

Abstract
Purpose of review
Systemic lupus erythematosus (SLE) is a serious autoimmune disease with a wide range of organ involvement. In addition to aberrant B-cell responses leading to autoantibody production, T-cell abnormalities are important in the induction of autoimmunity and the ensuing downstream organ damage. In this article, we present an update on how subsets of CD8+ T cells contribute to SLE pathogenesis.
Recent findings
Reduced cytolytic function of CD8+ T cells not only promotes systemic autoimmunity but also accounts for the increased risk of infections. Additional information suggests that effector functions of tissue CD8+ T cells contribute to organ damage. The phenotypic changes in tissue CD8+ T cells likely arise from exposure to tissue microenvironment and crosstalk with tissue resident cells. Research on pathogenic IL-17-producing double negative T cells also suggests their origin from autoreactive CD8+ T cells, which also contribute to the induction and maintenance of systemic autoimmunity.
Summary
Reduced CD8+ T-cell effector function illustrates their role in peripheral tolerance in the control of autoimmunity and to the increased risk of infections. Inflammatory cytokine producing double negative T cells and functional defects of regulatory CD8+ T cell both contribute to SLE pathogenesis. Further in depth research on these phenotypic changes are warranted for the development of new therapeutics for people with SLE.
Keywords: CD8T cells, lupus, systemic lupus erythematosus
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by autoantibody production with the presentation of inflammation and damage in multiple organs with serious life-threatening complications [1]. The cause of autoimmunity is multifactorial and various immune cell type abnormalities are involved in the pathogenesis of the disease (reviewed in [2]). Besides the aberrant expansion of autoreactive B cells, abnormalities in numerous T-cell subsets also contribute to the development of autoimmunity and systemic inflammation through direct action or cytokine production (reviewed in [3]). In this article, we focus on the role of CD8+ T cells in SLE pathogenesis and review how each of the various subsets contributes to the progression of autoimmunity (Fig. 1).
FIGURE 1.
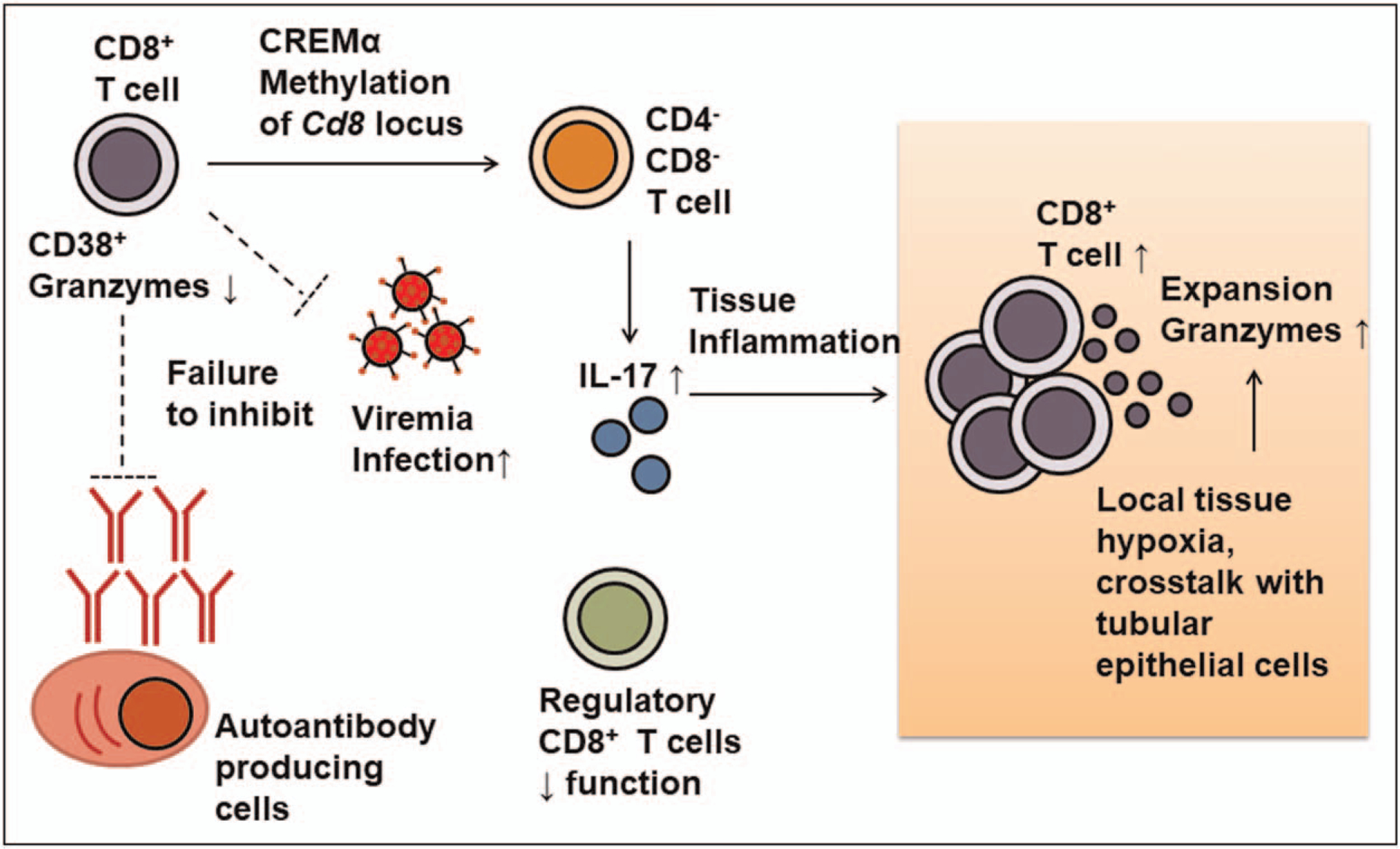
