

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder caused by dopamine deficiency. To elucidate network-level changes through the cortico-basal ganglia pathways in PD, we recorded neuronal activity in PD monkeys treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. We applied electrical stimulation to the motor cortices and examined responses in the internal (GPi) and external (GPe) segments of the globus pallidus, the output and relay nuclei of the basal ganglia, respectively. In the normal state, cortical stimulation induced a triphasic response composed of early excitation, inhibition, and late excitation in the GPi and GPe. In the PD state, cortically evoked inhibition in the GPi mediated by the cortico-striato-GPi “direct” pathway was largely diminished, whereas late excitation in the GPe mediated by the cortico-striato-GPe-subthalamo (STN)-GPe pathway was elongated. l-DOPA treatment ameliorated PD signs, particularly akinesia/bradykinesia, and normalized cortically evoked responses in both the GPi and GPe. STN blockade by muscimol injection ameliorated the motor deficit and unmasked cortically evoked inhibition in the GPi. These results suggest that information flow through the direct pathway responsible for the initiation of movements is largely reduced in PD and fails to release movements, resulting in akinesia/bradykinesia. Restoration of the information flow through the direct pathway recovers execution of voluntary movements.
Keywords: dopamine replacement therapy, extracellular neuronal recording, monkey, pathophysiology, subthalamic blockade
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder that affects around 1% of the population over 60 years of age (Reeve et al. 2014). PD is caused by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNc), a nucleus in the basal ganglia, and is characterized by motor and non-motor symptoms, such as bradykinesia, rigidity, tremor, cognitive impairments, depression, and autonomic dysfunctions (Fahn et al. 2011; Seppi et al. 2011; Obeso et al. 2017). These PD symptoms are manifested by malfunctions of the basal ganglia caused by dopamine deficiency (Marsden 1982; Albin et al. 1989; DeLong 1990; Gerfen et al. 1990).
In the normal state, basal ganglia play a pivotal role in controlling voluntary movements by way of receiving cortical inputs, processing information through the cortico-basal ganglia pathways, and returning processed information to the original cortices through the thalamus (Alexander et al. 1986; Alexander and Crutcher 1990). The striatum and subthalamic nucleus (STN) are input stations of the basal ganglia, and the internal segment of the globus pallidus (GPi) and substatia nigra pars reticulta (SNr) are output stations. On the other hand, the external segment of the globus pallidus (GPe) is a relay nucleus. There exist 3 cortico-basal ganglia pathways that convey distinct information from the cortex to the GPi/SNr: The cortico-STN-GPi/SNr “hyperdirect”, cortico-striato-GPi/SNr “direct,” and cortico-striato-GPe-STN-GPi/SNr “indirect” pathways (Nambu et al. 2000, 2002b; Nambu 2008). Signals through the direct pathway facilitate movements by GPi/SNr-thalamic disinhibitory mechanism, whereas signals thorough the hyperdirect and indirect pathways suppress voluntary movements.
The pathophysiology of PD has been extensively studied using non-human primate models, and the following representative 2 mechanisms were proposed: Firing rate increase/decrease in the basal ganglia (Miller and DeLong 1987, 1988; DeLong 1990) and abnormal oscillatory/synchronized activity in the basal ganglia (Bergman et al. 1994, 1998a; Nini et al. 1995; Raz et al. 2000; Soares et al. 2004; Tachibana et al. 2011). However, these previous studies mostly targeted the spontaneous activity changes in one or a few specific nuclei of the basal ganglia, and few studies have been conducted to examine network-level changes.
In the present study, we investigated network-level changes through the cortico-basal ganglia pathways underlying PD symptoms by using a Japanese monkey model of PD generated by the injection of a dopaminergic neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; Burns et al. 1983). We recorded neuronal activity of GPi and GPe neurons in the awake state, and analyzed not only spontaneous activity, but also activity changes in response to electrical stimulation of the motor cortices. Cortical stimulation induces a triphasic response composed of early excitation, inhibition and late excitation in the GPi and GPe in normal monkeys, and we have shown that each response component in the GPi is mediated by the hyperdirect, direct, and indirect pathways, respectively (Nambu et al. 2000, 2002b; Tachibana et al. 2008). Thus, by examining cortically evoked response patterns of GPi and GPe neurons, we can comprehend changes in the signal transmission through the cortico-basal ganglia pathways (Nambu et al. 2000; Chiken et al. 2008, 2015; Sano et al. 2013). We also performed dopamine replacement therapy (Cotzias et al. 1969; Fahn 2008) and STN blockade (Wichmann et al. 1994; Levy et al. 2001; Baron et al. 2002) to alleviate motor signs in the PD monkeys, and examined the causal relationship between changes in PD signs and cortically evoked responses in the GPi and GPe. We have found that reduced signal transmission through the direct pathway has a crucial role in the manifestation of PD symptoms and that dopamine replacement therapy and STN blockade restore this signal transmission. The present study has added a new perspective to the pathophysiology of PD: Alteration of dynamic information flow through the cortico-basal ganglia pathways is the fundamental pathophysiological mechanism underlying PD symptoms, and its restoration is a key mechanism to treat PD symptoms.
Materials and Methods
Animals
Two female Japanese monkeys (Macaca fuscata; Monkey A, 5.0-kg body weight; Monkey P, 6.5 kg) were used to produce an MPTP-induced hemi-parkinsonian monkey model. Additionally, 2 monkeys, one female Japanese monkey (Monkey M, 6.2 kg) and one male Rhesus monkey (Macaca mulatta; Monkey G, 7.2 kg), were used for analyses of spontaneous neuronal firings in the normal state. The experimental protocols were approved by the local Institutional Animal Care and Use Committee, and experiments were conducted according to the guidelines of the National Institutes of Health “Guide for the Care and Use of Laboratory Animals.” Prior to experiments, monkeys were trained daily to sit in a monkey chair quietly. During the experimental session, their body weight and food intake were monitored daily.
