

Abstract
X-linked dystonia-parkinsonism (XDP) is a monogenic neurodegenerative disorder of the basal ganglia, which presents as a combination of hyperkinetic movements and parkinsonian features. The underlying genetic mechanism involves the insertion of a SINE-VNTR-Alu retrotransposon within the TAF1 gene. Interestingly, alterations of TAF1 have been involved in multiple neurological diseases. In XDP, the SINE-VNTR-Alu insertion in TAF1 has been proposed to result in alternative splicing defects, including the decreased incorporation of a neuron-specific microexon annotated as 34′. This mechanism has become controversial as recent studies failed to provide support. In order to resolve this conundrum, we examined the alternative splicing patterns of TAF1 mRNAs in XDP and control brains. The impact of the disease-associated SINE-VNTR-Alu on alternative splicing of microexon 34′ was further investigated in cellular assays. Subsequently, microexon 34′ incorporation was explored by RT-PCR and Nanopore long-read sequencing of TAF1 mRNAs from XDP and control brains tissues. Using cell-based splicing assays, we demonstrate that presence of the disease-associated SINE-VNTR-Alu does not affect the inclusion of microexon 34′. In addition, we show that (1) microexon 34′-containing TAF1 mRNAs are detected at similar levels in XDP as in controls and that (2) the architecture of TAF1 transcripts is remarkably similar between XDP and controls brains. These results indicate that microexon 34′ incorporation into TAF1 mRNA is not affected in XDP brains. Our findings shift the current paradigm of XDP by discounting alternative splicing of TAF1 microexon 34′ as the molecular basis for this disease.
Keywords: X-linked dystonia-parkinsonism, motor disorders, neuronal microexon inclusion, alternative splicing, SVA retrotransposon
Capponi et al.19 propose a paradigm shift for the pathogenesis of XDP by demonstrating that microexon 34′ is incorporated within TAF1 mRNA in XDP brain specimens.
Graphical Abstract
Graphical abstract.

Introduction
X-linked Dystonia-Parkinsonism (XDP) is a hereditary neurodegenerative movement disorder, which presents in individuals with Philippines ancestors, and has an average onset at the age of ∼40 years.1 The nationwide prevalence of the disease on the Philippines is 0.31 per 100 000, rising to 0.57 on the island of Panay.2 The natural history of XDP is heterogeneous with the majority of patients presenting hyperkinetic movements at an early stage of the disease, to be accompanied or replaced by parkinsonism features over time.3 Together with well-documented motor symptoms, XDP patients present a series of nonmotor features, including anxiety and depression.4 Neuropathological analyses of ‘post-mortem’ XDP brains revealed striatal atrophy with a progressive loss of striosomal medium spiny neurons.5 Alteration of white matter (WM) microstructures, atrophy of the pallidum and reduced thickness of frontal and temporal cortex have also been reported.6,7
In recent years, XDP research has benefitted from inputs of different disciplines such as genetics, neurobiology, pathophysiology and clinical neurology, leading to significant advances in understanding of both the clinical manifestations and molecular pathomechanisms associated with the disease. To date, the most accredited genetic lesion underlying XDP is the insertion of a SINE-VNTR-Alu (SVA) retrotransposon within intron 32 of the TAF1 gene.8,9 Interestingly, hexamer repeat length within the XDP-SVA inversely correlates with the age at disease onset, corroborating the involvement of the retrotransposon in XDP pathogenesis.10,11 Genetic perturbations of the TAF1 locus have been linked to several neurological disorders. Whereas TAF1 mRNA expression is affected in XDP,9,12 point mutations and duplications have been associated with intellectual disability13,14 and reduced TAF1 expression has been reported in Huntington’s disease (HD).15 These observations indicate that while TAF1 is ubiquitously expressed, its function is particularly important for brain physiology.
TAF1 encodes the TATA-binding protein associated factor 1, the largest subunit of the basal transcription factor TFIID, required for transcription initiation by RNA polymerase II.16 Among the annotated TAF1 isoforms, the transcript named TAF1 34′ is restricted to neuronal tissues. This isoform differs from canonical TAF1 (cTAF1) by incorporation of the 6-nucleotide microexon 34′.8 Splicing of microexons of 3- to 27-nucleotides constitutes a temporally defined and conserved program during brain development, which promotes fine regulation of protein-interaction networks.17 Its relevance for neurophysiology is underlined by the link between microexon misregulation and autism spectrum disorder.17 The neuron-specific splicing factor Serine/Arginine Repetitive Matrix 4 (SRRM4) is the main driver of brain-specific microexon inclusion in general18 and of microexon 34′ incorporation into TAF1 mRNAs in particular.19
The current paradigm for the pathomechanism of XDP is a downregulation of TAF1 isoforms containing microexon 34′, which would be due to the XDP-specific SVA insertion within intron 32.8 This paradigm depends on TAF1 mRNA analysis of one XDP brain versus one control, showing 40-folds reduction in the striatum (ST) and about 5-fold reduction in the cortex.8 Similar observations were reported for XDP iPSCs-derived neural stem cells (NSCs).20 While this model has been used to explain XDP for many years, it has been challenged recently by in vitro differentiation studies of XDP NSCs, in which no significant difference in microexon 34′ incorporation was observed between XDP and control cell lines.9,12 In addition, transcriptome assembly indicated a reduced abundance of TAF1 exons downstream of the SVA, which would result in lower levels of full-length transcripts. The XDP-SVA insertion also affected intron 32 splicing resulting in intron retention and incorporation of intronic exons.9,12 Thus, it has become uncertain whether alterations in microexon 34′ splicing are key to understand XDP pathology.
