

Abstract
Half of all human transcripts are thought to be regulated by microRNAs. Therefore, quantifying microRNA expression can reveal underlying mechanisms in disease states and provide therapeutic targets and biomarkers. Here, we detail how to accurately quantify microRNAs. Briefly, this method describes isolating microRNAs, ligating them to adaptors suitable for high-throughput sequencing, amplifying the final products, and preparing a sample library. Then we explain how to align the obtained sequencing reads to microRNA hairpins, and quantify, normalize, and calculate their differential expression. Versatile and robust, this combined experimental workflow and bioinformatic analysis enables users to begin with tissue extraction and finish with microRNA quantification.
Keywords: microRNAs, small RNAs, high-throughput sequencing, bioinformatics, library preparation, sequence alignment
SUMMARY:
Here we describe a step-by-step strategy for isolating small RNAs, enriching for microRNAs, and preparing samples for high-throughput sequencing. We then describe how to process sequence reads and align them to microRNAs, using open source tools.
INTRODUCTION:
First discovered in 19931, it is now estimated that nearly 2000 microRNAs are present in the human genome2. MicroRNAs are small non-coding RNAs that are typically 21-24 nucleotides long. They are post-transcriptional regulators of gene expression, often binding to complementary sites in the 3′-untranslated region (3′-UTR) of target genes to repress protein expression and degrade mRNA. Quantifying microRNAs can give valuable insight into gene expression and several protocols have been developed for this purpose3.
We have developed a defined, reproducible, and long-standing protocol for small RNA sequencing, and for analyzing normalized reads using open source bioinformatics tools. Importantly, our protocol enables the simultaneous identification of both endogenous microRNAs and exogenously delivered constructs that produce microRNA-like species, while minimizing reads that map to other small RNA species, including ribosomal RNAs (rRNAs), transfer RNA-derived small RNAs (tsRNAs), repeat-derived small RNAs, and mRNA degradation products. Fortunately, microRNAs are 5′-phosphorylated and 2′-3′ hydroxylated4, a feature that can be leveraged to separate them from these other small RNAs and mRNA degradation products. Several commercial options exist for microRNA cloning and sequencing that are often quicker and easier to multiplex; however, the proprietary nature of kit reagents and their frequent modifications makes comparing sample runs challenging. Our strategy optimizes collecting only the correct size of microRNAs through acrylamide and agarose gel purification steps. In this protocol, we also describe a procedure for aligning sequence reads to microRNAs using open source tools. This set of instructions will be especially useful for novice informatics users, regardless of whether our library preparation method or a commercial method is used.
This protocol has been used in several published studies. For example, it was used to identify the mechanism by which the Dicer enzyme cleaves small hairpin RNAs at a distance of two nucleotides from the internal loop of the stem-loop structure – the so-called “loop-counting rule”5. We also followed these methods to identify the relative abundance of delivered small hairpin RNAs (shRNA) expressed from recombinant adeno-associated viral vectors (rAAVs), to identify the threshold of shRNA expression that can be tolerated prior to liver toxicity associated with excess shRNA expression6. Using this protocol, we also identified microRNAs in the liver that respond to the absence of microRNA-122 – a highly expressed hepatic microRNA – while also characterizing the degradation pattern of this microRNA7. Because we have used our protocol consistently in numerous experiments, we have been able to observe sample preparations longitudinally, and see that there are no discernible batch effects.
In sharing this protocol, our goal is to enable users to generate high quality, reproducible quantification of microRNAs in virtually any tissue or cell line, using affordable equipment and reagents, and free bioinformatics tools.
PROTOCOL:
Animal experiments were authorized by the Institutional Animal Care and Use Committee of the University of Washington.
Small RNA library preparation
1. RNA isolation
1.1) Isolate RNA from a biological source using a standard RNA isolation reagent, or a kit that enriches for microRNAs. For tissues, it is best to start with samples snap-frozen in liquid nitrogen and ground to a powder using a pre-chilled mortar and pestle.
1.2) Measure each sample’s RNA integrity on an instrument that can quantify RNA and provide an RNA integrity number (RIN). RINs should be >7.
2. 3′ adaptor ligation
2.1) Prepare a ligation reaction in PCR strip tubes, by combining: 11 μL RNA (1-3 μg, using the same amount for each sample), 1.5 μL 10x T4 RNA ligase reaction buffer, ATP-free, 1 μL poly-ethylene glycol (PEG), and 0.5 μL 3′-linker (100 μM Universal miRNA cloning linker).
