

Abstract
Immunomodulatory therapeutics represent a unique class of drug products that have tremendous potential to rebalance malfunctioning immune systems and are quickly becoming one of the fastest growing areas in the pharmaceutical industry. For these drugs to become mainstream medicines, they must provide more therapeutic benefit than the currently used treatments without causing severe toxicities. Immunomodulators, cell-based therapies, antibodies, and viral therapies have all achieved varying amounts of success in the treatment of cancers and/or autoimmune diseases. However, many challenges related to precision dosing, off-target effects, and manufacturing hurdles will need to be addressed before we see widespread adoption of these therapies in the clinic. This review provides a perspective on the progress of immunostimulatory and immunosuppressive therapies to date and discusses the opportunities and challenges for clinical translation of the next generation of immunomodulatory therapeutics.
Keywords: Immunomodulation, immunotherapy, regulatory considerations, drug delivery, cell therapy, antibodies, cytokines
Graphical abstract
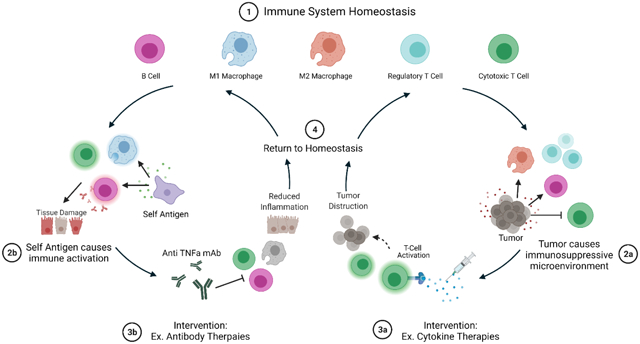
Schematic representation of immune system cycles. State 1 represents immune system homeostasis where the balance between activation and proliferation of cytotoxic cells and regulatory cells is maintained naturally and does not require intervention. State 2a represents local tumor development causing an immunosuppressive microenvironment in which cytotoxic cells such as CD8+ T cells, are repressed. Immunosuppressive microenvironments can also cause an increase in development and proliferation of M2 macrophages and regulatory T cells which contribute to the immunosuppression by secreting anti-inflammatory molecules. State 3a represents a potential therapeutic intervention: cytokine therapies. In this example, pro-inflammatory cytokines such as interleukin-2 can be administered to boost cytotoxic T cell activation and proliferation which can lead to increased anti-tumor effects. As the tumor cells are destroyed, the extent of the immunosuppressive microenvironment is reduced, and the immune system returns to homeostasis as shown in State 4. On the other hand, State 2b represents misidentification of self-antigens as foreign which is common during autoimmune diseases. In this state, antigen-presenting cells such as macrophages mark self-antigens for destruction by the immune system which leads to activation and proliferation of M1 macrophages, cytotoxic T cells, and B cells causing prolonged immune system activation and tissue damage. State 3b represents a potential therapeutic intervention: antibody therapies. In this example, antibodies against major pro-inflammatory cytokine TNFα are administered. Blockade of pro-inflammatory cytokine signaling reduces immune system activation by decreasing the number binding of these signaling molecules to additional immune cells and thus reducing the inflammation and allowing the immune system to return to homeostasis (State 4). Schematic was made using BioRender.com
1. Introduction
In the past decade, there have been major advances in synthetic biology, protein engineering, and clinical oncology leading to safer and more efficacious immunomodulatory therapeutics. Researchers have continued to think outside of the box to develop new drugs and delivery systems that learn from and build on previous generations. These developments include new clinical targets as well as repurposing previously approved drugs to extending the short serum half-life of unstable proteins. For example, the 2010s saw incredible advances in cancer therapeutics including many FDA-approved therapeutics. These include the first approved cancer vaccine, sipuleucel-T (Provenge®), for the treatment of prostate cancer [1], the first PD-1 inhibitor, pembrolizumab (Keytruda®), for advanced or unresectable melanoma [2], the first monoclonal antibody for patients with multiple myeloma, daratumumab (Darzalex®) [3], the first CAR T-cell immunotherapy, tisagenlecleucel (Kymriah®), to treat B-ALL patients [4], and the first cell-based immunotherapy for patients with relapsed or refractory Mantel cell lymphoma, brexucabtagene autoleucel (Tecartus™) [5].
Advances in cancer therapeutics were not alone, however. Critical steps forward were also made for autoimmune diseases like multiple sclerosis (MS), rheumatoid arthritis (RA), ulcerative colitis, and Crohn’s disease during the last decade. These include FDA approval of the following drugs: the first oral treatment to treat relapsing forms of MS, fingolimod (Gilenya™)[6], vedolizumab (Entyvio™) for the treatment of moderately to severely active ulcerative colitis and Crohn’s disease [7], ocrelizumab (Ocrevus®) for patients with primary progressive MS (PPMS) [8], sarilumab (Kevzara®) for the treatment of RA [9], tofacitinib (XELJANZ®) an inhibitor of JAK for the treatment of ulcerative colitis [10] and baricitinib (Olumiant®), a JAK inhibitor for the treatment of RA [11]. These innovations have been transformative for developing the next generation of clinically available immunomodulatory drugs.
To be marketed to patients in the U.S., immunomodulatory drugs must be approved by the United States Food and Drug Administration (FDA) [12]. In total, there are currently 14 FDA-approved immunomodulators for cancer therapy [13], 20 cellular and gene therapies [14], 85 vaccines [15], 98 antibody therapeutics [16], and 1 oncolytic virus therapeutic [17] which was approved for melanoma treatment in 2015. To keep up with the rapidly evolving clinical landscape, there is an urgent need for newer and more advanced therapeutics.
The immune system is a complex and dynamic system composed of cells and proteins that work together to defend the body against infections [18–20]. To keep us healthy, the immune system cycles between states of activation and suppression to fight off pathogens and repair damaged cells and tissues. Successful implementation of this cycle is necessary each time a pathogen or foreign particle is introduced into the body. Immunomodulation is any regulatory adjustment of the immune system that causes initiation continuation, or termination of an immune response. These changes can be naturally produced through cell-cell communication and signaling cascades or induced through exogenous administration of therapeutics [18]. When functioning properly, immune cells can sense the difference between self and non-self and naturally “self-modulate” or become activated when foreign particles or pathogens are identified and deactivate when the threat is no longer present. On-demand cycling through these activation states is critical for healthy immune system balance. Unfortunately, there are many ways that the immune system can fail or become misaligned or unregulated which can lead to diseases such as cancer or auto-immune disorders [21, 22]. To help compensate, immunomodulatory drugs have been developed and employed in the clinic for many years to assist in re-balancing a malfunctioning immune system. As we learn more about the immune system and how the individual cells function and work together, we can begin to identify and isolate new and more effective targets for diseases. Immunomodulatory therapeutics are uniquely poised to revolutionize the way we think about treating patients in the future.
The immune system is implicated in both pro-inflammatory diseases such as cancer and infectious diseases as well as autoimmune diseases such as RA and systemic lupus erythematosus (SLE). Two major classes of immunomodulators exist: immunostimulants and immunosuppressants [23]. As described in Figure 1, immunostimulatory drugs function to enhance or increase the extent of the immune system’s response to a particular pathogen and are often prescribed to patients with pro-inflammatory diseases. In contrast, immunosuppressive drugs function to reduce the immune response during chronic inflammation, organ transplants, and autoimmune diseases [23]. There are also four other main drug classes that are classified as immunotherapies: cell-based immunotherapy, vaccines, antibody therapies, and oncolytic viruses [24]. Many of these drug classes can be considered immunostimulants or immunosuppressants depending on the specific drug. For example, antibodies that block immune cell activation pathways are considered immunosuppressants while antibody therapies that block regulatory or deactivation pathways are considered immunostimulants.
Figure 1.
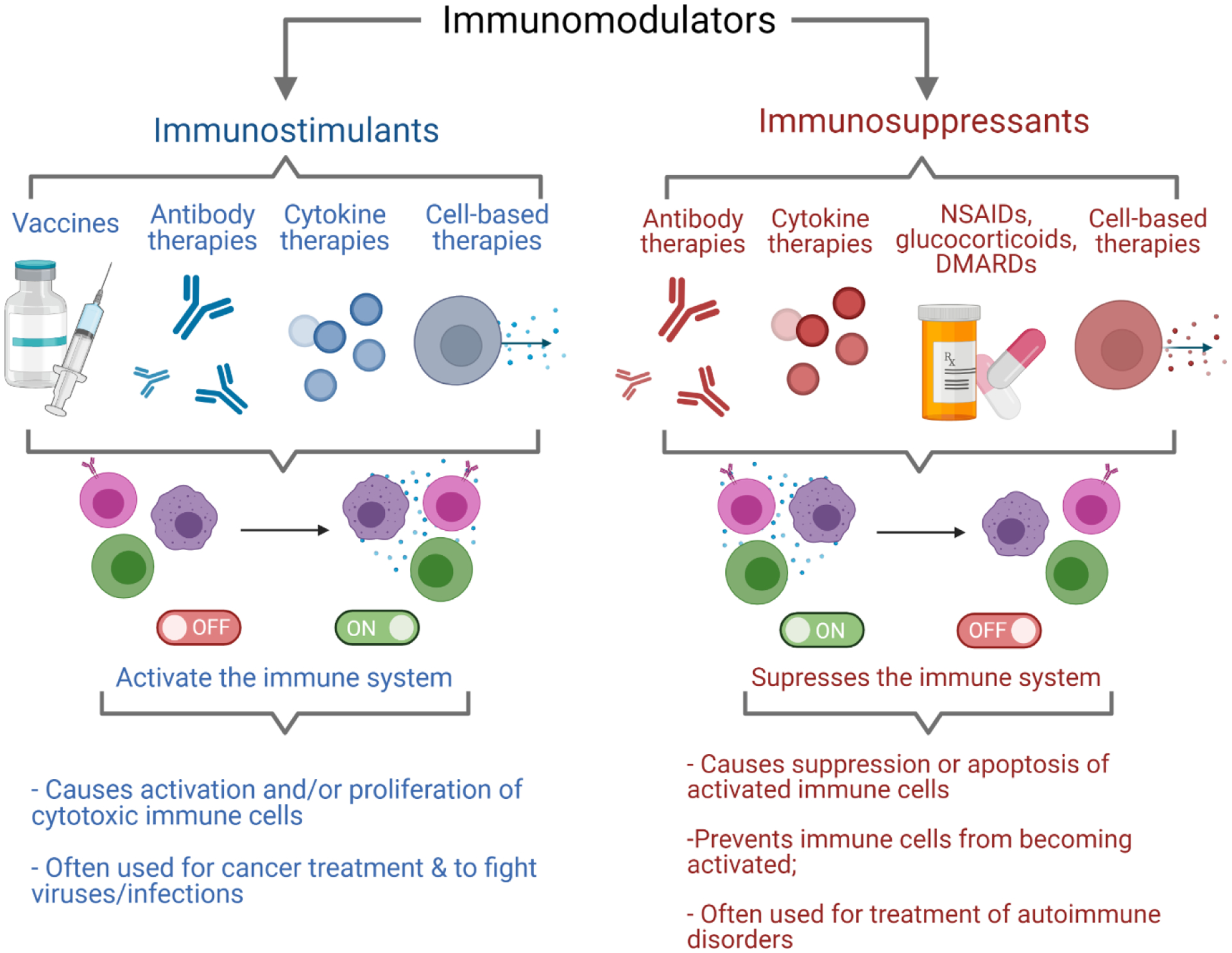
Diagram highlighting the main types of immunomodulators. Immunosuppressants (left) including vaccines, antibodies, cytokines, and cell-based therapies have been used to exogenously activate the immune system for treatment of diseases such as cancer. Immunosuppressants (right) including antibodies, cytokines, NSAIDs, glucocorticoids, DMARDs, and cell-based therapies have been used to repress overly active immune systems for treatment of autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, irritable bowel disease, systemic lupus erythematosus, and more. Schematic was made using BioRender.com
Innovative developments for the next generation of immunotherapeutic drugs require utilization of the plasticity of the cells and branches of the immune system, a deeper understanding of existing cellular and non-cellular targets, and more advanced tools to identify targets and therapeutic strategies. The next generation of therapeutics must expand on and synergize with the current immunotherapeutic modalities to build on their efficacy. In order to easily and efficiently translate into the clinic, the next generation of biologics must also consider manufacturing challenges and FDA requirements for approval during the research and development stage. At the forefront of these new ideas are engineered cytokine therapies that are less toxic and more bioactive than current products, targeted delivery platforms that allow for deeper penetration and more predictable drug release kinetics, and combination therapies that work in concert and build on the efficacies of each monotherapy to function as a more efficient combination therapy that can be personalized on a per patient basis. Herein, we will review the progress of immunostimulatory and immunosuppressive therapies to date and discuss the opportunities and challenges for clinical translation of the next generation of immunomodulatory therapeutics. We will review cytokine therapy, cell-based therapy, antibody therapy, and viral therapies in the context of clinical translation. Vaccine-based immunotherapies have been expertly reviewed by Hollingsworth and Jansen [25] as well as Dalgleish [26] and will not be discussed here.
1.1. The healthy immune system
1.1.1. Innate Immunity
The immune system is a complex, dynamic system made up of cells and chemicals that keep us healthy by responding to and eliminating harmful materials and pathogens that enter the body[27]. The mechanisms that allow the recognition of these hazards are innate and adaptive immunity [20, 27]. The first line of defense is innate immunity which are nonspecific defense mechanisms that take place once a pathogen is identified in the body. Major immune populations for innate immunity include macrophages, neutrophils, eosinophils, basophils, mast cells, monocytes, dendritic cells, and natural killer (NK) cells [28, 29]. The induction period for the innate system is much faster (minutes-hours) than the adaptive system (hours-days) and recognizes common pathogen-associated microbial patterns (PAMPs) as well as other foreign materials that enter the body [28].
1.1.2. Adaptive Immunity
The second wave of the immune system is called adaptive immunity. This mechanism recognizes unique epitopes on pathogens and causes antigen-specific immune responses once an antigen has been recognized, processed, and presented by antigen-presenting cells (APCs) [20, 27]. Adaptive immunity is more specific than the innate system, but also requires extensive cell proliferation to induce cytotoxicity, and is much slower to respond to threats [28]. The difference in the “time-to-action” and specificity of the two branches necessitates the involvement of both systems to efficiently identify and eliminate harmful materials. Adaptive immunity plays a major role in clinical oncology as well as autoimmune disease and its major immune cell populations include T cells and B cells [28, 29]. Figure 2 highlights the major immune cell population in each branch and a general time frame for activation of the associated cells [30]. Understanding the major cell populations, functions, and timescales of each branch of the immune system is critical for developing appropriate and efficacious new immunomodulators.
Figure 2.
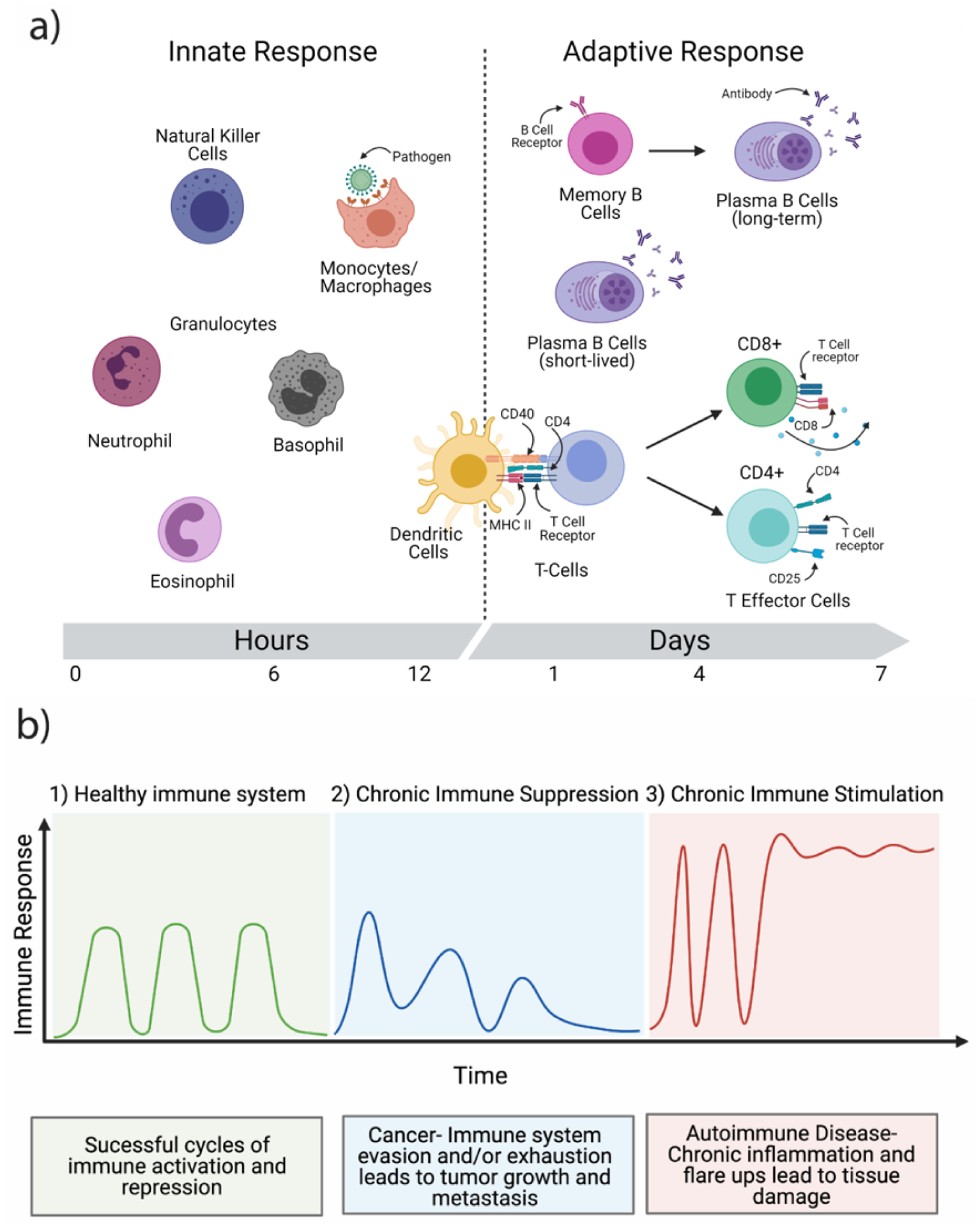
Schematic of the branches and function of the immune system. a) The innate immune system consists primarily of natural killer cells, monocytes, macrophages, neutrophils, basophils, eosinophils, and dendritic cells. Macrophages can identify and phagocytose foreign particles and pathogens. Upon stimulation, antigen presenting cells such as dendritic cells can activate T cells of the adaptive branch of the immune system via MHC II and CD40 receptors. The adaptive branch of the immune system consists of T cells and their associated subsets, and memory and plasma B cells. Memory B cells express B cell receptors and can differentiate into long-lived plasma B cells upon binding of the B cell receptor (BCR). Plasma B cells are short-lived B cells that rapidly produce pathogen-specific antibodies to neutralize the effects of a pathogen. Naïve T cells, upon binding to antigen presenting cells, can divide into CD4+ or CD8+ T cell lineages. CD8+ T cells are commonly referred to as cytotoxic T cells and typically last the duration of a particular immune response and then undergo apoptosis. The CD4+ T cells can further differentiate into a number of helper T cell subsets discussed below. b) Graphical depiction of the cyclical nature of a healthy immune system (1), a chronically suppressed immune system that is unable to mount a full immune response (2), and a chronically stimulated immune system subject to random “flare-ups” which may lead to tissue damage. Schematic was made using BioRender.com
1.2. Immune system failures: Cancer
The phenomenon of resistance, recurrence, and immune evasion by cancer cells continues to challenge scientists and oncologists and thus remains an active area of research [31]. Some of the most common types of cancer in the U.S. include bladder, breast, colorectal, endometrial, kidney, liver, lung, pancreatic, prostate, leukemia, Non-Hodgkin lymphoma, and melanoma [32, 33]. Survival rates and treatment availability vary substantially for patients and often depend on where the cancer is located and the severity of the disease at the time of diagnosis. For example, the current standard of care for patients with advanced pancreatic cancer is gemcitabine monotherapy [34, 35], while sunitinib [36, 37], and pazopanib [37, 38] are the standard of care for patients with advanced renal cell carcinoma, and debulking surgery followed by paclitaxel administration is frequently prescribed to patients with advanced ovarian cancer [39–41]. Many of these chemotherapy treatments, although generally successful, can cause severe side effects and toxicities that lessen the patient’s quality of life [40, 42, 43]. In addition to the severe toxicities associated with many traditionally used chemotherapeutic drugs, the heterogeneity of cancer cells, their cellular, and non-cellular microenvironments, and their remarkable ability to survive and rapidly evolve remain major hurdles for the field of oncology.
Various types of cancer cells arise and interface with the cells of our bodies almost every day but are usually rapidly identified and eliminated by the collaborative efforts of the branches of the immune system [44, 45]. However, in some cases, cancer cells evolve and can circumvent recognition by the immune system, thus evading activation of the pro-inflammatory immune response and allowing cancer cells to develop into tumors [44]. Since the immune system is responsible for fighting infection and eliminating pathogens, immune-oncology represents a unique approach and opportunity to boost or reactivate the immune response and ultimately destroy and eliminate tumors. In this section, we review recent clinical advancements in immunomodulatory drugs in the context of immune-oncology (Figure 3) and discuss potential future directions for immunostimulatory therapeutics.
Figure 3.
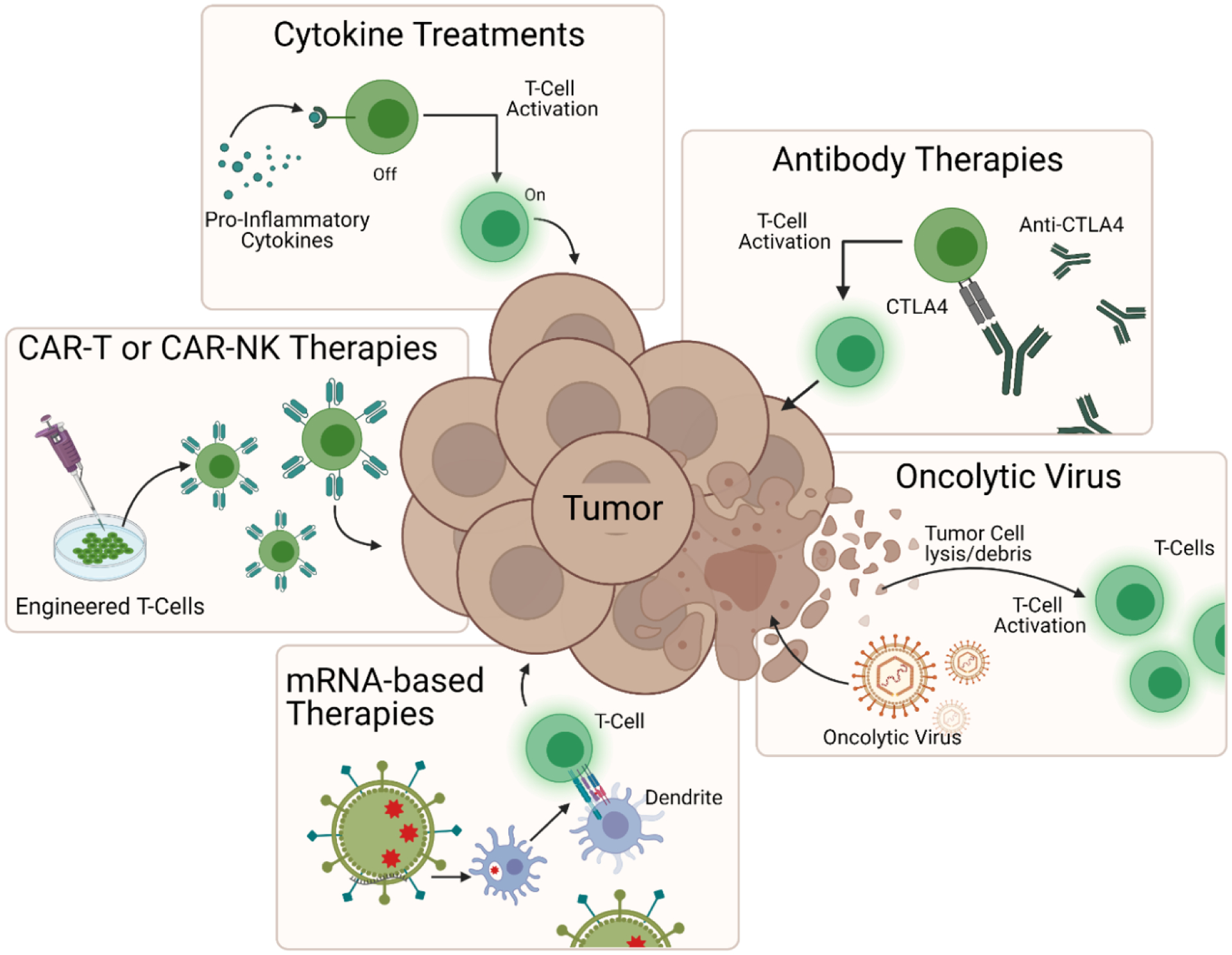
Major immunomodulatory strategies used to induce tumor regression. These include pro-inflammatory cytokine treatments such as interleukin-2 administration for T cell activation, antibody therapies such as anti-CTLA4 checkpoint blockade to reverse cytotoxic T cell repression, oncolytic virus therapy to induce tumor cell lysis and subsequent T cell activation, mRNA-based therapies to activate antigen presenting cells to induce T cell proliferation and engineered cell therapies such as CAR-T or CAR-NK therapy to enhance cell targeting and tumor destruction. Schematic was made using BioRender.com
1.2.1. Recent advances in cancer immunotherapies
Many new clinical trials and FDA-approved drugs have been evaluated recently. For example, a phase I clinical trial found Nivolumab (BMS-936558/MDX-1106), an immune checkpoint inhibitor, stimulated tumor regression in patients with advanced melanoma, renal, colon, and lung cancer and a phase III clinical trial found ipilimumab (Yervoy®) extends survival in advanced melanoma patients [46–48]. Researchers also found patients with metastatic melanoma had a significant improvement when they received the high dose of IL-2 and gp100 peptide vaccine [49] and a separate study found clinical responses in patients with chronic leukemia when they received genetically modified T cells for adoptive transfer [50]. In 2013, adult patients with relapsed B cell acute lymphoblastic leukemia had clinical responses when treated with CAR T cell transfer [51]. In 2014, the FDA approved ramucirumab (Cyramza®) to treat advanced stomach cancer and gastroesophageal junction adenocarcinoma [52] and in 2015, the FDA approved a genetically engineered virus, talimogene laherparepvec (Imlygic™), to kill cancer cells for treatment of advanced melanoma [53].
