

Summary
Neutrophil extracellular traps (NETs) have been implicated in the pathogenesis of acute respiratory distress syndrome (ARDS) driven by viruses or bacteria, as well as in numerous immune-mediated disorders. Histone citrullination by the enzyme peptidylarginine deiminase 4 (PAD4) and the consequent decondensation of chromatin are hallmarks in the induction of NETs. Nevertheless, additional histone modifications that may govern NETosis are largely overlooked. Herein, we show that histone deacetylases (HDACs) play critical roles in driving NET formation in human and mouse neutrophils. HDACs belonging to the zinc-dependent lysine deacetylases family are necessary to deacetylate histone H3, thus allowing the activity of PAD4 and NETosis. Of note, HDAC inhibition in mice protects against microbial-induced pneumonia and septic shock, decreasing NETosis and inflammation. Collectively, our findings illustrate a new fundamental step that governs the release of NETs and points to HDAC inhibitors as therapeutic agents that may be used to protect against ARDS and sepsis.
Subject areas: Molecular biology, Immunology, Cell biology, Functional aspects of cell biology
Graphical abstract
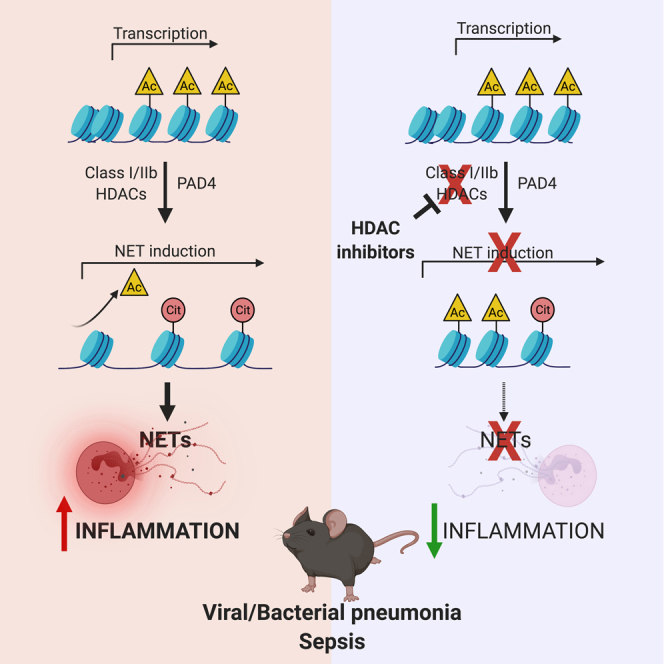
Highlights
-
•
Zn-dependent lysine deacetylases govern neutrophil extracellular trap (NET) formation
-
•
Class I/IIb HDACs deacetylate histone H3 allowing its citrullination by PAD4
-
•
HDAC inhibition with ricolinostat protects against viral and bacterial pneumonia
-
•
In a model of endotoxic shock, ricolinostat inhibits NETosis and dampens inflammation
Molecular biology; Immunology; Cell biology; Functional aspects of cell biology
Introduction
The immune system evolved to sense microbial and endogenous cues that alter cellular or organismal homeostasis. When infection and tissue damage coincide, the immune system reacts rapidly to avoid the spread of the infection, even at the cost of an overexuberant reaction that may cause further tissue damage. Neutrophils are key players in this process (Nathan, 2002). These polymorphonuclear cells orchestrate the innate immune response by neutralizing pathogens via several effector mechanisms, which also cause tissue damage (Jaillon et al., 2013). Given their tissue damaging capabilities, neutrophils serve central roles in many inflammatory disorders and favor immune-mediated coagulation and disseminated clot formation (Gomez-Moreno et al., 2018). Central for both pathogen containment as well as development of immune-mediated disorders is the formation of neutrophil-derived extracellular traps (NETs) through a cellular process termed NETosis (Brinkmann et al., 2004; Saitoh et al., 2012; Thiam et al., 2020). Despite the key functions played by NETs against viruses, bacteria, and fungi (Thiam et al., 2020), the dysregulated formation of NETs has been linked to a variety of diseases such as acute respiratory distress syndrome (ARDS) and sepsis. ARDS and sepsis are characterized by an exaggerated elevation of pro-inflammatory cytokines in the lungs and/or bloodstream, and also by coagulopathy, thrombosis, and multiorgan dysfunction (Broggi et al., 2020; Channappanavar and Perlman, 2017; Giannis et al., 2020; Guan et al., 2020; Iba et al., 2019; Lee et al., 2020; Lucas et al., 2020; Mehta et al., 2020; Ruan et al., 2020; Shin et al., 2019; Short et al., 2014; Tang et al., 2019; Wu et al., 2019; Yang et al., 2019; Zhou et al., 2020). The central roles played by neutrophils and NETosis in driving these life-threatening immune disorders provide a strong mandate to reveal new steps in the regulation of NET induction that can be used as novel therapeutic targets.
Akin to transcriptional activation that is driven by local chromatin remodeling, epigenetic modifications of histones that decondense chromatin are a prerequisite for NET release (Kenny et al., 2017). In particular, histone citrullination by the peptidylarginine deiminase 4 (PAD4) has been shown to facilitate NET formation and release (Thiam et al., 2020; Wang et al., 2009). Granule-resident serine proteases, neutrophil elastase, as well as gasdermin D also participate to chromatin decondensation, nuclear expansion, and formation of NETs (Chen et al., 2018; Papayannopoulos et al., 2010; Sollberger et al., 2018). Whether other epigenetic modifications also participate in NETosis, and how they may be linked to the above-mentioned processes that alter the structure of chromatin, remains largely overlooked. Based on the importance of histone acetylation in determining chromatin structure and accessibility, we hypothesized that enzymes involved in histone acetylation also contribute to NETosis. Histone acetylation is dynamically controlled by two counteracting protein families, the histone acetyltransferases and the histone deacetylases (HDACs) as “writers” and “erasers” (Grunstein, 1997; Zhao et al., 2018). HDACs belong to two families, based on the dependency on zinc or nicotinamide adenine dinucleotide (NAD+) for their activities (Seto and Yoshida, 2014). In this study, we found that class I and IIb HDACs, which belong to the family of zinc-dependent lysine deacetylases, regulate NETosis in human and mouse cells. Also, these zinc-dependent HDACs can be used as therapeutic targets in mouse models of ARDS and septic shock. In particular, the class I/IIb HDAC inhibitor ricolinostat dampens NET formation and pro-inflammatory cytokine production, reduced morbidity, and improved lung functionality in mouse models of microbial pneumonia and septic shock.