Immunopathology of various CD8+ T-cell subtypes and contribution to pathogenesis. Reduced cytolytic function of systemic CD8+ is related to the expanded CD38+ subpopulation and correlates with the increased risk of infection. Functional defects of cytotoxic T cells also result in failure to remove autoreactive B cells and thus increase autoantibody producing cells. Functional defects in regulatory CD8+ T cells also contribute to the loss of peripheral tolerance. Meanwhile, methylation of Cd8 locus through cAMP-responsive element modulator α and the inflammatory milieu lead to generation and expansion of IL-17-producing CD4−CD8−double negative T cells. Tissue infiltrating CD8+ T-cell cause tissue damage as a result of exposure to changes in tissue metabolic factors, such as hypoxia and arginine levels.
CYTOTOXIC CD8+ T CELL
Cytotoxic T lymphocytes (CTL), the most abundant type of CD8+ T cells, are characterized by their cytolytic activity through the expression of perforin and granzymes, as well as cytokine production, including IFN-γ and TNF-α. With specific T-cell receptors recognizing foreign antigens presented by antigen presenting cells, such as dendritic cells or macrophages, naïve CD8+ T cells differentiate into effector cells and expand exponentially. These expanded activated effector CTLs are cytotoxic against cells expressing nonself antigens – either virus-infected cells or cancer cells. After controlling the infection, the majority of these effector T cells undergo apoptosis, while a small portion of activated CD8+ T cells differentiate into memory T cells, a resting state that can be promptly reactivated upon demand.
CD8+ T cells from the peripheral blood of SLE patients frequently display a reduction in effector function, including attenuated granzyme B and perforin production [4]. The impaired cytolytic defect in CD8+ T cells likely contributes to the pathogenesis of autoimmunity [5]. Genetic elimination of perforin production in lupus-prone mice results in accelerated disease progression and confirms the role of CTLs in halting autoimmunity [6]. These data likely suggest the role of CD8+ T cells in the establishment and maintenance of peripheral tolerance, and defects in their cytolytic function results in failure to remove autoreactive B cells. In graft-vs.-host murine lupus models, both perforin-mediated and Fas-ligand-mediated cytotoxicity are required for optimal control of autoreactive B cells [7,8]. Enhancing cytolytic function of CTL suppresses B-cell autoreactivity and limits disease progression [9,10], but whether such treatment could apply to help reduce autoreactive B cells in patients remains to be investigated.
