

Abstract
Neuronal communication is typically mediated via synapses and gap junctions. New forms of intercellular communication, including nanotubes (NTs) and extracellular vesicles (EVs), have been described for non‐neuronal cells, but their role in neuronal communication is not known. Recently, transfer of cytoplasmic material between donor and host neurons (“material transfer”) was shown to occur after photoreceptor transplantation. The cellular mechanism(s) underlying this surprising finding are unknown. Here, using transplantation, primary neuronal cultures and the generation of chimeric retinae, we show for the first time that mammalian photoreceptor neurons can form open‐end NT‐like processes. These processes permit the transfer of cytoplasmic and membrane‐bound molecules in culture and after transplantation and can mediate gain‐of‐function in the acceptor cells. Rarely, organelles were also observed to transfer. Strikingly, use of chimeric retinae revealed that material transfer can occur between photoreceptors in the intact adult retina. Conversely, while photoreceptors are capable of releasing EVs, at least in culture, these are taken up by glia and not by retinal neurons. Our findings provide the first evidence of functional NT‐like processes forming between sensory neurons in culture and in vivo.
Keywords: extracellular vesicle, intercellular communication, material transfer, photoreceptor transplantation, tunnelling nanotube
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membranes & Trafficking; Neuroscience
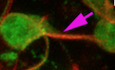
Mammalian photoreceptor neurons form open‐end nanotubes that mediate the exchange of cytoplasmic and membrane‐bound molecules, or material transfer, which can result in gain of function in the acceptor cells.
Introduction
Intercellular communication is an essential process for the development and maintenance of all tissues, including the nervous system. Typically, cells employ two ways of communication, either via contacting directly (e.g. synaptic transmission) or by releasing molecular information in the extracellular fluid. Recently, new mechanisms of molecular exchange between cells have been described and include, respectively, the formation of membranous tubes between cells, called nanotubes (NTs), and the release and uptake of extracellular vesicles (EVs) (Rajendran et al, 2014; Cordero Cervantes & Zurzolo, 2021; Ljubojevic et al, 2021).
Previously regarded as part of the cell’s ‘garbage disposal system’, EVs can carry cytosolic and membrane proteins and potentially even genetic material, which have been reported to alter acceptor cell function in culture and in vivo (Kowal et al, 2016; Pastuzyn et al, 2018; van Niel et al, 2018). EVs are released by almost every cell type (van Niel et al, 2018), including neurons and glia (Faure et al, 2006; Kramer‐Albers et al, 2007; Chivet et al, 2013; Ibanez et al, 2019), and may originate from the endosomal pathway or by simply budding off the plasma membrane (Kowal et al, 2016; Verweij et al, 2018). Similarly, a variety of membranous processes have been described in diverse organisms, including echinoids (where they have been termed as specialized filopodia) (Gustafson & Wolpert, 1967), flies (cytonemes) (Ramirez‐Weber & Kornberg, 1999), birds (cytoplasmic bridges) (Teddy & Kulesa, 2004; George et al, 2016) and mammals (nanotubes) (Rustom et al, 2004; Chinnery et al, 2008). These processes can facilitate the exchange of molecules in culture and in vivo during early embryo development (for reviews, see (Korenkova et al, 2020; Ljubojevic et al, 2021)). Existing either as actin‐enriched open‐end tubes or closed‐tip filopodia‐like protrusions, these processes have been reported to transfer Ca2+ (Alarcon‐Martinez et al, 2020), morphogens (Chen et al, 2017), fluorescent reporters (Kulesa et al, 2010; McKinney & Kulesa, 2011), vesicles (Gradilla et al, 2014), mRNA (Haimovich et al, 2017) and even organelles (Rustom et al, 2004; Alarcon‐Martinez et al, 2020) between cells.
Investigations into NT function in mammals have largely been limited to in vitro studies, mostly due to the technical challenges associated with their visualization. These have led to postulated roles in many pathological conditions, including viral infection, cancer, neuropathies and prion‐associated disease (Gerdes & Carvalho, 2008; Gousset et al, 2009; Gerdes et al, 2013; Peralta et al, 2013; Tardivel et al, 2016). However, to our knowledge, there is only one, very recent, study reporting the presence of mammalian NT‐like processes in vivo, which were shown to form between retinal pericytes and mediate the exchange of Ca2+ signals and coordinate vascular contraction (Alarcon‐Martinez et al, 2020). While there are in vitro reports of NT‐mediated coupling from astrocytes to neurons (Wang et al, 2012b) and within mammalian neuronal cell lines (Sun et al, 2012; Tardivel et al, 2016), it is still not known whether similar structures can form between neurons in vivo.
Neuronal replacement by transplantation is proposed as a treatment for several neurodegenerative disorders. Previous studies, by us and others, have demonstrated the rescue of visual function following the transplantation of healthy photoreceptors into animal models of retinal disease (MacLaren et al, 2006; Lamba et al, 2009; Pearson et al, 2012; Barber et al, 2013; Zhu et al, 2017; Mahato et al, 2020). In end‐stage retinal disease, this rescue is achieved by donor cells forming new synaptic connections with host inner retinal neurons (Ribeiro et al, 2021). However, in partial degeneration, where some/all host photoreceptor cells remain, the transplantation of healthy donor photoreceptors into recipient eyes results in the specific and surprisingly efficient transfer of a wide array of both endogenous and transgenic molecules from donor to recipient photoreceptors in both normal and diseased retina, a phenomenon that has been termed ‘material transfer’ (Pearson et al, 2016; Santos‐Ferreira et al, 2016; Singh et al, 2016; Ortin‐Martinez et al, 2017). This extraordinary finding prompted us to determine the cellular mechanism(s) underlying this exchange and to explore whether this represents a novel mechanism of non‐synaptic intercellular communication between neurons, both within the normal and the diseased (treated) nervous system. We explored two leading but currently untested hypotheses (Pearson et al, 2016; Santos‐Ferreira et al, 2016; Singh et al, 2016; Ortin‐Martinez et al, 2017; Nickerson et al, 2018; Gasparini et al, 2019): (i) targeted release and uptake of extracellular vesicle (EVs) by photoreceptors, and (ii) physical connections between individual photoreceptor pairs, in the form of cytoplasmic bridges. Here, we used a combination of primary cell cultures, photoreceptor transplantation and the generation of chimeric retinae to elucidate the cellular mechanisms underlying material transfer between photoreceptors in culture and in vivo.
Results
EVs do not mediate material transfer between photoreceptors
We first explored the possibility that material transfer might be mediated by the release and uptake of EVs. We examined the ultrastructure of early postnatal wildtype retinae and found that the photoreceptors often presented with multivesicular bodies (MVBs; ˜ 500 nm diameter; 1–2 MVBs/photoreceptor at postnatal day 7) containing intraluminal vesicles (ILVs) (Fig 1A), which can be released as EVs. To study EV release and other photoreceptor–photoreceptor interactions in detail, we next established a primary culture system (see Methods and Kalargyrou et al, bioRxiv) that could support purified postnatal rod photoreceptors for many days, allowing them to extend processes and release vesicles into the surrounding media (Fig 1B). EVs were enriched from the cell culture media using the standard methodology of differential ultracentrifugation (Kowal et al, 2016; Thery et al, 2018). The 100 K pellet contained vesicles of ˜ 120 nm diameter, as determined by electron microscopy (EM) (Fig 1C) and dynamic light scattering (DLS) (Fig 1D and E) analysis, and presented with markers of the endocytic machinery, including LAMP1 (Fig 1F). The classical EV tetraspanins CD81, and CD9 were also present (Fig 1F), albeit not enriched, consistent with recent reassessments of EV composition (Jeppesen et al, 2019). Conversely, the Golgi marker GM130 was absent from the 100 K sample, confirming the lack of contamination from other organelles (Fig 1F).
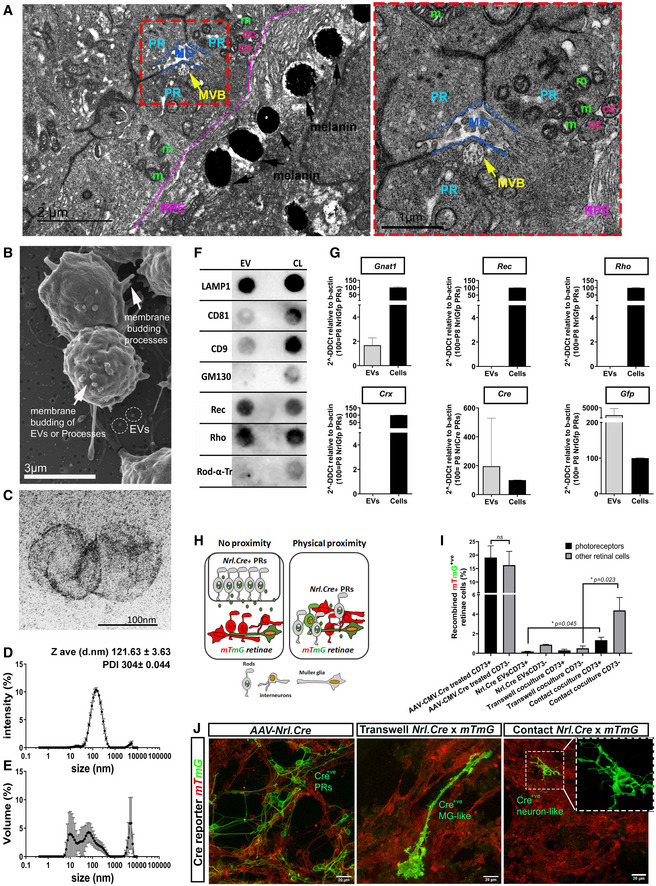
Figure 1. Primary photoreceptors release EVs that exert their function in Müller glial cells but not photoreceptors.
-
ATEM analysis of P7 wt retinae (N = 4 eyes) showing a (photoreceptor‐ Multivesicular Body) PR‐MVB in close proximity to (Muller glia) MG (red dashed box). PR‐photoreceptor cell, MG (blue dashed lines) ‐muller glia cell, MVB (yellow and yellow arrow)‐ multivesicular body, m (green)‐mitochondria, cc (pink)‐connecting cilium, RPE (purple and purple dashed line) ‐retina pigment epithelium, RPE melanin‐black arrows. Scale bar = 2 µm left, 1 µm right.
-
BRepresentative SEM microphotograph of cultured P8 Nrl.Gfp+/+ photoreceptors; arrows indicate membrane budding of processes or EVs; dashed circles denote EVs; Scale bar = 3 µm.
-
CRepresentative TEM microphotograph of 100 K EV pellet derived from Nrl.Gfp+/+ photoreceptor cultures (N = 10 independent preparations); Scale bar = 100 nm.
-
D, ERepresentative dynamic light scatter (DLS) plot of 100 K EV sample (N = 13 samples), showing (D) average intensity and (E) volume against diameter (13 DLS measurements per sample).
-
FRepresentative Dot blot of 100 K EV pellet (each dot represents three pooled EV isolations, derived from 60*106 cells; N = 8 experiments) vs cell lysate (CL) from P8 Nrl.Gfp+/+ photoreceptors. EV markers = LAMP1, CD8, CD9; Golgi marker = GM130; phototransduction markers = Recoverin, Rhodopsin, Rod α‐Transducin.
-
GRT–qPCR analysis of 100 K P8 photoreceptor pellets for Gnat1, Rec, Rho, Crx, Cre and Gfp, relative to β‐actin, comparing EVs (N = 8) against the appropriate (Nrl.Gfp+/+ or Nrl.Cre+/− ) photoreceptor cell lysate (N = 3). Gnat1, Cre and Gfp mRNA were present in all relevant samples.
-
HSchematic representation of Cre‐LoxP system to assess EV function in co‐culture of Nrl.Cre+/− photoreceptors (PRs) with mTmGfloxed reporter retinal cells in non‐proximity (trans‐well) versus physical proximity (contact).
-
IRepresentative flow cytometry analysis of Nrl.Cre+/− and mTmGfloxed co‐cultures. Samples were analysed for expression of myrGFP (recombination) and CD73 (photoreceptor identity) versus CD73−ve fraction (other retinal cells). N > 3 independent cultures for each condition with technical triplicate for each sample; one‐way ANOVA, non‐parametric, Kruskal–Wallis with Dunns’ multiple comparisons test.
-
JRepresentative maximum intensity projection (MIP) confocal images of mTmGfloxed reporter retinal cells, (left) treated with AAV‐Nrl.Cre virus (positive control), (middle) co‐culture with Nrl.Cre+/− photoreceptors separated by trans‐well (non‐proximity) or (right) in contact; Scale bar = 20 µm; Red = myrRFP‐expressing mTmGfloxed reporter, no recombination; green = myrGFP‐expressing mTmGfloxed reporter, cells recombined upon acquiring Cre; blue = nuclei. N = 7 cultures.
Data information: Graphs show mean ± SD.
We next examined whether photoreceptor‐derived EVs contained cell‐type specific, functionally relevant cargo, alongside reporter molecules that would permit tracing of their transfer in vitro and in vivo. Specifically, we assessed molecules previously shown to be exchanged during material transfer following photoreceptor transplantation, including the fluorescent reporter GFP, Cre and Rod α‐transducin (Pearson et al, 2016; Waldron et al, 2018). Cell cultures were established from three transgenic mouse lines: Nrl.Gfp+/−, Nrl.Cre+/− and Nrl.Cre+/− x mTmGfloxed , which express cytoplasmic cGFP, Cre and myristoylated (myr)GFP, respectively. Use of the Nrl promoter ensures that expression is specific restricted to post‐mitotic rod photoreceptors in the retina (Akimoto et al, 2006). Dot blot analysis demonstrated the presence of the phototransduction‐related proteins Recoverin and Rhodopsin, but not Rod α‐transducin (Fig 1F). Cytoplasmic GFP, myrGFP and Cre protein were detected in the 100 K pellets derived from Nrl.Gfp+/+ , Nrl.Cre+/− x mTmG (membrane‐tagged GFP; Appendix Fig S1A and B) and Nrl.Cre+/− photoreceptors, respectively, but varied between samples (Fig EV1A–C). Conversely, Gnat1 (rod α‐transducin), Gfp and Cre mRNA was readily detectable by qRT–PCR, while Rhodopsin, Recoverin and the photoreceptor‐specific transcription factor, Crx, were not detected (Fig 1G). Together, these data demonstrate that photoreceptor‐derived EVs contain cargo that differs from the composition of the cytoplasm of these cells.
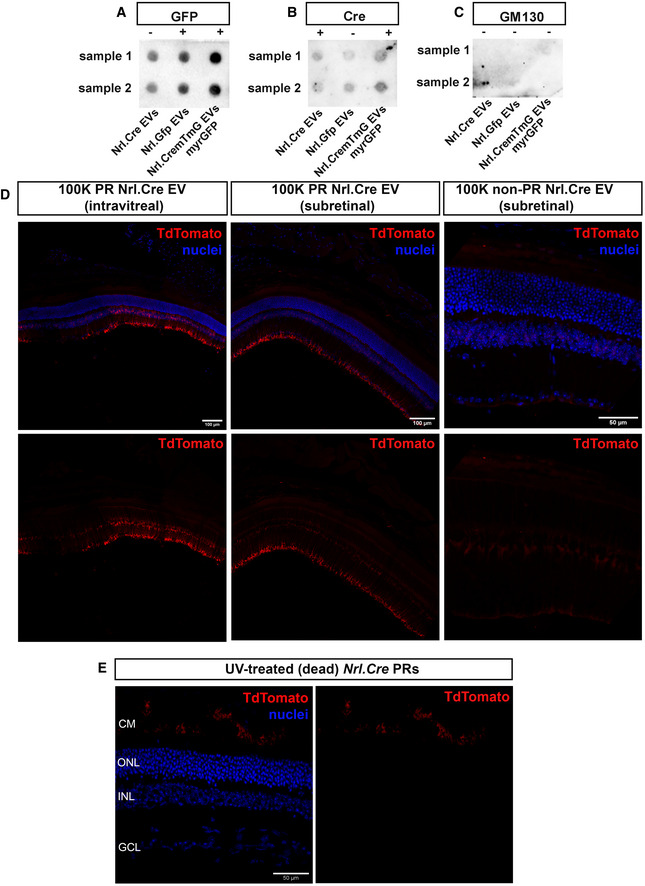
Figure EV1. Photoreceptor‐derived EV subpopulations contain Cre and GFP protein.
-
A–CRepresentative dot blots of GFP (A), Cre (B) and GM130 (C) expression in P8 Nrl.Cre+/− , Nrl.Gfp+/+ , Nrl.Cre+/− × mTmG +/+ (myrGFP) photoreceptor‐derived 100 K EV pellets, as appropriate (N = 8 experiments, each dot represents a pool from three independent EV isolations, derived from 60*106 cells from two samples). The lack of GM130 staining confirms the absence of contamination from Golgi within the EV preparations. Positive GM130 staining from whole cell lysate is shown in Fig 1.
-
DRepresentative tile‐scan images following (left) subretinal and (middle) intravitreal injection of EVs (100 K fraction) derived from P8 Nrl.Cre+/− photoreceptors, compared with (right) subretinal injection of EVs (100 K fraction) derived from non‐photoreceptors (Nrl.Cre+/− ); red = TdTomato recombined cells; blue = nuclei; Scale bar = 100 µm (left & middle) and 50 µm (right).
We next examined the potential for EV‐mediated transfer by employing a Nrl.Cre+/− and mTmGfloxed reporter retinal co‐culture system (Fig 1H–J) followed by confocal imaging and flow cytometry analysis. Here, rod photoreceptors were enriched from Nrl.Cre+/− mice using the cell surface marker, CD73 (Eberle et al, 2011; Lakowski et al, 2015) and co‐cultured with mixed retinal cells from mTmGfloxed mice a floxed Cre reporter line that ubiquitously expresses myristoylated RFP (myrRFP), but switches expression to myrGFP (“red‐to‐green”) upon acquisition of Cre and undergoing Cre‐mediated recombination (Appendix Fig S1). No spontaneous recombination was observed in untreated mTmGfloxed ‐only cultures, while mTmGfloxed cultures treated with AAV‐CMV.Cre virus exhibited widespread recombination (Fig 1I and J). When Nrl.Cre+/− photoreceptors were co‐cultured above mTmGfloxed cells and physical proximity between the two is prevented by use of a trans‐well (see schematic, Fig 1H, left; and (Zomer et al, 2016)), rare examples of recombination were seen after 21 days in culture (DIC) (Fig 1I and J). The majority of recombined cells were CD73‐ve (Fig 1I), indicating a non‐photoreceptor identity (Eberle et al, 2011; Lakowski et al, 2011) and exhibited glial‐like morphologies (Fig 1J). Others have reported that EVs may get trapped within trans‐well pores (Thayanithy et al, 2017), but direct application of Nrl.Cre+/− EVs to mTmGfloxed retinal cultures also yielded very low levels of recombination, again predominantly in CD73−ve cells (Fig 1I). However, when cells were grown in physical contact with one another (see schematic, Fig 1H, right), significantly higher levels of recombination were observed. Some of these cells presented neuronal‐like morphologies (Fig 1J, right) and included CD73+ve photoreceptors (Fig 1I). Together, these data indicate that cultured photoreceptors release bioactive EVs that are taken up predominantly by non‐photoreceptor populations, at least in vitro.