Here, we revisit the defective microexon-splicing paradigm by examining the exonic structure of TAF1 mRNAs from XDP and control brain samples. We find that microexon 34′ inclusion is not affected in XDP brains and that splicing of TAF1 alternative exons across the XDP-SVA insertion site is not different between XDP and control brains. Our findings motivate a reconsideration of the molecular basis for XDP pathogenesis.
Materials and Methods
Plasmids and cell lines generation
The minigene constructs included the genomic sequence of human TAF1 from exon 31, GRCh38 chrX:71423980, to exon 35, GRCh38 chrX:71458366. TAF1 intron 32 (29.9 kb) containing the insertion site of the XDP SVA element was split into three segments: the 5′-end (GRCh38 chrX:71424239-71425356), the region including the XDP-SVA insertion site (GRCh38 chrX:71439499-71441543) and the 3′-end (GRCh38 chrX:71453716-71454169). The XDP-SVA sequence was amplified from a bacteria artificial chromosome partially containing the XDP locus with 35 hexamer repeats (provided by the Collaborative Center for X-linked Dystonia Parkinsonism—CCXDP, Boston, MA, USA) using PrimeSTAR GXL DNA polymerase (Takara Bio). Both control and SVA-containing minigenes were cloned into the N-terminal GFP-tagged vector pcDNA5/FRT/TO using the GATEWAY system (Thermo Fisher Scientific). The control minigene was verified by Sanger sequencing. Sequence coverage of the XDP-SVA was complete with the exception of 76 bp within the VNTR region and the hexamer repeat. Restriction enzyme analysis indicates that hexamer repeat length is compatible with the original size of 35 (Supplementary Fig. S1). The pcDNA5/FRT/TO/GFP-SRRM4 expression vector has been described.19
Human embryonic kidney cells 293T, mouse neuroblastoma cells N2a and human cervical cancer cells HeLa were cultured in DMEM containing 4.5 g/l of glucose (Lonza), supplemented with 10% (v/v) fetal bovine serum (Lonza). Transient transfection was performed using FuGENE HD Transfection Reagent (Promega). Cells were lysed 48 h after transfection. Doxycyclin (DOX)-inducible HeLa derivatives expressing GFP-SRRM4 were previously described.19 DOX-inducible N2a were a kind gift from Dr Blencowe (University of Toronto, Canada) and N2a derivatives were generated and induced as described.19
Immunoblot and antibodies
Cells were lysed in E1A buffer (1% Nonidet P-40, 20 mM Hepes–KOH pH 8.0, 250 mM NaCl, 1 mM EDTA, 2 mM DTT, cOmplete protease inhibitor cocktail—Roche) and equal amounts of protein were analysed by SDS-PAGE followed by immunoblotting. The cTAF1- and TAF1-34′-specific antibodies were generated as described.19 The SRRM4 antibody was a kind gift from Dr Blencowe (University of Toronto, Canada).18 The α-vinculin antibody was purchased from Santa Cruz (sc-73614). Secondary antibodies were: α-mouse-HRP conjugate and α-rabbit-HRP conjugate (Bio-Rad).
Processing of post-mortem tissues
The analysis of post-mortem tissues included eight XDP and three control brains. Specimens from six XDP brains were provided by the CCXDP (IDs 17.01, 17.06, 17.10, 17.12, 17.13 and 17.17). All procedures related to donor informed consent, tissue collection and distribution for research have been reported.21 The other two brains were provided by Dr C. Klein (IDs L-10.322 and L-7.995). Specimens were used in accordance with the ‘Gesetz über das Leichen-, Bestattungs- und Friedhofswesen (Bestattungsgesetz) des Landes Schleswig-Holstein vom 04.02.2005, Abschnitt II, § 9 (Leichenöffnung, anatomisch)’. The control samples were obtained by Dr M. Creyghton (IDs c1 to c3) from the Netherlands Brain Bank. Sample c1 is included with a technical replicate named c1.1, consisting of an independent dissection and further processing. Informed consent was acquired as described.19 Frozen tissue was pulverized using CellCrusher in liquid nitrogen. XDP subjects had a mean age of 47.4 years (SD: ±10.01) while control subjects had a mean age of 72.3 years (SD: ±15.94). Additional information are listed in Supplementary Table S1.