NOTE: The absence of ATP helps enrich for miRNAs and minimizes cloning of mRNA degradation products. PEG acts as a molecular crowding agent, enhancing successful ligation. The Universal miRNA cloning linker has a 3′ blocking group (amine) to prevent self-ligation, circularization, and ligation to RNA at the 5′ end.
2.2) Heat samples at 95°C on thermocycler for 30-40 s. Cool on ice for 1 min. Add 1 μl T4 RNA ligase 2 and incubate at room temperature for 2 h. Prepare the gel (step 2.3) while samples are incubating.
NOTE: Incubation at room temperature helps prevent linker-linker ligation. We have also successfully used T4 RNA ligase 1.
2.3) Prepare 30 mL of a 15% polyacrylamide gel with 8 M urea (for a 20x20 cm gel): 14.4 g urea, 3 mL 10x Tris-buffered ethylenediaminetetraacetic acid (TBE), 11.2 mL 40% 19:1 acrylamide, and H2O to 30 mL. Solution is best dissolved at 42°C. Immediately before casting, add 150 μl of 10% ammonium persulfate (APS) and 30 μl of tetramethylethylenediamine (TEMED) for polymerization.
2.4) Pour between 0.8 mm separated glass plates in a plastic cast and insert comb. Once the gel is solidified (about 20 min), add 0.5x TBE to tank and wash wells of residual urea by pipetting vigorously.
2.5) Pre-run gel at a constant 375 volts for 25 min without samples so urea can enter gel, then wash the wells again.
NOTE: The amount of voltage may need to be reduced depending on the type of power supply and electrophoresis system that is used.
2.6) Once samples are done ligating, add 15 μL acrylamide loading dye to samples (for a 1:1 ratio), then denature for 5 min at 95°C on a thermocycler.
2.7) Prepare 25 ng/μL of 37 and 44 bp size markers, diluted with one part acrylamide loading dye. Sequences are listed in Table 1.
Table 1.
List of primers.
| Primer | Sequence |
|---|---|
| 3′ linker 1 | rAppCTGTAGGCACCATCAAT–NH2 |
| lower size marker | rArUrCrGrCrArUrGrCrUrGrArCrGrUrArCrUrArGGTAACCGCATCATGCGTC |
| upper size marker | rArArUrCrArGrCrGrGrArUrUrGrCrArUrGrArArCrGrUrArCrArUrArGGTAACCGCATCATGCGTC |
| barcode1 | /5AmMC6/ACGCTCTTCCGATCTrArGrCrG |
| barcode2 | /5AmMC6/ACGCTCTTCCGATCTrCrGrUrC |
| barcode3 | /5AmMC6/ACGCTCTTCCGATCTrCrUrGrG |
| barcode4 | /5AmMC6/ACGCTCTTCCGATCTrArCrUrU |
| barcode5 | /5AmMC6/ACGCTCTTCCGATCTrGrGrGrU |
| barcode6 | /5AmMC6/ACGCTCTTCCGATCTrGrUrUrA |
| barcode7 | /5AmMC6/ACGCTCTTCCGATCTrUrArUrG |
| barcode8 | /5AmMC6/ACGCTCTTCCGATCTrUrCrGrC |
| barcode9 | /5AmMC6/ACGCTCTTCCGATCTrGrCrArG |
| barcode10 | /5AmMC6/ACGCTCTTCCGATCTrArUrArC |
| barcode11 | /5AmMC6/ACGCTCTTCCGATCTrUrUrCrU |
| barcode12 | /5AmMC6/ACGCTCTTCCGATCTrCrArArU |
| barcode13 | /5AmMC6/ACGCTCTTCCGATCTrArArGrA |
| barcode14 | /5AmMC6/ACGCTCTTCCGATCTrUrGrArA |
| barcode15 | /5AmMC6/ACGCTCTTCCGATCTrUrGrGrG |
| barcode16 | /5AmMC6/ACGCTCTTCCGATCTrArUrUrG |
| barcode17 | /5AmMC6/ACGCTCTTCCGATCTrUrCrArU |
| barcode18 | /5AmMC6/ACGCTCTTCCGATCTrGrUrArU |
| RT primer | ATTGATGGTGCCTACAG |
| PCR primer F | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| PCR primer R | CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTATTGATGGTGCCTACAG |
2.8) Load samples on the polyacrylamide gel, leaving at least one lane in between each sample. Load 20 μL of at least two sets of markers, in an asymmetrical pattern to keep track of gel orientation.