Reduced cytolytic function of CTLs in lupus patients also poses another unfavorable outcome in lupus patients, because this reduction of T-cell effector function correlates with higher risk of infection [11] including a defect in controlling latent Epstein–Barr virus (EBV) [12,13]. Prescription of immunosuppressive medications could explain part of these defects, but T-cell function is still greatly diminished even in those taking low doses of immunosuppressive drugs [14]. Further, lupus-prone mice are highly susceptible to infections [15]. Poor latent EBV control also poses another unfavorable effect on lupus pathogenesis, since molecular mimicry of latent viral protein EBV nuclear antigen-1 (EBNA-1) progressively contributes to the production of autoantibodies [16]. Consistent with the above findings, reduced EBNA-1 specific T-cell response correlates with higher disease activity measured by SLEDAI score in SLE patients [17]. The functional defects in lupus CD8+ T cells are also linked to changes in the expression of several surface proteins. Signaling lymphocytic activation molecule family member 4 (CD244), also known as natural killer (NK) cell receptor 2B4 and thought to modulate cytolytic activity, is downregulated in CD8+ T cells from SLE patients [18]. CD38, cell surface expressing cyclic ADP ribose hydrolase, is a major regulator of cellular NAD+ levels through its catalytic function of NAD+ to synthesize ADP ribose and cyclic ADP-ribose. Studies of tumor infiltrating lymphocytes reveal that the expression of CD38 represents an irreversibly dysfunctional group of CD8+ T cells, which fail to recover cytokine production after ex vivo stimulation, and such fixed dysfunctional state of the CD38+ CD8+ T cell is strongly associated with a discrete chromatin landscape [19]. Similarly, dysfunctional CD38+ CD8+ T cells are also found expanded in the peripheral blood of SLE patients, with features of reduced granzyme and perforin production, and this functional defective state represents the result of CD38-dependent activation of histone methyl-transferase EZH2 in limiting chromatin accessibility of several key gene loci responsible for the regulation of T-cell effector function including RUNX3, EOMES and TBX21 [20■■]. Increased frequencies of these dysfunctional CD38+ CD8+ T cells correlate with a high risk of infection in SLE patients. Inhibition of CD38 could potentially reverse these adverse effects on CTL function. Administration of daratumumab, an anti-CD38 antibody, not only suppresses autoreactive plasma cells and reduces autoantibody titers, but also restored cytotoxicity of peripheral CD8+ T cells in two SLE patients [21].
CD8+ T CELL IN DAMAGED ORGAN
In SLE patients with class III or IV nephritis, CD8+ T cells are one of the predominant infiltrating immune cells, and their accumulation in periglomerular areas correlates with the degree of disease activity [22]. These CD8+ T cells presenting with effector memory phenotype are also found in the urine sediment, and are thought to contribute to tissue damage [23]. T cell receptor sequencing of infiltrating T cells shows clonal expansion and, interestingly, the same clonotype persists in the kidney years later in the repeat kidney biopsy tissue [24]. Taken together, these data suggest that the infiltrating CD8+ T cells in nephritic kidney are tissue-resident and likely expand locally as disease exacerbates. However, whether this whole process involves antigen-specific T-cell response and the nature of the local autoantigen remains to be investigated. Several therapeutic approaches have been designed to target these tissue resident memory cells. The Janus kinase inhibitor tofacitinib not only blocks cytokine receptors necessary for aberrant T-cell activation, but also effectively prevents the expansion of tissue-resident memory T cells in murine lupus nephritis [25]. These memory CD8+ T cells in nephritic kidneys also express high voltage-dependent Kv1.3 potassium channels, and targeted knockdown of Kv1.3 suppress CD40L expression and IFNγ production in a humanized mouse model of lupus nephritis [26■]. These tissue resident memory suppressing therapeutics also improve the outcome of kidney damage in lupus-prone mice, suggesting the role of tissue-resident memory CD8+ T cells in organ damage pathogenesis.