The second half of the decade also saw the first FDA approved anti-PD-1/PD-L1 checkpoint inhibitor that targets the PD-L1 ligand, atezolizumab (Tecentriq®), to treat bladder cancer and olaratumab (Lartruvo™), a monoclonal antibody directed against platelet-derived growth factor receptor alpha (PDGFRα), to treat patients with soft tissue sarcoma [54, 55]. In 2017, the FDA also approved axicabtagene ciloleucel (Yescarta®), a CD19-directed genetically modified autologous T cell immunotherapy, for the treatment of non-Hogdkin large B cell lymphoma [56]. The following year, cemiplimab (Libtayo®), a programmed death receptor-1 (PD-1) blocking antibody was approved for patients with advanced forms of cutaneous squamous cell carcinoma [57]. In 2019, the FDA approved the first systemic treatment for patients with tenosynovial giant cell tumor, pexidartinib (Turalio™) [58] and most recently, the FDA approved the first cell-based gene therapy, idecabtagene vicleucel, for adult multiple myeloma patients earlier this year [59]. As more disease and symptom-related targets are identified, we expect to see even better, faster, and smarter therapeutics in the next several decades.
1.3. Immune system failures: Autoimmune diseases
The immune system acts as the first line of defense against invading pathogens, but it also has important regulatory functions in the body [60, 61]. Without properly timed and implemented negative feedback mechanisms, the pro-inflammatory state of an active immune system can lead to chronic inflammation [62]. Autoimmune diseases can also occur when the self-recognition capabilities of the immune system break down, causing initiation of immune system attacks on self-molecules, cells, or tissues [30, 61, 63]. Typical treatments commonly prescribed for autoimmune diseases include non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying antirheumatic drugs (DMARDs) [64–66]. NSAIDs are potent inhibitors of neutrophil activation and are effective in the alleviation of pain and inhibition of inflammation [66, 67]. Glucocorticoids bind to specific cell receptors and often result in the inhibition of cellular signaling pathways and regulate the immune cell expression of cytokines and chemokines [64, 65] and DMARDs have the capacity of reducing tissue and organ damage caused by inflammatory responses [64]. There are also other classes of drugs such as the anti-interleukin-1 receptor antagonist, anakinra, which was approved for the treatment of rheumatoid arthritis (RA) in 2001 but due to lower efficacy than other biologics and the need for daily subcutaneous administration, anakinra is seldomly used for the treatment of RA today [68–70]. This drug recently regained attention in January 2013 when it was also approved for treatment of a severe form of Cryopyrin-Associated Periodic Syndromes (CAPS), known as NOMID and in 2020 when it was evaluated for its potential use in patients with COVID-19 and reduced the need for invasive mechanical ventilation in the ICU and mortality among patients with severe forms the virus without serious side-effects [71–73].
Autoimmune diseases represent the third most common cause of chronic illness in the United States and there are more than 80 types of autoimmune diseases that span a broad range of immunological dysfunctions [74]. The most common of these include type 1 diabetes, multiple sclerosis, RA, lupus, Crohn’s disease, psoriasis, and scleroderma [74–76]. Each of these diseases can be caused by defective immune system regulation, environmental stimuli, or genetic susceptibility and many of the currently available treatments function to reduce inflammation and provide symptom management for patients but do not address the fundamental problems responsible for the initiation and progression of the autoimmune process [75]. In most cases, this necessitates continued therapy, resulting in decreased quality of life and patient compliance and increased risk of infectious complications [75]. Future success at treating these diseases will require a deeper understanding of the mechanisms of a healthy immune system, how abnormal immune responses begin, and how they are maintained over time. Augmenting regulatory mechanisms and establishing robust disease resolution is a priority for new immunotherapeutic drugs [75].
1.3.1. Recent advances in autoimmune disease immunotherapies
There have also been several examples of advances in immunosuppressive therapeutics spanning the past decade. For example, in the early 2010s, a monoclonal antibody that binds to BLyS, belimumab (Benlysta®) for the treatment of SLE was approved [77] along with the second oral treatment for relapsing forms of MS, teriflunomide (Aubagio®) [78], dimethyl fumarate (Tecfidera™) as the first in line treatment for relapsing MS [79], and eluxadoline (Viberzi®) for the treatment of IBS-D [80].
In the second half of the decade, the FDA approved daclizumab (Zinbryta®) for the treatment of MS and a monoclonal antibody that targets IL-12 and IL-23, ustekinumab (Stelara®), for the treatment of moderately to severely active Crohn’s disease and ulcerative colitis [81, 82]. In 2019, the FDA approved upadacitinib (Rinvoq®), a Janus kinase inhibitor, for the treatment of RA and infliximab-axxq (Avsola™), a tumor necrosis factor (TNF) blocker, for the treatment of Crohn’s disease and ulcerative colitis [83, 84]. A clinical trial found low dose IL-2 might be effective in treating SLE [85]. The following year, the FDA approved ozanimod (Zeposia®), a sphingosine 1-phosphate receptor modulator, for the treatment of relapsing MS [86]. Most recently, a 2021 phase II and III clinical trials found anifrolumab enabled glucocorticoid reduction and SLE flare reduction for patients [87].
2. Immunomodulatory platforms
A well-balanced immune system is critical for maintaining human health. As discussed above, the branches of the immune system must coordinate to toggle through phases of activation and repression in order to accurately identify, process, and eliminate pathogens and foreign materials in the body [18]. Because immune system dysregulation can result in pro- or anti-inflammatory diseases, researchers have developed immunomodulatory drugs that function to boost or repress the immune system [21, 22]. Some of the most impressive advancements for next-generation therapeutics have been in the form of more sophisticated drug delivery platforms. The past decade has seen drugs delivered via nanoparticles [88], micelles [89], microparticles[90], liposomes [91], fusion-proteins [92], PEGylated proteins [93], engineered receptors [94], cell backpacks [95], viruses [96], combination systems, and more. In this section, we will discuss the clinical translation of cytokine therapies, antibodies, and pain/symptom management drugs for various disease settings. We will also discuss advances in cell-based therapeutics and combination therapies as well as the challenges and future directions of each.
2.1. Cytokine therapies
Cytokines are low molecular weight, soluble proteins that are secreted primarily by immune cells and function to relay instructions and mediate cell-cell communication in the body [97, 98]. These proteins act in concert with specific soluble, or membrane-bound, cytokine receptors to regulate the human immune response [98] and have been used as biological drugs for over 30 years [99]. In this section, we will discuss pro- and anti-inflammatory cytokines and highlight their utility as immunomodulators.
2.1.1. Pro-inflammatory cytokines
Pro-inflammatory cytokines are produced predominantly by activated immune cells and are involved in the up-regulation of inflammatory reactions [98]. Two pro-inflammatory cytokines, interleukin-2 (IL-2) and interferon-alpha (IFN-α) have been approved by the FDA for the treatment of several malignancies [100]. Other pro-inflammatory cytokines including granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-gamma (IFNγ), IL-7, IL-12, and IL-21 have also been evaluated in clinical trials and remain part of certain ongoing clinical trials [101]. Cytokines are usually released by a specific cell type in response to a stimulus, and the extent of their action is short-lived due to limited half-life in the circulation [100]. Many cytokines only circulate in the blood stream for minutes to hours[102]. Cytokine-based immunotherapy is a promising indication for cancer treatment because cytokines can modulate the immune response towards cancer cells, thus enabling enhanced cytotoxicity [101, 103, 104]. The biggest drawback of pro-inflammatory cytokine therapy is the short half-life which necessitates high and frequent doses that elicit treatment-limiting off-target effects and toxicities [99, 101, 105–107].
Although there are still only two FDA-approved cytokine therapies for cancer treatment, other pro-inflammatory cytokine therapies have been evaluated in pre-clinical and clinical studies as monotherapies and in combination with other treatments. Here we will discuss cytokine monotherapies and later we will discuss cytokines in combination with antibodies, and more.
2.1.2. Cytokine modifications
Like with the administration of any drug, careful consideration must be given to selecting the most appropriate dosing scheme for cytokine administration. In the body, various cytokines are rapidly produced by cells on an “as needed” basis to initiate, propagate, or terminate an immunological response [100]. It would, therefore, be evolutionarily disadvantageous for a particular cytokine to remain active in the body for long periods of time as this could potentially disrupt the cyclical nature of the immune system and be detrimental to the survival of an organism. However cytokines have notoriously short half-lives and are either utilized rapidly after production or degrade and are naturally excreted if not needed in order to maintain immune system homeostasis. Unfortunately for researchers, the short half-life of these proteins represents a major challenge for accurate and appropriate administration of exogenous cytokines for clinical immunotherapy.
Another crucial consideration for cytokine administration and half-life disparities is the toggle between safety and efficacy, especially for cancer immunotherapy. Pro-inflammatory cytokines, in particular, are often ineffective at low concentrations and elicit (sometimes life-threatening) toxicities when the concentration is too high [105]. However, when the concentration is within an appropriate therapeutic window, the treatment efficacy is remarkable [105]. Because the therapeutic window depends on the potency and mechanism of action of a particular cytokine, careful consideration for dosing must be given for each cytokine as well as each combination of cytokines administered to patients. For this reason, extensive time and effort have gone into developing second-generation cytokine products that have reduced toxicity or increased half-lives and work is ongoing to develop cytokine products that exhibit both characteristics. One of the most promising and prolific approaches has been utilizing advances in drug delivery platforms and chemistry techniques to create cytokine modifications and fusion proteins. A few of the most common cytokine modifications are depicted in Figure 4. Examples of these modifications have been developed and tested in pre-clinical studies and are described in the sections below (sections 2.1.3, 2.1.4, and 2.1.5).
Figure 4.
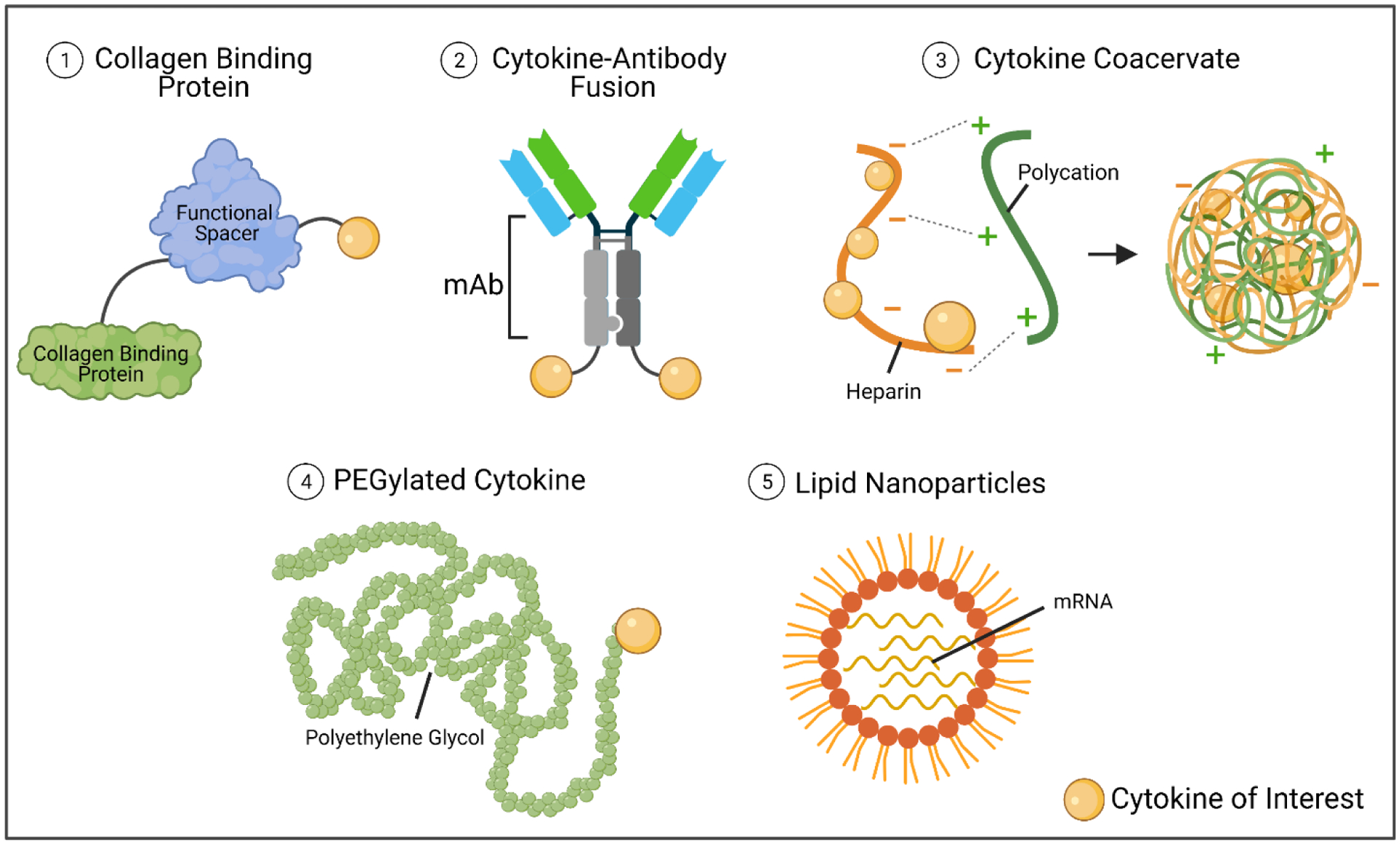
Common cytokine modifications. Cytokines are low molecular weight proteins and can be attached to other proteins or molecules easily. Here we describe cytokines attached to collagen-binding proteins via glycineserine linkers (commonly), fused to one or more chains of a monoclonal antibody, protected within charged molecules, pegylated, or administered as mRNA from within lipid nanoparticles. Schematic was made using BioRender.com
2.1.3. IL-2 modifications
Due to its therapeutic potential, IL-2 is one of the most widely studied cytokines. High-dose bolus administration of recombinant IL-2 treatment has been FDA approved for use in melanoma and renal cancers under the trade names Aldesleukin® or Proleukin® since 1992 [108–111]. IL-2 is of particular interest for immuno-oncology it plays a critical role in the activation, proliferation, and regulation of immune cells such as T cells and NK cells [108, 112–114]. There are three IL-2 receptor chains: IL-2 receptor α-chain (IL-2Rα, encoded by IL2RA; also known as CD25), the β-chain (IL-2Rβ, encoded by IL2RB; also known as CD122) and the γ-chain (IL-2Rγ, encoded by IL2RG; also known as CD132) and IL-2 molecules interact with each of the three classes of IL-2 receptor with different affinities [112]. IL-2 binds with low affinity (Kd ~10−8 M) to receptors containing only IL-2Rα, with intermediate affinity (Kd ~10−9 M) to receptors containing IL-2Rβ and IL-2Rγ, or with high affinity (Kd ~10−11 M) to receptors containing IL-2Rα, IL-2Rβ and IL-2Rγ (Figure 5). Intermediate-affinity receptors are expressed primarily on resting NK cells and CD8+ T cells, but immune cell activation induces IL-2Rα expression which converts intermediate-affinity receptors to high-affinity receptors and allows for cytotoxic immune cells to undergo proliferation [112].
Figure 5.
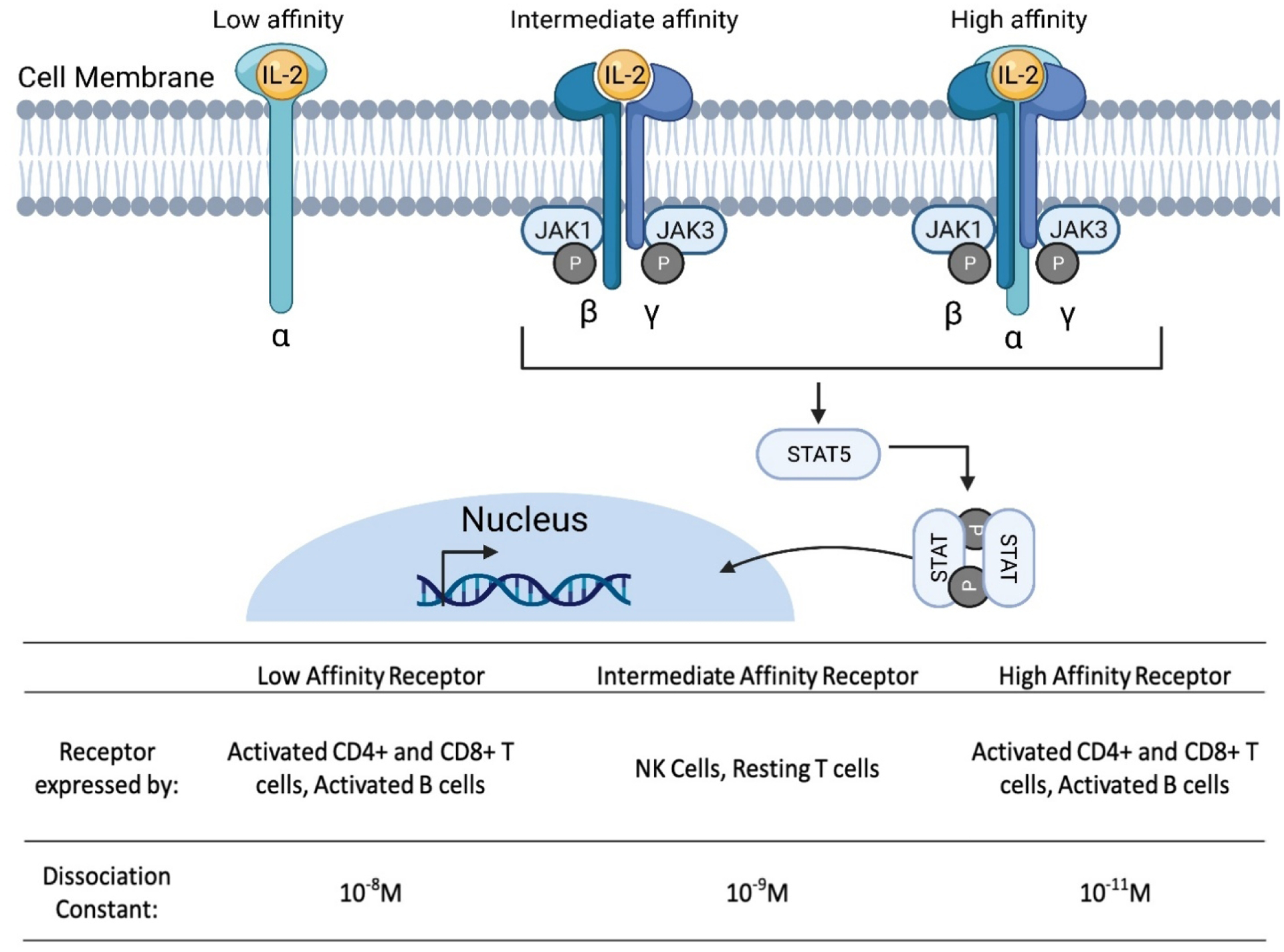
Interaction of interleukin-2 (IL-2) with its receptors to induce downstream signaling. Secreted IL-2 binds to IL-2Rα and subsequently forms the high affinity heterotrimeric receptor complex allowing the activation of downstream signaling. IL-2 receptor binding triggers the phosphorylation of the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway. Phosphorylated STAT molecules dimerize and then translocate into the nucleus to regulate the transcription of target genes. Different immune cells express various versions of the IL-2 receptor, each with different dissociation constants affecting the ability of IL-2 to bind and activate the associated cells. Schematic was made using BioRender.com
IL-2 has different effects on CD4+ and CD8+ T cells so it is imperative to understand the immunological landscape of the tumor of interest when developing new biologics [112]. Naive CD4+ T cells, for example, have the potential to differentiate into various functional populations, including Th1, Th2, Th17, Th9, Tregs, and T follicular helper (Tfh) cells, depending on the antigen and cytokine signals they receive during development [112]. Studies have demonstrated that IL-2 plays a role in Th1 [115], Th2 [116], and Treg cell differentiation, and aids in T helper 9 (Th9) cell generation [117] but inhibits the differentiation of Th17 cells [118], and T follicular helper (Tfh) cells [119] suggesting that the presence of IL-2 is critical to the fate of T cell subsets. The functions and implications of activation and repression of the various T cell subsets have been expertly reviewed by Ross and Cantrel [113] and will not be discussed in detail here. As for CD8+ T cells, early in vitro and in vivo work established a critical role for IL-2 as a T cell growth factor in driving the expansion of CD8+ T cells subsequent to TCR stimulation [114, 120]. The various T cell subsets play a major role in contributing to the activation cycles of the immune system by ramping up immune cell proliferation during an immune response and inducing cell death and regulation to end an immune response when the threat is no longer present. The plasticity of these immune cells is imperative for immune system homeostasis.
Unfortunately for researchers, the half-life of IL-2 in the blood is on the order of minutes and the needed high dose regimens often elicit life-threatening toxicities such as Vascular Leak Syndrome (VLS) which hinders the widespread adoption of this treatment [106, 121]. For this reason, various IL-2 analogs and fusion proteins have been created to reduce toxicity and extend the half-life. Major progress has been made in designing and implementing IL-2 analogs and fusion proteins. However, many of these modifications also decrease the therapeutic efficacy and thus future modification strategies are still needed.
Cytokine modifications have become a staple for immunotherapeutics. For example, Nektar Therapeutics created a PEGylated IL-2, NKTR-214, which binds to CD122. This recombinant cytokine has an increased half-life provides anti-tumor efficacy [93], and has been tested in rodents and non-human primates. Additionally, several clinical trials are underway testing NKTR-214 with a combination of immune checkpoint inhibitors (NCT03138889, NCT02983045, NCT03282344, NCT03435640, and NCT02983045) [122–124]. Another pharmaceutical company, Hoffman-La Roche, developed a mutant IL-2, cergutuzumab amunaleukin (CEA-IL2v), that reduces binding to IL-2Rα in order to reduce binding and activation of regulatory T cells and instead preferentially bind cytotoxic T cells to boost anti-tumor efficacy. This engineered mutated IL-2 variant also has an extended half-life and has been shown to increase tumor reduction in mice [125]. Recombinant IL-2 has also been fused to a single-chain monoclonal antibody L19 by Philogen S.p.A. The L19-IL2 fusion was expected to have high IL-2 concentrations and longer circulation because it was delivered intralesionally to patients in a phase II clinical trial. Unfortunately, the objective response rate recorded in this study was lower for patients treated with L19-IL2 than the untargeted IL-2 treatments suggesting that additional work needs to be done to boost the efficacy without increasing toxicity [126]. Children’s Oncology Group also generated hu14.18-IL2, a fusion protein consisting of the humanized 14.18 anti-GD2 monoclonal antibody linked to IL-2. This system localizes to GD2-positive tumor cell surfaces through the anti-GD2 mAb component, then binds to and activates cytotoxic lymphocytes through the IL-2 component. Hu14.18-IL2 caused minor toxicity 5 patients out of 23 had a complete response [127]. Finally, a hyperstable de novo mimic of IL-2 with reduced affinity to α-chain subunit of the IL-2 receptor was created in order to limit its pleotropic bioactivity [128]. This molecule has great potential for increasing the use of IL-2 and other cytokine therapies in the future, but additional work must be done to address the bioactivity of de novo mimics as well as the other modified constructs in order to translate into the clinic and be widely adapted for clinical use.