2.9) Run gel at a constant 375 volts for the first 15 min and then increase to a constant 425 volts for the remainder of the run. Run until bromophenol blue is about 1-4 cm from the bottom, which takes approximately 2 h.
NOTE: If necessary, the gel may be run at a lower constant voltage for a longer period of time until the bromophenol blue is about 1-4 cm from the bottom.
2.10) Remove gel from glass plates using a plate separator, and place gel on a plastic page protector. Dilute 5 μL of ethidium bromide in 500 μL distilled water, and pipette onto marker lanes just above the top light blue marker (see Figure 1A). CAUTION: Use gloves for ethidium bromide and dispose of waste in accordance with local regulations. Let sit for 5 min.
Figure 1. Extraction of small RNAs from an acrylamide gel.
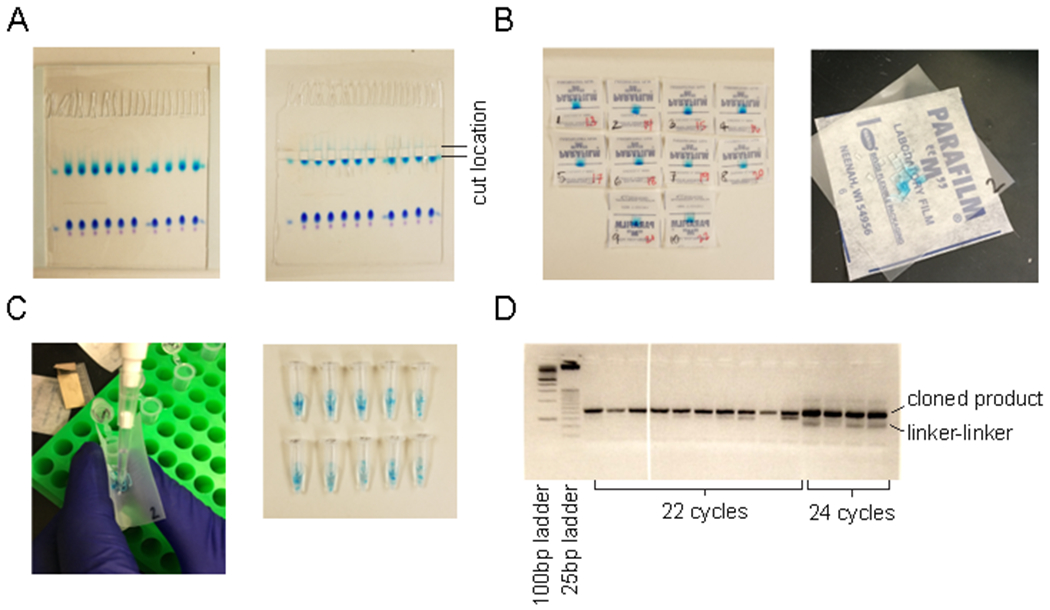
(A) Acrylamide gel and region that is cut corresponding to the size of microRNAs. (B) Gel pieces before and after cutting into smaller fragments. (C) Process for transferring gel fragments into siliconized tubes. (D) PCR reaction on low-melt agarose gel demonstrating correct cloned product compared with linker-linker product and unsaturated (22 cycles) versus saturated (24 cycles) samples.
2.11) Under ultraviolet (UV) light, cut gels from upper to lower marker in each lane using clean razor blade (see Figure 1A). Transfer to a 4 x 4 cm square of laboratory sealing film, then cut gel with about 4 cuts horizontally and 3 vertically to produce 12 small squares (see Figure 1B).
2.12) Pipette 400 μL of 0.3 M sodium chloride (NaCl) on to the sealing film square, and funnel gel pieces into 1.5 mL siliconized tubes (see Figure 1C). Agitate samples on a nutator at 4°C overnight.
NOTE: Other low-retention 1.5 mL tubes can be substituted for siliconized tubes.
2.13) After at least 12 hours of agitation at 4°C, retrieve samples and put them on ice, along with a conical tube of 100% ethanol.
