8.21–12.08 μM) and (R)‐d2‐ketamine with an IC50 of 7.73 μM (95% confidence intervals: 6.14–9.71 μM; Figure 1d,e), indicating no functional difference between the ability of these compounds to inhibit NMDA receptor‐mediated responses. To compare their action in an in vivo measure of (R,S)‐ketamine activity mediated by NMDA receptor inhibition (Irifune, Shimizu, Nomoto, & Fukuda, 1995), an OFT was performed where (R)‐ and (R)‐d2‐ketamine induced equivalent hyperlocomotion in mice (Figure 1f,g).
Figure 1.
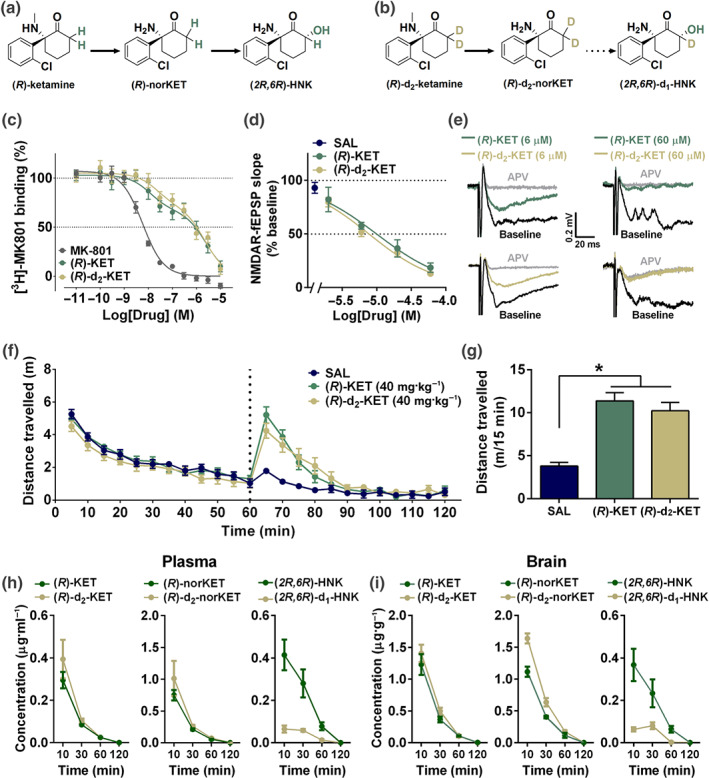
C6‐deuterated (R)‐ketamine attenuates metabolism to (2R,6R)‐hydroxynorketamine without modifying NMDA receptor binding or functional inhibition properties. Metabolic transformations of (a) (R)‐ketamine (KET) and its (b) deuterated isotope at the C6 position, (R)‐d2‐KET. (R)‐KET is demethylated to give (R)‐norketamine (norKET), which in turn is hydroxylated to result in (2R,6R)‐hydroxynorketamine (HNK). (R)‐d2‐KET is hydroxylated to (R)‐d2‐norKET. (R)‐d2‐norKET cannot readily be transformed to (2R,6R)‐d1‐HNK. (c) Both (R)‐KET and (R)‐d2‐KET displace [3H]MK‐801 binding with similar affinity. (d) Concentration–response relationships of NMDA receptor‐mediated fEPSP slope as a function of compound concentration for (R)‐KET and (R)‐d2‐KET. (e) Typical traces of NMDA receptor‐mediated fEPSPs in slices from mouse CA1 stratum radiatum in response to stimulation of the Schaffer collateral axons before and after perfusion of either 6‐ or 60‐μM (R)‐KET and (R)‐d2‐KET. (f) After recording baseline activity for 60 min, mice received drug (vertical dashed line), and locomotor activity was monitored for another 60 min. (g) Total distance moved during the first 15 min following drug exposure. (R)‐KET and (R)‐d2‐KET induced equivalent hyperlocomotor responses at the dose of 10 mg·kg−1. (h) Plasma and (i) brain levels of KET, norKET, and HNK following intraperitoneal administration of (R)‐KET or (R)‐d2‐KET (10 mg·kg−1) in mice. Data are the mean ± SEM. *P < .05, significantly different as indicated. See Tables 2 and S1 for statistical analyses and precise group sizes
The pharmacokinetic profile of (R)‐ketamine and (R)‐d2‐ketamine was evaluated in the plasma (Figure 1h) and brain (Figure 1i) of mice following administration of equivalent doses (10 mg·kg−1; intraperitoneal administration) to assess for differences in their metabolism in vivo. Maximum concentration (Cmax), AUC, and half‐lives of (R)‐ketamine and (R)‐d2‐ketamine and their respective metabolites are listed in Table 1. These data reveal that while brain tissue ketamine levels are not different, and norketamine levels are slightly higher, (2R,6R)‐HNK peak levels are ~4.5‐fold lower, and total exposure is reduced by ~7‐fold following administration of (R)‐d2‐ketamine compared with (R)‐ketamine. These findings support the use of (R)‐d2‐ketamine as a tool compound to assess the role of (2R,6R)‐HNK in the antidepressant‐relevant behavioural actions of (R)‐ketamine.
Table 1.
Pharmacokinetic analysis of ketamine, norketamine, and hydroxynorketamine in the plasma and brain of mice following a single administration of (R)‐ketamine or (R)‐d2‐ketamine
| Tissue | Cmax (μg·ml−1 for plasma; μg·g−1 for brain) | AUC0 → ∞ (μg·ml−1·hr−1 for plasma; μg·g−1·hr−1 for brain) | AUC0 → last (μg·ml−1·hr−1 for plasma; μg·g−1·hr−1 for brain) | t 1/2 (hr) | ||||
|---|---|---|---|---|---|---|---|---|
| (R)‐KET | (R)‐d2‐KET | (R)‐KET | (R)‐d2‐KET | (R)‐KET | (R)‐d2‐KET | (R)‐KET | (R)‐d2‐KET | |
| Plasma | 0.30a | 0.39a | 0.14 | 0.16 | 0.13 | 0.16 | 0.37 | 0.25 |
| Brain | 1.23a | 1.41a | 0.52 | 0.62 | 0.49 | 0.58 | 0.24 | 0.22 |
| (R)‐norKET | (R)‐d2‐norKET | (R)‐norKET | (R)‐d2‐norKET | (R)‐norKET | (R)‐d2‐norKET | (R)‐norKET | (R)‐d2‐norKET | |
| Plasma | 0.75a | 1.01a | 0.33 | 0.42 | 0.33 | 0.42 | 0.34 | 0.23 |
| Brain | 1.12a | 1.64a | 0.53 | 0.78 | 0.48 | 0.72 | 0.27 | 0.26 |
| (2R,6R)‐HNK | (2R,6R)‐d1‐HNK | (2R,6R)‐HNK | (2R,6R)‐d1‐HNK | (2R,6R)‐HNK | (2R,6R)‐d1‐HNK | (2R,6R)‐HNK | (2R,6R)‐d1‐HNK | |
| Plasma | 0.41a | 0.07a | 0.34 | 0.05 | 0.33 | 0.04 | 0.56 | 0.31 |
| Brain | 0.37a | 0.08b | 0.23 | ND | 0.21 | 0.03 | 0.32 | ND |
Note. Parameters for ketamine (KET), norketamine (norKET), and hydroxynorketamine (HNK) were calculated from five sampling time points following a single intraperitoneal injection of 10‐mg·kg−1 (R)‐KET or C6‐deuterated (R)‐KET ((R)‐d2‐KET; n = 4 per treatment per time point).
Abbreviations: Cmax, maximum concentration; ND, not determined.
Cmax achieved at 10 min after dosing.
Cmax achieved at 30 min after dosing.
Table 2.