The available regions from XDP brains included prefrontal cortex (PFC, n = 7), cerebellum (CB, n = 7), anterior putamen (AP, n = 6), ST (n = 1), substantia nigra (SN, n = 1), motor cortex (MC, n = 1), pituitary gland (PG, n = 1), external globus pallidus (eGP, n = 1), internal glubus pallidus (iGP, n = 1), nucleus (NC, n = 1), WM (n = 1) and grey substances (GS, n = 1). Control tissues included PFC (n = 3), MC (n = 1), CB (n = 2), AP (n = 1), caudate nuclei (CN, n = 3) and thalamus (TH, n = 1). Hexamer repeat length for the processed XDP samples was genotyped by Fernandez-Cerado et al.21 and Reyes et al.22 and repeat numbers are as follows: 17.01, 44; 17.06, 41; 17.10, 34; 17.12, 42; 17.13, 46; 17.17, 54; L-7.995, 45; L-10.332, 41.
Expression analysis with RT-PCR and RT-qPCR
Total RNA from brain specimens was isolated using the RNeasy Lipid Tissue kit (Qiagen) and treated with TURBO DNase (Thermo Fisher Scientific). RNA integrity was assessed with Bioanalyser using the RNA 6000 Nano kit (Agilent Technologies). RNA Integrity Numbers (RINs) were above 3 for 23/29 XDP and 5/13 control samples. Nevertheless, a successful PCR amplification was obtained for all samples, confirming that RNA quality does not impact PCR efficiency of amplicons smaller than 250 bp.23 Total RNA was isolated from transiently transfected 293T using the RNeasy kit (Qiagen), including on-column DNase treatment. Total RNA was used for cDNA synthesis (Superscript III, Thermo Fisher Scientific) using random hexamer primers.
RT-PCR analysis on brains was performed amplifying a region spanning TAF1 exon 34 and exon 35 (97 bp) for analysis on polyacrylamide gels.19 The incorporation of microexon 34′ is shown as an additional band of 103 bp. Percentage-Spliced-In (PSI) was calculated as the fraction of TAF1 cDNA containing microexon 34′ to the total. Relative band densities were calculated using ImageJ. The analysis of minigene-derived cDNA was performed using a nested PCR approach, in which the first PCR enriches for minigene-derived cDNA while the nested PCR generates the 97/103 bp amplicon described above.
RT-qPCR experiments were performed on CFX384 Real-Time system (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). The relative expression level of SRRM4 mRNA was evaluated with the ΔΔCt method. The normalization was performed using BestKeeper24 and TBP, GAPDH, HMBS, SDHA, HPRT1 and ACTB as reference mRNAs. Primers used for RT-PCR and RT-qPCR are listed in Supplementary Table S2.
Nanopore sequencing
Nanopore sequencing was performed using the cDNA SQK-LSK109 sequencing kit (Oxford Nanopore Technologies, ONT). The protocol consists of two amplification steps. In the first step, cDNA fragments of interest were amplified using primers tailed with the universal Nanopore tags (primers are listed in Supplementary Table S2). A total of 0.5 nM products were used as a template for the barcoding reaction using the PCR barcoding kit 96 (EXP-PBC096, ONT). Barcoded libraries were pooled (total of 1 µg DNA) and prepared for sequencing. The flow cell was loaded with 40–50 fmol of library pool and run for 48 h on MinION equipped with R9.4.1 flow cells. Control and XDP samples were divided into four sets and run independently. In addition, one sample from each set was repeated in the last run to investigate inter-run variability. The data from these sentinels were consistent with the data from the original run (data not shown).
Bioinformatic analysis of Nanopore data
Base calling of Nanopore data was performed using Guppy version 3.6.1. De novo assembly was performed aligning the on-target reads to the genome reference hg38 with minimap2,25 adapting the splice preset for the alignment of small exons (-B3 -O3.6). The average number of on-target reads used for further analysis was 611347 (min: 2315, max: 1197298). To gear up Nanopore sequencing for efficient microexon detection, we used a training set composed of cDNA from inducible GFP-SRRM4 HeLa cells as the ectopic expression of SRRM4 in HeLa allows the incorporation of microexon 34′ into endogenous TAF1 mRNA19 (Supplementary Fig. S2). Different splicing isoforms were detected, quantified and plotted using FLAIR.26 Isoforms below 1% usage in at least one sample were not included in further analyses.
Statistical analyses
Statistical analyses were performed focusing on the PFC as for this region data were collected for the higher number of XDP patients (n = 7) and controls (n = 4). Mann–Whitney U two-tailed test was used for the statistical analyses of RT-PCR and RT-qPCR. The analysis of differential transcript usage (DTU) derived from long-read sequencing was performed using drimseq, a statistical method based on Dirichlet-multinomial model.27 Statistical significance was considered with P-value of <0.05.
Data availability
The Nanopore long-read sequencing data have been deposited in the NIH Biosample database under the submission ID PRJNA666449.
