

Abstract
We describe association of olfactory bulb and olfactory tract abnormalities in a child with acrocallosal syndrome caused by kinesin family membrane 7 (KIF7) mutation in sonic hedgehog pathway. The child also had fontanellar bone in the anterior fontanelle, short sagittal suture, sagittal synostosis, hippocampal malrotation and Joubert malformation. Fontanellar bone has been described in GLI3 mutation and mutant mice models but has not been reported in KIF7 mutation. We briefly review the role of sonic hedgehog pathway and its components KIF7 and GLI3 in forebrain and olfactory system development and also describe olfactory system abnormality in a child with GLI3 mutation.
Keywords: Acrocallosal syndrome, Greig cephalopolysyndactyly syndrome, Infant, Magnetic resonance imaging, Olfactory bulb, Olfactory tract, Sonic hedgehog pathway
Introduction
Sonic hedgehog signaling (Shh) pathway is important for forebrain development including olfactory bulb and olfactory tract development [1]. Mutations in the Shh gene lead to holoprosencephaly [1], which can be associated with olfactory system abnormality. KIF7 and GLI3 are important components of the Shh signaling pathway (Fig. 1) [2], which plays an important role in many developmental processes. KIF7, a gene on chromosome 15q26.1 [2], encodes a cilium-associated kinesin family motor protein that moves antegrade along microtubule by adenosine triphosphate hydrolysis. It is involved in cellular functions such as axonal transport, mitosis and meiosis [1, 2]. GLI3, a gene on chromosome 7p14.1, encodes a zinc finger protein with an essential role in neural tube patterning via the regulation of Shh signaling [1, 2]. Shh signaling in the neural tube results in KIF7 translocation of GLI3 to the cilium tip, where it is sequestered and prevented from being cleaved, causing relief of GLI3-mediated transcription repression [1, 2]. Hence KIF7 mutation can lead to abnormal GLI3 processing and the disruption of Shh signaling [2].
Fig. 1.
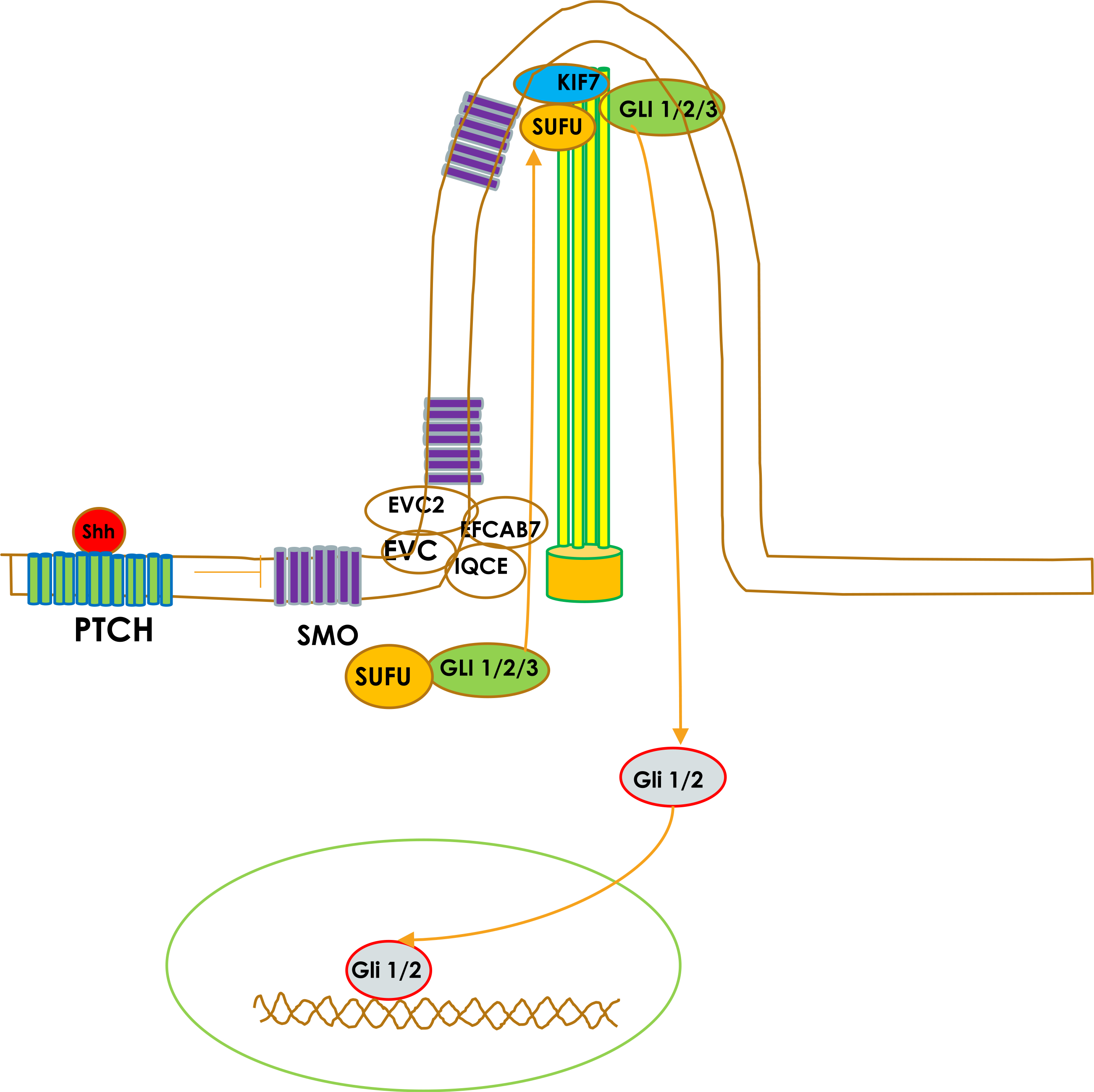
Sonic hedgehog signaling pathway. Binding of Shh ligands to patched (PTCH) derepresses smoothened protein (SMO). SMO translocates onto tip of primary cilium. This leads to transcriptional activation of GLI1/ 2. Activation and nuclear translocation of GLI1/2 involve dissociation of GLI1/2/3 from its endogenous inhibitor, SUFU. KIF7 interacts with SUFU and causes dissociation of the SUFU–GLI complex
In this study we report observations of brain abnormalities in an infant with acrocallosal syndrome involving KIF7 mutation and another child with Greig cephalopolysyndactyly syndrome associated with a GLI3 mutation. Acrocallosal syndrome [2] is characterized by agenesis or partial agenesis of corpus callosum, and craniofacial anomalies, namely macrocephaly, frontal bossing, hypertelorism, large anterior fontanel and everted nostrils. These children sometimes have polydactyly and psychomotor retardation. Greig cephalopolysyndactyly syndrome [3] is characterized by polydactyly, macrocephaly, hypertelorism and intellectual disability. This syndrome has been associated with corpus callosum partial agenesis and it is associated with mutation in GLI3 [3]. Although mutations in KIF7 cause disease in an autosomal-recessive manner and mutations in GLI3 cause disease in an autosomal-dominant manner, they have phenotypic similarities, likely given their involvement in regulating the same signaling pathway.