Relevant statistical analyses
| Figure/Experiment | Sample size (figure order) | Statistical test | Statistical analysis effect | ||||
|---|---|---|---|---|---|---|---|
| Figure 1 | |||||||
| (c) | [3H]MK‐801 binding | n = 12 per group | F test (two‐site fit)—(R)‐KET versus (R)‐d2‐KET | F(2, 280) = 0.36; P > .05 | |||
| (d) | NMDAR‐fEPSP | No statistical test | |||||
| ● SAL (control) | n = 17 | ||||||
| ● −5.5 Log[Drug] (M) | n = 4, 5 | ||||||
| ● −5.0 Log[Drug] (M) | n = 5, 6 | ||||||
| ● −4.5 Log[Drug] (M) | n = 6, 7 | ||||||
| ● −4.0 Log[Drug] (M) | n = 6, 6 | ||||||
| Treatment effect | Time effect | Treatment × Time interaction | |||||
| (f) | Open‐field test (timeline) | n = 10 per group | Two‐way RM ANOVA | F(2, 27) = 1.815; P > .05 | F(23, 621) = 88.04; P < .05 | F(46, 621) = 5.67; P < .05 | |
| (g) | Open‐field test (cumulative) | n = 10 per group | One‐way ANOVA | F(2, 27) = 24.90; P < .05 | |||
| (i) | Pharmacokinetics | n = 4 per group | |||||
| Figure 2 | Treatment effect | ||||||
| (b) | Forced‐swim test | n = 8 per group | One‐way ANOVA | F(6, 49) = 5.90; P < .05 | |||
| (d) | Escapable shock test/24‐hr test | n = 25, 15, 14, 15, 16, 12, 14, 16 | One‐way ANOVA | F(7, 119) = 11.65; P < .05 | |||
| (e) | Sucrose preference | n = 25, 16, 14, 15, 16, 14, 16, 13 | One‐way ANOVA | F(7, 121) = 18.46; P < .05 | |||
| (f) | Open‐field test | n = 25, 16, 14, 15, 16, 14, 16, 16 | One‐way ANOVA | F(7, 124) = 0.61; P > .05 | |||
| (g) | Escapable shock test/5‐day retest | n = 25, 15, 12, 15, 12, 11, 16, 16 | One‐way ANOVA | F(7, 114) = 13.12; P < .05 | |||
| Figure 3 | Treatment effect | ||||||
| (a) | Escapable shock test/24‐hr test | n = 13, 10, 10, 10, 10, 10 | One‐way ANOVA | F(5, 57) = 11.69; P < .05 | |||
| (b) | Sucrose preference | n = 15, 10, 10, 10, 10, 10 | One‐way ANOVA | F(5, 59) = 11.73; P < .05 | |||
| (c) | Open‐field test | n = 15, 10, 9, 10, 10, 10 | One‐way ANOVA | F(5, 58) = 1.45; P > .05 | |||
| (d) | Escapable shock test/5‐day retest | n = 14, 10, 10, 10, 8, 10 | One‐way ANOVA | F(5, 56) = 7.09; P < .05 | |||
| Figure 4 | |||||||
| (a) | Anaesthesia assessment—Loss of righting reflex | n = 7 per group | |||||
| Figure 4: Assessment for sex differences | Sex effect | Treatment effect | Sex × Treatment interaction | ||||
| (c) | Open‐field test |
Males: n = 8, 8, 8, 7, 8, 7 Females: n = 8, 8, 8, 7, 8, 8 |
Two‐way ANOVA | F(1, 84) = 3.10; P > .05 | F(5, 84) = 19.56; P < .05 | F(5, 84) = 0.77; P > .05 | |
| Sex effect | Treatment effect | Phase effect | Sex × Treatment × Phase interaction | ||||
| (e) | Conditioned‐place preference |
Males: n = 8, 8, 9 Females: n = 8, 9, 9 |
Three‐way RM ANOVA | F(1, 45) = 1.43; P > .05 | F(2, 45) = 3.19; P = .05 | F(2, 45) = 4.26; P < .05 | F(2, 45) = 2.43; P > .05 |
| Sex effect | Treatment effect | Intensity effect | Sex × Treatment × Intensity interaction | ||||
| (f) | % PPI |
Males: n = 12, 11, 11, 11, 10 Females: n = 11, 11, 11, 11, 11 |
Three‐way RM ANOVA | F(1, 202) = 0.69; P > .05 | F(4, 202) = 4.32; P < .05 | F(2, 202) = 52.23; P < .05 | F(8, 202) = 1.56; P > .05 |
| Sex effect | Treatment effect | Sex × Treatment interaction | |||||
| (g) | Rotarod training |
Males: n = 10, 9, 10, 9 Females: n = 10, 9, 10, 10 |
Two‐way ANOVA | F(1, 69) = 1.24; P > .05 | F(3, 69) = 0.002; P > .05 | F(3, 69) = 0.002; P > .05 | |
| Sex effect | Treatment effect | Sex × Treatment interaction | |||||
| (h) | Rotarod test |
Males: n = 10, 9, 10, 9 Females: n = 10, 9, 10, 10 |
Two‐way RM ANOVA | F(1, 84) = 3.10; P > .05 | F(5, 84) = 19.55; P < .05 | F(5, 84) = 0.77; P > .05 | |
| Figure 4: Statistical analysis with male and female combined data | Treatment effect | ||||||
| (c) | Open‐field test | n = 16, 16, 16, 14, 16, 15 | One‐way ANOVA | F(5, 87) = 21.69; P < .05 | |||
| Treatment effect | Conditioning effect | Treatment × Conditioning interaction | |||||
| (e) | Conditioned‐place preference | n = 16, 17, 18 | Two‐way RM ANOVA | F(2, 48) = 3.05; P = .06 | F(1, 48) = 4.27; P < .05 | F(2, 48) = 4.76; P < .05 | |
| Treatment effect | Intensity effect | Treatment × Intensity interaction | |||||
| (f) | % PPI | n = 23, 22, 22, 22, 21 | Two‐way RM ANOVA | F(4, 105) = 4.51; P < .05 | F(2, 210) = 51.25; P < .05 | F(8, 210) = 0.47; P > .05 | |
| Treatment effect | |||||||
| (g) | Rotarod training | n = 20, 18, 20, 19 | One‐way ANOVA | F(3, 73) = 0.004; P > .05 | |||
| Treatment effect | Time effect | Treatment × Time interaction | |||||
| (h) | Rotarod test | n = 20, 18, 20, 19 | Two‐way RM ANOVA | F(3, 73) = 42.09; P < .05 | F(4, 292) = 30.93; P < .05 | F(12, 292) = 19.81; P < .05 | |
Note: Raw data are provided in Table S1. Male mice used unless otherwise stated in the table. Variable n values between groups exist in our dataset for three reasons, dependent upon the specific experiment: (a) The numbers of susceptible and resilient mice yielded by our inescapable shock paradigm vary between experimental cohorts (on average approximately 47% of mice meet the criteria for susceptibility); thus, group sizes varied depending on the number of susceptible mice within a given experiment. For the resilient mice, we randomly selected a subset of mice to be administered drug. (b) For the electrophysiology experiments (NMDA receptor‐fEPSP), vehicle‐infused control slices were included within each recording session. Thus, for each concentration of (R)‐ketamine and (R)‐d2‐KET, separate vehicle controls were included for blinding purposes. At the completion of the experiment, data for all control slices were combined resulting in larger numbers of control slices. (c) We have used a priori exclusion criteria for identifying outliers by applying the ROUT outlier test.
Abbreviations: HNK, hydroxynorketamine; KET, ketamine;
norKET, norketamine; PPI, prepulse inhibition; RM, repeated measures; SAL, saline.
Male and female mouse data were pooled becauuse no sex interactions were observed.
3.2. Role of (R)‐ketamine's metabolism in its antidepressant‐relevant behavioural actions
3.2.1. Effect of (R)‐ketamine versus (R)‐d2‐ketamine administration in the FST
To test whether metabolism of (R)‐ketamine to its (2R,6R)‐HNK metabolite is involved in its antidepressant‐relevant behavioural actions, we administered 2.5, 5, or 10 mg·kg−1 of either (R)‐ketamine or (R)‐d2‐ketamine and tested mice 24 hr later in the FST (see timeline: Figure 2a). We note that the 24‐hr time point predicts sustained antidepressant‐like actions of rapidly acting antidepressants but is not sensitive to traditional antidepressants (Ramaker & Dulawa, 2017; Zanos, Thompson, et al., 2018). (R)‐Ketamine reduced immobility time at the doses of 5 and 10 mg·kg−1, whereas only 10 mg·kg−1 of (R)‐d2‐ketamine decreased immobility time compared with the control group (Figure 2b). At the dose of 5 mg·kg−1, (R)‐ketamine‐treated mice showed significantly less immobility time compared with mice receiving the same dose of (R)‐d2‐ketamine, indicating that (R)‐ketamine's metabolism to (2R,6R)‐HNK contributes to its sustained (24 hr post‐injection) actions in the FST.
Figure 2.