Results
The presence of the XDP-SVA within TAF1 intron 32 does not affect SRRM4-mediated microexon 34′ incorporation
The presence of the XDP-SVA within TAF1 intron 32 has been proposed to reduce the incorporation of microexon 34′ into TAF1 mRNA up to 40-fold.8,20 Since this microexon is spliced exclusively in neuronal tissues, misregulation would account for the neuronal specificity of XDP. We demonstrated previously that the neuronal-specific SRRM4 splicing factor is responsible for the incorporation of microexon 34′ into TAF1 mRNA. When SRRM4 is ectopically expressed in non-neuronal cells, it activates a microexon splicing program, which among others, results in the inclusion microexon 34′ in TAF1 mRNA. Conversely, when Srrm4 is downregulated in neuronal cells, the incorporation of Taf1 microexon 34′ is drastically reduced.19
To investigate whether the XDP-SVA could reduce microexon 34′ inclusion by affecting SRRM4-mediated splicing, we designed a TAF1 minigene reporter assay (Fig. 1A). These reporters were transiently co-transfected with a human SRRM4 expression vector into human embryonic kidney 293T cells. In our RT-PCR assay, the ratio between the alternatively spliced mRNAs is quantified with the PSI metric,17 which determines microexon 34′ abundance in all TAF1 mRNAs. Co-transfection of the minigene and SRRM4 expression vectors revealed that both control and XDP-SVA reporters support efficient microexon 34′ incorporation (Fig. 1B and C). As indicated by both mRNA and protein analysis, co-expression of SRRM4 mediates the switch from canonical TAF1 to the neuronal TAF1-34′, which is not influenced by the presence of the XDP-SVA. We confirmed these results in mouse neuroblastoma N2a cells (Fig. 1D), which endogenously express low levels of Srrm4. In this cell system, the TAF1 reporters are stably integrated in the host genome and human SRRM4 was transiently overexpressed. As expected28TAF1 mRNA analysis showed that microexon 34′ incorporation is already detectable in non-transfected N2a cells. Overexpression of human SRRM4 resulted in increased microexon 34′ incorporation. Either with endogenous Srrm4 or with overexpressed human SRRM4, the inclusion of microexon 34′ is not affected by the presence of XDP-SVA (Fig. 1D). These results indicate that the presence of the XDP-SVA does not impact the SRRM4-mediated inclusion of microexon 34′ into TAF1 mRNAs.
Figure 1.

The XDP-SVA does not affect SRRM4-mediated incorporation of microexon TAF1 34′ in human cells. The impact of the XDP SVA element on the alternative splicing of microexon 34′ was investigated using a TAF1 minigene reporter assay. These minigenes include the GFP-tagged genomic sequence of TAF1 from exon 31 to exon 35. The GFP N-terminal tag allows discrimination of minigene-derived splicing from processing of endogenous TAF1 mRNA. The two minigene constructs, excluding (MG, top) or including the XDP-SVA (MG SVA, bottom) sequences are depicted in panel A. The primer sets used for the RT-PCR to analyse minigene-derived mRNAs are indicated in red (PCR1) and blue (PCR2). The effect of the SVA on SRRM4-mediated alternative splicing was examined in 293T cells by co-transfection of minigene constructs (MG or MG SVA) with a plasmid encoding GFP-tagged SRRM4 (panels B and C). RT-PCR data are depicted in panel B, where the non-transfected control is indicated as NT. PSI values for minigene-derived mRNAs are calculated as described,19 by dividing the intensity of the 103 bp band by the total of the 97 and 103 bp bands. The protein products derived from the transfected plasmids are analysed in panel C. Immunoblot analysis includes the probing for SRRM4, canonical TAF1 (cTAF1), TAF1 34′ and vinculin as loading control. The SRRM4 antibody recognizes different phosphorylation statuses of the protein, therefore producing multiple bands. The two bands depicted in the cTAF1 immunoblot result from different splicing event on the minigene mRNA, where the fully spliced mRNA translates into the higher molecular weight band, indicated by a star. To validate the splicing assay in a different cell system, we used DOX-inducible mouse neuroblastoma cells N2a stably expressing the chromosomally integrated MG or MG SVA reporters (panel D). Both lines were transiently transfected with the GFP-SRRM4 plasmid. As expected, the PSI of microexon 34′ significantly increases upon overexpression of GFP-SRRM4 (lanes 2 and 4). N2a cells endogenously express mouse Srrm4, which accounts for the low PSI values obtained in the induced non-transfected cells (lanes 1 and 3).
Microexon 34′ is incorporated into TAF1 mRNA in XDP and control brains
The findings with the TAF1 reporters in the splicing assays motivated us to revisit the question of the TAF1-34′ mRNA expression in XDP brains.8 To account for inter-individual variability, we examined a series of post-mortem brain specimens from eight XDP patients and four controls. The inclusion of microexon 34′ into TAF1 mRNA was assessed with our RT-PCR assay, detecting TAF1 mRNAs including or excluding microexon 34′.19 It is important to note that previous studies relied on a TaqMan assay, which could not make this distinction and only allowed inter-brain comparisons.8,20 Applying our RT-PCR assay to different brain regions, we detected microexon 34′ inclusion in different XDP specimens (Fig. 2A). In addition, microexon 34′-containing TAF1 mRNAs were observed in all patients regardless of the hexamer repeat genotype (Supplementary Fig. S3). The PSI quantification showed a significant degree of variability in TAF1 34′ incorporation within different brain regions, ranging from below 10 to over 90. In particular, while the CB displayed the highest incorporation rate, the AP scored in the lower range. This tissue distribution correlates with SRRM4 expression levels, again corroborating the dependency of microexon 34′ on this neuronal splicing factor.19 A significant degree of variability was also observed among the PSI of different individuals within the same brain region. To assess whether microexon 34′ incorporation rate was quantitatively different in XDP patients compared with controls, we compared PFC PSIs. The selected tissue is relevant for XDP pathogenesis as decreased cortical thickness has been documented in XDP patients, challenging the concept of XDP as a pure striatal disease.7 The analysis of microexon 34′ inclusion did not reveal significant differences between XDP patients and controls (Fig. 2B). Previous TaqMan data suggested about five-fold reduction in the cortex from one XDP brain versus one control.8 In our RT-PCR setup, we do not observe this difference. Taken together, these results show that microexon 34′ is detectable in XDP brains and that its inclusion efficiency is not affected in XDP compared with controls.