2.1.4. IFN-α modifications
Cytokine efficacy for cancer treatment can also be improved by extending the serum half-life and increasing cytokine concentration within the tumor microenvironment. In 1986, IFN-α was the first biotherapeutic to be FDA approved [129]. It has been approved and evaluated for the treatment of hairy cell leukemia [130], follicular non-Hodgkin lymphoma [131], melanoma [132], and AIDS-related Kaposi’s sarcoma [133]. Similar to IL-2, discussed above, widespread use of IFN-α has declined due to extensive toxicities. For this reason, protein and delivery modifications have also been developed for IFN- α. These modifications includes advanced drug delivery systems such as nanoparticles, PEG-modified cytokines [134], and construction of fusion protein with antibodies [135], Fc domains, apolipoprotein A-I [136], albumin, or the latent peptide of TGF-β. For instance, Herndon and colleagues found that PEGylating IFN-α increases the half-life in circulation and exposes tumor cells to high IFN-α concentrations to treat patients with melanoma [134]. Fioravanti and colleagues found by connecting IFN-α to ApoA-I, the half-life increased and promoted anti-tumor activity [136]. This study showed that the fusion protein achieved immunostimulatory activity at low toxicity. IFN-α has also been successfully fused to single-domain antibodies targeting Clec9A+ in dendritic cells and displayed anti-tumor activity in several murine cancer models without detectable toxic side effects [135]. The progress that has been made is tremendous, but future work in this area is urgently needed to establish modified cytokines and delivery systems capable of operating within a defined therapeutic window to ensure safe and effective treatments for a wide range of cancer patients.
2.1.5. IL-12 modifications
Another pro-inflammatory cytokine, IL-12, has been evaluated numerous times in phase I-II clinical trials, starting in the 1990s but has not been found to be therapeutically relevant due to associated toxicities [137–140]. For this reason, similar to IL-2 and IFN-α, researchers have returned to the drawing board to develop IL-12 therapies that are effective without causing toxicities. These include protein modifications, alternative delivery strategies, localized administration, and more [141]. For example, in 2020, Hwang et al. investigated the antitumor effect of recombinant IL-12 delivered with complex coacervates, for treatment of mice with B16-F10 melanoma. The authors developed IL-12-heparin complexes and cationic poly(ethylene argininylaspartate diglyceride), which were mixed to induce coacervation and injected peritumorally in mice inoculated with bilateral injections of B16-F10 cells. Tumor-bearing animals were dosed with 1, 10, or 30μg IL-12 per complex coacervate. Higher concentrations of IL-12 delivered resulted in more efficient tumor growth inhibition without significant signs of cytokine-induced toxicity. Importantly, the IL-12-heparin complexes conferred better antitumor efficacy compared to free recombinant IL-12 injection treatment suggesting that the complexes were able to persist longer in vivo and cause increased cytotoxic immune cell activation. Immunofluorescence imaging was used to understand the mechanism of IL-12 coacervate therapy, and significant accumulation of lymphocytes was observed in the tumor microenvironment of mice after receiving IL-12 coacervate treatment. This work demonstrated heparin-based complex coacervates delivering recombinant IL-12 as an effective therapy for murine melanoma model and rationalized further development for translation into the clinic [142].
Intratumoral retention is beneficial for increasing anti-tumor effects and reducing systemic exposure. To attempt to increase the retention of IL-12 in the tumor microenvironment, Momin and colleagues developed a cytokine fusion complex using the collagen-binding protein lumican which is abundantly expressed in tumors. The authors found that their fusion protein was most effective when administered in combination with other marginally efficacious monotherapies such as CAR-T cells, checkpoint inhibitors or tumor-targeting antibodies. The authors also found that intratumoral administration of IL12-MSA-Lumican did not cause weight loss which is a proxy for IL-12 related toxicity in mice. Unfortunately, however, the fusion IL-12 treatment delayed tumor outgrowth but was not curative as a monotherapeutic. However, durable tumor regression was seen when a CAR-T cell therapy was combined with IL12-MSA-Lumican. This suggests that the partial benefit of CAR T cells alone and of IL12-MSA-Lumincan alone were able to act in concert to elicit the required level of immune activation and tumor cell destruction [143]. Future studies with additional combinations of immunotherapies are needed to determine which combinations of drugs are most effective for different tumor types and cancer stages. In another IL-12 related work, Li and colleagues developed self-replicating RNAs encapsulateded in lipid nanoparticles for intratumoral treatment of melanoma. The nanoparticles were used to protect the RNA and facilitate entry into the target cells. Additionally, similar to the study described above, the IL-12 was fused to the collagen-binding protein lumican. This system functions to transfect tumor cells in vivo which indirectly leads to immune cell activation and infiltration into the tumor microenvironment. The authors reported that a single injection of their system was able to induce tumor reduction in mice but also caused transient weight loss in mice which suggests treatment-related toxicity [144]. Unfortunately for the studies described here, the necessity to inject treatment directly into the tumor significantly reduces the types of cancers that can feasibly be treated with this method.
Algazi and colleagues studied the safety and effectiveness of different dosing regimens of IL-12 for treatment of patients with malignant melanoma. In this study, patients received 0.5 mg/mL plasmid encoding IL-12 (tavokinogene telseplasmid; tavo) intratumorally followed by electroporation on days 1, 5 and 8 for 90 days. The authors found the best overall response for treated lesions to be 43.8% and 25% for untreated lesions [145]. This treatment could be helpful in eliciting local and global immune responses following pro-inflammatory cytokine treatment. Future studies with IL-12 could benefit from the development of alternative administration strategies, such as electroporation of plasmid-based treatments, without causing off-target effects. Several clinical trials focused on local delivery of IL-12 through electroporation (NCT02345330, NCT01579318) were terminated due to company resource constraints, but local IL-12 administration still remains a promising area of research for future product development.
2.1.6. Pro-inflammatory cytokine combination therapies
The complexity of the immune system suggests that combination approaches may be necessary for the success of future treatments. As scientists develop a better understanding of the many feedback mechanisms and overlapping signaling pathways, the idea that a heterogeneous disease may require multiple drugs to achieve the right ‘balance’ for efficacy begins to emerge. Many groups have also evaluated the effects of administering cytokines in combination with other cytokines, chemotherapeutic drugs, and antibodies. In a study of the antitumor effects of interleukin-2 and interferon-β combination gene therapy for the treatment of colorectal cancer in BALB/C mice, scientists developed a cytokine fusion expression plasmid (pcDNA3.1A-IL-2/IFN-β) and a cytokine fusion, carcinoembryonic antigen (CEA) expression plasmid (pcDNA3.1ACEA-IL-2/IFN-β). The fusion genes exhibited antitumor tumor effects and an 8%−10% increase in apoptosis after being injected into the tumor-bearing mice. These results suggest that IFN-β and IL-2 combination gene therapy has potential for development into a clinical therapeutic strategy for colorectal cancer [146]. More recently, Gong and colleagues also investigated the effects of using a recombinant mutant human tumor necrosis factor-α (rmhTNF) combined with raltitrexed (RTX) for treatment of colorectal cancer in athymic male BALB/c nude mice. The authors combined rmhTNF, RTX, and hyperthermic intraperitoneal chemotherapy (HIPEC) and found that rmhTNF+RTX+HIPEC was the most effective treatment for in vitro and in vivo. This work highlights the importance of elucidating the right combination of therapies to improve the treatment of colorectal cancer [147].
In a phase II clinical trial, high dose IL-2 (720,000 IU/kg) has been tested in combination with MAGE-A3 cancer immunotherapeutic (MAGE-A3 CI) to evaluate the safety and anti-tumor efficacy in eighteen patients with unresectable or metastatic melanoma. The authors reported responses in 4/16 (25%) patients and stable disease in 6/16 (38%) patients. Unfortunately, the combination treatment had similar toxicities to high dose IL-2 monotherapy suggesting that other combination therapies may prove more effective and less toxic than high dose IL-2 and MAGE-A3 CI [148]. Further, a more recent study reported on the collective results of three clinical trials that evaluated adoptive T cell therapy in combination with IL-2 for safety and anti-tumor efficacy in patients with metastatic melanoma. In these studies, an objective response rate of 38% and a 3-year survival rate of 29% were achieved [149]. Although future iterations are needed to increase objective response rates and survival percentages, these studies highlight the potential of combination therapies.
2.1.7. Anti-inflammatory cytokines
Pro-inflammatory cytokines only represent one part of the cytokine story, however. Anti-inflammatory cytokines can also be produced by most immune cells and may either inhibit pro-inflammatory cytokine synthesis or control pro-inflammatory cytokine-mediated cellular activities in order to suppress inflammation [150, 151]. Although there are no current FDA approved anti-inflammatory cytokine treatments, cytokines such as IL-10 have been and are being tested in clinical trials for treatment of autoimmune diseases such as IBS, rheumatoid arthritis and more [152]. For example, Georgescu et al. studied the effects of IL-10 on systemic lupus erythematosus (SLE) outcomes in human patients. The authors studied the role of IL-10 in the induction of apoptosis in lymphocytes and the effects of neutralizing anti-IL-10 monoclonal antibodies on human peripheral blood mononuclear cells (PBMCs). The authors found significantly higher concentrations of IL-10 in the PBMCs isolated from patients with active SLE and that these high levels of IL-10 contributed to defective T cell immune responses and the generation of self-antigens characteristic of SLE patients. Further, the group treated with anti-IL-10 showed a significant reduction in spontaneous cell death, which is a cellular outcome of SLE, from 27.5% to 19.8% which provides rationale for further study of the IL-10 pathway in the development of new therapies for SLE [152].
IL-2, as discussed above, is a pleiotropic cytokine that is required for activation, growth, and differentiation of T cells, B cells, and natural killer (NK) cells. This cytokine is even more interesting because it has distinct functions at different concentrations [153]. Low dose IL-2 contributes to T cell survival and regulatory T cell proliferation which is beneficial for immune system repression [153], but high doses of IL-2 stimulate activation and proliferation of cytotoxic T cells that are needed for immune activation in situations like cancer as discussed above [106, 153]. For these reasons, however, IL-2 cytokine therapy has also been evaluated in the context of autoimmune diseases such as SLE. He and colleagues studied the effects of low-dosage IL-2 therapy in treatment of systemic lupus erythematosus (SLE) in human patients. The authors developed a recombinant human IL-2 from E. coli and demonstrated its safety and efficacy through flow cytometry and intracellular cytokine assays. Dosages were administered as seven subcutaneous injections over two weeks. The treatment group showed an improvement of disease symptoms (such as skin lesions, fever, joint pain) with 76.92% of patients achieving partial remission and 53.85% of patients achieving complete remission [85].
Anti-inflammatory cytokines have also been evaluated for immunomodulatory efficacy in the context of neurological diseases such as multiple sclerosis (MS) which is characterized as a chronic inflammatory disease resulting in central nervous system (CNS) lesions that can lead to severe physical or cognitive disability and neurological defects [154]. In untreated MS, excessive monocyte, T helper-1 (Th1), T helper-17 (Th17), and B cell activity is associated with improper regulatory/suppressor cell function in the brain. These cells penetrate the blood-brain barrier, give rise to CNS inflammation and demyelination, and thus represent a major challenge for immunologists and neurobiologists [154–156]. In a 2019 study, Feng and colleagues evaluated the immunoregulatory and neuroprotective effects of IFN-β in the treatment of multiple sclerosis (MS) in human patients. The authors studied IFN-β-induced transcriptome shifts from patients in the following four categories: IFN-β-treated MS Complete Responders (CR) who were stable for five years, stable and active Partial Responders (PR), stable and active untreated MS, and healthy patients. The authors used recombinant IFN-β (self-injected by the patients via two injections of the regular dose of 250μg or one double dose of 500μg of recombinant IFN-β in the clinic) and studied gene expression at 4 hours, 24 hours, and 4 days post injection. Short-term expression (4 hours post-injection) of 1,233 coding and 664 non-coding genes was altered in response to IFN-β injection. Additionally, genes related to immune regulation and inflammation processes were significantly altered. The authors also found that long-term IFN-β treatment led to a profound reversal of approximately 6,000 protein-coding genes dysregulated in untreated MS. This work uncovers dysregulated genes in MS and provides potential new targets for MS treatment [157]. Much of the anti-inflammatory cytokine work to date has been focused on dampening the immune response for symptom or pain management and therefore long-term solutions to address the physiological cause of the dysregulation are needed. Additionally, future work will also be needed to identify the immunological causes of various autoimmune diseases in order to make progress in treating the cause of the disease and develop curative treatments instead of symptom management.
2.2. Antibody-based therapies
Monoclonal antibodies (mAbs) are produced by B cells and specifically target antigens. This class of drugs is highly effective at binding to, and interfering with, molecular pathways that contribute to disease progression. The first monoclonal antibody, muromonab-CD3 (Orthoclone OKT3), was approved by the United States Food and Drug Administration (US FDA) in 1986 as an immunosuppressant for overcoming transplant rejection [158]. This was followed by Abciximab in 1994 for the prevention of blood clots in angioplasty and rituximab in 1997 for treatment of non-Hodgkin lymphoma. Since then, an additional 95 antibodies have been approved for treatment of several human diseases [16]. Because mAbs are highly specific and can be modified for increased binding as new targets continue to be discovered, we expect to see the development and evaluation of new mAbs for generations to come.
2.2.1. Antibody therapies for cancer treatment
Several types of monoclonal antibodies have been evaluated for antitumor effects. In this section we will highlight clinical and pre-clinical studies centered on cytokine signaling pathways and in the next section we will discuss immune checkpoint inhibitors for cancer treatment. Dominguez and colleagues studied the antitumor effects of an anti-IL-8 monoclonal antibody, HuMax-IL8, for treatment of triple negative breast cancer in C.B-17 SCID mice. The mice received either 2 IP injections of the control hIgG or HuMax-IL8 at 200 ug/mouse every two days. The authors found that there was an increase in epithelial markers E-cadherin and ZO-1 and a reduction of mesenchymal vimentin, fibronectin, and ALDH1A1 suggesting that HuMax-IL8 treatment has the potential to revert tumor phenotype to an epithelial-like state [159]. This antibody is also being evaluated in a phase I clinical trial testing the safety and efficacy of HuMax-IL8 (BMS-986253) in patients with metastatic or unresectable solid tumors (NCT02536469) and an ongoing clinical trial testing HuMax-IL8 (BMS-986253) in combination with immune checkpoint inhibitors nivolumab or nivolumab plus ipilimumab in advanced cancers (NCT03400332).
Clinical studies focused on neutralizing signaling pathways through antibodies targeting transforming growth factor-beta (TGF-β) and TNF-α have also been conducted. In a phase I clinical trial, Morris and colleagues studied the safety and activity of fresolimumab, a monoclonal antibody directed against TGF-β for treatment of patients with renal cell carcinoma or malignant melanoma. The authors reported that no dose-limiting toxicity was observed and that there was a median progression-free survival of 24 weeks for these 7 patients [160]. In a placebo-controlled, double-blind trial of infliximab, a monoclonal antibody directed against TNF-α, for cancer-associated weight loss, patients received either infliximab/docetaxel or placebo/docetaxel [161]. Unfortunately, the authors found no statistical difference between the groups in terms of tumor response rate or overall survival. The clinical trial was closed because infliximab did not prevent cancer-associated weight loss. Further studies are needed to determine if this treatment has the potential to lessen the side effects of chemotherapy in other cancer types or treatment regimens.
Ishida and colleagues studied the efficacy, safety, and pharmacodynamic profile of mogamulizumab, a monoclonal antibody directed against C-C chemokine receptor 4 (CCR4), for treatment of patients with CCR4-positive adult T-cell leukemia-lymphoma (ATL). Patients received intravenous infusions of 1 mg/kg mogamulizumab (KW-0761) once per week for 8 weeks. The authors report an objective response from 13 out of 26 evaluable patients and a complete response from 8 out of the 48 patients enrolled [162]. These results highlight the clinical antitumor activity of mogamulizumab in patients with relapsed ATL. Finally, Shanafelt and colleagues studied the efficacy of administering ofatumumab, a monoclonal antibody directed against CD20, with chemotherapeutic drugs pentostatin and cyclophosphamide for treatment of patients with untreated chronic lymphocytic leukemia or small lymphocytic lymphoma. Patients received a combination of chemotherapy and ofatumumab treatment every 21 days for a total of 6 cycles. The overall response rate was 96% out of the 48 patients enrolled, and the CR rate was 46%. The authors report that ofatumumab-based chemoimmunotherapy was more efficacious and less toxic than rituximab-based chemoimmunotherapy suggesting further development and translation of ofatumumab.
Antibody-drug conjugates (ADC) have also been a major area of research in the field of immunology and were as recently expertly reviewed by Criscitiello and colleagues. Briefly, an antibody-drug conjugate consists of a targeted antibody, a drug payload, and a linker connecting the antibody and the payload [163]. A few of the most effective ADCs include trastuzumab emtansine (anti-HER2 positive), trastuzumab deruxtecan (anti-HER2 positive), enfortumab vedotin (anti-nectin-4) and sacituzumab govitecan (anti-trop2). For example, Hurvitz and colleagues studied trastuzumab emtansine safety and efficacy of treating patients with HER2-positive metastatic breast cancer and found trastuzumab emtansine treatment showed a 41% reduction in progressive disease compared to standard treatment and significant improvement in progression-free survival for these patients [164]. Tamura and colleagues studied trastuzumab deruxtecan (DS-8201a) antitumor efficacy in patients with HER2-positive advanced-stage breast cancer that had previous trastuzumab emtansine treatment. The authors found 93% of the 110 patients had measurable tumor shrinkage, suggesting that trastuzumab deruxtecan is effective for advanced HER2-positive breast cancer [165]. Similar clinical results have also been seen in advanced HER2-expressing colorectal cancer earlier this year (NCT03384940) and clinical trials for HER2-mutated non-small cell lung cancer are underway (NCT03505710). Further, clinical trials evaluating enfortumab vedotin (NCT03474107, NCT03288545) and sacituzumab govitecan (NCT01631552, NCT02574455) have also shown promising results including prolonged survival in treatment groups [166, 167]. These studies highlight the benefit of ADCs and importance of developing combination strategies specific to various cancer types in the future.
2.2.2. Immune checkpoint inhibitors for cancer treatment
A transformative new type of antibody was discovered and characterized in the late 1990s [168]. The T cell immune checkpoint is located on the cell surface and functions as a safety mechanism to inhibit the over-activation of T cells. Under normal circumstances, this mechanism helps prevent autoimmune disease or inflammation-based tissue damage, but during cancer the immune checkpoint can prevent T cells from attacking the tumor, thus weakening the ability of the immune system to recognize and destroy tumor cells [169]. To overcome this challenge, immune checkpoint inhibitors were developed and have revolutionized cancer immunotherapy. The discovery of the CTLA-4 antibody blockade led to a Nobel Prize in Physiology or Medicine awarded to James P. Allison and Tasuku Honjo in 2018 [170]. These molecules prevent repression of T cell activation thus re-establishing the T-cell mediated anti-tumor immune responses [169, 171, 172]. Clinical immunotherapy with monoclonal antibodies to block the CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) or the programmed cell death protein 1 (PD-1)–PD-1 ligand (PD-L1) axes have been FDA-approved for the treatment of cancers such as melanoma, non-small cell lung cancer, Hodgkin lymphoma, Merkel cell carcinoma, carcinoma of the bladder and a few others [100]. In a 2017 phase II clinical trial with unresectable stage III or IV malignant melanoma, Geoerger and colleagues studied the efficacy and safety of ipilimumab (anti-CTLA-4 monoclonal antibody) in adolescents between 12–17 years of age. They developed a human IgG1 monoclonal antibody, anti-CTLA-4, which has been approved for the treatment of advanced melanoma in adults and tested the safety and efficacy of this treatment on adolescents. Two ipilimumab dose groups, 3 or 10 mg/kg every 3 weeks, were established. The authors found that 11 of 12 patients produced serum ipilimumab concentrations that reached the threshold to inhibit CTLA-4 binding to CD80 and CD86 previously seen in adults [173]. This work demonstrates that ipilimumab has a similar safety profile as previously seen in adults and could be a viable treatment option for younger patients with advanced melanoma [173] [NCT01696045]. Unfortunately, this trial was eventually stopped due to slow accrual which is a major challenge with developing therapeutics for rare diseases.
In a four-year phase 3 clinical trial, Ascierto and colleagues report on the efficacy of nivolumab (anti-PD-1 monoclonal antibody) when compared to ipilimumab (anti-CTLA-4 monoclonal antibody) in the prevention of recurrence of melanoma after complete resection of stage IIIb/c or stage IV melanoma. Human IgG4 monoclonal antibodies against PD-1 have been previously approved for the treatment of metastatic melanoma as either a monotherapy or combination therapy. Patients received intravenous (IV) injections of nivolumab at a dose of 3 mg/kg every 2 weeks or IV injections of ipilimumab at a dose of 10 mg/kg every 3 weeks for four doses, and then every 12 weeks for 1 year of treatment, disease recurrence, unacceptable toxicity, or withdrawal of consent. The authors found that after 4 years, the relapse-free survival (RFS) benefit was approximately 10% higher in patients treated with nivolumab but several adverse events were reported for both treatment groups. Diarrhea, diabetic ketoacidosis, and pneumonitis were reported for the nivolumab group, and pneumonitis, colitis (two patients), and two treatment-related deaths were reported for the ipilimumab group. These findings suggest that adjuvant therapy using nivolumab for resected stage IIIb/c or IV melanoma patients was safer and more effective than with ipilimumab [174] [NCT02388906].
Recent animal studies have also tested combinations of checkpoint inhibitors and shRNA therapy to determine if the sum individual therapeutic potentials provide a greater overall antitumor response. Imbert et al. investigated the effect of combined checkpoint inhibitor therapies anti-CTLA-4 or anti-PD-1 with SK1 silencing in treating melanoma models in C57BL/6 mice. The authors developed SK1 silenced Yumm1.7 melanoma cells by transfecting with SK1 shRNA and demonstrated that intraperitoneal injection of 3 doses of checkpoint inhibitor (200 μg per mouse on day 7 and 100 μg per mouse on day 10 and 13 with anti-CTLA-4, or 200 μg per mouse on day 5, 7 and 10 with anti-PD-1) significantly increased tumor rejection rate and overall survival in mice inoculated intradermally with SK1 silenced melanoma cells. Compared to the checkpoint inhibitor treatment of control melanoma, the treatment of SK1 silencing melanoma reached 100% and 67% tumor rejection rate for anti-CTLA-4 and anti-PD-1 treatment, respectively. The authors also found that the therapy also resulted in the decrease of Treg cell infiltration, higher CD8+/Treg ratio in the melanoma tumors, and long-term memory immune response in treated mice using flow cytometry and RT-qPCR. This work rationalized the development of SK1 targeting combined with checkpoint inhibitor immune therapies to increase the therapy response rate [175].
Although immune checkpoint inhibitors are commonly tested for treatment of melanoma, other inclinations have also been evaluated. In a 2010 study, Chung and colleagues reported on the safety and efficacy of tremelimumab (anti-CTLA-4 monoclonal antibody) in patients with treatment-refractory colorectal cancer. Patients enrolled in this study received 15 mg/kg tremelimumab injected IV every 90 days until progression. Unfortunately, 44 out of 45 response-evaluable patients did not reach the second dose (43 due to progressive disease and one due to discontinuation). Further, only mild adverse events were reported including diarrhea (n = 5; 11%), ulcerative colitis (n = 1; 2%), fatigue (n = 1; 2%), autoimmune thrombocytopenia (n = 1; 2%), and hypokalemia (n = 1; 2%). Although the authors found that tremelimumab did not demonstrate meaningful antitumor activity in colorectal cancer, because very few toxicities were reported, this treatment could still be promising for combination therapies with chemotherapies or other drugs in patients with solid tumors [176] [NCT00313794].