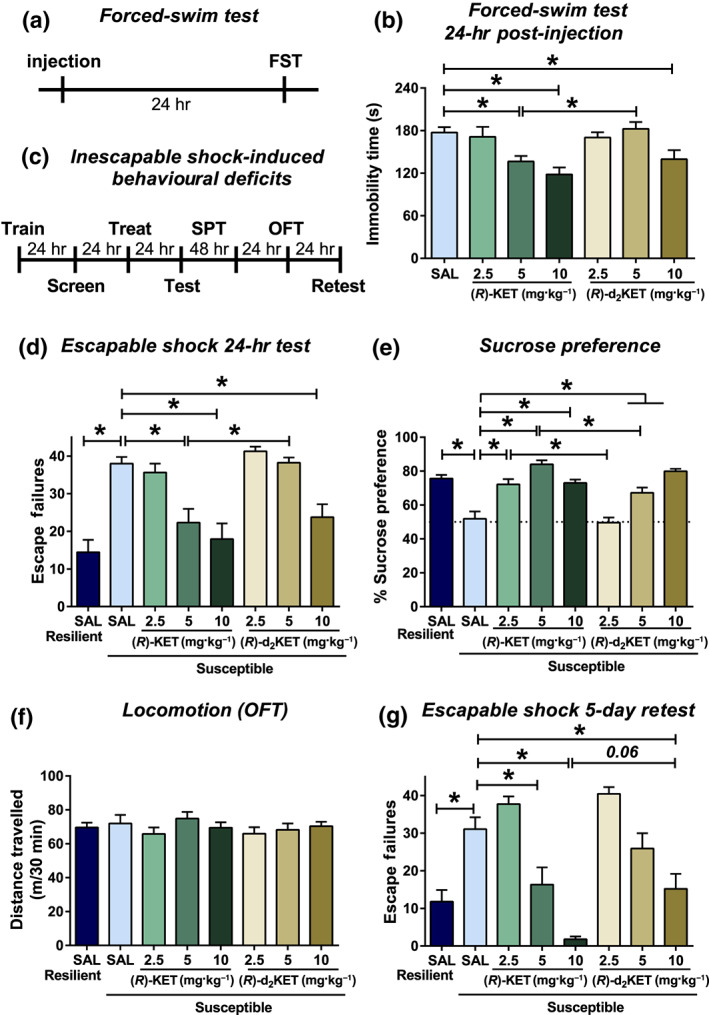
Metabolism of (R)‐ketamine to (2R,6R)‐HNK is involved in its antidepressant‐relevant actions. (a) Timeline for the forced‐swim test (24 hr post‐injection) paradigm. Mice received injection of vehicle or different doses of (R)‐ketamine and (R)‐d2‐ketamine, and 24 hr later, they were assessed in the forced‐swim test. (b) (R)‐Ketamine manifested greater potency to decrease immobility time in the forced‐swim test compared with (R)‐d2‐ketamine. (c) Timeline for the inescapable shock‐induced behavioural deficits. (d) Twenty‐four hours following treatment, mice were tested for helpless (escape deficits) behaviour, where (R)‐ketamine manifested greater potency to decrease escape deficits compared with (R)‐d2‐ketamine. (e) Similarly, (R)‐ketamine manifested greater potency to reverse inescapable shock‐induced sucrose preference deficits in susceptible mice. (f) No differences in locomotor activity of mice were observed between any treatment groups. (g) To assess long‐term antidepressant‐relevant actions of (R)‐ketamine and (R)‐d2‐ketamine, mice were retested for escape deficits 5 days post‐injection. (R)‐Ketamine exhibited longer lasting antidepressant‐relevant actions compared to its (R)‐d2‐ketamine isotope. Data are the mean ± SEM. *P < .05, significantly different as indicated. See Tables 2 and S1 for statistical analyses and precise group sizes. FST, forced‐swim test; OFT, open‐field test; SPT, sucrose preference test
3.2.2. Effect of (R)‐ketamine versus (R)‐d2‐ketamine administration following inescapable shock‐induced behaviours
To further assess the role of (2R,6R)‐HNK in the antidepressant‐relevant actions of (R)‐ketamine, we evaluated the effect of (R)‐d2‐ketamine on the reversal of inescapable shock‐induced escape and sucrose preference deficits (see timeline: Figure 2c). This model is sensitive to acute administration of (R,S)‐ketamine, but not traditional antidepressants, when tested 24 hr following drug exposure (see Ramaker & Dulawa, 2017). Here, mice are screened for susceptibility 24 hr following inescapable shock. Susceptible mice that received saline injections exhibited significantly more escape failures compared to resilient saline‐treated mice (Figure 2d). (R)‐Ketamine administration significantly reduced escape failures of susceptible mice at the dose of 5 and 10 mg·kg−1, whereas (R)‐d2‐ketamine was only active at the dose of 10 mg·kg−1 (Figure 2d). Notably, the 5‐mg·kg−1 dose of (R)‐ketamine induced significantly fewer escape failures compared with the same dose of (R)‐d2‐ketamine (Figure 2d), indicating greater potency of (R)‐ketamine and a critical role of the (2R,6R)‐HNK metabolite in this effect of (R)‐ketamine.
Forty‐eight hours after drug administration, anhedonia was assessed by sucrose preference measurements over a period of 48 hr. Susceptible mice that received saline exhibited significantly lower sucrose preference compared to resilient mice receiving saline (Figure 2e). (R)‐Ketamine administration significantly increased sucrose preference of susceptible mice at all doses tested (i.e., 2.5, 5, and 10 mg·kg−1), whereas (R)‐d2‐ketamine's anti‐anhedonic actions appeared only at the doses of 5 and 10 mg·kg−1 (Figure 2e). Notably, the 5‐mg·kg−1 doses of (R)‐ketamine induced greater reversal of sucrose preference deficits in susceptible mice compared with the same dose of (R)‐d2‐ketamine (Figure 2e), indicating greater potency of (R)‐ketamine. One day later, mice were assessed in the OFT, which revealed no differences in distance travelled between any treatment groups (Figure 2f).
To assess the role of (R)‐ketamine's metabolism in the long‐lasting antidepressant‐relevant actions of the drug, mice were retested for escape failures 5 days post‐treatment. Susceptible mice that received saline treatment continued to manifest increased escape failures compared with resilient mice (Figure 2g). Susceptible mice pretreated with (R)‐ketamine displayed reduced escape failures at the dose of 5 and 10 mg·kg−1. (R)‐d2‐ketamine was only active at the dose of 10 mg·kg−1 (Figure 2g). Administration of (R)‐ketamine at the dose of 10 mg·kg−1 showed a trend towards lower escape failures compared with the same dose of (R)‐d2‐ketamine (Figure 2g), indicating greater potency of (R)‐ketamine.
3.2.3. Effect of intracerebroventricular administration of (2R,6R)‐HNK on inescapable shock‐induced behaviours
We administered (2R,6R)‐HNK directly into the brain of mice following inescapable shock stress in order to determine whether its effects are mediated by direct brain actions (timeline identical to Figure 2a). Susceptible mice that received PBS exhibited significantly more escape failures compared to the resilient mice that received vehicle (Figure 3a). (2R,6R)‐HNK administration significantly decreased escape failures at the doses of 3 and 10 nmol, while a 1‐nmol infusion of (2R,6R)‐HNK showed a trend towards fewer escape failures of susceptible mice and 30‐nmol infusion had no effect (Figure 3a).
Figure 3.