Figure 2.
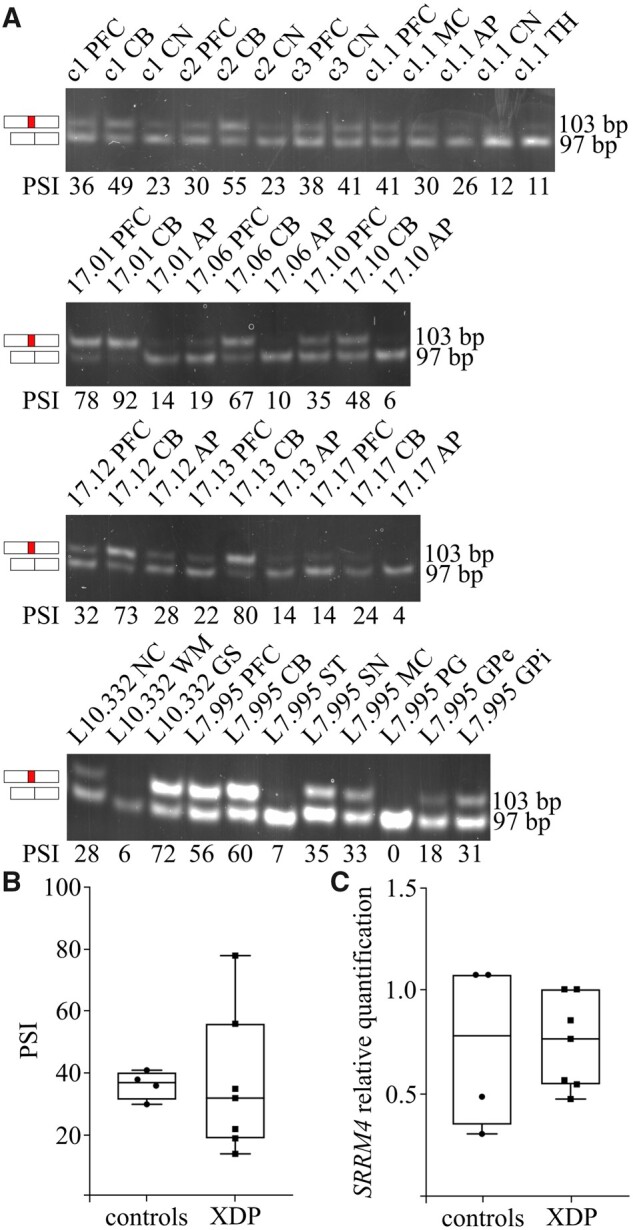
Microexon 34′ is incorporated into TAF1 mRNA in XDP brain specimens. The incorporation of microexon 34′ is investigated in a set of brain specimens from four controls (panel A—upper gel) and eight XDP patients (panel A—second, third and fourth gel). The PSI metric used to quantify microexon 34′ incorporation is depicted below each lane. The analysed regions from XDP patients included PFC (n = 7), CB (n = 7), AP (n = 6), CN (n = 5), ST (n = 1), SN (n = 1), MC (n = 1), PG (n = 1), eGP (n = 1), internal globus pallidus (iGP, n = 1), NC (NC, n = 1), WM (n = 1) and GS (n = 1). Control tissues included PFC (n = 3), MC (n = 1), CB (n = 2), AP (n = 1), CN (n = 3) and TH (n = 1). PSI from PFC for both controls and XDP samples were plotted in panel B. No statistical difference was observe between the two groups (Mann–Whitney test, p-value = 0.5273). SRRM4 expression level in PFC is shown in panel C and no statistically significant difference was observed between patients and controls (Mann–Whitney test, P-value = 0.6190).
The exonic structure of TAF1 3′-end is preserved in XDP brain specimens
The majority of the number of alternative splicing events for TAF1 are located at the 3′-part of the gene (9/13 events between exon 30 and 38).8,9,29,30 While only microexon 34′-containing isoforms display neuronal specificity,8,9 alternative exons 32′, 35b, 35′ and the skipping of exon 33 can also be detected in non-neuronal tissues.29,30 It is currently unclear how many TAF1 isoforms can be detected in the brain and what is their relative prevalence. In the light of the site of the XDP-SVA insertion, the characterization of 3′ exonic structures of TAF1 mRNAs in the brain could reveal an SVA-induced difference between XDP patients and controls.
