

SUMMARY
Epigenetic mechanisms contribute to the regulation of cell differentiation and function. Vascular smooth muscle cells (SMCs) are specialized contractile cells that retain phenotypic plasticity even after differentiation. Here, by performing selective demethylation of histone H3 lysine 4 di-methylation (H3K4me2) at SMC-specific genes, we uncovered that H3K4me2 governs SMC lineage identity. Removal of H3K4me2 via selective editing in cultured vascular SMCs and in murine arterial vasculature led to loss of differentiation and reduced contractility due to impaired recruitment of the DNA methylcytosine dioxygenase TET2. H3K4me2 editing altered SMC adaptative capacities during vascular remodeling due to loss of miR-145 expression. Finally, H3K4me2 editing induced a profound alteration of SMC lineage identity by redistributing H3K4me2 towards genes associated with stemness and developmental programs, thus exacerbating plasticity. Our studies identify the H3K4me2-TET2-miR145 axis as a central epigenetic memory mechanism controlling cell identity and function, whose alteration could contribute to various pathophysiological processes.
Keywords: epigenetics, gene regulation, cell differentiation, histone modifications, DNA methylation, microRNA, vascular injury
Graphical Abstract
eTOC Blurb (In brief):
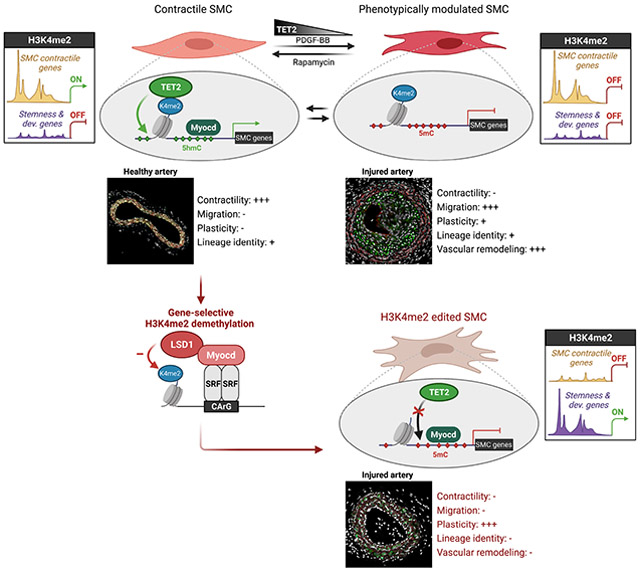
Regulating the contractile states of smooth muscle cells (SMCs) is critical for vascular homeostasis. Liu et al. find that the H3K4me2 modification is required to maintain SMC lineage identity and contractile state, serving as a genomic footprint for TET2-mediated DNA demethylation. H3K4me2 alteration in SMCs may contribute to cardiovascular disease.
INTRODUCTION
Unlike adult cardiac and skeletal myocytes, which are terminally differentiated, adult SMCs retain remarkable plasticity in response to acute and chronic changes in their environment (Owens, 1995; 2007). Recent fate mapping and transcriptomic studies have provided compelling evidence of SMC phenotypic modulation and the complex fate of this cell type in vascular diseases. In the context of major vascular diseases including atherosclerosis (Alencar et al., 2020; Pan et al., 2020; Wirka et al., 2019) and aortic aneurysm (Chen et al., 2020; Pedroza et al., 2020), SMCs profoundly alter their specialized phenotype by losing the expression of their contractile gene repertoire including smooth muscle α-actin (ACTA2) and smooth muscle myosin heavy chain (MYH11). Moreover, SMCs partition into multiple transcriptionally distinct subpopulations and can acquire the expression of genes associated with alternative cell types and functions like osteochondrogenic cells (RUNX2, SOX9), phagocytic cells (LGALS3, CD68), or mesenchymal stem cells (SCA1) (Dobnikar et al., 2018; Shankman et al., 2015). Although consensus regarding a definitive categorization of SMC populations has not been reached, these studies have markedly enhanced our understanding of SMC phenotypic diversity and plasticity (Liu and Gomez, 2019).
It is less appreciated that, besides its implication in vascular disease, SMC phenotypic modulation is an evolutionarily conserved mechanism under physiological conditions, which plays a key role in the efficient adaptation of SMCs to changes in environmental cues driving vascular network development, growth, and remodeling as well as maintenance of vascular homeostasis (e.g., extracellular matrix production in response to increased mechanical strain) (Owens, 2007; Ross and Klebanoff, 1971). This adaptive vascular remodeling implies that: 1) SMCs transiently decrease the expression of contractile genes, 2) activate genes associated with cell proliferation, migration, or extracellular matrix synthesis, and 3) re-differentiate into contractile SMCs and re-express their SMC marker genes. This process of reversible dedifferentiation has been well characterized in vitro. SMCs transiently exposed to Platelet Derived Growth Factor-BB (PDGF-BB), a potent inducer of SMC dedifferentiation, fully regain expression of the SMC contractile genes Acta2 and Myh11 upon removal of the growth factor (McDonald et al., 2006). In vivo, SMC-lineage tracing studies identified populations of SMCs with a differentiated phenotype (i.e., ACTA2+, MYH11+) after their investment in atherosclerotic lesions (Misra et al., 2018; Shankman et al., 2015), or pulmonary capillaries (Sheikh et al., 2014; Sheikh et al., 2015), a process involving their dedifferentiation, proliferation, and migration. These studies support the concept of reversible dedifferentiation, reacquisition of SMC contractile gene expression, and retention of SMC lineage identity during vascular remodeling.
Such reversible modulation of SMC contractile gene expression requires tight yet dynamic regulation by master regulators responsible for gene activation or repression. It also implies the participation of stable lineage maintenance mechanisms granting retention of their lineage identity during transient dedifferentiation. Many transcriptional and epigenetic mechanisms have been implicated in the dynamic and specific regulation of the contractile gene repertoire in SMC (Alexander and Owens, 2012). The CArG box-SRF-myocardin complex has been identified as a central master regulator of the SMC contractile gene activation. Myocardin is a potent myogenic coactivator exclusively expressed in SMC and cardiomyocytes (Chen et al., 2002; Du et al., 2003; Wang et al., 2001). Myocardin functions by forming a complex with SRF (Serum Response Factor) recruited to the cis-element CArG box CC(A/T)6GG commonly present in SMC marker genes including Acta2, Myh11, Tagln, and Cnn1. Myocardin is required for proper SMC differentiation during vascular development as demonstrated by in vivo loss-of-function studies (Huang et al., 2008; Li et al., 2003). Myocardin also restrains SMC phenotypic plasticity by counteracting transcription factors promoting differentiation into other lineages, including Sox9 for chondrocyte differentiation (Xu et al., 2012). Besides myocardin, the DNA methylcytosine dioxygenase TET2 promotes SMC differentiation by preventing DNA methylation at SMC contractile genes (Liu et al., 2013). Although such transcriptional or epigenetic control of SMC-specific gene expression has been extensively studied, factors responsible for upstream priming of these genes and maintenance of SMC lineage identity during reversible dedifferentiation have not been identified.
We have previously characterized the enrichment of Histone 3 Lysine 4 di-methylation (H3K4me2) on the CArG regions of myocardin-regulated SMC genes as a stable “epigenetic signature” of SMC lineage. This H3K4me2 epigenetic signature presents important characteristics (Gomez et al., 2013; McDonald et al., 2006). First, the enrichment of H3K4me2 on myocardin-regulated genes, including SMC gene markers Acta2, Myh11, and Tagln, is restricted to the SMC lineage. Second, the H3K4me2 epigenetic signature appears in SMC precursors during all trans retinoic acid (ATRA)-induced SMC differentiation. Third, H3K4me2 enrichment on the SMC contractile genes occurs independently of binding of the SRF-myocardin complex on CArG boxes. CArG box mutations in Acta2 promoter-enhancer transgenes did not prevent enrichment of H3K4me2. Fourth, the H3K4me2 signature is stably retained on poised SMC marker genes during SMC transient phenotypic modulation in vitro and in vivo. Collectively, these observations suggest that the presence of H3K4me2 on the SMC gene repertoire is an SMC-lineage specific epigenetic signature present on these genomic loci irrespective of gene activation status. Given H3K4me2 signature lineage selectivity and stability, it appears that H3K4me2 does not act as a conventional activating histone modification but instead provides a mechanism for the maintenance of lineage identity during reversible dedifferentiation (Gomez et al., 2015).
Contemporary to the development of the “histone code” paradigm (Jenuwein and Allis, 2001), the concepts of epigenetic control of lineage determination and maintenance of lineage epigenetic memory have been proposed (Cheedipudi et al., 2014; Reik, 2007; Zhu et al., 2013). In support of these concepts, several reports have shown that H3K4me2 is preferentially distributed on genes related to lineage identity and cell-specific functions in multiple cell types and tissues (Pekowska et al., 2010; Popova et al., 2012; Zhang et al., 2012). Cardiac-specific knockout (KO) of the H3K4 methyltransferase KMT2D led to an average decrease in H3K4me2 levels at promoter regions and functional defects during cardiac development (Ang et al., 2016). Together, these studies indicate that H3K4me2 might play a central role in cell differentiation and lineage-specific epigenetic programming. However, they also present experimental limitations precluding the direct and unambiguous investigation of H3K4me2 functions (Gomez et al., 2015). First, the comparison of genome-wide epigenetic landscape by ChIP sequencing is inherently descriptive and does not inform on the causality between histone modification enrichment, gene expression and cellular phenotype. Second, global genetic or pharmacologic inhibition of histone-modifying enzymes is often difficult to interpret due to the general lack of specificity of these enzymes for a single histone residue and their impact on a vast number of genomic loci. Thus, despite assumptions to the contrary, characterization of the role of H3K4me2 is lacking and there are critical unresolved questions: What is the role of H3K4me2 in regulating vascular SMC differentiated state? Does H3K4me2 serve as a mechanism for maintenance of SMC lineage identity? To address these questions, we performed gene-selective H3K4me2 loss-of-function studies by inducing demethylation of H3K4me2 selectively on myocardin-dependent genes in SMC. H3K4me2 editing on this gene repertoire led to a profound loss of SMC identity and contractile function in vitro and in vivo. Overall, our studies demonstrate a central role of H3K4me2 for maintenance of an epigenetically controlled lineage identity in adult cells.
RESULTS
Selective demethylation of H3K4me2 on Myocardin-regulated genes
To assess the functional relevance of H3K4me2 in controlling SMC phenotype and function, we developed a targeted epigenome editing strategy consisting of gene-specific demethylation of H3K4me2 on the family of myocardin-dependent gene repertoire. We engineered a fusion protein linking the nuclear localization signals and the SRF binding domain of myocardin and the Lysine Specific Demethylase 1 (LSD1) to serve as recruitment and catalytic subunits, respectively (Figure 1A). The myocardin fragment excluded the Leucine zipper and transactivation domains. This design allows for the selective binding of Myocd-LSD1 to the large family of CArG/SRF/Myocardin-regulated genes characteristic of differentiated SMC while preventing cytoplasmic sequestration and myocardin-dependent transactivation (Pipes et al., 2006). To control for potential H3K4me2 demethylation-independent effects of Myocd-LSD1, we designed a non-functional editing system (Myocd-LSD1NF) containing the catalytically inactive LSD1K661A (Figure 1A) (Lee et al., 2005). We generated rat aortic SMCs stably expressing Myocd-LSD1 and Myocd-LSD1NF (Figure S1A). Myocd-LSD1 and Myocd-LSD1NF were recruited to the CArG box regions of myocardin-regulated genes including the SMC contractile genes Acta2, Tagln and Myh11 (Figures 1B and S1B). We measured a significant decrease in H3K4me2 levels on these genes in SMCs expressing Myocd-LSD1 specifically while no noticeable effect was observed in SMCs expressing Myocd-LSD1NF, demonstrating that Myocd-LSD1-induced H3K4me2 demethylation is dependent on LSD1 demethylase activity. Importantly, there was no Myocd-LSD1 recruitment or H3K4me2 alteration on CArG/SRF-dependent but myocardin-independent early response genes (Figures 1B and S1B).
Figure 1: Myocd-LSD1 selectively performs H3K4me2 demethylation on myocardin-regulated SMC gene repertoire.

A. Schematic representation of Myocd-LSD1-mediated H3K4me2 editing. Functional (Myocd-LSD1) and non-functional (Myocd-LSD1K661A or Myocd-LSD1NF) constructs of Myocd-LSD1 were generated, both containing a Flag sequence (F) in the linker region. B. Myocd-LSD1/LSD1NF and H3K4me2 enrichment on CArG box regions of Acta2, Tagln and cfos in control non-transduced SMC, and SMC transduced with either Myocd-LSD1 or Myocd-LSD1NF. C. H3K4me2 and Myocd-LSD1 CUT&Tag sequencing tracks in CArG box regions of Acta2, Tagln and cfos in control, Myocd-LSD1, and Myocd-LSD1NF SMC. D. Heatmap of H3K4me2 enrichment around gene transcription start sites (TSS). E. Top 20 GO pathways enriched in genes with significant loss of H3K4me2 in Myocd-LSD1 SMC vs Myocd-LSD1NF SMC. F. H3K4me1 and H3K4me3 enrichment on CArG box regions of Myh11 and Acta2 in control, Myocd-LSD1 and Myocd-LSD1NF SMC. G. Measurement of SRF and KLF4 binding on CArG box region of Myh11 in control, Myocd-LSD1 and Myocd-LSD1NF SMC. H. Luciferase activity assay using Acta2 promoter-enhancer construct with intact (Acta2-CArGWT) or mutated CArG boxes (Acta2-promoCArGMut and Acta2-promo+intCArGMut). Data are represented as mean ± s.e.m of 3-5 independent experiments. Groups were compared by One-Way ANOVA or Two-way ANOVA. * p<0.05, ** and ## p<0.001, *** p<0.0001.
Genome-wide evaluation of H3K4me2 distribution and Myocd-LSD1 occupancy confirmed the concomitant binding of Myocd-LSD1 and the loss of H3K4me2 on myocardin-regulated contractile genes in Myocd-LSD1 SMC specifically, while no difference was observed on cfos or the myocardin-independent contractile gene Smtn (Figures 1C and S1C). Importantly, H3K4me2 abundance was not altered in Mycod-LSD1 SMC compared to control and Myocd-LSD1NF SMC (Figures 1D and S1D). However, we identified subsets of genes with either statistically higher or lower H3K4me2 enrichment, demonstrating that Myocd-LSD1 induces a redistribution of H3K4me2 (Figures 1E and 1F). Loci with lower H3K4me2 were located in gene promoters and 1st introns, consistent with CArG box location (Figure S1G) (Sun et al., 2006). By comparing H3K4me2 and Myocd-LSD1 CUT&Tag sequencing, we found that more than 75% of the genes with lower H3K4me2 contained Myocd-LSD1 peaks (Figure S1H). Gene Ontology (GO) Pathway analysis revealed that genes with lower H3K4me2 abundance in Myocd-LSD1 SMC participate in biological processes related to vascular development, SMC differentiation and SMC function demonstrating the demethylation of H3K4me2 on key SMC gene repertoire by Myocd-LSD1 (Figures 1E and S1I).