The nature of kidney infiltrating T cells is long thought to be functionally active and through their cytolytic function contribute to tissue damage. However, contrasting evidence by Tilstra et al. [27] surprisingly demonstrated near complete abolishment of the effector function in kidney-infiltrating of both CD4+ and CD8+ T cells. These data raises the questions about the functionality of kidney infiltrating T cells, and whether chronic antigen exposure can lead to exhaustion locally at the site of inflammation. However, single-cell RNA-sequencing of lupus nephritis biopsy samples did not reveal features of exhausted cell subsets, and the clusters of NK and CD8+ T cells were noted to express high numbers of GZMB and GZMK transcripts [28■]. These seemingly discrepant data likely suggest the complex nature of lupus, with the result of heterogeneous features of kidney infiltrating T cells. Another intriguing cause of T-cell functional changes may stem from tissue microenvironment as the result of tissue inflammation and damage. These alterations of in situ cues instruct phenotypic changes of infiltrating T cells, and could be used to selectively suppress the local inflammatory response. Local tissue hypoxia was found to result from organ damage in lupus nephritis followed by the subsequent activation of the transcription factor hypoxia-inducible factor-1 (HIF-1) which controls cell survival and adequate glycolytic metabolism required for effector function in kidney-infiltrating T cells [29■]. These data indicate the therapeutic potential of HIF-1 inhibition to block microenvironmental cues to restore tissue infiltrating T-cell functionality and reverse organ damage. Changes in local metabolite concentrations could also serve as an important route to control the crosstalk between resident tissue cells and infiltrating T cells. Renal tubular epithelial cells downregulate arginine degrading enzyme arginase 1 through the response of IL-23 receptor and calcium/calmodulin kinase IV (CaMK4). The resulting increase in free L-arginine promotes proliferation of infiltrating T cells, and targeted CaMK4 inhibition in renal tubular epithelial cells prevents the expansion of T cells locally [30■]. These data suggest phenotypic difference between systemic and local CD8+ T cells could arise from metabolic changes in the damaged tissue, and these phenotypic alterations could serve as potential therapeutic targets.
DOUBLE NEGATIVE T CELLS
Double negative T cells, a particular group of αβ T cells without surface expression of either CD4 or CD8, are thought to contribute to SLE pathogenesis as one of the major sources of IL-17 production [31]. Despite our limited knowledge about this population, current information suggests its derivation from self-reactive CD8+ T cells in tissues expressing autoantigens [32]. Transcriptome analysis has confirmed gene expression profile similarities between double negative T cells and CD8+ T cells [33], and double negative T cells share a skewed oligoclonal T-cell receptor repertoire with CD8+ T cells in patients with autoimmune lymphoproliferative syndrome [34]. These data strongly suggest the possibility of lineage transition from CD8+ T cells to double negative T cells, and this process involves downregulation of CD8 surface expression through cAMP-responsive element modulator α (CREMα)-dependent transcriptional silencing of CD8A and CD8B [35]. CREMα suppression of CD8 is mediated through histone methylation of the Cd8 locus mediated by the recruitment of DNA methyltransferase 3a and histone methyltransferase G9a [36]. Double negative T cells are expanded in lupus mice carrying lpr or gld variants as disease progresses [37,38]. These murine lupus models have defects in T-cell apoptosis due to loss of function of the Fas mutation, and prevent activation induced cell death despite repeated T-cell receptor stimulation. These murine models are especially useful to depict the pathogenic role of double negative T cells, as T-cell receptor stimulation of CD8+ T cells also promotes loss of surface CD8 expression to form double negative T cells [39]. Taken together, these double negative T cells in lupus arise likely from autoreactive CD8+ T cells as a result of chronic stimulation by autoantigen presented from apoptotic cells [40■■]. Furthermore, changes of cytokines in the inflammatory milieu, including reduction of TGF-β and elevation of IL-23 also contribute to the loss of tolerance and expansion of IL-17 producing double negative T cells [40■■]. Double negative T cells can also provide aberrant B cell help to augment anti-DNA autoantibody production [41]. In addition to the murine models, these double negative T cells are highly expanded in the peripheral blood and inflamed kidneys of lupus patients, further indicating their pathogenic role in the development of systemic autoimmunity and organ damage [42]. In summary, unlike the regulatory double negative T cell that can inhibit activation and proliferation of antigen-specific CD8+ T cells [43], double negative T cells in autoimmune diseases predominantly present with pathogenic and proinflammatory phenotypes. These phenotypic differences likely stem from the presence of autoantigens from apoptotic cells and cytokines that promote the inflammatory phenotype.