2.14) Transfer 400 μL supernatant to a new tube, and then add 1 mL 100% ethanol and 1 μL of 15 mg/mL glycogen coprecipitant. Be sure to collect as much supernatant as possible, spinning down at 4°C and pipetting more as necessary. Place at −80°C for 1 h, or −20°C for 2 h or longer.
NOTE: Glycogen coprecipitant improves pellet visibility and recovery.
2.15) Spin at 4°C at 17,000 x g for 20-30 min. Remove all traces of ethanol and let pellet air dry for 5 min.
3. 5′ linker ligation
3.1) Re-suspend pellet by pipetting in 6.5 μL nuclease-free water. Letting pellet sit in water for a few minutes first will help with re-suspension.
3.2) After spinning down the pellet and resuspending in water, add 0.5 μL of 100 μM 5′-linker (with barcodes; see Table 1), 1 μL of T4 RNA ligase buffer, 1 μL 10 mM ATP, and 1 μL of PEG. Heat at 90°C for 30 s, then place on ice. Add 1 μL of T4 RNA ligase 1 and let incubate at room temperature for 2 h.
3.3) Add 400 μL of 0.3 M NaCl, followed by 400 μL of acid phenol/chloroform. Vortex 30 s - 1 min (solution will look cloudy), and then spin at 4°C for 10-15 min at max speed in a microcentrifuge (~17,000 x g). Draw off the top layer and place in new 1.5 mL tube. NOTE: Avoid pipetting any of the bottom layer.
3.4) Add 350 μL chloroform, vortex briefly, and then spin at 4°C for 10 min at max speed (~17,000 x g). Draw off the top later and place in new 1.5 mL tube. Add 1.5 μL glycogen coprecipitant and 1 mL 100% ethanol.
NOTE: Again, avoid pipetting any of the bottom layer.
3.5) Vortex briefly, then place at −80°C for at least 1 h, or −20°C overnight.
4. Reverse transcription (RT)
4.1) Turn on 42°C heat block. Spin samples at 4°C at (~17,000 x g) for 20-30 min. Remove all supernatant and let pellet air dry for 5 min.
4.2) Re-suspend pelleted sample in 8.25 μL nuclease-free water, then add: 0.5 μL 100 μM RT primer (Table 1), and 5 μL 2x RT Reaction Mix from a cDNA synthesis kit. Incubate at 42°C for 3 min.
4.3) Add 1.5 μL of 10x RT enzyme to each sample and incubate at 42°C for 30 min in a thermocycler. Place at −20°C or continue with hydrolysis and neutralization.
NOTE: Several RT kits can be utilized for steps 4.2 and 4.3.
4.4) Perform alkaline hydrolysis and neutralization: Make 1 mL of 150 mM potassium hydroxide (KOH) solution (150 μL of 1 M KOH, 20 μL 1 M Tris Base pH 7.5, and 830 μL H2O) and 1 mL of 150 mM hydrochloric acid (HCl) (150 μl of 1 M HCl and 850 μl H2O).
4.5) Before adding to samples, determine the amount of HCl needed to neutralize the KOH solution. Generally, around 20-24 μl of HCl will neutralize the 25 μl of KOH. Check the combination on a pH strip to ensure it is in the right range (7.0 to 9.5 pH).
4.6) Hydrolyze samples by adding 25 μL of 150 mM KOH solution and incubate for 10 min at 95°C.
4.7) Neutralize samples by adding the amount of 150 mM HCl determined in step 4.4, to obtain a final sample pH between 7.0 and 9.5.
5. PCR amplification
5.1) After neutralization, prepare a PCR reaction with: 29.5 μL water, 5 μL 10x Taq buffer, 1 μL dNTP, 2 μL 25 μM forward primer (Table 1), 2 μL 25 μM reverse primer (Table 1), 0.5 μL Taq, and 10 μL of the reverse transcribed cDNA from step 4.6.
5.2) Run the following PCR reaction: 94°C for 2 min, then 20 cycles of 94°C for 45 s, 50°C for 75 s and 72°C for 60 s.
5.3) Run a second PCR reaction of about 2-4 more cycles using 5 μL of product from 5.2. Mix: 34.8 μL water, 5 μL 10x Taq buffer, 1 μL dNTP, 1 μL 25 μM forward primer (Table 1), 1 μL 25 μM reverse primer (Table 1), and 0.2 μL Taq polymerase. Follow the same thermocycler parameters outlined in step 5.2.