Results
Class I/IIb HDACs drive NET formation
To test the capacity of HDACs to modulate NET formation, we assembled a focused collection of class and isoform specific inhibitors of HDACs with high structural and functional diversity (Park and Kim, 2020). This included pan-HDAC inhibitors that selectively target HDAC class I (HDAC1/2/3/8), class IIa (HDAC4/5/7/9), class IIb (HDAC6/10), and class III (sirtuins), as well as an inhibitor targeting bromodomain proteins, which recognize acetylated lysine on histones. Initially, we investigated the capacity of these inhibitors to alter NETosis by measuring extracellular DNA release by primary human neutrophils activated with phorbol myristate acetate (PMA), a well-known inducer of NETs. DNA release is often used as a proxy for NETosis and, indeed, inhibition of NET induction by using the PAD4 inhibitor GSK484 resulted in significant decrease of extracellular DNA by PMA-treated cells (Figure S1A). Compounds that inhibited either class I or class I/IIb HDACs also significantly diminished extracellular DNA secretion from PMA-treated neutrophils (Figure S1B), whereas class IIa, pan-HDAC inhibitors, other HDACs inhibitors, or bromodomain inhibition did not affect DNA release. These data demonstrate that inhibition of certain, but not all, classes of HDACs efficiently decreases the release of extracellular DNA from PMA-treated human neutrophils and suggest that targeting HDACs may effectively prevent NETosis. Thus, we focused on the compounds that significantly reduced DNA release to further assess their activity on NETosis. To confirm the capacity of specific HDAC inhibitors to interfere with NETosis, we investigated whether HDAC inhibition also prevented the release of citrullinated histone H3 (CitH3), a hallmark of NET formation (Thiam et al., 2020; Wang et al., 2009), that peaked 3–5 hours upon PMA administration (Figure S1C). In agreement with previous findings (Lewis et al., 2015), pretreatment with the NET-disrupting PAD4 inhibitor GSK484 dramatically reduced NET induction, measured as CitH3 staining, in PMA-stimulated human neutrophils (Figures 1A and S1D). Similar to extracellular DNA release, class I or class I/II HDAC inhibitors entinostat and ricolinostat prevented NET release in a dose-dependent manner with an efficacy comparable with GSK484 (Figures 1A and S1D). Next, we tested if HDACs regulate NETosis not only in human but also in mouse neutrophils. We found that, similarly to what we observed using human cells, the class I/IIb HDAC inhibitor ricolinostat efficiently decreased the release of CitH3 in PMA-treated mouse neutrophils (Figure 1B). In contrast to the human cell data, the class I HDAC-specific inhibitor entinostat had no effect on murine neutrophils. In keeping with a key role of class I/II HDACs in regulating NET release, we also found that ricolinostat significantly reduced the expansion of nuclei, another hallmark of NETosis in PMA-treated cells (Sollberger et al., 2018) (Figures 1C and 1D). Moreover, ricolinostat inhibited NET induction in response to nigericin, a bacterial toxin known to induce NETosis in neutrophils (Chen et al., 2018; Sollberger et al., 2018) (Figure S1E). Overall, these data imply that zinc-dependent lysine deacetylases represented by class I/IIb HDACs are required to induce NETosis both in human and mouse neutrophils, and that the class I/IIb HDAC inhibitor ricolinostat, a phase II clinical trial drug, efficiently inhibits NETosis both in humans and mouse neutrophils.
Figure 1.

Class I/II HDACs drive NET formation
(A and B) Freshly purified human (A) or mouse (B) neutrophils were treated with the indicated HDAC inhibitors at different doses (10, 2, 0.4, 0.08 μM), or with GSK484 (10 μM), for 1 h and then stimulated with 1 μM PMA for 3 h. Cells were fixed and stained with anti-citrullinated histone H3 (CitH3) antibody. NETosis induction was measured as the level of anti-CitH3 fluorescent signal. PMA alone represents 100% of NET induction. “-”: Cells treated with the highest dose of the indicated drug but not activated with PMA.
(C and D) Human neutrophils were treated, or not, with ricolinostat or GSK484 and then stimulated with 1 μM PMA. NETosis induction was measured as area of nuclei stained with DAPI. (C) 3D reconstruction of nuclei in each treatment (grid size 7.8 μm). (D) Quantification of DAPI signal from multiple fields of view (D) (n = 9 for unstimulated, not pretreated with any drug [NT] and ricolinostat, n = 7 for GSK484).
(E and F) Human neutrophils were treated as in (C-D), and immunoblot analysis of the indicated proteins was performed. Relative quantification of band intensity was analyzed with ImageJ software (F). White violins plots depict the quantification of histone H3 acetylation; red violin plots depict the quantification of histone H3 citrullination. Immunoblot in E is representative of four independent experiments. Gasdermin D NT, gasdermin D N terminal; pMLKL, phospho-MLKL.
(G) Human (left) or murine (right) neutrophils were treated, or not, with ricolinostat (10 μM) and then stimulated with 1 μM PMA for 3 h. ROS production was measured by luminescence with luminol-HRP. Histograms represent the total amount of ROS produced overtime.
(H) Murine neutrophils were treated, or not, with ricolinostat (10 μM) and then stimulated with fluorescent E. coli at the indicated multiplicity of infection (MOI) for 1 h. The histogram represents the percentage of neutrophils positive for E. coli staining. Graphs show mean ± SEM. Statistics were calculated using one-way ANOVA (A, B, D) and two-way ANOVA (F, G, H) (∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001. n.s., not significant). Statistics refers to the comparison with neutrophils stimulated with PMA alone (A, B). Gray stars represent comparison of all experimental groups with the unstimulated control, and black stars represent comparison between drug-treated groups and cells treated with PMA only (NT) (D). See also Figure S1.
A previous study suggested that pan-HDAC inhibition switches neutrophil cell death from NETosis to apoptosis (Hamam and Palaniyar, 2019). To assess whether ricolinostat reduced NETosis by favoring other types of cell death, we tested the levels of apoptosis, pyroptosis, and/or necrosis induction in PMA-treated cells exposed to either ricolinostat or GSK484. We excluded the activation of apoptosis or necroptosis under our experimental conditions (Figure 1E). In agreement with previous reports (Chen et al., 2018; Sollberger et al., 2018), we found that PMA induced gasdermin D (GSDMD) cleavage, but, notably, this process was not altered by either ricolinostat or GSK484 (Figure 1E). Overall, these data confirm the capacity of ricolinostat, as well as GSK484, to prevent NETosis without affecting the induction of other forms of cell death.
Next, we assessed how HDAC inhibition prevents NETosis. Given that inhibition of class I/IIb HDACs decreased the release of citrullinated histone H3, we hypothesized that ricolinostat exerts its functions by increasing H3 acetylation, which in turn may prevent PAD4-dependent citrullination. Indeed, our data demonstrated that, upon ricolinostat, but not GSK484, administration, histone H3 acetylation was boosted in untreated as well as PMA-treated cells (Figures 1E and 1F). When cells were treated with PMA, and citrullination was increased to induce NETosis, ricolinostat significantly decreased H3 citrullination compared with cells that were treated with PMA only. As expected, the PAD4-inhibitor GSK484 completely abrogated H3 citrullination (Figures 1E and 1F). In keeping with the capacity of ricolinostat to affect neutrophil responses by preventing histone deacetylation, we found that ricolinostat treatment did not alter reactive oxygen species (ROS) production, another key step in NETosis (Douda et al., 2015; Fuchs et al., 2007; Remijsen et al., 2011), either in human or mouse cells (Figure 1G). Similarly, we excluded that ricolinostat administration altered other important functions of neutrophils, such as bacterial phagocytosis (Figure 1H). These data support that the capacity of ricolinostat to prevent histone H3 deacetylation impedes its citrullination and the release of NETs. Therefore, our data support a model in which acetylation and citrullination modifications on histone H3 are divergently regulated in the context of NETosis.