CD8+ REGULATORY T CELLS
Numerous subsets of CD8+ T cells, either natural or inducible, possess a cell suppressive activity similar to that of regulatory CD4+ T cells. The natural CD8+ CD25+ T regulatory cells express high levels of Foxp3 and exert their inhibitory function mainly through the secretion of transforming growth factor β1 (TGF-β1) and contact-dependent suppression through the cytotoxic T-lymphocyte-associated antigen 4 [44]. CD8+ T regulatory T cells can also be induced by continuous antigen stimulation with CD14+ monocytes [45], or coculture with CD40 ligand-activated plasmacytoid dendritic cells [46], or CD40-activated B cells [47]. The suppressive activity of these inducible CD8+ T regulatory T cells depends on IL-10 production [46,48] and TGF-β secretion [49].
The immune suppressive function of regulatory CD8+ T cells can also alleviate autoimmunity in lupus. In lupus prone NZB × NZW F1 mice, the immune tolerance induced by administering artificial peptide pConsensus depends on the activity of TGF-β secreting CD8+ T cells [49], and the inhibitory activity of these CD8+ suppressor cells depends on the expression of the transcription factor Foxp3 [50]. CD8+ regulatory T cells can have been reported during the induction of tolerance by nucleosomal histone peptides in lupus prone SWR × NZB F1 mice, and their suppressive function depended upon TGF-β secretion [51]. In another tolerogenic model induced by complementarity-determining region-1 peptide for NZB × NZW F1 lupus mice, Foxp3-expressing CD8+ cells promoted the expansion and suppressive function of CD4+ CD25+ regulatory T cells [52]. In addition to these tolerogenic models, ex-vivo-generated autologous CD4+ and CD8+ regulatory T cells help ameliorate autoimmunity in a graft-vs.-host model presenting with lupus-like syndrome [53]. Studies of peripheral blood mononuclear cells from SLE patients have reported that CD8+ suppressive cells are functionally impaired with reduced IL-10 and TGF-β production [54,55]. In patients with refractory disease who receive autologous hemopoietic stem cell transplantation, immunological remission depends on the suppressive activity of TGF-β producing CD8+ regulatory T cells [56]. These data imply the potential therapeutic exploitation of CD8+ regulatory T cells in SLE patients, but the key to bolster their suppressive activity to induce immune tolerance remains to be investigated.
CONCLUSION
Reduced effector function of peripheral blood CD8+ T cells promotes autoimmunity by failing to induce peripheral tolerance by removing autoreactive B cells, and by controlling the risk of infection. In contrast, at least part of the effector function of tissue CD8+ T cells remain intact, and correlates with the extent of organ damage. The difference in effector phenotype likely stems from metabolic changes in the tissue microenvironment and from the crosstalk with tissue resident cells. IL-17-producing double negative T cells and dysfunctional regulatory CD8+ T cell also contribute to disease pathogenesis. Recent studies have helped us understand better the immunopathology of CD8+ T cell and their subsets but more information is needed for the development of new therapeutic strategies.