NOTE: The purpose of doing two PCR reactions – with the first for 20 cycles and the second for just 2-4 more – is to ensure that the amount of cDNA amplified is in a dynamic range, i.e. not a saturated amount.
6. Agarose gel purification
6.1) Prepare a 4% agarose gel with low-melting agarose. Load 40 μL or more of the PCR product on the gel, along with loading dye. Load 100 bp and 25 bp size markers.
NOTE: The 25 bp ladder helps to distinguish amplified product from linker-linker ligation products. Low-melting agarose gels must be cast with greater care than traditional agarose gels, so closely follow the instructions from the manufacturer.
6.2) For gel extraction, select the cycle number that is visible on the gel but is not saturated (usually 22-24 cycles). Choose similar intensity bands when running multiple samples.
6.3) Cut the band that is above the 125 bp band (the darker band on 25 bp ladder; see Figure 1D). Using a gel extraction kit, follow the manufacturer’s instructions for adding buffer based on a 4% gel, then shake to dissolve agarose in buffer at room temperature.
NOTE: Dissolving at 55°C increases the potential for linker-linker ligation.
6.4) Follow the manufacturer’s gel extraction instructions and elute in 30 μL of elution buffer or water. If the product looked weak on the gel, then reduce elution to 20 μL.
6.5) Measure cDNA concentration using a sensitive technique, and prepare sample library for sequencing. Preparation will depend on the type of sequencing used.
NOTE: Minimum requirements for a sequencing library are typically a 10 μL volume of 10 μM product. If concentrations are too low, pool and ethanol precipitate samples to bring library to the desired concentration.
6.6) Sequence samples using available equipment. A common example would be to run samples using a kit for 50 bp single reads, to obtain approximately 15-25 million reads in a FASTQ output format.
Small RNA sequence alignment and bioinformatics
7. Data upload
7.1) Download FASTQ files generated from each sequencing run. Download a list of microRNA hairpin sequences from miRbase.org8–10.
7.2) Generate a Galaxy account at www.usegalaxy.org and upload a FASTQ file of sequence reads to this account.
7.3) Upload a text file of barcode sequences to the Galaxy account, such as barcodes.txt, which is available as a text file (Supplementary Table 1).
7.4) Upload a FASTA file of microRNA hairpins to the Galaxy account from a database like miRBase.org. Examples of mouse (mousehairpins.fa) or human (humanhairpins.fa) microRNA precursors are provided in Supplementary Tables 2 and 3.
8. Adaptor removal, barcode sort, and trim
8.1) In the left-hand tab, navigate to Genomic file manipulation > FASTA/FASTQ > Clip adapter sequences.
8.2) In “Input file in FASTA or FASTQ format,” enter FASTQ file from the drop-down list. Change “Minimum sequence length” to 18. Change “Source” to “Enter custom sequence.” Enter “CTGTAGGC.” Keep all other default parameters. Click “Execute.”
NOTE: Sequence reads that are shorter than 18 nucleotides are difficult to map uniquely to microRNAs and contain many degradation products.
8.3) In the left-hand tab, navigate to Genomic file manipulation > FASTA/FASTQ > Barcode Splitter.
NOTE: Galaxy functions and headers are updated periodically, so the search function may be necessary to find an equivalent tool or its location. Commercial kits using indexed primers are often already sorted by barcode. Therefore, this step and the barcode trim step are not necessary if starting from a commercial kit.
8.4) For “barcodes to use,” point to “barcodes.txt.” For “library to split,” use “Clip on data” file produced in the previous step. In “number of allowed mismatches,” enter “1.” Click “Execute.”
8.5) Trim the first 4 nucleotides: navigate to Text Manipulations > Trim leading or trailing characters. For “Input dataset” click on the folder icon, which is a dataset collection. Select the batch file of samples, which includes the label “Barcode splitter on data.” In “Trim from the beginning up to this position,” enter “5.” In “Is input dataset in FASTQ format?” enter “Yes.” Click “Execute.”
NOTE: Execution may take several minutes.
9. Alignment of reads to microRNAs
9.1) In Galaxy, navigate to Genomics Analysis > RNA-Seq > Sailfish transcript quantification11.
9.2) For the question “Select a reference transcriptome from your history or use a built-in index?” select “Use one from the history.” Enter the uploaded file mousehairpins.fa from the drop-down list. In FASTA/Q file, click the folder icon to use a dataset collection and select the file that includes “Trim on collection.” Click “Execute.”