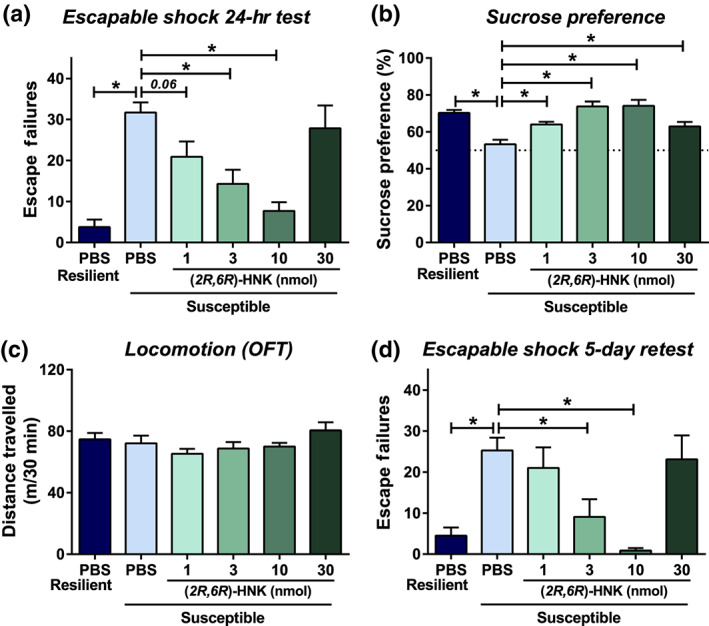
Intracerebroventricular administration of (2R,6R)‐hydroxynorketamine exerts rapid and long‐lasting antidepressant‐relevant behavioural actions in mice. Following inescapable shock and screening for escape deficits, mice were given intracerebroventricular infusions of vehicle or different doses of (2R,6R)‐hydroxynorketamine (HNK). For a timeline, see Figure 2c. (a) Twenty‐four hours following treatment, mice were tested for helpless (escape deficits) behaviour, where (2R,6R)‐HNK dose dependently reduced escape deficits. (b) (2R,6R)‐HNK reversed inescapable shock‐induced sucrose preference deficits in susceptible mice at all doses. (c) No differences in locomotor activity of mice were observed in any treatment group. (d) To assess for long‐term antidepressant‐relevant actions, mice were retested for shock‐induced escape deficits 5 days post‐injection. (2R,6R)‐HNK administration manifested long‐lasting antidepressant‐relevant actions in this paradigm. Data are the mean ± SEM. *P < .05, significantly different as indicated. See Tables 2 and S1 for statistical analyses and precise group sizes
Forty‐eight hours after (2R,6R)‐HNK infusion, mice were assessed for sucrose preference over a 48‐hr time period. PBS‐treated susceptible mice exhibited significantly lower sucrose preference compared to PBS‐treated resilient mice (Figure 3b). (2R,6R)‐HNK administration significantly increased sucrose preference of susceptible mice at all doses tested (Figure 3b). The following day, mice were tested in an open‐field arena, which revealed no difference in distance travelled between any of the treatment groups (Figure 3c).
To assess the sustained actions of brain‐infused (2R,6R)‐HNK, mice were retested for escape deficits 5 days post‐(2R,6R)‐HNK infusion. Susceptible mice that received vehicle continued to manifest increased escape failures compared to resilient mice (Figure 3d). (2R,6R)‐HNK administration significantly decreased escape failures of susceptible mice at the doses of 3 and 10 nmol, while there was no effect of 1‐ or 30‐nmol doses (Figure 3d).
3.3. (R)‐Ketamine exerts locomotor stimulant effects, induces CPP, and disrupts PPI performance of mice at sub‐anaesthetic doses
To determine the minimally effective anaesthetic dose, (R)‐ketamine was administered to mice at doses ranging from 90 to 180 mg·kg−1, and the duration of complete loss of righting reflex was measured. None of the mice receiving 90 mg·kg−1 of (R)‐ketamine lost their righting reflex, while six out of seven mice lost reflex at the dose of 120 mg·kg−1. Thus, we determined 120 mg·kg−1 to be the minimally effective anaesthetic dose of (R)‐ketamine in CD‐1 mice, while 90 mg·kg−1 is a sub‐anaesthetic dose (Figure 4a). All mice receiving 150 and 180 mg·kg−1 of (R)‐ketamine lost their righting reflex (Figure 4a).
Figure 4.
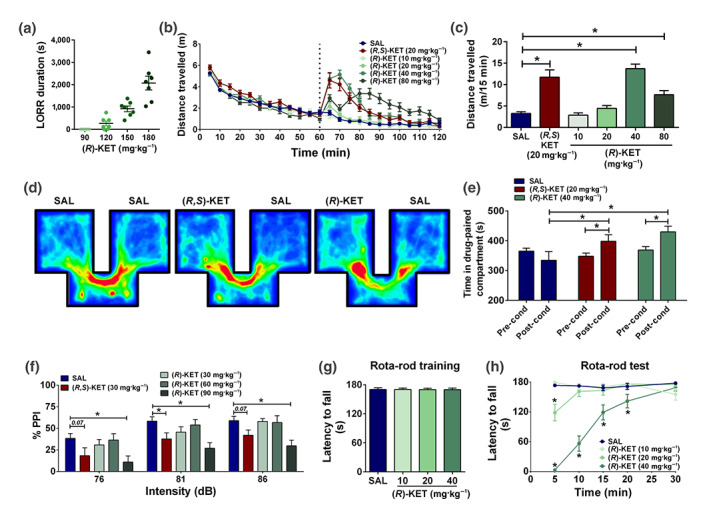
Sub‐anaesthetic doses of (R)‐ketamine induce acute stimulant effects, conditioned‐place preference, prepulse inhibition deficits, and motor incoordination. (a) (R)‐ketamine induced loss of righting reflex (LORR), with a minimally effective dose of 120 mg·kg−1. (b) Following recording baseline locomotor activity for 60 min, mice were given injections (dashed line) of vehicle, (R,S)‐ketamine (KET), or different doses of (R)‐KET and assessed for locomotor responses for another 60 min. (c) Similar to (R,S)‐KET, (R)‐KET dose dependently induced hyperlocomotor responses as measured during the first 15 min following drug administration. (d) Representative heat density maps during post‐conditioning sessions in the conditioned‐place preference paradigm in mice treated with vehicle, (R,S)‐KET (20 mg·kg−1) and (R)‐KET (40 mg·kg−1). (e) Similar to (R,S)‐KET, (R)‐KET induced a conditioned‐place preference. (f) Similar to (R,S)‐KET, (R)‐KET dose dependently disrupted prepulse inhibition deficits. (g) Following 5 days of training, there were no differences in the latency of the mice to fall from the rotating rod between groups. (h) (R)‐KET induced dose‐dependent motor coordination deficits. Data are the mean ± SEM. *P < .05, significantly different as indicated; panel (h), *P < .05, significantly different from saline (SAL) at the corresponding time point. See Tables 2 and S1 for statistical analyses and precise group sizes
Although (R)‐ketamine has been shown to exert more potent antidepressant‐relevant actions than the (S)‐ketamine enantiomer in rodents (Fukumoto et al., 2017; Yang, Qu, Fujita, et al., 2017; Yang et al., 2018; Yang et al., 2015; Zanos et al., 2016; Zhang et al., 2014), (R)‐ketamine is also an NMDA receptor antagonist, which predicts that it would induce NMDA receptor inhibition‐mediated effects. As (R,S)‐ketamine‐induced hyperlocomotion is attributed to NMDA receptor inhibition in rodents (Irifune et al., 1995), the effects of (R)‐ketamine and (R,S)‐ketamine were compared in the OFT. (R)‐Ketamine induced hyperlocomotion at the doses of 40 and 80 mg·kg−1 (Figure 4b,c), indicating stimulant effects. These doses are higher than those required for the antidepressant‐relevant actions of (R)‐ketamine (see Figure 2), but much lower than its minimally effective anaesthetic dose (Figure 4a). Similar hyperlocomotion was identified following a 20‐mg·kg−1 dose of (R,S)‐ketamine (Figure 4b,c).
To examine the abuse potential of (R)‐ketamine, the drug was assessed in the CPP paradigm, in which the rewarding properties of a drug conditions animals to prefer the environment in which this drug was previously administered. (R)‐Ketamine at 40 mg·kg−1, similar to (R,S)‐ketamine at 20 mg·kg−1, induced a robust CPP, as illustrated by an increase in the time spent in the drug‐paired compartment during the post‐conditioning phase compared to the preconditioning phase (Figure 4d,e). We note that a full dose–response determination of the effect of (R)‐ketamine in the CPP paradigm is warranted in light of our findings obtained using only a single sub‐anaesthetic dose.
Another important feature of (R,S)‐ketamine administration in humans, which limits its widespread use for the treatment of depression, is that it induces dissociative effects at sub‐anaesthetic doses (Krystal et al., 1994). In rodents, disruption of sensorimotor gating, measured using the PPI paradigm, is associated with the ability of drugs to induce dissociative/psychotomimetic effects. We observed that (R,S)‐ketamine administration disrupted sensorimotor gating at 30 mg·kg−1 (3 times higher than the mouse antidepressant‐relevant dose; Zanos et al., 2016), 30 min after administration (Figure 4f). At the dose of 90 mg·kg−1 (threefold higher than the effective dose of (R,S)‐ketamine in this paradigm), (R)‐ketamine also disrupted PPI (Figure 4f), without altering baseline startle responses (saline: 271.8 ± 45.49, (R,S)‐ketamine: 192.3 ± 43.38, (R)‐ketamine 30 mg·kg−1: 441.7 ± 81.60, (R)‐ketamine 60 mg·kg−1: 295.5 ± 45.18, (R)‐ketamine 90 mg·kg−1: 145.9 ± 33.33; one‐way ANOVA: F(4, 105) = 4.59; P < .05; post hoc comparisons: all treatments versus saline P > .05).