More recently, Liu et al. studied anti-PD-1 therapy in BALB/c and C57BL/6J mice with colorectal cancer. The authors identified miR-15b-5p as a target that retards tumorigenesis and further discovered IL-17A as a down regulator for miR-15b-5p. The combined administration of anti-IL-17A antibodies and anti-PD-1 antibodies significantly slowed tumor growth and prolonged survival in mice inoculated with either CT26 or MC38 cells. The authors found an increase in CD8+ T cells and a decrease in MDSCs in mice treated with combined therapy, suggesting that targeting IL-17A is a promising strategy to sensitize colorectal cancer to immune checkpoint inhibitors and encourages further studies to translate IL-17A antibodies into clinical treatments [177]. Antibody blockade treatments have the potential to inhibit the suppression of immune cells caused by anti-inflammatory molecules present in the tumor microenvironment as well as provide partial therapeutic efficacy in combination therapeutics. Interestingly, Wu and colleagues studied how sex correlates with the effectiveness of patients treated with immune checkpoint inhibitors. The authors found a significant sex-related efficacy difference between female and male melanoma patients suggesting that dose regimens, monotherapies, and combination treatments should be closely evaluated for males and females in the future [178]. These results also suggest that grouping the results of clinical and pre-clinical studies may be affecting the results of cancer treatments.
2.2.3. Antibody therapies for autoimmune diseases
Apart from immune checkpoint inhibitor treatments for cancer patients, several antibody therapeutics have also been developed and tested in the context of autoimmune diseases such as systemic lupus erythematosus (SLE). For example, Wallace et al. studied the effects of neutralizing IL-6 in treatment of SLE in human patients in a 2016 phase II clinical trial. The authors developed a recombinant human monoclonal antibody expressed in Chinese hamster ovary cells (CHO) binding to human IL-6 and demonstrated its safety and efficacy through electrochemiluminescence immunoassays to visualize antidrug antibodies (ADAbs) and neutralizing antibodies. Enrolled patients received either placebo (n=45), 10 mg hIL-6 (n=45), 50 mg hIL-6 (n=47), or 200 mg hIL-6 (n=46) administered as two subcutaneous injections over a 24-week treatment period. The 200 mg dose was discontinued due to toxicity findings and was not included in the primary efficacy analysis. Three deaths occurred in patients that were administered 200 mg hIL-6. The 10 mg and 50 mg hIL-6 treatment groups combined showed a reduction in SFI flares (a clinical outcome that increases mortality in SLE patients) when compared to the placebo group. Additionally, these treatments resulted in improvements in SF-36 physical component summary scores, which is a standardized measure of health status, but no significant improvement in the SLE Responder Index, which was the primary efficacy endpoint. Although this monoclonal antibody trial did not show sufficient efficacy in this SLE trial, the results demonstrated in this study support the need for further study of IL-6 targeting pathways and potential combination treatments for increased therapeutic efficacy in SLE patients in the future [179].
Several combination administration methods were also evaluated for RA patients. Lipsky and colleagues studied infliximab, a chimeric monoclonal antibody against TNF-α, in combination with methotrexate (DMARD) on 428 patients who had active arthritis despite methotrexate therapy. The combination treatment of infliximab + methotrexate reduced serum rheumatoid factor values by approximately 40% at 54 weeks and approximately 20% increase in the physical and social component of the SF-36. However, certain adverse effects occurred more frequently in the combination treatment such as upper respiratory tract infection, sinusitis, pharyngitis, and headache. Eight deaths also occurred in the methotrexate and the combination groups of this trial. Overall, this work supports further studies of infliximab + methotrexate treatment that targets TNF-α to treat RA, but extensive improvement in safety considerations is needed [180].
A 2008 phase III clinical trial studied the efficacy and safety of tocilizumab (anti-IL-6 receptor monoclonal antibody) in patients with rheumatoid arthritis refractory to tumor necrosis factor antagonist therapy. The authors used a combination treatment of tocilizumab (humanized anti-IL-6R monoclonal antibody) and methotrexate (DMARD) on patients with moderate to severely active RA and intolerance or failure to respond to TNF antagonists in a phase III, randomized, double-blind, placebo-controlled, parallel-group study. Over half of the patients that received 8 mg/kg Tocilizumab + Methotrexate achieved DAS28 < 3.2, and a third achieved DAS28 < 2.6 at 24 weeks. DAS28 is a measure of disease activity RA. DAS stands for ‘disease activity score’ and a DAS28 of greater than 5.1 implies active disease, less than 3.2 implies low disease activity, and less than 2.6 implies remission. This work provides rationale for further studies of Tocilizumab + Methotrexate combination treatment for rheumatoid arthritis with inadequate response to TNF antagonists, and further studies can examine the potential of using combination therapies with TNF antagonists to achieve remission for RA patients earlier and increase their quality of life [181].
Clinical studies have also evaluated the ability of antibodies treatments to provide disease improvement for patients with MS. Hauser and colleagues studied the efficacy and safety of ocrelizumab, a monoclonal antibody directed against CD20 which depletes B cells, in patients with relapsing MS. The patients were given 600 mg of ocrelizumab every 24 weeks intravenously. The authors found that the percentage of patients with disability progression confirmed at 12 weeks and 24 weeks were significantly lower with ocrelizumab than with interferon beta-1a treatment [182]. A similar study found that ocrelizumab is also more cost-effective than subcutaneous interferon beta-1a for the treatment of relapsing MS [183]. Additionally, Mellion and colleagues studied the efficacy of various doses of opicinumab, a monoclonal antibody that targets LINGO-1, a protein that suppresses the redevelopment of axons, in patients with MS. In this study, opicinumab was given concurrently with intramuscular interferon (IFN) beta-1a. The authors found improvement responses of 51.6% for placebo, 51.1% for 3 mg/kg opicinumab, 65.6% for 10 mg/kg, 68.8% for 30 mg/kg, and 41.2% for 100 mg/kg [184]. These results suggest that there is a dose responsiveness between the 10–30 mg/kg range, but the criteria of the clinical trial were not met and thus also highlights the need for further studies focused on development of effective MS treatments.
Finally, Furie et al. studied the effects of belimumab, a recombinant human IgG-1λ monoclonal antibody that works to inhibit B-cell activating factors, in treatment of active lupus nephritis in human patients. The authors assayed safety and efficacy in a phase III clinical trial. Patients received either IV injections of belimumab at a dose of 10 mg per kilogram of body weight or placebo, in addition to standard therapy (mycophenolate mofetil or cyclophosphamide-azathioprine injections). Dosages were administered on days 1, 15, 29, and then every 28 days until week 100. The belimumab treatment group showed an improvement of renal-related disease symptoms with 43% of patients having a primary efficacy renal response and 30% of patients having a complete renal response. Unfortunately, a total of 11 patients died during the trial (6 out of 224 in the belimumab group and 5 out of 224 in the placebo group). These results are consistent with previous clinical trials involving belimumab [185, 186] and provide rationale for further study of belimumab in conjunction with standard therapy (cyclophosphamide or mycophenolate mofetil) as an overall therapy for SLE [187]. Antibody therapies have been revolutionary for managing flare ups of inflammation in autoimmune diseases and newer and more specific antibodies are continuously being developed and evaluated.
2.3. Oncolytic virus therapies
Oncolytic viruses represent a novel drug class of genetically engineered or naturally occurring viruses that selectively replicate in and kill cancer cells without harming normal cells or tissues [188]. There is only one currently FDA-approved oncolytic virus therapy: T-Vec (talimogene laherparepvec), a second-generation oncolytic herpes simplex virus type 1 (HSV-1) engineered with GM-CSF, which was approved in 2015 for treatment of advanced melanoma [189, 190]. T-VEC selectively replicates in tumor cells and is designed to express GM-CSF, which promotes the maturation and function of dendritic cells, which activate anti-tumor T cells through antigen presentation of tumor derived antigens. Activated T cells specifically attack and eliminate tumor cells with matching antigen profiles, leading to enhanced anti-tumor effects [191]. The concept of oncolytic virus therapy has been around for many years and tumor regression has also been observed during or after a systemic viral infection [192]. Many clinical trials were performed in attempts to treat cancer with viruses but were not deemed effective as therapeutic reagents because there was no known method to simultaneously control the virulence and retain viral replication in cancer cells [193]. Modern technology has afforded the development of cancer cell-specific replication by engineering the virus genome [188].
Anti-tumor efficacy of ZD55-IL-24 (an oncolytic adenovirus ZD55 harboring IL-24) in treating pancreatic cancer was evaluated in C57BL/6 mice by He et al. in 2013 [194]. The authors constructed ZD55-IL-24 recombinant virus and infected panc-o2 cells, which were used to inoculate C57BL/6 mice by subcutaneous injection. The ZD55-IL-24 infected panc-o2 cells displayed significantly less tumor growth than a control group where panc-o2 cells were infected with ZD55-EGFP control virus. The authors further evaluated the effect of the therapy in inducing tumor apoptosis and increasing CTL secretion of γ-IFN and IL-6 using flow cytometry, cytotoxicity assay, and ELISA. Overall, this work demonstrated the feasibility of an oncolytic IL-24 virus for reducing the survival rate of tumor cells in vivo and highlights a first step towards clinical translation of oncolytic virus therapies. However, a demonstration of safety in mice does not readily transfer to safety in humans so a larger animal model is needed to justify dosing and treatment feasibility, which rationalizes this therapy as a candidate therapeutic approach for treating pancreatic cancer [194]. Another example of pre-clinical oncolytic viral therapy for immunotherapy was described by Wang and colleagues in 2017. This group evaluated the efficacy of a newly designed IL-12 in decreasing cytokine-mediated toxicity and reducing pancreatic cancer tumor burden in Syrian hamsters. The authors developed this IL-12 platform by deleting the N-terminal signal peptide and delivered the therapy using a tumor-targeted oncolytic adenovirus (Ad-TD-nsIL-12) via intratumoral or intraperitoneal injection. Six doses of the therapy were needed to successfully eradicate subcutaneous HPD1NR pancreatic tumors in the hamsters. The Ad-TD-nsIL-12 also significantly increased the survival rate in murine SHPC6 dissemination and HapT1 pancreatic cancer orthotopic models without causing severe toxicity. The authors further explored the effect of Ad-TD-nsIL-12 on various T-cell functions and activation states as well as the ability to reduce the release of pro-inflammatory cytokines in vivo using flow cytometry, and immunohistochemical staining. This work demonstrated a potential means of delivering IL-12 as an immunomodulatory therapy for pancreatic cancer and supports further analysis of the long-term effects and toxicology of the drug as it is potentially translated into a clinical-stage product [195].
A major hurdle for the field of oncotherapies is the inability to overcome the immunosuppressive tumor microenvironment of many tumor types. Researchers have developed many creative strategies in efforts to overcome this barrier including mechanical stimulation and targeted proteases. This topic has been recently reviewed by Aghlara-Fotovat and colleagues [196]. Controlled modulation of the tumor microenvironment has the potential to revolutionize oncotherapeutics in the future.
2.4. Cell-based therapeutics
Autologous cell therapies are one of the most rapidly expanding sectors of immuno-oncology and are leading the next generation of immunotherapy approaches. However, one of the biggest limitations of these therapies is their inability to persist within the body for extended periods of time [197, 198]. Even in the case of cancers where the immune system is repressed, cell therapies are often rejected by the host immune system within a few days, rendering them ineffective as long-term therapeutics [198]. Nevertheless, there have been some major cell-engineering and delivery feats that have proved beneficial for modulating the immune system.
2.4.1. CAR-T cell therapies
Among these advances is the development of engineered T-cells with tumor-targeting receptors, the chimeric antigen receptor (CAR)-T cell therapies [199, 200]. Briefly, T cells are isolated from a patient and modified ex-vivo. During this process, the T cell receptor (TCR) is replaced by CAR which includes an extracellular and an intracellular domain. The extracellular domain is typically a single-chain antibody fragment (scFv) that is against a specific cell surface antigen, while the intracellular domain includes fused signaling domains from a natural TCR complex and costimulatory molecules [201]. Different intracellular domains correspond to the various CAR-T cell generations [197, 202, 203]. The structure ranges from CD3z signaling domain alone in first-generation CARs (lack of costimulatory signal) to those that possess the signaling endo-domains of costimulatory molecules like CD28, CD134 (OX40) or CD137 (4–1BB), which are fused with CD3z, in second and third generation CARs [197, 203]. These engineered T cells are then expanded ex-vivo and then infused back into the patient with enhanced tumor-targeted capabilities. There are currently four CAR-T cell therapies FDA approved for the treatment of B cell malignancies based on their efficacy in clinical trials [204]. These include tisagenlecleucel (Kymriah), axicabtagene ciloleucel (Yescarta), brexucabtagene autoleucel (Tecartus), and, most recently, lisocabtagene maraleucel (Breyanzi) [204, 205]. These CAR-T cell products target CD19 and have induced durable clinical responses in refractory B cell malignancies.
Many groups have tested CAR-T cells in clinical trials and seen varying degrees of success. In 2014, Maude and colleagues studied the efficacy of CAR-modified T cells targeting CD19 in patients with refractory disease (relapsed acute lymphoblastic leukemia). The authors infused autologous T cells with CD19-directed CAR lentiviral vector (CTL019) and demonstrated the efficacy of the treatment using flow cytometry and PCR assays. CTL019 treatment led to complete remission in 27 patients (90%) including 2 with blinatumomab-refractory disease and 15 who had stem-cell transplantation. The sustained remission was also achieved with an overall survival rate of 78% up to 24 months [206].
In a 2018 phase I/IIa clinical trial, Enblad et al. studied the safety and efficacy of third-generation CAR T cells targeting CD19 in a Phase I/IIa trial for fifteen patients with relapsed, or refractory, B-cell lymphoma or leukemia. Of the fifteen, eleven patients had lymphoma and the remaining four had acute lymphoblastic leukemia (ALL). In phase I, the patients were treated with increasing doses of CAR-T cells ranging from 2 × 107 to 2 × 108 cells/m2. If no serious toxicity occurred at lower dose levels, patients were treated with 2 × 108 cells/m2 in phase IIa. The CAR-T cells were moderately toxic and a total of 154 related adverse events (between grade 1 and 4) were recorded, and four patients were hospitalized due to adverse reactions. Also, there was no apparent correlation between the CAR-Tcell dose and patient response. CAR-T treatment showed an initial complete response in 6 of the 15 treated patients (4 of 11 for lymphoma patients and 2 of 4 for ALL) but this trend did not persist throughout the study. Two of the lymphoma patients that did not respond to CAR-T treatment despite persisting CAR-T cells in blood were given one dose of anti-PD1 antibody therapy (nivolumab), but this did not convert progression. Overall, 2 of the 15 patients are surviving long-term but with persistent disease (>27–36 months). It is worth noting that the remission rates of this third-generation CAR-T clinical trial are very similar to second-generation CAR-T cells. Additional studies are needed to determine what, if any, appropriate dose/combination of immunotherapies will work in combination with CAR-T cell treatment to provide a robust and reproducible anti-tumor response for patients with lymphoma or ALL [207].
The development of new and improved generations of CAR-T cells is continuously evolving. In 2020, Gu and colleagues reported on the safety and feasibility of chimeric antigen receptor-modified T cell therapy in both pediatric and adult patients with relapsed/refractory acute lymphoblastic leukemia. The authors developed a new CD19 CAR T cell (HI19α-4-1BB-ζ CAR T) called CNCT19 and demonstrated the anti-leukemic activities in a human clinical study (open-label, single-center, and single-arm pilot study) by using flow cytometry, surface marker staining, Lee’s grading system for cytokine release syndrome, and complete remission (CR) rate and relapse measured by National Comprehensive Cancer Network (NCCN) guidelines. CNCT19 showed potent anti-leukemic activities with 90% of 20 patients reaching complete remission or complete remission with incomplete count recovery (CRi) within 28 days. A patients’ disease status was classified as CRi if blood counts met the requirements of complete remission with the exception of platelet and neutrophil counts below normal thresholds. The study results (Figure 6) provide a strong rationale for the ability of CAR-T cells to function as potent immunostimulants and suggest that further development of CNCT19 modified CAR-T cell therapy for patients with relapsed/refractory acute lymphoblastic leukemia could show increased patient survival [208]. CAR-T therapy has also been tested in combination with other treatments such as pro-inflammatory cytokines. For example, the antitumor CAR-T cell response in combination with mesenchymal stem cells (MSCs) engineered to release pro-inflammatory cytokines IL-7 and IL-12 for treatment of colorectal cancer in NOD-scid gamma (NSG) mice. The authors developed a gamma-retroviral vector to generate a human IL-7/IL-12 construct, engineered CARs with a modified CD28-CD3ζ signaling domain, and modified T cells for CAR expression in a murine colorectal cancer model. They found that CAR-T cells and IL-7/IL-12 MSCs were tumor-free and prolonged survival in comparison to CAR-T cells in the presence or absence of MSCs and IL-7/IL-12 secreting MSCs. This work shows that combination cell therapies can be used to modulate the inflammatory tumor microenvironment and can form an interaction between modified MSCs and CAR-T cells for anti-tumor effects for colorectal cancer [209].
Figure 6.
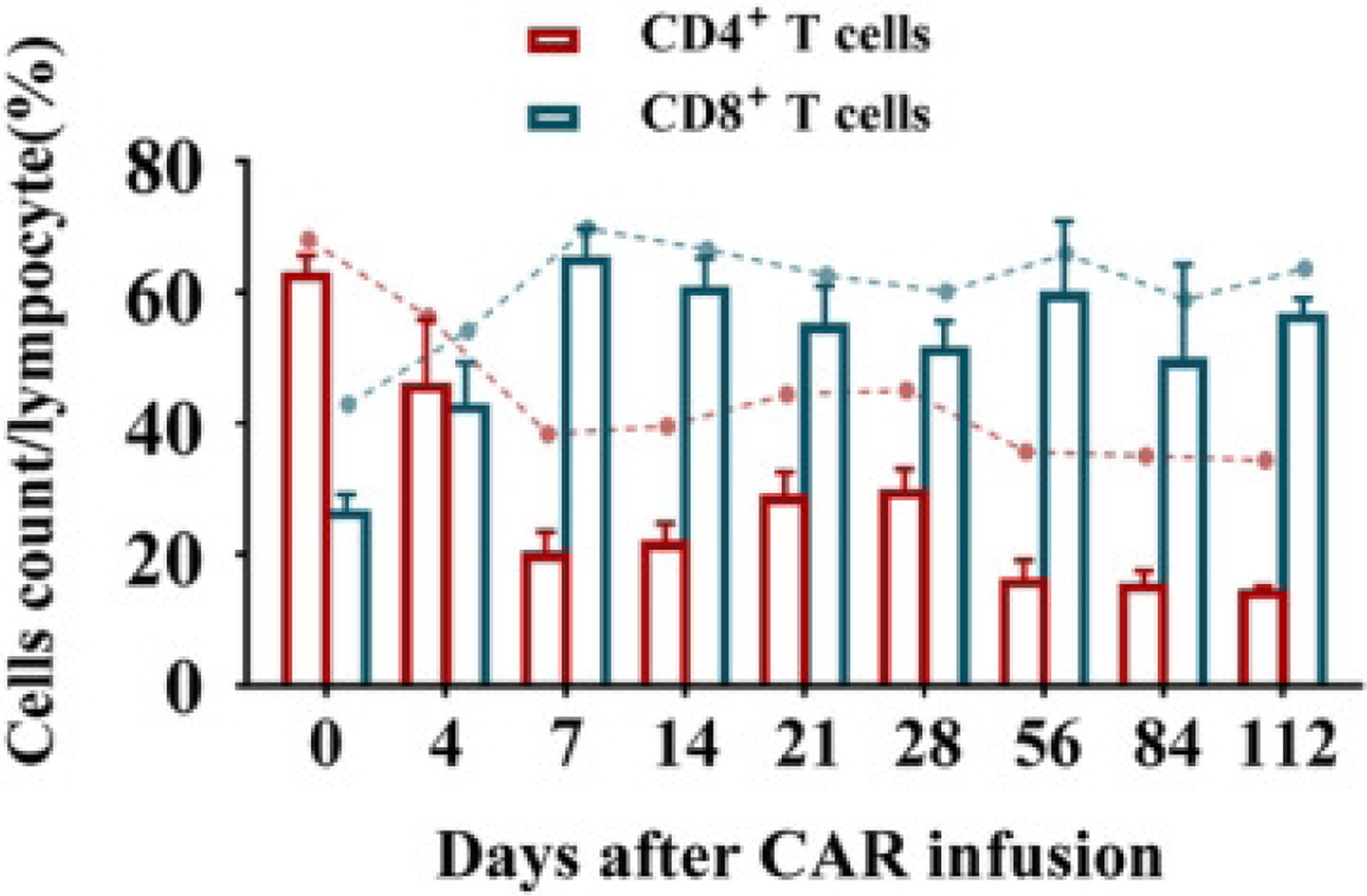
The expansion and percentage of CD4+ or CD8+ T cells over time in peripheral blood after CAR infusion. Reprinted with permission from ref. [190].
2.4.2. CAR-NK cell therapies
Similar to CAR-T cells discussed above, NK cells can also be engineered to express receptors that enable enhanced recognition and cytotoxicity towards specific cells or antigens using CAR technology [210]. Recent CAR constructs designed for NK cells have shown increased cytotoxicity and pro-inflammatory cytokine production, which highlights their potential for use as a cancer therapeutic [210]. The engineered receptors allow for CAR-NK cells to recognize the absence of HLA-proteins, which allows them to specifically identify malignant or virally infected cells and become activated [211]. Once stimulated, CAR-NK cells produce interferon-gamma (IFN-γ) and granulocyte-macrophage colony-stimulating factor (GM-CSF) to activate the immune response [210]. However, there are some limitations, such as safety and efficacy concerns. Scientists have attempted to mitigate these limitations through the integration of suicide genes into CAR constructs, or silencing NK inhibitory receptors, which will make the CAR-NK cells more resistant to tumors [212]. Although CAR-NK cells are most commonly interrogated for the anti-tumor potential, these engineered cells have also shown promise as a therapeutic intervention in autoimmune diseases like lupus to specifically target autoreactive immune cells [213]. To function as an immune cell attacker, CAR-NK cells can be equipped with specific ligands to bind to and deplete autoreactive B cells via cytolysis [213].
Oelsner and colleagues studied the effects of CAR-NK cells on pre-B cell acute lymphoblastic leukemia (B-ALL) using NSG mice. The authors engineered NK-92 cells by lentiviral gene transfer to express an FMS-like tyrosine kinase 3 (FLT3)-specific CAR with a composite CD28-CD3ζ signaling domain (overexpression of FLT3 has been reported in B-ALL). Mice were treated using an intravenous injection of 1 × 107 CAR-NK or control cells once a week for 4 weeks. The authors reported inhibited disease progression in mice treated with CAR-NK cells, which suggests that this treatment is feasible for future studies and development. To increase the safety and address any toxicity concerns or potential off-target effects, the authors also incorporated and evaluated an inducible caspase-9 (iCasp9) suicide switch. Excitingly, after the administration of iCasp9 chemical dimerizer AP20187 to the CAR-NK- cells, rapid iCasp9 activation, and cell death were observed, highlighting the feasibility of the suicide switch safety feature in cell therapy applications [214].
Liu and colleagues studied the antitumor effects of NK cells modified to express anti-CD19 CAR for the treatment of B-cell cancers such as non-Hodgkin’s lymphoma and CLL. The authors engineered NK cells to express anti-CD19 CAR, IL-15, and inducible caspase 9 in 11 adult patients. Patients received a single infusion of one of the three doses, 1×105, 1×106, or 1×107 cells. They found that CAR-NK cells were not associated with the development of cytokine release syndrome, neurotoxicity, or graft-versus-host disease and that 8 of 11 patients (73%) had an objective response as early as 30 days after infusion. This work showed that the engineered NK cells could help treat patients with relapsed or refractory CD19-positive cancers [215]. Prior to this work, Liu and colleagues also evaluated cord blood NK (CB-NK) cells modified to express iC9/CAR.19/IL-15 in a xenograft Raji lymphoma murine model. The authors transduced CB-NK cells to incorporate the genes for iC9, CAR-CD19, and IL-15 and demonstrated the efficacy and safety of using their modified NK cells using IVIS, flow cytometry, and SNP microarray analysis. They found they could eliminate the potential toxicities with the inducible suicide gene and could prolong the survival of murine models with their modified NK cells [216]. NK cell therapies have rapidly emerged and shown promise as immunotherapeutics, but there is much work left to do in order to improve the safety and efficacy of these cell-based treatments.
2.4.3. Dendritic cell therapies for cancer treatment
Dendritic cells (DCs) have also been targeted for the development of novel anti-cancer therapeutics for nearly two decades [217]. DCs are antigen-presenting cells that are essential in the initiation of an immune response [217, 218]. In a normal steady condition, DCs concomitantly process and present antigens on major histocompatibility complex (MHC) class I and II [218]. Due to their superior ability to activate CD8+ T-cells, DCs have recently been used to elicit powerful immune responses against tumor cells. In such DC therapies, DCs are obtained from a patient and presented with cancer antigens ex vivo. The primed cells are then re-administered in order to provoke an immune response toward tumor elimination, acting as a therapeutic vaccine [219].