NOTE: Execution may take several minutes.
9.3) In the history tab on the right, click on “Sailfish on collection…” which is a list with 19 items. Click on each individual file and click the disk icon to save to your local computer.
NOTE: Individually downloaded files first need to be uncompressed. They also may need to be re-saved with a .txt extension for the purpose of importing into a spreadsheet.
9.4) Open each spreadsheet file in and re-name the “NumReads” column to the treatment condition. Merge the columns together to generate a matrix of microRNAs in the first column and read counts per condition in subsequent columns.
9.5) To calculate differentially expressed microRNAs for each treatment condition, use the file with raw microRNA read counts as input for programs such as DESeq212. DESeq2 is present in Galaxy in the Genomics Analysis > RNA-seq > DESeq2 tab.
9.6) Convert raw reads to normalized microRNA read counts. Counts are normalized to the library sequence depth of the library through the following calculation: [(raw reads/total microRNA reads) * (1,000,000 − number of microRNAs counted) + 1].
NOTE: This calculation provides a normalized reads per million (RPM) mapped microRNAs that can be compared across datasets and biological conditions. The output is a tab delimited file. Sailfish provides a “tpm” output column, though this value is normalized by microRNA hairpin length, which is not necessary in this context.
9.7) If relevant, repeat alignment with custom input sequences (e.g. a vector) to identify reads that map to delivered constructs, like shRNAs.
REPRESENTATIVE RESULTS:
Schematic of steps involved in library preparation
An overall schematic of small RNA extraction, sequencing, and alignment is outlined in Figure 2. Liver samples from one male and one female mouse were collected and snap frozen in liquid nitrogen. Total RNA was extracted and evaluated for quality and concentration.
Figure 2. Schematic of protocol.
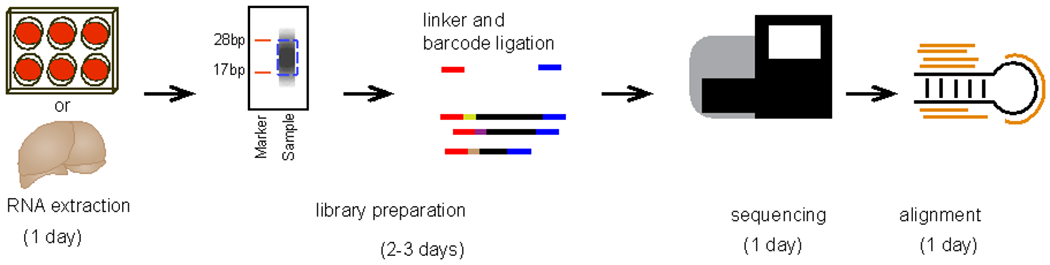
A timeline showing the major steps involved in the procedure.
Small RNA sequencing yields sufficient RNA for sequencing
3 μg of RNA from two independent RNA extractions were used as starting material for small RNA sequencing. Samples were run on an acrylamide gel and cut out between size markers corresponding to 17-28 nt of RNA (Figure 1A). Samples were chopped into fragments for RNA isolation (Figure 1B) and transferred to a low-retention 1.5 mL centrifuge tube (Figure 1C). Barcodes bc7 and bc17 (Table 1) were ligated to the 5’ end of the small RNA. Small RNA libraries were PCR amplified using 22 cycles of PCR to yield 8.0 and 11.2 ng/μL product respectively. Samples were pooled and a 10 nM pooled sample was submitted for high-throughput sequencing using a 50 bp read length.
MiR-122 is the most abundant microRNA in the mouse liver
After barcode sorting, 851,931 reads contained barcodes from liver sample 1 and 650,154 from liver sample 2. Of the reads, 83.5% and 90.0% mapped to microRNAs respectively, with the remaining reads mapping to rRNAs (1.8% and 0.6% respectively), tRNAs and mRNA degradation fragments. After alignment to human microRNA hairpins, we observed strong concordance between microRNA read counts in each replicate (R2 = 0.998; Figure 3). A total of 306 microRNA species were detected, with the greatest number of reads mapping to miR-122 (Supplementary Table 4). MicroRNA abundance was similar between male and female liver samples.
Figure 3. Reproducibility of results from two independent RNA extractions.