We next focused on the specificity of action of ricolinostat. Ricolinostat inhibits both class I and class IIb HDACs, with a preference for the latter. We, thus, compared the activity of ricolinostat with other inhibitors specific for class IIb HDACs (tubastatin A and nexturastat) as well as with tubacin, that similarly to ricolinostat targets not only class IIb HDACs but also class I HDACs (Bergman et al., 2012; Butler et al., 2010; Haggarty et al., 2003). The siderophore deferoxamine and the hydroxamate-based metalloprotease inhibitor batimastat were used as negative controls to exclude bystander activities of the drugs utilized on other divalent metals. We found that class I/IIb HDAC inhibition prevented NETosis, whereas selective class IIb targeting did not (Figure S1F). In agreement with these data, neutrophils that lack HDAC6, the major class IIb HDAC, behaved similarly to wild-type cells and remained sensitive to ricolinostat administration (Figure S1G). Finally, human neutrophils derived from two independent donors as well as murine neutrophils were treated, or not, with ricolinostat or its chlorinated analog citarinostat (or GSK484, used as a positive control) and the level of CitH3 was assessed. Our data revealed both compounds inhibited NET formation to a comparable extent (Figures S1H–S1L). Overall, these data demonstrate that the capacity of zinc-dependent class I/IIb HDACs to drive NET formation is conserved in humans and mice.
Inhibition of class I/IIb HDACs in vivo dampens NETosis and protects against pneumonia induced by exposure to viral ligands
The conserved activity of class I/IIb HDACs in regulating NETosis in human and mouse cells prompted us to assess how HDAC inhibition with ricolinostat affects the development of pathological inflammation in vivo in murine models. To determine if the capacity of ricolinostat to inhibit NETosis as assessed in vitro also impacts viral pneumonia, we used a well-characterized mouse model that mimics the immune response to RNA respiratory viruses and that we previously successfully used to unveil some features of RNA viral infections (Broggi et al., 2020). The synthetic analog of double-stranded RNA polyinosine:polycytidylic acid (poly(I:C)) was intratracheally instilled to mice daily for 4 days to induce NET formation (Figure S2A). Poly I:C was administered in the presence or absence of ricolinostat, and PAD4 inhibition with GSK484 was used as a comparison (Figure 2A). Administration of either ricolinostat or GSK484 significantly reduced morbidity in poly(I:C)-treated mice, measured as temperature drop (Figure 2B). Most importantly, lung permeability and functionality, measured as previously described in mouse models based on the use of synthetic viral ligands and/or influenza A viral infection (Broggi et al., 2020; Harb et al., 2020, 2021; Jamieson et al., 2013), were significantly preserved in mice that received either ricolinostat or GSK484 and poly(I:C), compared with mice that were treated with poly(I:C) only (Figures 2C and 2D). Poly(I:C)-treated mice showed a significant increase of NETosis in the bronco-alveolar lavage fluid (BALF) and in the lung tissue, measured as levels of CitH3 (Figures 2E and 2G), as well as by levels of myeloperoxidase (MPO)-DNA complexes (Figures 2F and 2H). Increased NET induction was abrogated upon drug treatment (Figures 2E–2H). Although neutrophil levels in the blood did not change significantly among different treatments (Figures S2B and S2C), in keeping with a reduced death of neutrophils by NETosis, we found a trend of accumulation of neutrophils in the BALF as well as in the lung of mice treated with ricolinostat and GSK484 compared with mice administered poly(I:C) only (Figures S2D–S2F). Of note, in all the conditions tested, ricolinostat phenocopied, and in some cases outcompeted (i.e., increased survival of neutrophils in the BALF), the activity of GSK484. These results demonstrate that class I/IIb HDACs play a role as important as the key enzyme PAD4 in regulating NETosis and that zinc-dependent HDACs can be targeted against viral-induced ARDS.
Figure 2.

Inhibition of class I/IIb HDACs protects against NETosis and ARDS in a mouse model of RNA viral lung infection
(A) Timeline of poly(I:C) instillation and drug treatments. Mice were intratracheally treated with 2.5 mg/kg poly(I:C) or saline daily for 4 days and euthanized 18 h post the last poly(I:C) instillation. Ricolinostat (30 mg/kg/dose) or GSK484 (25/mg/kg/dose) was administered intraperitoneally at day 2, day 4, and 3 h before the endpoint.
(B) Body temperature measurements at the endpoint.
(C) Lung permeability was measured as albumin content level in the BALF (n = 5).
(D) Measure of airway hyperreactivity (AHR) in the indicated groups in response to increasing concentration of methacholine. RI, responsiveness index.
(E–H) CitH3 and MPO-DNA levels were analyzed by ELISA in BALFs (E, F) and in lungs (G, H) of mice treated as indicated. The saline group represents mice that received saline instead of poly(I:C). NT group represents mice that were challenged with poly(I:C) without receiving any drug. Statistics were calculated using one-way ANOVA (B, C, E-H) and two-way ANOVA with Tukey's analysis (D) (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. n.s., not significant). n = 10 (B, D) and 15 (E-H) for each group except for the GSK484-treated group (n = 5). Gray stars represent comparison of all experimental groups with the saline-treated control. Black stars represent comparison between drug-treated groups and mice treated with poly (I:C) alone. Violin plots represent median (dashed line) with quartiles (dotted line). See also Figure S2.
Class I/IIb HDACs increase NETosis and ARDS in a mouse model of bacterial pneumonia
To further assess whether the capacity of ricolinostat to prevent NETosis can be used against ARDS, we employed a bacterial lung pneumonia mouse model based on the intratracheal injection of S. aureus (Figure 3A). As before, in this set of experiments we compared the activity of ricolinostat with the PAD4 inhibitor GSK484. When class I/IIb HDACs were inhibited with ricolinostat, morbidity was significantly decreased compared with mice injected with S. aureus only, and also with S. aureus and GSK484 (Figure 3B). Lung barrier alteration was prevented both by ricolinostat or GSK484 treatment, compared with mice exposed to S. aureus alone, whereas bacterial control was not affected by these treatments (Figures 3C and 3D). These data suggest the capacity of these drugs to affect tissue tolerance without changing immune resistance (Medzhitov et al., 2012). NETs are well known to alter tissue functionality and, in fact, have been shown to dampen tissue tolerance during lung infections (Iwasaki and Pillai, 2014; Pillai et al., 2016). Indeed, we found that administration of either ricolinostat or GSK484 potently decreased NETosis as measured by levels of citrullinated histone H3 as well as MPO-DNA complexes in the BALF of mice treated with S. aureus (Figures 3E and 3F). The capacity of these drugs to inhibit NETosis was further demonstrated by cytofluorimetric quantification of citrullinated histone H3-positive neutrophils in the BALF (Figure 3G). In keeping with our previous findings in the poly(I:C)-driven pneumonia model, we found no major differences in the levels of neutrophils circulating in the blood, whereas reduction of NETosis led to the presence of increased numbers of neutrophils in the BALF (Figures 3H and 3I, and S3A). Pro-inflammatory cytokines IL-6 and IL-1β, but not TNF, were also significantly decreased in the BALF of mice administered ricolinostat and GSK484, compared with mice that received S. aureus only (Figures S3B–S3D). Collectively, these results demonstrate that zinc-dependent HDAC inhibition is as efficacious as PAD4 inhibition in preventing bacterial-induced NETosis and ARDS. Also, treatment with ricolinostat was more efficient in terms of morbidity reduction compared with GSK484.
Figure 3.

Class I/IIb HDACs increase NETosis and ARDS in a mouse model of bacterial pneumonia
(A) Timeline of drug treatment and S. aureus infection. Mice were intraperitoneally injected with two doses of ricolinostat or GSK484 and were intratracheally treated with S. aureus (6.5 × 107 CFU). Six hours later, BALFs, lungs, and blood were collected and analyzed.
(B) Body temperature measurements at the endpoint.