Dendritic cell vaccines have been used to treat several aggressive cancers including pancreatic ductal adenocarcinoma (PDAC) [217]. PDAC is a metastatic cancer with a 5-year survival rate of less than 5% [220]. In 2019, Yang et al conducted a study to improve the therapeutic efficacy of dendritic immunotherapy in a transgenic mouse model of PDAC [217]. The authors extracted immature dendritic cells from harvested bone marrow cells, which they then primed with pancreatic tumor cell lysates. The cells were subsequently injected intraperitoneally in tumor-bearing mice. The authors reported that their dendritic cell vaccine resulted in an overall reduction in tumor volume, and extension of survival in both transgenic KPC and orthotropic KPC models of PDAC. Additionally, they concluded that intraperitoneal injection of the DC vaccine improved migration to the draining lymph nodes, which is critical for induction of cytotoxic T lymphocytes. Despite their therapeutic results, the authors were not able to achieve complete tumor eradication and observed tumor metastasis to the lungs, liver, and pancreatic draining lymph nodes indicating that improvements still need to be made to this therapy before it can be considered curative [217].
Prostate cancer is another aggressive cancer that has the potential to benefit from dendritic cell immunotherapy [221]. In 2000, Lodge et al. reported a phase II clinical trial where 107 patients were infused with autologous dendritic cells, pulsed with two human leukocyte antigens (HLA)-A2 binding prostate cancer derived peptides, once every week for six weeks. The authors evaluated the immune status of the patients to both non-specific and peptide specific antigens. They found that based on both delayed type hypersensitivity responses to recall antigens, and cytokine secretion, their dendritic cell infusion did not cause a significant peptide specific response. However, based on the non-specific response, they were able to conclude that low IFN-γ production in response to antigen presentation correlated with poor clinical response, highlighting the importance of a functional immune system in the treatment of cancer. Of all the participants included in the study, the authors reported one complete responder who demonstrated a Th-1 type immune response to the immunizing peptides. Overall, the vaccine elicited poor therapeutic efficacy; however, this may be a result of the antigen selected. At the time of this publication, there were no reports on the T-cell reactivity against prostate-specific membrane antigen-derived peptides. This highlights one of the drawbacks of dendritic cell therapy, which is the identification of the optimal anti-tumor antigen [221].
Since 1998 when Nestle et al. first reported the efficacy of dendritic cell vaccines in treating melanoma, several studies and clinical trials have been conducted [222]. In 2012, a phase 2 clinical trial reported that injection of DCs pulsed HLA-A24 or A2 melanoma-associated synthetic peptides could be an effective therapy against metastatic melanoma. In the study, pulsed dendritic cells were injected subcutaneously in the inguinal region of 24 patients expressing either HLA-A24 or A2 melanoma. Patients received up to 10 injections over the span of 5 months. At the end of the study, of the 24 participants, 1 had partial remission, 7 had stable disease, and 16 had progressive disease. 75% of the participants had positive ELISPOT reactions against all melanoma antigen-related peptides, which is indicative of the number of positive cytotoxic T cell (CTL) responses. Overall, compared to non-vaccinated patients, vaccinated patients had significantly longer average survival times (13.6 months vs 7.3 months respectively). However, despite the ability of the vaccine to prolong survival, several questions remain unanswered including information regarding prognostic factors. Interestingly, the authors found that the anti-MAGE-A1 antibody titer before vaccination was a good prognostic factor, while dendritic cell processing-related parameters such as the number of injected DCs, and the presence of surface markers, such as CD83 and CCR7, was not. CD8 and IL-17 were also reportedly not involved in prognosis, in contrast to other studies that found close involvement in the prognosis of solid cancer patients. Additional studies that address these questions are critical in the improvement of dendritic cell therapy [223].
Glioblastoma, which is one of the most aggressive primary malignant brain tumors, has also reportedly been treated with dendritic cell therapy. In a phase III clinical trial conducted in 2018, autologous dendritic cells pulsed with tumor lysate were injected into patients who had previously undergone surgical resection and chemotherapy, which is the standard of care for glioblastoma [224]. The vaccine was administered intradermally 6 times in the first year and twice a year thereafter and was co-administered with a temozolomide adjuvant. At the time of publication, the study was ongoing, but the median overall survival for patients was 23.1 months, suggesting that patients are living longer than expected; reported median overall survival of 16 and 17.4 months has been previously observed in standard of care clinical trials. Despite being incomplete, the data at the time of publication indicates the potential of this vaccine to extend the survival of patients with glioblastoma. Importantly, only 7 out of 331 patients experienced any adverse events related to the treatment, which highlights the safety profile of this technique. The combination of dendritic cell vaccine and temozolomide therapy utilized in this clinical trial also indicates the potential for the therapeutic efficacy of dendritic cell vaccines to be improved by combining treatment with immune checkpoint inhibitors or other immunotherapies, an area that researchers should explore further [224].
2.4.4. Dendritic cell therapies for autoimmune diseases
Though there are several applications of dendritic cell therapy in cancer, autoimmune diseases have also seen benefits from DC therapy. Specifically, tolerogenic DCs (tolDCs) are essential in maintaining immune tolerance [225]. Studies have shown that complete depletion of DCs can result in fatal autoimmunity [226]. It is thought that tolDCs exercise their function through several characteristics, including their lack of co-stimulatory molecules [227]. Without co-stimulatory molecules, tolDC interactions with T-cells result in T-cell anergy and decreased proliferation [225]. Thus, delivery of tolDCs is being investigated for the treatment of various autoimmune diseases.
T1D was the first autoimmune disease to be targeted via tolDCs in a clinical trial in 2007 [228]. Patients in the trial were dosed with either control DCs or immunosuppressive DCs-grown ex vivo with antisense oligonucleotides targeting CD40, CD80, and CD86 co-stimulatory molecules. Overall, the study results showed that tolDCs are tolerated well, with no adverse events observed, outlining the safety profile of this novel therapy. Additionally, the authors noted that in 57% of participants (4/7), there was an emergence of detectable C-peptide, a surrogate marker for functional beta cells [229]. This suggests that tolDC therapy may restore insulin production, outlining a promising area for further research.
Rheumatoid arthritis is another promising target for tolDC therapy. Approximately 70% of RA patients have anti-citrullinated peptide autoantibodies in their serum that target citrullinated proteins, causing chronic inflammation [230]. In 2012, the first-in-human clinical trial using tolDCs for RA was reported whereby autologous DCs were modified with a nuclear factor kb (NF-kB) inhibitor and exposed to various citrullinated peptide antigens before administration. Eighteen participants received intradermal injections of either a low dose or high dose of the therapy, Rheumavax. Aside from the excellent safety profile, the results of the study also showed promising biological activity. The authors reported a 25% decrease in T effector cells and a 25% increase in T regulatory cells one month after treatment [231]. Other clinical trials have also reported utilizing dexamethasone (Dex), a synthetic glucocorticoid with anti-inflammatory effects, to reduce symptoms associated with RA (NCT01352858). Specifically, tolDCs cultured with Dex are characterized by a decrease in the expression of co-stimulatory molecules CD40, CD86, CD83, as well as MHC II expression. Additionally, they express high concentrations of anti-inflammatory cytokine IL-10.
Clinical trials have also began to evaluate the feasibility and safety of myelin-derived peptide-pulsed tolDCs in MS patients (NCT02618902). In a 2015 study, the myelin peptide pre-loaded tolDCs are treated with vitamin D3, which induces resistance to maturation (low expression of co-stimulatory molecules) [232]. The manipulated cells are then injected intradermally every two weeks for a total of three injections. The study is set to be completed by November 2021; however, their pre-clinical results show that T-cells stimulated with myelin-derived peptide-pulsed vitamin D3-treated tolDCs are unresponsive to the myelin peptide but maintain responsiveness to unrelated antigens such as CMV, highlighting the potential mechanism for therapeutic efficacy [233–235].
Crohn’s disease, a gastrointestinal autoimmune disease, has similarly been targeted by tolDC therapy. In 2015, a phase I clinical trial aimed at determining the safety and tolerability of autologous tolDCs was reported. Participants were injected intraperitoneally with either a single dose of tolDCs generated ex vivo at escalating doses or bi-weekly injections at the same escalating doses. The patients were evaluated for up to 1 year for adverse effects. In this instance, tolDCs were cultured with Dex and Vitamin A, along with a cocktail of cytokines to develop semi-mature tolDCs with high IL-10 production in response to inflammatory stimuli (i.e., lipopolysaccharide). Vitamin A is normally converted to retinoic acid by retinaldehyde dehydrogenase (RALDH) positive DCs, which helps maintain tolerance to GI cells and tissue, which may explain its inclusion in the tolDC culture conditions [236]. Overall, tolDC therapy was not associated with any adverse events, and of participants treated, clinical remission was seen in 11%, and clinical response was seen in 22%.
Finally, tolDCs are also being utilized to improve transplantation outcomes and reduce the risk of allograft rejection. The primary immunological mechanism through which allograft rejection occurs is the innate non-specific reaction and the donor-specific adaptive response [237]. Donor APCs migrate to the lymphoid organs and interact with T-cells to promote differentiation into effector T-cells, which travel to the graft and elicit their immunological effect [238]. The inflammation that occurs during transplant helps to maintain the adaptive T-cell response, perpetuating the risk of rejection. Though immunosuppressive drugs have significantly improved 1-year graft survival, long-term immunosuppression can increase the risk of developing, outlining a space for alternatives [239]. Cell-based therapies are an innovative approach that may promote the long-term acceptance of transplanted grafts.
At the University of Pittsburgh, a phase I, dose-escalation trial that aims to evaluate the effect of a single infusion of autologous tolDCs along with a immunosuppressive drug regimen prior to renal transplant is on-going [240]. Two other phase I/II trials are investigating the safety and efficacy of a single infusion of tolDCs one week before liver transplant with primary data collection set for 2023 (NCT03164265, NCT04208919).
Overall, there are several advantages of using dendritic cells as a form of immunotherapy. Considerable progress has been made in understanding the immune system and the involvement of dendritic cells in T cell activation. However, several challenges remain that must be considered before dendritic cell vaccines can take the place of conventional treatment approaches. Some of these challenges include the identification of optimal anti-tumor antigens and adjuvants, determining the best route of administration, and the timing of treatment to achieve therapeutic efficacy. Addressing these limitations will help improve the overall translation of dendritic cell immunotherapy into the clinic.
2.4.5. Cells for payload delivery
In addition to being engineered to express CAR receptors, cells can also be modified to “carry” payloads to a destination. One of the biggest advantages of using non-T cell-based cell delivery is that a specific target antigen is not required. Antigen presenting cells, for example, survey many types of cells and pathogens and can interact with their surroundings more efficiently than T-cells. In a 2020 report, scientists developed soft discoidal particles referred to as cell “backpacks”, composed of a cell-adhesive poly(lactic-co-glycolic) acid (PLGA) layer, a polyvinyl alcohol (PVA) layer, and a second PLGA layer which allowed for adherence to macrophage cell surfaces without being phagocytosed for several days. These backpacks encased the pro-inflammatory cytokine IFN-γ and were as reported to cause preferential maintenance of M1 macrophage phenotypes in healthy and tumor-bearing mice demonstrating precise immunomodulation (Figure 7). Treatment of 50ng of IFN-γ worth of backpacks in mice with metastatic breast cancer led to delays in tumor growth when compared with control mice. The authors report that 37 days after inoculation, tumors of mice receiving IFN-γ backpack therapy were 51.9% smaller than those of mice receiving injections of saline [95].
Figure 7.
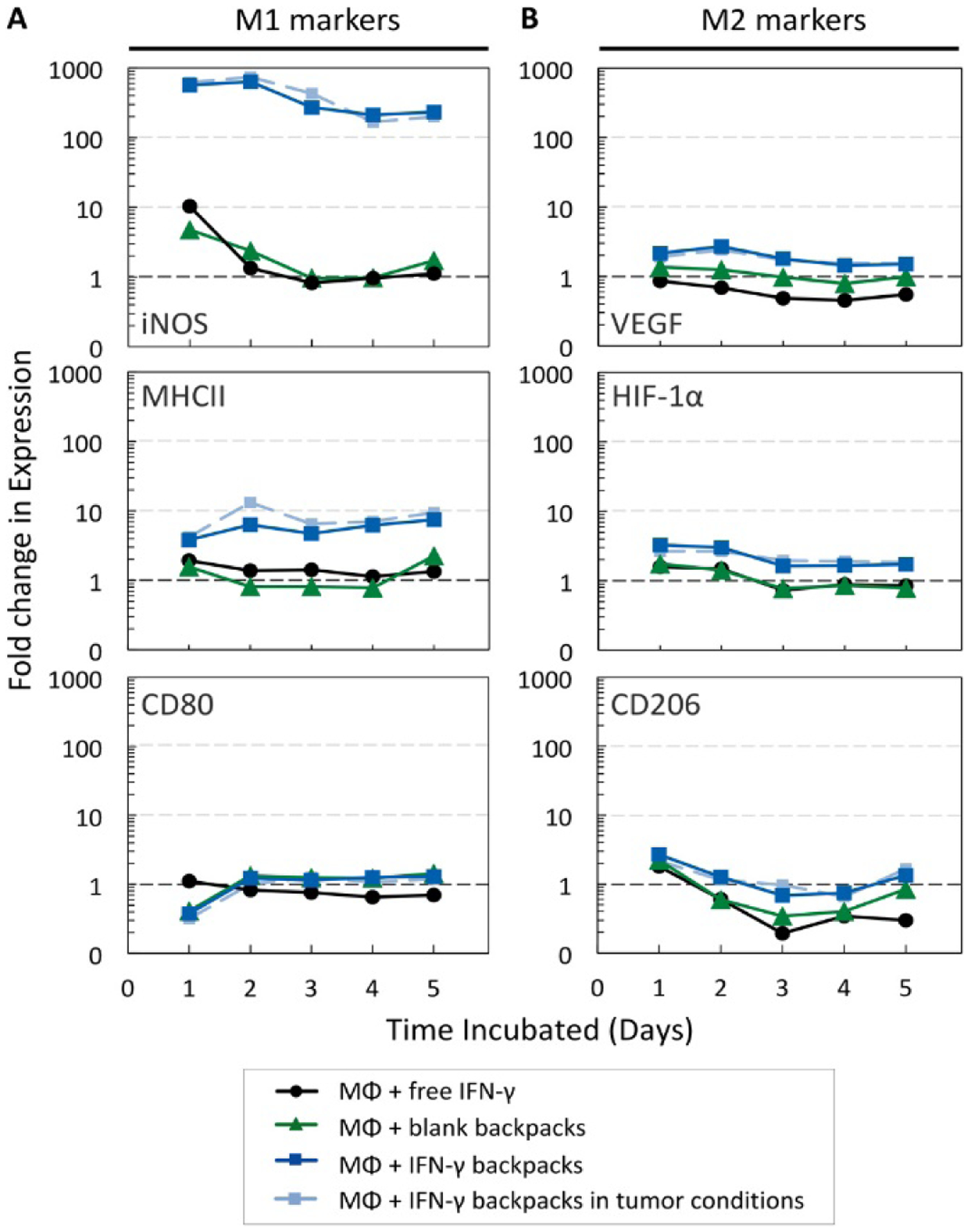
Bone marrow–derived macrophages were cultured for 5 days with free IFN-γ, blank backpacks, or IFN-γ backpacks. Expression of representative (A) M1 markers (iNOS, MHCII, and CD80) and (B) M2 markers [vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1α (HIF-1α), and CD206], relative to unpolarized macrophages. Graphs are logarithmic (n = 10,000 events per data point). Reprinted with permission from ref. [96].
The future of autologous cell therapy lies in their ability to persist within the body long enough to impart the necessary therapeutic effect and minimize toxicities such as treatment-related cytokine release syndrome and neurologic toxicity, which limit patients’ ability to complete treatment regimens [200, 204, 241]. These challenges will be essential to overcome for future generations of CAR-T, CAR-NK, dendritic cells therapies, and more as they move towards clinical translation.
2.5. Multiple therapies in combination
Combination therapies have tremendous potential to revolutionize the future of immunotherapies, but it is imperative to determine the correct existing therapies to administer together and the most appropriate dosing scheme for a particular ailment. A few recent studies have shown early success. Hu et al. designed a triple combination regimen to enable long-term survival in mice bearing colorectal cancer. The therapy was composed of relaxin (RLN), FOLFOX (combination of 5-fluorouracil, leucovorin, and oxaliplatin), and IL-12 plasmid DNA. Lipid nanoparticles surface modified with aminoethyl anisamide were used to encapsulate RLN and the IL-12 plasmid, which were delivered intravenously. The FOLFOX, however, was delivered intraperitoneally. The RLN gene therapy was administered first, followed by the combination of FOLFOX and IL-12 gene therapy in a span of 17 days. The authors discovered that triple combination therapy resulted in tumor regression and completely blocked liver metastases in 50% of the mice. The triple combination therapy was able to preferentially increase the percentage of CD3+CD8+ cytotoxic T cells without causing Treg cell expansion. This work proposed a rational combination strategy that results in tumor regression after colorectal cancer metastasis and thus provides a framework for future development of combination therapies for colorectal cancer in future clinical translations [242].
In a recent combination therapeutic study, Hu and colleagues developed a hyaluronic-acid-based reservoir encapsulating a triple combination treatment. These gels consisted of human chondroitin sulfate proteoglycan 4-targeted CAR-T cells (CSPG4; CSPG4.CAR), IL-15 loaded nanoparticles, and human platelets conjugated with the anti-PDL1 blocking antibody (aPDL1). The authors explain that surgery-induced inflammation triggers the activation of platelets which leads to the release aPDL1 antibodies. This combination treatment was tested for anti-tumor efficacy in NOD-scid Il2rgnull (NSG) mice inoculated with subcutaneous WM115 tumors. The luminescent signal (which was used as an in vivo surrogate for tumor burden) was found to be ~60-fold lower in mice treated with the triple combination therapy than control mice when quantified 3 weeks post-treatment [243]. Although relatively complex, these combination studies are imperative for elucidation of treatment modalities that function well together for various tumor types.
In a phase Ib clinical trial, Montfort and colleagues studied the safety and efficacy of Nivolumab and Ipilimumab (immune checkpoint inhibitors) in combination with Infliximab or Certolizumab (monoclonal anti-TNFα antibodies) for treatment of patients with advanced melanoma. Patients received nivolumab (1 mg/kg) and ipilimumab (3 mg/kg) combined with infliximab (5mg/kg) or certolizumab (400/200 mg). The authors reported four complete responses and three partial responses in the certolizumab combination treatment and one complete response and two partial responses for the infliximab combination treatment [244]. These results further highlight the importance of determining the most effective combination of drugs for a particular disease.
3. The Transition from Discovery to FDA-regulated Preclinical Studies and Clinical Grade Manufacturing
Before a new therapy can be tested in people, an Investigational New Drug (IND) Application must first be submitted, containing manufacturing and nonclinical data to support the safety of the proposed First-in-Human (FIH) trial [246]. The development of immunomodulatory therapies may present unique challenges for meeting FDA expectations, as outlined by the examples below.
3.1. Challenges for Preclinical Pharmacology and Toxicology Studies
The path to a FIH clinical trial can be complex for the translation of new immunotherapies. Small molecules and some of the first biologics developed, such as monoclonal antibodies, often have a single, clearly defined, mechanism of action and a well-established regulatory path to the clinic. By contrast, many new immunotherapies are increasingly complex and impact biology by multiple mechanisms and overlapping signaling pathways [247–249]. This complexity can present challenges in demonstrating pharmacology and safety either in vitro or in animal models.
For example, development of human-derived biologics, such as human cytokines and cellular therapies, can face hurdles in preclinical development because these therapies are often not compatible with immune competent animal models and require immune suppression or immunodeficient species for in vivo studies [250]. Therefore, preclinical development of complex human-derived biologics often requires (1) extensive preclinical characterization of cross-reactivity with multiple species; (2) generation of a suitable ‘surrogate’ product that is compatible with rodents or other species; and/or (3) testing the intended clinical product in immunocompromised animals [251]. These approaches naturally come with challenges. Preliminary assessment of species cross-reactivity may be overlooked during early-stage development when research efforts are focused on identifying a good model to demonstrate Proof-of-Concept. The use of immunocompromised animals may allow for in vivo testing of the intended clinical product but could generate data that is not clinically relevant, especially for products that rely on interactions with an intact immune system for their biological activity. The use of surrogate products (e.g., murine-derived cells or proteins) may help demonstrate proof-of-mechanism in relevant disease models, but the surrogates may be significantly different from the intended clinical product such that it is difficult to obtain relevant safety data in preclinical models. A solid regulatory strategy is therefore needed early in the transition from research to IND-enabling studies in addition to well-timed discussions with the FDA. Sponsors should take advantage of the opportunity to discuss the nonclinical studies at both an INTERACT meeting [252] and a pre-IND meeting [253] if possible. The Office of Combination Products can also be consulted to obtain feedback on whether a novel therapy will be regulated as a drug, biologic, device, or any combination thereof [254].
Since biologics have the potential to induce an immune response that is species-specific, the immune system response to a new drug product must be carefully characterized in animal models. While preclinical assessment of immunotoxicity may be more straightforward for small molecules and recombinant proteins [255], there are limitations to the assessment of immunotoxicity of complex biologics such as cell and gene therapies. A xenogeneic immune response to human proteins or cells may limit the use of a particular species in preclinical studies, and may not be predictive of the potential immune response expected in human patients [256].
3.2. Transitioning to Clinical Grade Manufacturing
In parallel to generating the required pharmacology and safety data, development of immunotherapies must also confront several challenges in ensuring tightly controlled manufacturing of clinical-grade material. Biologics in particular, such as recombinant proteins or more complex cellular therapies require much more specialized manufacturing processes [257, 258]. The use of cell lines or cell-culture systems to generate biologics is also time-consuming and labor-intensive and carries a high regulatory burden for the control of materials used in the manufacturing process. Manufacturing that requires genetic engineering, through viral vectors or other newer methods such as CRISPR/Cas9, also must be tightly controlled from start to finish.
Regardless of the specific manufacturing process, a set of release criteria must also be established for the clinical-grade product [246]. Release criteria for the clinical product should be considered early in development when preclinical material is being manufactured for pharmacology and safety studies. Release criteria must be designed to address potential issues of impurities or variability in the clinical grade manufacturing process. Addressing variability in manufacturing may be particularly difficult with autologous cell therapy products since the starting material for each lot is patient specific. Release criteria must also include a potency assay, which is often tied to the primary mechanism of action of the product. For complex immunotherapies with multiple mechanisms of action, potency assay development can be a major challenge, and this is also an area of evolving regulatory requirements [259].
While early preclinical development can occur in parallel with manufacturing development, every therapeutic program will reach a point where the manufacturing process should be locked down and accurately represented in pivotal safety studies to be included in the IND. Changes in manufacturing, even during preclinical development, may require additional studies to support a future marketing application [260]. Preclinical planning must therefore consider both manufacturing issues (such as the use of clinical-grade reagents and the development of the clinical formulation) as well as the availability of material for pivotal nonclinical studies that are sufficiently representative of the intended clinical product.
4. Considerations for Clinical Development and Marketing Approval
4.1. Patient Monitoring and Biomarkers
Once clinical trials are started under an IND, immunomodulatory therapies require extensive monitoring of patients as a key component of understanding both the safety profile and efficacy of the product. Some immunotherapies are very effective in only a portion of patients, and therefore the success of new immunomodulatory therapies will be enhanced by development of relevant biomarkers for the prediction of which patients will exhibit a beneficial response [261].
Monitoring the immune response during clinical trials is also key to supporting the safety of the product. For example, the measurement of cytokine levels in patients is a critical tool for measuring immune response during immunotherapy [262]. Additionally, testing for anti-drug antibody products during early and late-stage clinical trials is imperative to understand the extent of the potential impact on the immune system [263]. Production of neutralizing anti-drug antibodies by the immune system in response to administration of biologic drugs remains a major hurdle for biologic drugs and thus must be carefully understood and assessed during development. Several groups are developing screening/identification methods as well as prediction models and clinical programs to test for, and hopefully develop strategies to avoid, the formation of anti-drug antibodies.
Bharadwaj and colleagues, for example, have implemented a high throughput, tiered assay approach to measure anti-drug antibodies. The authors optimized their high throughput approach using five antibodies (3NC117, 3BNC117-LS, 10–1074, PGT121, and PGDM1400) and demonstrate modularity and sufficient sensitivity to be used as an identification method for a range of biologics that are advancing to the clinic. The authors show that a positivity cut point, competition threshold, and dilution step size can be implemented to define response titers for each drug product according to established guidance [264]. Another group developed a prediction model using machine learning methods based on quantified serum metabolites and lipids for patients with MS that were treated with IFNβ. The authors showed that their method could predict future anti-drug antibody status at baseline or month 3 with F1 score (a measure of accuracy) > 0.735 and specificity > 0.83, suggesting that this technique, if able to translate to other diseases, could be used as a low invasive method for predicting drug efficacy [265]. The success of these efforts, and others like it, have the potential to provide the field with a tool for predicting the success of biologics early on to reduce attrition of critical therapeutic drugs.