Surgery
Each monkey received a surgical operation to fix its head painlessly in a stereotaxic frame attached to a monkey chair. After anesthesia with ketamine hydrochloride (5–8-mg/kg body weight, i.m.) and xylazine hydrochloride (0.5–1 mg/kg, i.m.), sodium thiopental (25 mg/kg, i.v.), or propofol (6–9 μg/mL blood concentration, i.v.; using target-controlled infusion pump, TE-371, Terumo) with fentanyl (2–5 μg/kg, i.m.) was injected. The monkey’s head was fixed in a stereotaxic apparatus. The skull was widely exposed, and small screws were attached to the skull as anchors. The exposed skull and screws were completely covered with transparent acrylic resin, and then 2 pipes for head fixation were mounted and fixed on the monkey’s head. All surgical procedures were performed under aseptic conditions, and arterial oxygen saturation and heart rate were continuously monitored. Antibiotics and analgesics were injected (i.m.) after surgery.
After recovery from the first surgery, the monkey was positioned in a stereotaxic apparatus with its head restrained painlessly using the pipes under anesthesia with ketamine hydrochloride (8 mg/kg, i.m.), and the skull over the primary motor cortex (M1) and supplementary motor area (SMA) was removed. The forelimb regions of the M1 and SMA were identified by electrophysiological mapping (for details, Nambu et al. 2000, 2002a). According to this mapping, 3 pairs of bipolar stimulating electrodes made of a 200 μm-diameter Teflon-coated stainless steel wire were implanted chronically into the forelimb regions of the M1 and SMA: 2 pairs into the M1, and 1 pair into the SMA. These electrodes and exposed areas were covered with transparent acrylic resin.
Recording of Neuronal Activity in the GPi/GPe and STN
Two or 3 days after implantation of the stimulating electrodes, the monkey was positioned in a stereotaxic apparatus with its head painlessly restrained under anesthesia with ketamine hydrochloride (8 mg/kg, i.m.), 2 holes (10–15-mm diameter) were made in the skull over the midline and the lateral region of the M1, and then 2 rectangular plastic chambers covering the holes were fixed with acrylic resin.
Extracellular recordings of GPi/GPe and STN neuronal activity were started on the next day in the awake state. A glass-coated Elgiloy-alloy microelectrode (0.8–1.2 MΩ at 1 kHz) was inserted obliquely (40° or 45° from vertical in the frontal plane) into the GPi/GPe or vertically into the STN through the dura mater using a hydraulic microdrive (Narishige Scientific Instrument Lab) with local application of lidocaine. Signals from the recording electrodes were amplified (×10 000) and filtered (300–5000 Hz). Waveforms of action potentials were continuously monitored with an oscilloscope. The GPi/GPe and STN were identified by the depth profile of microelectrode penetration and their firing patterns. Unit activity was isolated and converted to digital pulses using a homemade time-amplitude window discriminator. Then, single pulse stimulation (monophasic, 300-μs pulse width, 0.5-mA strength) was applied every 1.4 s through the bipolar stimulating electrodes implanted in the M1 and SMA. Responses of GPi/GPe and STN neurons to cortical stimulation and their spontaneous activity were sampled at 2 kHz, and stored on the computer. Cortical stimulation typically induces a triphasic response composed of early excitation followed by inhibition and late excitation in GPi/GPe neurons and biphasic excitation in STN neurons (Nambu et al. 2000; Kita et al. 2004; Tachibana et al. 2008; Iwamuro et al. 2017; Polyakova et al. 2020). Thus, the forelimb regions of the GPi, GPe, and STN were confirmed by the response patterns evoked by cortical stimulation. The GPe and GPi were distinguished by their characteristic firing patterns and GPe/GPi border: GPe neurons fire at high frequencies with pauses, whereas GPi neurons fire continuously without any pauses (DeLong 1971); and the GPe/GPi border was identified by border neurons with middle-frequency regular firings and silent zones corresponding to the medial medullary lamina.
MPTP Treatment
After recordings of neuronal activity in the normal state, MPTP treatment was conducted. Under general anesthesia with ketamine hydrochloride (5–8 mg/kg, i.m.), xylazine hydrochloride (0.5–1 mg/kg, i.m.), and sodium thiopental (25 mg/kg, i.v.), the common carotid artery together with the internal and external carotid arteries were dissected at its bifurcation point from the neck region ipsilateral to the side of neuronal recordings. MPTP was dissolved in saline (2 mg/mL) and applied through the common carotid artery (2.1 mg/kg for Monkey A and 0.7 mg/kg for Monkey P) with the external carotid artery clumped (Tachibana et al. 2011). Monkey A exhibited clear PD signs with akinesia and rigidity on the contralateral side to the carotid arterial injection. Monkey P exhibited only mild bradykinesia after the carotid arterial injection and received additional intravenous MPTP injection through the great saphenous vein (0.3 mg/kg, 3 times, every 3 days; 0.9 mg/kg in total). Two weeks after the final MPTP treatment, monkey’s behavior was clinically assessed by a monkey parkinsonian rating scale (Smith et al. 1993; score, 0–20), which is a sum of the following scores: Tremor (0–3), posture (0–2), gait (0–4), bradykinesia (0–4), balance (0–2), gross motor skills (0–3), and defense reaction (0–2). More serious parkinsonian stages are represented by higher scores. Then, recordings of GPi/GPe and STN activity were performed in the PD state by the same methods as used in the normal state for 5 months in Monkey A and 9 months in Monkey P.
l-DOPA Treatment of PD Monkeys
Prior to l-DOPA treatment, activity of a single isolated GPi/GPe or STN neuron was recorded, and spontaneous firings and cortically evoked responses were examined. Then, l-DOPA (DOPASTON; 2.0–2.5 mg/kg) was intravenously injected through the great saphenous vein, while recording of the neuronal activity was continued. Changes in PD motor signs were carefully observed, and spontaneous firings and cortically evoked responses of the same neuron were continuously monitored. In case obvious improvement of PD signs was not observed in 10 min, additional l-DOPA injections were made (up to 5.0 mg/kg in total).