To explore this in vivo, Nrl.Cre+/− photoreceptor‐derived EVs were injected into the subretinal or intravitreal space of TdTomatofloxed reporter mice (“no reporter‐to‐red” following Cre‐mediated recombination) and compared to subretinal transplantation of Nrl.Cre+/− photoreceptors or injection of either recombinant Cre or AAV‐Nrl.Cre virus (Fig 2). As we have reported previously (Pearson et al, 2016), transplantation of viable Cre+ve photoreceptors in the subretinal space results in recombination of host photoreceptors (Fig 2A and B). In notable contrast, when 100 K Nrl.Cre+/− photoreceptor‐derived EVs were injected into either the subretinal space (Figs 2C and D, and EV1, 2) or intravitreally (Figs 2E and F, and EV1D) we observed striking levels of recombination but only in Müller glia cells (Fig 2D and F). Injection of the 10 K and 2 K fractions, which may additionally contain microvesicles and/or apoptotic bodies (Thery et al, 2006, 2018), also yielded Müller glia‐specific labelling (Fig 2D) throughout the injection site. No other cell types were labelled. Importantly, transplantation of Nrl.Cre+/− photoreceptors that were pre‐treated with UV light to induce cell death yielded no recombination (Figs 2G and EV1E), nor did subretinal injection of recombinant Cre protein (2 μg; Fig 2F, right) or non‐photoreceptor‐derived EVs (Fig EV1D). Transduction with AAV‐Nrl.Cre virus yielded widespread recombination in the photoreceptors and some labelling of RPE cells (Fig 2B), most likely due to non‐specific expression of Cre within the RPE, or possibly recombination resulting from uptake of small amounts of Cre protein. Taken together, these data demonstrate that photoreceptor cells have the capacity to release EVs, at least in culture, and if present in the intact retina, these are specifically taken up by Müller glial cells. However, EVs do not mediate material transfer between photoreceptors.
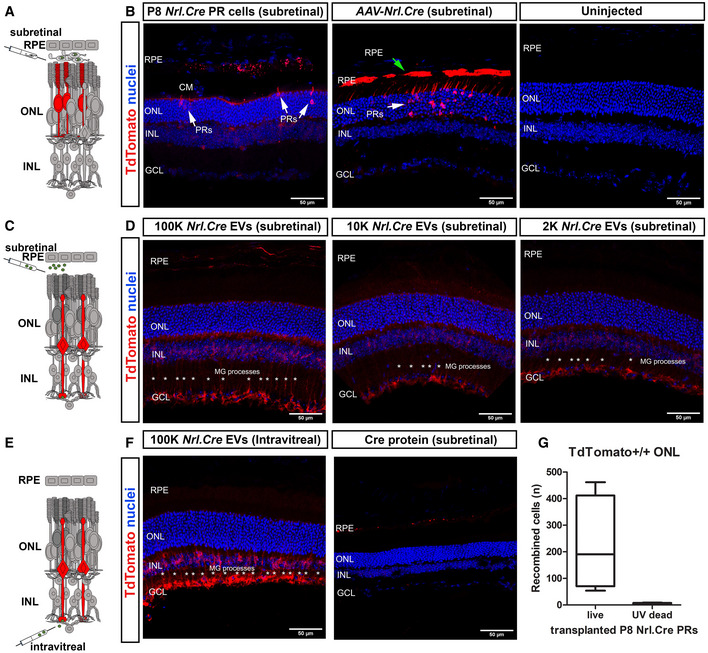
Figure 2. Nrl.Cre+/− photoreceptor‐derived EVs are taken up by Müller glia cells when injected in vivo .
-
ASchematic representation of photoreceptor transplantation, shown in (B).
-
B(left) P8 Nrl.Cre+/− photoreceptors transplanted into subretinal space of TdTomatofloxed reporter mice (N = 5 eyes), compared to (middle) subretinal injection of AAV‐Nrl.Cre subretinal injection (1012 vp/ml), (N = 10 eyes) and (right) contralateral uninjected control (N = 10 eyes). Arrows indicate recombined host photoreceptors (white arrows) or RPE (green arrows). Note clearly recombined RPE cells upon AAV transduction.
-
CSchematic representation of subretinal EV injection, shown in (D). Red indicates host cells undergoing Cre‐mediated recombination and expressing TdTomato (red).
-
DP8 Nrl.Cre+/− PR ‐derived (left) 100 K, (middle) 10 K and (right) 2 K EV pellets injected into the subretinal space of TdTomatofloxed reporter mice (N = 4 eyes per EV preparation). Recombination was restricted to Müller Glia (asterisks).
-
ESchematic representation of intravitreal EV injection, shown in (F).
-
F(left) intravitreal injection of P8 Nrl.Cre+/− photoreceptor‐derived 100 K EV pellets (N = 2 eyes). Recombination was restricted to Müller Glia (asterisks) with little or no recombination in either photoreceptors or the RPE; (right) subretinal injection of recombinant Cre protein (control) into TdTomatofloxed reporter mice. Blue = nuclei, red = TdTomato+ve recombined cells. N = 4 retinae per group. Representative MIP confocal images.
-
GQuantification of no. of TdTomato+ve host photoreceptor cells seen following transplantation of live (224 ± 181) versus UV‐treated (dead) (3 ± 4) Nrl.Cre+/− P8 photoreceptors (N = 4 eyes per condition). Graph shows mean ± SD.
Data information: Scale bars = 50 µm; PR—photoreceptor; MG—Müller Glia; RPE—retinal pigment epithelium; ONL—outer nuclear layer; INL—inner nuclear layer; GCL—Ganglion cell layer.
Photoreceptors form NT‐like processes in vitro
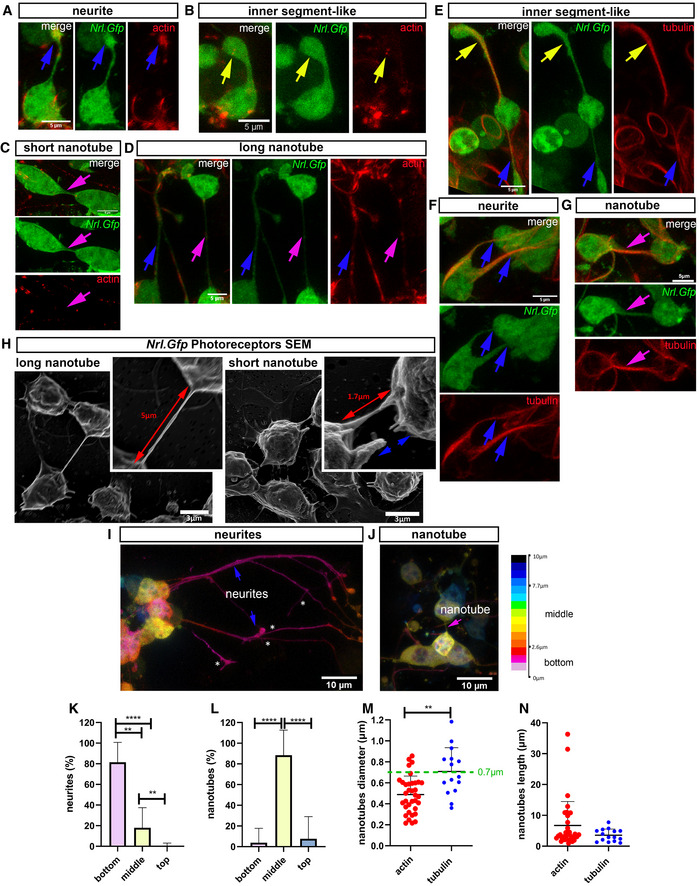
In the original descriptions of material transfer in photoreceptor transplantation, donor cells were often, but not always, in close physical proximity with acceptor host cells (Pearson et al, 2016; Santos‐Ferreira et al, 2016; Singh et al, 2016), sometimes apparently extending processes towards the host acceptor cells (Singh et al, 2016; Ortin‐Martinez et al, 2017). We therefore considered whether physical cell–cell connections underlie photoreceptor material transfer (Fig 3). First, we performed confocal live imaging of Nrl.Gfp+/+ photoreceptor cultures labelled with the cytoskeletal probes SiR‐actin or SiR‐tubulin, to explore and characterize the different processes extended by photoreceptors (Fig 3A–G; Appendix Table S1; Fig EV2); these include (i) neurite‐like extensions, (ii) nascent inner segment‐like processes, and (iii) nanotubes. Analysis of the actin cytoskeleton revealed neurite‐like extensions that terminate in actin‐rich exploratory growth cones (Fig 3A), as well as thicker processes ending in bulbous inner segment‐like terminals, which could be distinguished by the additional presence of tubulin (Fig 3B and E; Movie EV6). These inner segment‐like processes often robustly expressed the photopigment Rhodopsin (Fig EV2A). Examining SiR‐tubulin, many tubulin‐rich processes were long and thick and often extended around other cells (Fig 3F). However, they only rarely appeared to form cell‐to‐cell connections (Fig 3G). Strikingly, however, using either probe, we also observed distinctive NT‐like processes that formed continuous structures connecting the somas of pairs of adjacent cells (Fig 3C, D and G; Movies EV1–EV3). These NTs were typically short (< 10 μm), although longer lengths were observed (Fig 3C, D and G) and often remarkably straight (Fig 3H). Membrane continuity between the connected cells was further confirmed by scanning electron microscopy of Nrl.Gfp+/+ photoreceptor cultures (Fig 3H). Live imaging showed that NTs extend freely between cells and are not attached to the substratum, in contrast with neurite‐like processes, which were usually attached to the substratum or other cells (Fig 3I–L); this free‐floating property also makes them sensitive to fixation (Fig 3H, right).
Figure 3. Photoreceptors form nanotube‐like processes to connect with neighbouring photoreceptors in culture.
-
A–DRepresentative 3D deconvolved MIP images from live imaging of Nrl.Gfp+/+ (green) P8 photoreceptors at DIV1–3, labelled with SiR‐actin (red) (N = 7 independent cultures). Photoreceptors exhibit a variety of processes, including (A) neurites (blue arrow), (B) nascent inner segment‐like protrusions (yellow arrow), and (C) short and (D) long (magenta arrows) actin+ve nanotube‐like (herein termed PhNTs) connections; Scale bar = 5 µm.
-
E–GRepresentative 3D deconvolved MIP images from live imaging of Nrl.Gfp+/+ (green) P8 photoreceptors at DIV1–3 labelled with SiR‐tubulin (red) (N = 7 independent cultures). Photoreceptors exhibit a variety of processes, including nascent inner segment‐like protrusions (yellow arrows), neurites (blue arrows) and rare tubulin+ve PhNTs (magenta arrows); Scale bar = 5 µm.
-
HRepresentative SEM images with digitally enhanced microphotographs of cultured P8 Nrl.Gfp+/+ photoreceptors. Red arrows indicate PhNT connections between neighbouring photoreceptors, while blue arrows indicate broken PhNT connections (N = 3 cultures). Scale bar = 5 µm.
-
I, JRepresentative images of depth colour coding (ImageJ) of x,y,z live imaging of Nrl.Gfp+/+ photoreceptor cultures showing (I) neurites (blue arrows) growing along the substrate (asterisks denote secondary branching of long GFP+ neurites), and in (J), a free‐floating PhNT (magenta arrow). Scale bar = 10 µm.
-
K, LQuantification of the localization of Nrl.Gfp+/+ processes in the z‐axis (depth) (N = 4 independent cultures, n = 1,096 cells, n neurites = 440, n nanotubes = 31) where (K) neurites. P‐values = ****P < 0.0001 (bottom vs top); **P = 0.002 (bottom vs middle); **P = 0.003 (middle vs top) and (L) PhNTs; P‐values = ****P < 0.0001 (bottom vs middle & middle vs top). One‐way ANOVA, non‐parametric, Kruskal–Wallis with Dunns’ multiple comparisons test shows means vary significantly.
-
M, NDiameter and length quantification of SiR‐actin+ve PhNTs (N = 7 cultures; n cells = 646 cells; n nanotubes = 35, red dots) compared to SiR‐tubulin+ve PhNTs (N = 7 cultures; n cells = 885 cells, n nanotubes = 15; blue dots). One‐way ANOVA, non‐parametric, Kolmogorov–Smirnov test shows non‐significant (ns) difference in length but significant difference in diameter between actin+ve versus tubulin+ve PhNTs. P‐values; **P = 0.001, and P = 0.245, in (M) and (N), respectively.
Data information: Graphs show mean ± SD.
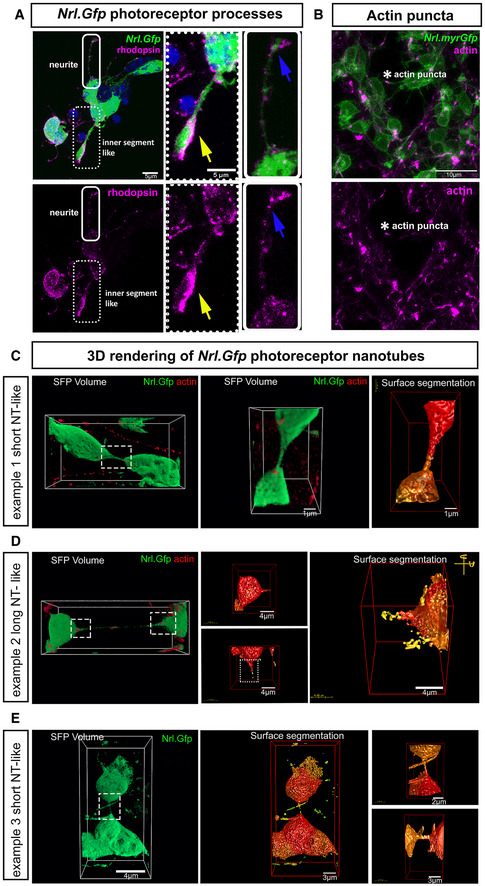
Figure EV2. Photoreceptors form nanotube‐like processes in culture.
-
ARepresentative MIP example of rhodopsin immunostaining of fixed Nrl.Gfp+/+ photoreceptors at 6 DIC. Digitally enhanced microphotographs of a neurite (solid line rectangle) and an inner segment‐like process (dotted rectangle), showing strong rhodopsin staining in the bulbous inner segment‐like structure (yellow arrow), compared to more dispersed staining in a neurite (blue arrows). Green = myrGFP, Magenta = Rhodopsin; Scale bar = 5 µm.
-
BRepresentative MIP example of live imaging of P8 Nrl.Cre+/− × mTmG +/+ (myrGFP) photoreceptor processes. SiR‐actin (magenta) reveals the presence of actin puncta (white asterisk) at the point of contact between a nanotube‐like process (herein termed PhNTs) and an adjacent photoreceptor. Green = myrGFP, Magenta = SiR‐actin; Scale bar = 10 µm.
-
C–ERepresentative 3D deconvolved images of PhNTs from Nrl.Gfp+/+ (green) P8 photoreceptor live‐imaged at DIV1–3 and labelled with SiR‐actin (red). (C) (left) Simulated Fluorescence Process (SFP) image of an example of a short PhNT and (middle, right) digitally enhanced 3D rendered surface per volume images of the same PhNT in rotation; Scale bar = 1 µm; (D) (left) SFP image of an example of a long PhNT and (middle, right) digitally enhanced 3D rendered surface per volume images of the same PhNT in rotation. Representative images from N = 7 independent cultures. Scale bar = 4 µm; (E) (left) SFP and (middle, right) digitally enhanced 3D rendered surface per volume images of the PhNT shown in Fig 3J. Scale bar = 4 µm; 3 µm; 2 µm; 3 µm, respectively, from left to right.
We were able to further classify photoreceptor NTs into Type I and Type II, based on their morphological appearance and molecular structure (Onfelt et al, 2006). Both types are usually short in length (< 10 μm) (Fig 3C and J) although some extend many tens of microns (Fig 3D and N). Type I are actin‐rich (Figs 3C, D, and J, and EV2C–E), the actin typically observed as plaques or puncta at the interface between soma and process, and along the process itself (Figs 3D and EV2B). In photoreceptor cultures, type I processes are typically very straight (Fig 3D) and < 0.7 μm diameter (Fig 3M). Type II processes additionally contain tubulin (Fig 3G) and are of thicker diameter (> 0.7 μm; Fig 3M). Neither type exhibited secondary branching in culture. At present, there is no single or panel of molecules that serve as definitive markers of NT‐like processes, but the above morphological properties of both types are consistent with published descriptions of NT‐like structures in other cell types (Korenkova et al, 2020). Both Type I and Type II NT‐like structures are relatively rare, comprising 5.4% and 1.7%, respectively, of all photoreceptor processes seen in culture. Herein, we refer to all photoreceptor–photoreceptor NT‐like processes as PhNTs.
Photoreceptor NT‐like processes (PhNTs) mediate material transfer in vitro
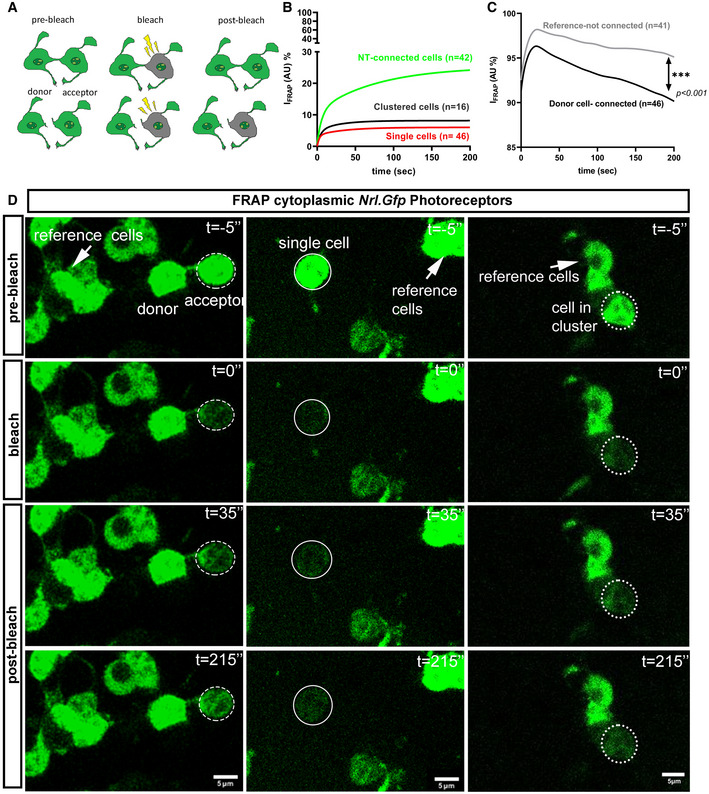
To assess the potential for cytoplasmic exchange of molecular information via PhNTs, pairs of PhNTs‐connected Nrl.Gfp+/+ photoreceptors were examined using an adapted fluorescence recovery after photobleaching (FRAP) protocol (Fig 4A) and compared to isolated photoreceptors or those in clusters (but without apparent cell–cell connections; see Appendix Table S1 for criteria used to pre‐define connected cells prior to FRAP). As expected, isolated cells and cells within a cluster exhibited only limited recovery of cGFP (cytoplasmic GFP), reaching a maximum of ˜ 5% and ˜ 8% recovery of pre‐bleaching fluorescence levels, respectively (Fig 4B–D) with recovery kinetics indicating the redistribution of small amounts of cGFP throughout the cell (n SINGLE = 46 cells, ti1/2 fast = 1.87 s, ti1/2 slow = 24.84 s; n CLUSTERED = 16 cells, ti1/2 fast = 3.18 s, ti1/2 slow = 19.32 s). Conversely, in cells that exhibited a PhNT‐like connection with an adjacent “donor” cell (PhNT‐connected cells; Movie EV4), we observed a slow but much larger recovery, achieving ˜ 25% of pre‐bleach fluorescence levels by the end of the imaging period (225 s) without having reached plateau (Fig 4B and C; n NT = 42 cells, ti1/2 fast = 4.19 s ti1/2 slow = 54.86 s). Notably, this was accompanied by a concomitant reduction in cGFP fluorescence in the non‐bleached donor cell (Fig 4D), indicating that these cells are connected via an open‐ended cytoplasmic bridge.
Figure 4. PhNTs facilitate recovery after photobleaching in culture.
-
ASchematic representation of cytoplasmic (c)GFP mobility during fluorescence recovery after photobleaching (FRAP).
-
BFRAP recovery curves for PhNT‐connected cells (green, n = 42, R 2 = 0.99), clustered cells (black, n = 16, R 2 = 0.94) and single cells (red, n = 46, R 2 = 0.97); Mann–Whitney analysis of fit for curves.