4.2. Regulatory Considerations in Late-Phase Development
Many of the challenges that arise during clinical development, which can impact the design of pivotal clinical trials, can be discussed at milestone meetings with the FDA. An End of Phase 1 meeting gives the sponsor an opportunity to discuss multiple development topics, which may include proposed Phase 2 controlled trials, optimal dose selection, potential biomarker development, and PK/PD. An End of Phase 2 meeting can be held to determine if it is safe to proceed to Phase 3 trials and gain FDA feedback on the Phase 3 plan and protocols [266].
Following the completion of human clinical trials, a new drug or biologic must be evaluated by the FDA in the form of a New Drug Application (NDA) or Biologic Licensing Application (BLA) [245] before it can be legally marketed. The role of the FDA is to ensure that new therapeutics are both safe and effective for the proposed indication. This evaluation begins with an in-depth review of the manufacturing process and controls, nonclinical data, and clinical data supporting the safety and effectiveness of the drug for the proposed indication and at the intended dose.
The FDA utilizes a structured framework for the drug approval process to ensure that drugs prescribed to human patients are both safe and effective. The clinical review for FDA approval takes into account the available treatments for an indication, the severity of the intended illness along with whether there is a current unmet medical need for the condition. Sponsors must provided clinic data to demonstrate both safety and effectiveness in human patients in order to allow the FDA to make an assessment of potential risks and benefits of the new product [245]. Unfortunately, this process is often very time and resource-intensive, which limits many products from becoming widely available to the general population. Figure 8 shows average timelines for product approval from research and development through FDA approval [267]. It has been estimated that it takes, on average, 14 years and $2.6 billion to develop a new medicine from the start of research and development to FDA approval [268, 269]. While this is generally beneficial for the overall safety of patients, it may also result in the rejection of products that could potentially save lives.
Figure 8.
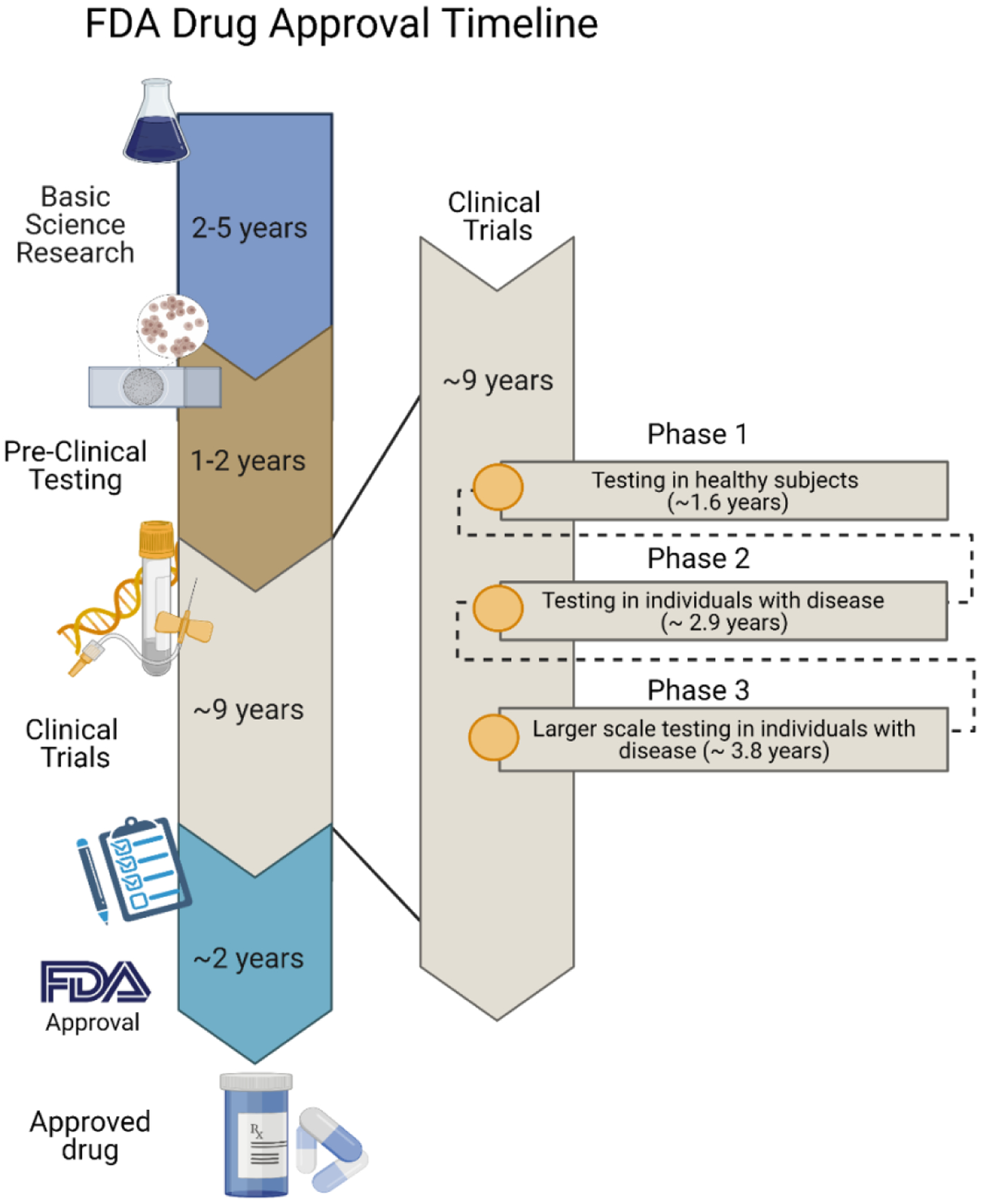
Approximate timelines for FDA drug approval. Based on information in ref. [212]. The average time spent on basic research is 2–5 years followed by 1–2 years of advanced clinical testing and nearly a decade of clinical trials. Average times for each stage of clinical trials are listed in the inset. Finally, the FDA approval process takes roughly 2 additional years. Schematic was made using BioRender.com
4.1. Opportunities for Expedited Drug Development
Recent trends in the development of complex biologics have often resulted in a much smaller nonclinical package compared to other product classes such as small molecules and mAbs, due to the limitations of what types of nonclinical studies can be conducted for these new product classes. As a result, some development timelines can be much shorter than described in the previous section. In addition, the FDA has established several opportunities for expedited development (Figure 9) of novel therapies that are for treatment of life-threatening conditions and show therapeutic benefit beyond products currently available [270].
Figure 9.
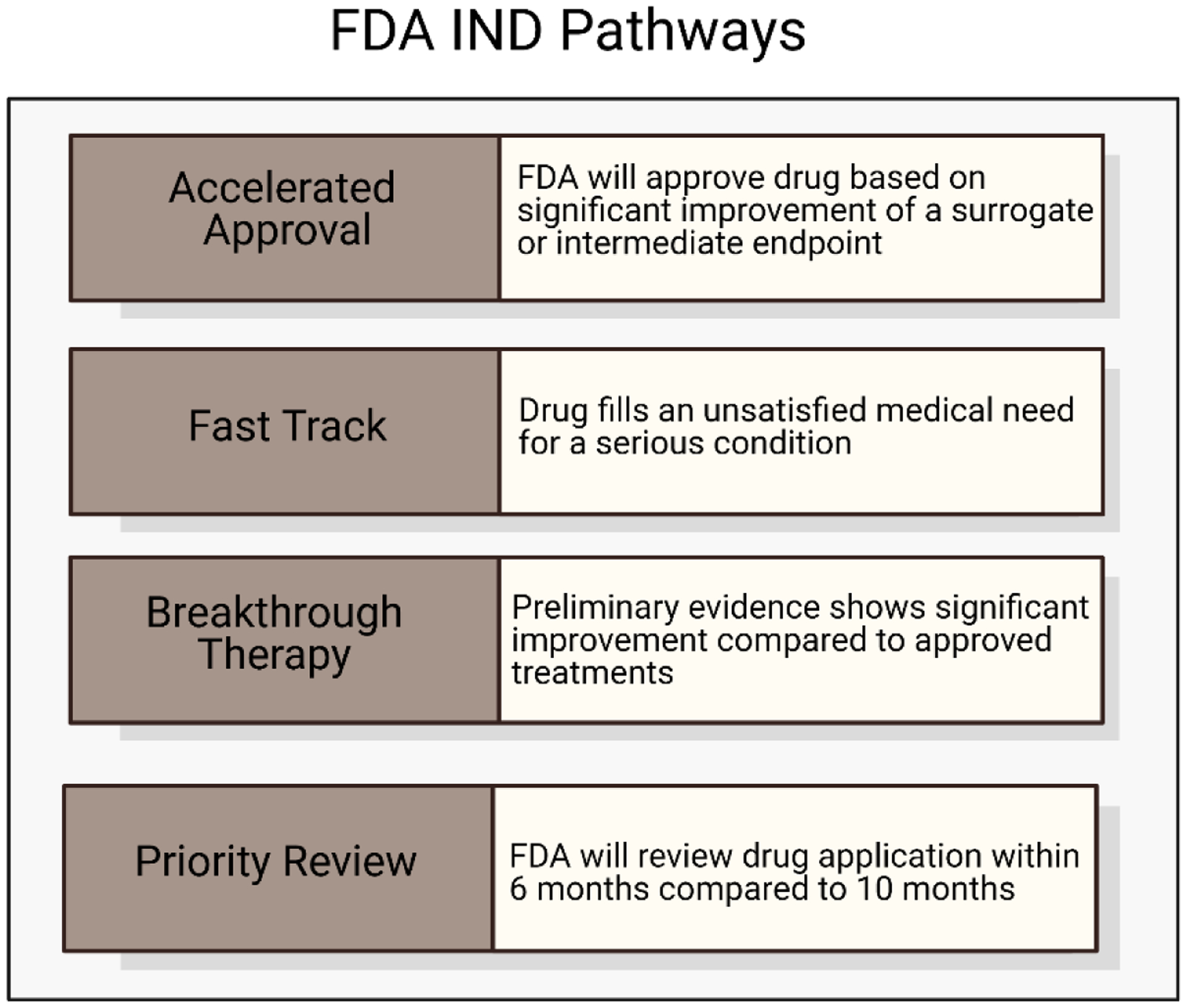
FDA IND Pathways. Four potential pathways for new drug products to be expedited during the FDA approval process. These include accelerated approval, fast track designation, breakthrough therapy designation, and priority review. Adapted from ref. [217]. Schematic was made using BioRender.com
4.1.1. Accelerated Approval
The first of these approval pathways was established in 1992 and is called Accelerated Approval [271–273]. In this pathway, a “surrogate endpoint” can be used by FDA reviewers as a measurement of clinical benefit to make a preliminary ruling on a drug that meets a clinically unmet need. This allows, for example, a drug that causes tumor reduction to be approved before data is collected to demonstrate extended survival time in clinical trial patients. The surrogate endpoint is used if the FDA agrees that it is “reasonably likely to predict the clinical benefit of the drug”. The clinical studies would still have to be continued and completed by the drug company to prove the long-term effects and benefits and the drug could still be withdrawn if it does not demonstrate sufficient clinical benefit to justify any identified risks.
4.1.2. Fast Track
The FDA fast track process was designed to advance the review of new drugs that treat serious conditions and fill an unmet medical need, based on promising animal or human data [245]. If a new drug product meets these criteria, a Fast Track designation can be assigned; however, the FDA can remove the designation if emerging data no longer supports the original designation [271]. Benefits of Fast Track designation include earlier and more frequent communication with the FDA, and the option for a rolling NDA/BLA submission to speed up the marketing application review.
4.1.3. Breakthrough Therapy
The Breakthrough Therapy designation applies to a new drug product if it “is intended, alone or in combination with 1 or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on 1 or more clinically significant endpoints” [271]. The FDA has specified “preliminary clinical evidence” to mean evidence from Phase 1 or 2 clinical trials that are “sufficient to indicate that the drug may demonstrate substantial improvement in effectiveness or safety over available therapies, but in most cases is not sufficient to establish safety and effectiveness for purposes of approval.” Benefits of this designation also include engagement with FDA reviewers on clinical trial designs as part of earlier and more frequent communications, as well as the option for rolling review. In most cases, a drug with Breakthrough Therapy designation is also eligible for the Fast Track process [271].
4.1.4. Priority Review
The FDA decides on either a standard or priority review designation for every application. Priority review is a designation for new products that are intended to treat a serious or life-threatening condition and “provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions compared to available therapies,” if approved [271] and applications assigned to priority review are reviewed four-months faster than products assigned to standard review. The FDA may consider evidence pertaining to increased effectiveness, substantial reduction of a treatment-limiting toxicity, or evidence of safety or effectiveness in a new subpopulation and decides whether a new drug product represents a significant improvement on a case-by-case basis [274]. A Priority Review application secures a commitment from FDA to shorten the review time to 6 months.
4.1.5. Regenerative Medicine Advanced Therapy (RMAT)
A newer opportunity for expedited development was established by the designation of Regenerative Medicine Advanced Therapies. RMAT products are cell therapies (or combination products with cell therapies) and can receive the same benefits as Breakthrough products. However, while clinical data is needed for an RMAT application, the eligibility criteria is different from Breakthrough designation in that RMAT products only need evidence that indicates the therapy has the potential to address an unmet medical need, whereas Breakthrough products also need to demonstrate a substantial improvement over existing therapies.
5. Emerging opportunities for future therapies
Pro- and anti-inflammatory diseases have a major impact on the lives of people around the world. Clinical advances and the next generation of immunotherapeutic drugs require a deeper understanding of existing targets and more advanced technologies for screening and identification of new immunological targets. One of the most promising opportunities for the future of targeted immunotherapies is to decrease host rejection during organ transplantation [275]. Decreasing the percentage of transplanted organs that are attacked by the host immune system using immunomodulatory drugs has the potential to save lives and increase the quality of the future of medicine. For cancer immunotherapy specifically, the possibilities are numerous. The heterogeneity of the tumor microenvironment suggests the need for the development of unique treatment regimens with a combination of immunostimulants and thus offers countless possibilities for future drug treatments. In-depth animal and human studies will be needed to determine which specific combinations are the most appropriate for different tumor microenvironments and different immune systems. Several recent reviews also highlight the potential of the field of ‘immunometabolism’ and suggest that because cancer cells rely heavily on metabolic pathways to fuel their rapid proliferation, in-depth studies to understand the complexities of many of these pathways will lead to novel targets for future intervention [276]. Additionally, a successful cytokine modification or delivery strategy that is efficacious for extended timeframes without causing toxicities would be transformative for immunoncology.
To translate new products into the clinic, the next generation of biologics must also consider manufacturing challenges and FDA requirements for approval during the research and development stage. Building on and enhancing previously FDA-approved products to make them more efficacious, less toxic, and/or last longer will allow for rapid testing and development. The development of advanced high-throughput in vitro and in vivo screening methods to assess the unique pharmacokinetic properties, biodistribution, and mechanisms of action of new therapies may offer a more efficient approach to evaluate as many combination treatments as possible. The success of these screening methods could aid in the selection of the drug candidates to test in clinical trials in the future.
6. Conclusion
In the future development of clinical therapeutics, two key aspects must be considered: therapeutic efficacy of immunomodulators must be increased without increasing the toxicities and large-scale manufacturing must not compromise the safety and efficacy of the products. We anticipate that as the understanding and investigation of the immunological landscape deepen, the next generation of immunomodulatory agents will be more sophisticated, less toxic, and therapeutically efficacious such that scientists can optimize kinetic properties and gain temporal and spatial control of dosing. Combination therapies, as well as collaborative efforts of interdisciplinary research, will likely drive a majority of the innovation seen in the next decades. We predict that solving biological problems that remain unaddressed by simpler treatments may require not complex treatment strategies, but instead, innovative delivery vehicles and dosing strategies.
Acknowledgements:
This work was supported by the Cancer Prevention Research Institute of Texas (Grant ID, RR160047), the National Institutes of Health (NIH Grant 1R01DK120459-01), and the National Science Foundation Graduate Research Fellowship Program (NSF GRFP Grant 1842494).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: Omid Veiseh is co-founder, holds equity and receives consulting payments from Sigilon Therapeutics, Pana Bio, and Avenge Bio. Veiseh also receives compensation for consulting from Establishment Labs and Auregen BioTherapeutics SA.
References
- [1].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS, Sipuleucel-T immunotherapy for castration-resistant prostate cancer, N Engl J Med, 363 (2010) 411–422. [DOI] [PubMed] [Google Scholar]
- [2].Raedler LA, Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma, Am Health Drug Benefits, 8 (2015) 96–100. [PMC free article] [PubMed] [Google Scholar]
- [3].McKeage K, Daratumumab: First Global Approval, Drugs, 76 (2016) 275–281. [DOI] [PubMed] [Google Scholar]
- [4].O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, Gavin D, Lee S, Liu K, George B, Bryan W, Theoret MR, Pazdur R, FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia, Clin Cancer Res, 25 (2019) 1142–1146. [DOI] [PubMed] [Google Scholar]
- [5].Mian A, Hill BT, Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma, Expert Opin Biol Ther, 21 (2021) 435–441. [DOI] [PubMed] [Google Scholar]
- [6].Faissner S, Gold R, Efficacy and Safety of the Newer Multiple Sclerosis Drugs Approved Since 2010, CNS Drugs, 32 (2018) 269–287. [DOI] [PubMed] [Google Scholar]
- [7].Scribano ML, Vedolizumab for inflammatory bowel disease: From randomized controlled trials to real-life evidence, World J Gastroenterol, 24 (2018) 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mulero P, Midaglia L, Montalban X, Ocrelizumab: a new milestone in multiple sclerosis therapy, Ther Adv Neurol Disord, 11 (2018) 1756286418773025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McCarty D, Robinson A, Efficacy and safety of sarilumab in patients with active rheumatoid arthritis, Ther Adv Musculoskelet Dis, 10 (2018) 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fernandez-Clotet A, Castro-Poceiro J, Panes J, Tofacitinib for the treatment of ulcerative colitis, Expert Rev Clin Immunol, 14 (2018) 881–892. [DOI] [PubMed] [Google Scholar]
- [11].Veeravalli V, Dash RP, Thomas JA, Babu RJ, Madgula LMV, Srinivas NR, Critical Assessment of Pharmacokinetic Drug-Drug Interaction Potential of Tofacitinib, Baricitinib and Upadacitinib, the Three Approved Janus Kinase Inhibitors for Rheumatoid Arthritis Treatment, Drug Saf, 43 (2020) 711–725. [DOI] [PubMed] [Google Scholar]
- [12].Avorn J, Learning about the safety of drugs--a half-century of evolution, N Engl J Med, 365 (2011) 2151–2153. [DOI] [PubMed] [Google Scholar]
- [13].Cancer Research Institute, Immunomodulators: Checkpoint Inhibitors, Cytokines, Agonists, and Adjuvants, 2020.
- [14].U.S. Food & Drug Administration, Approved Cellular and Gene Therapy Products, 2021.
- [15].U.S. Food & Drug Administration, Vaccines Licensed for Use in the United States, 2021.
- [16].The Antibody Society, Antibody therapeutics approved or in regulatory review in the EU or US, 2021.