Finally, we tested the effect of (R)‐ketamine in the rotarod test for motor coordination disturbances. Motor incoordination is a common effect in many patients receiving (R,S)‐ketamine infusion (Wan et al., 2015). Mice were trained over 5 days to reach performance criteria in the task (Figure 4g). At the 5‐min time point following a 20‐mg·kg−1 dose, (R)‐ketamine induced motor incoordination (Figure 4h). At 40 mg·kg−1, (R)‐ketamine induced a robust motor incoordination effect lasting for 20 min post‐injection (Figure 4h).
4. DISCUSSION
Here, we demonstrated that modifying (R)‐ketamine by substituting deuterium at the C6 position of the carboxyl ring did not alter the ability of the drug to inhibit NMDA receptors but did hinder its metabolism to the (2R,6R)‐HNK metabolite and decreased its potency to exert long‐lasting antidepressant‐relevant behavioural actions in mice. Our experiments also reveal that direct administration of (2R,6R)‐HNK in the brain of mice dose dependently abolished inescapable shock‐induced escape deficits and reversed sucrose preference deficits following inescapable shock in the susceptible mice. We also show that (R)‐ketamine, at sub‐anaesthetic doses, mimicked (R,S)‐ketamine's actions in rodent predictors of human adverse effects including abuse liability and sensorimotor gating deficits.
Although both the (2S,6S)‐ and (2R,6R)‐HNK enantiomers exert dose‐dependent actions in rodent tests predicting antidepressant efficacy, the (2R,6R)‐HNK metabolite was shown to be more potent (Chou et al., 2018; Zanos et al., 2016). This is consistent with data showing that (R)‐ketamine (parent compound of (2R,6R)‐HNK) is a more potent antidepressant than (S)‐ketamine (Fukumoto et al., 2017; Yang, Qu, Fujita, et al., 2017; Yang et al., 2018; Yang et al., 2015; Zanos et al., 2016; Zhang et al., 2014). The results of a recent study revealed that preventing metabolism of (R)‐ketamine to (2R,6R)‐HNK by using cytochrome P450 inhibitors does not prevent the antidepressant‐relevant actions of (R)‐ketamine, administered at a single dose, in the FST 3 and 24 hr post‐treatment in LPS‐treated mice (Yamaguchi et al., 2018). In that study, while (2R,6R)‐HNK production was reduced, the brain levels of (R)‐ketamine were dramatically increased (~13‐fold), which may explain the increased potency of (R)‐ketamine observed. The results of another recent study showed similar antidepressant‐relevant actions of both (R)‐ketamine and (R)‐d2‐ketamine (identical to the compound used here) at the single dose of 10 mg·kg−1 (Zhang, Toki, et al., 2018). Consistent with these findings, at a 10‐mg·kg−1 dose, we also observed statistically indistinguishable antidepressant‐relevant actions between (R)‐ketamine and (R)‐d2‐ketamine (see Figure 2), indicating similar efficacy of these compounds. Nevertheless, our experiments revealed that lower doses of (R)‐ketamine compared to (R)‐d2‐ketamine are able to exert antidepressant‐relevant actions. Thus, we conclude that metabolism of (R)‐ketamine to (2R,6R)‐HNK increases the potency of (R)‐ketamine to exert antidepressant‐relevant actions in mice. Our findings do not determine whether (R)‐ketamine and its (2R,6R)‐HNK metabolite engage the same or different target(s) to exert independent antidepressant‐relevant behavioural actions. (2R,6R)‐HNK‐independent actions of (R)‐ketamine are supported by reports that (R)‐ketamine exerts antidepressant‐relevant effects when administered intracerebroventricularly (Zhang, Fujita, & Hashimoto, 2018), or bilaterally into the medial prefrontal cortex or CA3 and dentate gyrus regions of the hippocampus in rats (Shirayama & Hashimoto, 2017), considering that ketamine is not readily metabolised in brain tissue (Moaddel et al., 2015). Our findings revealing that when (R)‐ketamine is converted to (2R,6R)‐HNK it requires lower doses to exert its antidepressant‐relevant actions indicate an involvement of this metabolite in (R)‐ketamine's behavioural effects.
We next sought to understand whether the involvement of (2R,6R)‐HNK in (R)‐ketamine's antidepressant‐relevant behavioural actions is due to direct actions of the metabolite in the brain. Our data reveal a U‐shaped dose response following brain infusion of (2R,6R)‐HNK in mice. We had not previously observed U‐shaped dose responses in antidepressant‐relevant tasks following intraperitoneal or oral administration of (2R,6R)‐HNK to mice (Highland et al., 2018; Zanos et al., 2016). At the highest dose tested (30 nmol), (2R,6R)‐HNK did not reverse escape deficits in susceptible mice, while 3 and 10 nmol did (Figure 3a,d). This is in line with a previous study that failed to identify antidepressant‐like actions of (2R,6R)‐HNK when it was administered intracerebroventricularly in social defeated mice at a single dose of 72 nmol (Zhang, Fujita, & Hashimoto, 2018), which is well beyond the U‐shaped dose response identified here. These data also highlight that low levels of (2R,6R)‐HNK in the brain are sufficient to exert potent and long‐lasting antidepressant‐relevant actions when administered intracerebroventricularly. Nevertheless, it should be noted that the dose of 30 nmol, while it did not reverse inescapable shock‐induced escape deficits, did attenuate the development of sucrose preference deficits. In line with our findings, several other reports have reported rapid antidepressant‐relevant behavioural actions of (2R,6R)‐HNK (Chou et al., 2018; Fukumoto et al., 2019; Highland et al., 2018; Pham et al., 2018; Zanos et al., 2016). Moreover, signalling and synaptic mechanisms considered critical for the antidepressant actions of (R,S)‐ketamine are similarly modulated by (2R,6R)‐HNK (Cavalleri et al., 2018; Collo, Cavalleri, Chiamulera, & Merlo Pich, 2018; Wray et al., 2018; Yao, Skiteva, Zhang, Svenningsson, & Chergui, 2018; Zanos et al., 2016). The failure of studies by Hashimoto and colleagues to identify antidepressant‐relevant actions of (2R,6R)‐HNK in animal tests (Shirayama & Hashimoto, 2018; Yamaguchi et al., 2018; Yang, Qu, Abe, et al., 2017) may be due to the single or limited range of doses used in these studies.
Although the rapid onset of antidepressant actions of (R,S)‐ketamine were considered a breakthrough for the treatment of depression, its use is limited due to its dissociative properties (Krystal et al., 1994), as well as its recreational use (Sassano‐Higgins et al., 2016). (R)‐ketamine has been proposed as a safer alternative to (R,S)‐ketamine due to recent reports of no rodent behavioural or neurochemical changes predictive of adverse effects in humans (Hashimoto et al., 2017; Tian et al., 2018; Yang et al., 2016; Yang et al., 2015). Yang et al. (2015) previously concluded that (R)‐ketamine lacks acute hyperlocomotor effects, does not disrupt sensorimotor gating in the PPI paradigm, and does not induce CPP at doses up to 20 mg·kg−1 in mice. Our data suggest that these previous conclusions were based on experiments not utilising a full range of relevant sub‐anaesthetic doses. Here, we demonstrate that a dose of 40 mg·kg−1 is required to induce acute stimulant hyperlocomotor effects and is also sufficient to induce CPP in mice. The locomotor and CPP data indicate that a twofold higher dose of (R)‐ketamine compared with the (R,S)‐ketamine is necessary to exert these effects. However, a threefold higher dose (i.e., 90 mg·kg−1 for (R)‐ketamine vs. 30 mg·kg−1 for (R,S)‐ketamine) was required to disrupt PPI. This discrepancy could be attributed to the different targets that (S)‐ketamine engages compared with the (R)‐ketamine enantiomer (see Zanos, Moaddel, et al., 2018), which may change the potency of the drug to affect PPI. Nevertheless, disruption of PPI was previously reported by administration of (R)‐ketamine at 12 mg·kg−1 in rats (Halberstadt, Slepak, Hyun, Buell, & Powell, 2016).