-
CFRAP recovery curves in unbleached reference cells (black, n = 41) versus “donor” PhNT‐connected photoreceptor cells (purple, n = 46) show significant slope difference. Mann–Whitney analysis of fit for curves, ***P < 0.001.
-
DRepresentative time‐lapse images of cGFP mobility between two PhNT‐connected Nrl.Gfp+/+ photoreceptors (dashed ellipse = bleached area) versus an isolated cell (solid ellipse = bleached area) and clustered cells (dot ellipse = bleached area). Images show pre‐bleach (t = −5’’), bleach (t = 0’’) and post‐bleach (t = 35’’ and 215’’). Unbleached reference cells also indicated (arrows); Green = cytoplasm; Scale bar = 5 µm.
We next sought to determine the levels of cytoplasmic fluorescent reporter exchange occurring spontaneously between cells in cocultures of sorted Nrl.Gfp +/+ and DsRed +/+ photoreceptors. As assessed by flow cytometry (Figs 5A and EV3A), a proportion of cells exhibited detectable levels of both labels (˜ 4% at 21DIV, increasing over time; Fig 5A), consistent with previous observations regarding NT‐like protrusions in other cell types (Rustom et al, 2004). Together, these results indicate that fluorescent cytoplasmic molecules can be exchanged between PhNT‐connected photoreceptor cells in vitro, but that this is likely to be a transient process occurring in a relative minority of cells at any given point in time, consistent with material transfer events occurring after photoreceptor transplantation (Pearson et al, 2016).
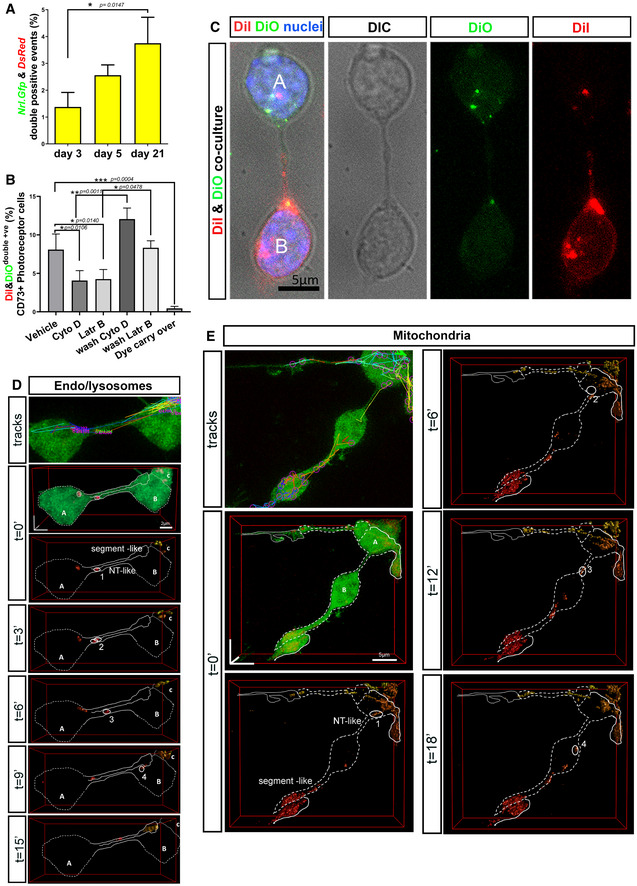
Figure 5. PhNTs enable the intercellular transfer of cytoplasmic and membrane reporters and, rarely, organelles, in culture.
-
AFlow cytometry analysis of cGFP and cDsRed following co‐culture of Nrl.Gfp and DsRed cells (1:1) and analysed at 3 DIC (N = 3 independent co‐cultures with a technical triplicate), 7 DIC (N = 5), 21 DIC (N = 4); one‐way ANOVA non‐parametric, Kruskal–Wallis.
-
BFlow cytometry analysis of DiI/DiO dual labelling of CD73+ cells following co‐culture of DiI‐ and DiO‐labelled photoreceptors (1:1) with pharmacological interventions applied at 5 DIC for 48 h, including vehicle control (DMSO), cytochalasin D (2 μM) and Latrunculin B (5 μM), after wash‐off of Cyto D and LatrB, and a final wash control for DiI/DiO carry over; N = 4 independent co‐cultures per condition; one‐way ANOVA non‐parametric, F‐test, Kolmogorov–Smirnov.
-
CRepresentative MIP of DiI/DiO co‐cultures after fixation, showing DiO+ve cell (A) and a DiI+ve cell (B) cell connected by a PhNT. Note DiI and DiO puncta in the respective acceptor cells. Blue = Dapi (nuclei), Green = DiO, Red = DiI. Scale bar = 5 µm.
-
DLysosomes can be exchanged, rarely, between PhNT‐connected cells. Lysosomes were labelled with SiR‐Lyso, and their movements were analysed by TrackMate software. Images show deconvolution of 3D reconstructions of lysosomes (surface; red) and cGFP (volume; green) from time‐lapse live imaging series of Nrl.Gfp+/+ photoreceptor cultures (green); the position of a transferred lysosome is marked as1 (t = 0), 2 (t = 3’), 3 (t = 6’), 4 (t = 9’); N.B. Deconvolution shows a segment‐like process extending from cell “A” and a PhNT connecting cells “A” and “B”; Scale bar = 2 µm.
-
EMitochondria can be exchanged, rarely, between PhNT‐connected cells. Mitochondria movements determined by TrackMate software. Images show deconvolution of 3D reconstructions of mitochondria (surface; red) and cGFP(volume; green) from time‐lapse live imaging series; position of transferred mitochondrion is marked as 1 (t = 0), 2 (t = 6’), 3 (t = 12’), 4 (t = 18’); Mitochondria labelled with mito‐Tracker‐Orange; Scale bar = 5 µm.
Data information: Graphs show mean ± SD.
Figure EV3. Pharmacological disruption of actin dynamics affects photoreceptor process formation and transfer of cytoplasmic and lipid reporters between photoreceptors in culture.
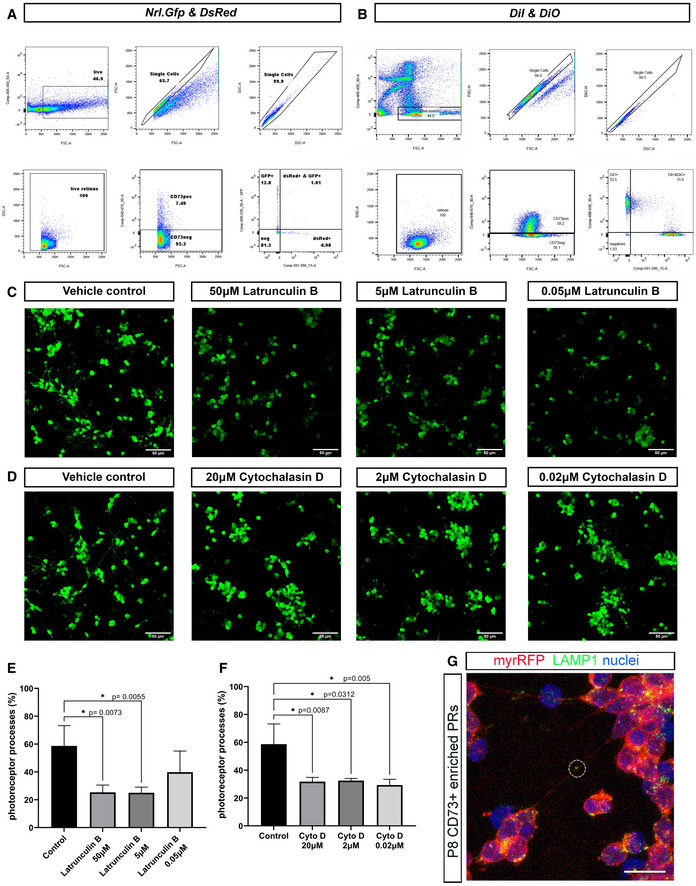
-
A, BFlow cytometry gating strategy of either (A) co‐cultures of Nrl.Gfp and DsRed cells, or (B) DiI and DiO labelled cells. In each case, panels reading top left to bottom right are: 1. Live cells selection, 2. Single events in FSC, 3. More single events in SSC, 4. Single live events in FSC/SSC, 5. CD73−APC versus FSC, 6. Nrl.Gfp+ vs DsRed+ within the CD73APC+ population (for A) or 6. DiI+ vs DiO+ within the CD73APC+ population (for B).
-
C, DRepresentative examples of confocal imaging of P8 Nrl.Gfp+/+ photoreceptors fixed after 48 h exposure to different concentrations of the actin polymerization inhibitors (C), Latrunculin B (50 µM, 5 µM and 0.05 µM) and (D), Cytochalasin D (20 µM, 2 µM and 0.02 µM), compared to vehicle controls (DMSO); Scale bars = 50 µm.
-
E, FStatistical analysis of the percentage of photoreceptors exhibiting processes following 48 h treatment with Latrunculin B (50 µM, 5 µM and 0.05 µM) or Cytochalasin D (20 µM, 2 µM and 0.02 µM), compared to vehicle control. One‐way ANOVA non‐parametric, two‐tailed analysis, F‐test, Kolmogorov–Smirnov, *P < 0.05 shows significant effect of both drugs on the ability of photoreceptors to elaborate processes, compared to vehicle control; (N = 3 independent cultures, n = 1 well per condition). Graph shows mean ± SD.
-
GRepresentative MIP of myrRFP+ve/CD73+ve photoreceptors (red) immunostained for LAMP1 (green) after 3DIC; Circle = LAMP1+ve vesicle in long PhNTs. Scale bar = 10 µm.
Next, we examined membrane exchange by co‐culture of wildtype photoreceptors pre‐labelled with either DiI or DiO membrane dyes (Figs 5B and C, and EV3B). Flow cytometry analysis showed that under control conditions (5DIC plus 48 h with DMSO vehicle control) ˜ 8% of rod photoreceptors were double labelled (Fig 5B), indicating lipid exchange. Of note, the fluorescence shift indicated that cells from each starting population acquired relatively small amounts of the other fluorescent label, rather than extensive membrane exchange (Fig EV3B). This was confirmed by live imaging, which showed pairs of PhNT‐connected photoreceptors, each containing puncta of the other label (Fig 5C). Puncta of both labels were also visible along the connecting process.
To examine the involvement of actin in PhNT formation and transfer, we combined DiI/DiO co‐culture with application of either Cytocholasin D (2 μM) or Latrunculin B (5 μM), which each prevent actin polymerization. Appropriate doses were selected based on the literature (Onfelt et al, 2006; Gurke et al, 2008; Bukoreshtliev et al, 2009) and a dose–response‐based assessment of process formation (all types) (Fig EV3C–F). The selected doses significantly impaired the formation of photoreceptor processes but did not affect cell viability. Each drug was therefore applied to DiI/DiO co‐cultures for 48 h at 5DIC; both inhibitors significantly reduced DiI/DiO exchange on photoreceptor cultures in a reversable manner (Fig 5B). Interestingly, DiI/DiO transfer was decreased by ˜ 2‐fold in the presence of actin inhibitors, which is similar to the observed ˜ 2‐fold reduction in PhNT formation.
We also assessed whether larger structures like lysosomes and mitochondria can be exchanged between PhNT‐connected photoreceptors. LAMP1+ve endosomal vesicles were observed within PhNT processes connecting neighbouring photoreceptors in fixed cultures (Fig EV3G). Moreover, live imaging of Nrl.Gfp+/+ photoreceptors labelled with the lysosomal label, SiR‐lyso (Fig 5D; Movie EV5) or mitochondrial label, Mito‐tracker Orange (Fig 5E; Movie EV6) showed rapid movements of lysosome/late endosomes and mitochondria within the cytoplasm and within a variety of processes, including segment‐like processes and PhNTs. Object tracking software and careful analysis of deconvolved images revealed examples where lysosomes or mitochondria were clearly transferred from one cell to another via PhNTs, although these events were relatively rare (1 and 2 confirmed exchange events, respectively, observed from imaging 50 pairs of PhNT‐connected cells, each pair imaged for ˜ 60 min; Fig 5D and E).
PhNT‐like processes facilitate material transfer in vivo
The existence of PhNT‐like processes capable of transferring membrane lipids, proteins and potentially even organelles between photoreceptors in culture is striking and we sought to determine whether similar structures exist and function in vivo. In the first instance, we examined interactions between transplanted donor and host photoreceptors. The outer segments of host photoreceptors project towards the RPE and those of GFP+ host acceptor cells are thus in very close proximity to the donor cell mass (Fig 6A). This is to be expected but makes it difficult to visualize donor–host connections using standard imaging. However, careful analysis of deconvolved confocal images of host wild‐type retinae following transplantation of Nrl.Gfp +/+ donor photoreceptors revealed, alongside with the expected presence of larger neurites, GFP+ PhNT‐like processes extending between individual donor–host pairs (Fig 6A; Movie EV7). Sensitivity to fixation and tissue processing constraints meant that verified examples were seen relatively infrequently but those observed were very thin (Fig 6B) and lacked secondary branching. They were also typically < 10 μm in length (Fig 6C), but we note that longer connections may be more prone to disruption by fixation and sectioning. We also transplanted donor cells derived from Nrl.Gfp+/+ x myrRfp+/+ mice, in which GFP+ rods additionally express membrane‐tagged RFP (myrRFP). While the transfer of cGFP was similar to that reported previously (Pearson et al, 2016; Santos‐Ferreira et al, 2016; Singh et al, 2016), fewer examples of myrRFP transfer were found, despite very robust fluorescence being observed in the donor cell mass (Fig 6D and E). RFP fluorescence was predominantly observed in the inner segments of host cells (Fig EV4A). This may reflect a more spatially limited exchange of membrane‐bound molecules, as was also observed for DiI/DiO exchange in photoreceptor cultures, and by others (Rustom et al, 2004). However, detection of myrRFP transfer may also be reduced due to its slightly weaker fluorescence, compared to cGFP. Regardless, transfer of both membrane and cytoplasmic reporters requires healthy viable donor cells and does not arise from uptake of debris; no transfer was seen in any host photoreceptors following transplantation of UV‐treated donors, at either 72 h (N = 4) or 21 days (N = 6) post‐transplantation (Fig 6E and Appendix Fig S2A and B).
Figure 6. Transplanted photoreceptors form PhNT processes with host photoreceptors and transfer cytoplasmic and membrane proteins but transfer of mRNA was not detected.
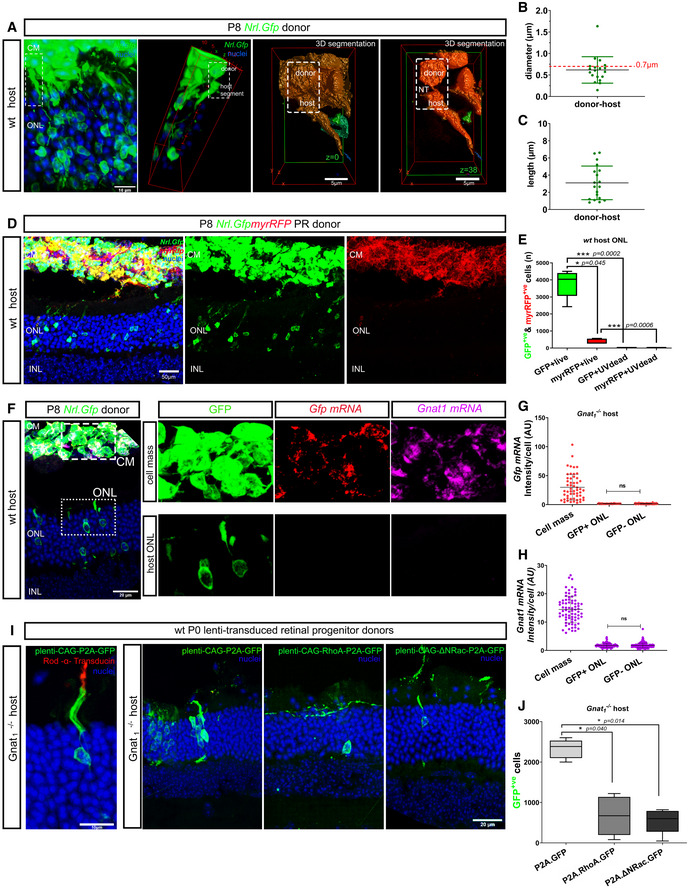
-
AMIP image of wild‐type (wt) retina transplanted with P8 Nrl.Gfp+/+ CD73+ MACS‐enriched photoreceptors; dashed box indicates ROI subjected to 3D deconvolution and volume reconstruction (right) and shows PhNT‐like connection between donor and host photoreceptor at the level of host inner segment. Additional images show 3D segmentation of z = 0 and z = 38 focal planes, and reveal the PhNT; green = GFP, blue = nuclei; Scale bar = 10 µm in MIP and 5 µm volume reconstruction.
-
B, CAssessment of properties of PhNT‐like processes between donor and host photoreceptors (n = 19 PhNT‐like processes; N = 10 eyes) showing (B) diameter and (C) length.
-
DMIP image of wt retina transplanted with P8 Nrl.Gfp+/+ x myrRFP/+ photoreceptors; showing wt host ONL photoreceptor labelled with cGFP (green) but not with myrRFP (red); Scale bar = 50 µm.
-
EQuantification and statistical analysis of material transfer events following subretinal transplantation of live (cGFP = 3,775 ± 804, myrRFP = 436 ± 145) or UV‐treated (cGFP = 1.3 ± 2.5, myrRFP = 4.3 ± 5.7) Nrl.Gfp+/+x myr‐Rfp +/+ photoreceptors into wt hosts (N = 8 and N = 4 retinae, respectively); one‐way ANOVA, parametric, two‐tail, Tukey’s multiple comparisons; graph shows mean ± SD.
-
F–HAssessment of potential for transfer of donor Gfp and Gnat1 mRNA during transplantation using RNAscope™. (F) Representative MIP images of Gnat1 −/− eyes transplanted with Nrl.Gfp+/+ donor cells and processed in situ for Gfp mRNA (red) and Gnat1 mRNA (magenta); blue = nuclei; ROIs indicated in (F) show GFP+ve donor cell mass (CM) and GFP+ve cells within Gnat1 −/− host outer nuclear layer (ONL). G, H Quantification of Gfp and Gnat1 mRNA (mean fluorescence intensity per cell), respectively (N = 3 retinae); one‐way ANOVA, non‐parametric, two‐tailed analysis showed no significant differences in signal intensity for either Gnat1 or Gfp mRNA between GFP+ve and GFP−ve host photoreceptors.
-
IManipulation of actin signalling alters material transfer in vivo: (far left) MIP image showing GFP+ve host photoreceptor (green) expressing Rod α‐transducin (red) after transplantation of retinal cells transduced with lenti‐CAG‐P2A.Gfp (transduction control) into Gnat1 −/− recipient; scale bar = 10 µm; adjacent images show Gnat1 −/− eyes transplanted with P2 retinal cells transduced with (left) plenti‐CAG‐P2A.GFP (green), or (middle) plenti‐CAG‐RhoA‐P2A.GFP (green), or (right) plenti‐CAG‐ΔΝRac1‐P2A.GFP (green); green = GFP, blue = nuclei; Scale bar = 20 µm.
-
JQuantification of the number of GFP+ cells in Gnat1 −/− host ONL, (N = 5 retinae per condition); plenti‐CAG‐P2A.GFP (2,328 ± 234), plenti‐CAG‐RhoA‐P2A.GFP (668 ± 481), plenti‐CAG‐ΔΝRac1‐P2A.GFP (547 ± 306); one‐way ANOVA, non‐parametric, Dunn’s multiple comparisons test. Graph shows mean ± SD.
Data information: CM = cell mass; all eyes were processed 3 weeks post‐transplantation. RPE—Retinal Pigment Epithelium; ONL—outer nuclear layer; INL—inner nuclear layer; GCL—Ganglion Cell Layer. Graphs show mean ± SD.
Figure EV4. Transplantation of Nrl.Gfp+/+ photoreceptors results in the transfer of cytoplasmic protein and some membrane‐bound protein, but little or no mRNA.