Injection of Muscimol into the STN of PD Monkeys
To examine the effects of STN blockade on PD signs and cortically evoked responses of GPi/GPe neurons, muscimol, gamma-aminobutyric acid type A (GABA A) receptor agonist, was injected into the STN. Based on the mapping of the STN, an injection electrode consisting of a needle of a microsyringe for muscimol injection and a 50-μm diameter Teflon-coated tungsten wire electrode for recording (attached 0.7 mm ahead the tip of the needle; see Nambu et al. 2000) was vertically inserted into the STN using a hydraulic microdrive. Neuronal activity was recorded, and responses to cortical stimulation were examined. The STN where neurons showed biphasic responses to cortical stimulation was sought, and the injection electrode was placed 0.7-mm ventral to the position. Before muscimol injection into the STN, activity of a single isolated GPi/GPe neuron was recorded, and spontaneous firings and cortically evoked responses were examined. Then, 0.5–2.0 μL of muscimol (0.5–1.0-μg/μL dissolved in saline) was injected into the STN through the injection electrode. Changes in spontaneous firings and cortically evoked responses of the same GPi/GPe neuron together with PD motor signs were continuously monitored.
Data Analysis
Neuronal responses to cortical stimulation were examined by constructing peri-stimulus time histograms (PSTHs; bin width of 1 ms) for 100 stimulus trials. The mean value and standard deviation (SD) of the discharge rate during the 100-ms period preceding the stimulation onset were calculated for each PSTH and considered the baseline discharge rate. Changes in neuronal activity were judged significant if the discharge rate during at least 2 consecutive bins (2 ms) reached a significance level of P < 0.05 (one-tailed t-test; Nambu et al. 2000; Chiken et al. 2008, 2015; Tachibana et al. 2008; Sano et al. 2013). The latency of each component was defined as the time of the first bin that exceeded this level. The responses were judged to end when 2 consecutive bins fell below the significance level. The end point was determined as the time at which the last bin exceeded this level. The duration of each component was defined as the time during the significant change (between the latency and the end point). The amplitude of each component of cortically evoked responses was defined as the difference between the number of spikes during the significant change and that of the baseline discharge in the PSTH (i.e., the area of the response). If no significant changes were found, the amplitude was set to zero. For population PSTHs, PSTHs of all neurons exhibiting significant responses to cortical stimulation were averaged and filtered with a Gaussian filter (σ = 1.6 ms).
Spontaneous discharge rates and patterns were examined in neurons that exhibited responses to cortical stimulation. Firing rates were calculated from continuous digitized recordings for 30 s or 50 s. Autocorrelograms (bin width of 0.5 ms) were constructed from the same spike trains, and shuffled autocorrelograms were also obtained after random shuffling of the spike trains. Then, they were filtered with a Gaussian filter (σ = 1.6 ms). The mean value and SD of autocorrelation coefficients were calculated between 0.1 and 0.6 s (1000 bins) of the shuffled autocorrelograms, and the peaks and troughs of the autocorrelograms were judged significant if the coefficient during at least 8 consecutive bins (4 ms) exceeded the level of P < 0.0005 (one-tailed t-test). The firing pattern of neurons was judged to be oscillatory if at least 2 peaks and one trough were observed (Chiken et al. 2008; Tachibana et al. 2008). The mean and coefficient of variation (CV) of interspike intervals (ISIs) and the burst index (Hutchison et al. 1998; Sano et al. 2013; Chiken et al. 2015) were also calculated to characterize firing patterns.
Data obtained from Monkeys A and P were separately analyzed, except that those of STN blockade were combined and analyzed due to small numbers of neurons. All values are expressed as mean ± SD unless otherwise stated.
Histology
In the final experiment, several sites of neuronal recordings were marked by passing a cathodal direct current (20 μA for 30 s) through a recording electrode. Each monkey was anesthetized deeply with sodium thiopental (50 mg/kg, i.v.), ketamine hydrochloride (10 mg/kg, i.m.), and xylazine hydrochloride (1–2 mg/kg, i.m.), and perfused transcardially with 0.1-M phosphate buffer (PB, pH 7.3) followed by 10% formalin in 0.1-M PB, and 0.1-M PB containing 10% sucrose. Brains were removed immediately and saturated with 0.1-M PB containing 30% sucrose. They were cut into frontal 60-μm thick sections on a freezing microtome. To confirm the recording sites, 2 of every 3 sections were mounted onto gelatin-coated glass slides, stained with 0.7% neutral red, dehydrated, and cover-slipped. Images of the brain sections were captured using a digital microscope (BZ-X710, Keyence), and the recording sites in the GPi/GPe and STN were reconstructed according to the lesions made by current injection and traces of electrode tracks. The sites of stimulation in the M1 and SMA were also confirmed histologically. Every 3 sections were used for tyrosine hydroxylase (TH) immunohistochemistry to evaluate the loss of dopaminergic neurons. Free-floating sections were incubated overnight with mouse anti-TH antiserum (Chemicon 1:2000 dilution) in 0.1-M phosphate buffered saline containing 1% normal horse serum and 0.5% Triton X-100 at 4°C. Subsequently, sections were incubated with biotinylated anti-mouse IgG antibody (1:200 dilution; Vector) for 2 h, and were then visualized by the ABC method (Vectastain Elite ABC kit; Vector) with intensification using NiCl2. Sections were mounted, dehydrated, and cover-slipped. Images of the brain sections were captured using a digital microscope (BZ-X710) with the constant exposure. Numbers of TH-positive cells in a region of interest (ROI, 500 μm × 500 μm) in the SNc was counted. Intensity of TH-immunoreactivity in a ROI (500 μm × 500 μm) in the putamen (Put, IPut) and optic tract (IOT) of the same slice was measured using ImageJ software (National Institute of Health), and the relative intensity in the Put was calculated as (IPut—IOT)/IOT.
Results
PD Signs and Loss of Dopaminergic Neurons after MPTP Treatment
Two Japanese monkeys were subjected to MPTP and exhibited prominent PD motor signs in their contralateral bodies to the carotid arterial injection of MPTP (Table 1): Monkey A treated with 2.1-mg/kg MPTP exhibited severe PD signs (17/20 in monkey parkinsonian rating scale; Smith et al. 1993) with akinesia and rigidity, and Monkey P treated with 1.6-mg/kg MPTP exhibited moderate PD signs (10/20) with bradykinesia, rigidity, and a decreased capacity to use its affected limbs. These motor signs were stable during the whole experimental period (5 months in Monkey A and 9 months in Monkey P), and no obvious motor recoveries were observed.
Table 1.