Scatterplot of microRNA read counts from a male mouse liver (x-axis) compared with a female mouse liver (y-axis) using a log10-based scale. Each point represents the reads per million (RPM) mapped microRNA count for each individual microRNA.
DISCUSSION:
Despite the identification of microRNAs over 20 years ago13, the process of microRNA sequencing remains laborious and requires specialized equipment, hindering laboratories from routinely adopting in-house protocols14. Other techniques can simultaneously evaluate microRNAs, like microRNA microarrays and multiplexed expression panels; however, these approaches are limited in that they only quantify the microRNAs present in their probe set. Because of this, they miss important features of small RNA sequencing, like the identification of novel microRNAs, and of microRNA isoforms – nucleoside changes that can have important biological function6,7,15.
When starting a new experiment, using a commercial vendor is often easiest because they offer technical support and ease of use. Several commercial options are available for microRNA sequencing, which can be multiplexed to reduce the workload when processing large numbers (>100) of samples. These commercial kits are continually being improved, which is both an advantage and disadvantage. On the one hand, the companies who make these kits have developed novel microRNA capture methods, for example through circularization of their 5′ and 3′ ends prior to sequencing, or use degenerate linkers with random sequences at each end to reduce ligation bias. They have also developed methods to remove adaptor-dimers, for example through ligation of double-stranded adaptors or hybridization of complementary oligonucleotides. On the other hand, commercial kits recommend against modification or alteration of any step. Therefore, if any updates are made to a kit, it is difficult or impossible to compare data derived from old and new versions of kits, as well as data derived from kits from different commercial vendors. Here we have described a protocol that has staying power in the face of commercial alternatives. Our focus on gel purification steps – while they add time to the protocol – enables consistent microRNA capture and reproducibility over the many years we have used it. Several evaluations of the reproducibility between commercial kits and in-house protocols have been made, and we refer the reader to a few of these studies16–19. Importantly, the steps we outline for bioinformatics analysis of microRNAs can be employed regardless of the choice of kit or in-house protocol.
MicroRNA sequencing is often troubled by the choice of barcodes: in some instances, the ligation efficiency of the various barcodes may not be equivalent, leading to biased distributions of sequences in the samples20. It is now recommended to use degenerate bases at the 5′ and 3′ end to minimize ligation biases of specific microRNAs21,22. In this protocol, we have not observed these ligation issues and have observed consistent readouts for technical and biological replicates evaluated with different barcodes5–7,23, but it is important to be aware of them. Methods to avoid ligation bias include the incorporation of index primers in the PCR primers, or to add one or more random RNA nucleosides at the 3′ end of the 5′ adaptor sequence (Table 1). Introducing one or more synthetic spike-in RNA, such as the C. elegans miR-39 microRNA24, is also an option for normalization purposes, which is critical for low-yield situations, like quantifying microRNAs from exosomes. Likewise, for RNA ligation, we have successfully used T4 RNA ligase 1, but ligation with less bias has been demonstrated for a truncated form of T4 RNA ligase 225. Finally, Superscripts III and IV are alternative reverse transcription enzymes that we have used without issue.
The choice of microRNA database can also influence the final normalized results. A challenge with curating microRNA databases is that several novel small RNAs listed as microRNAs are actually fragments of repeat elements and not bona fide microRNAs2. Efforts have been made to retire microRNAs that do not conform to standard criteria, so that the next version of a microRNA database is more refined; however, the next iteration also contains new candidates that need confirmation. When repeat-derived microRNAs are included in alignments, they can skew the results and overwhelm data from existing microRNAs. Therefore, the use of well-curated datasets of microRNAs from different species is essential26,27. We have experienced greatest reproducibility when aligning small RNA sequencing reads to curated lists of conserved microRNAs and have included these hairpins in Supplementary Tables 2 and 3. These lists match estimates of about 500 high confidence microRNAs in the human genome2,26.
As with any technique, results should be confirmed with an orthogonal approach. We have successfully reproduced small RNA sequencing results with small RNA northern blots that incorporate radiolabeled probes to confirm the size and relative abundance of candidate microRNAs6,7,23. Quantitative PCR of microRNAs using split-read sequencing, and confirmation of target mRNA changes using qPCR and western blotting are other options for validation.