LSD1 induces demethylation of H3K4me2, as well as H3K4me1 and H3K9me. Expression of Myocd-LSD1 was not associated with differences in activating (H3K4me3, panH3ac) or repressive histone modifications (H3K9me3, H3K27me3), as well as expression of epigenetic modifiers (Figures 1F, S2A, S2B, and S2C). We observed an increase in H3K4me1 in Myocd-LSD1 SMCs which can be due to the active H3K4me2 demethylation (Figure 1F). Expression of Myocd-LSD1 did not impact the expression, recruitment, or transcriptional activity of the endogenous SRF-myocardin complex (Figures 1G, 1H, S2D, and S2E). Moreover, we found that more than 50% of genes with loss of H3K4me2 in Myocd-LSD1 SMC were those with enrichment in SRF binding (Figure S1J) (Yao et al., 2018). Analysis of these intersected genes was enriched in pathways associated with SMC differentiation and function (Figure S1K). Overall, these data demonstrate the specificity and efficiency of Myocd-LSD1 in performing targeted H3K4me2 demethylation on myocardin-regulated genes and validate our strategy to address the biological relevance of H3K4me2 in SMC.
H3K4me2 editing impairs SMC contractile function
SMC expressing Myocd-LSD1 presented a marked decrease in expression of the myocardin-regulated contractile genes at the transcript and protein levels (Figures 2A, 2B, 2C, and 2D). These alterations in SMC contractile gene expression were observed specifically in Myocd-LSD1 SMC as compared to Myocd-LSD1NF and control SMCs. Moreover, Myocd-LSD1 SMC presented a defect in contractility in 3D-collagen culture (Figures 2E, S2F, and S2G). SMC contractility ex vivo in response to potassium was decreased by 25-30% in vessels transduced with Myocd-LSD1 (Figure 2F). Interestingly, overexpression of full-length myocardin in Myocd-LSD1 infected SMCs failed to rescue expression of contractile genes (Figure 2G). Similarly, rapamycin, a potent SMC differentiation inducer, did not increase SMC contractile gene expression or SMC contractility (Figures 2H and 2I). Finally, Myocd-LSD1 SMCs failed to re-differentiate to a contractile state after transient PDGF-BB treatment (Figure S2H).
Figure 2: H3K4me2 editing induces loss of SMC contractility in vitro.

A. Transcript expression of contractile genes in Myocd-LSD1 and Myocd-LSD1NF SMC. Expressed as fold change compared to control SMC. B. ACTA2 and DAPI immunofluorescent staining in Myocd-LSD1 and Myocd-LSD1NF SMC. Scale bar = 100 μm. C. SMC-related gene protein expression in control, Myocd-LSD1/LSD1NF SMC. D. Protein expression quantification normalized to GAPDH. E. Collagen contraction assay using control, Myocd-LSD1 and Myocd-LSD1NF SMC. Scale bar = 1mm. Measurement of diameter over time. F. Ex-vivo wire myography combined with potassium chloride infusion in thoracodorsal artery rings incubated with Myocd-LSD1 or control lentivirus for 48h (n=4 mice per group). G. mRNA expression of myocardin, Acta2 and Tagln in Myocd-LSD1 and Myocd-LSD1NF SMC transduced with Adv-GFP or Adv-Myocardin. H. Acta2 transcript expression in Myocd-LSD1 and Myocd-LSD1NF SMC treated with rapamycin (100 nM). I. Collagen contraction assay using control, Myocd-LSD1NF, Myocd-LSD1 and Myocd-LSD1 SMC treated with rapamycin (100 nM). Gel area variation at day 5 post-gelation normalized to baseline area. Scale bar = 1 mm. Data are represented as mean ± s.e.m of 3-6 independent experiments. Groups were compared by Student t-test, One-way ANOVA, Two-way ANOVA, or Mann-Whitney (F). * p<0.05, ** p<0.001, *** p<0.0001.
Next, we locally delivered lentivirus encoding Myocd-LSD1 or Myocd-LSD1NF on the right carotid arteries of Myh11 CreERT2-YFP mice by Pluronic gel application (Figure 3A). Upon tamoxifen treatment, Myh11+ SMC are permanently and specifically labeled with YFP. Consequently, SMC identity can be traced regardless of conventional SMC marker gene expression (e.g., ACTA2, MYH11), which could be altered upon H3K4me2 editing. Two weeks after virus delivery, Myocd-LSD1 was highly expressed in transduced carotid arteries and SMCs (Figures S3A, S3B, S3C, and S3D). While Myocd-LSD1 did not induce significant changes in vessel morphology (Figures S3E and S3F), a marked reduction in ACTA2 and MYH11 expression was observed in YFP+ SMCs (Figures 3B and 3C). Importantly, there was a direct association between expression of Myocd-LSD1 (mCherry+) and reduced ACTA2 expression (Figure 3D). Expression of Myocd-LSD1 was not associated with a change in SMC proliferation, survival, apoptosis rate (Figures S3G, S3H, S3I, and S3H). Our studies provide evidence that the presence of H3K4me2 on the CArG box regions of SMC contractile genes is essential for maintenance of SMC contractile phenotype and function in vitro and in vivo.
Figure 3: H3K4me2 editing impairs SMC contractile function in vivo.

A. Schematic of lentiviral-mediated H3K4me2 editing in vivo. B. Quantification YFP+, ACTA2+ and MYH11+ Integrated Optical Density (IOD) normalized to medial area in carotid cross-sections of Myh11-CreERT2 YFP mice locally infected with Myocd-LSD1 or Myocd-LSD1NF lentivirus (n=4 mice per group). C. YFP, ACTA2, and MYH11 staining in non-infected left carotid arteries (LCA), Myocd-LSD1 and Myocd-LSD1NF infected right carotid arteries (RCA). Scale bar = 100 μm. D. YFP, mCherry and Acta2 staining in non-infected left carotid or Myocd-LSD1 infected right carotid arteries. Scale bar = 100 μm. Data are represented as mean ± s.e.m of 4 independent biological replicates. Groups were compared by Student t-test (B). * p<0.05, ** p<0.001, *** p<0.0001.
H3K4me2 mediates TET2 recruitment on myocardin-regulated contractile genes
H3K4me2 enrichment on myocardin-regulated genes has been reported irrespective of their activation status (Gomez et al., 2013; McDonald et al., 2006) which suggests that H3K4me2 may not exhibit intrinsic gene activation functions but rather control transcriptional and epigenetic factor recruitment. We found that loss of H3K4me2 induced marked changes in DNA methylation. Myocd-LSD1 expression induced an increase in DNA methylation (5mC) and a simultaneous reduction in DNA hydroxymethylation (5hmC) on Acta2 and Myh11 CArG box promoter regions (Figure 4A). Correspondingly, we observed a striking loss of TET2 recruitment to these loci in Myocd-LSD1 SMC, while expression of TET2 was not altered (Figures 4B and S2C). It has been reported that TET2 promotes SMC differentiation by converting 5mC into 5hmC on SMC marker genes (Liu et al., 2013). Overexpression of TET2 rescued Acta2 and Myh11 expression in Myocd-LSD1 SMC, suggesting that H3K4me2 editing-mediated defect in TET2 recruitment causes loss of SMC contractile gene expression (Figure 4C).
Figure 4: H3K4me2 interacts with TET2 and is required for its recruitment on the SMC contractile genes.

A. 5mC and 5hmC levels on Myh11 and Acta2 CArG box regions in control, Myocd-LSD1, and Myocd-LSD1NF SMC. B. TET2 enrichment on Acta2 and Myh11 CArG box regions in Myocd-LSD1NF and Myocd-LSD1 SMC. C. Correlation between Myh11, Acta2 and Tet2 mRNA levels in Myocd-LSD1 SMC transduced with Adv-TET2. R2 is calculated by linear regression. D. Heatmap of HA-TET2 enrichment around transcription start sites (TSS) measured by CUT&Tag sequencing in control SMC and Myocd-LSD1 SMC transduced with Adv-HA-TET2. E. Visualization of HA-TET2 enrichment peaks on the Acta2 gene in control SMC and Myocd-LSD1 SMC. F. GO pathway analysis of genes with decreased TET2 enrichment in Myocd-LSD1. G. Venn diagrams of annotated genes with HA-TET2 enrichment and H3K4me2 enrichment in control SMC or Myocd-LSD1 SMC. H. Venn Diagram comparing H3K4me1, H3K4me2, H3K4me3, and 5hmC distribution in human SMC. I. Proximity Ligation Assay (PLA) detecting proximity between H3K4me2 and TET2. Scale bar = 25μm. J. Quantification of H3K4me2/TET2 PLA signals in control SMC, Myocd-LSD1NF, and Myocd-LSD1 SMC. K. HA-tagged TET2 and biotin-H3K4me2 peptide co-immunoprecipitation (IP). IP: anti-HA antibody or IgG control; Blotting: anti-biotin antibody. L. HA-tagged TET2 and biotin-H3K4me1/2/3 peptide co-immunoprecipitation. IP: anti-HA antibody; Blotting: anti-biotin antibody. M. Quantification of HA-TET2 and Biotin-H3K4me1/2/3 peptide co-immunoprecipitation. IP signals normalized with INPUT Biotin-H3K4me1/2/3 signals. Data are represented as mean ± s.e.m of 3-5 independent experiments. Groups were compared by One-Way ANOVA. * p<0.05, ** p<0.001, *** p<0.0001.
The mechanisms by which TET2 selectively binds to given genomic regions have not been fully characterized. Unlike the other TET enzyme isoforms, TET1 and TET3, TET2 lacks a defined CXXC DNA-binding domain to mediate its recruitment (Williams et al., 2011). By performing HA-tag CUT&Tag sequencing in control or Myocd-LSD1 SMCs expressing HA-TET2, we found that H3K4me2 editing was associated with a partial loss of TET2 recruitment in Myocd-LSD1 SMC (Figures 4D, S4A, and S4B), including in the Acta2 CArG box region (Figure 4E). Genes exhibiting a loss of TET2 occupancy in Myocd-LSD1 are associated with several biological processes related to SMC differentiation and functions (Figure 4F). Genome-wide, we found that 86.6% of TET2 occupancy overlapped with H3K4me2 in control SMCs, while the number of genes with TET2 and H3K4me2 co-distribution was reduced by more than 50% in Myocd-LSD1 SMCs (Figure 4G). We next examined the co-distribution between H3K4me2 and 5hmC in human SMC using publicly available H3K4me1/me2/me3 datasets (Figure 4H) (Consortium, 2012). 73% of genes enriched in 5hmC in human SMC also presented an enrichment for H3K4me2 (Figures 4H, S4C, and S4D). Remarkably, we found that 5hmC is preferentially distributed with H3K4me2 compared to H3K4me1 and H3K4me3 (Figure 4H, and S4D). While 73% of 5hmC-enriched genes overlapped with H3K4me2-enriched genes, the co-enrichment with 5hmC dropped to 36% and 51% for H3K4me1 or H3K4me3 respectively. If we analyze the co-distribution of 5hmC with only one type of H3K4 methylation, 13.4% of 5hmC distribution overlaps with genes enriched in H3K4me2 only, compared to 2.7% and 1% for H3K4me1 and H3K4me3 respectively.
Proximity Ligation Assay (PLA) showed the proximity between H3K4me2 and TET2 in cultured SMCs (Figures S4E, S4F, and S4G) and mouse aorta (Figure S4H). We found a decrease in the number of H3K4me2/TET2 interactions in Myocd-LSD1 SMCs, suggesting that a significant subset of these interactions occurs on myocardin-regulated genes impacted by Myocd-LSD1 (Figures 4I and 4J). We next performed co-immunoprecipitation (Co-IP) between biotinylated H3K4me2 peptides and nuclear extracts of 293T cells expressing HA-tagged TET2. We found that H3K4me2 was immunoprecipitated with HA-TET2 (Figures 4K and S4I). Co-IP with Biotin-H3K4me1, me2, or me3 peptides revealed a preferential interaction between TET2 and H3K4me2 (Figures 4L and 4M). Together, these data provide evidence that H3K4me2 serves as a hub for dynamic TET2 recruitment to SMC gene repertoire in SMC.
H3K4me2 editing induces profound transcriptomic changes consistent with loss of SMC lineage identity
Bulk RNAseq revealed profound changes in the transcriptomic profile of SMC expressing Myocd-LSD1 as compared to Myocd-LSD1NF (Figure 5A). GO pathway analysis showed that H3K4me2 editing impaired pathways associated with contractility, vascular development and differentiation (Figures 5B, S5A, and S5B). Down-regulated genes with loss of H3K4me2 enrichment (RNAseq/CUT&Tag seq comparison) were also associated with SMC differentiation and contraction (Figure 5C). There was significant enrichment in SRF binding motifs in genes downregulated in Myocd-LSD1 SMC, providing further evidence of the specificity of H3K4me2 editing (Figure S5C).
Figure 5: H3K4me2 editing is associated with a profound loss of SMC lineage identity.

Bulk RNAseq performed on control, Myocd-LSD1, Myocd-LSD1NF, and PDGF-BB-treated SMC. A. Volcano plot representing genes differentially expressed in Myocd-LSD1 vs. Myocd-LSD1NF SMC. B. GO Pathway analysis showing the most significantly downregulated pathways in Myocd-LSD1 vs. Myocd-LSD1NF SMC. C. GO pathway analysis on intersected gene sets with transcript downregulation and H3K4me2 loss in Myocd-LSD1 SMC vs Myocd-LSD1NF SMC. D. Principal component analysis. E. Venn diagram representing the overlap between differentially expressed genes in Myocd-LSD1 and PDGF-BB treated SMC. F. Heat map of differential transcript expression in Myocd-LSD1, PDGF-BB treated, Myocd-LSD1NF, and control SMC. G. Expression levels of SMC contractile genes (cluster 1, top graphs) and SMC master differentiation regulators (cluster 2, bottom graphs) in Myocd-LSD1 and PDGF-BB treated SMC. Expression normalized to the expression in pooled control and Myocd-LSD1NF SMC. H. Normalized transcript expression in control SMC transiently transfected with Myocd-LSD1 or control plasmid. I. Transcript expression in Myocd-LSD1 SMC treated with vehicle or all-trans retinoic acid (ATRA). J. ChIP-qPCR of H3K4me2 on CArG box region of Acta2, Tagln and Myh11 in Myocd-LSD1 SMC treated with vehicle or ATRA. Data are represented as mean ± s.e.m of 3-4 independent biological replicates. Groups were compared by multiple Student t-test. * p<0.05, ** p<0.001, *** p<0.0001.