To annotate the different composition of TAF1 mRNAs at the 3′-end, we applied Nanopore sequencing of single cDNA molecules to obtain long-reads crossing multiple splicing junctions. After defining the exon composition of different isoforms, the relative abundance of each structure was calculated as percentage to the total number of aligned isoforms. We designed a RT-PCR amplicon spanning from exon 30 to the last exon 38 to analyse stable TAF1 mRNAs and to exclude premature terminations. We first validated that Nanopore sequencing could accurately detect microexon 34′-containing TAF1 mRNAs (Supplementary Fig. S2). Next, Nanopore sequencing was applied to mRNAs from XDP and control brains, which revealed the presence of 17 different TAF1 exonic structures for PFC (Fig. 3A and Supplementary Table S3). Isoform usage was scored above the 1% cut-off value for constitutive and alternative exons reported in literature within the selected region, with the exception of exons 30b, 31b, 33′ and 35′. TAF1 mRNA pools could be stratified into three core scaffolds with mutually exclusive sequences. These include: canonical (#1), skipping of exon 33 (#2) and inclusion of 35b (#3, where exon 35 is extended to a downstream splice site). From these three scaffolds, TAF1 architecture includes the alternative exons 32′, 34′ and 35′, which are present singularly and in composite combinations. No novel junctions were identified above the cut-off value of 1%. When analysing control samples for PFC, microexon 34′ was incorporated within different exonic structures and canonical TAF1 + 34′ and canonical TAF1 + 34′ +35′ are the most abundant (Supplementary Fig. S4). Upon comparison of TAF1 structures containing microexon 34′ of patient versus PFC controls, no statistically significant difference was observed (drimseq test, P-value = 0.991) (Fig. 3B). The single data points for isoforms excluding (Fig. 3C) or including (Fig. 3D) microexon 34′ show a degree of variability within both control and XDP PFCs. Taken together, long-read sequencing data for TAF1 mRNAs highlight a complex exonic structure and the presence of the XDP-SVA element does not affect this distribution. Isoforms including microexon 34′ were the second most abundant species, which were similarly represented in XDP samples and controls. In conclusion, the Nanopore sequencing results support the conclusion that the XDP-SVA does not impact microexon 34′ incorporation into TAF1 mRNA.
Figure 3.

The exonic structure of TAF1 mRNA is preserved in XDP brains. The structure of the 10 most abundant TAF1 isoforms detected using long-read sequencing is depicted in panel A. Three different TAF1 scaffolds are indicated: 1 = canonical TAF1, including all canonical exons; 2 = skipping of exon 33, including all canonical exons but exon 33; 3=35b, in which all canonical TAF1 exons are present but exon 35 is substituted by 35b. The DTU estimating isoform proportion in the PFC of XDP versus controls is shown as percentage of the total mapped reads in panel B. The color-coding is adherent with the composition of the 10 most abundant isoforms depicted in panel A. The remaining isoforms are represented in grey scale. No significant difference was observed when comparing 34′-containing TAF1 mRNAs between XDP patients and controls (drimseq test, p-value = 0.991). Panels C and D represent single data points for the percentage of isoform excluding (panel C, exon34—exon35) or including (panel D, exon 34—exon 34’) microexon 34′ for each control or XDP sample.
Discussion
The molecular mechanism of XDP has eluded researchers in the field of neurodegeneration for a long time. The current paradigm for XDP pathogenesis is that the neuron-specific isoform TAF1-34′ is dramatically reduced in patients compared with controls, due to the presence of an SVA retrotransposon within intron 32 of the TAF1 gene.8,20 While evidence is accumulating for the SVA as the XDP disease-responsible element, data regarding microexon 34′ splicing are conflicting and the resulting molecular mechanism and XDP neuronal specificity is unclear.
In this study, we provide strong evidence against an involvement of reduced microexon 34′ splicing in XDP pathogenesis. Our findings mark a paradigm shift in the understanding of this neurological disorder. Using cell-based assays, we find that the presence of the XDP-SVA does not affect SRRM4-mediated inclusion of microexon 34′ into TAF1 mRNAs. Next, we demonstrate that microexon 34′ is included in TAF1 mRNAs isolated from different regions of XDP brains and that the exonic structure of TAF1 transcripts is preserved when compared with controls. Taken together, our data demonstrate that microexon 34′ is included into TAF1 mRNAs in brain specimens irrespective of the presence of the XDP-SVA element. This provides strong evidence that the XDP-SVA does not impact this neuron-specific splicing event. The validation of mRNA studies with the analysis of TAF1-34′ protein expression pattern in XDP brain would further corroborate this result. Our data indicate that alterations in splicing of this alternative microexon is not a signature for XDP pathology, shifting XDP research and possible future therapeutic interventions from microexon 34′ splicing to other SVA-dependent pathomechanisms.