(C) Lung permeability was measured as albumin content in the BALF (n = 5).
(D) Numbers of S. aureus CFU were measured in lung homogenates (n = 5).
(E and F) CitH3 and MPO-DNA levels in BALFs were detected by ELISA in the indicated groups and normalized to saline controls.
(G) NETosis levels in BALFs were quantified by cytofluorimetry as neutrophils that were positive for the CitH3 staining (n = 5).
(H and I) Neutrophil numbers were analyzed in blood (H) and BALF (I) by cytofluorimetry. The saline group represents healthy mice. The NT group represents mice that were challenged with S. aureus and did not receive any drug. Statistics were calculated using one-way ANOVA (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.01; ∗∗∗∗p < 0.0001. n.s., not significant). n = 10 (B, H, I) and 15 (E, F) for each group except for the GSK484-treated group (n = 5). Gray stars represent comparison of all experimental groups with the saline-treated healthy control. Black stars represent comparison between drug-treated groups and mice treated with S. aureus alone. Violin plots represent median (dashed line) with quartiles (dotted line). See also Figure S3.
Class I/IIb HDAC inhibition protects against systemic inflammation
Finally, we investigated whether inhibition of class I/IIb HDACs dampens systemic inflammation in a mouse model of septic shock. Septic shock is characterized by systemic inflammation and disseminated intravascular coagulation (van der Poll et al., 2017), and neutrophils play a critical role in driving this lethal syndrome. We hypothesized that inhibition of NET formation using ricolinostat may yield beneficial effects in reducing sepsis-associated symptoms. Thus, we employed a well-established lipopolysaccharide (LPS)-driven model of septic shock (Zanoni et al., 2017) in mice treated, or not, with ricolinostat or GSK484, as a comparison (Figure 4A). Mice that received either ricolinostat or GSK484 showed no temperature loss, compared with untreated animals, and were significantly protected compared with mice treated with LPS only (Figure 4B). Previous studies demonstrated that decreased platelet counts are a hallmark of disseminated intravascular coagulation during sepsis (Yang et al., 2019). It is also worth noting that NETs are major drivers of disseminated coagulation in sepsis (Xu et al., 2009) and that recent studies underscore that interactions between platelets and neutrophils are critical in sustaining disseminated intravascular coagulation (Clark et al., 2007; McDonald et al., 2012, 2017). Of note, platelet counts were significantly higher in mice that received ricolinostat or GSK484, compared with mice treated with LPS alone (Figure 4C). Although more data will be needed to directly link HDAC inhibition to the reduction of disseminated coagulation during septic shock, these data suggest that immunothrombosis can be similarly prevented upon administration of inhibitors that target either HDAC or PAD4. Indeed, also in this septic shock mouse model we confirmed the efficacy of class I/IIb HDAC or PAD4 inhibition in preventing NETosis. We found not only reduced release of NETs by neutrophils in the peritoneal lavage (Figure 4D) but also increased neutrophils levels (Figures S4A–S4C). As before, these data confirm the efficacy of the used drugs in preventing NETosis and death of neutrophils. In keeping with the systemic nature of this sepsis model, and with the positive feedback loop recently described between NETosis and increased cytokine production (Apel et al., 2021), we found that NET inhibition by either ricolinostat or GSK484 significantly decreased pro-inflammatory cytokine production (Figures 4E–4G).
Figure 4.

Class I/IIb HDAC inhibition protects against systemic inflammation
(A) Time line of drug treatment and LPS priming and challenge. Mice were intraperitoneally injected, or not, with two doses of ricolinostat or GSK484 and were then intraperitoneally administered with two doses of LPS (-5 h, 1 mg/kg and 0 h, 2 mg/kg) to induce lethal sepsis. Peritoneal lavage and blood were collected 2 h after the LPS challenge.
(B) Body temperature was measured 2 h post challenge.
(C) Concentration of platelets in the blood was measured by cytofluorimetry.
(D) NETosis was measured as neutrophils (Ly6G+/CD11b+) positive for citrullinated histone H3 staining in the peritoneal lavage.
(E–G) Serum cytokines were measured by ELISA. The NT group represents mice that were challenged with LPS and did not receive any drug. Statistics were calculated using one-way ANOVA (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; n.s., not significant). n = 15 for each group except the GSK484-treated group (n = 10). Gray stars represent comparison of all experimental groups with the saline-treated healthy control. Black stars represent comparison of drug-treated groups with LPS-stimulated mice without drug treatment. Violin plots represent median (dashed line) with quartiles (dotted line). See also Figure S4.
Since dexamethasone is widely used to dampen inflammation, we also compared the therapeutic effects of ricolinostat with dexamethasone. We did not observe a significant difference in platelet numbers when comparing the dexamethasone-treated group with the group treated with LPS only (Figure S4D), whereas both drugs efficiently reduced pro-inflammatory cytokine production (Figures S4E and S4F). Overall, our results demonstrate that inhibition of NETosis by ricolinostat protects against systemic inflammation in a mouse model of septic shock.
Discussion
When infection and tissue damage coincide, the immune system potently reacts to avoid the spread of the infection, even at the cost of further damage. Neutrophils are central to this process, thanks to the release of NETs that contain and restricts viral, bacterial, as well as fungal infections (Brinkmann et al., 2004; Saitoh et al., 2012; Thiam et al., 2020). Nevertheless, excessive tissue damage caused by NET release has been often associated with immune-driven disorders (Thiam et al., 2020). Moreover, NETosis has been shown to play both protective as well as detrimental roles during tissue repair and resolution (Hahn et al., 2019; Kang et al., 2020; Schauer et al., 2014). Thus, a better understanding of the mechanisms that regulate NET induction is fundamental to develop future possible therapeutic ways of intervention to either potentiate or block the induction of NETs.
Herein, we show that class I/IIb HDACs drive the release of NETs by human and mouse neutrophils. Their inhibition in mice dampens inflammation and protects against microbial pneumonia or septic shock. These data establish a new biological feature of a group of HDACs whose activity is well known to regulate gene transcription and open to the use of drugs that inhibit HDACs to restrain the tissue-damaging functions of neutrophils. Although targeting of PAD4 with the chemical compound GSK484 was previously shown to be effective in inhibiting CitH3 and NET formation, we demonstrated that ricolinostat, a drug currently utilized in a phase II clinical trial, can also be used to efficiently inhibit NETosis and inflammation. Using multiple in vivo mouse models mimicking bacterial- or viral-induced infections and septic shock, we show that pharmacological inhibition of HDACs ameliorates lung functionality, reduces systemic inflammation, and decreases thrombosis via inhibition of NETosis.
Viral or bacterial-induced pneumonia, as well as COVID-19, have a strong correlation to elevated levels of circulating neutrophils, NETs, and inflammatory cytokines that hint at a chronic inflammatory storm in patients (Mehta et al., 2020). NET formation is intimately linked to thrombosis that drives microvasculature damages in lung and other organs (Ackermann et al., 2020; Al-Samkari et al., 2020; Tang et al., 2020). Neutrophils and NETosis also participate in the most severe cases of COVID-19 (Zuo et al., 2020a, 2020b). NETs infiltrate lungs of patients with severe COVID-19, and SARS-CoV-2 has been shown to trigger the release of NETs that mediate lung pathology (Middleton et al., 2020; Radermecker et al., 2020; Veras et al., 2020). As dysregulated production of NETs is known to exacerbate inflammation and accelerate the pathogenesis of ARDS and sepsis caused by other coronaviruses (Blondonnet et al., 2016; Perlman and Dandekar, 2005), these findings support the hypothesis that targeting NET formation represents a viable strategy to alleviate tissue damage and hyperinflammation. Therefore, our results imply that pharmacological inhibition of HDAC activity may be beneficial for patients with COVID-19 by blocking NETosis.