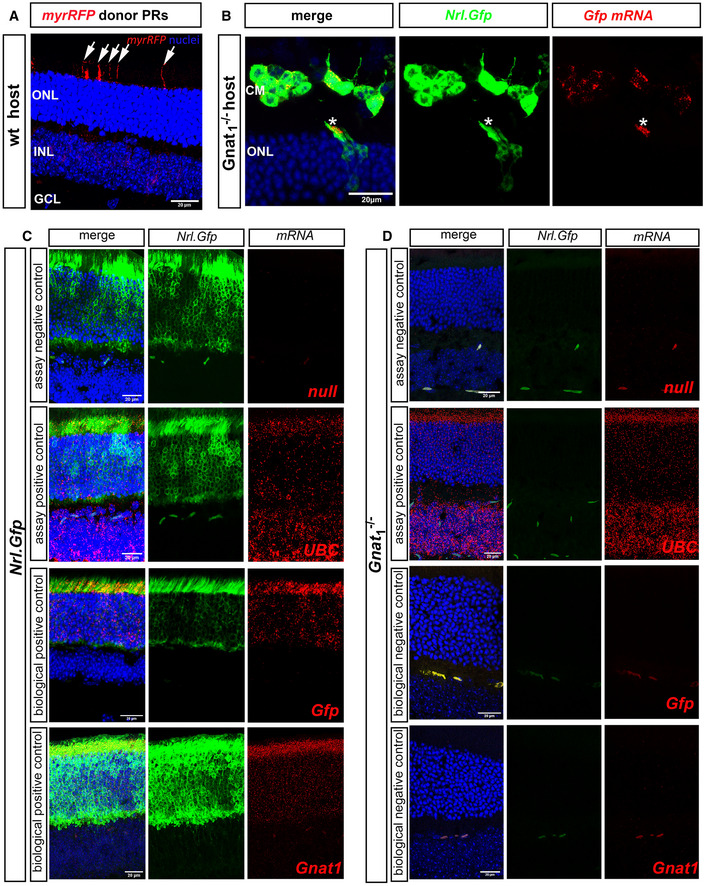
-
ARepresentative MIP confocal images of wild‐type (wt) eye cups fixed 21 days post‐transplantation with P8 myrRFP+ve CD73+ve MACS‐enriched photoreceptors. Note the presence of RFP+ host inner segments (arrows). myr‐RFP (red), nuclei (blue).
-
BRepresentative MIP confocal images of Gnat1 −/− eye cups fixed and stained in situ (RNAScope) with Gfp mRNA probe (red) 21 days post‐transplantation with P8 Nrl.Gfp+/+ photoreceptors (green). Blue = Dapi (nuclei); CM: cell mass; ONL: Outer Nuclear Layer. Asterisks denote a rare example of an integrated donor photoreceptor located within the host ONL and presenting robust staining for Gnat1 mRNA in the inner segment.
-
C, DRepresentative MIP confocal images of (C), Nrl.Gfp+/+ and (D), Gnat1 −/− eye cups fixed and stained in situ (RNAScope) with a null probe (assay negative control, red), UBC mRNA probe (assay positive control, red), Gfp mRNA probe (biological positive control, red), Gnat1 mRNA probe (biological positive control, red), and Gnat1 −/− tissue stained with either Gfp mRNA probe (biological negative control, red) or Gnat1 mRNA probe (biological negative control, red). Nrl.Gfp+/+ photoreceptors (green), nuclei (blue); mRNA (red).
Data information: Scale bars = 20 µm.
We have previously shown that transplantation of Nrl.Gfp +/+ photoreceptors into the Gnat1 −/− model of stationary night blindness leads to the presence of both GFP and rod α‐transducin (Gnat1) in host photoreceptors (Pearson et al, 2012; Warre‐Cornish et al, 2014). The efficiency of transfer appears similar for both molecules as rod α‐transducin is found in > 95% of GFP+ host acceptor cells (Warre‐Cornish et al, 2014; Pearson et al, 2016) and the extent of transfer is sufficient to restore visual function (Pearson et al, 2012, 2016). Whether this rescue occurs by the transfer of RNA and/or protein is not yet known but is important for our understanding of material transfer in the transplantation setting and any potential role PhNT‐like extensions might play in intercellular communication more broadly. To examine this, we transplanted Nrl.Gfp+/+ photoreceptors into Gnat1 −/− hosts and employed RNA in situ hybridization (ISH), using RNAscope™, to label either Gfp or Gnat1 mRNA (Figs 6F–H, and EV4B–D). RNAscope uses an RNA ISH method, where single‐molecule visualization in individual cells is achieved through use of a novel probe design strategy and a hybridization‐based signal amplification system to simultaneously amplify signals and suppresses background. As expected, Nrl.Gfp+/+ retinae show robust labelling in the ONL with both Gfp and Gnat1 probes, while the same probes yielded no labelling in Gnat1 −/− retinae (Fig EV4C and D). Similarly, after transplantation, Nrl.Gfp+/+ donor cells in the subretinal space of Gnat1 −/− recipients showed robust labelling for both probes (Fig 6F). Conversely, while numerous GFP+ acceptor cells were present within the host ONL, little or no labelling for either Gfp (Fig 6F and G) or Gnat1 (Fig 6F and H). Rarely (2/246 GFP+ host cells), we observed GFP+ cells in the host ONL that showed positive labelling for Gfp mRNA (Fig EV4B). Both cells were within the first 1–2 rows of the host ONL and their nuclei exhibited a multi‐chromocenter pattern of staining, consistent with that of immature rods (Solovei et al, 2009), or cones. These therefore likely represent rare true integration events, as we have described previously (Pearson et al, 2016). Thus, we suggest that material transfer between donor and host is predominantly mediated as protein, consistent with the rapid exchange of cGFP observed in vitro (Fig 4A). It also supports the notion that such transfer is transient, limited by the half‐life of the protein in question and sustained by the continued presence of, and physical interaction with, donor cells, as we had previously predicted (Pearson et al, 2016).
We next sought to manipulate actin dynamics in donor cells in vivo to examine whether this impeded cGFP transfer, like the effects of pharmacological inhibition of actin in cultures. Rho GTPases are important for actin stress fibre formation and actomyosin contractility and the activity of RhoA is incompatible with membrane protrusion (Raftopoulou & Hall, 2004). Conversely, Rac1 activity is required for membrane protrusion at lamellipodia tips (Mehidi et al, 2019) and, of relevance here, has been linked to a fundamental role in the biogenesis of NTs (Hanna et al, 2017). Wild‐type P0–2 cells were transduced with lentivirus over‐expressing RhoA (RhoA.GFP) or a dominant negative form of Rac1 (DNRac1.GFP) or empty vector expressing only GFP (Ctrl.GFP). Expression was confirmed by GFP fluorescence, Western blotting and F‐actin staining (Fig EV5A–C). Donor cells were transplanted into Gnat1 −/− adult eyes and examined 3 weeks later. Similarly sized, large cell masses were seen in the subretinal space of all retinae transplanted with RhoA.GFP+, DNRac1.GFP+ or Ctrl.GFP donor cells (Fig EV5D). However, both displayed significantly reduced transfer of cGFP (Fig 6I and J) and rod α‐transducin (Fig EV5E–G) to host acceptor photoreceptors, indicating that actin polymerization is required for material transfer and the concomitant gain of function (rod‐α‐transducin), in vivo.
Figure EV5. Manipulation of actin dynamics by overexpression of RhoA or DNRac1 in transplanted photoreceptors correlates with reduced GFP and rod α‐transducin transfer.
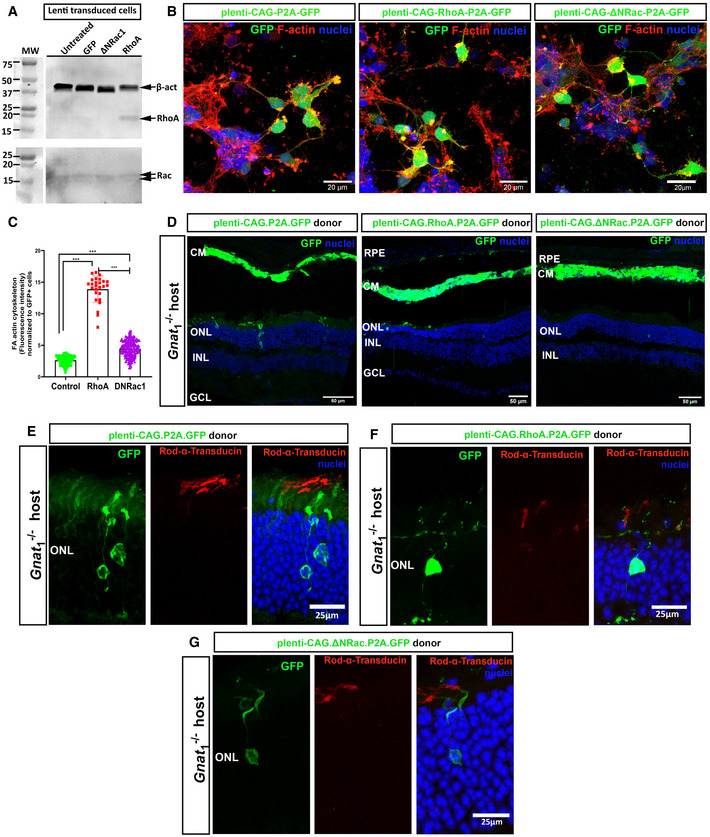
-
ARepresentative Western blots of RhoA and Rac1 expression in mixed P0–2 retinal cultures transduced with plenti‐CAG‐P2A.GFP, plenti‐CAG‐ΔΝRac1‐P2A.GFP or plenti‐CAG‐RhoA‐P2A.GFP compared to β‐actin and assessed after 8 DIC.
-
BRepresentative MIP confocal images of P0–2 retinal cultures transduced with plenti‐CAG‐P2A.GFP (green) or plenti‐CAG‐RhoA‐P2A.GFP (green) or plenti‐CAG‐ΔΝRac1‐P2A.GFP (green), fixed and stained with F‐actin (red) (N = 2 independent cultures, n = 4 wells per condition); Scale bars = 20 µm.
-
CQuantification and statistical analysis of the effect of overexpression of RhoA and ΔΝRac1, versus control, on actin in P0–2 retinal cultures at 7DIC. Effect assessed by measurement of mean intensities (Image J) of F‐actin plaques (red) in GFP+ cells (green), normalized to GFP+ cell number; control 2.6 ± 0.7; RhoA 13.9 ± 2.1, ΔΝRac 4.5 ± 1.3. Mean ± SD. One‐way ANOVA non‐parametric two‐tail, Kruskal–Wallis post‐test ***P < 0.001 (N = 2 independent cultures, n = 4 wells per condition).
-
DRepresentative tile‐scan images of Gnat1 −/− eyes transplanted with P2 retinal cells transduced with plenti‐CAG‐P2A.GFP (green) or plenti‐CAG‐RhoA‐P2A.GFP (green) or plenti‐CAG‐ΔΝRac1‐P2A.GFP (green). Green = GFP, blue = Dapi (nuclei); Scale bars = 50 µm.
-
E–GRepresentative MIP images of Gnat1 −/− eyes transplanted with P2 retinal cells transduced with (E), plenti‐CAG‐P2A.GFP (green) or (F), plenti‐CAG‐RhoA‐P2A.GFP (green) or (G), plenti‐CAG‐ΔΝRac1‐P2A.GFP (green). Immunostaining for Rod α‐transducin indicates that inhibition of actin polymerization impairs transfer of Rod α‐transducin alongside that of GFP. Green = GFP, blue = Dapi (nuclei), red = Rod α‐transducin; Scale bars = 50 µm.
Data information: RPE = retinal pigment epithelium, CM = cell mass, ONL = outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer. All eyes were fixed and examined 21 days post‐transplantation.
Evidence of material transfer between photoreceptors in the normal retina
Lastly, since transplantation involves a degree of stress to both donor and host cells, we asked whether material transfer between photoreceptors occurs in the normal mammalian retina. For this purpose, chimeric embryos were generated by aggregation of Nrl.Cre+/− (in which expression of Cre is tightly restricted to rod photoreceptors; Appendix Fig S1A and B) and TdTomato+/− morulae. In Nrl.Cre+/−/TdTomato+/− chimeras, rod photoreceptors will carry either Cre or TdTomatofloxed , but not both; only TdTomato+/− cells acquiring Cre from neighbouring Cre+ rods could potentially express TdTomato and recombination is not possible in any other chimera outcome (Fig 7A). Strikingly, we observed many TdTomato+ photoreceptors—and only photoreceptors—in Nrl.Cre+/−/TdTomato+/− adult chimera (Fig 7B–D). These cells bore all the morphological characteristics of rod photoreceptors and only ever exhibited a single nucleus that displayed a single chromocenter, typical of rods. They presented both as single cells (Fig 7B, left dashed box) and more typically as small clusters (Fig 7B, right dashed box, C and D) and were most numerous towards the peripheral retina. Conversely, no spontaneous recombination was seen in any control TdTomato+/− eyes (n = 20). Administration of AAV‐Nrl.Cre virus, in which expression of Cre is under the control of the Nrl promoter, was used to confirm photoreceptor‐specific recombination of the Nrl.Cre+/−/TdTomato+/− genotype (Fig 7E and F; see also Appendix Fig S1B). To our knowledge, this is the first demonstration of material transfer between photoreceptors in the intact adult neuroretina in vivo.
Figure 7. Generation of Nrl.Cre+/−/TdTomatofloxed aggregate chimeras reveals intercellular exchange of Cre between photoreceptors in vivo .
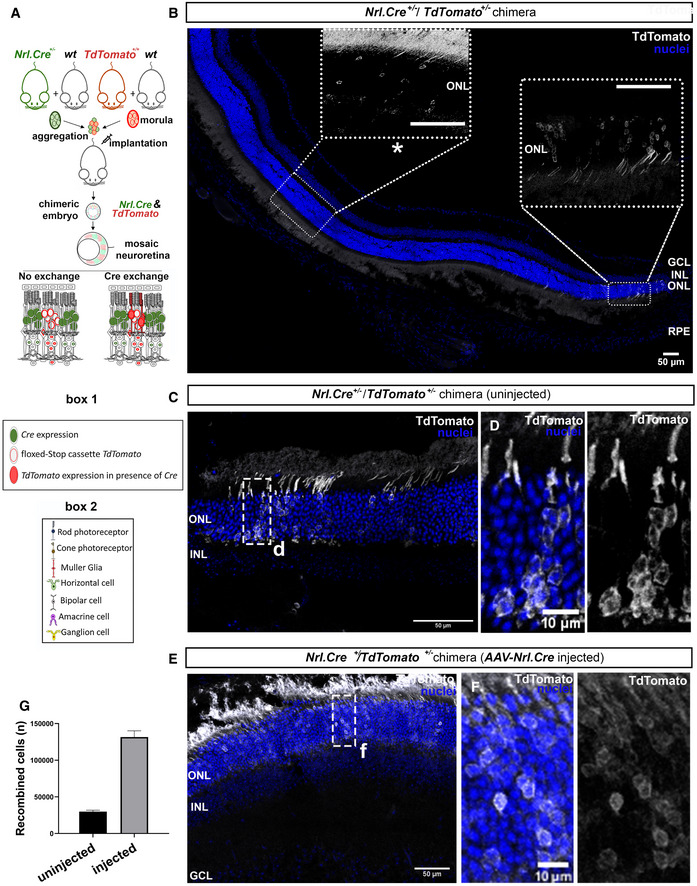
-
ASchematic representation of Nrl.Cre+/−/TdTomato+/− floxed chimera generation via morula aggregation, which results in chimeric mosaicism in the neural retina. Recombination and expression of TdTomato is only possible if Nrl.Cre+/− photoreceptors exchange material (Cre) with TdTomatofloxed cells via intercellular communication.
-
BRepresentative tile scan of uninjected Nrl.Cre+/− /TdTomato+/− chimera showing endogenous recombination (white); ROIs, as indicated with dashed or dot boxes; Scale bars = 50 µm.
-
C, DROI from uninjected Nrl.Cre+/− /TdTomato+/− chimera showing spontaneous recombination presenting in mosaic stripe patterning; Scale bar = 50 µm in (C) and 10 µm in (D).
-
E, FRepresentative images of Nrl.Cre+/− /TdTomato+/− chimera after receiving a subretinal injection of AAV‐Nrl.Cre. The injected eyes show widespread recombination, which also shows the anticipated mosaic stripe patterning; Scale bar = 50 µm in (E) and 10 µm in (F).
-
GQuantification of number of recombined cells in uninjected (29,825 cells ± 2,031) and AAV‐Nrl.Cre‐injected (131,562 ± 8,478) Nrl‐Cre+/− /TdTomato+/− chimeras, compared with uninjected Nrl‐Cre −/− (wt)/TdTomato+/− chimera (0 ± 0). Graph shows mean ± SD.
Data information: white = TdTomato+ve recombined cells; blue = nuclei. RPE‐ Retinal Pigment Epithelium; ONL—outer nuclear layer; INL—inner nuclear layer; GCL—Ganglion Cell Layer.
Discussion
Neuronal communication is mediated by chemical and electrical synapses and by paracrine signals, such as neurotrophic factors and neuropeptides. Increasingly, however, other mechanisms including EVs, TNTs and other NT‐like processes are being proposed to play important roles in neuro‐glial networks in processes as diverse as synaptic pruning and neurodegeneration (Agnati & Fuxe, 2014). There is mounting evidence to suggest that EVs might mediate neuron–neuron and neuron‐glia signalling, but evidence of the existence of NT‐like structures in primary mammalian neurons is very limited, and whether they form in vivo has not been shown.
During our investigations into photoreceptor transplantation, we, and others, discovered the unusual phenomenon of material transfer (Pearson et al, 2016; Santos‐Ferreira et al, 2016; Singh et al, 2016; Decembrini et al, 2017; Ortin‐Martinez et al, 2017), where transplanted donor photoreceptors exchange a wide range of molecules, both endogenous and transgenic, with photoreceptors in the host retina. Where the host acceptor cells are diseased, this process can be sufficient to restore function (MacLaren et al, 2006; Lamba et al, 2009; Pearson et al, 2012). Here, using aggregate chimeras, we report the important further discovery that material transfer may also occur between photoreceptors in the intact retina.
In the first instance, we considered EVs as the most likely mediators of material transfer between photoreceptors, since they fulfil many of the characteristics of transfer seen both in chimeric retinae and in transplantation. We found that postnatal rod photoreceptors form MVBs, which are necessary for EV maturation, in vivo and can release EVs, at least in culture. Photoreceptor‐derived EVs display a molecular signature specific to photoreceptors, but distinct from the cytoplasmic contents of the same cells. These EVs contained either protein or mRNA for components of the phototransduction machinery, amongst others, but not the transcription factor, Crx, in contrast to recent reports (Zhou et al, 2018). Interestingly, some molecules were present as mRNA while others were detectable as protein. This is consistent with other reports of EV‐derived cargo (Jeppesen et al, 2019; Murillo et al, 2019) although it remains to be determined why these differences arise. One potential explanation may be that membrane‐bound and cytosolic proteins might become incorporated within an EV in different forms (i.e. as mRNA or as protein) but this is beyond the scope of the current study.
Perhaps surprisingly, photoreceptor‐derived EVs are not taken up by other photoreceptors either in culture or following injection into the adult but are instead taken up specifically by Müller Glia. Elsewhere, EV‐mediated neuron‐glia communication has been linked to synaptic activity and plasticity (Chivet et al, 2014; Bahrini et al, 2015; Ashley et al, 2018; Pastuzyn et al, 2018) and myelination (Fruhbeis et al, 2013). It is perhaps surprising therefore that we did not see recombination in Müller Glia in Nrl.Cre+/−/TdTomato+/− chimeric mice, indicating that, if photoreceptors do indeed release EVs in the normal retina, they do not represent a significant mode of neuron‐glia communication in the healthy retina. An alternative explanation may be that EVs are indeed released by photoreceptors in vivo, but they are immediately cleared by local immune cells under normal conditions, as recently shown in zebrafish (Verweij et al, 2019). Indeed, other studies indicate that EVs are involved in various stages of the injury process, including propagating inflammation, mediating neuroprotection (Sun et al, 2020), and modulating regeneration (Peng et al, 2018). As such, they are proposed to play active roles in the pathogenesis of several neurodegenerative diseases (Hill, 2019). It will therefore be of significant future interest to determine what (if any) role photoreceptor‐derived EVs play in neuron‐glia signalling in retinal degeneration.