It is important to note that the analysis was performed using bulk tissues and does not account for differential cell type composition in the investigated tissues. From the analysis of available single cell sequencing data, we observed mutually exclusive usage of canonical or 34′ exon junctions, with only a minority of cells showing both transcripts (Supplementary Fig. S5). Based on this evidence, the analysis of bulk tissues could mask more subtle variations in microexon 34′ incorporation caused by a potential decrease in the fraction of TAF1 microexon 34′ expressing neurons in XDP brains. In addition, the hexamer repeat in the XDP-SVA has displayed mosaicism in different brain regions11; therefore, a detailed characterization of Nanopore sequencing combined with the specification of hexamer repeat length at single cell level could dissect this possibility. In addition, a possible limitation of our study relates to the age difference between XDP samples and controls (mean age XDP samples: 47.4 years [SD: ±10.01], mean age control samples: 72.3 years [SD: ±15.94]). To date, there is no evidence for specific SRRM4-dependent microexon regulation during ageing, but we cannot exclude that the process might affect the comparison of microexon 34′ PSI between the two groups.
While our data suggest that the presence of the XDP-SVA element per se does not constitute a direct obstruction to microexon 34′ incorporation, it remains possible that a decrease in TAF1-34′ mRNAs is a secondary process of XDP pathogenesis. It has been proposed that the XDP-SVA can form G-quadruplexes,10 which might represent an obstacle for transcribing RNA polymerase II by slowing transcription elongation and/or promoting premature termination. This scenario would be supported by the fact that there is a reduced expression level of TAF1 exons downstream of the SVA in XDP-derived cell systems.9 A working hypothesis could include the possibility that only a minority of RNA polymerase II molecules is able to transcribe the TAF1 gene beyond the XDP-SVA element in intron 32. This would reduce the transcription of downstream exons, which includes the neuron-specific microexon 34′. Additional studies focused on the transcriptional and epigenetic environment across the XDP-SVA are needed to examine this mechanism further.
Together with providing key evidence for XDP research, our study also characterizes for the first time the distribution of TAF1 isoforms in the brain. With the application of Nanopore sequencing technology to XDP and control brains it was possible to reveal the complex neuronal architecture of TAF1 transcripts spanning exon 30 to exon 38. These data show that microexon 34′ is abundant in TAF1 mRNAs and it is present in at least four different transcripts. This re-defines the concept of canonical TAF1 versus TAF1-34′, for which the latter was solely including microexon 34′ in the canonical mRNA.8 Based on this, it will be very intriguing to investigate the function of brain-specific TAF1 isoforms containing microexon 34′. Microexons are known to activate tissue-specific protein interaction networks, therefore modulating protein functions17 and possibly shaping cell type identity. Recently, it has been proposed that XDP and HD share a common molecular denominator, converging to TAF1 decreased expression. The proposed mechanism for HD implicates the Serine and arginine Rich Splicing Factor 6 (SRSF6), which becomes sequestered in HD-inclusion bodies.15 Interestingly, SRSF6 protein interacts with SRRM4.19 Determining whether TAF1 downregulation contributes to the pathogenesis of HD or is rather a consequence remains to be elucidated. Framing the presented data in the light of the communalities between these two monogenic striatal disorders, it would be relevant to expand the investigation of TAF1 exonic structures and SRRM4 contribution to HD. These data would open a possible scenario where different TAF1 alternative splicing events could be linked to striatal degeneration, making TAF1 a central hub for basal ganglia pathology.
Supplementary Material
Acknowledgements
We are deeply grateful to the CCXDP for their financial support and their inspiring support to understand this complex and devastating disease. We would like to thank the CCXDP biobank, Imke Wyers, MD, Norbert Brüggemann, MD, and Charles Reyes, MS, for their assistance with brain preparation. Furthermore, we would like to thank Dr Omid Fotouhi for critical reading of the manuscript and all the members of the Timmers group for helpful discussions and suggestions. At last, we would like to thank the XDP patients and their families for their participation in the consortium activities and research.
Funding
The presented work was supported by the Collaborative Center for X-linked Dystonia-Parkinsonism (H.T.M.T., N.S. and S.C.), the Deutsche Forschungsgemeinschaft (DFG—FOR 2488: K.G. and C.K.; DFG—192904750-SFB 992: H.T.M.T.) and the Stichting Parkinson Fonds (M.P.C., M.W.V. and B.C.).
Supplementary Material
Supplementary material is available at Brain Communications online.
Competing interest
The authors report no competing interests.