Overall, (R)‐ketamine induced locomotor stimulant and conditioning effects at approximately half the potency of (R,S)‐ketamine, as predicted by their respective potencies to inhibit the NMDA receptor in vitro (Ebert et al., 1997; Moaddel et al., 2013; Zanos et al., 2016; Zeilhofer et al., 1992); also see Zanos, Moaddel, et al. (2018). In contrast with (R,S)‐ketamine and both its enantiomers, (2R,6R)‐HNK has a low potency to bind to NMDA receptors and to block NMDA receptor function (Lumsden et al., 2019; Moaddel et al., 2013; Morris et al., 2017; Suzuki, Nosyreva, Hunt, Kavalali, & Monteggia, 2017; Zanos et al., 2016). This metabolite lacked locomotor stimulant effects at doses up to 125 mg·kg−1 when administered intraperitoneally (Zanos et al., 2016) and 450 mg·kg−1 following oral administration (Highland et al., 2018), to be devoid of self‐administration properties at doses where (R,S)‐ketamine was self‐administered and to not disrupt PPI at doses up to 375 mg·kg−1 in mice (Zanos et al., 2016).
Although the doses required to induce adverse effects of (R)‐ketamine observed in the present study, and also those needed for (R,S)‐ketamine (Cilia, Hatcher, Reavill, & Jones, 2007; Zanos et al., 2016), are higher than the doses required to exert antidepressant‐relevant actions in rodents, the abuse liability properties of both (R)‐ketamine (present data) and (R,S)‐ketamine (Cilia et al., 2007; Zanos et al., 2016) occur below their anaesthetic doses in mice (80–100 mg·kg−1 for (R,S)‐ketamine, Xu, Ming, Dart, & Du, 2007; 120 mg·kg−1 for (R)‐ketamine, Figure 4a). Similarly, (R,S)‐ketamine is used as a human recreational drug at doses significantly higher than those required for human antidepressant actions but less than those used to induce anaesthesia (Sassano‐Higgins et al., 2016), indicating the relevance of our findings to human abuse potential. While preclinical data predict greater potency of (R)‐ketamine compared to (R,S)‐ketamine (Fukumoto et al., 2017; Yang, Qu, Fujita, et al., 2017; Yang et al., 2018; Yang et al., 2015; Zanos et al., 2016; Zhang et al., 2014), our present data, as well as previous literature (Fukumoto et al., 2017; Halberstadt et al., 2016; Ryder, Way, & Trevor, 1978), raise questions regarding potential safety issues with (R)‐ketamine when administered as an outpatient treatment. Overall, our findings highlight the need for better prospective treatments for depression that will retain the potent antidepressant actions of (R,S)‐ketamine but lack its abuse liability and adverse effects.
CONFLICT OF INTEREST
R.M. is listed as a co‐inventor on a patent for the use of (2R,6R)‐hydroxynorketamine, (S)‐dehydronorketamine, and other stereoisomeric dehydro‐ and hydroxylated metabolites of (R,S)‐ketamine metabolites in the treatment of depression and neuropathic pain. P.Z., P.J.M., C.J.T., R.M., and T.D.G. are listed as co‐authors in patent applications related to the pharmacology and use of (2R,6R)‐hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post‐traumatic stress disorder. T.D.G. has received research funding from Allergan, Janssen, and Roche Pharmaceuticals and served as a consultant for FSV7 during the preceding 3 years. J.N.H., X.L., T.A.T., P.G., J.L., B.W.S., and S.M.T. declare no conflict of interest.
AUTHOR CONTRIBUTIONS
P.Z., S.M.T., R.M., and T.D.G. designed the experimental details. P.Z., J.N.H., X.L., P.G., B.W.S. (supervised by T.D.G.), T.A.T. (supervised by S.M.T.), and J.L. (supervised by R.M.) performed the experiments. P.Z., J.N.H., P.G., B.W.S., and R.M. analysed the data. P.J.M. and C.J.T. contributed reagents/materials/analysis tools. P.Z., J.N.H., P.G., and T.D.G. wrote the manuscript. All the authors reviewed the final draft of the manuscript and provided critical comments.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organisations engaged with supporting research.
Supporting information
Table S1 Raw data for all the figures.
ACKNOWLEDGEMENTS
We thank Dr. Amy Q. Wang (NCATS, NIH) for performing the WinNonlin pharmacokinetic analysis and Ms. Carleigh E. Jenne for assisting with the forced‐swim test. This work was supported by a National Institute of Mental Health Grant MH107615, U.S. Department of Veterans Affairs Merit Award 1I01BX004062, and a Harrington Discovery Institute Scholar‐Innovator grant to T.D.G., a National Institute of Mental Health Grant MH086828 to S.M.T., and a NARSAD (Brain and Behavior Research Foundation; 26826) Young Investigator Grant to P.Z. R.M. is supported by the National Institute on Aging Intramural NIH Research Program. P.J.M. and C.J.T. are supported by the National Center for Advancing Translational Sciences Intramural NIH Research Program. Receptor binding profiles and Ki determinations were supported by the National Institute of Mental Health Psychoactive Drug Screening Program, Contract HHSN‐271‐2008‐025C, to Bryan Roth in conjunction with Ms. Jamie Driscoll (NIMH, Bethesda, MD, USA). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Zanos P, Highland JN, Liu X, et al. (R)‐Ketamine exerts antidepressant actions partly via conversion to (2R,6R)‐hydroxynorketamine, while causing adverse effects at sub‐anaesthetic doses. Br J Pharmacol. 2019;176:2573–2592. 10.1111/bph.14683
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Peters, J. A. , Kelly, E. , Marrion, N. V. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. British Journal of Pharmacology, 174, S130–S159. 10.1111/bph.13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry, A. E. , Adachi, M. , Nosyreva, E. , Na, E. S. , Los, M. F. , Cheng, P. F. , … Monteggia, L. M. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature, 475(7354), 91–95. 10.1038/nature10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalleri, L. , Merlo Pich, E. , Millan, M. J. , Chiamulera, C. , Kunath, T. , Spano, P. F. , & Collo, G. (2018). Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC‐derived dopaminergic neurons via AMPAR‐driven BDNF and mTOR signaling. Molecular Psychiatry, 23(4), 812–823. 10.1038/mp.2017.241 [DOI] [PubMed] [Google Scholar]
- Chou, D. , Peng, H. Y. , Lin, T. B. , Lai, C. Y. , Hsieh, M. C. , Wen, Y. C. , … Ho, Y. C. (2018). (2R,6R)‐hydroxynorketamine rescues chronic stress‐induced depression‐like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology, 139, 1–12. 10.1016/j.neuropharm.2018.06.033 [DOI] [PubMed] [Google Scholar]
- Cilia, J. , Hatcher, P. , Reavill, C. , & Jones, D. N. C. (2007). (±) Ketamine‐induced prepulse inhibition deficits of an acoustic startle response in rats are not reversed by antipsychotics. Journal of Psychopharmacology, 21(3), 302–311. 10.1177/0269881107077718 [DOI] [PubMed] [Google Scholar]
- Collo, G. , Cavalleri, L. , Chiamulera, C. , & Merlo Pich, E. (2018). (2R,6R)‐Hydroxynorketamine promotes dendrite outgrowth in human inducible pluripotent stem cell‐derived neurons through AMPA receptor with timing and exposure compatible with ketamine infusion pharmacokinetics in humans. Neuroreport, 29(16), 1425–1430. 10.1097/WNR.0000000000001131 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , and Ahluwalia, A. (2018) Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, B. , Mikkelsen, S. , Thorkildsen, C. , & Borgbjerg, F. M. (1997). Norketamine, the main metabolite of ketamine, is a non‐competitive NMDA receptor antagonist in the rat cortex and spinal cord. European Journal of Pharmacology, 333(1), 99–104. 10.1016/S0014-2999(97)01116-3 [DOI] [PubMed] [Google Scholar]