Material transfer between photoreceptors instead appears to be mediated by physical connections in the form of PhNT processes. TNTs and NT‐like processes have been regarded with scepticism by some parts of the scientific community (Ariazi et al, 2017; Baker, 2017) as there is relatively limited information regarding their structural identity and if, or how, they differ both amongst each other and from other cellular protrusions, such as filopodia. Nonetheless, in the chick embryo NT‐like processes can form between migrating neural crest cells and mediate directional changes in following cells (Teddy & Kulesa, 2004). Perhaps the most compelling evidence for a functional role in vivo comes from Drosophila, where cytonemes are responsible for the distribution of morphogens, including Wnt and Hedgehog (Hg), in the development of the wing imaginal disk and eye disk (Roy et al, 2011; Bischoff et al, 2013; Stanganello et al, 2015). To date, the only evidence that such structures exist in mammalian tissues in vivo is between immune cells in the mouse cornea (Chinnery et al, 2008) and, recently, between pericytes (Alarcon‐Martinez et al, 2020).
Here, we show that, at least in culture, photoreceptors can form PhNT structures that fall into one of two groups, Type I (thin, typically straight, actin‐rich) or Type II (thicker, containing both actin and microtubules) processes, with Type I being more numerous; both were distinct from exploratory neurite‐like processes. These characteristics are consistent with the descriptions of TNTs between macrophages in culture (Onfelt et al, 2006) and differ from the purely actin‐containing TNTs described in PC12 (Rustom et al, 2004) and CAD (Gousset et al, 2009; Vargas et al, 2019) cells. Earlier studies indicated that actin‐only and actin/microtubule‐containing structures may support unidirectional (Onfelt et al, 2006; Arkwright et al, 2010) and bidirectional (Rustom et al, 2004; Gousset et al, 2009; Domhan et al, 2011) transfer, respectively. Moreover, a recent elegant fluorescent EM analysis of NTs formed by CAD cells in vitro showed bundles of individual actin‐rich NTs facilitating the movement of vesicles and organelles along their length using motor proteins such as Myo10 (Sartori‐Rupp et al, 2019). Regardless of type, the biosynthesis of NT‐like protrusions is attributed to the activity of Rac1 and Cdc42 (Hanna et al, 2017) and F‐actin polymerization (Onfelt et al, 2006; Gurke et al, 2008; Bukoreshtliev et al, 2009; Ljubojevic et al, 2021). These pathways obviously perform many different functions within the cell, but in keeping with a role in NT formation, we found that F‐actin depolymerizing drugs significantly impaired material transfer between photoreceptors in culture, while a dominant negative form of Rac1 and overexpression of RhoA each significantly impaired the transfer of the cytoplasmic fluorescent reporter, GFP, and the phototransduction protein, rod α‐transducin, from GFP+ wildtype donors to dysfunctional host photoreceptors in vivo, following transplantation.
Precisely what is exchanged via NT‐like processes is likely determined by the cells in question and the type of processes formed. Some include gap junction proteins and permit the propagation of electrical and calcium signals, but may exclude proteins of molecular weights > 1 kD, like GFP (Watkins & Salter, 2005; Hase et al, 2009; Wang et al, 2012b; Alarcon‐Martinez et al, 2020). Paradoxically, open‐ended TNTs formed by PC12 cells are reported to permit the exchange of small diameter organelles but not the passive diffusion of cGFP (Rustom et al, 2004). Conversely, cytonemes are thought to be close‐end tubes and can establish signalling gradients by releasing molecules, such as Hg, packaged in EVs, from their terminal tips (Gradilla et al, 2014; Chen et al, 2017). Here, we found that cytoplasmic proteins can be exchanged between connected photoreceptors with surprising speed and efficacy, as assessed by FRAP in culture and in transplantation in vivo, while membrane labels were exchanged but to a more limited extent. Organelles (lysosomes, mitochondria) were also observed to pass, at least in culture, albeit rarely. We suggest that organelle exchange may be mediated by Type II PhNTs, which are thicker than Type I and additionally contain tubulin and may thus be able to facilitate large cargo transfer. Type II PhNTs were rarely present in culture, so the chances of directly observing organelle transfer may be correspondingly limited. Type I PhNTs were more numerous, and we consider these to be the most likely mediators of protein transfer between photoreceptors, both in culture and in transplantation.
Along with proteins and organelles, we also addressed the potential for mRNA transfer. Despite the robust presence of proteins such as cGFP and rod‐α‐transducin in acceptor cells, at least in transplantation, mRNA of these molecules was not observed using a sensitive RNA in situ hybridization technique (RNAscope™). We cannot exclude the possibility of small amounts of mRNA transferring between connected cells, as reported recently between stem cells in culture (Haimovich & Gerst, 2019). Together, the above indicate that material transfer between photoreceptors can occur via two types of open‐end PhNTs. These thus appear distinct from the recently reported closed‐end pericyte NTs that regulate neurovascular coupling in the retina (Alarcon‐Martinez et al, 2020). Our data show that, at least for cytoplasmic molecules, the transfer mechanism involves an actin‐dependent network, both in culture and in vivo, in line with in vitro reports by others for other cell types (Rustom et al, 2004; Hanna et al, 2017). Elucidating the sorting mechanisms that determine what cargo can/cannot be exchanged and how this relates to different types of NT‐like processes will be of major importance as we explore their role in tissue homeostasis.
Perhaps one of the most striking findings of our investigations was the demonstration of material transfer between photoreceptors in the intact chimeric retina. Further work is required to conclusively show that this is mediated by PhNTs but it nonetheless raises fundamental questions regarding the role of material transfer in retinal function, and how this might sit within the framework of intercellular communication more broadly. Here, we show that PhNT‐like processes mediate the transfer of molecules that, at least within the transplantation paradigm, have been shown to support gain of function in the receiving cell. Rod α‐transducin can be transferred from transplanted donor cells to non‐functional Gnat1 −/− host photoreceptors in sufficient quantities to render those cells light responsive (Pearson et al, 2012, 2016). This raises the possibility that PhNT‐like structures may be important for maintaining function under stress and/or dysfunction. Consistent with this notion, transplanted hematopoietic progenitor stem cells appeared to partially rescue the corneal defect seen in cystinosin knockout (Ctns −/−) mice by local, unidirectional transfer of cystinosin, from healthy donors to Ctns −/− macrophages in vivo (Rocca et al, 2015). Indeed, the role of NT‐like processes in disease and tissue homeostasis is still unclear as, at least in culture, the transfer of organelles such as mitochondria and lysosomes has been reported to occur both to and from the diseased/dying cells (Spees et al, 2006; Wang et al, 2011; Rocca et al, 2015; Wang & Gerdes, 2015; Rustom, 2016). In our hands, material transfer occurs between apparently healthy neurons in vitro, in transplantation and in the intact retina, and has the potential to bring about gain‐of‐function where the acceptor cell is dysfunctional.
In summary, we show that photoreceptors can form open‐ended membranous NTs that mediate the transfer of cytoplasmic and lipid‐bound material to other photoreceptors, both in vitro and in transplantation. Of note, a similar set of observations have been described in a parallel study conducted by Ortín‐Martínez et al (2021); Wallace and colleagues also show that material transfer between photoreceptors is mediated via membranous processes whose formation is regulated via Rho‐GTPases. These processes, which they also term photoreceptor nanotubes (PhNTs), facilitate the transfer of fluorescent reporter molecules in culture and in transplantation. Moreover, we further show, for the first time, that material transfer can occur between photoreceptors in the intact retina. Both studies provide new insights into the mechanisms underlying transplant‐mediated cellular repair in the nervous system and raise important questions about the potential roles played by NTs and material transfer in both the healthy nervous system and in disease.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental Models | ||
| C57BL/6J (M. musculus) | Jackson Lab | B6.129P2Gpr37tm1Dgen/J |
| Nrl.gfp (M. musculus) | Kind gift of Dr Anand Swaroop, now available through Jackson Lab | B6. Cg‐Tg(Nrl‐EGFP)1Asw/J |
| Ai9(RCL‐tdT) or TdTomato (M. musculus) | Jackson Lab | B6. Cg‐Gt(ROSA)26Sortm9(CAG‐tdTomato)Hze/J |
| mTmG or myrRFP (M. musculus) | Jackson Lab | Gt(ROSA)26Sortm4(ACTB‐tdTomato,‐EGFP)Luo/J |
| Gnat1−/− (M. musculus) | kind gift of Dr Janis Lem, Tufts Medical College | Gnat1−/−; Gnat1KO |
| Nrl.Cre (M. musculus) | Jackson Lab | C57BL/6J‐Tg(Nrl‐cre)1Smgc/J |
| Recombinant DNA | ||
| Pd10.Nrl.Cre | This paper, based on Akimoto et al, 2006 and Brightman et al, 2016 sequences | N/A |
| AAVPk | Addgene | N/A |
| pHGTI helper | Plasmid factory | PF1810 |
| pLenti‐CAG‐GFP‐P2A | Generated by Dr Ortín‐Martínez Arturo and Professor Valerie Wallace, University of Toronto | N/A |
| pLenti‐CAG‐RhoA‐GFP‐P2A | Generated by Dr Ortín‐Martínez Arturo and Professor Valerie Wallace, University of Toronto | N/A |
| pLenti‐CAG‐ΔNRac‐GFP‐P2A | Generated by Dr Ortín‐Martínez Arturo and Professor Valerie Wallace, University of Toronto | N/A |
| pMD2.G | Addgene | N/A |
| pCMVΔR8.74 | Addgene | N/A |
| Antibodies | ||
| CD81 | Cell Signalling | 10037 |
| TSG101 | BD Transd | 612696 |
| CD9 | EDM Millipore | CBL162 |
| Alix | Cell Signalling | 2171 |
| Cre recomb | EDM Millipore | MAB3120 |
| GM130 | Cell Signalling | 12480 |
| Opsin | Sigma | O4886 |
| G‐alpha‐t1 | Santa Cruz INC | Sc389 |
| Recoverin | MERCK | AB5585 |
| GFP/CFP/RFP | Clontech | 632380 |
| Anti‐LAMP1 | Abcam | [1D4B] (ab25245) |
| Beta actin | R&D Systems | MAB8929 |
| Beta actin | Sigma | A2228 |
| Rac | Cytoskeleton Inc | ARC03‐S |
| RhoA | Cell Signaling | (67B9) Rabbit mAb #2117 |
| CD73 APC | Miltenyl | 130‐103‐052 |
| Oligonucleotides and sequence‐based reagents | ||
| rtQPCR primers | This study | Expanded View‐ Materials and Methods |
| Gfp: GAAGCGCGATGACATGGT/CCATGCCGAGAGTGATCC/ probe 67 | This study | Expanded View‐ Materials and Methods |
| Cre: ATCTGGCATTTCTGGGGATTG/ GCAACACCATTTTTTCTGACCC/ probe 20 | This study | Expanded View‐ Materials and Methods |
| Gnat1: AGAGCTGGAGAAGAAGCTGAAA/ TAGTGCTCTTCCCGGATTCA/ probe 89 | This study | Expanded View‐ Materials and Methods |
| Rcvrn: CAATGGGACCATCAGCAAA/ CCTCAGGCTTGATCATTTTGA/ probe 67 | This study | Expanded View‐ Materials and Methods |
| Rho: ACCTGGATCATGGCGTT/ TGCCCTCAGGGATGTACC/probe 32 | This study | Expanded View‐ Materials and Methods |
| Crx: CCCCAATGTGGACCTGAT/GGCTCCTGGTGAATGTGGT/ probe 64 | This study | Expanded View‐ Materials and Methods |
| Nrl; TTCTGGTTCTGACAGTGACTACG/ TGGGACTGAGCAGAGAGAGG/ probe 53 | This study | Expanded View‐ Materials and Methods |
| Actb:AAGGCCAACCGTGAAAAGAT/GTGGTACGACCAGAGGCATAC/probe 56 | This study | Expanded View‐ Materials and Methods |
| Chemicals, enzymes and other reagents | ||
| RNase | Sigma | |
| Super RNAse Out 20 U/µl, | Thermo Fisher | |
| TaqMan® Universal PCR Master Mix | Roche UK | |
| FAM RTQPCR probe | Universal Probe Library; Roche, Germany | |
| Mitotracker‐Orange | (Invitrogen/Molecular Probes, M7512) | |
| SiR‐actin | Cytoskeleton | |
| SiR‐tubulin | Cytoskeleton | |
| SiR700‐lysosome | Cytoskeleton | |
| verapamil | Cytoskeleton | |
| DiI or DiO lipophilic tracers | Thermo Fisher | |
| Cytochalasin D | Sigma | |
| ROCK inhibitor Y‐27632 | Sigma | |
| Latrunculin B | Sigma | |
| Fibronectin | Sigma | |
| Poly‐D‐lysine hydrobromide | Sigma | |
| Software | ||
| GraphPad Prism 8 | GraphPad | |
| Adobe Photoshop version 22.4 | Adobe | |
| Image J (FIJI) | NIH | |
| HyVolution software (Scientific Volume Imaging Huygens) | Scientific Volume Imaging | |
| LEICA LASX | Leica | |
| Chemidoc‐Gel Lab | Bio‐Rad | |
| Fast Real‐time Sequence Detection System (Applied Bioscience, USA) | Applied Bioscience | |
| Other | ||
| QuantiTect® Whole Transcriptome | Qiagen | |
| Qiagen RNeasy® Mini kit | Qiagen | |
| Immobilon™ Western Chemiluminescent HRP substrate kit | Millipore, USA | |
| CD73 APC and anti APC microbeads and magnetic rack | Miltenyl | |
| Worthington Papain Dissociation System | Worthington Biochemical Corporation, USA | |
Methods and Protocols
Animals
Animal lines used: C57BI/6J (wildtype, wt) (Harlan), Nrl.Gfp+/+ (kind gift of A. Swaroop, University of Michigan, USA) (Akimoto et al, 2006), Ai9(RCL‐tdT) or TdTomatofloxed (Madisen et al, 2010) Gt(ROSA)26Sortm4(ACTB‐tdTomato,‐EGFP)Luo or “mTmG” or “myrRFP” (Muzumdar et al, 2007) Gnat1tm1Clma or Gnat1−/− (kind gift of J. Lem, Tufts University School of Medicine, USA) (Calvert et al, 2000) and C57BL/6J‐Tg(Nrl‐cre)1Smgc/J or Nrl.Cre+/− (Brightman et al, 2016), except for Nrl.Cre, which was maintained as hemizygote, due to lethality issues. Mice were maintained in the animal facilities at University College London or King’s College London. All experiments have been conducted in accordance with the Policies on the Use of Animals and Humans in Neuroscience Research, revised and approved by the ARVO Statement for Use of Animals in the Ophthalmic Research, under the regulation of the UK Home Office Animals (Scientific Procedures) Act 1986. Briefly, rodents were maintained on a standard 12/12‐h light dark cycle, housed in same sex groups or sustained breeding pairs wherever possible and provided with fresh bedding and nesting material and food and water ad libitum.
Transplantation surgery
Transplantation was conducted as we have described previously (Pearson et al, 2012) All recipients were adult (6–8 weeks) at time of transplantation. Briefly, recipient mice were anaesthetized with a single intra‐peritoneal injection of a mixture of Dormitor (1 mg/ml medetomidine hydrochloride, Pfizer Pharmaceuticals, Kent, UK), ketamine (100 mg/ml, Fort Dodge Animal Health, Southampton, UK) and sterile water for injections in the ratio of 5:3:42. Prior to cell transplantation, the tip of 34‐G needle was inserted to the sclera and slowly removed. The needle with the cell suspension was then inserted tangentially in through the sclera and RPE, injecting slowly the 1 μl cell suspension, or 2 µl (2 μg) of concentrated EV fractions, or 2 μl (2 μg) of Cre protein (ab134845) underneath the RPE in the superior retina resulting in small retinal detachment.
Generation of Chimeras by morula aggregation
De novo chimeric mice were generated by mixing two genetic populations Nrl.Cre and the TdTomatofloxed reporter lines, both of C57BI/6J background, by morula reaggregation. Briefly, 3–4 weeks old wt females were super ovulated with PMSG; 5 IU [International Unit] in 0.1 ml IP on day 1 followed by a second injection of hCG; 5 IU on day 3, after which they are immediately placed in the cage with the mutant male. Embryos were collected at the eight‐cell morula‐stage from the two genotypes by flushing oviducts at 2.5 dpc. All embryos were collected and washed with FHM medium and KSOM. The sandwich aggregation technique was performed by Dr Signore, UCL Institute of Child Health. Briefly, the removal of embryo’s zona pellucida was achieved using Tyrode’s acid solution and transferred to FHM medium, and finally, the zona‐free embryos were transferred back into KSOM. Once all the embryos are isolated, the embryos of the first genotype were placed individually into the indentations of the aggregation plate, and the process was repeated for the second genotype making sure that the embryos in each indentation are physically attached to each other. The aggregation plate was incubated for 19–24 h at 37°C/ 5% CO2. The next day, the aggregates should have formed a single embryo at the late morula or blastocyst stage. The embryos were implanted by microinjection in pseudo‐pregnant recipient females and the females checked for pregnancy several days post‐implantation. Chimeras were collected at P45.
Chimera genotyping
Genomic DNA was extracted from ear clips following manufacture’s protocol (Extracta™DNA Prep for PCR, QuantaBio), and 5 μl of extract was used per 50 μL PCR reaction. PCR reactions were performed following the manufacture’s protocol (Promega) following a cycle of: Denaturation (2 min 95°C), Reaction (30 s 95°C, 30 s 60°C, 45 s 72°C), and Final extension (5 min 72°C), with a final hold at 10°C.
Primers sequences: Nrl.Cre; 5'‐GCATTACCGGTCGATGCAACGAGTGATGAG‐3' and 5’GAGTGAACGAACCTGGTCGAAATCAGTGCG‐3’ with a product of 400 bps. TdTomato (wt); 5’‐AAGGGAGCTGCAGTGGAGTA‐3’ and 5’‐CCGAAAATCTGTGGGAAGTC‐3’ (mutant) 5’‐GGCATTAAAGCAGCGTATCC‐3’ and 5’‐CTGTTCCTGTACG GCATGG‐3’ with products; tdTomato−/− 297 bp, tdTomato−/+ 196 bp 297 bp tdTomato+/+ 196 bp accordingly.
Tissue harvesting fixation and cryo‐sectioning
Animals were sacrificed by cervical dislocation. Eyes were removed, and the cornea punctured to improve the efficiency of the fixation. The eyes were fixed in 4% PFA (PBS) for 1 h at RT (conventional IHC), or 4% PFA (dH2O) 24 h (RNAScope), eye cups dissections performed, followed by cryopreservation with 20% sucrose (Sigma, USA) in PBS for 1 h at RT or for 24 h at 4°C (RNAScope). To preserve nanotube‐like structures in vivo punctured eyes were washed in 5% Sucrose and fixed with 7.5% sucrose, 2% PFA (dH2O) overnight in dark humid chambers at 4–8°C. Eyes embedded in OCT and rapidly frozen in liquid Nitrogen. The frozen samples were stored at −80°C until cryo‐sectioning on a Bright OTF5000 cryostat (Bright Instruments Co Ltd, UK) set at 12–17 μm thickness. Sections were serially transferred onto 2–10 sets of SuperFrost‐TM ultra Plus Adhesion glass slides (Thermo Fisher, USA) and stored at −20°C for staining, and at −80°C for RNAScope.