KEY POINTS.
Reduction in systemic CD8+ T-cell effector function contributes to the induction of autoimmunity as a result of failure to remove autoreactive B cells, and the control of infections.
IL-17-producing double negative T cells originate likely from autoreactive CD8+ T-cell and promote systemic autoimmunity.
Functional impairment in regulatory CD8+ T cells contributes to disease pathogenesis.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011; 365:2110–2121. [DOI] [PubMed] [Google Scholar]
- 2.Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol 2020; 21:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen PM, Tsokos GC. T cell abnormalities in the pathogenesis of systemic lupus erythematosus: an update. Curr Rheumatol Rep 2021; 23:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comte D, Karampetsou MP, Yoshida N, et al. Signaling lymphocytic activation molecule family member 7 engagement restores defective effector CD8+ T cell function in systemic lupus erythematosus. Arthritis Rheumatol 2017; 69:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stohl W. Impaired polyclonal T cell cytolytic activity. A possible risk factor for systemic lupus erythematosus. Arthritis Rheum 1995; 38:506–516. [DOI] [PubMed] [Google Scholar]
- 6.Peng SL, Moslehi J, Robert ME, Craft J. Perforin protects against autoimmunity in lupus-prone mice. J Immunol 1998; 160:652–660. [PubMed] [Google Scholar]
- 7.Soloviova K, Puliaiev M, Puliaev R, et al. Both perforin and FasL are required for optimal CD8 T cell control of autoreactive B cells and autoantibody production in parent-into-F1 lupus mice. Clin Immunol 2018; 194:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shustov A, Luzina I, Nguyen P, et al. Role of perforin in controlling B-cell hyperactivity and humoral autoimmunity. J Clin Invest 2000; 106:R39–R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen V, Rus H, Chen C, Rus V. CTL-promoting effects of IL-21 counteract murine lupus in the parent!F1 graft-versus-host disease model. J Immunol 2016; 196:1529–1540. [DOI] [PubMed] [Google Scholar]
- 10.Puliaev R, Puliaeva I, Welniak LA, et al. CTL-promoting effects of CD40 stimulation outweigh B cell-stimulatory effects resulting in B cell elimination and disease improvement in a murine model of lupus. J Immunol 2008; 181:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 2016; 12:716–730. [DOI] [PubMed] [Google Scholar]
- 12.Kang I, Quan T, Nolasco H, et al. Defective control of latent Epstein–Barr virus infection in systemic lupus erythematosus. J Immunol 2004; 172: 1287–1294. [DOI] [PubMed] [Google Scholar]
- 13.Larsen M, Sauce D, Deback C, et al. Exhausted cytotoxic control of Epstein–Barr virus in human lupus. PLoS Pathog 2011; 7:e1002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassaniti I, Cavagna L, Calarota SA, et al. Evaluation of EBV- and HCMV-specific T cell responses in systemic lupus erythematosus (SLE) patients using a normalized enzyme-linked immunospot (ELISPOT) assay. J Immunol Res 2019; 2019:4236503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieberman LA, Tsokos GC. Lupus-prone mice fail to raise antigen-specific T cell responses to intracellular infection. PLoS One 2014; 9:e111382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClain MT, Heinlen LD, Dennis GJ, et al. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 2005; 11:85–89. [DOI] [PubMed] [Google Scholar]
- 17.Draborg AH, Jacobsen S, Westergaard M, et al. Reduced response to Epstein–Barr virus antigens by T-cells in systemic lupus erythematosus patients. Lupus Sci Med 2014; 1:e000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kis-Toth K, Comte D, Karampetsou MP, et al. Selective loss of signaling lymphocytic activation molecule family member 4-positive CD8+ T cells contributes to the decreased cytotoxic cell activity in systemic lupus erythematosus. Arthritis Rheumatol 2016; 68:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Philip M, Fairchild L, Sun L, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017; 545:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.■■.Katsuyama E, Suarez-Fueyo A, Bradley SJ, et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep 2020; 30:112–123.e114. [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed mechanistic insight about how CD38 affect CD8 T cell in systemic lupus erythematosus, and how their dysfunction incerases the risk for infection.