- Ette, E. I. , Kelman, A. W. , Howie, C. A. , & Whiting, B. (1995). Analysis of animal pharmacokinetic data: Performance of the one point per animal design. Journal of Pharmacokinetics and Biopharmaceutics, 23(6), 551–566. 10.1007/BF02353461 [DOI] [PubMed] [Google Scholar]
- Fukumoto, K. , Fogaça, M. V. , Liu, R.‐J. , Duman, C. , Kato, T. , Li, X.‐Y. , & Duman, R. S. (2019). Activity‐dependent brain‐derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)‐hydroxynorketamine. Proceedings of the National Academy of Sciences, 116(1), 297–302. 10.1073/pnas.1814709116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto, K. , Toki, H. , Iijima, M. , Hashihayata, T. , Yamaguchi, J.‐i. , Hashimoto, K. , & Chaki, S. (2017). Antidepressant potential of (R)‐ketamine in rodent models: Comparison with (S)‐ketamine. Journal of Pharmacology and Experimental Therapeutics, 361(1), 9–16. 10.1124/jpet.116.239228 [DOI] [PubMed] [Google Scholar]
- Halberstadt, A. L. , Slepak, N. , Hyun, J. , Buell, M. R. , & Powell, S. B. (2016). The novel ketamine analog methoxetamine produces dissociative‐like behavioral effects in rodents. Psychopharmacology, 233(7), 1215–1225. 10.1007/s00213-016-4203-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, K. , Kakiuchi, T. , Ohba, H. , Nishiyama, S. , & Tsukada, H. (2017). Reduction of dopamine D2/3 receptor binding in the striatum after a single administration of esketamine, but not R‐ketamine: A PET study in conscious monkeys. European Archives of Psychiatry and Clinical Neuroscience, 267(2), 173–176. 10.1007/s00406-016-0692-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highland, J. , Morris, P. , Zanos, P. , Lovett, J. , Gosh, S. , Wang, A. , … Gould, T. D. (2018). Mouse, rat, and dog bioavailability and mouse oral antidepressant efficacy of (2R,6R)‐hydroxynorketamine. Journal of Psychopharmacology, 33, 12–24. 10.1177/0269881118812095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irifune, M. , Shimizu, T. , Nomoto, M. , & Fukuda, T. (1995). Involvement of N‐methyl‐D‐aspartate (NMDA) receptors in noncompetitive NMDA receptor antagonist‐induced hyperlocomotion in mice. Pharmacology, Biochemistry, and Behavior, 51(2–3), 291–296. 10.1016/0091-3057(94)00379-W [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal, J. H. , Karper, L. P. , Seibyl, J. P. , Freeman, G. K. , Delaney, R. , Bremner, J. D. , … Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of General Psychiatry, 51(3), 199–214. 10.1001/archpsyc.1994.03950030035004 [DOI] [PubMed] [Google Scholar]
- Li, N. , Lee, B. , Liu, R. J. , Banasr, M. , Dwyer, J. M. , Iwata, M. , … Duman, R. S. (2010). mTOR‐dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science, 329(5994), 959–964. 10.1126/science.1190287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden, E. , Troppoli, T. A. , Myers, S. J. , Zanos, P. , Aracava, Y. , Kehr, J. , … Gould, T. D. (2019). Antidepressant‐relevant concentrations of the ketamine metabolite (2R,6R)‐hydroxynorketamine do not block NMDA receptor function. Proceedings of the National Academy of Sciences, 116, 5160–5169. 10.1073/pnas.1816071116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng, S. , Zarate, C. A. Jr. , Du, J. , Schloesser, R. J. , McCammon, J. , Chen, G. , & Manji, H. K. (2008). Cellular mechanisms underlying the antidepressant effects of ketamine: Role of α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptors. Biological Psychiatry, 63(4), 349–352. 10.1016/j.biopsych.2007.05.028 [DOI] [PubMed] [Google Scholar]
- Mathisen, L. C. , Skjelbred, P. , Skoglund, L. A. , & Oye, I. (1995). Effect of ketamine, an NMDA receptor inhibitor, in acute and chronic orofacial pain. Pain, 61(2), 215–220. 10.1016/0304-3959(94)00170-J [DOI] [PubMed] [Google Scholar]
- Moaddel, R. , Abdrakhmanova, G. , Kozak, J. , Jozwiak, K. , Toll, L. , Jimenez, L. , … Wainer, I. W. (2013). Sub‐anesthetic concentrations of (R,S)‐ketamine metabolites inhibit acetylcholine‐evoked currents in α7 nicotinic acetylcholine receptors. European Journal of Pharmacology, 698(1–3), 228–234. 10.1016/j.ejphar.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moaddel, R. , Sanghvi, M. , Dossou, K. S. S. , Ramamoorthy, A. , Green, C. , Bupp, J. , … Wainer, I. W. (2015). The distribution and clearance of (2S,6S)‐hydroxynorketamine, an active ketamine metabolite, in Wistar rats. Pharmacology Research & Perspectives, 3(4), e00157–e00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, P. J. , Moaddel, R. , Zanos, P. , Moore, C. E. , Gould, T. D. , Zarate, C. A. Jr. , & Thomas, C. J. (2017). Synthesis and N‐methyl‐d‐aspartate (NMDA) receptor activity of ketamine metabolites. Organic Letters, 19(17), 4572–4575. 10.1021/acs.orglett.7b02177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky, H. J. , & Brown, R. E. (2006). Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics, 7, 123. 10.1186/1471-2105-7-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough, J. W. , Iosifescu, D. V. , Chang, L. C. , Al Jurdi, R. K. , Green, C. E. , Perez, A. M. , … Mathew, S. J. (2013). Antidepressant efficacy of ketamine in treatment‐resistant major depression: A two‐site randomized controlled trial. The American Journal of Psychiatry, 170(10), 1134–1142. 10.1176/appi.ajp.2013.13030392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport, D. J. , Carpenter, L. L. , McDonald, W. M. , Potash, J. B. , Tohen, M. , & Nemeroff, C. B. (2015). Ketamine and other NMDA antagonists: Early clinical trials and possible mechanisms in depression. The American Journal of Psychiatry, 172(10), 950–966. 10.1176/appi.ajp.2015.15040465 [DOI] [PubMed] [Google Scholar]
- Pham, T. H. , Defaix, C. , Xu, X. , Deng, S.‐X. , Fabresse, N. , Alvarez, J.‐C. , … Gardier, A. M. (2018). Common neurotransmission recruited in (R,S)‐ketamine and (2R,6R)‐hydroxynorketamine‐induced sustained antidepressant‐like effects. Biological Psychiatry, 84(1), e3–e6. 10.1016/j.biopsych.2017.10.020 [DOI] [PubMed] [Google Scholar]
- Portmann, S. , Kwan, H. Y. , Theurillat, R. , Schmitz, A. , Mevissen, M. , & Thormann, W. (2010). Enantioselective capillary electrophoresis for identification and characterization of human cytochrome P450 enzymes which metabolize ketamine and norketamine in vitro. Journal of Chromatography. A, 1217(51), 7942–7948. 10.1016/j.chroma.2010.06.028 [DOI] [PubMed] [Google Scholar]
- Ramaker, M. J. , & Dulawa, S. C. (2017). Identifying fast‐onset antidepressants using rodent models. Molecular Psychiatry, 22(5), 656–665. 10.1038/mp.2017.36 [DOI] [PubMed] [Google Scholar]
- Rush, A. J. , Trivedi, M. H. , Wisniewski, S. R. , Nierenberg, A. A. , Stewart, J. W. , Warden, D. , … Fava, M. (2006). Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. The American Journal of Psychiatry, 163(11), 1905–1917. 10.1176/ajp.2006.163.11.1905 [DOI] [PubMed] [Google Scholar]
- Ryder, S. , Way, W. L. , & Trevor, A. J. (1978). Comparative pharmacology of the optical isomers of ketamine in mice. European Journal of Pharmacology, 49(1), 15–23. 10.1016/0014-2999(78)90217-0 [DOI] [PubMed] [Google Scholar]
- Sassano‐Higgins, S. , Baron, D. , Juarez, G. , Esmaili, N. , & Gold, M. (2016). A review of ketamine abuse and diversion. Depression and Anxiety, 33(8), 718–727. 10.1002/da.22536 [DOI] [PubMed] [Google Scholar]
- Shirayama, Y. , & Hashimoto, K. (2017). Effects of a single bilateral infusion of R‐ketamine in the rat brain regions of a learned helplessness model of depression. European Archives of Psychiatry and Clinical Neuroscience, 267(2), 177–182. 10.1007/s00406-016-0718-1 [DOI] [PubMed] [Google Scholar]