MPTP injection and parkinsonian rating scale in Monkeys A and P
| MPTP injection | Monkey parkinsonian rating scale (Smith et al. 1993) | |
|---|---|---|
| Monkey A | 2.1 mg/kg (Carotid arterial injection, 2.1 mg/kg) | 17/20 (Tremor, 0/3; posture, 2/2; gate, 4/4; bradykinesia, 4/4; balance, 2/2; gross motor skills, 3/3; and defense reaction, 2/2) |
| Monkey P | 1.6 mg/kg (Carotid arterial injection, 0.7 mg/kg; Venous injection, 0.9 mg/kg) | 10/20 (Tremor, 0/3; posture, 1/2; gate, 2/4; bradykinesia, 3/4; balance, 1/2; gross motor skills, 2/3; and defense reaction, 1/2) |
TH immunohistochemistry confirmed loss of dopaminergic neurons in the SNc and dopaminergic axonal terminals in the Put and caudate nucleus (Cd) of the ipsilateral side to the carotid arterial injection of MPTP (Fig. 1). In the ipsilateral side, TH immunoreactivity in the SNc, Cd, and Put was almost completely lost in Monkey A (Fig. 1A,D) and greatly diminished in Monkey P (Fig. 1B,E). On the other hand, TH immunoreactivity in the ventral tegmental area (VTA) was rather preserved (Fig. 1A,B). In the contralateral side to the MPTP injection, TH immunoreactivity was robustly observed in the VTA, SNc, Cd, and Put (Fig. 1C,F). Quantitative analysis (see Supplementary Fig. S1A) revealed that the numbers of TH-positive cells in the ipsilateral SNc of Monkeys A and P were significantly smaller than that in the contralateral SNc (P < 0.0001, one-way analysis of variance [ANOVA] with Tukey’s post hoc test). The relative intensities of TH-positive terminals in the ipsilateral Put of both Monkeys A and P were significantly lower than that in the contralateral Put (see Supplementary Fig. S1B; P < 0.0001, one-way ANOVA with Tukey’s post hoc test), and that in the ipsilateral Put of Monkey A was lower than that of Monkey P (P < 0.0001). In summary, TH immunoreactivity was almost completely lost in Monkey A exhibiting severe PD signs, and greatly diminished in Monkey P exhibiting moderate PD signs.
Figure 1 .
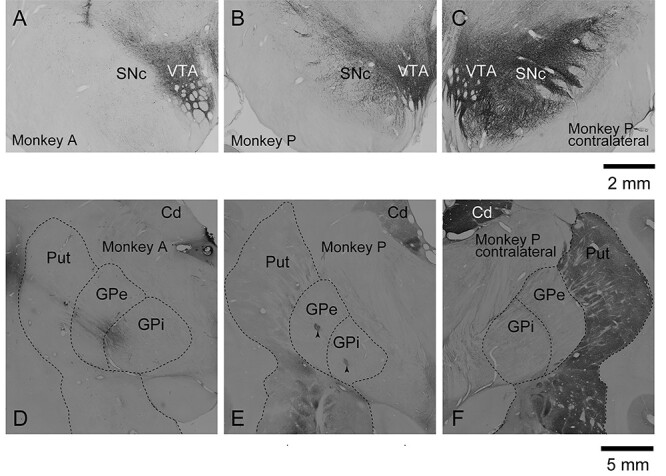
Photomicrographs showing immunoreactivity of TH in the basal ganglia in Monkeys A and P. TH immunoreactivity in the SNc and VTA (A–C) and in the Cd and Put (D–F) in the hemisphere ipsilateral to the carotid arterial injection of MPTP in Monkeys A (A, D) and P (B, E), and that in the contralateral hemisphere in Monkey P (C, F). Arrowheads (E) indicate lesion sites in the GPe and GPi made by current injection (see Materials and Methods).
Spontaneous Firings of GPi and GPe Neurons in the Normal and PD States
We examined spontaneous activity of GPi and GPe neurons exhibiting responses to cortical stimulation in the normal and PD states (Fig. 2A,B). In the normal state, the mean firing rates of GPi and GPe neurons of Monkey A were in the range of normal monkeys (Monkeys M and G). The mean firing rates of GPi neurons of Monkeys A and P in the PD state were not significantly different from those of normal monkeys (Fig. 2A; P = 0.53; one-way ANOVA). However, the mean firing rates of their GPe neurons in the PD state were significantly lower than those of normal monkeys (Fig. 2B; Monkey A, P < 0.0001; and Monkey P, P = 0.0012; one-way ANOVA with Dunnett’s post hoc test).
Figure 2 .
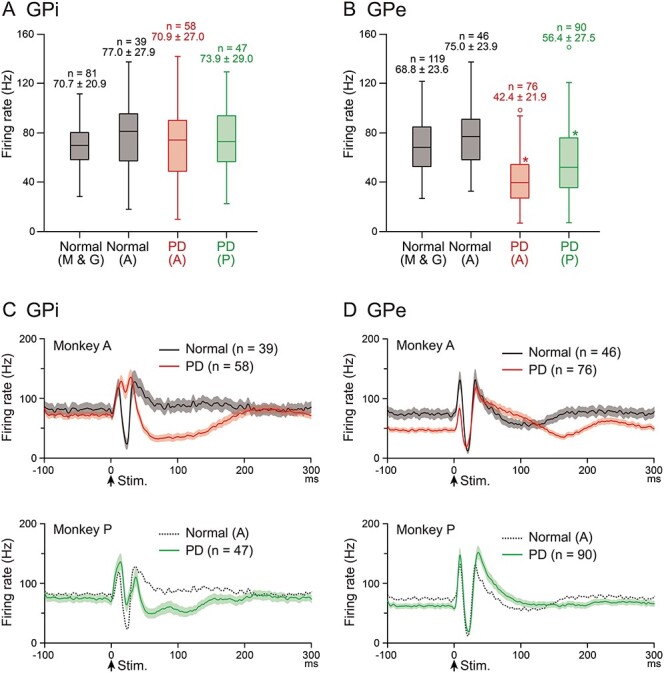
Spontaneous firing rates and cortically evoked responses of GPi and GPe neurons in the normal and PD states. (A, B) Spontaneous firing rates of GPi (A) and GPe (B) neurons in the normal (Monkeys M and G, and A) and PD (Monkeys A and P) states are shown in box plots (center lines, median; top and bottom of boxes, the first and third quartiles; whiskers, the minimum and maximum values excluding outliers; and open circles, outliers outside 1.5 times the interquartile range from the first and third quartiles). Numbers of neurons (n) and firing rates (mean ± SD in Hz) are also indicated. *P < 0.01 significantly different from those in normal monkeys (Monkeys M and G). (C, D) Population PSTHs (with Gaussian filter, σ = 1.6 ms) of GPi (C) and GPe (D) neurons in Monkey A in the normal (black lines in upper 2 panels) and PD (red lines) states, and in Monkey P in the PD state (green lines in lower 2 panels). Population PSTHs in the normal state in Monkey A are also shown in the lower panels (gray dotted lines) as reference. The number of neurons used for each population PSTH is indicated by n, and the shaded areas represent ± standard error of the mean. Stimulation was delivered at time 0 (arrows).