We found remarkable differences in the transcriptomic profiles of Myocd-LSD1 SMC and SMC stimulated by PDGF-BB (Figures 5D, 5E, 5F, and S5E). Interestingly, while both groups showed comparable levels of SMC contractile gene downregulation, H3K4me2 edited SMC displayed a selective downregulation of regulators of SMC differentiation and lineage determination (e.g., Mef2c, Notch3, Gata6, Rbpms) suggesting that H3K4me2 editing causes a loss of SMC lineage identity (Figure 5G). Interestingly, transient expression of Myocd-LSD1 in SMC only induced downregulation of myocardin-dependent contractile genes while expression of Smtn (myocardin-independent gene) and SMC master regulators (Mef2c, Tet2) was unchanged (Figure 5H). This discrepancy suggests that constitutive H3K4me2 editing in Myocd-LSD1 expressing SMC may induce secondary downregulation of a cohort of genes responsible for maintenance of SMC lineage identity. Finally, we treated Myocd-LSD1 SMC with all-trans retinoic acid (ATRA), which has been widely used to induce SMC differentiation from SMC precursors, embryonic stem cells or iPS cells (Blank et al., 1995; Manabe and Owens, 2001; Shen et al., 2021; Spin et al., 2004). ATRA-treatment induced a significant recovery of early contractile genes (Acta2, Tagln) and Gata6 (Figure 5I). This recovery was associated with a gain in H3K4me2 enrichment on myocardin-regulated gene CArG box regions (Figure 5J).
H3K4me2 editing exacerbates SMC phenotypic plasticity
Remarkably, genes with gain of H3K4me2 enrichment in Myocd-LSD1 were associated with stemness, developmental programs, and lineage specification and differentiation pathways (Figure 6A). This result was confirmed at the transcript level with increased expression of genes involved in multiple lineages differentiation (Figures 6B and S6A). Interestingly, we found that H3K4me2 editing led to the upregulation of genes associated with atherosclerosis-related SMC plasticity and phenotypic modulation, including genes associated with synthetic SMC (s100a4, fn1), mesenchymal stem cells (Eng, Nt5e), phagocytic cells (Lgals3, CD68) and osteochondrogenic cells (Runx2, Sox9) (Figures 6C, S6B). Surprisingly, genes associated with SMC transition to an atheroprotective fibromyocyte state (lum, tnfrsf11b) were down-regulated in Myocd-LSD1 SMC (Figure 6C) (Wirka et al., 2019). Notably, the increase in expression of plasticity markers was significantly higher in Myocd-LSD1 SMC as compared to PDGF-BB treated SMC, supporting again a deeper loss of SMC lineage identity induced by H3K4me2 editing (Figure S6C).
Figure 6: Loss of H3K4me2 on myocardin-regulated genes exacerbates SMC plasticity.

A. Top20 Gene Ontology pathways of genes with enhanced H3K4me2 enrichment in Myocd-LSD1 SMC compared with Myocd-LSD1NF SMC. B. Significantly upregulated GO pathways related with lineage plasticity from RNAseq in Myocd-LSD1 vs Myocd-LSD1NF SMC. C. Expression of markers of SMC phenotypic transitions in Myocd-LSD1 SMC. D. Oil Red O staining in Myocd-LSD1 and Myocd-LSD1NF SMC treated with cholesterol (40 μg/ml) or vehicle for 48h and quantification of Oil Red O+ area normalized to cellular area. Arrow: example of intracellular Oil Red O+ lipid vacuoles. Scale bar: 100μm. E. FABP4 and lipid droplets staining (left) and quantification of FABP4+ SMC (right) in Myocd-LSD1 and Myocd-LSD1NF SMC cultured in adipogenic differentiation media. Scale bar: 100μm. F. Immunofluorescent staining with Aggrecan in SMC cultured in chondrogenic differentiation media. Scale bar: 100μm. Data are represented as mean ± s.e.m of 3-4 independent experiments. Groups were compared by One-Way ANOVA (D) and Student t-test (C, E). * p<0.05, ** p<0.001, *** p<0.0001.
When exposed to cholesterol, Myocd-LSD1 SMC exhibited higher lipid uptake capacities and increased expression of phagocytosis markers compared with Myocd-LSD1NF SMC (Figures 6D, S6D, and S6E). We then performed lineage differentiation assays on Myocd-LSD1 and Myocd-LSD1NF SMC by treatment with adipogenic and chondrogenic differentiation supplements. After 14 days of culture in adipogenic differentiation medium, 30% of Myocd-LSD1 SMC were positive for both AdipoRed (lipid droplets) and FABP4 (adipocyte marker) (Figure 6E). In contrast, the ability of Myocd-LSD1NF SMC to express FABP4 was extremely limited. We also found that Myocd-LSD1 SMC cells formed chondrogenic structures and expressed Aggrecan (chondrogenesis marker) after 28 days in differentiation media (Figure 6F). Together, our data show that H3K4me2 is a key regulator of SMC lineage identity and restrains phenotypic plasticity.
H3K4me2 editing impairs SMC participation in adaptive vascular remodeling
Based on the impact of H3K4me2 editing on SMC phenotype, we sought to investigate the functional consequences of alteration of H3K4me2 distribution on SMC participation in vascular remodeling. We combined local Myocd-LSD1 lentivirus delivery and unilateral ligation of the right carotid. Three weeks after injury, ligated vessels displayed high expression of Myocd-LSD1 constructs (Figure S7A). Surprisingly, a significant decrease in neointima formation in ligated carotids transduced with Myocd-LSD1 was observed (Figures 7A and 7B). SMC investment in the neointima was nearly abolished after Myocd-LSD1 delivery (Figures 7C and 7D). Mechanistically, we found that expression of Myocd-LSD1 in SMC induced loss of migration capacity in response to PDGF-BB (Figures 7E and 7F), despite unaltered PDGF-β receptor (PDGFβR) expression and PDGFβR-dependent downstream signaling cascade (Figures S7B, S7C, and S7D). Defective migration is rather due to disorganization of the SMC cytoskeleton network (including actin polymerization and Talin distribution) (Figure 7G). These results further demonstrate that H3K4me2 controls SMC contractility but also the intrinsic physiological properties of SMC adaptation in response to vascular injury.
Figure 7: H3K4me2 editing inhibits SMC investment in the neointima after vascular injury.

A. Mason staining of ligated carotid cross sections. Scale bar = 100 μm. B. Morphometric analysis of neointima and media area in Myocd-LSD1 and Myocd-LSD1NF infected carotids. N = 9-12 mice per group. C. Immunofluorescent staining for YFP, ACTA2, MYH11, and DAPI on cross-sections from ligated right carotids infected with Myocd-LSD1 or Myocd-LSD1NF. Scale bar = 100 μm. D. Percentage of neointimal lesion populated by YFP+ SMC. N = 9-12 mice per group. E. Scratch wound assay on Myocd-LSD1NF and Myocd-LSD1 SMC. Representative images at baseline or after 24h treatment with PDGF-BB (30 ng/ml). Scale bars: 100 μm. F. Quantification of SMC migration: percentage closure normalized to the wound area at baseline. G. Immunofluorescent staining of cytoskeleton components: F-actin (phalloidin), ACTA2, and Talin in Myocd-LSD1NF and Myocd-LSD1 SMC. Scale bar: 50 μm. Data are represented as mean ± s.e.m of 9-12 independent biological replicates. Groups were compared by unpaired Student t-test, Fisher’s exact test, or Two-Way ANOVA. * p<0.05, ** p<0.001, *** p<0.0001.
H3K4me2/TET2 complex impairment leads to miR145 repression and loss of miR145-dependent cytoskeleton dynamics.
Remarkably, the impaired participation of SMC in remodeling observed in Myocd-LSD1 transduced vessels is a feature shared with miR-145 deficient mice (Xin et al., 2009). We found that miR-145 was markedly downregulated in Myocd-LSD1 SMC compared to Myocd-LSD1NF and PDGF-BB treated counterparts (Figures 8A and S8A) and the expression of miR-145 target genes involved in plasticity and migration inhibition was increased (Figures 8B, S8B, and S8C). GO analysis on upregulated miR-145 target genes showed enrichment in several pathways associated with negative regulation of cell motility (Figure S8D). The increased expression of migration inhibitory genes (Srgap1, Ssh2, Sema3a) was specifically observed in H3K4me2-edited SMC, but not in PDGF-BB treated SMC (Figure 8C).
Figure 8: H3K4me2/TET2 complex impairment leads to miR145 repression and loss of miR145-dependent cytoskeleton dynamics.

A. miR145 expression in Myocd-LSD1 and PDGF-BB treated SMC (30 ng/ml, 24h). B. Expression of miR145 target genes associated with cell plasticity (yellow) and migration inhibition (orange) in Myocd-LSD1 SMC vs Myocd-LSD1NF SMC. C. Transcript levels of Srgap1, Ssh2, and Sema3a in Myocd-LSD1NF, Myocd-LSD1 SMC and SMC treated with PDGF-BB (normalized transcript counts). D. ChIPseq track showing H3K4me2 distribution on the miR143/miR145 gene cluster in human SMC. Source: ENCODE Project. E. CUT&Tag sequencing tracks for H3K4me2, HA-TET2 and Myocd-LSD1 occupancy at the miR143/miR145 gene in control and Myocd-LSD1 SMC. F. Enrichment of H3K4me2, TET2 and 5hmC level on miR145 CArG region Myocd-LSD1NF and Myocd-LSD1 SMC. G. Transwell assay using Myocd-LSD1 SMC transfected with miR-Control (10 nM) or miR-145 at different doses (1 nM, 3 nM, and 10 nM) for 24 hours. Scale bar = 0.5 mm. Data are represented as mean ± s.e.m of 3-4 independent biological replicates. Groups were compared by unpaired Student t-test or One-Way ANOVA. * p<0.05, ** p<0.001, *** p<0.0001.
The miR145 locus contains a distal functional CArG box (Xin et al., 2009). By analyzing ENCODE datasets, we found that the CArG region of the miR145/miR143 cluster was enriched in H3K4me2 in human SMC, suggesting that these genes could be primary targets of Myocd-LSD1 (Figure 8D). In line with this hypothesis, a marked loss of H3K4me2 enrichment, TET2 recruitment and decreased 5hmC level were observed at the miR145 CArG region in Myocd-LSD1 SMC (Figures 8E, 8F, and S8E). Interestingly, we observed a dose-dependent rescue of PDGF-BB-dependent migration in Myocd-LSD1 SMC after miR-145 overexpression (Figure 8G). Meanwhile, higher expression of miR-145 inhibited migration, which can be due to the loss of expression of other key miR-145 targets like KLF4, potent inducer of SMC migration (Pidkovka et al., 2007). Together, these results provide evidence for the regulation of miR-145 expression by the H3K4me2/TET2 complex and that loss of the SMC-specific H3K4me2 signature leads to miR-145-dependent disorganization of cytoskeleton dynamics, explaining, at least in part, the defect in participation in vascular remodeling of H3K4me2 edited SMC.
DISCUSSION
It has been widely assumed that H3K4 methylation is a mechanism of gene activation based on the correlation between the distribution of H3K4 methylated residues and actively transcribed genes (Buratowski and Kim, 2010; Sims et al., 2003). However, our studies and others provide evidence contradicting this paradigm. H3K4me2 enrichment was also found on transcriptionally poised and inactive genes, suggesting that presence of H3K4me2 is not sufficient to induce gene activation (McDonald et al., 2006; Orford et al., 2008; Schneider et al., 2004). Moreover, H3K4me2 enrichment on the SMC gene repertoire is stably retained during SMC dedifferentiation and repression of these genomic loci in vitro and in vivo (Gomez et al., 2013; McDonald et al., 2006; Shankman et al., 2015). The present functional studies support a model in which H3K4me2 serves as a lineage memory mechanism by priming lineage-specific genes for dynamic activation and ensuring appropriate gene re-expression after transient repression.
Our studies uncovered that H3K4me2 serves as a stable hub for selective recruitment of the methylcytosine dioxygenase TET2 on SMC gene repertoire. H3K4me2-mediated TET2 binding promotes activation of SMC pro-differentiation gene program via DNA demethylation and hydroxymethylation (Cakouros et al., 2019; Liu et al., 2013; Montagner et al., 2016). We found a strong genome-wide co-distribution of TET2 and H3K4me2 and a marked alteration of TET2 genome occupancy after H3K4me2-editing. The property of H3K4me2 in regulating factor recruitment may be broader since H3K4me2 distribution closely overlaps with transcription factor binding regions (Wang et al., 2014). TET2 recruitment may also be dependent on the cooperation with other factors. Indeed, studies have shown that TET2 also interacts with transcription factors such as SNIP1 (Chen et al., 2018), TFCP2L1, KLF4, C/EBPa (Sardina et al., 2018) and histone methylations, such as H3K36me2 (Yamagata and Kobayashi, 2017) in other cell types. Whether these mechanisms play a role in TET2-mediated SMC differentiation will need to be empirically evaluated. H3K4me2 mediates TET2 recruitment on SMC contractile genes and miR145 (Cordes et al., 2009; Xin et al., 2009; Zhong et al., 2018), highlighting the complexity and interdependency of several epigenetic mechanisms, namely, histone modifications, DNA methylation, and non-coding RNAs.
H3K4me2 appears as a key epigenetic mediator of SMC lineage identity maintenance. We hypothesize that this mechanism is required for retention of a transcriptional memory in SMC undergoing transient dedifferentiation and acts in concert with other mechanisms associated with this process (D'Urso and Brickner, 2014). For example, the histone variant H2A.Z mediates the subnuclear localization of genes to confer memory of previous activation and promote reactivation (Brickner et al., 2007). The establishment of lineage-specific DNA methylation patterns throughout the genome has also been implicated in cell differentiation and lineage commitment by determining the expression or repression of differentiation programs. Moreover, there is evidence that these methylated or demethylated DNA patterns are stably retained in adult cells and represent an epigenetic memory of the cell lineage origin (Kim and Costello, 2017). It has been shown, for example, that MSCs from different tissues and different origins present distinct DNA methylation patterns while similarly expressing MSC markers such as CD106, CD146 (de Almeida et al., 2016). Our data showed that H3K4me2 editing led to a profound loss of lineage identity, not only characterized by the repression of the SMC contractile genes, but also by the redistribution of H3K4me2. Loss of H3K4me2 in myocardin-regulated genes induces a gain of H3K4me2 on genes involved in embryonic development and differentiation programs associated with other lineages. This suggests that after “erasure” of the SMC lineage H3K4me2 signature, the cell regains poising and priming of other lineage-specific gene subsets for context-dependent de novo lineage determination. This hypothesis is consistent with the increased plasticity and the remarkable ability of H3K4me2 edited SMC to transdifferentiate into other lineages upon adequate treatment.
Importantly, the upstream mechanisms responsible for the acquisition of the H3K4me2 signature during vascular development remain to be identified. Several histone methyltransferases are potential candidates for performing H3K4 di-methylation (Hyun et al., 2017). In cardiomyocytes, the lysine methyltransferase, KMT2D, contributes to H3K4me2 appearance and is required for proper cardiac development (Ang et al., 2016). Yet, our understanding of H3K4me2 contribution during SMC differentiation and which histone modifiers “write” the H3K4me2 signature is remarkably limited. Our data obtained in adult SMC have also potential implications for cell reprogramming. Indeed, it raises the question of the persistence of H3K4me2 signature from the original cell type during induced pluripotent stem (iPS) cell reprogramming. Seminal studies have demonstrated that iPS cells retain, at least partially, the epigenetic memory of the original cell type and tissue (Kim et al., 2010). Conversely, one could wonder if reprogrammed iPS cells recapitulate stable epigenetic signatures of the desired somatic lineage. Further identification and functional characterization of stable lineage-specific epigenetic programming associated with targeted epigenome editing could increase cell reprogramming efficiency.