Case report
A 2-month-old girl presented with macrocephaly and abnormal head shape to the neurosurgery clinic. On clinical examination she had scaphocephaly, large open flat anterior fontanelle, and open metopic suture. There was posterior sagittal ridging, bilateral frontal bossing, hypertelorism, occipital bulleting and occult submucous cleft palate, and clinical findings were suggestive of sagittal craniosynostosis. A noncontrast CT demonstrated short sagittal suture and ridging of sagittal sutures, even though there was no osseous bridging of suture on bone window images, suggesting early sagittal craniosynostosis (Fig. 2). There was a large bone in the anterior fontanelle (fontanellar bone), which had partially fused with both parietal bones posteriorly (Fig. 2). The coronal, metopic and lambdoid sutures were patent and there was frontal bossing and hypertelorism. In addition, CT demonstrated corpus callosum agenesis and brainstem dysplasia. MRI was obtained to further characterize brain malformation. High-resolution fast imaging employing steady-state acquisition (FIESTA) images demonstrated absent olfactory bulb and absent olfactory tracts (Fig. 3). There were corpus callosum agenesis and hippocampal malrotation (Fig. 3). MRI also demonstrated isthmic thinning, batwing configuration of 4th ventricle (although the roof of the 4th ventricle was not horizontally oriented), thickened and abnormally oriented superior cerebellar peduncle, and mild vermian hypoplasia from Joubert malformation (Fig. 3). Diffusion tensor imaging demonstrated absent superior cerebellar peduncle decussation and thickened superior cerebellar peduncle (Fig. 3). The pituitary gland was normal on MRI. The girl developed seizures at 3 months of age and had multiple subsequent follow-up MRIs; molar tooth abnormality was obvious on follow-up MRI obtained at 4 years of age and images showed only partial agenesis of corpus callosum. A skeletal survey was normal except for macrocephaly. The girl underwent sagittal strip craniectomy, bilateral temporoparietal barrel stave osteotomies, bilateral frontal osteotomies and fronto-nasal wedge osteotomy for sagittal craniosynostosis. The girl had epilepsy and was being managed with antiepileptics. The girl had global developmental delay and speech delay, and she was beginning to take few steps with support at 3 years of age; she was receiving physical therapy and speech therapy. The girl underwent targeted exome sequencing, which revealed two pathological mutations in KIF7, one being a novel mutation c.2364del (p. R789fs), and one known pathogenic mutation, c.2593–3C>G, indicating this girl might be compound heterozygous and likely to have acrocallosal syndrome. Of note, the parents did not undergo genotyping.