- 21.Ostendorf L, Burns M, Durek P, et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N Engl J Med 2020; 383: 1149–1155. [DOI] [PubMed] [Google Scholar]
- 22.Couzi L, Merville P, Deminière C, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum 2007; 56:2362–2370. [DOI] [PubMed] [Google Scholar]
- 23.Dolff S, Abdulahad WH, van Dijk MC, et al. Urinary T cells in active lupus nephritis show an effector memory phenotype. Ann Rheum Dis 2010; 69:2034–2041. [DOI] [PubMed] [Google Scholar]
- 24.Couzi L, Merville P, Deminiere C, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum 2007; 56:2362–2370. [DOI] [PubMed] [Google Scholar]
- 25.Zhou M, Guo C, Li X, et al. JAK/STAT signaling controls the fate of CD8(+)CD103(+) tissue-resident memory T cell in lupus nephritis. J Autoimmun 2020; 109:102424. [DOI] [PubMed] [Google Scholar]
- 26.■.Khodoun M, Chimote AA, Ilyas FZ, et al. Targeted knockdown of Kv1.3 channels in T lymphocytes corrects the disease manifestations associated with systemic lupus erythematosus. Sci Adv 2020; 6:eabd1471. [DOI] [PMC free article] [PubMed] [Google Scholar]; Therapeutic potential of targeting resident memory T cells in lupus nephritis.
- 27.Tilstra JS, Avery L, Menk AV, et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J Clin Invest 2018; 128:4884–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.■.Arazi A, Rao DA, Berthier CC, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol 2019; 20:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive single Cell RNAseq data of the immune landscape in lupus nephritis.
- 29.■.Chen PM, Wilson PC, Shyer JA, et al. Kidney tissue hypoxia dictates T cell-mediated injury in murine lupus nephritis. Sci Transl Med 2020; 12:eaay1620. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tissue hypoxia in the microenvironment could affect T-cell phenotype and contribute to organ damage.
- 30.■.Li H, Tsokos MG, Bhargava R, et al. Interleukin-23 reshapes kidney resident cell metabolism and promotes local kidney inflammation. J Clin Invest 2021; 131:e142428. [DOI] [PMC free article] [PubMed] [Google Scholar]; Changes in local metabolism shaped by resident tubular epithelial cells serve as crosstalk with the infiltrating T cells.
- 31.Crispin JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol 2008; 181:8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez-Rodríguez N, Apostolidis SA, Penaloza-MacMaster P, et al. Programmed cell death 1 and Helios distinguish TCR-αβ+ double-negative (CD4−CD8−) T cells that derive from self-reactive CD8 T cells. J Immunol 2015; 194:4207–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crispín JC, Tsokos GC. Human TCR-alpha beta+ CD4− CD8− T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol 2009; 183:4675–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bristeau-Leprince A, Mateo V, Lim A, et al. Human TCR alpha/beta+ CD4− CD8− double-negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vbeta TCR diversity and are clonally related to CD8+ T cells. J Immunol 2008; 181:440–448. [DOI] [PubMed] [Google Scholar]
- 35.Hedrich CM, Rauen T, Crispin JC, et al. cAMP-responsive element modulator α (CREMα) trans-represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4−CD8− T cells in health and disease. J Biol Chem 2013; 288:31880–31887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedrich CM, Crispín JC, Rauen T, et al. cAMP responsive element modulator (CREM) α mediates chromatin remodeling of CD8 during the generation of CD3+ CD4− CD8− T cells. J Biol Chem 2014; 289:2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morse HC 3rd, Davidson WF, Yetter RA, et al. Abnormalities induced by the mutant gene Ipr: expansion of a unique lymphocyte subset. J Immunol 1982; 129:2612–2615. [PubMed] [Google Scholar]
- 38.Roths JB, Murphy ED, Eicher EM. A new mutation, gld, that produces lymphoproliferation and autoimmunity in C3H/HeJ mice. J Exp Med 1984; 159:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehal WZ, Crispe IN. TCR ligation on CD8+ T cells creates double-negative cells in vivo. J Immunol 1998; 161:1686–1693. [PubMed] [Google Scholar]
- 40.■■.Li H, Adamopoulos IE, Moulton VR, et al. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat Commun 2020; 11:2859. [DOI] [PMC free article] [PubMed] [Google Scholar]; Double negative T cells are expaneded in the inflammatory millieu with persisent autoantigen stimulation.