H3K4me2 signature on the SMC gene repertoire appears as a fundamental mechanism engraving SMC lineage identity and restraining SMC plasticity. Our unbiased transcriptional profiling revealed notable differences between SMC treated with PDGF-BB and SMC subjected to Myocd-LSD1-mediated chromatin editing. Moreover, H3K4me2 editing profoundly altered SMC participation in adaptive vascular remodeling in vivo. Interestingly, a recent study showed a similar lack of neointima formation in a model of wire-induced femoral artery injury in SMC-specific TET2 deficient mice (Ostriker et al., 2021). These findings motivate the reevaluation of central SMC biology paradigm. First, SMC phenotypic switching has classically been described as a dedifferentiation process due to the loss of expression of SMC contractile genes. However, our data and previous studies suggest that phenotypically modulated SMC (i.e., ACTA2− and MYH11−) do not lose their H3K4me2 epigenetic signature, nor their lineage identity (Gomez et al., 2013). Second, our results demonstrate that the SMC ability to undergo reversible loss of contractility is a fundamental and epigenetically programmed property of SMC. SMC reversible phenotypic modulation in response to modifications of the environment is an evolutionary-conserved process necessary for the maintenance of vascular homeostasis. H3K4me2 plays a key role in allowing this reversible loss of contractility.
We anticipate that H3K4me2 programming biases SMC phenotype and behavior in chronic vascular diseases by promoting the re-expression of the contractile gene repertoire and limiting SMC plasticity. For example, in atherosclerosis, we and others have reported that the H3K4me2 signature on contractile gene promoters was retained during SMC phenotypic modulation, including dedifferentiated SMC (ACTA2−), and SMC expressing phagocytosis markers (LGALS3+, CD68+) (Gomez et al., 2013; Shankman et al., 2015; Wang et al., 2019). Interestingly, there is evidence that SMC-derived foam cells have defective and limited phagocytosis capacities compared with their myeloid-derived counterparts (Vengrenyuk et al., 2015; Wirka et al., 2019). There is a possibility that retention of SMC-lineage H3K4me2 programming restrains the full transition to a phagocytic macrophage-like cell. It remains to be determined how loss of the H3K4me2 signature if occurring during development or progression of vascular diseases, would impact SMC fate and contribution to the disease. Epigenetic programming can be subject to alterations induced by various environmental influences relevant to cardiovascular disease pathogenesis, including metabolic disorders (Jufvas et al., 2013; Miao et al., 2007) and aging (Cheung et al., 2018; Zhang et al., 2018). Further investigation of the persistence of H3K4me2 signature in the context of vascular disease and comorbidity (aging, obesity) would greatly enhance our understanding of SMC behavior and offer new strategies to restore SMC functions and modulate SMC phenotype and fate in these pathological contexts.
Limitations of the study
Although our approach consisting of utilizing myocardin as a recruitment system for the H3K4me2 demethylase is proven to be effective for the selective and coordinate editing of the SMC lineage related genes, the striking lack of reliable tools (e.g., antibodies) to study endogenous myocardin leads to a possible partial understanding of myocardin recruitment mechanisms and target genes (Miano, 2015). Further analysis of Myocd-LSD1 and H3K4me2 CUT&Tag datasets could help identify new CArG-dependent or CArG-independent regulatory roles for Myocardin. Myocd-LSD1 potently induced the decrease in expression of a large cohort of SMC lineage-related genes, leading to loss of lineage identity and specialized functions. Interestingly, single mutations in some of these key genes have been associated with vascular diseases. For example, mutations in ACTA2 or MYH11 are associated with aortic dilation and SMC dysfunction (Guo et al., 2007; Zhu et al., 2006). Although impossible with our experimental system, it would be interesting to determine the minimal and core H3K4me2-enriched genes necessary for maintenance of the lineage identity.
Star Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Delphine Gomez (gomezd@pitt.edu).
Materials availability
All unique materials generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
RNA-seq and CUT&Tag-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contract upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-dimethyl-Histone H3 (Lys4) Antibody | Millipore | Cat# 07-030, RRID:AB_310342 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Recombinant Anti-Histone H3 (tri methyl K9) antibody | Abcam | Cat# ab176916, RRID:AB_2797591 |
| Anti-trimethyl-Histone H3 (Lys27) Antibody | MIllipore | Cat# 07-449, RRID:AB_310624 |
| SRF (H-300) antibody | Santa Cruz Biotechnology | Cat# sc-13029, RRID:AB_2302440 |
| GKLF (H-180) antibody | Santa Cruz Biotechnology | Cat# sc-20691, RRID:AB_669567 |
| Anti-Tet2 Antibody | Sigma-Aldrich | Cat# ABE364 |
| Anti-acetyl-Histone H3 Antibody | Millipore | Cat# 06-599, RRID:AB_2115283 |
| Rabbit IgG, polyclonal - Isotype Control (ChIP Grade) | Abcam | Cat# ab171870, RRID:AB_2687657 |
| Mouse IgG - Isotype Control | Abcam | Cat# ab37355, RRID:AB_2665484 |
| Histone H3K4me2 antibody (pAb) | Active Motif | 39141, RRID:AB_2614985 |
| ANTI-FLAG® antibody produced in rabbit | Sigma-Aldrich | Cat# F7425, RRID:AB_439687 |
| Anti-HA tag antibody - ChIP Grade | Abcam | Cat# ab9110, RRID:AB_307019 |
| 5-hydroxymethylcytosine (5-hmC) monoclonal antibody (mouse) | Diagenode | C15200200-50 |
| 5-methylcytosine (5-mC) Antibody - clone 33D3 | Diagenode | C15200081-100 |
| Monoclonal Anti-Actin, α-Smooth Muscle | Sigma-Aldrich | Cat# A2547, RRID:AB_476701 |
| Rat Anti-Smooth Muscle Myosin Heavy Chain (sml) Monoclonal Antibody, Unconjugated, Clone KM3669 | Kamiya Biomedical Company | Cat# MC-352, RRID:AB_1241986 |
| Anti-TAGLN/Transgelin antibody | Abcam | Cat# ab14106, RRID:AB_443021 |
| Anti-GAPDH antibody [6C5] - Loading Control | Abcam | Cat# ab8245, RRID:AB_2107448 |
| Anti-GAPDH antibody - Loading Control | Abcam | Cat# ab9485, RRID:AB_307275 |
| Anti-Histone H3 antibody - Nuclear Marker and ChIP Grade | Abcam | Cat# ab1791, RRID:AB_302613 |
| H3K4me1 | Millipore | Cat# 07-436, RRID:AB_310614 |
| Anti-Dimethyl Histone H3 (Lys4) Antibody, clone CMA303 | Sigma Aldrich | Cat# 05-1338, RRID:AB_1977248 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4060, RRID:AB_2315049 |
| Akt1/2/3 Antibody (H-136) | Santa Cruz Biotechnology | Cat# sc-8312, RRID:AB_671714 |
| IRDye® 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat# 926-32211, RRID:AB_621843 |
| IRDye® 680RD Donkey anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat# 926-68072, RRID:AB_10953628 |
| Anti-GFP antibody | Abcam | Cat# ab6673, RRID:AB_305643 |
| Anti-Ki67 antibody | Abcam | Cat# ab15580, RRID:AB_443209 |
| Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| DYKDDDDK Tag (D6W5B) Rabbit mAb | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| Anti-mCherry antibody | Abcam | Cat# ab167453, RRID:AB_2571870 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Molecular Probes | Cat# A-21447, RRID:AB_141844 |
| Donkey Anti-Rat IgG H&L (Alexa Fluor® 555) | Abcam | Cat# ab150154, RRID:AB_2813834 |
| Monoclonal Anti-Actin, α-Smooth Muscle - FITC antibody produced in mouse | Sigma-Aldrich | Cat# F3777, RRID:AB_476977 |
| Anti-Actin, α-Smooth Muscle - Cy3™ antibody, Mouse monoclonal | Sigma-Aldrich | Cat# C6198, RRID:AB_476856 |
| Anti-Tet2 antibody | Abcam | Cat# ab124297, RRID:AB_2722695 |
| Anti-Biotin antibody | Abcam | Cat# ab53494, RRID:AB_867860 |
| Bacterial and Virus Strains | ||
| Ad-m-MYOCD-GFP | Vector Biolabs | Cat# ADV-265349 |
| Ad-mTET2-HA | Applied Biological Materials | Cat# 465200540200 |
| Lenti-Myocd-LSD1 | This paper | N/A |
| Lenti-Myocd-LSD1NF | This paper | N/A |
| Biological Samples | ||
| Human Coronary Artery Smooth Muscle Cells | LONZA | Cat# CC-2583 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Corning® Fetal Bovine Serum, 500 mL, Premium, United States Origin | Corning | Cat# 35-015-CV |
| L-Glutamine (200 mM) | Gibco | Cat# 25030081 |
| L-Ascorbic acid | Sigma-Aldrich | Cat# A4403; CAS: 50-81-7 |
| apo-Transferrin human | Sigma-Aldrich | Cat# T5391; CAS: 11096-37-0 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261; CAS: 10102-18-8 |
| PDGF-BB human | Sigma-Aldrich | Cat# SRP3138 |
| Rapamycin from Streptomyces hygroscopicus | Sigma-Aldrich | Cat# R0395; CAS: 53123-88-9 |
| Retinoic acid | Sigma-Aldrich | Cat# R2625; CAS: 302-79-4 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat# L3000015 |
| Polybrene Infection / Transfection Reagent | Sigma-Aldrich | Cat# TR-1003-G |
| Recombinant Human EGF (Animal-Free) | Biolegend | Cat# 713008 |
| Recombinant Human FGF-basic (146 aa) (Animal-Free) | Biolegend | Cat# 713304 |
| Dynabeads™ Protein G for Immunoprecipitation | Invitrogen | Cat# 10004D |
| ACCUTASE™ Cell detachment solution | Stem cell technologies | Cat# 07922 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat# A25742 |
| mirVana® miRNA mimic miR-145-5p | ThermoFisher | Cat# MC11480 |
| mirVana® miRNA mimic negative control | ThermoFisher | Cat# 4464058 |
| Lipofectamine RNAiMax Reagent | ThermoFisher | Cat# 13778075 |
| Invitrogen™ CellLight™ Talin-GFP, BacMam 2.0 | Invitrogen | Cat# C10611 |
| Invitrogen™ Alexa Fluor™ 647 Phalloidin | Invitrogen | Cat# A22287 |
| PureCol® Type I Collagen Solution, 3 mg/ml (Bovine) | Advanced BioMatrix | Cat# 5005 |
| Duolink® In Situ PLA Probe anti-rabbit PLUS | Sigma-Aldrich | Cat# DUO92002 |
| Duolink® In Situ PLA Probe anti-mouse MINUS | Sigma-Aldrich | Cat# DUO92004 |
| Duolink® In Situ Detection reagents Orange | Sigma-Aldrich | Cat# DUO92007 |
| Duolink® In Situ mounting medium with DAPI | Sigma-Aldrich | Cat# DUO82040 |
| Histone H3K4me1 Peptide - biotinylated | Active Motif | Cat# 81040 |
| Histone H3K4me2 Peptide - biotinylated | Active Motif | Cat# 81041 |
| Histone H3K4me3 Peptide - biotinylated | Active Motif | Cat# 81042 |
| IRDye 800CW Streptavidin | LI-COR Biosciences | Cat# 926-32230 |
| Methyl-β-cyclodextrin (Cholesterol) | Sigma-Aldrich | Cat# C4555; CAS: 128446-36-6 |
| AdipoRed™ Assay Reagent | LONZA | Cat# PT-7009 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648; CAS: 10540-29-1 |
| Pluronic® F-127 | Sigma-Aldrich | Cat# P2443; CAS: 9003-11-6 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade, Ampoule 10 ML | Electron Microscopy Sciences | Cat# 15710 |
| Crystal violet solution | Sigma-Aldrich | Cat # V5265; CAS: 548-62-9 |
| Antigen Unmasking Solution, Citrate-Based | Vector Laboratories | Cat# H-3300 |
| Critical Commercial Assays | ||
| Qubit™ RNA BR Assay Kit | Invitrogen | Cat# Q10210 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| RNeasy FFPE Kit | Qiagen | Cat# 73504 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| TaqMan™ MicroRNA Reverse Transcription Kit | Applied Biosystems | Cat# 4366596 |
| Histone Extraction Kit | Abcam | Cat# ab113476 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat# 78833 |
| Oil Red O staining kit | Sigma-Aldrich | Cat# MAK194 |
| Rat Mesenchymal Stem Cell Functional Identification Kit | R&D Systems | Cat# SC020 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# 7570 |
| CUT&Tag-IT™ Assay Kit | Active Motif | Cat# 53160 |
| MeDIP kit | Diagenode | Cat# C02010010 |
| hMeDIP kit | Diagenode | Cat# C02010031 |
| Luciferase assay kit | Promega | Cat# E1500 |
| Deposited Data | ||
| H3K4me3 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR515PKY/ |
| VSMC_SRF | NCBI's Gene Expression Omnibus | GSM3069844 |
| H3K4me1 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR130IMV/ |
| H3K4 di-methylation controls smooth muscle cell lineage identity and vascular homeostasis | NCBI's Gene Expression Omnibus | GSE179220 |
| H3K4me2 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR783AXV/ |
| Experimental Models: Cell Lines | ||
| Human coronary artery smooth muscle cells (hCASMCs) | LONZA | Cat# CC-2583 |
| Experimental Models: Organisms/Strains | ||
| Mouse: YFP: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | (Srinivas et al., 2001) | MGI: J:80963 |
| Mouse: Myh11-CreERT2: B6.FVB-Tg(Myh11-cre/ERT2)1Soff/J | (Wirth et al., 2008a) | MGI: J:141641 |
| Oligonucleotides | ||
| See Supplementary Table 1 | ||
| Recombinant DNA | ||
| pLVX-Myocd-LSD1-IRES-mCherry | This paper | N/A |
| pLVX-Myocd-LSD1NF-IRES-mCherry | This paper | N/A |
| pGL3-Acta2-CArGWT | (Hendrix et al., 2005) | N/A |
| pGL3-Acta2-promoCArGMut | (Hendrix et al., 2005) | N/A |
| pGL3-Acta2-promo+intCArGMut | (Hendrix et al., 2005) | N/A |
| Software and Algorithms | ||
| Prism 9 | Graph Pad | https://www.graphpad.com/ |
| Adobe Illustrator 2021 | Adobe | https://www.adobe.com/products/illustrator.html |
| Fiji | http://fiji.sc | RRID:SCR_002285 |
| Office 365 | Microsoft | https://www.microsoft.com/en-us/microsoft-365 |
| Adobe Photoshop 2021 | Adobe | https://www.adobe.com/products/photoshop.html |
| R version 4.0.4 | http://www.r-project.org/ | RRID:SCR_001905 |
| Galaxy | http://galaxyproject.org/ | RRID:SCR_006281 |
| Bowtie2 | (Langmead and Salzberg, 2012) | RRID:SCR_016368 |
| Deeptools | (Ramirez et al., 2016) | RRID:SCR_016366 |
| Integrative Genomics Viewer | http://www.broadinstitute.org/igv/ | RRID:SCR_011793 |
| MACS | https://github.com/macs3-project/MACS | RRID:SCR_013291 |
| Diffind | http://bioconductor.org/packages/release/bioc/html/DiffBind.html | RRID:SCR_012918 |
| ChIPseeker | https://bioconductor.org/packages/ChIPseeker/ | RRID:SCR_021322 |
| ClusterProfiler | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | RRID:SCR_016884 |
| QIAGEN CLC Genomics Workbench | Qiagen | https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/informatics-and-data/analysis-and-visualization/clc-genomics-workbench/ |
| BaseSpace Correlation Engine | Illumina | https://www.illumina.com/products/by-type/informatics-products/basespace-correlation-engine.html |
| Targetscan | http://targetscan.org/ | RRID:SCR_010845 |
| Ocular Advanced Scientific Camera Control software | Digital Opitcs Limited | https://www.photometrics.com/products/ocular |
| Image Pro Premier | Media Cybernetics | https://www.mediacy.com/support/imagepropremier |
| Other | ||
| Multi Wire Myograph System | DMT | Cat# 620M |
| LEICA Dmi8 Inverted Fluorescent Microscope | LEICA | https://www.leica-microsystems.com/products/light-microscopes/p/leica-dmi8-id/ |
| Nikon Instruments A1 Confocal Laser Microscope | Nikon | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/a1hd25-a1rhd25 |
| Synergy-HTX multi-mode reader | BioTek | https://www.biotek.com/products/detection-multi-mode-microplate-readers/synergy-htx-multi-mode-reader/ |
| 6.5 mm Transwell® with 8.0 μm Pore Polycarbonate Membrane Insert, Sterile | Corning | Cat# 3422 |
| Bioruptor® Pico sonication device | Diagenode | Cat# B01060010 |
| CFX Connect Realtime System | Bio-Rad | Cat# 1855201 |
| Odyssey® CLx Imaging System | LI-COR Biosciences | https://www.licor.com/bio/odyssey-dlx/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The animal protocols including all listed mouse strains and procedures were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Mice were housed and all experimental procedures were performed in an American Association for Accreditation of Laboratory Animal Care-accredited laboratory animal facility at University of Pittsburgh. All mice were housed in routinely sanitized cages at a controlled temperature (20 - 26°C) and humidity (50% - 60%) environment with a 12-hour light/dark cycle. Mice had unlimited access to standard rodent chow and water supply. Myh11-CreERT2 (Wirth et al., 2008b) and R26R-EYFP (Srinivas et al., 2001) were crossed to obtain Myh11-CreERT2-YFP lineage tracing mice as previously described (Gomez et al., 2013). All strains are on a C57BL/6J background and mice were genotyped by PCR. Male mice were exclusively used as experimental animals due to the location of the Myh11-CreERT2 transgene on the Y chromosome. At 6 weeks of age, male mice were injected daily with 1mg of tamoxifen in peanut oil (10 mg/mL). After 10 injections over the course of two weeks, mice recovered for one week to allow tamoxifen to be fully metabolized. Littermates received 106-7 lentiviral particles encoding for Myocd-LSD1 or Myocd-LSD1NF which were delivered unilaterally to the right carotid by application of Pluronic gel (20% Wt/Vol). Briefly, a 1 cm incision was made along the midline of the cervical region. The cranial bifurcation of the right carotid was exposed and separated from the surrounding fascia. Pluronic gel was applied directly to the right carotid, using retractors to keep the artery exposed while the gel solidified. A similar procedure was employed in association with permanent carotid ligation. In this case, the right carotid was ligated caudal to the bifurcation using 7-0 silk suture. Pluronic gel was then applied at the site of the ligation. 14- or 21-days following surgery, mice were euthanized by CO2 asphyxiation and perfused with PBS and 4% paraformaldehyde (PFA) using a gravity perfusion system via the left ventricle. Left and right carotid arteries, starting at the cranial bifurcation, were excised and fixed overnight in 4% PFA at 4°C. Tissues were processed and embedded in paraffin vertically and serial 10 μm sections were collected from the cranial bifurcation or the ligation site.