In our mouse models, we show that drugs that inhibit class I/IIb HDACs can partially reduce the levels of pro-inflammatory cytokines including IL-1β, TNF, and IL-6. Whether in our model IL-1β levels are directly controlled by HDAC activity, as recently shown in a different LPS-driven sepsis model (Magupalli et al., 2020), or they are a consequence of NET formation will require further studies.
Overall, our data demonstrate that zinc-dependent lysine deacetylase inhibition prevents NET formation by neutrophils, which in turn reduces systemic inflammation. This strongly suggests that HDAC inhibition represents a new potential therapeutic strategy for the treatment of a variety of diseases mediated by the overproduction of NETs such as COVID-19, pulmonary diseases, autoimmune diseases (e.g., rheumatoid arthritis and systemic lupus erythematosus), diabetes, and cancers (Arpinati et al., 2020; Lee et al., 2017; Porto and Stein, 2016; Wong et al., 2015).
Limitations of the study
A previous study suggested that abrogation of HDAC activities using pan-HDAC inhibitors may impact NETosis by favoring neutrophil apoptosis (Hamam and Palaniyar, 2019). Our data excluded that class I/IIb HDAC inhibition alters other forms of cell death in neutrophils and instead point to deacetylation of histone H3 as a step necessary to allow histone citrullination and NET release. In another report, the same group suggested pan-HDAC inhibition increases NETosis (Hamam et al., 2019); however, it failed to address whether inhibition of a specific HDAC class has a differential impact and if in vitro data can be translated in vivo. In our hands, pan-HDAC inhibition does not alter the release of NET compared with specific targeting of class I/IIb HDAC activity. Whether this is due to cross talk between and/or opposing effects of different classes of HDACs or specific HDAC complexes, as well as the dose and/or type of drugs utilized, remains to be investigated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-citrullinated histone H3 | Abcam | Cat# ab5103 RRID:AB_304752 |
| Anti-rabbit Alexa 568 | Invitrogen | Cat# A11011 RRID:AB_143157 |
| Anti-Ly6G APC | BioLegend | Cat# 127613 RRID:AB_1877163 |
| Anti-CD11b FITC | BioLegend | Cat# 101206 RRID:AB_312789 |
| Anti-CD45 Brilliant Violet 510 | BioLegend | Cat# 103138 RRID:AB_2563061 |
| Anti-acetyl-histone H3 | Sigma/Millipore | Cat# 06-599 RRID:AB_2115283 |
| Anti-histone H3 | Cell Signaling Technology | Cat# 4499 RRID:AB_10544537 |
| Anti-caspase 8 | Cell Signaling Technology | Cat# 4790 RRID:AB_10545768 |
| Anti-cleaved caspase 8 | Cell Signaling Technology | Cat# 9496 RRID:AB_561381 |
| Anti-caspase 3 | Cell Signaling Technology | Cat# 9662 RRID:AB_331439 |
| Anti-phospho-MLKL | Cell Signaling Technology | Cat# 91689 RRID:AB_2732034 |
| Anti-MLKL | Cell Signaling Technology | Cat# 14993 RRID:AB_2721822 |
| Anti-GSDM-D | Cell Signaling Technology | Cat# 93709 RRID:AB_2800210 |
| Anti-β-actin | Sigma | Cat# A2228 RRID:AB_476697 |
| Anti-Myeloperoxidase | Sigma/Millipore | Cat# 07-496-I RRID:AB_11212795 |
| Anti-neutrophil | Abcam | Cat# ab2557 RRID:AB_303154 |
| Bacterial and virus strains | ||
| Staphylococcus aureus subsp. aureus Rosenbach | ATCC | Cat# 25904 |
| Escherichia coli (O111) | ATCC | Cat# BAA-2440 |
| Chemicals, peptides, and recombinant proteins | ||
| Ricolinostat (ACY-1215) | Cayman Chemical | Cat# 21531 |
| Citarinostat | Cayman Chemical | Cat# 26173 |
| GSK848 (PAD4 inhibitor) | Cayman Chemical | Cat# 17488 |
| Entinostat | Sellckchem | Cat# S1053 |
| Givinostat | Sellckchem | Cat# S2170 |
| TMP195 | Sellckchem | Cat# S8502 |
| Belinostat | Sellckchem | Cat# S1085 |
| Panobinostat | Sellckchem | Cat# S1030 |
| FK866 | Cayman Chemical | Cat# 13287 |
| Apabetalone | Sellckchem | Cat# S7295 |
| Deferoxamine | Cayman Chemical | Cat# 14595 |
| Batimastat | Cayman Chemical | Cat# 14742 |
| Tubastatin A | Cayman Chemical | Cat# 10559 |
| Nexturastat A | Cayman Chemical | Cat# 16874 |
| Tubacin | Cayman Chemical | Cat# 13691 |
| Phorbol 12-myristate 13-acetate | Cayman Chemical | Cat# 10008014 |
| Poly(I:C) HMW | Invivogen | Cat# tlrl-pic |
| LPS from E. coli, Serotype O55:B5 (TLRGRADE™) | Enzo Lifesciences | Cat# ALX-581-013-L002 |
| Nigericin | Invivogen | Cat# tlrl-nig |
| Peroxidase from horseradish (HRP) | Sigma | Cat# P8375-1KU |
| Luminol | Sigma | Cat# A8511 |
| DAPI | Invitrogen | Cat# D1306 |
| Cell Proliferation Dye eFluor™ 670 | Invitrogen | Cat# 65-0840-85 |
| FACS Lysing Solution | BD Biosciences | Cat# 349202 |
| Intracellular Staining Permeabilization Wash buffer | BioLegend | Cat# 421002 |
| Diisopropylfluorophosphate | Sigma | Cat# D0879 |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| Phosphatase inhibitors | A. G. Scientific | Cat# T-2494 |
| Critical commercial assays | ||
| Quant-iT PicoGreen | Invitrogen | Cat# P11495 |
| Citrullinated histone H3 ELISA kit | Cayman Chemical | Cat# 501620 |
| Cell Death Detection ELISA | Sigma | Cat# 11544675001 |
| TNF ELISA kit | BioLegend | Cat# 430901 |
| IL-6 ELISA kit | BioLegend | Cat# 431301 |
| IL-1β ELISA kit | BioLegend | Cat# 432601 |
| Albumin Assay Kit | Abcam | Cat# ab235628 |
| Experimental models: organisms/strains | ||
| Mouse C57BL/6J | Jackson Laboratory | Cat# 000664 |
| Mouse C57BL/6J-Hdac6em2Lutzy/J | Jackson Laboratory | Cat# 029318 |
| Software and algorithms | ||
| GraphPad Prism 8.0.2 | GraphPad | Prism - GraphPad RRID:SCR_002798 |
| FlowJo software | Tree star | FlowJo v10.8 RRID:SCR_008520 |
| ImageJ | NIH | ImageJ (nih.gov) RRID:SCR_003070 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ivan Zanoni (ivan.zanoni@childrens.harvard.edu; @Lo_Zanzi).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
6–8 week-old female C57BL/6J (Jax 000664) and C57BL/6J-Hdac6em2Lutzy/J (Jax 029318) mice were purchased from The Jackson Laboratory. Mice were housed under specific pathogen-free conditions at Boston Children’s Hospital, and all the procedures were approved under the Institutional Animal Care and Use Committee (IACUC) and operated under the supervision of the department of Animal Resources at Children’s Hospital (ARCH). Sex as a biological variable was not analyzed in this work.