The percentage of neurons exhibiting oscillatory activity was very small in the PD state: Only 8.6% of GPi neurons (Monkey A, 1/58 and Monkey P, 8/47) and 1.2% of GPe neurons (Monkey A, 0/76 and Monkey P, 2/90) exhibited oscillation in the PD state (see Supplementary Fig. S2), whereas no GPi and GPe neurons exhibited oscillation in normal monkeys (Monkeys M and G). The burst index was increased in the PD state in both the GPi and GPe of Monkeys A and P (Table 2). The CV of ISIs in the GPi and mean ISIs in the GPe were increased in the PD state in Monkey A.
Table 2.
Spontaneous firing patterns of GPi and GPe neurons in the normal and PD states
| Normal (Monkeys M and G) | PD Monkey A | PD Monkey P | ||
|---|---|---|---|---|
| GPi | No. of neurons | 81 | 58 | 29 |
| Mean ISIs (ms) | 15.6 ± 6.2 | 18.3 ± 15.2 | 15.1 ± 7.7 | |
| CV of ISIs | 0.79 ± 0.62 | 1.18 ± 0.50* | 0.81 ± 0.16 | |
| Burst index | 1.83 ± 0.92 | 4.60 ± 2.64* | 2.96 ± 1.57* | |
| GPe | No. of neurons | 119 | 76 | 28 |
| Mean ISIs (ms) | 16.7 ± 7.1 | 30.8 ± 20.7* | 23.8 ± 24.4 | |
| CV of ISIs | 1.33 ± 0.89 | 1.39 ± 0.59 | 1.17 ± 0.31 | |
| Burst index | 1.85 ± 0.68 | 4.90 ± 3.97* | 5.46 ± 8.52* |
Notes: Values are mean ± SD. *P < 0.01, significantly different from the values in normal monkeys (Monkeys M and G) (one-way ANOVA with Dunnett’s post hoc test).
Cortically Evoked Responses of GPi and GPe Neurons in the Normal and PD States
Cortically evoked responses of GPi and GPe neurons were examined before and after MPTP treatment, that is, in the normal and PD states in Monkey A, and in the PD state in Monkey P (Fig. 2C,D). In the normal state in Monkey A, the most common response pattern of GPi neurons was a triphasic response composed of early excitation followed by inhibition and late excitation (23/39 neurons, 59%), as observed in population PSTHs (black line in Fig. 2C, upper), which is in accordance with our previous reports in normal monkeys (Nambu et al. 2000; Tachibana et al. 2008; Iwamuro et al. 2017). On the other hand, in the PD state in Monkey A, cortically evoked inhibition in the GPi was mostly lost as observed in population PSTHs (red line in Fig. 2C, upper). Among 58 GPi neurons recorded in the PD state in Monkey A, only 6 neurons (10%) exhibited a triphasic response, and the great majority (47/58, 81%) exhibited biphasic or monophasic excitation, which was often followed by long-lasting inhibition. Similarly, in the PD state in Monkey P, cortically evoked inhibition in the GPi was much smaller than that in normal Monkey A, as observed in population PSTHs (green line in Fig. 2C, lower), and the number of GPi neurons showing a triphasic response was smaller (18/47, 38%) than that showing biphasic or monophasic excitation (19/47, 40%). The proportion of GPi neurons exhibiting the responses with inhibition, such as excitation-inhibition-excitation, excitation-inhibition, inhibition-excitation, and monophasic inhibition, was significantly smaller in the PD state than that in the normal state (Normal, 97% in Monkey A; PD, 19% in Monkey A, and 60% in Monkey P; chi-square test with Bonferroni correction, P < 0.0001). Quantitative analysis (Table 3) revealed that the duration and amplitude of inhibition became significantly smaller, and those of early excitation became significantly larger in the PD state in both Monkeys A and P. In the monkey with severer PD motor signs (Monkey A), cortically evoked inhibition was more profoundly lost, suggesting causality between the loss of cortically induced inhibition and PD signs.
Table 3.
Cortically evoked responses of GPi and GPe neurons in the normal and PD states
| Normal Monkey A | PD Monkey A | PD Monkey P | |||
|---|---|---|---|---|---|
| GPi | No. of responding neurons | 39 | 58 | 47 | |
| Early excitation | |||||
| Duration (ms) | 2.8 ± 2.8 | 10.4 ± 10.0** | 7.9 ± 7.4** | ||
| Amplitude (spikes) | 28.6 ± 36.0 | 93.2 ± 110.4** | 83.0 ± 90.1* | ||
| Inhibition | |||||
| Duration (ms) | 11.7 ± 4.6 | 2.3 ± 7.0** | 10.7 ± 18.0* | ||
| Amplitude (spikes) | 87.5 ± 35.9 | 11.6 ± 35.6** | 65.1 ± 79.7** | ||
| Late excitation | |||||
| Duration (ms) | 8.5 ± 9.5 | 10.1 ± 10.9 | 12.7 ± 18.1 | ||
| Amplitude (spikes) | 104.2 ± 152.2 | 89.3 ± 126.7 | 133.4 ± 205.9 | ||
| GPe | No. of responding neurons | 46 | 76 | 90 | |
| Early excitation | |||||
| Duration (ms) | 3.5 ± 2.5 | 2.5 ± 2.9** | 6.1 ± 6.7 | ||
| Amplitude (spikes) | 39.8 ± 35.2 | 22.5 ± 30.0** | 68.8 ± 61.2 | ||
| Inhibition | |||||
| Duration (ms) | 11.6 ± 5.4 | 9.7 ± 7.1 | 9.5 ± 6.6 | ||
| Amplitude (spikes) | 79.9 ± 36.2 | 46.1 ± 45.6** | 57.7 ± 45.7** | ||
| Late excitation | |||||
| Duration (ms) | 9.9 ± 12.1 | 47.7 ± 37.3** | 33.6 ± 33.6** | ||
| Amplitude (spikes) | 106.2 ± 119.7 | 350.9 ± 362.2** | 316.2 ± 357.7** | ||
Notes: Values are mean ± SD.