Cell culture
Male rat aortic smooth muscle cells constitutively expressing Myocd-LSD1 or Myocd-LSD1NF were generated by lentiviral transduction. Uninfected control SMC, Myocd-LSD1 and Myocd-LSD1NF SMC were routinely cultured in growth medium (DMEM:F12, Gibco, 11320-033) supplemented with fetal bovine serum (10%, Corning, 35-015-CV), L-glutamine (1.6 mM, Gibco, 25030081), and penicillin-streptomycin (100 U/mL, Gibco, 15140122) at 37°C with 5% CO2. Before stimulation and for all baseline measurements, SMC were starved in a serum-free, insulin-free medium supplemented with L-glutamine (1.6 mM, Gibco, 25030081), L-ascorbic acid (0.2 mM, Sigma Aldrich, A4403), Apo-Transferrin (5 μg/ml, Sigma Aldrich, T5391) and Na Selenite (6.25 ng/ml, Sigma-Aldrich, S5261) for 48-72 hours. SMC from 3-5 constitutive passages were used to repeat independent experiments. Human recombinant PDGF-BB (Sigma Aldrich, SRP3138) was reconstituted in 10 mM Acetic Acid at 30 ng/ml for treatment. Rapamycin (Sigma Aldrich, R0395) was reconstituted in DMSO at 100 nM for treatment. SMC were treated with PDGF-BB or Rapamycin for 24h before being harvested for analysis. All-trans Retinoic Acid (Sigma, R2625) was reconstituted in DMSO at 1 μM for treatment. Transient overexpression of Myocd-LSD1 was performed by transfecting pLVX-Myocd-LSD1 with Lipofectamine 3000 Transfection Reagent (Invitrogen, L3000015). Myocardin and TET2 overexpression was achieved by transduction of Ad-m-Myocd-GFP (Vector Biolabs, ADV-265349) or Ad-HA-mTET2 (Applied Biological Materials, 465200540200) adenovirus with 10% polybrene infection/transfection reagent (Millipore, TR1003) at 2x106 to 107IFU per well in 6-well-plates.
Human coronary artery smooth muscle cells (hCASMCs, Lonza) were propagated in M199 medium (Gibco, 11150-067) supplemented with 10% FBS, 100 U/ml each penicillin-streptomycin, 2.7 ng/ml rhEGF (Biolegend, 713008), and 2ng/ml rhFGF (Biolegend, 713034). Cells were starved in 2% serum medium for 24 hours before treatment with 50 nM rapamycin for 48 hours to enhance SMC differentiation.
METHOD DETAILS
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) was performed as previously described (Dahl and Collas, 2008; Gomez et al., 2013). In brief, passage-matched control, Myocd-LSD1, Myocd-LSD1NF SMC were fixed with 1% PFA for 10 min at room temperature. Cells were sonicated with a Bioruptor Pico (Diagenode) to obtain chromatin fragments of 200-500 base pairs. Chromatin was incubated with Protein G Dynabeads (Invitrogen, 10004D) and one of the following antibodies: H3K4me2 (2μg; #07-030, Millipore), Flag (4;g #F3165, Sigma Aldrich), H3K9me3 (2μg; ab176916, Abcam), H3K27me3 (2μg; 07-449, Sigma Aldrich), SRF (2μg; sc-13029, Santa Cruz), KLF4 (2μg; sc-20691, Santa Cruz), TET2 (5μg; ABE364, Millipore), H3ac (2μg; 06-599, Millipore), rabbit IgG (Abcam, ab171870) or mouse IgG (Abcam, ab37355). Genomic DNA was extracted with phenol-chloroform from immunoprecipitated (IP) and non-immunoprecipitated (INPUT) samples. Histone modification and protein enrichment was measured by qPCR using primer sets targeting CArG regions of the SMC contractile genes. Results were expressed as IP/INPUT. Primers used for ChIP-qPCR are listed in Key Resources Table.
CUT & Tag sequencing and data analysis
CUT&Tag seq (Kaya-Okur et al., 2020) was performed using CUT&Tag-IT™ Assay Kit (Active motif, 53160) following manufacturer’s protocol. Briefly, 5 x 105 cells were harvested using ACCUTASE™ cell detachment solution (Stem Cell Technologies, 07920). After binding with Concanavalin A beads, cells were incubated with primary antibody targeting H3K4me2 (Active motif, 39141), FLAG (Sigma Aldrich, F7425) or HA-tag (Abcam, ab9110) overnight at 4°C. Samples were then incubated with Guinea Pig anti-Rabbit antibody, followed by CUT&Tag-IT Assembled pA-Tn5 Transposomes. After tagmentation in tagmentation buffer at 37°C for 1 hour, released DNA was extracted by DNA purification column for library preparation and SPRI Beads clean-up. Paired-end sequencing was performed with illumina MiSeq (MS v2 50 cycle kit 12-15M reads) at Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh.
Paired-end raw reads were aligned to Rn6 using Bowtie2 (Langmead and Salzberg, 2012).Tracks were generated by deepTools2 Bamcoverage (Ramirez et al., 2016) with normalized counts as bins per million (BPM) and visualized with IGV (Robinson et al., 2011). Peaks were called using MACS2 (Feng et al., 2012) callpeak with cut off FDR (q-value) at 0.05. Global heatmap was produced with deepTools2 (Ramirez et al., 2016). Peaks were annotated by ChIPseeker with rn6.ncbiRefSeq as the annotation source (Yu et al., 2015). Differential binding analysis of H3K4m2 enrichment was performed by DiffBind with significant threshold at FDR<0.05 (Ross-Innes et al., 2012). Geno-ontology analysis for annotated peaks were performed by ClusterProfiler (Yu et al., 2012). Reduced H3K4me2 peaks in Myocd-LSD1 SMC was compared with published SRF ChIPseq dataset in mouse vascular SMC (GSM3069844) (Yao et al., 2018). Bioinformatic analysis was performed on Galaxy platform (Nekrutenko et al., 2020) and data visualization was performed in R.
Methylated/Hydroxymethylated DNA Immunoprecipitation (MeDIP/hMeDIP)
MeDIP/hMeDIP was performed using MeDIP kit (Diagenode, C02010010) and hMeDIP kit (Diagenode, C02010031) following manufacturer’s protocol. Briefly, genomic DNA was extracted from control, Myocd-LSD1 and Myocd-LSD1NF SMC starved in serum-free medium for 48 hours. DNA was sheared into fragment around 400 bp on Bioruptor Pico (Diagenode) prior to immunoprecipitation. 10% - 20% of sheared DNA was used as the INPUT control. Hydroxymethylated DNA or methylated DNA was captured by 5-hmC monoclonal antibody (Diagenode, C15200200) or anti-5meC antibody (Diagenode, MAb-081-100) with Protein G-coated magnetic beads at 4 °C overnight respectively. Captured DNA was isolated with DNA isolation buffer supplemented with proteinase K (Diagenode). 5hmC/5mC enrichment levels were determined by qRT-PCR following manufacturer’s protocol. Primers used for MeDIP/hMeDIP-qPCR are listed in Key Resources Table.
hMeDIP Sequencing and Data Processing
Genomic DNA was isolated from human coronary SMC. hMeDIP was performed as described previously (Liu et al., 2013). Briefly, DNA with 5-hmC modification was immunoprecipitated with the hydroxymethylated DNA IP kit (Diagenode, C02010031) following manufacturer’s instructions. Precipitated genomic DNA was sonicated to 200 – 500 bp using the Covaris sonicator (Covaris). 1 μg of fragmented genomic DNA was immunoprecipitated with a 5-hmC monoclonal antibody (Diagenode, C15200200) provided in hMeDIP kit. The DNA-antibody mixture was then incubated with magnetic beads overnight at 4°C, washed and DNA purified using purification kit (Qiagen, 69504). The purified DNA was adapter ligated using NEBNext DNA Library Prep Master Mix Set for Illumina 6040 (New England Biolabs) and NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) E7335 (New England Biolabs). Each library was layered on one of the eight lanes of the Illumina flow cell at appropriate concentration and bridge amplified to get around 180 million raw reads. The DNA reads on the flow cell were then sequenced on HiSeq 2000 using appropriate base pair sequencing recipe. The quality of the sequence and its alignment to reference genome was carried out by the Illumina supported Consensus Assessment of Sequence and Variation (CASAVA) software program. For sequencing data analysis, raw reads were aligned to hg38 using Bowtie2 (Langmead and Salzberg, 2012). Peaks were called using MACS2 (Feng et al., 2012) callpeak with cut off FDR (q-value) at 0.05. Peaks were annotated by ChIPseeker with hg38.ncbiRefSeq as the annotation source(Yu et al., 2015). Annotated peaks were compared to H3K4me1 (ENCSR130IMV_2), H3K4me2 (ENCSR783AXV_2) and H3K4me3 (ENCSR515PKY_2) enriched peaked derived from ENCODE project (Davis et al., 2018). Bioinformatic analysis was performed on Galaxy platform (Nekrutenko et al., 2020).
Real-time Quantitative PCR (RT-qPCR)
Passage-matched control, Myocd-LSD1, Myocd-LSD1NF SMC were collected and total RNA was extracted by TRIzol Reagent (Invitrogen, 15596026) according to manufacturer’s protocol. Total RNA was quantified by Qubit RNA Broad Range Assay kit (Invitrogen, Q10210). cDNA was synthesized from 1μg of RNA using iScript cDNA Synthesis Kit (Bio-Rad, 1708891) and real-time quantitative PCR was performed with PowerUp™ SYBR™ Green Master Mix (Applied Biosystems, A25742) using CFX Connect Realtime System (Bio-Rad, 1855201). mRNA levels of target genes were normalized to 18s/Gapdh expression. Primers used for qPCR are listed in Key Resources Table.
RNA extraction from FFPE sections
Tissue was scraped from 5 μm thick FFPE tissue slides and deparaffinized with Xylene solution in 1.5 ml tubes, followed by a 100% EtOH wash. Protein contamination was removed by incubation with Proteinase K and genomic DNA was removed using DNase I (Qiagen). The remained pellet was in 100% EtOH and RNA was isolated with RNeasy FFPE Kit (Qiagen, 73504) following manufacturer’s instructions.