- [17].Pol J, Kroemer G, Galluzzi L, First oncolytic virus approved for melanoma immunotherapy, Oncoimmunology, 5 (2016) e1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee SJ, Chinen J, Kavanaugh A, Immunomodulator therapy: monoclonal antibodies, fusion proteins, cytokines, and immunoglobulins, J Allergy Clin Immunol, 125 (2010) S314–323. [DOI] [PubMed] [Google Scholar]
- [19].Nicholson LB, The immune system, Essays Biochem, 60 (2016) 275–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chaplin DD, Overview of the immune response, J Allergy Clin Immunol, 125 (2010) S3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Agha-Mohammadi S, Lotze MT, Immunomodulation of cancer: potential use of selectively replicating agents, J Clin Invest, 105 (2000) 1173–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Isaacs JD, Burmester GR, Smart battles: immunosuppression versus immunomodulation in the inflammatory RMDs, Ann Rheum Dis, 79 (2020) 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bascones-Martinez A, Mattila R, Gomez-Font R, Meurman JH, Immunomodulatory drugs: oral and systemic adverse effects, Med Oral Patol Oral Cir Bucal, 19 (2014) e24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stanculeanu DL, Daniela Z, Lazescu A, Bunghez R, Anghel R, Development of new immunotherapy treatments in different cancer types, J Med Life, 9 (2016) 240–248. [PMC free article] [PubMed] [Google Scholar]
- [25].Hollingsworth RE, Jansen K, Turning the corner on therapeutic cancer vaccines, NPJ Vaccines, 4 (2019) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dalgleish AG, Vaccines versus immunotherapy: overview of approaches in deciding between options, Hum Vaccin Immunother, 10 (2014) 3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Marshall JS, Warrington R, Watson W, Kim HL, An introduction to immunology and immunopathology, Allergy Asthma Clin Immunol, 14 (2018) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hansson GK, Libby P, Schonbeck U, Yan ZQ, Innate and adaptive immunity in the pathogenesis of atherosclerosis, Circ Res, 91 (2002) 281–291. [DOI] [PubMed] [Google Scholar]
- [29].Simon AK, Hollander GA, McMichael A, Evolution of the immune system in humans from infancy to old age, Proc Biol Sci, 282 (2015) 20143085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bachmann MF, Kopf M, On the role of the innate immunity in autoimmune disease, J Exp Med, 193 (2001) F47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Adjiri A, Identifying and Targeting the Cause of Cancer is Needed to Cure Cancer, Oncol Ther, 4 (2016) 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Combalia A, Carrera C, Squamous Cell Carcinoma: An Update on Diagnosis and Treatment, Dermatol Pract Concept, 10 (2020) e2020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, Kohler BA, Jemal A, Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics, Cancer, 124 (2018) 2785–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mohammad AA, Advanced pancreatic cancer: The standard of care and new opportunities, Oncol Rev, 12 (2018) 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD, Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial, J Clin Oncol, 15 (1997) 2403–2413. [DOI] [PubMed] [Google Scholar]
- [36].Le Tourneau C, Raymond E, Faivre S, Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST), Ther Clin Risk Manag, 3 (2007) 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gallardo E, Mendez-Vidal MJ, Perez-Gracia JL, Sepulveda-Sanchez JM, Campayo M, Chirivella-Gonzalez I, Garcia-Del-Muro X, Gonzalez-Del-Alba A, Grande E, Suarez C, SEOM clinical guideline for treatment of kidney cancer (2017), Clin Transl Oncol, 20 (2018) 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cella D, Beaumont JL, Pazopanib in the treatment of advanced renal cell carcinoma, Ther Adv Urol, 8 (2016) 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mikula-Pietrasik J, Witucka A, Pakula M, Uruski P, Begier-Krasinska B, Niklas A, Tykarski A, Ksiazek K, Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells, Cell Mol Life Sci, 76 (2019) 681–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kumar S, Mahdi H, Bryant C, Shah JP, Garg G, Munkarah A, Clinical trials and progress with paclitaxel in ovarian cancer, Int J Womens Health, 2 (2010) 411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Della Pepa C, Tonini G, Pisano C, Di Napoli M, Cecere SC, Tambaro R, Facchini G, Pignata S, Ovarian cancer standard of care: are there real alternatives?, Chin J Cancer, 34 (2015) 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nurgali K, Jagoe RT, Abalo R, Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae?, Front Pharmacol, 9 (2018) 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ramirez LY, Huestis SE, Yap TY, Zyzanski S, Drotar D, Kodish E, Potential chemotherapy side effects: what do oncologists tell parents?, Pediatr Blood Cancer, 52 (2009) 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bockhorn M, Jain RK, Munn LL, Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed?, Lancet Oncol, 8 (2007) 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segre AV, Branton PA, Polak P, Ardlie KG, Getz G, RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues, Science, 364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL, Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates, J Clin Oncol, 28 (2010) 3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ, Improved survival with ipilimumab in patients with metastatic melanoma, N Engl J Med, 363 (2010) 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, Safety, activity, and immune correlates of anti-PD-1 antibody in cancer, N Engl J Med, 366 (2012) 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, Kendra KL, White RL, Gonzalez R, Kuzel TM, Curti B, Leming PD, Whitman ED, Balkissoon J, Reintgen DS, Kaufman H, Marincola FM, Merino MJ, Rosenberg SA, Choyke P, Vena D, Hwu P, gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma, N Engl J Med, 364 (2011) 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH, T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia, Sci Transl Med, 3 (2011) 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M, CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia, Sci Transl Med, 5 (2013) 177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Casak SJ, Fashoyin-Aje I, Lemery SJ, Zhang L, Jin R, Li H, Zhao L, Zhao H, Zhang H, Chen H, He K, Dougherty M, Novak R, Kennett S, Khasar S, Helms W, Keegan P, Pazdur R, FDA Approval Summary: Ramucirumab for Gastric Cancer, Clin Cancer Res, 21 (2015) 3372–3376. [DOI] [PubMed] [Google Scholar]
- [53].Greig SL, Talimogene Laherparepvec: First Global Approval, Drugs, 76 (2016) 147–154. [DOI] [PubMed] [Google Scholar]
- [54].Markham A, Atezolizumab: First Global Approval, Drugs, 76 (2016) 1227–1232. [DOI] [PubMed] [Google Scholar]
- [55].Shirley M, Olaratumab: First Global Approval, Drugs, 77 (2017) 107–112. [DOI] [PubMed] [Google Scholar]
- [56].Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, Blumenthal GM, Bryan W, McKee AE, Pazdur R, FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma, Clin Cancer Res, 25 (2019) 1702–1708. [DOI] [PubMed] [Google Scholar]
- [57].Markham A, Duggan S, Cemiplimab: First Global Approval, Drugs, 78 (2018) 1841–1846. [DOI] [PubMed] [Google Scholar]
- [58].Lamb YN, Pexidartinib: First Approval, Drugs, 79 (2019) 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, Raje N, Lin Y, Siegel D, Oriol A, Moreau P, Yakoub-Agha I, Delforge M, Cavo M, Einsele H, Goldschmidt H, Weisel K, Rambaldi A, Reece D, Petrocca F, Massaro M, Connarn JN, Kaiser S, Patel P, Huang L, Campbell TB, Hege K, San-Miguel J, Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma, New England Journal of Medicine, 384 (2021) 705–716. [DOI] [PubMed] [Google Scholar]
- [60].Ronnblom L, Pascual V, The innate immune system in SLE: type I interferons and dendritic cells, Lupus, 17 (2008) 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith DA, Germolec DR, Introduction to immunology and autoimmunity, Environ Health Perspect, 107 Suppl 5 (1999) 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lawrence T, Gilroy DW, Chronic inflammation: a failure of resolution?, Int J Exp Pathol, 88 (2007) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Waldner H, The role of innate immune responses in autoimmune disease development, Autoimmun Rev, 8 (2009) 400–404. [DOI] [PubMed] [Google Scholar]
- [64].Li P, Zheng Y, Chen X, Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics, Front Pharmacol, 8 (2017) 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Barnes PJ, Anti-inflammatory actions of glucocorticoids: molecular mechanisms, Clin Sci (Lond), 94 (1998) 557–572. [DOI] [PubMed] [Google Scholar]
- [66].Blumenthal KG, Lai KH, Huang M, Wallace ZS, Wickner PG, Zhou L, Adverse and Hypersensitivity Reactions to Prescription Nonsteroidal Anti-Inflammatory Agents in a Large Health Care System, J Allergy Clin Immunol Pract, 5 (2017) 737–743 e733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Scheinberg MA, Santoro J, Sanchez ML, Effect of tenoxicam on inflammation and immune cellular function, Braz J Med Biol Res, 23 (1990) 1143–1148. [PubMed] [Google Scholar]
- [68].Cohen SB, The use of anakinra, an interleukin-1 receptor antagonist, in the treatment of rheumatoid arthritis, Rheum Dis Clin North Am, 30 (2004) 365–380, vii. [DOI] [PubMed] [Google Scholar]
- [69].Fleischmann RM, Schechtman J, Bennett R, Handel ML, Burmester GR, Tesser J, Modafferi D, Poulakos J, Sun G, Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial, Arthritis Rheum, 48 (2003) 927–934. [DOI] [PubMed] [Google Scholar]
- [70].Dinarello CA, Simon A, van der Meer JW, Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases, Nat Rev Drug Discov, 11 (2012) 633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, Oltolini C, Castiglioni B, Tassan Din C, Boffini N, Tomelleri A, Farina N, Ruggeri A, Rovere-Querini P, Di Lucca G, Martinenghi S, Scotti R, Tresoldi M, Ciceri F, Landoni G, Zangrillo A, Scarpellini P, Dagna L, Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study, Lancet Rheumatol, 2 (2020) e325–e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Khan NA, Anakinra for severe forms of COVID-19, Lancet Rheumatol, 2 (2020) e586–e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, Sacco E, Naccache JM, Bezie Y, Laplanche S, Le Berre A, Le Pavec J, Salmeron S, Emmerich J, Mourad JJ, Chatellier G, Hayem G, Anakinra for severe forms of COVID-19: a cohort study, Lancet Rheumatol, 2 (2020) e393–e400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fairweather D, Rose NR, Women and autoimmune diseases, Emerg Infect Dis, 10 (2004) 2005–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rosenblum MD, Remedios KA, Abbas AK, Mechanisms of human autoimmunity, J Clin Invest, 125 (2015) 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rosenthal KS, Carambula R, Zimmerman DH, Why Don’t We Have a Vaccine Against Autoimmune Diseases? - A Review, J Clin Cell Immunol, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sanz I, Yasothan U, Kirkpatrick P, Belimumab, Nature Reviews Drug Discovery, 10 (2011) 335–336. [DOI] [PubMed] [Google Scholar]
- [78].Oh J, O’Connor PW, Teriflunomide, Neurol Clin Pract, 3 (2013) 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].English C, Aloi JJ, New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis, Clin Ther, 37 (2015) 691–715. [DOI] [PubMed] [Google Scholar]
- [80].Garnock-Jones KP, Eluxadoline: First Global Approval, Drugs, 75 (2015) 1305–1310. [DOI] [PubMed] [Google Scholar]
- [81].Gold R, Radue EW, Giovannoni G, Selmaj K, Havrdova E, Stefoski D, Sprenger T, Montalban X, Cohan S, Umans K, Greenberg SJ, Ozen G, Elkins J, Safety and efficacy of daclizumab in relapsing-remitting multiple sclerosis: 3-year results from the SELECTED open-label extension study, BMC Neurol, 16 (2016) 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Deepak P, Loftus EV Jr., Ustekinumab in treatment of Crohn’s disease: design, development, and potential place in therapy, Drug Des Devel Ther, 10 (2016) 3685–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Duggan S, Keam SJ, Upadacitinib: First Approval, Drugs, 79 (2019) 1819–1828. [DOI] [PubMed] [Google Scholar]
- [84].Kaplon H, Muralidharan M, Schneider Z, Reichert JM, Antibodies to watch in 2020, MAbs, 12 (2020) 1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, Liu J, Zhang X, Zhang X, Jin Y, Wang Y, Zhang S, Zhu L, Jacob A, Jia R, You X, Li X, Li C, Zhou Y, Yang Y, Ye H, Liu Y, Su Y, Shen N, Alexander J, Guo J, Ambrus J, Lin X, Yu D, Sun X, Li Z, Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial, Ann Rheum Dis, 79 (2020) 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lamb YN, Ozanimod: First Approval, Drugs, 80 (2020) 841–848. [DOI] [PubMed] [Google Scholar]
- [87].Tummala R, Abreu G, Pineda L, Michaels MA, Kalyani RN, Furie RA, Morand EF, Safety profile of anifrolumab in patients with active SLE: an integrated analysis of phase II and III trials, Lupus Sci Med, 8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Witwer KW, Wolfram J, Extracellular vesicles versus synthetic nanoparticles for drug delivery, Nature Reviews Materials, 6 (2021) 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Stephen ZR, Kievit FM, Veiseh O, Chiarelli PA, Fang C, Wang K, Hatzinger SJ, Ellenbogen RG, Silber JR, Zhang M, Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors, ACS Nano, 8 (2014) 10383–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Finbloom JA, Sousa F, Stevens MM, Desai TA, Engineering the drug carrier biointerface to overcome biological barriers to drug delivery, Adv Drug Deliv Rev, 167 (2020) 89–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dimov N, Kastner E, Hussain M, Perrie Y, Szita N, Formation and purification of tailored liposomes for drug delivery using a module-based micro continuous-flow system, Sci Rep, 7 (2017) 12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Skrombolas D, Sullivan M, Frelinger JG, Development of an Interleukin-12 Fusion Protein That Is Activated by Cleavage with Matrix Metalloproteinase 9, J Interferon Cytokine Res, 39 (2019) 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, Rubas W, Doberstein SK, Eldon M, Hoch U, Zalevsky J, Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy, PLoS One, 12 (2017) e0179431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Feins S, Kong W, Williams EF, Milone MC, Fraietta JA, An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer, Am J Hematol, 94 (2019) S3–S9. [DOI] [PubMed] [Google Scholar]
- [95].Shields C.W.t., Evans MA, Wang LL, Baugh N, Iyer S, Wu D, Zhao Z, Pusuluri A, Ukidve A, Pan DC, Mitragotri S, Cellular backpacks for macrophage immunotherapy, Sci Adv, 6 (2020) eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lockney D, Franzen S, Lommel S, Viruses as nanomaterials for drug delivery, Methods Mol Biol, 726 (2011) 207–221. [DOI] [PubMed] [Google Scholar]
- [97].Arango Duque G, Descoteaux A, Macrophage cytokines: involvement in immunity and infectious diseases, Front Immunol, 5 (2014) 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang JM, An J, Cytokines, inflammation, and pain, Int Anesthesiol Clin, 45 (2007) 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rider P, Carmi Y, Cohen I, Biologics for Targeting Inflammatory Cytokines, Clinical Uses, and Limitations, Int J Cell Biol, 2016 (2016) 9259646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, Rodriguez-Ruiz ME, Ponz-Sarvise M, Castanon E, Melero I, Cytokines in clinical cancer immunotherapy, Br J Cancer, 120 (2019) 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Conlon KC, Miljkovic MD, Waldmann TA, Cytokines in the Treatment of Cancer, J Interferon Cytokine Res, 39 (2019) 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Whiteside TL, Cytokines and cytokine measurements in a clinical laboratory, Clin Diagn Lab Immunol, 1 (1994) 257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chulpanova DS, Kitaeva KV, Green AR, Rizvanov AA, Solovyeva VV, Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy, Front Cell Dev Biol, 8 (2020) 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Waldmann TA, Cytokines in Cancer Immunotherapy, Cold Spring Harb Perspect Biol, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ramani T, Auletta CS, Weinstock D, Mounho-Zamora B, Ryan PC, Salcedo TW, Bannish G, Cytokines: The Good, the Bad, and the Deadly, Int J Toxicol, 34 (2015) 355–365. [DOI] [PubMed] [Google Scholar]
- [106].Pachella LA, Madsen LT, Dains JE, The Toxicity and Benefit of Various Dosing Strategies for Interleukin-2 in Metastatic Melanoma and Renal Cell Carcinoma, J Adv Pract Oncol, 6 (2015) 212–221. [PMC free article] [PubMed] [Google Scholar]
- [107].Schwartz RN, Stover L, Dutcher JP, Managing toxicities of high-dose interleukin-2, Oncology (Williston Park), 16 (2002) 11–20. [PubMed] [Google Scholar]
- [108].Choudhry H, Helmi N, Abdulaal WH, Zeyadi M, Zamzami MA, Wu W, Mahmoud MM, Warsi MK, Rasool M, Jamal MS, Prospects of IL-2 in Cancer Immunotherapy, Biomed Res Int, 2018 (2018) 9056173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Jiang T, Zhou C, Ren S, Role of IL-2 in cancer immunotherapy, Oncoimmunology, 5 (2016) e1163462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Waldmann TA, The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design, Nat Rev Immunol, 6 (2006) 595–601. [DOI] [PubMed] [Google Scholar]
- [111].McDermott DF, Atkins MB, Interleukin-2 therapy of metastatic renal cell carcinoma--predictors of response, Semin Oncol, 33 (2006) 583–587. [DOI] [PubMed] [Google Scholar]
- [112].Spolski R, Li P, Leonard WJ, Biology and regulation of IL-2: from molecular mechanisms to human therapy, Nat Rev Immunol, 18 (2018) 648–659. [DOI] [PubMed] [Google Scholar]
- [113].Ross SH, Cantrell DA, Signaling and Function of Interleukin-2 in T Lymphocytes, Annu Rev Immunol, 36 (2018) 411–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Smith KA, Interleukin-2: inception, impact, and implications, Science, 240 (1988) 1169–1176. [DOI] [PubMed] [Google Scholar]
- [115].Liao W, Lin JX, Wang L, Li P, Leonard WJ, Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages, Nat Immunol, 12 (2011) 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE, Interleukin 2 plays a central role in Th2 differentiation, Proc Natl Acad Sci U S A, 101 (2004) 3880–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, Leonard WJ, Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression, Proc Natl Acad Sci U S A, 111 (2014) 3508–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ, Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation, Immunity, 26 (2007) 371–381. [DOI] [PubMed] [Google Scholar]
- [119].Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, Randall TD, Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation, Immunity, 36 (2012) 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R, Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo, Nat Med, 9 (2003) 540–547. [DOI] [PubMed] [Google Scholar]
- [121].Kammula US, White DE, Rosenberg SA, Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer, Cancer, 83 (1998) 797–805. [PubMed] [Google Scholar]
- [122].Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS, Curti BD, Tagliaferri MA, Zalevsky J, Hannah AL, Hoch U, Aung S, Fanton C, Rizwan A, Iacucci E, Liao Y, Bernatchez C, Hurwitz ME, Cho DC, Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02), Cancer Discov, (2020). [DOI] [PubMed] [Google Scholar]
- [123].Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, Fanton C, Aung S, Hwu P, Curti BD, Tannir NM, Sznol M, Diab A, A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rbetagamma-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors, Cancer Discov, 9 (2019) 711–721. [DOI] [PubMed] [Google Scholar]
- [124].Parisi G, Saco JD, Salazar FB, Tsoi J, Krystofinski P, Puig-Saus C, Zhang R, Zhou J, Cheung-Lau GC, Garcia AJ, Grasso CS, Tavare R, Hu-Lieskovan S, Mackay S, Zalevsky J, Bernatchez C, Diab A, Wu AM, Comin-Anduix B, Charych D, Ribas A, Persistence of adoptively transferred T cells with a kinetically engineered IL-2 receptor agonist, Nat Commun, 11 (2020) 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, Lang S, Roemmele M, Hofer T, van Puijenbroek E, Wittig D, Moser S, Ast O, Brunker P, Gorr IH, Neumann S, de Vera Mudry MC, Hinton H, Crameri F, Saro J, Evers S, Gerdes C, Bacac M, van Dongen G, Moessner E, Umana P, Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines, Oncoimmunology, 6 (2017) e1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, Giovannoni L, Ruffini PA, Elia G, Neri D, Gutzmer R, Becker JC, Garbe C, Intralesional treatment of stage III metastatic melanoma patients with L19-IL2 results in sustained clinical and systemic immunologic responses, Cancer Immunol Res, 2 (2014) 668–678. [DOI] [PubMed] [Google Scholar]
- [127].Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, Gan J, Eickhoff J, DeSantes KB, Cohn SL, Hecht T, Gadbaw B, Reisfeld RA, Maris JM, Sondel PM, Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study, J Clin Oncol, 28 (2010) 4969–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, Biary T, Crowley SJ, Marcos E, Walkey CD, Weitzner BD, Pardo-Avila F, Castellanos J, Carter L, Stewart L, Riddell SR, Pepper M, Bernardes GJL, Dougan M, Garcia KC, Baker D, De novo design of potent and selective mimics of IL-2 and IL-15, Nature, 565 (2019) 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Pestka S, The human interferon alpha species and receptors, Biopolymers, 55 (2000) 254–287. [DOI] [PubMed] [Google Scholar]
- [130].Golomb HM, Jacobs A, Fefer A, Ozer H, Thompson J, Portlock C, Ratain M, Golde D, Vardiman J, Burke JS, et al. , Alpha-2 interferon therapy of hairy-cell leukemia: a multicenter study of 64 patients, J Clin Oncol, 4 (1986) 900–905. [DOI] [PubMed] [Google Scholar]
- [131].Solal-Celigny P, Lepage E, Brousse N, Reyes F, Haioun C, Leporrier M, Peuchmaur M, Bosly A, Parlier Y, Brice P, et al. , Recombinant interferon alfa-2b combined with a regimen containing doxorubicin in patients with advanced follicular lymphoma. Groupe d’Etude des Lymphomes de l’Adulte, N Engl J Med, 329 (1993) 1608–1614. [DOI] [PubMed] [Google Scholar]
- [132].Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH, Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684, J Clin Oncol, 14 (1996) 7–17. [DOI] [PubMed] [Google Scholar]
- [133].Groopman JE, Gottlieb MS, Goodman J, Mitsuyasu RT, Conant MA, Prince H, Fahey JL, Derezin M, Weinstein WM, Casavante C, et al. , Recombinant alpha-2 interferon therapy for Kaposi’s sarcoma associated with the acquired immunodeficiency syndrome, Ann Intern Med, 100 (1984) 671–676. [DOI] [PubMed] [Google Scholar]
- [134].Herndon TM, Demko SG, Jiang X, He K, Gootenberg JE, Cohen MH, Keegan P, Pazdur R, U.S. Food and Drug Administration Approval: peginterferon-alfa-2b for the adjuvant treatment of patients with melanoma, Oncologist, 17 (2012) 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cauwels A, Van Lint S, Paul F, Garcin G, De Koker S, Van Parys A, Wueest T, Gerlo S, Van der Heyden J, Bordat Y, Catteeuw D, Rogge E, Verhee A, Vandekerckhove B, Kley N, Uze G, Tavernier J, Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments, Cancer Res, 78 (2018) 463–474. [DOI] [PubMed] [Google Scholar]
- [136].Fioravanti J, Gonzalez I, Medina-Echeverz J, Larrea E, Ardaiz N, Gonzalez-Aseguinolaza G, Prieto J, Berraondo P, Anchoring interferon alpha to apolipoprotein A-I reduces hematological toxicity while enhancing immunostimulatory properties, Hepatology, 53 (2011) 1864–1873. [DOI] [PubMed] [Google Scholar]
- [137].Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzone PD, Mier JW, Canning CM, Battiato L, Tahara H, Sherman ML, Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies, Clin Cancer Res, 3 (1997) 409–417. [PubMed] [Google Scholar]
- [138].Younes A, Pro B, Robertson MJ, Flinn IW, Romaguera JE, Hagemeister F, Dang NH, Fiumara P, Loyer EM, Cabanillas FF, McLaughlin PW, Rodriguez MA, Samaniego F, Phase II clinical trial of interleukin-12 in patients with relapsed and refractory non-Hodgkin’s lymphoma and Hodgkin’s disease, Clin Cancer Res, 10 (2004) 5432–5438. [DOI] [PubMed] [Google Scholar]
- [139].Bajetta E, Del Vecchio M, Mortarini R, Nadeau R, Rakhit A, Rimassa L, Fowst C, Borri A, Anichini A, Parmiani G, Pilot study of subcutaneous recombinant human interleukin 12 in metastatic melanoma, Clin Cancer Res, 4 (1998) 75–85. [PubMed] [Google Scholar]
- [140].Weiss GR, O’Donnell MA, Loughlin K, Zonno K, Laliberte RJ, Sherman ML, Phase 1 study of the intravesical administration of recombinant human interleukin-12 in patients with recurrent superficial transitional cell carcinoma of the bladder, J Immunother, 26 (2003) 343–348. [DOI] [PubMed] [Google Scholar]
- [141].Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, Zaharoff DA, Localized Interleukin-12 for Cancer Immunotherapy, Front Immunol, 11 (2020) 575597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hwang MP, Fecek RJ, Qin T, Storkus WJ, Wang Y, Single injection of IL-12 coacervate as an effective therapy against B16-F10 melanoma in mice, J Control Release, 318 (2020) 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, Wittrup KD, Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy, Sci Transl Med, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Li Y, Su Z, Zhao W, Zhang X, Momin N, Zhang C, Wittrup KD, Dong Y, Irvine DJ, Weiss R, Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity, Nature Cancer, 1 (2020) 882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Algazi A, Bhatia S, Agarwala S, Molina M, Lewis K, Faries M, Fong L, Levine LP, Franco M, Oglesby A, Ballesteros-Merino C, Bifulco CB, Fox BA, Bannavong D, Talia R, Browning E, Le MH, Pierce RH, Gargosky S, Tsai KK, Twitty C, Daud AI, Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients, Ann Oncol, 31 (2020) 532–540. [DOI] [PubMed] [Google Scholar]
- [146].Wang Y, Wang M, Li Y, Anti-colorectal cancer effect of interleukin-2 and interferon-beta fusion gene driven by carcinoembryonic antigen promoter, Onco Targets Ther, 9 (2016) 3259–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Gong Q, Song C, Wang X, Wang R, Cai G, Liang X, Liu J, Hyperthermic intraperitoneal chemotherapy with recombinant mutant human TNF-alpha and raltitrexed in mice with colorectal-peritoneal carcinomatosis, Exp Biol Med (Maywood), 245 (2020) 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].McQuade JL, Homsi J, Torres-Cabala CA, Bassett R, Popuri RM, James ML, Vence LM, Hwu WJ, A phase II trial of recombinant MAGE-A3 protein with immunostimulant AS15 in combination with high-dose Interleukin-2 (HDIL2) induction therapy in metastatic melanoma, BMC Cancer, 18 (2018) 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Borch TH, Andersen R, Ellebaek E, Met O, Donia M, Marie Svane I, Future role for adoptive T-cell therapy in checkpoint inhibitor-resistant metastatic melanoma, J Immunother Cancer, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF, Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction--a novel mechanism for maintaining vascular function, J Hematol Oncol, 7 (2014) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kany S, Vollrath JT, Relja B, Cytokines in Inflammatory Disease, Int J Mol Sci, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Georgescu L, Vakkalanka RK, Elkon KB, Crow MK, Interleukin-10 promotes activation-induced cell death of SLE lymphocytes mediated by Fas ligand, J Clin Invest, 100 (1997) 2622–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Mahmoudpour SH, Jankowski M, Valerio L, Becker C, Espinola-Klein C, Konstantinides S, Quitzau K, Barco S, Safety of low-dose subcutaneous recombinant interleukin-2: systematic review and meta-analysis of randomized controlled trials, Sci Rep, 9 (2019) 7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ghasemi N, Razavi S, Nikzad E, Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy, Cell J, 19 (2017) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hemmer B, Kerschensteiner M, Korn T, Role of the innate and adaptive immune responses in the course of multiple sclerosis, Lancet Neurol, 14 (2015) 406–419. [DOI] [PubMed] [Google Scholar]