* P < 0.05, **P < 0.01, significantly different from the values in the normal state in Monkey A (one-way ANOVA with Dunnett’s post hoc test).
On the other hand, in the GPe, the most common response pattern was triphasic in both the normal and PD states (Fig. 2D). However, in the PD state, late excitation was elongated in both Monkeys A and P (red line in Fig. 2D, upper, and green line in Fig. 2D, lower). Quantitative analysis (Table 3) confirmed that the duration and amplitude of late excitation became significantly larger in the PD state in both monkeys. In addition, the amplitude of inhibition became smaller in the PD state in both monkeys, and the amplitude and duration of early excitation were diminished in the PD state in Monkey A.
Effects of l-DOPA Treatment on Cortically Evoked Responses of GPi and GPe Neurons
To examine causality between changes in cortically evoked responses in the GPi/GPe and PD signs, we performed dopamine replacement therapy, which is commonly used to alleviate motor symptoms in PD patients (Cotzias et al. 1969; Fahn 2008). In both Monkeys A and P, intravenous l-DOPA injection (2.0–2.5 mg/kg) improved akinesia/bradykinesia, gross motor skills, and rigidity of their affected forelimbs within 5–10 min. In 35–50 min after injection, PD motor signs returned to the original levels before injection in most cases. During l-DOPA treatment, dyskinesia was rarely observed (oral dyskinesia, twice; 2/29 sessions in Monkey A and 0/32 in Monkey P), and these data during dyskinesia were not used for analyses. We examined activity of one and the same GPi and GPe neurons in the PD monkeys before and during dopamine replacement therapy (Fig. 3).
Figure 3 .
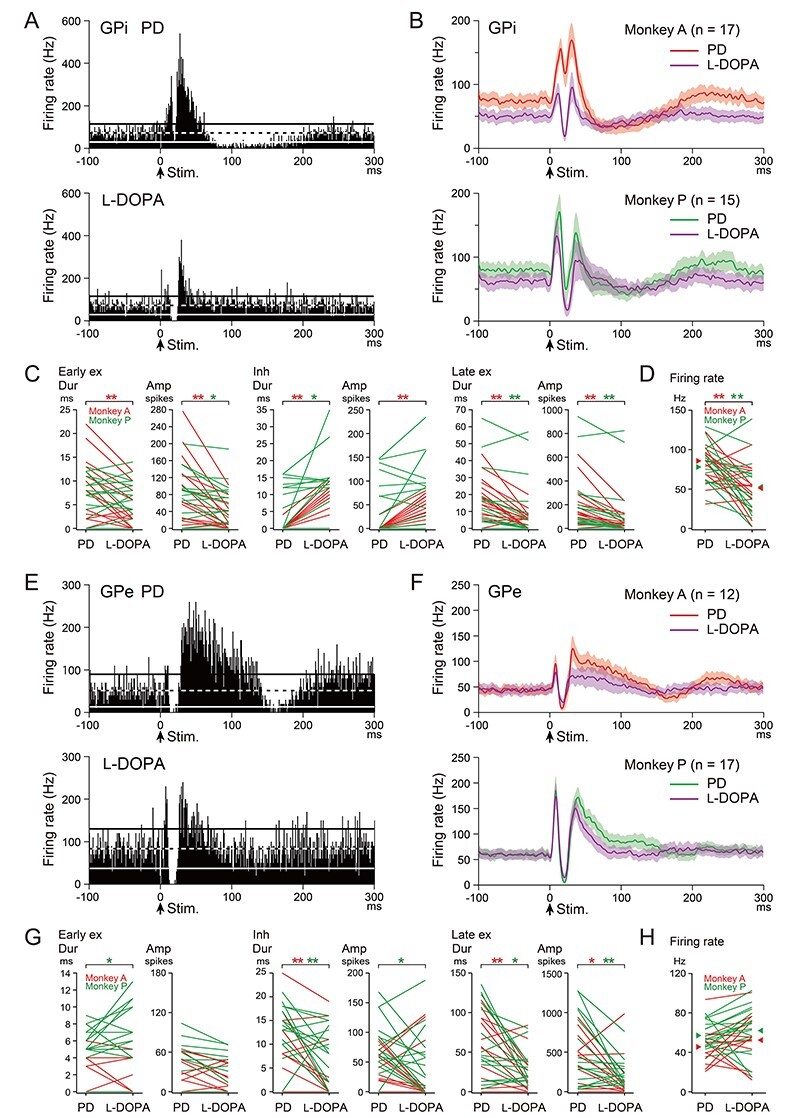
Effects of l-DOPA treatment on cortically evoked responses and spontaneous firing rates of GPi (A–D) and GPe (E–H) neurons in the PD monkeys. (A, E) Typical examples of PSTHs (100 trials, bin width of 1 ms) of one and the same GPi (A) and GPe (E) neurons in the PD state (upper) and during l-DOPA treatment (lower) in Monkey A. Stimulation was delivered at time 0 (arrows). The mean firing rate and statistical levels of P < 0.05 (one-tailed t-test) are indicated in PSTHs by a black and white dashed line (mean), and black (upper limit) and white (lower limit) solid lines, respectively. (B, F) Population PSTHs of GPi (B) and GPe (F) neurons in the PD state (red and green lines) and during l-DOPA treatment (purple lines) in Monkeys A (upper) and P (lower). (C, G) Durations (Dur) and amplitudes (Amp) of cortically evoked early excitation (Early ex), inhibition (Inh), and late excitation (Late ex) in the PD state (PD) and during l-DOPA treatment (l-DOPA) of GPi (C) and GPe (G) neurons. Data from the same neurons are connected in Monkeys A (red lines) and P (green lines). Difference was examined and indicated separately in Monkeys A (red asterisk) and P (green asterisk). *P < 0.05, **P < 0.01, significantly different between the PD state and during l-DOPA treatment (one-tailed paired t-test). (D, H) Spontaneous firing rates of GPi (D) and GPe (H) neurons in the PD state and during l-DOPA treatment in Monkeys A (red lines) and P (green lines). Mean firing rates in the PD state and during l-DOPA treatment are indicated separately in Monkeys A (red arrowheads) and P (green arrowheads).