RNA-sequencing and Data Analysis
Total RNA was extracted and column purified from control SMC (n = 4), Myocd-LSD1 SMC (n = 4), Myocd-LSD1NF SMC (n = 4), and SMCs treated with PDGF-BB (30 ng/ml, Sigma Aldrich, SRP3138) for 24 hours (n = 3) using RNeasy Mini Kit (Qiagen, 74104) following manufacture’s protocol. Genomic DNA was removed using DNase I (Qiagen). RNA quality control, library preparation and sequencing were performed by Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh. Briefly, RNA quality was checked with Agilent RNA Screen Tape Assay Tape Station System. RNA library was prepared by TruSeq Stranded mRNA (PolyA+) according to manufacturer’s protocol. Libraries were sequenced with illumina NextSeq High Output 150 cycle kit (2 x 75 bp) with reading depth of 20 million reads per sample. The 75 bp paired-end reads were mapped to Rattus Norvegicus reference genome and gene ID were mapped to Rattus Norvegicus ensembl_v91 using default setting by CLC Genomic Workbench (Qiagen). All mapped reads were normalized by Trimmed Mean of M values (TMM normalization) for differential gene expression analysis in CLC Genomic Workbench. Principle Component Analysis (PCA), volcano plots and heatmaps were created by CLC Genomic Workbench at default setting. Global heatmap was generated with statistic parameters as Euclidean distance measure and complete linkage cluster. Cutoff value for data filtration is minimum fold-change of 1.2. Venn Diagram of differentially expressed genes was plotted with cutoff value at minimum fold-change of 1.5. Corrected FDR p-value < 0.05 is considered statistically significant. Pathway enrichment analysis was performed using the Biogroups app from Correlation Engine (IIlumina) clustering the different biosets taking into account the direction of each gene (Kupershmidt et al., 2010). Gene Ontology (GO) (Ashburner et al., 2000), and the microRNA target database Targetscan (Agarwal et al., 2015) were used to analyze the up-regulated and down-regulated genes separately. P value ≤ 0.05 was considered as statistical significance.
MicroRNA Isolation and Quantification
microRNA-enriched RNA was isolated using the miRNeasy Mini Kit (Qiagen, 217004) based on Qizol/Chloroform extraction (Espinosa-Diez et al., 2015). Reverse transcription of 100 ng RNA was performed using TaqMan™ MicroRNA Reverse Transcription Kit (Applied Biosystems, 4366596) and specific microRNA RT primers according to the manufacturer’s instructions. qPCR was performed using specific microRNA TaqMan primers (miR-145 assay ID: 002278; U6 assay ID: 001973, Applied Biosystems) (Espinosa-Diez et al., 2018). The relative quantification of microRNA expression was determined using the 2-ΔΔCt method, obtaining the fold changes in gene expression normalized to the internal control small nuclear RNA U6. Primers used for qPCR are listed in Key Resources Table.
MicroRNA Transfection
SMC was seeded at 1.5 × 105 cells per well in routine growth medium into 6-well plates. When cells reached 70-80% confluency, miR-145-5p mimic (ThermoFisher, mirVana, MC11480) or the negative control mimic (ThermoFisher, miRVana, 4464058) were prepared in designated concentration and transfected into cells using Lipofectamine RNAiMax Reagent (ThermoFisher, 13778075) according to manufacturer’s instructions.
Western Blot
Total proteins were extracted using CHAPS buffer (1% CHAPS hydrate, 150 mM NaCl, 25 mM HEPES Buffer). Extracts were separated by electrophoresis on NuPAGE 4-12% Bis-Tris Gels (Invitrogen, NP0321PK2) or 3-8% Tris-Acetate Gels (Invitrogen, EA0375PK2), followed by transfer onto 0.45 μm Nitrocellulose Membranes (Bio-Rad, 1620115). Histone proteins were extracted by Histone Extraction Kit (Abcam, ab113476), separated on NuPAGE 4-12% Bis-Tris Gels (Invitrogen, NP0321PK2) and transferred on 0.2 μm Nitrocellulose Membranes (Thermo Scientific, 77012). Membranes were incubated with antibodies specific to ACTA2 (Sigma A2547), MYH11 (Kamiya Biomedical Company KM3669), TAGLN (Abcam ab14106), GADPH (Abcam ab8245/ab9485), unmodified H3 (Abcam ab1791), H3K4me1 (Millipore, 07-436), H3K4me2 (clone CMA303, Millipore 05-1338), H3K9me3 (Abcam ab176916), p-Akt-S473 (Cell Signaling Technology 4060), AKT1/2 (Santa Cruz sc8312). IRDye® anti-rabbit IgG secondary antibody (LI-COR, RRID AB_621843) or anti-mouse IgG secondary antibody (LI-COR, RRID AB_10953628) were used for immune-blotting. Membranes were imaged on the LI-COR Odyssey CLx imaging system.
Ex-vivo Myography
Thoracodorsal artery (TDA) from C57BL/6J mice were excised after euthanasia and perfusion with PBS. TDA were cleaned of fat, cut into 2 mm rings and incubated in supplemented smooth muscle cell media (Lonza) containing 106 lentiviral particles encoding for Myocd-LSD1 or Myocd-LSD1NF. Following 48 hours of incubation, two 25 μm wires were passed through the lumen of the rings and placed on a small vessel myograph (DMT 620M) filled with physiological salt solution (PSS) containing (in mM): NaCl 119, KCl 4.7, MgSO 4 1.17, KH2 PO4 1.18, D-glucose 5.5, NaHCO 3 25, EDTA 0.027, CaCl2 2.5, pH 7.4 when bubbled with 95% O2 5% CO 2 at 37 °C. Following a 30 min rest, vessels were stretched to an internal diameter equivalent to 80mmHg. Vessels were constricted by the addition of 60 mM potassium solution (KPSS) for 5 min
Immunofluorescent Staining
Tissues were embedded vertically and serial 10μm sections were collected beginning at the ligation site. For immunofluorescent staining, sections at 180μm from the ligation site were selected. Following deparaffinization, antigen retrieval was performed (Vector Laboratories H-3300). Staining was performed to evaluate gene expression in cells using primary antibodies for GFP (Abcam ab6673 1:250), MYH11 (Kamiya Biomedical Company MC-352 1:250, Abcam ab683 1:250), Ki67 (Abcam ab15580), cleaved Caspase 3 (Cell Signaling Technology 9661), FLAG tag (Cell Signaling Technology 14793s), mCherry (Abcam ab167453) or IgG as a control. Secondary antibodies included donkey anti-goat 647 (Life Technologies A21447) and donkey anti-rat 555 (Abcam ab150154). Along with the secondary antibodies, sections were also stained for ACTA2 (Sigma-Aldrich F3777 1:500) and DAPI (1:1000). Slides were mounted using Prolong Gold Antifade Reagent (Fisher P36930). For immunofluorescent staining on cultured cells, cells were fixed with 4% Paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.5% Triton-X-100 (Sigma). Smooth muscle α-actin was stained by FITC-conjugated ACTA2 antibody (Sigma F3777). For cytoskeleton dynamic studies, SMC were transduced with Talin-GFP (Cell Light, Invitrogen, C10611) for 24h, and stained with phalloidin (conjugated with Alexa-647; A22287, Invitrogen) and ACTA2 (conjugated with Cy3; C6198, Sigma Aldrich).
Images were acquired on a fluorescent microscope (Leica, DMi8) using the Ocular Advanced Scientific Camera Control software (Digital Optics Limited) or Nikon A1 Confocal microscope using NIS-Elements software (Nikon). Image processing was performed using ImageJ and Image Pro Premier (Media Cybernetics).
Collagen Contraction Assay
Control, Myocd-LSD1, and Myocd-LSD1NF SMC were resuspended in a 10% FBS F12:DMEM media at the density 5x105 cells/mL after starvation in serum-free, insulin-free media for 48 to 72 hours. The cell suspension was incorporated in a collagen solution [bovine collagen I solution (PurCol®, Advanced Biomatrix, #5005), 10 mM NaOH, 1X PBS] and the cell-collagen solution was deposed on non-treated tissue culture 12-well plates. Plates were incubated at 37°C for 2 to 3 hours to allow gelation. Then, gels were immersed in 10% FBS culture medium and lifted from the bottom of the plate. Gel images were acquired immediately (baseline) and every 24 hours under a dissection microscope. Gel diameter and area were calculated using Image Pro Premier software. Gels were stained with ACTA2-FITC and DAPI (overnight incubation) after Triton-X-100 permeabilization and blocking in 5% BSA 10% horse serum PBS. Cell density was evaluated by counting the number of DAPI+ cells in 3 individual fields/gel and normalized to the field area (cells/mm2). Estimation of the cell number per gel was done as follow: number of cells = Area x Cell density.
Proximity Ligation Assay
Proximity Ligation Assay (PLA) was performed using Duolink In Situ PLA reagents according to the manufacturer’s instructions (Sigma Aldrich). Cultured rat SMC were fixed after treatment with PDGF-BB, rapamycin, or control vehicle with 4% PFA for 15 min and permeabilized with 0.25% Triton X-100 PBS for 15 min at room temperature. Aorta cryosections were post-fixed with pre-cooled acetone for 10 min. The same protocol was then used for cultured cells and cryosections. Incubation with antibodies against H3K4me2 (clone CMA303, Millipore, 05-1338) and TET2 (Abcam, ab124297) was done overnight at 4°C. Cells and sections were incubated with Duolink® In Situ PLA Probe anti-rabbit PLUS (Sigma Aldrich, DUO92002) and anti-mouse MINUS (Sigma Aldrich, DUO92004) secondary antibodies, followed by ligation and amplification with Duolink® In Situ Detection reagents Orange (555nm) (Sigma Aldrich, DUO92007). Finally, staining with ACTA2-FITC antibody (4.4 μg/ml, Clone 1A4, Sigma Aldrich, F3777) was performed on cryosection. Slides were mounted using Duolink® In Situ mounting medium with DAPI (Sigma Aldrich, DUO82040). Images were acquired on a fluorescent microscope (Leica, DMi8) using the Ocular Advanced Scientific Camera Control software (Digital Optics Limited). Image processing and PLA dot quantification was performed using ImageJ.
Peptide Binding Assay
HA-tagged mTET2 was overexpressed in 293T cells by adenoviral transduction. Nuclear proteins were extracted from transduced 293T cells using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, 78833) following manufacturer’s instructions. 200 μg of nuclear protein extract from 293T cells overexpressing HA-tagged mTET2 was incubated with 20 μg of synthetic biotinylated H3K4me1 (Active Motif, 81040), H3K4me2 (Active Motif, 81041) and H3K4me3 peptide (Active motif, 81042) in binding buffer (150 mM NaCl, 50 mM HEPES, 0.1% Tween, 10% Glycerol, protease inhibitor cocktail, 1 mM PMSF) at least 4 hours at 4°C. 7 μg of rabbit anti-HA antibody (Abcam, ab9110) and anti-Biotin antibody (Abcam, ab53494) was used to Immunoprecipitate HA-TET2 or Biotinylated H3K4me2 peptide respectively in presence of Dynabeads Protein G (Invitrogen, 10004D) overnight at 4°. The immunoprecipitation complex was washed 3 times with binding buffer. After discarding Dynabeads, immunoprecipitated or INPUT control biotinylated histone peptides were run on 4-12% Bis-Tris Gel (NuPAGE, Invitrogen, NP0321PK2) and followed by Western Blotting with IRDye 800CW Streptavidin (LI-COR, 926-32230). Immunoprecipitated or INPUT control HA-tagged Tet2 were run on 3-8% Tris-Acetate Gel (NuPAGE, Invitrogen, EA0375PK2) and followed by Western Blotting with Conjugated secondary antibodies (Li-COR). One-twelves the amount of immunoprecipitation was run in parallel as the INPUT control and 7 μg of rabbit IgG was used as negative control for immunoprecipitation. Immunoblotting was imaged by the LI-COR Odyssey CLx imaging system.
Plasticity Assays
SMC were loaded with Cholesterol–methyl-β-cyclodextrin (Sigma Aldrich, C4555) for 24 to 72h as previously described(Rong et al., 2003; Shankman et al., 2015). Cholesterol was used at 20, 40, or 80μg/ml. After cholesterol loading, SMC were either harvested for RT-qPCR analysis as described above or stained for lipid uptake with Oil Red O staining kit (Sigma Aldrich, MAK194), following manufacturer’s instructions. Oil Red O staining was quantified using Image Pro Premier. SMC phenotypic plasticity was characterized by Rat Mesenchymal Stem Cell Functional Identification Kit (R&D Systems, SC020) following manufacture’s protocol. Briefly, for adipogenic differentiation assay, control, Myocd-LSD1, and Myocd-LSD1NF SMC were plated on 6-well-plates and 2-well chamber slides and cultured until confluency in F12:DMEM containing 10% FBS. Culture media was then replaced with Adipogenic Differentiation Media (αMEM, 10% FBS, adipogenic supplement, R&D Systems) and changed every 4 days for 2 weeks. Cells were incubated with AdipoRed Assay (Lonza, PT-7009) and immunofluorescent stained for mFABP4 (R&D Systems) following manufactures’ protocols. For chondrogenic differentiation assay, 106 Myocd-LSD1 and Myocd-LSD1NF SMC were transferred and centrifuged down in 15 ml Falcon tubes to form cell pellets. Chondrogenic differentiation media (DMEM/F12, ITS supplement, chondrogenic supplement, R&D Systems) were used and replaced every 3 days for 28 days without disturbing cell pellets. Cell pellets were then harvested and immunofluorescent stained for hAggrecan (R&D Systems) following manufactures’ protocol. Images were acquired by Ocular Advanced Scientific Camera Control software (Digital Optics Limited) on a fluorescent microscope (Leica, DMi8). Cell counting was performed on acquired images using ImageJ.
Cell Viability Assay
Uninfected control, Myocd-LSD1, and Myocd-LSD1NF SMC were seeded in a 96-well-plate at a confluency of 1000 cells/well and were cultured in growth medium (DMEM:F12, Gibco) supplemented with fetal bovine serum (10%, Corning), L-glutamine (1.6 mM, Gibco), and penicillin-streptomycin (100 U/mL, Gibco). Cell viability was assessed by measuring ATP content at 24, 48 and 72 hours by using the Cell Titer-Glo luminescent cell viability assay following manufacture’s recommendations (Promega, G7570). Luminesce measurements were performed in a Synergy-HTX multi-mode reader (Biotek).
Transwell Assay
Myocd-LSD1 SMC were seeded (8 × 105 cells/ml, 150ul) with serum-free insulin-free media in 8 μm pore size polycarbonate transwell inserts (Corning, 3422). Serum-free insulin-free media supplemented with PDGF-BB (Sigma-Aldrich, SRP3138) was added to the lower compartment in a final concentration of 30 ng/ml as the chemo-attractant. After 24 hours incubation, cells were fixed with 4% PFA (Electron Microscopy Science, 15710) and stained with 0.1% crystal violet (Sigma-Aldrich, V5265). Cells on the upper compartment were carefully removed by a cotton swab. The membrane was excised and mounted onto a glass slide with Prolong Gold Mounting Media (ThermoFisher, P10144). Images were taken by microscope (Leica DM500) with the Leica LAS EZ software and processed using ImageJ software.
Luciferase Assay
SMCs were seeded at 1.5 × 105 cells per well in growth medium into 6-well plates and transfected at 70-80% confluency, with control pGL3, pGL3-Acta2-CArGWT (−2.6/+2.8kb with intact CArG boxes), pGL3-Acta2-promoCArGMut (−2.6/+2.8kb with mutated CArG boxes within the promoter region), and pGL3-Acta2-promo+intCArGMut (−2.6/+2.8kb with mutated CArG boxes within the promoter and the first intron region) (Hendrix et al., 2005). After 24 hours post transfection, SMCs were cultured in serum-free media for 24h. Cells were collected and luciferase activity was quantified using Firefly Luciferase Assay System (Promega, E1500) according to manufacturer’s instructions. The luciferase signal results were normalized to total protein level.