In situ branched hybridization method (RNA Scope)
Multi‐plex RNAscope assay was performed according to manufacturer’s protocol with sterile solutions and enzymes in a specific HybEZ™ Oven with EZ‐Batch™ Slide holder all provided by Advanced Cell Diagnostics (ACD) (Wang et al, 2012a). Briefly, samples were air‐dried, washed with PBS, baked for 30 min at 60°C and post‐fixed with 4% PFA 15 min at 4°C. Slides were then dehydrated through a series of Ethanol washes (50%, 70%, 100%), air‐dried and treated with Hydrogen Peroxidase for 10 min, washed at RT and subjected to antigen retrieval solution (ACD) for 3 min at 88°C, before immediately washing with dH2O, 100% Et‐OH, and air‐dried again prior to protease III treatment for 30 min at 40°C. The slides then were washed with dH2O and hybridized with RNAscope probe mix (Gnat1 probe (524881‐C2) diluted 1:50 in EGFP probe (400281‐C1) solution; see ACD bank link for sequences: https://acdbio.com/catalog‐probes) for 3 h 40°C, before being washed with Wash Buffer (ACD) and stored overnight in 5XSSC solution at RT. Slides were then subjected in three subsequent series of amplification with Multiplex FL v2 Amp 1, Amp2 and Amp3 for 30 min at 40°C each and subsequent wash in wash buffer. For visualizing both EGFP_C1 and Gnat1_C2, HRP signal was developed in two steps for HRP_C1 and then HRP C2 for 15 min at 40°C followed by a 5 min wash in wash buffer. Fluorochromes were assigned by using Opal™ dyes (Akoya’s Opal™ Multiplex IHC kits diluted 1;1,500 in TSA probe diluent buffer, ACD) and incubation for 30 min at 40°C followed by a 5 min wash. Finally, HRP signal was blocked by incubation for 15 min at 40°C with Multiplex FL v2 HRP blocker (ACD) and slides washed with wash buffer and counterstained with DAPI and subjected to IHC for anti‐GFP immunostaining. Negative and positive multiplex control probes were provided by ACD and tested in parallel with main probes following either a single or multiplex protocol (shown in Fig EV4).
Immunohistochemistry (IHC) and Immunocytochemistry (ICC)
Tissue sections were air‐dried for 30 min, rehydrated with PBS and incubated in blocking buffer at RT for 1 h. ICC blocking buffer composed of 0.1% Saponin, 1% BSA and 2% NGS or the appropriate serum depending on the secondary used. IHC blocking buffer composed of 2% NGS, 1% BSA and 0.05–1% Triton X‐100. Primary antibodies used in the range of 1:100–1:1,000. Primary antibodies used; Anti‐LAMP1 (rat monoclonal, 1:500, [1D4B] (ab25245), Abcam), anti‐Gat1 (rabbit polyclonal, 1:1,000, K‐20, SC‐389 Santa Cruiz), anti‐Rhodopsin (mouse, 1:2,000, O4886, Sigma) and anti‐GFP, (goat polyclonal, 1:1,000, ab6673, Abcam). Sections were then incubated in primary antibody overnight at 4°C, washed with PBS and incubated with secondary antibody for 2 h at RT in blocking solution. Sections were then washed and counterstained with DAPI (4’,6‐Diamidino‐2‐Phenylindole, Dihydrochloride; 1:1,000 dilution in 1× PBS (Molecular ProbesTM, USA)). Subsequently, sections mounted under coverslips with fluorescence mounting medium (Dako, Denmark) and stored covered at 4°C to prevent exposure to light. Immunocytochemistry followed the same procedure as immunohistochemistry. Negative controls omitted the primary antibody.
Confocal microscopy of fixed tissue
Images of fixed tissue sections were acquired on a Leica TCS SPE upright confocal laser scanning microscope (Leica, Germany) that was configured to a scanning frequency of 400 Hz, fitted with 5× (air, numerical aperture or NA = 0.15), 40× (oil immersion, NA = 1.15) and 63× (oil immersion, NA = 1.3) objectives and photomultiplier tubes to detect fluorescence emission. DAPI was excited with a 405 nm laser source. The 488 nm and 532 nm laser sources were used for the fluorophores Alexa Fluor® 488 and Alexa Fluor® 546, respectively. When acquiring the fixed images, xyz confocal stacks of representative fields of view were captured at a resolution of 1,024 × 1,024 pixels at a step size of 1 μm for retinae tissue, 0.5 μm for cell cultures. To maintain consistency, tissue sections within a given experiment were imaged with the same laser power, detector gain and offset settings. Identical settings allowed for semi‐quantitative comparison of protein expression levels between developmental time points. LAS AF image browser software was used to compile and export the acquired images. Lif data files were then processed in Fiji/ImageJ (National Institutes of Health, USA) (Schindelin et al, 2012).
Primary photoreceptors, retinal cultures and cocultures
Retinal dissociation was performed as previously described (Pearson et al, 2012) with some modifications. Briefly, neuroretinas were removed and placed in fresh dissection medium were the neuroretina was removed (DMEM/F12 of PBS or HBSS or EBSS no phenol red (Invitrogen) supplemented with 15 mM HEPES, 1 mM Taurine and 0.5 U/ml DNAase) and then placed in prewarm papain/DNAase solution (˜ 100 μl/retinae) for dissociation using the Papain Dissociation System (Huettner & Baughman, 1986). The solution was then incubated for ˜ 20 min in a water bath at 37°C with discontinuous agitation, following centrifugation at 260 × g for 5 min at RT. The supernatant was discarded, and the pelleted cells were reconstituted with resuspension buffer (2.7 ml EBSS, 300 µls reconstituted albumin‐ovomucoid inhibitor, and 50 µl of DNase solution), 250 µl of resuspension buffer was used per 10 retinae and layered over 1 ml of albumin‐inhibitor solution in a centrifuge tube, to form a discontinuous gradient, then centrifuged at 70 × g for 10 min at RT. Dissociated retinae cells pellet at the bottom of the tube and transferred in chemically defined medium (CD), cell viability, assessed with Trypan blue and a ViCellXR Cell Counter and for retinae cultures cells plated in a final density not exceeding 70,000 cells/mm2 in 1 mg/ml PDL (Sigma) /0.04 mg/ml fibronectin (Sigma) pre‐coated glass bottom plates. For mixed population retinal cocultures, cells from two different genotypes were isolated in the same day and mixed in a ratio 1:1.
In the experiments involving primary photoreceptor cultures, photoreceptors were enriched using APC‐conjugated CD73 (Eberle et al, 2011; Lakowski et al, 2015), which immuno‐reacts with anti‐APC magnetic beads (Miltenyl) and sorted with a MACSQuant® Analyzer (Miltenyl). Briefly, after dissociation, cells were resuspended into MACS buffer (Miltenyl) supplemented with 15 mM HEPES, 1 mM Taurine and 0.5 U/ml DNAase at a concentration of 107 cells per 100 µl and Primary anti‐mouse‐CD73 APC‐conjugated antibody (Miltenyl) was added to the cells at a concentration of 1 μl Ab/106 cells for 20–30 min at 4°C. Cells were then centrifuged for 10 min at 260 × g at 4°C, and the cell pellet was resuspended in anti‐APC solution (20 µl anti‐APC‐Microbeads and 80 µl MACS buffer) and incubated for 20 min at 4°C. The samples were subsequently loaded into the MACSQuant® Analyzer, selecting the “Positive selection sensitive” programme for magnetic separation. CD73+APC Photoreceptor precursors are found in the second eluate, with the first (negative) eluate comprising the fraction of all other retinae cells. The positive fraction was centrifuged immediately at 160 × g for 20 min at 4°C. Viability was then assessed as for retinal cultures and photoreceptors were plated in a final density not exceeding 85,000 cells/mm2 in 1 mg/ml PDL (Sigma)/0.04 mg/ml fibronectin (Sigma) pre‐coated glass bottom plates.
Both photoreceptor and retinal primary cultures were maintained in CD medium; BrainPhys™ Neuronal Medium, (STEMCELL Technologies Inc) supplemented with 15 mM HEPES (Invitrogen), 0.01 mg/ml recombinant human insulin, 0.0055 mg/ml human transferrin (substantially iron‐free) and 0.005 μg/ml sodium selenite, 1 mM Taurine, 3% Tissue culture grade BSA solution (Sigma) and antibiotics Pen/ Strep 10 mg/ml (Sigma) and incubated at 37°C, 5% CO2. Medium was changed every 3–4 days.
For photoreceptor‐retinal cell trans‐well co‐cultures, a trans‐well insert system was used where retinal cells were plated in the bottom of a 24‐well plate pre‐coated with PDL at a density of 3–4 × 105 cells per well. The retinae cells that plated at the bottom of the dish were allowed to recover for 8–19 h, before the medium was changed, and photoreceptor cells were added on top of them at a trans‐well insert (0.4 μm pore diameter, Millipore) at a density of 1 × 106 cells/insert. For these experiments, CD medium was supplemented with 5% BSA (Sigma) instead of 3% and medium was not changed but topped up to adequate volumes every 5 days for 21 days.
DiI/DiO co‐cultures and pharmacological interventions
After retinal cells were dissociated, cells were labelled with DiI or DiO lipophilic tracers (Thermofisher, 1 μl per 106 cells) in a suspension at 37°C for 15 min in CD medium and then simultaneously washed by centrifugation (300 × g, 5 min, RT) for four times with PBS every time in new clean tubes. Supernatant of the final wash was kept and used as control for dye carry over in wt retinal cultures. DiI+ and DiO+ labelled populations were mixed in a ratio 1:1 and plated as stated above for 5 days and on day 5 in culture pharmacological inhibitors were added in appropriate concentrations of Cytochalasin D (Hanna et al, 2017) or Latrunculin B (Osteikoetxea‐Molnar et al, 2016) for 48 h (Fig EV3). Dose range was based on the published literature and confirmed by a dose–response assessment and determined as the concentration required to significantly decrease the proportion of photoreceptors extending actin+ processes but without causing cell detachment and loss of viability. As the inhibitors were dissolved in DMSO, controls were supplemented with the equivalent dose of DMSO (vehicle control).
Flow cytometry analysis
After the incubation of co‐cultures with the appropriate drugs/conditions, the cells were detached with pre‐warmed papain/DNAase solution and layered over 1 ml of albumin‐inhibitor solution, centrifuged as above and resuspended in FACS buffer (PBS, 0.05% BSA, 50 mM HEPES and DNAse) containing CD73PE‐Cy7 BioLegend antibody in a dilution 1/300 μl (1,000,000 cells) on ice. Single stain controls and compensation controls were also used to set up the lasers of the cytometer (Fortessa BD). Gating strategy is provided in Fig EV2. The samples acquired with FACS Diva software and further analysed with FlowJo (LLC) program.
Live imaging of organelles and cytoskeleton
Cells were grown in 12 mm glass bottom dishes, and after 2 days in culture, the culture medium was replaced with staining solution, which comprised the culture medium and the diluted fluorescent probe, supplemented with the verapamil inhibitor. Live imaging of the dynamics of endosomes/lysosomes and the cytoskeleton of primary photoreceptor cultures performed with the use of SiR‐actin, SiR‐tubulin, SiR700‐lysosome, described in (Lukinavicius et al, 2013, 2014), and purchased from Cytoskeleton, Inc. SiR‐actin & SiR‐tubulin and SiR700‐lysosome probes were dissolved in anhydrous DMSO to make a 1 mM stock solution. This was further diluted in the cell medium (phenol red free) in the dilution of 1:10,000 (SiR‐actin & SiR‐tubulin) or 1:5,000 (SiR‐lysosome) supplemented with 10 μM verapamil. Serial dilutions were performed ranging from 1:1,000 to 1:10,000 according to manufacturer’s protocol. The selected dilutions of SiR‐probes could stain the cells without affecting their mobility/viability. Similarly, for mitochondria visualization Mitotracker‐Orange (Invitrogen/Molecular Probes, M7512) was diluted in 1 mM stock in DMSO and then further diluted in 0.01 mM working solution in culture medium from which a final dilution of 1:1,000 was used to stain the primary cells. Post‐labelling the cells were incubated for 3 h of incubation at 37°C at 5%CO2 prior live video recording with Leica TCS SP8 confocal microscope equipped with a temperature‐controlled chamber and CO2 supply. When acquiring live tissue, xyzt confocal stacks of representative fields of view were captured at resolution ranging from 1,024 × 1,024 to 2,046 × 2,046, step size 0.2–0.5 μm, bidirectional scanning, time‐lapse duration 60 min with 3 min interval using Leica SP8 inverted confocal laser scanning microscope, 63× (oil immersion, NA = 1.3) objectives and photomultiplier tubes to detect fluorescence emission. For SiR‐actin and SiR‐tubulin, the cells were imaged with standard Cy5 filter sets with optimal excitation 650 nm and emission 670 nm, but for SiR700‐lysosome cells were imaged with standard Cy7 filter sets. Excitation was 630–640 nm for SiR700 and SiR emission 650–700 nm (SiR channel) and 700–770 nm (SiR700 channel). For Mitotracker Orange, the 543‐nm line of a green He:Ne laser was used. Images were acquired with the Leica LASX software followed by post hoc analysis using ImageJ.
Post‐acquisition confocal image analysis
3D reconstruction was demonstrated as surface (organelles/cytoskeleton) per volume (cGFP) for culture live video recordings. For the visualization of NT‐like structures in culture and in vivo, Otsu watershed segmentation of appropriately thresholded cGFP surface was performed. 3D reconstructions and applied deconvolution post‐acquisition was performed with HyVolution software (Scientific Volume Imaging Huygens). xyz drift correction post‐acquisition from live video recordings was performed with object stabilizer with HyVolution software. Movies or live video recordings or 3D reconstruction rotations were generated as mp4 files via Movie maker plugin of HyVolution software and were further edited with Photoshop 2020 (version 21.2).
Object tracking analysis
Lysosomes and mitochondria movement was imaged as described above and was analysed with TrackMate plugin of ImageJ Software (Tinevez et al, 2017). Resolution 1,024 × 1,024, z step size 0.5 μm, bidirectional scanning, time‐lapse duration 60 min with 3‐min interval using Leica SP8 inverted confocal laser scanning microscope, 63× (oil immersion, NA = 1.3) objectives and photomultiplier tubes to detect fluorescence emission. Briefly, lif files of single cells or connected cell pairs were loaded as maximum z projections of 8 bit colour xyt images. Tracks analysed with DoG detector segmentation in organelle channel; for lysosomes blob diameter = 0.6 microns/threshold = 2.5, for mitochondria blob diameter=1 microns/threshold = 2.5, with enabled median filter and sub‐pixel localization. Initial threshold was further manually adjusted to eliminate artificial generated tracks. Hyperstack displayer view was selected, and appropriate filters on spots were adjusted appropriately where needed. Simple LAP tracker algorithm was selected, and linking max distance/ gap closing max distance was the length between the outer edge of the cell cytoplasm of both interconnected cells or the single cell length composed from the cell body and the processes (ex 15 microns) gap closing max frame=2. Track feature analyses: Branching: N spots, Gaps, Longest gap, Splits, Merges, Complex.; Track duration provides: Duration, T start, T stop, Displacement.; Track index provides: Index, ID.; Track location provides: X, Y, Z.; Velocity provides: Mean V, Max V, Min V, Median V, V std.; TRACK_SPOT_QUALITY provides: Mean Q, Max Q, Min Q, Median Q, Q std. Analysis was further assessed by the Track‐Scheme analyser and methodology/data/images of tracks exported as doc, xls and tiff files, respectively.
NT Depth localization analysis
The cells were imaged as above after 2–3 days in culture and temporal colour‐coding hyper‐stacks were generated with ImageJ to analyse their localization within the z dimension (Fig 3). 16 colour grade scale was selected and according to this scale, blue shades (7.7–10 μm) indicate free floating processes/cells, yellow to orange shades (2.6–7.7 μm) indicate depths in the middle of the dish, while purple to white colours (0–2.6 μm) indicate proximity to the dish surface. Neurites versus nanotube‐like processes were assessed according to the criteria laid out in Appendix Table S1, manually quantified and plotted as percentages normalized to cell numbers.
cGFP Fluorescent recovery after photobleaching (FRAP) of Nrl.Gfp Photoreceptor primary cultures
FRAP is the most commonly used bulk kinetics approach to study the dynamics of protein mobility. The assay was performed at 37°C and with 5% CO2 in PR culture medium and followed the general guidelines of FRAP experiment described by Koskinen and Hotulainen (Koskinen & Hotulainen, 2014). For the purposes of revealing the mobility of cGFP between two interconnecting cells, cell clusters or single cells, the cGFP was photobleached within the round area that surrounds the whole cell body cytoplasm (ROI). For the confirmation of nanotube‐connected cells, xyz imaging in 1,024 × 1,024 resolution as described above occurred prior photobleaching and nanotube processes vs neurites were classified according to the criteria of Appendix Table S1. The frame including the ROI (diameter ˜ 8 μm) was imaged five times before bleaching with 512 × 512, 2× digital zoom, 63× oil immersion inverted lens, resolution settings and frame average = 3 with bidirectional on, power of 700 Hz and pinhole = 2 Airy Units. Photobleaching was achieved with three scans (total bleach time 2.589 s) of the region of interest with 100% laser power at the sample (442/405 diode). Imaging of the area was resumed immediately after photo‐bleaching and continued every ti = 0.863 ms for a duration t = 215 s with normal scanning with approximately 10% power from image acquisition settings. The recording of the last frames was used based on FRAP in single cells to ensure a plateau is reached. All the post‐bleach values were double normalized hence, divided by the values from the non‐bleached area of the cells and normalized to the first pre‐bleach value. The first post‐bleach measurement was set to 0 s. The analysis of the FRAP recovery data was performed as described in the text using LAS AF (Leica Microsystems, Germany), ImageJ, Excel (Microsoft) software. Double normalized values were then fitted with double association exponential over a single exponential equation, as required, providing a set of two t 1/2 values (fast and slow) as shown below in GraphPad prism 8 software, and fitted curves are represented of an average of n ˜ 40 cells (Fig 4) per condition.
Primary photoreceptor culture SEM
Sample preparation was performed by Dr Matt Hayes, UCL Institute of Ophthalmology Imaging Facility. For this purpose, photoreceptors were cultured in glass coverslips or cultured in 12 mm, hydrophilic PTFE, 0.4 µm pore culture inserts for 3 days in culture, washed with HBSS wCa/Mg (Invitrogen) and then fixed overnight with in Karnovsky’s Glutaraldehyde solution (3% glutaraldehyde / 1% PFA (sigma, UK) buffered to pH 7.4 with 0.08 sodium cacodylate‐HCl). The samples then osmicated and dehydrated as described above and further dehydrated by rapid immersion in hexamethyldisilazane (Sigma‐Aldrich, USA) for 2 min and then were allowed to dry on a conductive carbon‐tab on an aluminium SEM stub. The sample was ‘back‐filled’ with silver paint to promote conductivity, allowed to dry for 2 h in a desiccator and platinum‐coated (1.5 nm) in a sputter‐coater (Cressington 108 auto, Cressington, UK). Samples were examined using a Zeiss Sigma SEM using both in‐lens and VP‐back scatter detectors (Zeiss, Germany). Imaging settings; InLens, aperture size 30 mm, WW 3.3 mm, 14.24 K X EHT = 3 kV.
EVs enrichment from primary cultures
EV enrichment from primary culture supernatants was based on the protocol described by Thery and colleagues (Thery et al, 2006), with the modifications proposed by Kowal and colleagues (Kowal et al, 2016) (see also (Thery et al, 2018)). To generate adequate numbers of 100 K small EVs for characterization and experimentation, a pool of three independent cultures was required >90*106 photoreceptor cells. Cells were cultured in absence of serum in a chemically defined medium (CM) that was collected every 48 h for 7 days from primary photoreceptor cultures. Briefly, the cell supernatant was centrifuged at 350 × g for 10 min at 4°C pellet was discarded, then at 2,000 × g for 20 min at 4°C (2 K pellet was retrieved from this step), and then 10,000 rpm for 60 min at 4°C (10 K pellet was retrieved from this step). Remaining supernatant was filtered with 0.22 µm PVDF filter (Millipore) and transfer into ultra‐centrifuge tubes (30 ml, Beckman), and spun at 4°C, for ˜ 3 h at 25,000 rpm on a SW 32Ti rotor 38 ml tubes. The supernatant was then discarded, and the tubes drained in tissue paper, the 100 K pellets retrieved from this step were then reconstituted in 100 μl PBS, allowed on ice for 1 h and vortexed thoroughly before aliquoted and stored at −80°C.