Cortically evoked responses of 32 GPi neurons (Monkey A, 17 and Monkey P, 15) were examined in the PD monkeys before and during l-DOPA treatment (Fig. 3A–C). A typical example is shown in Figure 3A. In the PD state, cortical stimulation induced biphasic excitation without inhibition followed by long-lasting inhibition (Fig. 3A, upper). However, during l-DOPA treatment, PD signs were relieved, and cortical stimulation induced a triphasic response composed of early excitation, inhibition, and late excitation in the same neuron (Fig. 3A, lower). Other GPi neurons showed similar changes as observed in population PSTHs: In the PD state, cortical stimulation induced biphasic excitation (Monkey A, red line in Fig. 3B, upper) or a triphasic response with small inhibition (Monkey P, green line in Fig. 3B, lower). However, during l-DOPA treatment, cortical stimulation induced a distinct triphasic response in both monkeys (purple lines in Fig. 3B, upper and lower). The majority of GPi neurons in the PD state exhibited biphasic or monophasic excitation without inhibition (Monkey A, 16/17 and Monkey P, 8/15), whereas most of these neurons exhibited responses with inhibition during l-DOPA treatment (Monkey A, 13/17 and Monkey P, 11/15). Quantitative analysis (Fig. 3C) also confirmed that inhibition was increased in the duration (Monkeys A and P) and amplitude (Monkey A) during l-DOPA treatment. In addition, early excitation (duration in Monkey A, amplitude in Monkeys A and P) and late excitation (duration and amplitude in Monkeys A and P) were diminished during l-DOPA treatment.
Spontaneous firing rates were also examined in the same GPi neurons (Monkey A, 17 and Monkey P, 15) before and during l-DOPA treatment in the PD monkeys (Fig. 3D). In the PD state, the mean firing rates of these GPi neurons were 85.3 ± 19.2 Hz (Monkey A) and 77.8 ± 27.4 Hz (Monkey P), whereas they were significantly decreased during l-DOPA treatment in both monkeys (Monkey A, 50.9 ± 26.8 Hz, P = 0.0003, and Monkey P, 52.6 ± 36.2 Hz, P = 0.029; one-tailed paired t-test).
Cortically evoked responses of 29 GPe neurons (Monkey A, 12 and Monkey P, 17) were also examined in the PD monkeys before and during l-DOPA treatment (Fig. 3E–G). A typical example is shown in Figure 3E. In the PD state, cortical stimulation induced a triphasic response with elongated late excitation (Fig. 3E, upper). However, during l-DOPA treatment, the elongated late excitation was markedly diminished (Fig. 3E, lower). Other GPe neurons showed similar changes as observed in population PSTHs of both monkeys (Fig. 3F). Most GPe neurons in the PD state exhibited responses with elongated late excitation (Monkey A, 10/12, red line in Fig. 3F upper; and Monkey P, 13/17, green line in Fig. 3F, lower), whereas the elongated late excitation was diminished in most of them during l-DOPA treatment (Monkey A, 8/10 and Monkey P, 9/13; purple lines in Fig. 3F, upper and lower). Quantitative analysis (Fig. 3G) confirmed that the duration and amplitude of late excitation were significantly diminished in both Monkeys A and P during l-DOPA treatment. In addition, inhibition was also slightly but significantly diminished during l-DOPA treatment (duration in Monkeys A and P, amplitude in Monkey P), whereas early excitation was increased (duration in Monkey P).
As for the spontaneous firing rates of these GPe neurons (Fig. 3H), the mean firing rates in the PD state were 45.7 ± 20.0 Hz (Monkey A, 12 neurons) and 57.5 ± 13.4 Hz (Monkey P, 17 neurons) and remained unchanged during l-DOPA treatment (Monkey A, 52.6 ± 27.0 Hz, P = 0.19, and Monkey P, 62.4 ± 24.0 Hz, P = 0.21; one-tailed paired t-test).
Effects of STN Blockade on Cortically Evoked Responses of GPi and GPe Neurons
To further examine causality between changes in cortically evoked responses in the GPi/GPe and PD signs, we next performed STN blockade in the PD monkeys. STN lesion or blockade is also known to effectively ameliorate PD symptoms in patients (Alvarez et al. 2001; Levy et al. 2001; Rodriguez-Rojas et al. 2018) and PD signs in a monkey model (Bergman et al. 1990; Wichmann et al. 1994; Baron et al. 2002). To block STN activity, muscimol (0.5–2.0 μL of 0.5–1.0 μg/μL) was injected into the STN of the PD monkeys. STN blockade ameliorated PD motor signs, such as akinesia/bradykinesia, deficits of gross motor skills, and rigidity of their forelimbs within 15–30 min after muscimol injection, and the effects usually continued over 80 min in both Monkeys A and P. We examined activity of one and the same GPi and GPe neurons in the PD monkeys before and during STN blockade (Fig. 4).
Figure 4 .