- Shirayama, Y. , & Hashimoto, K. (2018). Lack of antidepressant effects of (2R,6R)‐hydroxynorketamine in a rat learned helplessness model: Comparison with (R)‐ketamine. International Journal of Neuropsychopharmacology, 21(1), 84–88. 10.1093/ijnp/pyx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, J. B. , Fedgchin, M. , Daly, E. J. , De Boer, P. , Cooper, K. , Lim, P. , … Van Nueten, L. (2016). A double‐blind, randomized, placebo‐controlled, dose‐frequency study of intravenous ketamine in patients with treatment‐resistant depression. The American Journal of Psychiatry, 173(8), 816–826. 10.1176/appi.ajp.2016.16010037 [DOI] [PubMed] [Google Scholar]
- Suzuki, K. , Nosyreva, E. , Hunt, K. W. , Kavalali, E. T. , & Monteggia, L. M. (2017). Effects of a ketamine metabolite on synaptic NMDAR function. Nature, 546(7659), E1–E3. 10.1038/nature22084 [DOI] [PubMed] [Google Scholar]
- Tian, Z. , Dong, C. , Fujita, A. , Fujita, Y. , & Hashimoto, K. (2018). Expression of heat shock protein HSP‐70 in the retrosplenial cortex of rat brain after administration of (R,S)‐ketamine and (S)‐ketamine, but not (R)‐ketamine. Pharmacology, Biochemistry, and Behavior, 172, 17–21. 10.1016/j.pbb.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Wan, L. B. , Levitch, C. F. , Perez, A. M. , Brallier, J. W. , Iosifescu, D. V. , Chang, L. C. , … Murrough, J. W. (2015). Ketamine safety and tolerability in clinical trials for treatment‐resistant depression. The Journal of Clinical Psychiatry, 76(3), 247–252. 10.4088/JCP.13m08852 [DOI] [PubMed] [Google Scholar]
- Wray, N. H. , Schappi, J. M. , Singh, H. , Senese, N. B. , & Rasenick, M. M. (2018). NMDAR‐independent, cAMP‐dependent antidepressant actions of ketamine. Molecular Psychiatry. 10.1038/s41380-018-0083-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Ming, Z. , Dart, A. M. , & Du, X. J. (2007). Optimizing dosage of ketamine and xylazine in murine echocardiography. Clinical and Experimental Pharmacology & Physiology, 34(5–6), 499–507. 10.1111/j.1440-1681.2007.04601.x [DOI] [PubMed] [Google Scholar]
- Yamaguchi, J.‐i. , Toki, H. , Qu, Y. , Yang, C. , Koike, H. , Hashimoto, K. , … Chaki, S. (2018). (2R,6R)‐Hydroxynorketamine is not essential for the antidepressant actions of (R)‐ketamine in mice. Neuropsychopharmacology, 43, 1900–1907. 10.1038/s41386-018-0084-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Han, M. , Zhang, J. C. , Ren, Q. , & Hashimoto, K. (2016). Loss of parvalbumin‐immunoreactivity in mouse brain regions after repeated intermittent administration of esketamine, but not R‐ketamine. Psychiatry Research, 239, 281–283. 10.1016/j.psychres.2016.03.034 [DOI] [PubMed] [Google Scholar]
- Yang, C. , Qu, Y. , Abe, M. , Nozawa, D. , Chaki, S. , & Hashimoto, K. (2017). (R)‐Ketamine shows greater potency and longer lasting antidepressant effects than its metabolite (2R,6R)‐hydroxynorketamine. Biological Psychiatry, 82(5), e43–e44. 10.1016/j.biopsych.2016.12.020 [DOI] [PubMed] [Google Scholar]
- Yang, C. , Qu, Y. , Fujita, Y. , Ren, Q. , Ma, M. , Dong, C. , & Hashimoto, K. (2017). Possible role of the gut microbiota‐brain axis in the antidepressant effects of (R)‐ketamine in a social defeat stress model. Translational Psychiatry, 7(12), 1294. 10.1038/s41398-017-0031-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Ren, Q. , Qu, Y. , Zhang, J. C. , Ma, M. , Dong, C. , & Hashimoto, K. (2018). Mechanistic target of rapamycin‐independent antidepressant effects of (R)‐ketamine in a social defeat stress model. Biological Psychiatry, 83(1), 18–28. 10.1016/j.biopsych.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Yang, C. , Shirayama, Y. , Zhang, J. C. , Ren, Q. , Yao, W. , Ma, M. , … Hashimoto, K. (2015). R‐ketamine: A rapid‐onset and sustained antidepressant without psychotomimetic side effects. Translational Psychiatry, 5, e632. 10.1038/tp.2015.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, N. , Skiteva, O. , Zhang, X. , Svenningsson, P. , & Chergui, K. (2018). Ketamine and its metabolite (2R,6R)‐hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Molecular Psychiatry, 23(10), 2066–2077. 10.1038/mp.2017.239 [DOI] [PubMed] [Google Scholar]
- Zanos, P. , & Gould, T. D. (2018). Mechanisms of ketamine action as an antidepressant. Molecular Psychiatry, 23(4), 801–811. 10.1038/mp.2017.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos, P. , Moaddel, R. , Morris, P. J. , Georgiou, P. , Fischell, J. , Elmer, G. I. , … Gould, T. D. (2016). NMDAR inhibition‐independent antidepressant actions of ketamine metabolites. Nature, 533(7604), 481–486. 10.1038/nature17998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos, P. , Moaddel, R. , Morris, P. J. , Riggs, L. M. , Highland, J. N. , Georgiou, P. , … Gould, T. D. (2018). Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacological Reviews, 70(3), 621–660. 10.1124/pr.117.015198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos, P. , Nelson, M. E. , Highland, J. N. , Krimmel, S. R. , Georgiou, P. , Gould, T. D. , & Thompson, S. M. (2017). A negative allosteric modulator for α5 subunit‐containing GABA receptors exerts a rapid and persistent antidepressant‐like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro, 4(1), ENEURO.0285–ENEU16.2017. 10.1523/ENEURO.0285-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos, P. , Piantadosi, S. C. , Wu, H. Q. , Pribut, H. J. , Dell, M. J. , Can, A. , … Gould, T. D. (2015). The prodrug 4‐chlorokynurenine causes ketamine‐like antidepressant effects, but not side effects, by NMDA/glycineB‐site inhibition. The Journal of Pharmacology and Experimental Therapeutics, 355(1), 76–85. 10.1124/jpet.115.225664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos, P. , Thompson, S. M. , Duman, R. S. , Zarate, C. A. Jr. , & Gould, T. D. (2018). Convergent mechanisms underlying rapid antidepressant action. CNS Drugs, 32(3), 197–227. 10.1007/s40263-018-0492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate, C. A. Jr. , Brutsche, N. , Laje, G. , Luckenbaugh, D. A. , Venkata, S. L. , Ramamoorthy, A. , … Wainer, I. W. (2012). Relationship of ketamine's plasma metabolites with response, diagnosis, and side effects in major depression. Biological Psychiatry, 72(4), 331–338. 10.1016/j.biopsych.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate, C. A. Jr. , Singh, J. B. , Carlson, P. J. , Brutsche, N. E. , Ameli, R. , Luckenbaugh, D. A. , … Manji, H. K. (2006). A randomized trial of an N‐methyl‐D‐aspartate antagonist in treatment‐resistant major depression. Archives of General Psychiatry, 63(8), 856–864. 10.1001/archpsyc.63.8.856 [DOI] [PubMed] [Google Scholar]
- Zeilhofer, H. U. , Swandulla, D. , Geisslinger, G. , & Brune, K. (1992). Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. European Journal of Pharmacology, 213(1), 155–158. 10.1016/0014-2999(92)90248-3 [DOI] [PubMed] [Google Scholar]
- Zhang, J. C. , Li, S. X. , & Hashimoto, K. (2014). R (−)‐ketamine shows greater potency and longer lasting antidepressant effects than S (+)‐ketamine. Pharmacology, Biochemistry, and Behavior, 116, 137–141. 10.1016/j.pbb.2013.11.033 [DOI] [PubMed] [Google Scholar]
- Zhang, K. , Fujita, Y. , & Hashimoto, K. (2018). Lack of metabolism in (R)‐ketamine's antidepressant actions in a chronic social defeat stress model. Scientific Reports, 8(1), 4007. 10.1038/s41598-018-22449-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , Toki, H. , Fujita, Y. , Ma, M. , Chang, L. , Qu, Y. , … Hashimoto, K. (2018). Lack of deuterium isotope effects in the antidepressant effects of (R)‐ketamine in a chronic social defeat stress model. Psychopharmacology, 235(11), 3177–3185. 10.1007/s00213-018-5017-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Raw data for all the figures.