- 41.Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4−/CD8−) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol 1989; 143:103–112. [PubMed] [Google Scholar]
- 42.Crispín JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol 2008; 181:8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang ZX, Yang L, Young KJ, et al. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med 2000; 6:782–789. [DOI] [PubMed] [Google Scholar]
- 44.Cosmi L, Liotta F, Lazzeri E, et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood 2003; 102:4107–4114. [DOI] [PubMed] [Google Scholar]
- 45.Mahic M, Henjum K, Yaqub S, et al. Generation of highly suppressive adaptive CD8(+)CD25(+)FOXP3(+) regulatory T cells by continuous antigen stimulation. Eur J Immunol 2008; 38:640–646. [DOI] [PubMed] [Google Scholar]
- 46.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med 2002; 195:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng J, Liu Y, Qin G, et al. Efficient induction and expansion of human alloantigen-specific CD8 regulatory T cells from naive precursors by CD40-activated B cells. J Immunol 2009; 183:3742–3750. [DOI] [PubMed] [Google Scholar]
- 48.Wei S, Kryczek I, Zou L, et al. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res 2005; 65:5020–5026. [DOI] [PubMed] [Google Scholar]
- 49.Hahn BH, Singh RP, La Cava A, Ebling FM. Tolerogenic treatment of lupus mice with consensus peptide induces Foxp3-expressing, apoptosis-resistant, TGFbeta-secreting CD8+ T cell suppressors. J Immunol 2005; 175:7728–7737. [DOI] [PubMed] [Google Scholar]
- 50.Singh RP, La Cava A, Wong M, et al. CD8+ T cell-mediated suppression of autoimmunity in a murine lupus model of peptide-induced immune tolerance depends on Foxp3 expression. J Immunol 2007; 178:7649–7657. [DOI] [PubMed] [Google Scholar]
- 51.Kang HK, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol 2005; 174:3247–3255. [DOI] [PubMed] [Google Scholar]
- 52.Sharabi A, Mozes E. The suppression of murine lupus by a tolerogenic peptide involves foxp3-expressing CD8 cells that are required for the optimal induction and function of foxp3-expressing CD4 cells. J Immunol 2008; 181:3243–3251. [DOI] [PubMed] [Google Scholar]
- 53.Zheng SG, Wang JH, Koss MN, et al. CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-βeta suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J Immunol 2004; 172:1531–1539. [DOI] [PubMed] [Google Scholar]
- 54.Filaci G, Bacilieri S, Fravega M, et al. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol 2001; 166:6452–6457. [DOI] [PubMed] [Google Scholar]
- 55.Tulunay A, Yavuz S, Direskeneli H, Eksioglu-Demiralp E. CD8+CD28−, suppressive T cells in systemic lupus erythematosus. Lupus 2008; 17:630–637. [DOI] [PubMed] [Google Scholar]
- 56.Zhang L, Bertucci AM, Ramsey-Goldman R, et al. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-βeta-producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol 2009; 183:6346–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]