Neutrophil purification
Human neutrophils from blood of healthy donors were purified with Polymorphoprep (Progen) according to manufacturer’s instruction. Murine neutrophils were purified over a 62.5% Percoll gradient (GE Healthcare) as previously described (Broggi et al., 2017). Purified neutrophils were resuspended in DMEM medium (ThermoFisher) and plated in a 96 multi-well plate previously coated with poly-D-lysine (ThermoFisher). Plate was spin at 200 x g for 1 minute without brake to facilitate neutrophils seeding.
Methods details
Extracellular DNA quantification
Neutrophils were treated with indicated drugs for 1 hour and then stimulated with 1 μM PMA for 3 hours. Extracellular DNA was quantified in the supernatant with Quant-iT PicoGreen (Invitrogen).
Citrullinated histone H3 in vitro quantification and imaging
Neutrophils pretreated with indicated HDAC inhibitors, or protein-arginine deaminase (PAD4) inhibitor GSK484, and then stimulated with 1 μM PMA for 3 hours, were fixed with equal volume of 10% formalin for 1 h at room temperature. Samples were blocked with 2% BSA in PBS for 1 hour at room temperature and stained with the anti-citrullinated histone H3 antibody overnight at 4°C, followed by anti-rabbit Alexa 568 secondary antibody. Nuclei were stained with DAPI. NETs formation was measured as induction of citrullinate histone H3 positivity and quantified with a plate-reader (Ex 568 nm, Em 603 nm). Percentage of NET induction was calculated using signal in un-treated cells as 0% of induction and signal in PMA-treated cells as 100% of induction. Alternatively, NETosis induction was quantified by imaging. Images were acquired with EVOS M7000 Imaging System (ThermoFisher Scientific). Number of citrullinated histone H3 positive cells and area of DAPI signal were quantified in multiple fields of view (FOV) using ImageJ software (NIH). For 3D nuclei reconstruction images were acquired with ZEISS 880 Fast Airyscan and analyzed with ZEISS ZEN Blue Software.
Immunoblotting
For western blotting, 2x106 neutrophils pretreated with indicated HDAC inhibitor, or protein-arginine deaminase (PAD4) inhibitor GSK484, and then stimulated with 1 μM PMA for 3 hours were lysed using RIPA buffer with protease and phosphatase inhibitors (T-2494, A.G. Scientific) and diisopropylfluorophosphate (D0879, Sigma). Immunoblotting was performed using standard molecular biology techniques. Band intensity was quantified using ImageJ software.
ROS quantification
Neutrophils plated in a white 96 multi-well plate were pretreated with indicated HDAC inhibitor, or protein-arginine deaminase (PAD4) inhibitor GSK484 for 1 hour and then were stimulated with 1 μM PMA in presence of luminol (Cat# A8511, Sigma) and HRP (Sigma). ROS production was monitored in real-time by luminescence. Luminescence was measured every 3 minutes for 3 hours in a plate-reader analyzer (Molecular Device).
Phagocytosis assay
E. coli (O111) was stained with Cell Proliferation Dye eFluor™ 670 (cat# 65-0840-85, Invitrogen) for 30 minutes and then opsonized with 20% mouse serum for 10 minutes. Neutrophils were stimulated with the indicated MOI for 1 hour at 37°C, washed, and then stained with anti-Ly6G PE antibody and DAPI. Ultimately, samples were acquired with BD FACS Fortessa and percentage of neutrophils positive for E. coli was quantified.
In vivo NET quantification
NETosis was measured in bronco-alveolar lavage fluid or lung tissue homogenates as quantity of citrullinated histone H3 with Citrullinated histone H3 ELISA kit (Cayman) and as quantity of myeloperoxidase (MPO)-DNA complexes by using a modified Cell Death Detection ELISA (Roche). Briefly, anti-myeloperoxidase antibody (ThermoFisher) was used to coat a high-binding 96 multi-well plate overnight at 4°C. Following a 1h blocking with 2% BSA/PBS solution, samples were added for an overnight incubation at 4°C. Finally, DNA was detected with Cell Death Detection kit according to manufacture instruction. Absorbance was measured at 405 nm and MPO-DNA complex levels were quantified as normalized relative to saline.
Alternatively, neutrophils positive for citrullinated histone H3 were quantified by cytofluorimetry. Briefly, neutrophils from bronco-alveolar lavage fluid or peritoneal lavage fluid were stained for Ly6G, CD11b, CD45 and Zombie dye for 20 minutes at room temperature and then fixed with BD FACS Lysing Solution (cat# 349202, BD Biosciences). Samples were washed twice with Intracellular Staining Permeabilization Wash buffer (cat# 421002, Biolegend), incubated with anti-citrullinated histone H3 antibody (Abcam) 1 hour at room temperature and subsequently with PE anti-rabbit antibody for 30 minutes. Finally, samples were acquired with BD FACS Fortessa and neutrophils (CD45+/Ly6G+/CD11b+) positive for citrullinated histone H3 were quantified with FlowJo software.
Poly(I:C) pulmonary NET induction
6-8 weeks-old female mice were treated daily with 2.5 mg/kg of poly (I:C) for 4 days and euthanized 18 hours post the last poly(I:C) intra-tracheal instillation, as previously described (Broggi et al., 2020). Temperatures were monitored with a rectal probe. NETosis was measured in bronco-alveolar lavage fluid and lung homogenates as quantity of citrullinated histone H3 and quantity of MPO-DNA complexes. Ricolinostat (30 mg/kg/dose) and GSK484 (25 mg/kg/dose) were administered intraperitoneally at day 2, day 4 and 3 hours prior the end point. To assess lung permeability, albumin in BALF was measured with Albumin Assay Kit (ab235628, Abcam). Number of neutrophils (CD45+/Ly6G+/CD11b+) in blood, BALF and lung homogenate was quantified by cytofluorimetry using CountBright Absolute Cell Counting Beads (Thermo Scientific). Paraffin-embedded lung sections were stained with DAPI for nuclei and with anti-neutrophil antibody (Abcam) for the presence of neutrophils.
Measurement of airway functional responses
Airway hyperreactivity (AHR) was measured, as previously described (Harb et al., 2020). Anesthetized mice were exposed to doubling concentrations of aerosolized acetyl-β-methacholine (Sigma-Aldrich) by using a Buxco small-animal ventilator (Data Sciences International). The relative peak airway resistance for each methacholine dose, normalized to the saline baseline (Score = 1), was calculated.
S. aureus pulmonary infection model
6-8-week-old female mice were pretreated with indicated HDAC inhibitors at −1 day and −3 hours prior in vivo NETs induction by intra-tracheal instillation of 65 × 106 CFU of Staphylococcus aureus subsp. aureus Rosenbach (ATCC 25904). Ricolinostat was used at 30 mg/kg/dose and GSK484 at 25 mg/kg/dose. Temperatures were monitored with a rectal probe. NETosis was measured in bronco-alveolar lavage fluid as quantity of citrullinated histone H3 and quantity of MPO-DNA complexes by ELISA. Alternatively, neutrophils positive for citrullinated histone H3 were analyzed by cytofluorimetry in BALFs samples. Albumin in BALF was measured with Albumin Assay Kit (ab235628, Abcam). Number of neutrophils (CD45+/Ly6G+/CD11b+) in blood and BALF was quantified by flow cytometry using CountBright Absolute Cell Counting Beads (Thermo Scientific).