Fig. 2.
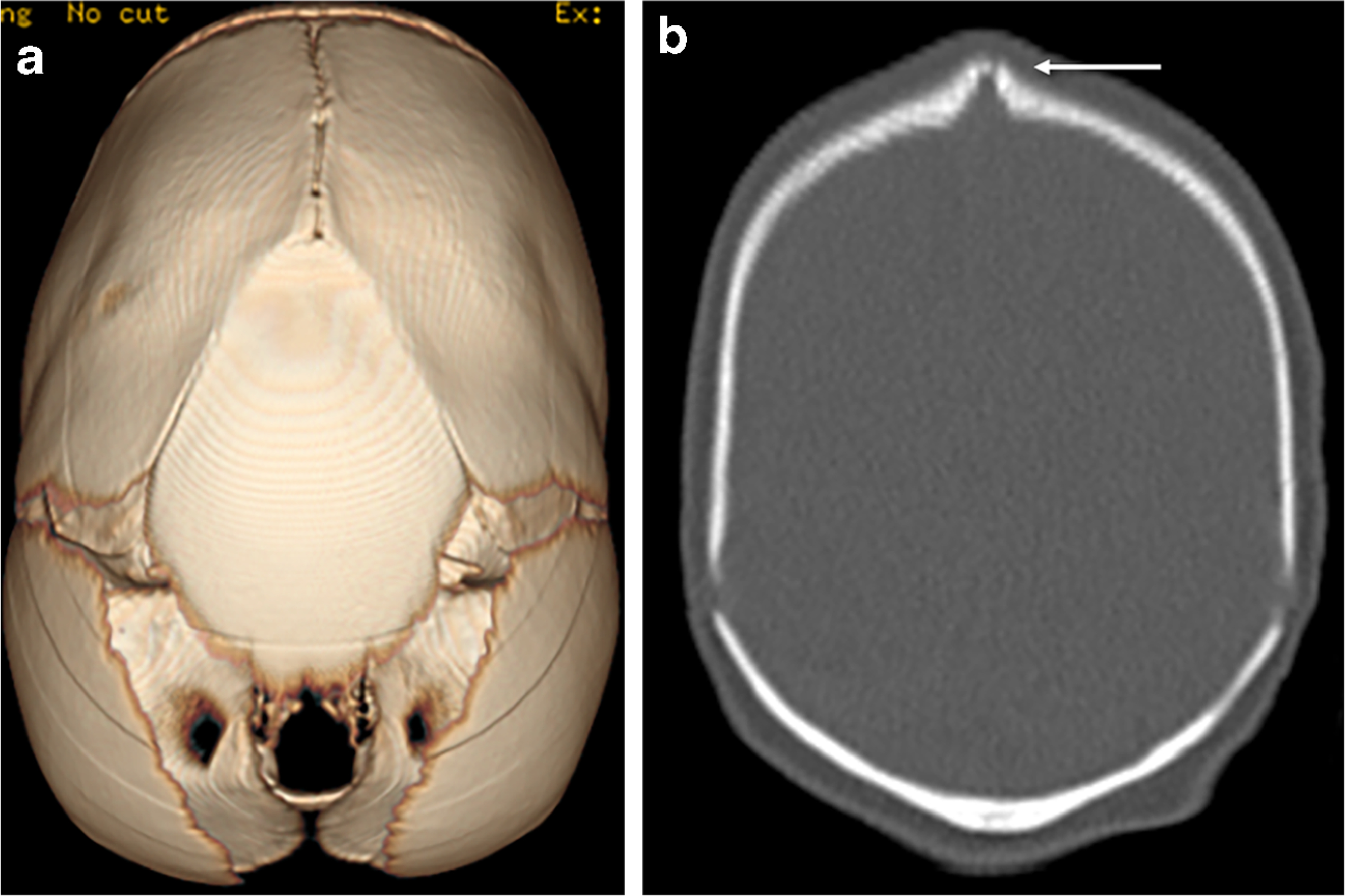
CT head in a 2-month-old girl with KIF7 mutation. a CT volume-rendered superior projection demonstrates fontanellar bone and short sagittal suture. b There is ridging of the sagittal suture without osseous bridging in the coronal bone window image (arrow) suggesting early sagittal craniosynostosis
Fig. 3.