Glossary
- AP =
anterior putamen
- CB =
cerebellum
- CCXDP =
Center for X-linked dystonia parkinsonism
- CN =
caudate nuclei
- DTU =
differential transcript usage
- eGP =
external globus pallidus
- GS =
grey substances
- HD =
Huntington’s disease
- iGP =
internal glubus pallidus
- MC =
motor cortex
- NC =
nucleus
- NSCs =
neural stem cells
- PG =
pituitary gland
- PFC =
pre-frontal cortex
- PSI =
Percent-Spliced-In
- SN =
substantia nigra
- ST =
striatum
- SVA =
SINE-VNTR-Alu retrotransposon
- TH =
thalamus
- WM =
white matter
- XDP =
X-linked dystonia parkinsonism
References
- 1. Lee LV, Pascasio FM, Fuentes FD, Viterbo GH.. Torsion dystonia in Panay, Philippines. Adv Neurol. 1976;14:137–151. [PubMed] [Google Scholar]
- 2. Lee LV, Rivera C, Teleg RA, et al. The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”). Int J Neurosci. 2011;121(Suppl 1):3–11. [DOI] [PubMed] [Google Scholar]
- 3. Pauly MG, Ruiz Lopez M, Westenberger A, et al. Expanding data collection for the MDSGene database: X-linked dystonia–parkinsonism as use case example. Mov Disord. 2020;35(11):1933–1938. [DOI] [PubMed] [Google Scholar]
- 4. Jamora RD, Ledesma LK, Domingo A, Cenina AR, Lee LV.. Nonmotor features in sex-linked dystonia parkinsonism. Neurodegener Dis Manag. 2014;4(3):283–289. [DOI] [PubMed] [Google Scholar]
- 5. Goto S, Lee LV, Munoz EL, et al. Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann Neurol. 2005;58(1):7–17. [DOI] [PubMed] [Google Scholar]
- 6. Bruggemann N, Heldmann M, Klein C, et al. Neuroanatomical changes extend beyond striatal atrophy in X-linked dystonia parkinsonism. Parkinsonism Relat Disord. 2016;31:91–97. [DOI] [PubMed] [Google Scholar]
- 7. Hanssen H, Heldmann M, Prasuhn J, et al. Basal ganglia and cerebellar pathology in X-linked dystonia–parkinsonism. Brain. 2018;141(10):2995–3008. [DOI] [PubMed] [Google Scholar]
- 8. Makino S, Kaji R, Ando S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80(3):393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aneichyk T, Hendriks WT, Yadav R, et al. Dissecting the causal mechanism of X-linked dystonia-parkinsonism by integrating genome and transcriptome assembly. Cell. 2018;172(5):897–909.e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bragg DC, Mangkalaphiban K, Vaine CA, et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci USA. 2017;114(51):E11020–E11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westenberger A, Reyes CJ, Saranza G, et al. A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann Neurol. 2019;85(6):812–822. [DOI] [PubMed] [Google Scholar]
- 12. Rakovic A, Domingo A, Grutz K, et al. Genome editing in induced pluripotent stem cells rescues TAF1 levels in X-linked dystonia-parkinsonism. Mov Disord. 2018;33(7):1108–1118. [DOI] [PubMed] [Google Scholar]
- 13. O'Rawe JA, Wu Y, Dorfel MJ, et al. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. 2015;97(6):922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng H, Capponi S, Wakeling E, et al. Missense variants in TAF1 and developmental phenotypes: Challenges of determining pathogenicity. Hum Mutat. 2019;41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hernandez IH, Cabrera JR, Santos-Galindo M, et al. Pathogenic SREK1 decrease in Huntington's disease lowers TAF1 mimicking X-linked dystonia parkinsonism. Brain. 2020;143(7):2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhuiyan T, Timmers HTM.. Promoter recognition: Putting TFIID on the spot. Trends Cell Biol. 2019;29(9):752–763. [DOI] [PubMed] [Google Scholar]
- 17. Irimia M, Weatheritt RJ, Ellis JD, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159(7):1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Calarco JA, Superina S, O’Hanlon D, et al. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138(5):898–910. [DOI] [PubMed] [Google Scholar]
- 19. Capponi S, Stoffler N, Irimia M, et al. Neuronal-specific microexon splicing of TAF1 mRNA is directly regulated by SRRM4/nSR100. RNA Biol. 2020;17(1):62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ito N, Hendriks WT, Dhakal J, et al. Decreased N-TAF1 expression in X-linked dystonia-parkinsonism patient-specific neural stem cells. Dis Model Mech. 2016;9(4):451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandez-Cerado C, Legarda GP, Velasco-Andrada MS, et al. Promise and challenges of dystonia brain banking: Establishing a human tissue repository for studies of X-linked dystonia-parkinsonism. J Neural Transm (Vienna). 2021;128(4):575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reyes CJ, Schaake S, Lüth T, et al. Brain regional differences in hexanucleotide repeat length in X-linked dystonia-parkinsonism using nanopore sequencing. Neurol Genet. 2021;7(4):e608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fleige S, Pfaffl MW.. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med. 2006;27(2-3):126–139. [DOI] [PubMed] [Google Scholar]
- 24. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP.. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–515. [DOI] [PubMed] [Google Scholar]
- 25. Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang AD, Soulette CM, van Baren MJ, et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun. 2020;11(1):1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nowicka M, Robinson MD.. DRIMSeq: A Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Res. 2016;5:1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quesnel-Vallieres M, Irimia M, Cordes SP, Blencowe BJ.. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015;29(7):746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nolte D, Niemann S, Muller U.. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci USA. 2003;100(18):10347–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herzfeld T, Nolte D, Muller U.. Structural and functional analysis of the human TAF1/DYT3 multiple transcript system. Mamma Genome. 2007;18(11):787–795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Nanopore long-read sequencing data have been deposited in the NIH Biosample database under the submission ID PRJNA666449.