DLS Analysis of 100 K small EVs
Dynamic light scattering (DLS) was used to measure particle biophysical properties (size) as describe by Van Der Pol and colleagues (van der Pol et al, 2010). All measurements were taken with Zetasizer Nano ZS (Malvern, UK). Briefly, samples allowed to equilibrate from 4°C to 25°C. Particles dispersed in sterile PBS EM grade in dilutions varying from 1:2 to 1:100. Liquid refractive index and viscosity values for 25°C are: viscosity of 0.8882 cPoise, refractive index of 1.33 and dielectric constant of 79.0. The intensity changes are analysed with a digital autocorrelator, which generates a correlation function. This curve was analysed to give the size and the size distribution. Due to the polydispersity of EV samples, results are presented in intensity and volume per size plots for further clarity. This is because in some cases small intensity peaks may disappear in the transformation from intensity to volume or number of particles (according to manufacturer Malvern Panalytical). All data generated (PDI, size intensity, volume with Mie Theory) were saved in excel files and plots were generated with GraphPad Prism 8. If PDI was high, (example > 0.7 measurement was omitted).
TEM analysis of photoreceptor derived EVs
Transmission Electron Microscopy (TEM) was used to visualize the various EV pellets and to confirm their identity. Briefly, fresh samples of 2 K, 10 K and 100 K pellets were resuspended in fixation buffer containing 2% PFA, 5% Sucrose. EV samples were allowed to be absorbed by Formvar‐carbon coated EM grids 20 min RT (Agar scientific), washed with PBS and then fixed with 1% Glutaraldehyde 5 min RT and washed with ddH2O. Negative staining performed with 1% uranyl‐acetate solution (UA) (pH = 7) for 5 min, followed by methyl‐cellulose UA incubation for 10 min on ice and the excess fluid was gently absorbed with a Whatman filter paper. Grids allowed to air dry for 5 min and stored in appropriate grid storage boxes. Grids were analysed using a JEOL 1010 Transmission Electron Microscope (80 kV), fitted with a digital camera for image capture.
Protein quantification of EV pellets or cells
EV pellets or cell pellets were resuspended in RIPA buffer (10 mM Tris‐Cl (pH 8.0), 1 mM EDTA, 1% Triton X‐100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF, Invitrogen, USA) supplemented with 1% v/v protease inhibitor cocktail. Further disruption occurred with sonication 4 × 10 s on ice. Lysates were allowed to recover on a rocking plate for 30 min at 4°C. 5 µl of supernatant was used for protein estimation with a DC Assay (Bio‐Rad, USA), according to manufacturer’s instructions. Samples that were used for dot blot analysis were further diluted in SDS lysis buffer (see below) to achieve a concentration of 2.5 μg/dot, whereas samples that used for Western blot analysis were diluted to a final concentration of 30 μg/lane.
Western blotting analysis
For Western blotting, further denaturation of cell lysates occurred with the addition of 5% beta‐mercaptoethanol in in 1× Laemmli sample buffer (40% Glycerol, 240 mM Tris/HCl pH 6.8, 8% SDS, 0.04% bromophenol blue, Bio‐Rad) and incubation at 95°C for 5 min. Samples were loaded into an acrylamide gel gradient of 4–15% for SDS–PAGE analysis. Protein separation took place in a voltage of 180–220 V for 40 min – 1 h, followed by wet‐transfer in pre‐activated with methanol PVDF membrane (Millipore) at 100 mA for 3 h at 4°C. Membranes were subsequently blocked in blocking buffer (5% milk in Tris‐buffered saline (TBS) with 0.05% Tween 20) for 1 h at RT with agitation. Primary antibodies (RhoA, (67B9) Rabbit mAb #2117 Cell Signaling; Rac, ARC03‐S, Cytoskeleton Inc; beta actin; A2228, Sigma) were diluted in the blocking solution, supplemented with 0.01% NaN3 and incubated overnight at 4°C with agitation. Blots were washed with TBS with Tween‐20 (TBS‐T). Horseradish peroxidase (HRP)‐conjugated secondary antibodies (DAKO) were diluted in TBS‐T (1:5000). Blots were then incubated in the secondary solution at RT with agitation for 1 h. Protein bands were visualized using Immobilon™ Western Chemiluminescent HRP substrate kit (Millipore, USA). The resulting blot was imaged with a ChemiDoc TM MP Imaging System (Bio‐Rad, USA).
Filter trap assay (Dot Blot)
EV pellets and cell pellets were resuspended in SDS lysis buffer [10 mM Tris–HCl buffer, pH 8.0, containing 150 mM NaCl and 2% (w/v) SDS] supplemented with 1% (v/v) Protease inhibitor cocktail (Sigma). Cell disruption was completed by sonication. Undiluted samples were loaded onto a 0.45 µm PVDF membrane activated with 100% Met‐OH and pre‐equilibrated with SDS wash buffer [10 mM Tris–HCl buffer, pH 8.0, containing 150 mM NaCl and 0.1% (w/v) SDS]. After loading, 0.1% (w/v) SDS wash buffer was added to each well. Samples allowed to flow through the membrane by gravity for 20 min at 22°C. Thereafter, membranes were washed three times in SDS wash buffer and subjected to a vacuum (Lai et al, 2015). Membranes were blocked with 5% milk in TBS‐T‐0.05% or 5% BSA in TBS‐T‐0.05% for 1 h RT and then immunoblotted with primary antibodies listed below. Primary antibodies were diluted in the blocking solution and supplemented with 0.01% NaN3. Primary antibodies for the following epitopes were used; Beta actin (MAB8929, R&D Systems,1:5,000), CD63 (MAB5417, R&D Systems, 1:1,000), CD81 (10037, Cell Signalling, 1:1,000), TSG101 (612696, BD Transd, 1:1,000), CD9 (CBL162, EDM Millipore, 1:1,000), Alix (2171, Cell Signalling, 1:1,000), Cre recombinase (MAB3120, EDM Millipore, 1:1,000), GM130 (12480, Cell Signalling, 1:1,000), Rhodopsin (O4886, Sigma, 1:5,000), G‐alpha‐t1 (Sc389, Santa Cruz INC, 1:500), Recoverin (AB5585, MERCK, 1:5,000) and GFP/CFP/RFP (632380, Clontech, 1:2,000). Blots were washed with TBS with Tween‐20 (TBS‐T) × 3, 5 min each, at RT with agitation. Horseradish peroxidase (HRP)‐conjugated secondary antibodies (Sigma) were diluted in TBS‐T (1:5,000). Results visualized using Immobilon™ Western Chemiluminescent HRP substrate kit (Millipore, USA) following the manufacturer’s instructions. Blots were imaged with a ChemiDoc TM MP Imaging System (Bio‐Rad, USA).
Real‐time quantitative PCR in small EVs
To assess the potential presence of RNA within EVs, 100 K EV pellets were treated as described by Valadi and colleagues (Valadi et al, 2007) with 0.4 μg/μl RNase (Sigma) for 10 min at 37°C in reactions of 50 µl in PBS. After incubation, 1 U/µl RNase Inhibitor (Super RNAse Out 20 U/µl, Thermo Fisher) was added to the mixture and incubated at RT for 2 min. Mixtures were additionally treated with 0.25% trypsin for 10 min at 37°C. After incubation, samples were diluted in 3 ml sterile EM grade PBS and ultra‐centrifuged for 2 h at 100 K (55 K A100, Beckman) at 10°C. Supernatant was discarded, and samples resuspended in 200 µl PBS to further purify RNA and left for 30 min at RT with discontinuous vortexing of 10 s. As a control, 5 μg cellular RNA was added to the control EVs before the RNase treatment. RNA extraction was performed with Qiagen RNeasy® Mini kit according to manufacturer’s instructions. Whole transcriptome Reverse transcription PCR performed with QuantiTect® Whole Transcriptome (Qiagen) kit according to manufacturer’s instructions. For the RTQ‐PCR analysis, cDNA 50 ng/µl were diluted 1:16 in TaqMan® Universal PCR Master Mix (Roche, UK), 2 µM forward primer, 2 µM reverse primer and 0.2 µM probe (Universal Probe Library; Roche, Germany). Primers for the target markers and endogenous reference control (β‐actin) were designed, and appropriate hydrolysis probes were chosen via the Universal Probe Library Design Centre (Roche, UK). RTQ‐PCR was run on an ABI Prism 7900HT Fast Real‐time Sequence Detection System (Applied Bioscience, USA). All samples were run in triplicates. The general conditions used had an initial denaturation at 94°C of 2 min. Reactions followed by 40 cycles of: Denaturing step at 95°C for 30 s, Annealing step at 42°C for 30 s, Extension step at 72°C for 1 min, and final extension of 5 min at 72°C was performed to finish the reaction. The relative expression between comparable samples in relation to the expression of the genes was normalized using the following methodology: signal thresholds were first manually determined and maintained for each individual experimental run on Sequence Detection Systems software 2.2.2 (Applied Biosystems, USA). To normalize target against β‐actin control expression levels, cycle numbers at threshold (Ct) were used to calculate ΔCt. With the formula; ΔCt = Ct (target) – Ct (β‐actin). To normalize sample (e.g. treated) against control levels of target expression (e.g. nontreated), ΔΔCt was calculated by the formula ΔΔCt = ΔCt (sample) – ΔCt (control). Finally, to obtain normalized relative target expression levels in ‘% of control levels’, the following formula was applied Normalized expression levels (%) = (2‐ΔΔCt) × 100. The following primer/probe combinations were used; Gfp: GAAGCGCGATGACATGGT/CCATGCCGAGAGTGATCC/ probe 67, Cre: ATCTGGCATTTCTGGGGATTG/ GCAACACCATTTTTTCTGACCC/ probe 20, Gnat1: AGAGCTGGAGAAGAAGCTGAAA/ TAGTGCTCTTCCCGGATTCA/ probe 89, Rho: ACCTGGATCATGGCGTT/ TGCCCTCAGGGATGTACC/probe 32, Rcvrn: CAATGGGACCATCAGCAAA/ CCTCAGGCTTGATCATTTTGA/ probe 67, Crx: CCCCAATGTGGACCTGAT/GGCTCCTGGTGAATGTGGT/ probe 64, Nrl; TTCTGGTTCTGACAGTGACTACG/ TGGGACTGAGCAGAGAGAGG/ probe 53, Actb:AAGGCCAACCGTGAAAAGAT/GTGGTACGACCAGAGGCATAC/probe 56.
AAV production
Nrl promoter‐driven expression constructs
Cre was subcloned into a pD10 expression and AAV packaging‐compatible construct downstream of either the CMV promoter or the NRL promoter region. A 2.5 kb segment upstream of the Nrl gene was cloned from mouse genome and used as the NRL promoter in this study, as per (Akimoto et al, 2006). Nrl.Cre and CMV.Cre plasmid constructs were used to produce AAVShH10‐Y445F (herein termed AAV‐Nrl.Cre and AAV‐CMV.Cre). Recombinant AAVShH10‐Y445F vectors were produced following the tripartite transfection method, which required the transfection of three plasmids (pD10 vector containing transgene with packaging signal, AAV capsid plasmid ShH10 and pDHelper) in HEK293T cells as previously described(Gonzalez‐Cordero et al, 2018). Briefly, HEK293T cells were grown in D10 cell culture medium (DMEM supplemented with 10% foetal calf serum (Gibco, USA) and antibiotics (Gibco, USA)) at 37°C / 5% CO2 until the cells reached 70% confluency. The transfection mix was prepared with the appropriate plasmids and polyethylenimine 2 mg/ml (PEI) transfection reagent. The cell factory was then left to incubate at 37°C / 5% CO2 for three days. Three days post‐transfection, the cells were harvested and centrifuged for 20 min at 3,500 × g. Cell pellets were then resuspended in TD buffer (140 mM NaCl / 5 mM KCl / 0.7 mM K2HPO4 / 3.5 mM MgCl2 / 25 mM Tris base in ddH2O, pH 7.5) and lysed by 4 freeze/thaw/vortex cycles to release the viral particles, followed by enzymatic lysis using 50 U/ml benzonase (Sigma, USA) for 30 min at 37°C. Subsequently, the solution was then centrifuged at 18,000 × g for 30 min. The supernatant was then filtered through 0.45‐μm syringe filters to remove cell debris and purified using affinity chromatography (AVB Sepharose column, GE Healthcare, USA). The final virus preparation was resuspended in PBS‐MK buffer (0.1 M phosphate‐buffered saline (PBS) / 2.5 mM KCl / 1 mM MgCl2) and concentrated with Vivaspin 6 columns to a final volume of 200–250 μl. The virus was then aliquoted and stored at −80°C. qPCR analysis indicated that the virus titre as viral particles/ml.
Lentivirus production
pLenti plasmids; pLenti‐CAG‐GFP‐P2A, pLenti‐CAG‐RhoA‐GFP‐P2A and pLenti‐CAG‐ΔNRac‐GFP‐P2A were a kind gift generated by Dr Ortín‐Martínez Arturo and Professor Valerie Wallace, University of Toronto. Lentivirus production performed by Dr Mark Basche and followed the tripartite transfection method in HEK293T cells similar to AAV production, which required the transfection of three plasmids (pLenti‐containing transgene with packaging signal, envelope plasmid pMD2.G, helper plasmid pCMVΔR8.74) diluted in OptiMEM. In this case, 80 µg DNA per 15 cm dish in DMEM+PEI were used for transfection in a total volume of ˜ 2.7 ml for 5 h. Cell medium was then replaced with D10, and the cells were incubated at 37°C, 5% CO2 for 48 h. Harvest of the cell supernatant containing the lentiviral particles took place 48 and 72 h post‐transfection. Simply the cell supernatant was then sequentially centrifuged at 350 × g for 10 min at 4°C, then at 2,000 × g for 20 min at 4°C and then 10,000 rpm for 60 min at 4°C to remove course cell debris, then filtered with 0.22 µm PVDF filter (Millipore) and transfer into ultra‐centrifuge tubes (30 ml, Beckman), and spun at 4°C, for ˜ 3 h at 23,000 rpm on a SW 32Ti rotor 38 ml tubes. The supernatant was then discarded, and the tubes drained in tissue paper, the pellets were then reconstituted in 100 μl OptiMEM, allowed on ice for 1 h and vortexed thoroughly before aliquoted and stored at −80°C. Quantification performed with infecting HEK293T in serial dilutions of virus and assessed via cell counting based on GFP expression levels.
Statistical analysis
All the data analysis is shown as means ± standard deviation (SD) of the mean, unless otherwise indicated. Generation of plots, curve fitting and statistical analyses were performed using GraphPad Prism 8 for Windows Version 10 (GraphPad Software Inc). Statistical significance was assessed using non‐parametric one‐way analysis of variance (ANOVA) with Dunnett's multiple comparison post hoc test (compared all groups against control group) or Bonferroni multiple comparisons post hoc test (compared all groups or selected groups) or as stated in figure legends. N and n numbers as stated below, unless indicated; cell cultures; N = number of experiments derived from an independent litter dissociation and/or isolation and consecutive cell culture, n = number of wells, chambers, coverslips used. Processes counts; n = number of processes counted from a pool of individual isolations/cultures (3 individual experiments or more, unless stated). Cell counts; n = number of cell’s somata/nuclei counted from a pool of individual isolations/cultures (three individual experiments or more, unless stated). Animal work: N = number of cohorts of independent injections/treatments, n = number of eyes of all cohorts pooled. Flow cytometry: N = number of experiments derived from an independent litter dissociation/ isolation and consecutive cell culture, n = number of wells used per condition. P‐values; P < 0.05 = statistically significant*, P < 0.01 = very significant**, P < 0.001 = highly significant***
Post hoc image processing
Figure panels were generated with Photoshop software (version 22.3.1). For better visualization, some automatically generated scale bars were covered and redrawn. Image stitching after tile scans acquired using 40× magnification were performed automatically in LasX software and ImageJ.
Author contributions
Conceptualization: AAK, RRA, RAP; Methodology: AAK, AJS, RRA, RAP; Formal analysis: AKK, RAP; Investigation: AAK, MB, AH, ELW; Resources: RRA, RAP; Writing—original draft: AAK, RAP; Writing—review & editing: AAK, AJS, RRA, RAP; Funding acquisition: RRA, RAP; Supervision: RAP.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Acknowledgements
We thank Professor V. Wallace and colleagues for the kind provision of lentiviral RhoA and DN‐Rac1 expression constructs. We thank O. Semenyuk, IoO vector production facility, for vector production, Drs M. Branch and R. Sampson, IoO FACS facility, Dr M. Hayes and D. Sefic Svara, UCL IoO Microscopy Unit, for technical support with FACS, imaging and electron microscopy, Professor F. Cordeiro for access to Dynamic Light Scatter and Dr A. Matsuyama for assistance with culture maintenance and Dr N. D. Aghaizu for cloning. We would like to thank all members of the Pearson and Ali groups for additional assistance and for constructive criticism and discussion of earlier drafts of the manuscript. This work was supported by Fight for Sight UK (1566/1567), Moorfields Eye Charity (E170004A; R180005A); Medical Research Council UK (MR/J004553/1 and MR/T002735/1); AKK was a Frankenburg Fight for Sight UK PhD Student. RAP was additionally supported by an unrestricted Alcon Research Institute Young Investigator Award.
EMBO reports (2021) 22: e53732.
Correction added on 20 November 2021, after first online publication: The copyright line was changed.
Contributor Information
Aikaterini A Kalargyrou, Email: aikaterini.kalargyrou@kcl.ac.uk.
Rachael A Pearson, Email: rachael.pearson@kcl.ac.uk.
Data availability
This study includes no data deposited in external repositories.