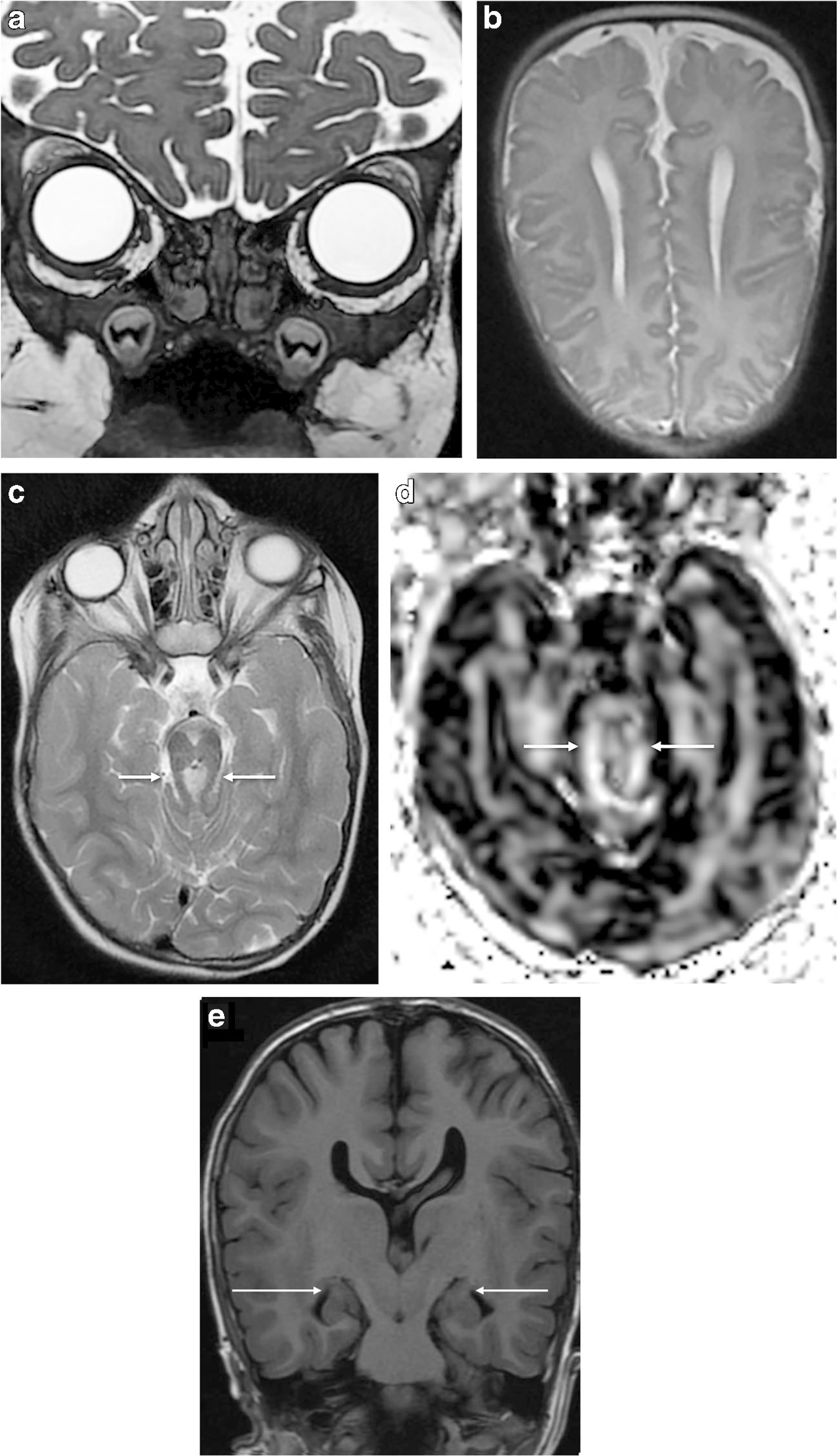
MRI brain in same girl. a Coronal fast imaging employing steady-state acquisition (FIESTA) MR head image at age 4 years demonstrates absent olfactory bulb and olfactory tract. b Axial T2-W image at 2 months of age demonstrates parallel lateral ventricles. c The molar tooth sign is demonstrated on axial T2-W image (arrows) in the same girl at age 4 years. d, e Axial diffusion tensor imaging (DTI) at 2 months of age (d) demonstrates absent superior cerebellar peduncle decussation and thickened superior cerebellar peduncle (arrows); coronal T1-weighted image at 4 years (e) demonstrates bilateral hippocampal malrotation, which has abnormal rounded configuration (arrows)
Discussion
Acrocallosal syndrome is an autosomal-recessive syndrome characterized by corpus callosum agenesis, occasional anencephaly or Dandy-Walker malformation, hypertelorism, postaxial polydactyly of hand, and pre-axial polydactyly of feet [2]. Our patient fulfilled three of the four minimal criteria required for diagnosing acrocallosal syndrome, namely corpus callosum agenesis, developmental delay and craniofacial abnormality, despite no polydactyly [4]. A similar syndrome, Greig cephalopolysyndactyly, was described in 1926 in a patient with unusual head shape, hypertelorism and limb anomalies [3]. It is a syndrome with multiple congenital anomalies characterized by macrocephaly, hypertelorism and polysyndactyly and has phenotypic similarity with acrocallosal syndrome.
The olfactory bulb and olfactory tract develop from telencephalon. The olfactory placodes develop from frontal prominence at 30 days post conception when complete closure of neural tube also occurs. The olfactory placodes invaginate to form olfactory pits and by 40 days axons from olfactory epithelium contact the olfactory area of telencephalon. By Carnegie stage 18 (44–48 days post conception) the olfactory bulb begins to appear [5]. Alteration in sonic hedgehog pathway can lead to abnormal development of the olfactory system and has been described in children with holoprosencephaly. We have shown olfactory system abnormality in our patient, with KIF7 mutation causing acrocallosal syndrome, which is component of sonic hedgehog signaling pathway.