Quantification and Statistical Analysis
All data are presented as the mean ± SEM. In vitro experiments were repeated independently at least 3 times with duplicated technical repeats. One single data symbol represented the mean value of technical repeats for one independent experiment. For in vivo experiments, 4 to 12 mouse littermates were used for each group. All statistics were performed using GraphPad Prism 8. Two-tailed unpaired Student’s t-tests with a confidence level of 95% were used to compare two groups with continuous variables with normal distribution and equal variances tested by Kolmogorov-Smirnov test. Two-tailed unpaired Student’s t-tests followed by a Welch’s correction with the confidence level of 95% were performed if two groups have unequal variances. Two-tailed unpaired Mann-Whitney U-tests with the confidence level of 95% were used if variables were non-normally distributed. For comparison between multiple groups with a single factor or two factors, we used one-way or two-way ANOVA, respectively. For categorial data, we used two-sided Fisher’s exact test. P ≤ 0.05 was considered as statistically significant.
Supplementary Material
Highlights.
Gene-selective epigenome editing reveals that H3K4me2 controls SMC lineage identity
H3K4me2 editing induces loss of contractility and exacerbates phenotypic plasticity
H3K4me2 interacts with TET2 and mediates TET-dependent gene activation
Loss of H3K4me2 impairs SMC-mediated vascular remodeling due to miR-145 deficiency
ACKNOWLEDGMENTS
We thank the Center for Biologic Imaging (supported by NIH 1S10OD019973-01), at the University of Pittsburgh, the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh, the Health Sciences Library System at the University of Pittsburgh, and the Yale Cancer Center Sequencing facility at Yale University. We thank Dr. Q. Gan, J. Krasner, R. Tripathi, J. Krzemien, and Dr. A. Ostriker for their help. Schematics were created with Biorender.
SOURCE OF FUNDING
This work is supported by the National Institute of Health R01HL146465 and the American Heart Association 15SDG25860021 grants to DG; the National Institute of Health R01 HL133864, R01 HL128304, R01 HL153532 and AHA Established Investigator Award to A.C.S; the National Institute of Health R01 HL142090-01, R01 HL146101 to K.A.M; and the National Institute of Health R01 HL136314 and R01 HL057353 to G.K.O.
NON-STANDARD ABBREVIATIONS:
- ACTA2
Smooth muscle alpha-actin
- ChIP
Chromatin Immunoprecipitation
- ChIPseq
Chromatin Immunoprecipitation sequencing
- GO
Gene Ontology
- H3K4me2
Histone 3 Lysine 4 di-methylation
- iPS
induced Pluripotent Stem
- KO
Knockout
- LSD1
Lysine Specific Demethylase 1
- MSC
Mesenchymal Stem Cell
- MYH11
Smooth muscle myosin heavy chain
- Myocd
Myocardin
- PDGF-BB
Platelet Derived Growth Factor-BB
- PDGFβR
Platelet Derived Growth Factor beta receptor
- SMC
Smooth Muscle Cells
- SRF
Serum Response Factor
- TET2
Ten Eleven Transformation 2
- YFP
Yellow Fluorescent Protein
Footnotes
DECLARATION OF INTEREST
The authors have no conflict of interest to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agarwal V, Bell GW, Nam JW, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, Williams CM, Shamsuzzaman S, Mokry M, Henderson CA, et al. (2020). Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 142, 2045–2059. 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MR, and Owens GK (2012). Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74, 13–40. 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- Ang SY, Uebersohn A, Spencer CI, Huang Y, Lee JE, Ge K, and Bruneau BG (2016). KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development 143, 810–821. 10.1242/dev.132688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank RS, Swartz EA, Thompson MM, Olson EN, and Owens GK (1995). A retinoic acid-induced clonal cell line derived from multipotential P19 embryonal carcinoma cells expresses smooth muscle characteristics. Circ Res 76, 742–749. 10.1161/01.res.76.5.742. [DOI] [PubMed] [Google Scholar]
- Brickner DG, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee PC, Widom J, and Brickner JH (2007). H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol 5, e81. 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S, and Kim T (2010). The role of cotranscriptional histone methylations. Cold Spring Harb Symp Quant Biol 75, 95–102. 10.1101/sqb.2010.75.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakouros D, Hemming S, Gronthos K, Liu R, Zannettino A, Shi S, and Gronthos S (2019). Specific functions of TET1 and TET2 in regulating mesenchymal cell lineage determination. Epigenetics Chromatin 12, 3. 10.1186/s13072-018-0247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheedipudi S, Genolet O, and Dobreva G (2014). Epigenetic inheritance of cell fates during embryonic development. Front Genet 5, 19. 10.3389/fgene.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Kitchen CM, Streb JW, and Miano JM (2002). Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34, 1345–1356. 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- Chen LL, Lin HP, Zhou WJ, He CX, Zhang ZY, Cheng ZL, Song JB, Liu P, Chen XY, Xia YK, et al. (2018). SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep 25, 1485–1500 e1484. 10.1016/j.celrep.2018.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Qin L, Li G, Malagon-Lopez J, Wang Z, Bergaya S, Gujja S, Caulk AW, Murtada SI, Zhang X, et al. (2020). Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 26, 542–557 e511. 10.1016/j.stem.2020.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, et al. (2018). Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397 e1314. 10.1016/j.cell.2018.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, and Srivastava D (2009). miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710. 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Urso A, and Brickner JH (2014). Mechanisms of epigenetic memory. Trends Genet 30, 230–236. 10.1016/j.tig.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, and Collas P (2008). A rapid micro chromatin immunoprecipitation assay (microChIP). Nat Protoc 3, 1032–1045. 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018). The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46, D794–D801. 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida DC, Ferreira MR, Franzen J, Weidner CI, Frobel J, Zenke M, Costa IG, and Wagner W (2016). Epigenetic Classification of Human Mesenchymal Stromal Cells. Stem Cell Reports 6, 168–175. 10.1016/j.stemcr.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, and Jorgensen HF (2018). Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun 9, 4567. 10.1038/s41467-018-06891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, and Parmacek MS (2003). Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol 23, 2425–2437. 10.1128/Mcb.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Diez C, Fierro-Fernandez M, Sanchez-Gomez F, Rodriguez-Pascual F, Alique M, Ruiz-Ortega M, Beraza N, Martinez-Chantar ML, Fernandez-Hernando C, and Lamas S (2015). Targeting of Gamma-Glutamyl-Cysteine Ligase by miR-433 Reduces Glutathione Biosynthesis and Promotes TGF-beta-Dependent Fibrogenesis. Antioxid Redox Signal 23, 1092–1105. 10.1089/ars.2014.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Diez C, Wilson R, Chatterjee N, Hudson C, Ruhl R, Hipfinger C, Helms E, Khan OF, Anderson DG, and Anand S (2018). MicroRNA regulation of the MRN complex impacts DNA damage, cellular senescence, and angiogenic signaling. Cell Death Dis 9, 632. 10.1038/s41419-018-0690-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JX, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nature Protocols 7, 1728–1740. 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D, Shankman LS, Nguyen AT, and Owens GK (2013). Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods 10, 171–177. 10.1038/Nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D, Swiatlowska P, and Owens GK (2015). Epigenetic Control of Smooth Muscle Cell Identity and Lineage Memory. Arterioscler Thromb Vasc Biol 35, 2508–2516. 10.1161/ATVBAHA.115.305044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39, 1488–1493. 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Hendrix JA, Wamhoff BR, McDonald OG, Sinha S, Yoshida T, and Owens GK (2005). 5′ CArG degeneracy in smooth muscle α -actin is required for injury-induced gene suppression in vivo. J Clin Invest 115, 418–427. 10.1172/jci22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JH, Cheng L, Li J, Chen M, Zhou DY, Lu MM, Proweller A, Epstein JA, and Parmacek MS (2008). Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest 118, 515–525. 10.1172/Jc133304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, and Kim J (2017). Writing, erasing and reading histone lysine methylations. Exp Mol Med 49, e324. 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, and Allis CD (2001). Translating the histone code. Science 293, 1074–1080. 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jufvas A, Sjodin S, Lundqvist K, Amin R, Vener AV, and Stralfors P (2013). Global differences in specific histone H3 methylation are associated with overweight and type 2 diabetes. Clin Epigenetics 5, 15. 10.1186/1868-7083-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Janssens DH, Henikoff JG, Ahmad K, and Henikoff S (2020). Efficient low-cost chromatin profiling with CUT&Tag. Nature Protocols 15, 3264–3283. 10.1038/s41596-020-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, and Costello J (2017). DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med 49, e322. 10.1038/emm.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmidt I, Su GJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, et al. (2010). Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5. 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–U354. 10.1038/Nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, and Shiekhattar R (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435. 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Li SJ, Wang DZ, Wang ZG, Richardson JA, and Olson EN (2003). The serum response factor coactivator myocardin is required for vascular smooth muscle development. P Natl Acad Sci USA 100, 9366–9370. 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, and Gomez D (2019). Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vase Biol, ATVBAHA119312131. 10.1161/ATVBAHA.119.312131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Jin Y, Tang WH, Gin L, Zhang X, Tellides G, Hwa J, Yu J, and Martin KA (2013). Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128, 2047–2057. 10.1161/CIRCULATIONAHA.113.002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe I, and Owens GK (2001). Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res 88, 1127–1134. 10.1161/hh1101.091339. [DOI] [PubMed] [Google Scholar]
- McDonald OG, Wamhoff BR, Hoofnagle MH, and Owens GK (2006). Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest 116, 36–48. 10.1172/Jci26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM (2015). Myocardin in biology and disease. J Biomed Res 29, 3–19. 10.7555/JBR.29.20140151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao F, Wu X, Zhang L, Yuan YC, Riggs AD, and Natarajan R (2007). Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem 282, 13854–13863. 10.1074/jbc.M609446200. [DOI] [PubMed] [Google Scholar]
- Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, Sheikh AQ, Ding L, Qin L, Fernandez-Hernando C, et al. (2018). Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun 9, 2073. 10.1038/s41467-018-04447-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner S, Leoni C, Emming S, Della Chiara G, Balestrieri C, Barozzi I, Piccolo V, Togher S, Ko M, Rao A, et al. (2016). TET2 Regulates Mast Cell Differentiation and Proliferation through Catalytic and Non-catalytic Activities. Cell Rep 15, 1566–1579. 10.1016/j.celrep.2016.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko A, Taylor J, Goecks J, Blankenberg D, Clements D, Gu Q, Afgan E, and Jalili V (2020). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res 48, W395–W402. 10.1093/nar/gkaa434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford K, Kharchenko P, Lai W, Dao MC, Worhunsky DJ, Ferro A, Janzen V, Park PJ, and Scadden DT (2008). Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev Cell 14, 798–809. 10.1016/j.devcel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostriker AC, Xie Y, Chakraborty R, Sizer AJ, Bai Y, Ding M, Song WL, Huttner A, Hwa J, and Martin KA (2021). TET2 Protects Against VSMC Apoptosis and Intimal Thickening in Transplant Vasculopathy. Circulation. 10.1161/CIRCULATIONAHA.120.050553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK (1995). Regulation of differentiation of vascular smooth muscle cells. Physiol Rev 75, 487–517. 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- Owens GK (2007). Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 283, 174–191; discussion 191-173, 238-141. [DOI] [PubMed] [Google Scholar]
- Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, et al. (2020). Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 142, 2060–2075. 10.1161/CIRCULATIONAHA.120.048378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, Nakamura K, Yokoyama N, Cui JZ, Iosef C, et al. (2020). Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol 40, 2195–2211. 10.1161/ATVBAHA.120.314670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Ferrier P, and Spicuglia S (2010). A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res 20, 1493–1502. 10.1101/gr.109389.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, and Owens GK (2007). Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res 101, 792–801. 10.1161/CIrCrESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- Pipes GC, Creemers EE, and Olson EN (2006). The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev 20, 1545–1556. 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- Popova EY, Xu X, DeWan AT, Salzberg AC, Berg A, Hoh J, Zhang SS, and Barnstable CJ (2012). Stage and gene specific signatures defined by histones H3K4me2 and H3K27me3 accompany mammalian retina maturation in vivo. PLoS One 7, e46867. 10.1371/journal.pone.0046867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432. 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong JX, Shapiro M, Trogan E, and Fisher EA (2003). Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 100, 13531–13536. 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R, and Klebanoff SJ (1971). The smooth muscle cell. I. In vivo synthesis of connective tissue proteins. J Cell Biol 50, 159–171. 10.1083/jcb.50.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–U177. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardina JL, Collombet S, Tian TV, Gomez A, Di Stefano B, Berenguer C, Brumbaugh J, Stadhouders R, Segura-Morales C, Gut M, et al. (2018). Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem Cell 23, 727–741 e729. 10.1016/j.stem.2018.08.016. [DOI] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, and Kouzarides T (2004). Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 6, 73–77. 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, et al. (2015). KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine 21, 628–637. 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh AQ, Lighthouse JK, and Greif DM (2014). Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep 6, 809–817. 10.1016/j.celrep.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh AQ, Misra A, Rosas IO, Adams RH, and Greif DM (2015). Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 7, 308ra159. 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Quertermous T, Fischbein MP, and Wu JC (2021). Generation of Vascular Smooth Muscle Cells From Induced Pluripotent Stem Cells: Methods, Applications, and Considerations. Circ Res 128, 670–686. 10.1161/CIRCRESAHA.120.318049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ 3rd, Nishioka K, and Reinberg D (2003). Histone lysine methylation: a signature for chromatin function. Trends Genet 19, 629–639. 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Spin JM, Nallamshetty S, Tabibiazar R, Ashley EA, King JY, Chen M, Tsao PS, and Quertermous T (2004). Transcriptional profiling of in vitro smooth muscle cell differentiation identifies specific patterns of gene and pathway activation. Physiol Genomics 19, 292–302. 10.1152/physiolgenomics.00148.2004. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology 1. 10.1186/1471-213x-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ Jr., and Miano JM (2006). Defining the mammalian CArGome. Genome Res 16, 197–207. 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, and Fisher EA (2015). Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 35, 535–546. 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, and Olson EN (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862. 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, Sin DD, Seidman MA, Leeper NJ, and Francis GA (2019). Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol 39, 876–887. 10.1161/ATVBAHA.119.312434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li X, and Hu H (2014). H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228. 10.1016/j.ygeno.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Williams K, Christensen J, and Helin K (2011). DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep 13, 28–35. 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, et al. (2019). Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 25, 1280–1289. 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, et al. (2008a). G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14, 64–68. 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, et al. (2008b). G(12)-G(13)-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension (vol 14, pg 64, 2008). Nature Medicine 14, 222–222. DOI 10.1038/nm0208-222. [DOI] [PubMed] [Google Scholar]
- Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, and Olson EN (2009). MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23, 2166–2178. 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZH, Ji GD, Shen JB, Wang XB, and Li L (2012). SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation. Biochem Bioph Res Co 422, 285–290. 10.1016/j.bbrc.2012.04.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, and Kobayashi A (2017). The cysteine-rich domain of TET2 binds preferentially to mono- and dimethylated histone H3K36. J Biochem 161, 327–330. 10.1093/jb/mvx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao F, Yu P, Li Y, Yuan X, Li Z, Zhang T, Liu F, Wang Y, Wang Y, Li D, et al. (2018). Histone Variant H2A.Z Is Required for the Maintenance of Smooth Muscle Cell Identity as Revealed by Single-Cell Transcriptomics. Circulation 138, 2274–2288. 10.1161/circulationaha.117.033114. [DOI] [PubMed] [Google Scholar]
- Yu GC, Wang LG, Han YY, and He QY (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omics 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GC, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- Zhang J, Parvin J, and Huang K (2012). Redistribution of H3K4me2 on neural tissue specific genes during mouse brain development. BMC Genomics 13 Suppl 8, S5. 10.1186/1471-2164-13-S8-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Song M, Qu J, and Liu GH (2018). Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ Res 123, 773–786. 10.1161/CIRCRESAHA.118.312497. [DOI] [PubMed] [Google Scholar]
- Zhong W, Li B, Xu Y, Yang P, Chen R, Wang Z, Shao C, Song J, and Yan J (2018). Hypermethylation of the Micro-RNA 145 Promoter Is the Key Regulator for NLRP3 Inflammasome-Induced Activation and Plaque Formation. JACC Basic Transl Sci 3, 604–624. 10.1016/j.jacbts.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654. 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, et al. (2006). Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 38, 343–349. 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and CUT&Tag-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contract upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-dimethyl-Histone H3 (Lys4) Antibody | Millipore | Cat# 07-030, RRID:AB_310342 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Recombinant Anti-Histone H3 (tri methyl K9) antibody | Abcam | Cat# ab176916, RRID:AB_2797591 |
| Anti-trimethyl-Histone H3 (Lys27) Antibody | MIllipore | Cat# 07-449, RRID:AB_310624 |
| SRF (H-300) antibody | Santa Cruz Biotechnology | Cat# sc-13029, RRID:AB_2302440 |
| GKLF (H-180) antibody | Santa Cruz Biotechnology | Cat# sc-20691, RRID:AB_669567 |
| Anti-Tet2 Antibody | Sigma-Aldrich | Cat# ABE364 |
| Anti-acetyl-Histone H3 Antibody | Millipore | Cat# 06-599, RRID:AB_2115283 |
| Rabbit IgG, polyclonal - Isotype Control (ChIP Grade) | Abcam | Cat# ab171870, RRID:AB_2687657 |
| Mouse IgG - Isotype Control | Abcam | Cat# ab37355, RRID:AB_2665484 |
| Histone H3K4me2 antibody (pAb) | Active Motif | 39141, RRID:AB_2614985 |
| ANTI-FLAG® antibody produced in rabbit | Sigma-Aldrich | Cat# F7425, RRID:AB_439687 |
| Anti-HA tag antibody - ChIP Grade | Abcam | Cat# ab9110, RRID:AB_307019 |
| 5-hydroxymethylcytosine (5-hmC) monoclonal antibody (mouse) | Diagenode | C15200200-50 |
| 5-methylcytosine (5-mC) Antibody - clone 33D3 | Diagenode | C15200081-100 |
| Monoclonal Anti-Actin, α-Smooth Muscle | Sigma-Aldrich | Cat# A2547, RRID:AB_476701 |
| Rat Anti-Smooth Muscle Myosin Heavy Chain (sml) Monoclonal Antibody, Unconjugated, Clone KM3669 | Kamiya Biomedical Company | Cat# MC-352, RRID:AB_1241986 |
| Anti-TAGLN/Transgelin antibody | Abcam | Cat# ab14106, RRID:AB_443021 |
| Anti-GAPDH antibody [6C5] - Loading Control | Abcam | Cat# ab8245, RRID:AB_2107448 |
| Anti-GAPDH antibody - Loading Control | Abcam | Cat# ab9485, RRID:AB_307275 |
| Anti-Histone H3 antibody - Nuclear Marker and ChIP Grade | Abcam | Cat# ab1791, RRID:AB_302613 |
| H3K4me1 | Millipore | Cat# 07-436, RRID:AB_310614 |
| Anti-Dimethyl Histone H3 (Lys4) Antibody, clone CMA303 | Sigma Aldrich | Cat# 05-1338, RRID:AB_1977248 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4060, RRID:AB_2315049 |
| Akt1/2/3 Antibody (H-136) | Santa Cruz Biotechnology | Cat# sc-8312, RRID:AB_671714 |
| IRDye® 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat# 926-32211, RRID:AB_621843 |
| IRDye® 680RD Donkey anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat# 926-68072, RRID:AB_10953628 |
| Anti-GFP antibody | Abcam | Cat# ab6673, RRID:AB_305643 |
| Anti-Ki67 antibody | Abcam | Cat# ab15580, RRID:AB_443209 |
| Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| DYKDDDDK Tag (D6W5B) Rabbit mAb | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| Anti-mCherry antibody | Abcam | Cat# ab167453, RRID:AB_2571870 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Molecular Probes | Cat# A-21447, RRID:AB_141844 |
| Donkey Anti-Rat IgG H&L (Alexa Fluor® 555) | Abcam | Cat# ab150154, RRID:AB_2813834 |
| Monoclonal Anti-Actin, α-Smooth Muscle - FITC antibody produced in mouse | Sigma-Aldrich | Cat# F3777, RRID:AB_476977 |
| Anti-Actin, α-Smooth Muscle - Cy3™ antibody, Mouse monoclonal | Sigma-Aldrich | Cat# C6198, RRID:AB_476856 |
| Anti-Tet2 antibody | Abcam | Cat# ab124297, RRID:AB_2722695 |
| Anti-Biotin antibody | Abcam | Cat# ab53494, RRID:AB_867860 |
| Bacterial and Virus Strains | ||
| Ad-m-MYOCD-GFP | Vector Biolabs | Cat# ADV-265349 |
| Ad-mTET2-HA | Applied Biological Materials | Cat# 465200540200 |
| Lenti-Myocd-LSD1 | This paper | N/A |
| Lenti-Myocd-LSD1NF | This paper | N/A |
| Biological Samples | ||
| Human Coronary Artery Smooth Muscle Cells | LONZA | Cat# CC-2583 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Corning® Fetal Bovine Serum, 500 mL, Premium, United States Origin | Corning | Cat# 35-015-CV |
| L-Glutamine (200 mM) | Gibco | Cat# 25030081 |
| L-Ascorbic acid | Sigma-Aldrich | Cat# A4403; CAS: 50-81-7 |
| apo-Transferrin human | Sigma-Aldrich | Cat# T5391; CAS: 11096-37-0 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261; CAS: 10102-18-8 |
| PDGF-BB human | Sigma-Aldrich | Cat# SRP3138 |
| Rapamycin from Streptomyces hygroscopicus | Sigma-Aldrich | Cat# R0395; CAS: 53123-88-9 |
| Retinoic acid | Sigma-Aldrich | Cat# R2625; CAS: 302-79-4 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat# L3000015 |
| Polybrene Infection / Transfection Reagent | Sigma-Aldrich | Cat# TR-1003-G |
| Recombinant Human EGF (Animal-Free) | Biolegend | Cat# 713008 |
| Recombinant Human FGF-basic (146 aa) (Animal-Free) | Biolegend | Cat# 713304 |
| Dynabeads™ Protein G for Immunoprecipitation | Invitrogen | Cat# 10004D |
| ACCUTASE™ Cell detachment solution | Stem cell technologies | Cat# 07922 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat# A25742 |
| mirVana® miRNA mimic miR-145-5p | ThermoFisher | Cat# MC11480 |
| mirVana® miRNA mimic negative control | ThermoFisher | Cat# 4464058 |
| Lipofectamine RNAiMax Reagent | ThermoFisher | Cat# 13778075 |
| Invitrogen™ CellLight™ Talin-GFP, BacMam 2.0 | Invitrogen | Cat# C10611 |
| Invitrogen™ Alexa Fluor™ 647 Phalloidin | Invitrogen | Cat# A22287 |
| PureCol® Type I Collagen Solution, 3 mg/ml (Bovine) | Advanced BioMatrix | Cat# 5005 |
| Duolink® In Situ PLA Probe anti-rabbit PLUS | Sigma-Aldrich | Cat# DUO92002 |
| Duolink® In Situ PLA Probe anti-mouse MINUS | Sigma-Aldrich | Cat# DUO92004 |
| Duolink® In Situ Detection reagents Orange | Sigma-Aldrich | Cat# DUO92007 |
| Duolink® In Situ mounting medium with DAPI | Sigma-Aldrich | Cat# DUO82040 |
| Histone H3K4me1 Peptide - biotinylated | Active Motif | Cat# 81040 |
| Histone H3K4me2 Peptide - biotinylated | Active Motif | Cat# 81041 |
| Histone H3K4me3 Peptide - biotinylated | Active Motif | Cat# 81042 |
| IRDye 800CW Streptavidin | LI-COR Biosciences | Cat# 926-32230 |
| Methyl-β-cyclodextrin (Cholesterol) | Sigma-Aldrich | Cat# C4555; CAS: 128446-36-6 |
| AdipoRed™ Assay Reagent | LONZA | Cat# PT-7009 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648; CAS: 10540-29-1 |
| Pluronic® F-127 | Sigma-Aldrich | Cat# P2443; CAS: 9003-11-6 |
| 16% Paraformaldehyde Aqueous Solution, EM Grade, Ampoule 10 ML | Electron Microscopy Sciences | Cat# 15710 |
| Crystal violet solution | Sigma-Aldrich | Cat # V5265; CAS: 548-62-9 |
| Antigen Unmasking Solution, Citrate-Based | Vector Laboratories | Cat# H-3300 |
| Critical Commercial Assays | ||
| Qubit™ RNA BR Assay Kit | Invitrogen | Cat# Q10210 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| RNeasy FFPE Kit | Qiagen | Cat# 73504 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| TaqMan™ MicroRNA Reverse Transcription Kit | Applied Biosystems | Cat# 4366596 |
| Histone Extraction Kit | Abcam | Cat# ab113476 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat# 78833 |
| Oil Red O staining kit | Sigma-Aldrich | Cat# MAK194 |
| Rat Mesenchymal Stem Cell Functional Identification Kit | R&D Systems | Cat# SC020 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# 7570 |
| CUT&Tag-IT™ Assay Kit | Active Motif | Cat# 53160 |
| MeDIP kit | Diagenode | Cat# C02010010 |
| hMeDIP kit | Diagenode | Cat# C02010031 |
| Luciferase assay kit | Promega | Cat# E1500 |
| Deposited Data | ||
| H3K4me3 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR515PKY/ |
| VSMC_SRF | NCBI's Gene Expression Omnibus | GSM3069844 |
| H3K4me1 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR130IMV/ |
| H3K4 di-methylation controls smooth muscle cell lineage identity and vascular homeostasis | NCBI's Gene Expression Omnibus | GSE179220 |
| H3K4me2 (Homo Sapiens smooth muscle cell originated from H9) | ENCODE project (Davis et al., 2018) | https://www.encodeproject.org/experiments/ENCSR783AXV/ |
| Experimental Models: Cell Lines | ||
| Human coronary artery smooth muscle cells (hCASMCs) | LONZA | Cat# CC-2583 |
| Experimental Models: Organisms/Strains | ||
| Mouse: YFP: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | (Srinivas et al., 2001) | MGI: J:80963 |
| Mouse: Myh11-CreERT2: B6.FVB-Tg(Myh11-cre/ERT2)1Soff/J | (Wirth et al., 2008a) | MGI: J:141641 |
| Oligonucleotides | ||
| See Supplementary Table 1 | ||
| Recombinant DNA | ||
| pLVX-Myocd-LSD1-IRES-mCherry | This paper | N/A |
| pLVX-Myocd-LSD1NF-IRES-mCherry | This paper | N/A |
| pGL3-Acta2-CArGWT | (Hendrix et al., 2005) | N/A |
| pGL3-Acta2-promoCArGMut | (Hendrix et al., 2005) | N/A |
| pGL3-Acta2-promo+intCArGMut | (Hendrix et al., 2005) | N/A |
| Software and Algorithms | ||
| Prism 9 | Graph Pad | https://www.graphpad.com/ |
| Adobe Illustrator 2021 | Adobe | https://www.adobe.com/products/illustrator.html |
| Fiji | http://fiji.sc | RRID:SCR_002285 |
| Office 365 | Microsoft | https://www.microsoft.com/en-us/microsoft-365 |
| Adobe Photoshop 2021 | Adobe | https://www.adobe.com/products/photoshop.html |
| R version 4.0.4 | http://www.r-project.org/ | RRID:SCR_001905 |
| Galaxy | http://galaxyproject.org/ | RRID:SCR_006281 |
| Bowtie2 | (Langmead and Salzberg, 2012) | RRID:SCR_016368 |
| Deeptools | (Ramirez et al., 2016) | RRID:SCR_016366 |
| Integrative Genomics Viewer | http://www.broadinstitute.org/igv/ | RRID:SCR_011793 |
| MACS | https://github.com/macs3-project/MACS | RRID:SCR_013291 |
| Diffind | http://bioconductor.org/packages/release/bioc/html/DiffBind.html | RRID:SCR_012918 |
| ChIPseeker | https://bioconductor.org/packages/ChIPseeker/ | RRID:SCR_021322 |
| ClusterProfiler | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | RRID:SCR_016884 |
| QIAGEN CLC Genomics Workbench | Qiagen | https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/informatics-and-data/analysis-and-visualization/clc-genomics-workbench/ |
| BaseSpace Correlation Engine | Illumina | https://www.illumina.com/products/by-type/informatics-products/basespace-correlation-engine.html |
| Targetscan | http://targetscan.org/ | RRID:SCR_010845 |
| Ocular Advanced Scientific Camera Control software | Digital Opitcs Limited | https://www.photometrics.com/products/ocular |
| Image Pro Premier | Media Cybernetics | https://www.mediacy.com/support/imagepropremier |
| Other | ||
| Multi Wire Myograph System | DMT | Cat# 620M |
| LEICA Dmi8 Inverted Fluorescent Microscope | LEICA | https://www.leica-microsystems.com/products/light-microscopes/p/leica-dmi8-id/ |
| Nikon Instruments A1 Confocal Laser Microscope | Nikon | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/a1hd25-a1rhd25 |
| Synergy-HTX multi-mode reader | BioTek | https://www.biotek.com/products/detection-multi-mode-microplate-readers/synergy-htx-multi-mode-reader/ |
| 6.5 mm Transwell® with 8.0 μm Pore Polycarbonate Membrane Insert, Sterile | Corning | Cat# 3422 |
| Bioruptor® Pico sonication device | Diagenode | Cat# B01060010 |
| CFX Connect Realtime System | Bio-Rad | Cat# 1855201 |
| Odyssey® CLx Imaging System | LI-COR Biosciences | https://www.licor.com/bio/odyssey-dlx/ |