References
- Agnati LF, Fuxe K (2014) Extracellular‐vesicle type of volume transmission and tunnelling‐nanotube type of wiring transmission add a new dimension to brain neuro‐glial networks. Philos Trans R Soc Lond B Biol Sci 369: 20130505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh ECT, Jing Y, Linares J‐L, Brooks M et al (2006) Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow‐sorted photoreceptors. Proc Natl Acad Sci USA 103: 3890–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon‐Martinez L, Villafranca‐Baughman D, Quintero H, Kacerovsky JB, Dotigny F, Murai KK, Prat A, Drapeau P, Di Polo A (2020) Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 585: 91–95 [DOI] [PubMed] [Google Scholar]
- Ariazi J, Benowitz A, De Biasi V, Den Boer ML, Cherqui S, Cui H, Douillet N, Eugenin EA, Favre D, Goodman S et al (2017) Tunneling nanotubes and gap junctions‐their role in long‐range intercellular communication during development, health, and disease conditions. Front Mol Neurosci 10: 333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkwright PD, Luchetti F, Tour J, Roberts C, Ayub R, Morales AP, Rodríguez JJ, Gilmore A, Canonico B, Papa S et al (2010) Fas stimulation of T lymphocytes promotes rapid intercellular exchange of death signals via membrane nanotubes. Cell Res 20: 72–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley J, Cordy B, Lucia D, Fradkin LG, Budnik V, Thomson T (2018) Retrovirus‐like gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172: 262–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrini I, Song JH, Diez D, Hanayama R (2015) Neuronal exosomes facilitate synaptic pruning by up‐regulating complement factors in microglia. Sci Rep 5: 7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M (2017) How the Internet of cells has biologists buzzing. Nature 549: 322–324 [DOI] [PubMed] [Google Scholar]
- Barber AC, Hippert C, Duran Y, West EL, Bainbridge JWB, Warre‐Cornish K, Luhmann UFO, Lakowski J, Sowden JC, Ali RR et al (2013) Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci USA 110: 354–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M, Gradilla AC, Seijo I, Andres G, Rodriguez‐Navas C, Gonzalez‐Mendez L, Guerrero I (2013) Cytonemes are required for the establishment of a normal Hedgehog morphogen gradient in Drosophila epithelia. Nat Cell Biol 15: 1269–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman DS, Razafsky D, Potter C, Hodzic D, Chen S (2016) Nrl‐Cre transgenic mouse mediates loxP recombination in developing rod photoreceptors. Genesis 54: 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukoreshtliev NV, Wang X, Hodneland E, Gurke S, Barroso JF, Gerdes HH (2009) Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett 583: 1481–1488 [DOI] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicolo M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL et al (2000) Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha ‐subunit. Proc Natl Acad Sci USA 97: 13913–13918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Huang H, Hatori R, Kornberg TB (2017) Essential basal cytonemes take up Hedgehog in the Drosophila wing imaginal disc. Development 144: 3134–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery HR, Pearlman E, McMenamin PG (2008) Cutting edge: Membrane nanotubes in vivo: a feature of MHC class II+ cells in the mouse cornea. J Immunol 180: 5779–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivet M, Javalet C, Hemming F, Pernet‐Gallay K, Laulagnier K, Fraboulet S, Sadoul R (2013) Exosomes as a novel way of interneuronal communication. Biochem Soc Trans 41: 241–244 [DOI] [PubMed] [Google Scholar]
- Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R (2014) Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles 3: 24722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero Cervantes D, Zurzolo C (2021) Peering into tunneling nanotubes‐the path forward. EMBO J 40: e105789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decembrini S, Martin C, Sennlaub F, Chemtob S, Biel M, Samardzija M, Moulin A, Behar‐Cohen F, Arsenijevic Y (2017) Cone genesis tracing by the Chrnb4‐EGFP mouse line: evidences of cellular material fusion after cone precursor transplantation. Mol Ther 25: 634–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domhan S, Ma L, Tai A, Anaya Z, Beheshti A, Zeier M, Hlatky L, Abdollahi A (2011) Intercellular communication by exchange of cytoplasmic material via tunneling nano‐tube like structures in primary human renal epithelial cells. PLoS One 6: e21283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle D, Schubert S, Postel K, Corbeil D, Ader M (2011) Increased integration of transplanted CD73‐positive photoreceptor precursors into adult mouse retina. Invest Ophthalmol Vis Sci 52: 6462–6471 [DOI] [PubMed] [Google Scholar]
- Fauré J, Lachenal G, Court M, Hirrlinger J, Chatellard‐Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V et al (2006) Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 31: 642–648 [DOI] [PubMed] [Google Scholar]
- Frühbeis C, Fröhlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, Kirchhoff F, Möbius W, Goebbels S, Nave K‐A et al (2013) Neurotransmitter‐triggered transfer of exosomes mediates oligodendrocyte‐neuron communication. PLoS Biol 11: e1001604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini SJ, Llonch S, Borsch O, Ader M (2019) Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog Retin Eye Res 69: 1–37 [DOI] [PubMed] [Google Scholar]
- George L, Dunkel H, Hunnicutt BJ, Filla M, Little C, Lansford R, Lefcort F (2016) In vivo time‐lapse imaging reveals extensive neural crest and endothelial cell interactions during neural crest migration and formation of the dorsal root and sympathetic ganglia. Dev Biol 413: 70–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes HH, Carvalho RN (2008) Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol 20: 470–475 [DOI] [PubMed] [Google Scholar]
- Gerdes HH, Rustom A, Wang X (2013) Tunneling nanotubes, an emerging intercellular communication route in development. Mech Dev 130: 381–387 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Cordero A, Goh D, Kruczek K, Naeem A, Fernando M, Kleine Holthaus S‐M, Takaaki M, Blackford SJI, Kloc M, Agundez L et al (2018) Assessment of AAV vector tropisms for mouse and human pluripotent stem cell‐derived RPE and photoreceptor cells. Hum Gene Ther 29: 1124–1139 [DOI] [PubMed] [Google Scholar]
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J et al (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11: 328–336 [DOI] [PubMed] [Google Scholar]
- Gradilla A‐C, González E, Seijo I, Andrés G, Bischoff M, González‐Mendez L, Sánchez V, Callejo A, Ibáñez C, Guerra M et al (2014) Exosomes as Hedgehog carriers in cytoneme‐mediated transport and secretion. Nat Commun 5: 5649 [DOI] [PubMed] [Google Scholar]
- Gurke S, Barroso JF, Hodneland E, Bukoreshtliev NV, Schlicker O, Gerdes HH (2008) Tunneling nanotube (TNT)‐like structures facilitate a constitutive, actomyosin‐dependent exchange of endocytic organelles between normal rat kidney cells. Exp Cell Res 314: 3669–3683 [DOI] [PubMed] [Google Scholar]
- Gustafson T, Wolpert L (1967) Cellular movement and contact in sea urchin morphogenesis. Biol Rev Camb Philos Soc 42: 442–498 [DOI] [PubMed] [Google Scholar]
- Haimovich G, Ecker CM, Dunagin MC, Eggan E, Raj A, Gerst JE, Singer RH (2017) Intercellular mRNA trafficking via membrane nanotube‐like extensions in mammalian cells. Proc Natl Acad Sci USA 114: E9873–E9882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovich G, Gerst JE (2019) Detection of mRNA transfer between mammalian cells in coculture by single‐molecule fluorescent in situ hybridization (smFISH). Methods Mol Biol 2038: 109–129 [DOI] [PubMed] [Google Scholar]
- Hanna SJ, McCoy‐Simandle K, Miskolci V, Guo P, Cammer M, Hodgson L, Cox D (2017) The role of Rho‐GTPases and actin polymerization during macrophage tunneling nanotube biogenesis. Sci Rep 7: 8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase K, Kimura S, Takatsu H, Ohmae M, Kawano S, Kitamura H, Ito M, Watarai H, Hazelett CC, Yeaman C et al (2009) M‐Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat Cell Biol 11: 1427–1432 [DOI] [PubMed] [Google Scholar]
- Hill AF (2019) Extracellular vesicles and neurodegenerative diseases. J Neurosci 39: 9269–9273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW (1986) Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci 6: 3044–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez F, Montesinos J, Urena‐Peralta JR, Guerri C, Pascual M (2019) TLR4 participates in the transmission of ethanol‐induced neuroinflammation via astrocyte‐derived extracellular vesicles. J Neuroinflammation 16: 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu QI, Evans R et al (2019) Reassessment of exosome composition. Cell 177: 428–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenkova O, Pepe A, Zurzolo C (2020) Fine intercellular connections in development: TNTs, cytonemes, or intercellular bridges? Cell Stress 4: 30–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen M, Hotulainen P (2014) Measuring F‐actin properties in dendritic spines. Front Neuroanat 8: 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal‐Bengtson B, Dingli F, Loew D, Tkach M, Thery C (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 113: E968–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer‐Albers EM, Bretz N, Tenzer S, Winterstein C, Mobius W, Berger H, Nave KA, Schild H, Trotter J (2007) Oligodendrocytes secrete exosomes containing major myelin and stress‐protective proteins: trophic support for axons? Proteomics Clin Appl 1: 1446–1461 [DOI] [PubMed] [Google Scholar]
- Kulesa PM, Bailey CM, Cooper C, Fraser SE (2010) In ovo live imaging of avian embryos. Cold Spring Harb Protoc 2010: pdb prot5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, Breakefield XO (2015) Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun 6: 7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski J, Han YT, Pearson RA, Gonzalez‐Cordero A, West EL, Gualdoni S, Barber AC, Hubank M, Ali RR, Sowden JC (2011) Effective transplantation of photoreceptor precursor cells selected via cell surface antigen expression. Stem Cells 29: 1391–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski J, Gonzalez‐Cordero A, West EL, Han Y‐T, Welby E, Naeem A, Blackford SJI, Bainbridge JWB, Pearson RA, Ali RR et al (2015) Transplantation of photoreceptor precursors isolated via a cell surface biomarker panel from embryonic stem cell‐derived self‐forming retina. Stem Cells 33: 2469–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba DA, Gust J, Reh TA (2009) Transplantation of human embryonic stem cell‐derived photoreceptors restores some visual function in Crx‐deficient mice. Cell Stem Cell 4: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubojevic N, Henderson JM, Zurzolo C (2021) The ways of actin: why tunneling nanotubes are unique cell protrusions. Trends Cell Biol 31: 130–142 [DOI] [PubMed] [Google Scholar]
- Lukinavičius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Corrêa Jr IR, Luo Z‐G et al (2013) A near‐infrared fluorophore for live‐cell super‐resolution microscopy of cellular proteins. Nat Chem 5: 132–139 [DOI] [PubMed] [Google Scholar]
- Lukinavičius G, Reymond L, D'Este E, Masharina A, Göttfert F, Ta H, Güther A, Fournier M, Rizzo S, Waldmann H et al (2014) Fluorogenic probes for live‐cell imaging of the cytoskeleton. Nat Methods 11: 731–733 [DOI] [PubMed] [Google Scholar]
- MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC, Ali RR (2006) Retinal repair by transplantation of photoreceptor precursors. Nature 444: 203–207 [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al (2010) A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahato B, Kaya KD, Fan Y, Sumien N, Shetty RA, Zhang W, Davis D, Mock T, Batabyal S, Ni A et al (2020) Pharmacologic fibroblast reprogramming into photoreceptors restores vision. Nature 581: 83–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney MC, Kulesa PM (2011) In vivo calcium dynamics during neural crest cell migration and patterning using GCaMP3. Dev Biol 358: 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehidi A, Rossier O, Schaks M, Chazeau A, Binamé F, Remorino A, Coppey M, Karatas Z, Sibarita J‐B, Rottner K et al (2019) Transient activations of Rac1 at the lamellipodium tip trigger membrane protrusion. Curr Biol 29: 2852–2866 [DOI] [PubMed] [Google Scholar]
- Murillo OD, Thistlethwaite W, Rozowsky J, Subramanian SL, Lucero R, Shah N, Jackson AR, Srinivasan S, Chung A, Laurent CD et al (2019) exRNA atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. Cell 177: 463–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double‐fluorescent Cre reporter mouse. Genesis 45: 593–605 [DOI] [PubMed] [Google Scholar]
- Nickerson PEB, Ortin‐Martinez A, Wallace VA (2018) Material exchange in photoreceptor transplantation: updating our understanding of donor/host communication and the future of cell engraftment science. Front Neural Circuits 12: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D'Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19: 213–228 [DOI] [PubMed] [Google Scholar]
- Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM, Davis DM (2006) Structurally distinct membrane nanotubes between human macrophages support long‐distance vesicular traffic or surfing of bacteria. J Immunol 177: 8476–8483 [DOI] [PubMed] [Google Scholar]
- Ortin‐Martinez A, Tsai EL, Nickerson PE, Bergeret M, Lu Y, Smiley S, Comanita L, Wallace VA (2017) A Reinterpretation of cell transplantation: GFP transfer from donor to host photoreceptors. Stem Cells 35: 932–939 [DOI] [PubMed] [Google Scholar]
- Ortin‐Martinez A, Yan NE, Tsai ELS, Comanita L, Gurdita A, Tachibana N, Liu ZC, Lu S, Dolati P, Pokrajac NT (2021) Photoreceptor nanotubes mediate the in vivo exchange of intracellular material. EMBO J e07264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteikoetxea‐Molnár A, Szabó‐Meleg E, Tóth EA, Oszvald Á, Izsépi E, Kremlitzka M, Biri B, Nyitray L, Bozó T, Németh P et al (2016) The growth determinants and transport properties of tunneling nanotube networks between B lymphocytes. Cell Mol Life Sci 73: 4531–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuzyn ED, Day CE, Kearns RB, Kyrke‐Smith M, Taibi AV, McCormick J, Yoder N, Belnap DM, Erlendsson S, Morado DR et al (2018) The neuronal gene arc encodes a repurposed retrotransposon Gag protein that mediates intercellular RNA transfer. Cell 172: 275–288.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RA, Barber AC, Rizzi M, Hippert C, Xue T, West EL, Duran Y, Smith AJ, Chuang JZ, Azam SA et al (2012) Restoration of vision after transplantation of photoreceptors. Nature 485: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RA, Gonzalez‐Cordero A, West EL, Ribeiro JR, Aghaizu N, Goh D, Sampson RD, Georgiadis A, Waldron PV, Duran Y et al (2016) Donor and host photoreceptors engage in material transfer following transplantation of post‐mitotic photoreceptor precursors. Nat Commun 7: 13029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Baulier E, Ke Y, Young A, Ahmedli NB, Schwartz SD, Farber DB (2018) Human embryonic stem cells extracellular vesicles and their effects on immortalized human retinal Muller cells. PLoS One 13: e0194004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta B, Gil‐Carton D, Castano‐Diez D, Bertin A, Boulogne C, Oksanen HM, Bamford DH, Abrescia NG (2013) Mechanism of membranous tunnelling nanotube formation in viral genome delivery. PLoS Biol 11: e1001667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pol E, Hoekstra AG, Sturk A, Otto C, van Leeuwen TG, Nieuwland R (2010) Optical and non‐optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost 8: 2596–2607 [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265: 23–32 [DOI] [PubMed] [Google Scholar]
- Rajendran L, Bali J, Barr MM, Court FA, Kramer‐Albers E‐M, Picou F, Raposo G, van der Vos KE, van Niel G, Wang J et al (2014) Emerging roles of extracellular vesicles in the nervous system. J Neurosci 34: 15482–15489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez‐Weber FA, Kornberg TB (1999) Cytonemes: cellular processes that project to the principal signaling center in Drosophila imaginal discs. Cell 97: 599–607 [DOI] [PubMed] [Google Scholar]
- Ribeiro J, Procyk CA, West EL, O’Hara‐Wright M, Martins MF, Khorasani MM, Hare A, Basche M, Fernando M, Goh D et al (2021) Restoration of visual function in advanced disease after transplantation of purified human pluripotent stem cell‐derived cone photoreceptors. Cell Rep 35: 109022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca CJ, Kreymerman A, Ur SN, Frizzi KE, Naphade S, Lau A, Tran T, Calcutt NA, Goldberg JL, Cherqui S (2015) Treatment of inherited eye defects by systemic hematopoietic stem cell transplantation. Invest Ophthalmol Vis Sci 56: 7214–7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Hsiung F, Kornberg TB (2011) Specificity of Drosophila cytonemes for distinct signaling pathways. Science 332: 354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH (2004) Nanotubular highways for intercellular organelle transport. Science 303: 1007–1010 [DOI] [PubMed] [Google Scholar]
- Rustom A (2016) The missing link: does tunnelling nanotube‐based supercellularity provide a new understanding of chronic and lifestyle diseases? Open Biol 6: 160057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Ferreira T, Llonch S, Borsch O, Postel K, Haas J, Ader M (2016) Retinal transplantation of photoreceptors results in donor‐host cytoplasmic exchange. Nat Commun 7: 13028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori‐Rupp A, Cordero Cervantes D, Pepe A, Gousset K, Delage E, Corroyer‐Dulmont S, Schmitt C, Krijnse‐Locker J, Zurzolo C (2019) Correlative cryo‐electron microscopy reveals the structure of TNTs in neuronal cells. Nat Commun 10: 342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MS, Balmer J, Barnard AR, Aslam SA, Moralli D, Green CM, Barnea‐Cramer A, Duncan I, MacLaren RE (2016) Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat Commun 7: 13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctôt C, Kösem S, Peichl L, Cremer T, Guck J, Joffe B (2009) Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137: 356–368 [DOI] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 103: 1283–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanganello E, Hagemann AI, Mattes B, Sinner C, Meyen D, Weber S, Schug A, Raz E, Scholpp S (2015) Filopodia‐based Wnt transport during vertebrate tissue patterning. Nat Commun 6: 5846 [DOI] [PubMed] [Google Scholar]
- Sun MK, Passaro AP, Latchoumane CF, Spellicy SE, Bowler M, Goeden M, Martin WJ, Holmes PV, Stice SL, Karumbaiah L (2020) Extracellular vesicles mediate neuroprotection and functional recovery after traumatic brain injury. J Neurotrauma 37: 1358–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang Y, Zhang J, Tu J, Wang XJ, Su XD, Wang L, Zhang Y (2012) Tunneling‐nanotube direction determination in neurons and astrocytes. Cell Death Dis 3: e438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardivel M, Bégard S, Bousset L, Dujardin S, Coens A, Melki R, Buée L, Colin M (2016) Tunneling nanotube (TNT)‐mediated neuron‐to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 10.1186/s40478-016-0386-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teddy JM, Kulesa PM (2004) In vivo evidence for short‐ and long‐range cell communication in cranial neural crest cells. Development 131: 6141–6151 [DOI] [PubMed] [Google Scholar]
- Thayanithy V, O'Hare P, Wong P, Zhao X, Steer CJ, Subramanian S, Lou E (2017) A transwell assay that excludes exosomes for assessment of tunneling nanotube‐mediated intercellular communication. Cell Commun Signal 15: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3 22 [DOI] [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin‐Smith GK et al (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7: 1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single‐particle tracking. Methods 115: 80–90 [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO (2007) Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9: 654–659 [DOI] [PubMed] [Google Scholar]
- Vargas JY, Loria F, Wu YJ, Cordova G, Nonaka T, Bellow S, Syan S, Hasegawa M, van Woerden GM, Trollet C et al (2019) The Wnt/Ca(2+) pathway is involved in interneuronal communication mediated by tunneling nanotubes. EMBO J 38: e101230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij FJ, Bebelman MP, Jimenez CR, Garcia‐Vallejo JJ, Janssen H, Neefjes J, Knol JC, de Goeij‐de Haas R, Piersma SR, Baglio SR et al (2018) Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J Cell Biol 217: 1129–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij FJ, Revenu C, Arras G, Dingli F, Loew D, Pegtel DM, Follain G, Allio G, Goetz JG, Zimmermann P et al (2019) Live tracking of inter‐organ communication by endogenous exosomes in vivo. Dev Cell 48: 573–589.e4 [DOI] [PubMed] [Google Scholar]
- Waldron PV, Di Marco F, Kruczek K, Ribeiro J, Graca AB, Hippert C, Aghaizu ND, Kalargyrou AA, Barber AC, Grimaldi G et al (2018) Transplanted donor‐ or stem cell‐derived cone photoreceptors can both integrate and undergo material transfer in an environment‐dependent manner. Stem Cell Reports 10: 406–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cui J, Sun X, Zhang Y (2011) Tunneling‐nanotube development in astrocytes depends on p53 activation. Cell Death Differ 18: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012a) RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn 14: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bukoreshtliev NV, Gerdes H‐H (2012b) Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS One 7: e47429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22: 1181–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warre‐Cornish K, Barber AC, Sowden JC, Ali RR, Pearson RA (2014) Migration, integration and maturation of photoreceptor precursors following transplantation in the mouse retina. Stem Cells Dev 23: 941–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SC, Salter RD (2005) Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 23: 309–318 [DOI] [PubMed] [Google Scholar]
- Zhou J, Benito‐Martin A, Mighty J, Chang L, Ghoroghi S, Wu H, Wong M, Guariglia S, Baranov P, Young M et al (2018) Retinal progenitor cells release extracellular vesicles containing developmental transcription factors, microRNA and membrane proteins. Sci Rep 8: 2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Cifuentes H, Reynolds J, Lamba DA (2017) Immunosuppression via loss of IL2rγ enhances long‐term functional integration of hESC‐derived photoreceptors in the mouse retina. Cell Stem Cell 20: 374–384.e5 [DOI] [PubMed] [Google Scholar]
- Zomer A, Steenbeek SC, Maynard C, van Rheenen J (2016) Studying extracellular vesicle transfer by a Cre‐loxP method. Nat Protoc 11: 87–101 [DOI] [PubMed] [Google Scholar]