In vivo LPS-induced septic shock
8-week-old female mice were primed with 1 mg/kg of LPS intra peritoneum (i.p.) for 5 hours and then challenged with 2 mg/kg of LPS i.p. Ricolinostat was used at 30 mg/kg/dose, GSK484 at 25 mg/kg/dose and dexamethasone at 10 mg/kg/dose. Drugs were administered i.p. at day −1 and 2 hours prior priming. Blood samples were collected 2 hours post-challenge and cytokines were measured by ELISA. The platelet concentrations and neutrophils numbers in the blood and peritoneal cavity were calculated by cytofluorimetry using CountBright Absolute Cell Counting Beads (Thermo Scientific) according to the manufacturer's instructions. NETosis was measured as quantity of neutrophils in the peritoneal lavage positive for citrullinated histone H3 by cytofluorimetry.
Quantification and statistical analysis
Results were analyzed with GraphPad Prism statistical software (version 8). One-way ANOVA and Two-way ANOVA were used to analyze statistically significant differences between the means of two or more independent groups, as indicated in the figure legends. Sample sizes for each experiment are provided in the figures and the respective legends. Asterisks were used as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Acknowledgments
I.Z. is supported by NIH grants 2R01AI121066, 5R01DK115217, 1R01AI165505, NIAID-DAIT-NIHAI201700100, and Lloyd J. Old STAR Program CRI3888 and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. V.P-Y.M. is supported by NCI-F99/K00 predoc-to-postdoc transition award (K00CA223074).
Author contributions
V.P., M.D.G., A.B., M.B., and Q.C. designed, performed, and analyzed the experiments; I.Z., V.P., and V.P-Y.M. wrote the paper; T.A.C., R.M., and S.J.H. contributed to design the experiments; I.Z. and J.M.K. conceived the project, designed the experiments, and supervised the study.
Declaration of interests
V.P., V.P-Y.M., R.M., S.J.H., J.M.K., and I.Z. submitted a patent related to the findings of this work. J.M.K. has been a paid consultant and/or equity holder for companies including Stempeutics, Sanofi, Celltex, LifeVaultBio, Camden Partners, Stemgent, Biogen, Pancryos, Element Biosciences, Frequency Therapeutics, and Mesoblast. J.M.K. is also an inventor on a patent that was licensed to Mesoblast. J.M.K. holds equity in Frequency Therapeutics, a company that has licensed IP generated by J.M.K. that may benefit financially if the IP is further validated. J.M.K. has been a paid consultant and or equity holder for multiple companies (listed here https://www.karplab.net/team/jeff-karp). The interests of J.M.K. were reviewed and are subject to a management plan overseen by his institutions in accordance with its conflict-of-interest policies. S.J.H. was or is a member of the scientific advisory board and equity holder in Rodin Therapeutics, Psy Therapeutics, Frequency Therapeutics, and Vesigen Therapeutics and has received speaking or consulting fees from AstraZeneca, Amgen, Merck, Biogen, Syros Pharmaceuticals, Regenacy Pharmaceuticals. S.J.H.’s laboratory has received sponsored research funding from the Tau Consortium, F-Prime Biomedical Research Initiative, AstraZeneca, JW Pharmaceuticals, ATAI, and Stealth BioTherapeutics, although none of these sponsors were involved in the present study. R.M. is a scientific advisory board (SAB) member and equity holder of Regenacy Pharmaceuticals, ERX Pharmaceuticals, and Frequency Therapeutics.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103256.
Contributor Information
Jeffrey M. Karp, Email: jmkarp@bwh.harvard.edu.
Ivan Zanoni, Email: ivan.zanoni@childrens.harvard.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., Vanstapel A., Werlein C., Stark H., Tzankov A. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. New Engl. J. Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Samkari H., Karp Leaf R.S., Dzik W.H., Carlson J.C.T., Fogerty A.E., Waheed A., Goodarzi K., Bendapudi P.K., Bornikova L., Gupta S. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. 2020;136:489–500. doi: 10.1182/blood.2020006520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel F., Andreeva L., Knackstedt L.S., Streeck R., Frese C.K., Goosmann C., Hopfner K.P., Zychlinsky A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021;14:eaax7942. doi: 10.1126/scisignal.aax7942. [DOI] [PubMed] [Google Scholar]
- Arpinati L., Shaul M.E., Kaisar-Iluz N., Mali S., Mahroum S., Fridlender Z.G. NETosis in cancer: a critical analysis of the impact of cancer on neutrophil extracellular trap (NET) release in lung cancer patients vs. mice. Cancer Immunol. Immunother. 2020;69:199–213. doi: 10.1007/s00262-019-02474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J.A., Woan K., Perez-Villarroel P., Villagra A., Sotomayor E.M., Kozikowski A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012;55:9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondonnet R., Constantin J.-M., Sapin V., Jabaudon M. A pathophysiologic approach to biomarkers in acute respiratory distress syndrome. Dis. Markers. 2016;2016:3501373. doi: 10.1155/2016/3501373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Broggi A., Ghosh S., Sposito B., Spreafico R., Balzarini F., Lo Cascio A., Clementi N., De Santis M., Mancini N., Granucci F. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369:706–712. doi: 10.1126/science.abc3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A., Tan Y., Granucci F., Zanoni I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 2017;18:1084–1093. doi: 10.1038/ni.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K.V., Kalin J., Brochier C., Vistoli G., Langley B., Kozikowski A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.W., Monteleone M., Boucher D., Sollberger G., Ramnath D., Condon N.D., von Pein J.B., Broz P., Sweet M.J., Schroder K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018;3:eaar6676. doi: 10.1126/sciimmunol.aar6676. [DOI] [PubMed] [Google Scholar]
- Clark S.R., Ma A.C., Tavener S.A., McDonald B., Goodarzi Z., Kelly M.M., Patel K.D., Chakrabarti S., McAvoy E., Sinclair G.D. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- Douda D.N., Khan M.A., Grasemann H., Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. U S A. 2015;112:2817–2822. doi: 10.1073/pnas.1414055112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannis D., Ziogas I.A., Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 2020;127:104362. doi: 10.1016/j.jcv.2020.104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Moreno D., Adrover J.M., Hidalgo A. Neutrophils as effectors of vascular inflammation. Eur. J. Clin. Invest. 2018;48:e12940. doi: 10.1111/eci.12940. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Guan W.-j., Ni Z.-y., Hu Y., Liang W.-h., Ou C.-q., He J.-x., Liu L., Shan H., Lei C.-l., Hui D.S.C. Clinical characteristics of coronavirus disease 2019 in China. New Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty S.J., Koeller K.M., Wong J.C., Grozinger C.M., Schreiber S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U S A. 2003;100:4389. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J., Schauer C., Czegley C., Kling L., Petru L., Schmid B., Weidner D., Reinwald C., Biermann M.H.C., Blunder S. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019;33:1401–1414. doi: 10.1096/fj.201800752R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamam H.J., Khan M.A., Palaniyar N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules. 2019;9:32. doi: 10.3390/biom9010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamam H.J., Palaniyar N. Histone deacetylase inhibitors dose-dependently switch neutrophil death from netosis to apoptosis. Biomolecules. 2019;9 doi: 10.3390/biom9050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb H., Benamar M., Lai P.S., Contini P., Griffith J.W., Crestani E., Schmitz-Abe K., Chen Q., Fong J., Marri L. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity. 2021;54:1186–1199.e7. doi: 10.1016/j.immuni.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb H., Stephen-Victor E., Crestani E., Benamar M., Massoud A., Cui Y., Charbonnier L.M., Arbag S., Baris S., Cunnigham A. A regulatory T cell Notch4-GDF15 axis licenses tissue inflammation in asthma. Nat. Immunol. 2020;21:1359–1370. doi: 10.1038/s41590-020-0777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba T., Levy J.H., Raj A., Warkentin T.E. Advance in the management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Clin. Med. 2019;8 doi: 10.3390/jcm8050728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., Pillai P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon S., Galdiero M.R., Del Prete D., Cassatella M.A., Garlanda C., Mantovani A. Neutrophils in innate and adaptive immunity. Semin. Immunopathol. 2013;35:377–394. doi: 10.1007/s00281-013-0374-8. [DOI] [PubMed] [Google Scholar]