In summary, we have provided a method to isolate and sequence microRNAs, and perform alignments against existing microRNA databases. The affordability of the reagents and equipment and the use of open source tools for analysis should make this protocol accessible to anyone. Finally, this protocol is can be used in any tissue or cell line to yield highly reproducible, high-quality reads.
Supplementary Material
Supplemental Table 1. List of barcode sequences.
Supplemental Table 2. Curated list of mouse microRNA precursor sequences.
Supplemental Table 3. Curated list of human microRNA precursor sequences.
Supplemental Table 4. Raw and normalized microRNA read counts.
ACKNOWLEDGMENTS:
We would like to thank members of the laboratories of Andrew Fire and Mark Kay for guidance and suggestions.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Lee RC, Feinbaum RL & Ambros V The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 75 (5), 843–854, (1993). [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP Metazoan MicroRNAs. Cell. 173 (1), 20–51, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau NC, Lim LP, Weinstein EG & Bartel DP An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 294 (5543), 858–862, (2001). [DOI] [PubMed] [Google Scholar]
- 4.Kim VN, Han J & Siomi MC Biogenesis of small RNAs in animals. Nature Reviews Molecular Cell Biology. 10 (2), 126–139, (2009). [DOI] [PubMed] [Google Scholar]
- 5.Gu S et al. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 151 (4), 900–911, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valdmanis PN et al. RNA interference-induced hepatotoxicity results from loss of the first synthesized isoform of microRNA-122 in mice. Nature Medicine. 22 (5), 557–562, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valdmanis PN et al. miR-122 removal in the liver activates imprinted microRNAs and enables more effective microRNA-mediated gene repression. Nature Communications. 9 (1), 5321, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths-Jones S The microRNA Registry. Nucleic Acids Research. 32 (Database issue), D109–111, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A & Enright AJ miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Research. 34 (Database issue), D140–144, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffiths-Jones S, Saini HK, van Dongen S & Enright AJ miRBase: tools for microRNA genomics. Nucleic Acids Research. 36 (Database issue), D154–158, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patro R, Mount SM & Kingsford C Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nature Biotechnology. 32 (5), 462–464, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 15 (12), 550, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fire A et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391 (6669), 806–811, (1998). [DOI] [PubMed] [Google Scholar]
- 14.Etheridge A, Wang K, Baxter D & Galas D Preparation of Small RNA NGS Libraries from Biofluids. Methods in Molecular Biology. 1740 163–175, (2018). [DOI] [PubMed] [Google Scholar]
- 15.Yamane D et al. Differential hepatitis C virus RNA target site selection and host factor activities of naturally occurring miR-122 3 variants. Nucleic Acids Research. 45 (8), 4743–4755, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giraldez MD et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nature Biotechnology. 36 (8), 746–757, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dard-Dascot C et al. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genomics. 19 (1), 118, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeri A et al. Evaluation of commercially available small RNASeq library preparation kits using low input RNA. BMC Genomics. 19 (1), 331, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coenen-Stass AML et al. Evaluation of methodologies for microRNA biomarker detection by next generation sequencing. RNA Biology. 15 (8), 1133–1145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baran-Gale J et al. Addressing Bias in Small RNA Library Preparation for Sequencing: A New Protocol Recovers MicroRNAs that Evade Capture by Current Methods. Frontiers in Genetics. 6 352, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayaprakash AD, Jabado O, Brown BD & Sachidanandam R Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Research. 39 (21), e141, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Nieuwerburgh F et al. Quantitative bias in Illumina TruSeq and a novel post amplification barcoding strategy for multiplexed DNA and small RNA deep sequencing. PLoS One. 6 (10), e26969, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valdmanis PN et al. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene. 34 (1), 94–103, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarzenbach H, da Silva AM, Calin G & Pantel K Data Normalization Strategies for MicroRNA Quantification. Clinical Chemistry. 61 (11), 1333–1342, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viollet S, Fuchs RT, Munafo DB, Zhuang F & Robb GB T4 RNA ligase 2 truncated active site mutants: improved tools for RNA analysis. BMC Biotechnology. 11 72, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang HR et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes & Development. 24 (10), 992–1009, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fromm B et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annual Review of Genetics. 49 213–242, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. List of barcode sequences.
Supplemental Table 2. Curated list of mouse microRNA precursor sequences.
Supplemental Table 3. Curated list of human microRNA precursor sequences.
Supplemental Table 4. Raw and normalized microRNA read counts.