We recently observed absent olfactory bulb and olfactory tract in another child, this one with GLI3 mutation, which is also a component of sonic hedgehog signaling pathway. A newborn boy presented with postaxial polydactyly in both upper and lower extremities, macrocephaly, and wide open anterior fontanelle. The boy also had undescended testes, laryngomalacia, and bronchus suis. A skeletal survey at birth revealed sutural widening and postaxial polydactyly in all extremities (Fig. 4). An MRI obtained at Day 1 after birth revealed macrocephaly, hypertelorism, and absent olfactory bulb and olfactory tract (Fig. 4). The pituitary gland and corpus callosum were normal. The boy had hypertrophic cardiomyopathy, and genetic testing confirmed heterozygous c4431dupT GLI3 pathogenic variant, confirming Greig cephalopolysyndactyly. In addition, the boy’s elder brother had the same pathogenic mutation. The elder brother’s radiograph revealed postaxial polydactyly, and CT revealed large wormian bone in the metopic suture. The boy’s elder brother had fast MRI, which was not adequate for evaluating olfactory bulb and olfactory tract abnormalities. His father had hypertrophic cardiomyopathy.
Fig. 4.

Anteroposterior radiograph in a newborn boy with GLI3 mutation of the right upper extremity demonstrates postaxial polydactyly in the right hand (a); similar findings were also observed in the left hand and both feet. b A 2-mm-thick coronal T2-W MR head image obtained at 1 day of age in the same boy demonstrates absent olfactory bulb and olfactory tract
Olfactory bulb agenesis has been described in holoprosencephaly; Kallman syndrome; and CHARGE syndrome (coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities and ear abnormalities) [6]. In a recent review of olfactory system abnormality, Booth et al. [6] identified olfactory system abnormalities in DiGeorge syndrome, Jacobsen syndrome and Johanson-Blizzard syndrome. In their report of 41 cases, no one had acrocallosal syndrome or Greig cephalopolysyndactyly syndrome. Children with olfactory bulb and olfactory tract abnormalities also had other brain malformation: 34% had pituitary dysfunction, 37% had supratentorial cortical dysplasia and 37% had posterior fossa anomalies. The author suggested that in children with olfactory and olfactory tract abnormalities screening for brain skull base and pituitary gland defects is required and genetic testing is warranted.