- Jamieson A.M., Pasman L., Yu S., Gamradt P., Homer R.J., Decker T., Medzhitov R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340:1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L., Yu H., Yang X., Zhu Y., Bai X., Wang R., Cao Y., Xu H., Luo H., Lu L. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020;11:2488. doi: 10.1038/s41467-020-16191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny E.F., Herzig A., Krüger R., Muth A., Mondal S., Thompson P.R., Brinkmann V., Bernuth H.v., Zychlinsky A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 2017;6:e24437. doi: 10.7554/eLife.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.S. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Kronbichler A., Park D.D., Park Y., Moon H., Kim H., Choi J.H., Choi Y., Shim S., Lyu I.S. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun. Rev. 2017;16:1160–1173. doi: 10.1016/j.autrev.2017.09.012. [DOI] [PubMed] [Google Scholar]
- Lewis H.D., Liddle J., Coote J.E., Atkinson S.J., Barker M.D., Bax B.D., Bicker K.L., Bingham R.P., Campbell M., Chen Y.H. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015;11:189–191. doi: 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magupalli V.G., Negro R., Tian Y., Hauenstein A.V., Di Caprio G., Skillern W., Deng Q., Orning P., Alam H.B., Maliga Z. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369:eaas8995. doi: 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B., Davis R.P., Kim S.J., Tse M., Esmon C.T., Kolaczkowska E., Jenne C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–1367. doi: 10.1182/blood-2016-09-741298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B., Urrutia R., Yipp B.G., Jenne C.N., Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12:324–333. doi: 10.1016/j.chom.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Schneider D.S., Soares M.P. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., Hlh Across Speciality Collaboration U.K. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton E.A., He X.-Y., Denorme F., Campbell R.A., Ng D., Salvatore S.P., Mostyka M., Baxter-Stoltzfus A., Borczuk A.C., Loda M. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V., Metzler K.D., Hakkim A., Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-Y., Kim J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020;52:204–212. doi: 10.1038/s12276-020-0382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Dandekar A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005;5:917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai P.S., Molony R.D., Martinod K., Dong H., Pang I.K., Tal M.C., Solis A.G., Bielecki P., Mohanty S., Trentalange M. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto B.N., Stein R.T. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. 2016;7:311. doi: 10.3389/fimmu.2016.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radermecker C., Detrembleur N., Guiot J., Cavalier E., Henket M., d'Emal C., Vanwinge C., Cataldo D., Oury C., Delvenne P. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020;217:e20201012. doi: 10.1084/jem.20201012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remijsen Q., Vanden Berghe T., Wirawan E., Asselbergh B., Parthoens E., De Rycke R., Noppen S., Delforge M., Willems J., Vandenabeele P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q., Yang K., Wang W., Jiang L., Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846–848. doi: 10.1007/s00134-020-05991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T., Komano J., Saitoh Y., Misawa T., Takahama M., Kozaki T., Uehata T., Iwasaki H., Omori H., Yamaoka S. Neutrophil Extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12:109–116. doi: 10.1016/j.chom.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Schauer C., Janko C., Munoz L.E., Zhao Y., Kienhofer D., Frey B., Lell M., Manger B., Rech J., Naschberger E. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014;20:511–517. doi: 10.1038/nm.3547. [DOI] [PubMed] [Google Scholar]
- Seto E., Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect. Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H.-S., Kim Y., Kim G., Lee J.Y., Jeong I., Joh J.-S., Kim H., Chang E., Sim S.Y., Park J.-S. Immune responses to middle east respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin. Infect Dis. 2019;68:984–992. doi: 10.1093/cid/ciy595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short K.R., Kroeze E.J.B.V., Fouchier R.A.M., Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis. 2014;14:57–69. doi: 10.1016/S1473-3099(13)70286-X. [DOI] [PubMed] [Google Scholar]
- Sollberger G., Choidas A., Burn G.L., Habenberger P., Di Lucrezia R., Kordes S., Menninger S., Eickhoff J., Nussbaumer P., Klebl B. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018;3:eaar6689. doi: 10.1126/sciimmunol.aar6689. [DOI] [PubMed] [Google Scholar]
- Tang B.M., Cootes T., McLean A.S. In: Annual Update in Intensive Care and Emergency Medicine 2019. Vincent J.-L., editor. Springer International Publishing; 2019. From influenza-induced acute lung injury to multiorgan failure; pp. 449–458. [Google Scholar]
- Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam H.R., Wong S.L., Wagner D.D., Waterman C.M. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020;36:191–218. doi: 10.1146/annurev-cellbio-020520-111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T., van de Veerdonk F.L., Scicluna B.P., Netea M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017;17:407–420. doi: 10.1038/nri.2017.36. [DOI] [PubMed] [Google Scholar]
- Veras F.P., Pontelli M.C., Silva C.M., Toller-Kawahisa J.E., de Lima M., Nascimento D.C., Schneider A.H., Caetite D., Tavares L.A., Paiva I.M. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020;217:e20201129. doi: 10.1084/jem.20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S.A. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.L., Demers M., Martinod K., Gallant M., Wang Y., Goldfine A.B., Kahn C.R., Wagner D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015;21:815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Lu W., Zhang Y., Zhang G., Shi X., Hisada Y., Grover S.P., Zhang X., Li L., Xiang B. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. 2019;50:1401–1411.e1404. doi: 10.1016/j.immuni.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Zhang X., Pelayo R., Monestier M., Ammollo C.T., Semeraro F., Taylor F.B., Esmon N.L., Lupu F., Esmon C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Cheng X., Tang Y., Qiu X., Wang Y., Kang H., Wu J., Wang Z., Liu Y., Chen F. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity. 2019;51:983–996.e986. doi: 10.1016/j.immuni.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Zanoni I., Tan Y., Di Gioia M., Springstead J.R., Kagan J.C. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity. 2017;47:697–709.e693. doi: 10.1016/j.immuni.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Zhang X., Li H. Beyond histone acetylation—writing and erasing histone acylations. Curr. Opin. Struct. Biol. 2018;53:169–177. doi: 10.1016/j.sbi.2018.10.001. [DOI] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y., Estes S.K., Ali R.A., Gandhi A.A., Yalavarthi S., Shi H., Sule G., Gockman K., Madison J.A., Zuo M. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl Med. 2020:eabd3876. doi: 10.1126/scitranslmed.abd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:e138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.