- [156].Ontaneda D, Thompson AJ, Fox RJ, Cohen JA, Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function, Lancet, 389 (2017) 1357–1366. [DOI] [PubMed] [Google Scholar]
- [157].Feng X, Bao R, Li L, Deisenhammer F, Arnason BGW, Reder AT, Interferon-beta corrects massive gene dysregulation in multiple sclerosis: Short-term and long-term effects on immune regulation and neuroprotection, EBioMedicine, 49 (2019) 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC, Development of therapeutic antibodies for the treatment of diseases, J Biomed Sci, 27 (2020) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Dominguez C, McCampbell KK, David JM, Palena C, Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer, JCI Insight, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP, Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma, PLoS One, 9 (2014) e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Jatoi A, Ritter HL, Dueck A, Nguyen PL, Nikcevich DA, Luyun RF, Mattar BI, Loprinzi CL, A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9), Lung Cancer, 68 (2010) 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, Saburi Y, Miyamoto T, Takemoto S, Suzushima H, Tsukasaki K, Nosaka K, Fujiwara H, Ishitsuka K, Inagaki H, Ogura M, Akinaga S, Tomonaga M, Tobinai K, Ueda R, Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study, J Clin Oncol, 30 (2012) 837–842. [DOI] [PubMed] [Google Scholar]
- [163].Criscitiello C, Morganti S, Curigliano G, Antibody-drug conjugates in solid tumors: a look into novel targets, J Hematol Oncol, 14 (2021) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V, Chu YW, Perez EA, Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer, J Clin Oncol, 31 (2013) 1157–1163. [DOI] [PubMed] [Google Scholar]
- [165].Tamura K, Tsurutani J, Takahashi S, Iwata H, Krop IE, Redfern C, Sagara Y, Doi T, Park H, Murthy RK, Redman RA, Jikoh T, Lee C, Sugihara M, Shahidi J, Yver A, Modi S, Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study, Lancet Oncol, 20 (2019) 816–826. [DOI] [PubMed] [Google Scholar]
- [166].Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Duran I, Lee JL, Matsubara N, Vulsteke C, Castellano D, Wu C, Campbell M, Matsangou M, Petrylak DP, Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma, N Engl J Med, 384 (2021) 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, Brufsky A, Sardesai SD, Kalinsky K, Zelnak AB, Weaver R, Traina T, Dalenc F, Aftimos P, Lynce F, Diab S, Cortes J, O’Shaughnessy J, Dieras V, Ferrario C, Schmid P, Carey LA, Gianni L, Piccart MJ, Loibl S, Goldenberg DM, Hong Q, Olivo MS, Itri LM, Rugo HS, Investigators ACT, Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer, N Engl J Med, 384 (2021) 1529–1541. [DOI] [PubMed] [Google Scholar]
- [168].Dobosz P, Dzieciatkowski T, The Intriguing History of Cancer Immunotherapy, Front Immunol, 10 (2019) 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Tan S, Li D, Zhu X, Cancer immunotherapy: Pros, cons and beyond, Biomed Pharmacother, 124 (2020) 109821. [DOI] [PubMed] [Google Scholar]
- [170].Guo ZS, The 2018 Nobel Prize in medicine goes to cancer immunotherapy (editorial for BMC cancer), BMC Cancer, 18 (2018) 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Lee L, Gupta M, Sahasranaman S, Immune Checkpoint inhibitors: An introduction to the next-generation cancer immunotherapy, J Clin Pharmacol, 56 (2016) 157–169. [DOI] [PubMed] [Google Scholar]
- [172].Azoury SC, Straughan DM, Shukla V, Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety, Curr Cancer Drug Targets, 15 (2015) 452–462. [DOI] [PubMed] [Google Scholar]
- [173].Geoerger B, Bergeron C, Gore L, Sender L, Dunkel IJ, Herzog C, Brochez L, Cruz O, Nysom K, Berghorn E, Simsek B, Shen J, Pappo A, Phase II study of ipilimumab in adolescents with unresectable stage III or IV malignant melanoma, Eur J Cancer, 86 (2017) 358–363. [DOI] [PubMed] [Google Scholar]
- [174].Ascierto PA, Del Vecchio M, Mandala M, Gogas H, Arance AM, Dalle S, Cowey CL, Schenker M, Grob JJ, Chiarion-Sileni V, Marquez-Rodas I, Butler MO, Maio M, Middleton MR, de la Cruz-Merino L, Arenberger P, Atkinson V, Hill A, Fecher LA, Millward M, Khushalani NI, Queirolo P, Lobo M, de Pril V, Loffredo J, Larkin J, Weber J, Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial, Lancet Oncol, 21 (2020) 1465–1477. [DOI] [PubMed] [Google Scholar]
- [175].Imbert C, Montfort A, Fraisse M, Marcheteau E, Gilhodes J, Martin E, Bertrand F, Marcellin M, Burlet-Schiltz O, Peredo AG, Garcia V, Carpentier S, Tartare-Deckert S, Brousset P, Rochaix P, Puisset F, Filleron T, Meyer N, Lamant L, Levade T, Segui B, Andrieu-Abadie N, Colacios C, Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1, Nat Commun, 11 (2020) 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, Criscitiello PJ, Healey DI, Huang B, Gomez-Navarro J, Saltz LB, Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer, J Clin Oncol, 28 (2010) 3485–3490. [DOI] [PubMed] [Google Scholar]
- [177].Liu C, Liu R, Wang B, Lian J, Yao Y, Sun H, Zhang C, Fang L, Guan X, Shi J, Han S, Zhan F, Luo S, Yao Y, Zheng T, Zhang Y, Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer, J Immunother Cancer, 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wu Y, Ju Q, Jia K, Yu J, Shi H, Wu H, Jiang M, Correlation between sex and efficacy of immune checkpoint inhibitors (PD-1 and CTLA-4 inhibitors), Int J Cancer, 143 (2018) 45–51. [DOI] [PubMed] [Google Scholar]
- [179].Wallace DJ, Strand V, Merrill JT, Popa S, Spindler AJ, Eimon A, Petri M, Smolen JS, Wajdula J, Christensen J, Li C, Diehl A, Vincent MS, Beebe J, Healey P, Sridharan S, Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: a phase II dose-ranging randomised controlled trial, Ann Rheum Dis, 76 (2017) 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN, Anti-Tumor G Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study, Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group, N Engl J Med, 343 (2000) 1594–1602. [DOI] [PubMed] [Google Scholar]
- [181].Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, Alecock E, Lee J, Kremer J, IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial, Ann Rheum Dis, 67 (2008) 1516–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, Lublin F, Montalban X, Rammohan KW, Selmaj K, Traboulsee A, Wolinsky JS, Arnold DL, Klingelschmitt G, Masterman D, Fontoura P, Belachew S, Chin P, Mairon N, Garren H, Kappos L, Opera I, Investigators OIC, Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis, N Engl J Med, 376 (2017) 221–234. [DOI] [PubMed] [Google Scholar]
- [183].Yang H, Duchesneau E, Foster R, Guerin A, Ma E, Thomas NP, Cost-effectiveness analysis of ocrelizumab versus subcutaneous interferon beta-1a for the treatment of relapsing multiple sclerosis, J Med Econ, 20 (2017) 1056–1065. [DOI] [PubMed] [Google Scholar]
- [184].Mellion M, Edwards KR, Hupperts R, Drulović J, Montalban X, Hartung H-P, Brochet B, Calabresi PA, Rudick R, Ibrahim A, Zhang Y, Xu L, Cadavid D, Efficacy Results from the Phase 2b SYNERGY Study: Treatment of Disabling Multiple Sclerosis with the Anti-LINGO-1 Monoclonal Antibody Opicinumab (S33.004), Neurology, 88 (2017) S33.004. [Google Scholar]
- [185].Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, Leon MG, Tanasescu C, Nasonov E, Lan JL, Pineda L, Zhong ZJ, Freimuth W, Petri MA, Group B-S, Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial, Lancet, 377 (2011) 721–731. [DOI] [PubMed] [Google Scholar]
- [186].Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzova D, Sanchez-Guerrero J, Schwarting A, Merrill JT, Chatham WW, Stohl W, Ginzler EM, Hough DR, Zhong ZJ, Freimuth W, van Vollenhoven RF, Group B-S, A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus, Arthritis Rheum, 63 (2011) 3918–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, Amoura Z, Yu X, Mok CC, Santiago MB, Saxena A, Green Y, Ji B, Kleoudis C, Burriss SW, Barnett C, Roth DA, Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis, N Engl J Med, 383 (2020) 1117–1128. [DOI] [PubMed] [Google Scholar]
- [188].Fukuhara H, Ino Y, Todo T, Oncolytic virus therapy: A new era of cancer treatment at dawn, Cancer Sci, 107 (2016) 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Bommareddy PK, Patel A, Hossain S, Kaufman HL, Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma, Am J Clin Dermatol, 18 (2017) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Conry RM, Westbrook B, McKee S, Norwood TG, Talimogene laherparepvec: First in class oncolytic virotherapy, Hum Vaccin Immunother, 14 (2018) 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Harrington KJ, Puzanov I, Hecht JR, Hodi FS, Szabo Z, Murugappan S, Kaufman HL, Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy, Expert Rev Anticancer Ther, 15 (2015) 1389–1403. [DOI] [PubMed] [Google Scholar]
- [192].Larson C, Oronsky B, Scicinski J, Fanger GR, Stirn M, Oronsky A, Reid TR, Going viral: a review of replication-selective oncolytic adenoviruses, Oncotarget, 6 (2015) 19976–19989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Kelly E, Russell SJ, History of oncolytic viruses: genesis to genetic engineering, Mol Ther, 15 (2007) 651–659. [DOI] [PubMed] [Google Scholar]
- [194].He B, Huang X, Liu X, Xu B, Cancer targeting gene-viro-therapy for pancreatic cancer using oncolytic adenovirus ZD55-IL-24 in immune-competent mice, Mol Biol Rep, 40 (2013) 5397–5405. [DOI] [PubMed] [Google Scholar]
- [195].Wang P, Li X, Wang J, Gao D, Li Y, Li H, Chu Y, Zhang Z, Liu H, Jiang G, Cheng Z, Wang S, Dong J, Feng B, Chard LS, Lemoine NR, Wang Y, Author Correction: Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent, Nat Commun, 9 (2018) 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Aghlara-Fotovat S, Nash A, Kim B, Krencik R, Veiseh O, Targeting the extracellular matrix for immunomodulation: applications in drug delivery and cell therapies, Drug Deliv Transl Res, (2021). [DOI] [PubMed] [Google Scholar]
- [197].Zhao Z, Chen Y, Francisco NM, Zhang Y, Wu M, The application of CAR-T cell therapy in hematological malignancies: advantages and challenges, Acta Pharm Sin B, 8 (2018) 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Musial-Wysocka A, Kot M, Majka M, The Pros and Cons of Mesenchymal Stem Cell-Based Therapies, Cell Transplant, 28 (2019) 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Caliendo F, Dukhinova M, Siciliano V, Engineered Cell-Based Therapeutics: Synthetic Biology Meets Immunology, Front Bioeng Biotechnol, 7 (2019) 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Larson RC, Maus MV, Recent advances and discoveries in the mechanisms and functions of CAR T cells, Nat Rev Cancer, 21 (2021) 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Enblad G, Karlsson H, Loskog AS, CAR T-Cell Therapy: The Role of Physical Barriers and Immunosuppression in Lymphoma, Hum Gene Ther, 26 (2015) 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S, Teaching an old dog new tricks: next-generation CAR T cells, Br J Cancer, 120 (2019) 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Srivastava S, Riddell SR, Engineering CAR-T cells: Design concepts, Trends Immunol, 36 (2015) 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Britten CM, Shalabi A, Hoos A, Industrializing engineered autologous T cells as medicines for solid tumours, Nat Rev Drug Discov, (2021). [DOI] [PubMed] [Google Scholar]
- [205].Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, McSweeney PA, Miklos DB, Pagel JM, Kersten MJ, Milpied N, Fung H, Topp MS, Houot R, Beitinjaneh A, Peng W, Zheng L, Rossi JM, Jain RK, Rao AV, Reagan PM, KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma, N Engl J Med, 382 (2020) 1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, Chimeric antigen receptor T cells for sustained remissions in leukemia, N Engl J Med, 371 (2014) 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Enblad G, Karlsson H, Gammelgard G, Wenthe J, Lovgren T, Amini RM, Wikstrom KI, Essand M, Savoldo B, Hallbook H, Hoglund M, Dotti G, Brenner MK, Hagberg H, Loskog A, A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia, Clin Cancer Res, 24 (2018) 6185–6194. [DOI] [PubMed] [Google Scholar]
- [208].Gu R, Liu F, Zou D, Xu Y, Lu Y, Liu B, Liu W, Chen X, Liu K, Guo Y, Gong X, Lv R, Chen X, Zhou C, Zhong M, Wang H, Wei H, Mi Y, Qiu L, Lv L, Wang M, Wang Y, Zhu X, Wang J, Efficacy and safety of CD19 CAR T constructed with a new anti-CD19 chimeric antigen receptor in relapsed or refractory acute lymphoblastic leukemia, J Hematol Oncol, 13 (2020) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Hombach AA, Geumann U, Gunther C, Hermann FG, Abken H, IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells, Cells, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J, CAR-NK cells: A promising cellular immunotherapy for cancer, EBioMedicine, 59 (2020) 102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Albinger N, Hartmann J, Ullrich E, Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany, Gene Ther, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Wang W, Jiang J, Wu C, CAR-NK for tumor immunotherapy: Clinical transformation and future prospects, Cancer Lett, 472 (2020) 175–180. [DOI] [PubMed] [Google Scholar]
- [213].Yang Y, Day J, Souza-Fonseca Guimaraes F, Wicks IP, Louis C, Natural killer cells in inflammatory autoimmune diseases, Clin Transl Immunology, 10 (2021) e1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Oelsner S, Waldmann A, Billmeier A, Roder J, Lindner A, Ullrich E, Marschalek R, Dotti G, Jung G, Grosse-Hovest L, Oberoi P, Bader P, Wels WS, Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth, Int J Cancer, 145 (2019) 1935–1945. [DOI] [PubMed] [Google Scholar]
- [215].Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K, Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors, N Engl J Med, 382 (2020) 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, Gagea M, Banerjee P, Cai R, Bdaiwi MH, Basar R, Muftuoglu M, Li L, Marin D, Wierda W, Keating M, Champlin R, Shpall E, Rezvani K, Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity, Leukemia, 32 (2018) 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Yang J, Hu S, Shangguan J, Eresen A, Li Y, Pan L, Ma Q, Velichko Y, Wang J, Hu C, Yaghmai V, Zhang Z, Dendritic cell immunotherapy induces anti-tumor effect in a transgenic mouse model of pancreatic ductal adenocarcinoma, Am J Cancer Res, 9 (2019) 2456–2468. [PMC free article] [PubMed] [Google Scholar]
- [218].Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, Abolghasemi M, Poursaei E, Asadi M, Zafari V, Aghebati-Maleki L, Shanehbandi D, Dendritic cell therapy in cancer treatment; the state-of-the-art, Life Sci, 254 (2020) 117580. [DOI] [PubMed] [Google Scholar]
- [219].Palucka K, Banchereau J, Dendritic-cell-based therapeutic cancer vaccines, Immunity, 39 (2013) 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Adamska A, Domenichini A, Falasca M, Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies, Int J Mol Sci, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Lodge PA, Jones LA, Bader RA, Murphy GP, Salgaller ML, Dendritic cell-based immunotherapy of prostate cancer: immune monitoring of a phase II clinical trial, Cancer Res, 60 (2000) 829–833. [PubMed] [Google Scholar]
- [222].Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D, Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells, Nat Med, 4 (1998) 328–332. [DOI] [PubMed] [Google Scholar]
- [223].Oshita C, Takikawa M, Kume A, Miyata H, Ashizawa T, Iizuka A, Kiyohara Y, Yoshikawa S, Tanosaki R, Yamazaki N, Yamamoto A, Takesako K, Yamaguchi K, Akiyama Y, Dendritic cell-based vaccination in metastatic melanoma patients: phase II clinical trial, Oncol Rep, 28 (2012) 1131–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D’Andre SD, Iwamoto FM, Dropcho EJ, Moshel YA, Walter KA, Pillainayagam CP, Aiken R, Chaudhary R, Goldlust SA, Bota DA, Duic P, Grewal J, Elinzano H, Toms SA, Lillehei KO, Mikkelsen T, Walbert T, Abram SR, Brenner AJ, Brem S, Ewend MG, Khagi S, Portnow J, Kim LJ, Loudon WG, Thompson RC, Avigan DE, Fink KL, Geoffroy FJ, Lindhorst S, Lutzky J, Sloan AE, Schackert G, Krex D, Meisel HJ, Wu J, Davis RP, Duma C, Etame AB, Mathieu D, Kesari S, Piccioni D, Westphal M, Baskin DS, New PZ, Lacroix M, May SA, Pluard TJ, Tse V, Green RM, Villano JL, Pearlman M, Petrecca K, Schulder M, Taylor LP, Maida AE, Prins RM, Cloughesy TF, Mulholland P, Bosch ML, First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma, J Transl Med, 16 (2018) 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Hasegawa H, Matsumoto T, Mechanisms of Tolerance Induction by Dendritic Cells In Vivo, Front Immunol, 9 (2018) 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D, Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity, J Exp Med, 206 (2009) 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Goronzy JJ, Weyand CM, T-cell co-stimulatory pathways in autoimmunity, Arthritis Res Ther, 10 Suppl 1 (2008) S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Hu J, Wan Y, Tolerogenic dendritic cells and their potential applications, Immunology, 132 (2011) 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M, Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients, Diabetes Care, 34 (2011) 2026–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Kurowska W, Kuca-Warnawin EH, Radzikowska A, Maslinski W, The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis, Cent Eur J Immunol, 42 (2017) 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, Pahau H, Lee BT, Ng J, Brunck ME, Hyde C, Trouw LA, Dudek NL, Purcell AW, O’Sullivan BJ, Connolly JE, Paul SK, Le Cao KA, Thomas R, Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients, Sci Transl Med, 7 (2015) 290ra287. [DOI] [PubMed] [Google Scholar]
- [232].Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, De Laere M, Wens I, Cras P, Parizel P, Van Hecke W, Ribbens A, Billiet T, Adams G, Couttenye MM, Navarro-Barriuso J, Teniente-Serra A, Quirant-Sanchez B, Lopez-Diaz de Cerio A, Inoges S, Prosper F, Kip A, Verheij H, Gross CC, Wiendl H, Van Ham MS, Ten Brinke A, Barriocanal AM, Massuet-Vilamajo A, Hens N, Berneman Z, Martinez-Caceres E, Cools N, Ramo-Tello C, consortium R, Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration, BMJ Open, 9 (2019) e030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Lee WP, Willekens B, Cras P, Goossens H, Martinez-Caceres E, Berneman ZN, Cools N, Immunomodulatory Effects of 1,25-Dihydroxyvitamin D3 on Dendritic Cells Promote Induction of T Cell Hyporesponsiveness to Myelin-Derived Antigens, J Immunol Res, 2016 (2016) 5392623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Raiotach-Regue D, Grau-Lopez L, Naranjo-Gomez M, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E, Borras FE, Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients, Eur J Immunol, 42 (2012) 771–782. [DOI] [PubMed] [Google Scholar]
- [235].Naranjo-Gomez M, Raich-Regue D, Onate C, Grau-Lopez L, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E, Borras FE, Comparative study of clinical grade human tolerogenic dendritic cells, J Transl Med, 9 (2011) 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N, Clinical Tolerogenic Dendritic Cells: Exploring Therapeutic Impact on Human Autoimmune Disease, Front Immunol, 8 (2017) 1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].LaRosa DF, Rahman AH, Turka LA, The innate immune system in allograft rejection and tolerance, J Immunol, 178 (2007) 7503–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Alegre ML, Lakkis FG, Morelli AE, Antigen Presentation in Transplantation, Trends Immunol, 37 (2016) 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Matas AJ, Kandaswamy R, Humar A, Payne WD, Dunn DL, Najarian JS, Gruessner RW, Gillingham KJ, McHugh LE, Sutherland DE, Long-term immunosuppression, without maintenance prednisone, after kidney transplantation, Ann Surg, 240 (2004) 510–516; discussion 516–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Thomson AW, Zahorchak AF, Ezzelarab MB, Butterfield LH, Lakkis FG, Metes DM, Prospective Clinical Testing of Regulatory Dendritic Cells in Organ Transplantation, Front Immunol, 7 (2016) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jager U, Jaglowski S, Andreadis C, Westin JR, Fleury I, Bachanova V, Foley SR, Ho PJ, Mielke S, Magenau JM, Holte H, Pantano S, Pacaud LB, Awasthi R, Chu J, Anak O, Salles G, Maziarz RT, Investigators J, Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma, N Engl J Med, 380 (2019) 45–56. [DOI] [PubMed] [Google Scholar]
- [242].Hu M, Zhou X, Wang Y, Guan K, Huang L, Relaxin-FOLFOX-IL-12 triple combination therapy engages memory response and achieves long-term survival in colorectal cancer liver metastasis, J Control Release, 319 (2020) 213–221. [DOI] [PubMed] [Google Scholar]
- [243].Hu Q, Li H, Archibong E, Chen Q, Ruan H, Ahn S, Dukhovlinova E, Kang Y, Wen D, Dotti G, Gu Z, Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets, Nat Biomed Eng, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Montfort A, Filleron T, Virazels M, Dufau C, Milhes J, Pages C, Olivier P, Ayyoub M, Mounier M, Lusque A, Brayer S, Delord JP, Andrieu-Abadie N, Levade T, Colacios C, Segui B, Meyer N, Combining Nivolumab and Ipilimumab with Infliximab or Certolizumab in Patients with Advanced Melanoma: First Results of a Phase Ib Clinical Trial, Clin Cancer Res, 27 (2021) 1037–1047. [DOI] [PubMed] [Google Scholar]
- [245].U.S. Food & Drug Administration, Development & Approval Process | Drugs, 2019.
- [246].U.s. Food & Drug Administration, Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived Products, 1995.
- [247].Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, Wherry EJ, Zhang NR, Ishwaran H, Hellmann MD, Wolchok JD, Kambayashi T, Minn AJ, Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade, Cell, 178 (2019) 933–948 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Shada AL, Molhoek KR, Slingluff CL Jr., Interface of signal transduction inhibition and immunotherapy in melanoma, Cancer J, 16 (2010) 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Ward RC, Kaufman HL, Targeting costimulatory pathways for tumor immunotherapy, Int Rev Immunol, 26 (2007) 161–196. [DOI] [PubMed] [Google Scholar]
- [250].Mak IW, Evaniew N, Ghert M, Lost in translation: animal models and clinical trials in cancer treatment, Am J Transl Res, 6 (2014) 114–118. [PMC free article] [PubMed] [Google Scholar]
- [251].Sanmamed MF, Chester C, Melero I, Kohrt H, Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies, Ann Oncol, 27 (2016) 1190–1198. [DOI] [PubMed] [Google Scholar]
- [252].U.S. Food & Drug Administration, SOPP 8214: INTERACT Meetings with Sponsors for Drugs and Biological Products, (2020).
- [253].U.S. Food & Drug Administration, Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products Guidance for Industry, 2017.
- [254].U.S. Food & Drug Administration, Requesting FDA Feedback on Combination Products Guidance for Industry and FDA Staff, 2020.
- [255].U.S. Food & Drug Administration, Nonclinical Safety Evaluation of the Immunotoxic Potential of Drugs and Biologics Guidance for Industry, 2020.
- [256].Brinks V, Jiskoot W, Schellekens H, Immunogenicity of therapeutic proteins: the use of animal models, Pharm Res, 28 (2011) 2379–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Morrow T, Felcone LH, Defining the difference: What Makes Biologics Unique, Biotechnol Healthc, 1 (2004) 24–29. [PMC free article] [PubMed] [Google Scholar]
- [258].Haddock R, Lin-Gibson S, Lumelsky N, McFarland R, Roy K, Saha K, Zhang J, and Zylberberg C., Manufacturing Cell Therapies: The Paradigm Shift in Health Care of this Century. NAM Perspectives. Discussion Paper. National Academy of Medicine, Washington, DC., 2017 [Google Scholar]
- [259].U.S. Food & Drug Administration, Biologics Guidances, 2021.
- [260].U.S. Food & Drug Administration, Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process, 2020.
- [261].Characiejus D, Jacobs JJ, Pasukoniene V, Kazlauskaite N, Danileviciute V, Mauricas M, Den Otter W, Prediction of response in cancer immunotherapy, Anticancer Res, 31 (2011) 639–647. [PubMed] [Google Scholar]
- [262].Siebert JC, Walker EB, Monitoring cytokine profiles during immunotherapy, Immunotherapy, 2 (2010) 799–816. [DOI] [PubMed] [Google Scholar]
- [263].U.S. Food & Drug Administration, Immunogenicity Assessment for Therapeutic Protein Products, 2018.
- [264].Bharadwaj P, Riekofski C, Lin S, Seaman MS, Garber DA, Montefiori D, Sarzotti-Kelsoe M, Ackerman ME, Weiner JA, Implementation of a three-tiered approach to identify and characterize anti-drug antibodies raised against HIV-specific broadly neutralizing antibodies, J Immunol Methods, 479 (2020) 112764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Waddington KE, Papadaki A, Coelewij L, Adriani M, Nytrova P, Kubala Havrdova E, Fogdell-Hahn A, Farrell R, Donnes P, Pineda-Torra I, Jury EC, Using Serum Metabolomics to Predict Development of Anti-drug Antibodies in Multiple Sclerosis Patients Treated With IFNbeta, Front Immunol, 11 (2020) 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].U.S. Food & Drug Administration, Best Practices for Communication Between IND Sponsors and FDA During Drug Development: Guidance for Industry and Review Staff, 2017.
- [267].Wong CH, Siah KW, Lo AW, Estimation of clinical trial success rates and related parameters, Biostatistics, 20 (2019) 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].DiMasi JA, Grabowski HG, Hansen RW, Innovation in the pharmaceutical industry: New estimates of R&D costs, J Health Econ, 47 (2016) 20–33. [DOI] [PubMed] [Google Scholar]
- [269].Wouters OJ, McKee M, Luyten J, Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018, JAMA, 323 (2020) 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].U.S. Food & Drug Administration, Expedited Programs for Serious Conditions – Drugs and Biologics, 2020.
- [271].Kepplinger EE, FDA’s Expedited Approval Mechanisms for New Drug Products, Biotechnol Law Rep, 34 (2015) 15–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].U.S. Food & Drug Administration, Accelerated Approval, 2018.
- [273].Cassidy C, Dever D, Stanbery L, Edelman G, Dworkin L, Nemunaitis J, FDA efficiency for approval process of COVID-19 therapeutics, Infect Agent Cancer, 15 (2020) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].U.S. Food & Drug Administration, Priority Review, 2018.
- [275].Nasr M, Sigdel T, Sarwal M, Advances in diagnostics for transplant rejection, Expert Rev Mol Diagn, 16 (2016) 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].DePeaux K, Delgoffe GM, Metabolic barriers to cancer immunotherapy, Nature Reviews Immunology, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]