Putoux et al. [2] identified KIF7 mutation as a cause for acrocallosal syndrome. They evaluated 12 cases with KIF7 mutation. All patients had acral abnormalities including preor postaxial polydactyly, and there was molar tooth abnormality in 58%, agenesis of the corpus callosum in 58%, thinning of the corpus callosum in 17%, hippocampal abnormality in 8%, anencephaly in 17%, hydrocephalus in 17%, ventriculomegaly in 25% and paracollosal cyst in 8% of cases. No olfactory bulb or olfactory tract abnormalities were reported. The authors suggested that KIF7 protein regulates ciliary length. KIF7 mutations are known to cause a spectrum of disorders — namely hydrolethalus, acrocallosal syndrome, open neural tube defects and Joubert syndrome. Ciliary disorders are commonly associated with cerebral cortical and cerebellar abnormalities but have not been described to cause olfactory bulb or olfactory tract defects. The girl in our case had no acral abnormalities, and Asadollahi et al. [7] observed limb abnormalities in only 63% of people with KIF7 mutation causing acrocallosal syndrome. We hypothesize that absence of olfactory bulb and olfactory tract associated with KIF7 mutation might arise from impaired axon navigation from bipolar neurons in olfactory epithelium and failure to induce olfactory bulb formation. In addition, we observed evidence of more widespread abnormal axon navigation leading to partial agenesis of corpus callosum, thickened and horizontally oriented superior cerebellar peduncle and failure of superior cerebellar peduncle decussation in acrocallosal syndrome.
Elson et al. [8] described extra bone in anterior fontanel in a child who had GLI3 mutation, pre-axial polysyndactyly in hands and feet, corpus callosum agenesis and craniofacial abnormality similar to our patient with KIF7 mutation.
In summary, we described unique imaging findings in acrocallosal syndrome caused by autosomal-recessive KIF7 mutation — namely olfactory bulb and olfactory tract agenesis, hippocampal malrotation leading to seizures, and presence of fontaneller bone leading to short sagittal suture and sagittal synostosis. Absence of olfactory bulb and olfactory tract in KIF7 mutation is likely from impaired axon navigation from bipolar neurons in olfactory epithelium and failure to induce formation of olfactory bulb, with more widespread abnormal axon navigation caused by ciliopathy. We also demonstrated olfactory bulb and olfactory tract abnormalities in a related syndrome, Greig cephalopolysyndactyly syndrome, caused by dominant GLI3 mutation, a gene required for Shh signaling.
Acknowledgments
This work was supported by the Department of Defense (W81XWH-16-1-0613), the National Heart, Lung and Blood Institute (R01 HL128818-03), the National Institute of Neurological Disorders and Stroke (K23-063371), the Pennsylvania Department of Health, the Mario Lemieux Foundation, and the Twenty-Five Club Fund of Magee Women’s Hospital.
Footnotes
Compliance with ethical standards
Conflicts of interest None
References
- 1.Yao E, Chuang P (2015) Hedgehog signaling: from basic research to clinical applications. JFMA 114:569–576 [DOI] [PubMed] [Google Scholar]
- 2.Putoux A, Thomas S, Coene KL et al. (2011) KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 43:601–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Debeer P, Peeters H, Driess S et al. (2003) Variable phenotype in Greig cephalopolysyndactyly syndrome: clinical and radiological findings in 4 independent families and 3 sporadic cases with identified GLI3 mutations. Am J Med Genet A 120A:49–58 [DOI] [PubMed] [Google Scholar]
- 4.Courtens W, Vamos E, Christophe C et al. (1997) Acrocallosal syndrome in an Algerian boy born to consanguineous parents: review of the literature and further delineation of the syndrome. Am J Med Genet 69:17–22 [DOI] [PubMed] [Google Scholar]
- 5.Azoulay R, Fallet-Bianco C, Garel C et al. (2016) MRI of olfactory bulbs and sulci in human fetuses. Pediatr Radiol 36:97–107 [DOI] [PubMed] [Google Scholar]
- 6.Booth TM, Rollins NK (2016) Spectrum of clinical and associated MR imaging findings in children with olfactory anomalies. AJNR Am J Neuroradiol 37:1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asadollahi R, Strauss JE, Zenker M et al. (2018) Clinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through sonic hedgehog signaling. Eur J Hum Genet 26: 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elson E, Perveen R, Donnai D et al. (2002) De novo GLI3 mutation in acrocallosal syndrome: broadening the phenotypic spectrum of GLI3 defects and overlap with murine models. J Med Genet 39: 804–806 [DOI] [PMC free article] [PubMed] [Google Scholar]