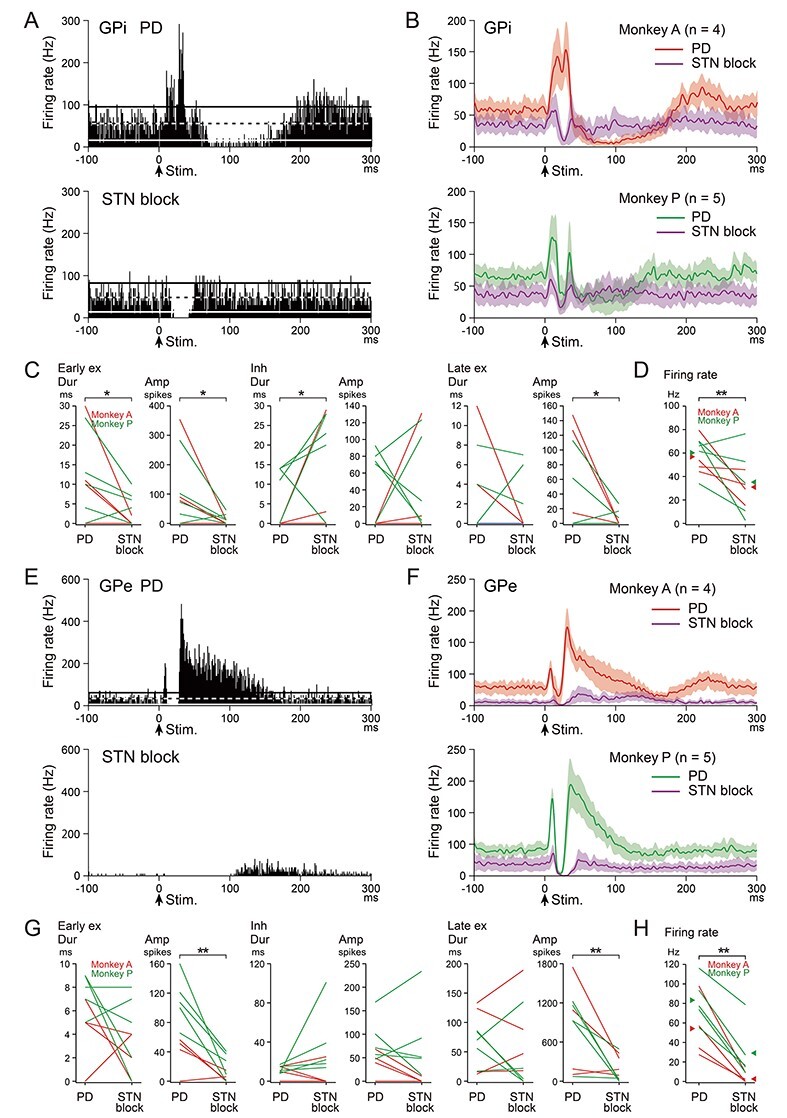
Effects of blockade of the STN on cortically evoked responses and spontaneous firing rates of GPi (A–D) and GPe (E–H) neurons in the PD monkeys. (A, E) Typical examples of PSTHs of one and the same GPi (A) and GPe (E) neurons in the PD state (upper) and during STN blockade (lower) in Monkey A. (B, F) Population PSTHs of GPi (B) and GPe (F) neurons in the PD state (red and green lines) and during STN blockade (purple lines) in Monkeys A (upper) and P (lower). (C, G) Durations and amplitudes of cortically evoked early excitation, inhibition, and late excitation in the PD state (PD) and during STN blockade (STN block) of GPi (C) and GPe (G) neurons in Monkeys A (red lines) and P (green lines). Data from Monkeys A and P were combined and examined. *P < 0.05, **P < 0.01, significantly different between the PD state and during STN blockade (one-tailed paired t-test). (D, H) Spontaneous firing rates of GPi (D) and GPe (H) neurons in the PD state and during STN blockade in Monkeys A (red lines) and P (green lines). Mean firing rates in the PD state and during STN blockade are indicated separately in Monkeys A (red arrowheads) and P (green arrowheads).
Cortically evoked responses of 9 GPi neurons (Monkey A, 4 and Monkey P, 5) were examined in the PD monkeys before and during STN blockade (Fig. 4A–C). A typical example is shown in Figure 4A. In the PD state, cortical stimulation induced biphasic excitation with following long-lasting inhibition (Fig. 4A, upper). During STN blockade, PD signs were relieved, and cortical stimulation induced inhibition without early and late excitation in the same neuron (Fig. 4A lower). Other GPi neurons showed similar changes as observed in population PSTHs (Fig. 4B). In the PD state, all GPi neurons in Monkey A (4/4) exhibited biphasic or monophasic excitation without inhibition (red line in Fig. 4B, upper), and most GPi neurons in Monkey P (4/5) exhibited a triphasic response (green line in Fig. 5B, lower). During STN blockade, early and late excitation was largely diminished, and clear inhibition appeared in both monkeys (purple lines in Fig. 4B, upper and lower). Quantitative analysis (Fig. 4C) revealed that STN blockade increased inhibition (duration), and diminished early excitation (duration and amplitude) and late excitation (amplitude).
Figure 5 .
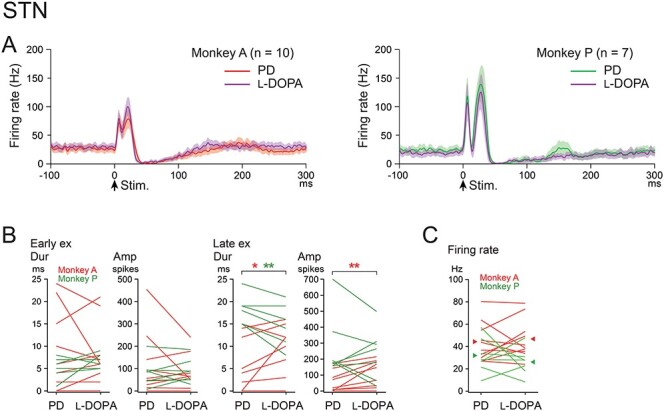
Effects of l-DOPA treatment on cortically evoked responses and spontaneous firing rates of STN neurons in the PD monkeys. (A) Population PSTHs of STN neurons in the PD state (red and green lines) and during l-DOPA treatment (purple lines) in Monkeys A (left) and P (right). (B) Durations and amplitudes of cortically evoked early excitation (Early ex) and late excitation (Late ex) in the PD state and during l-DOPA treatment in Monkeys A (red lines) and P (green lines). Difference was examined and indicated separately in Monkeys A (red asterisk) and P (green asterisk). *P < 0.05, **P < 0.01, significantly different between the PD state and during l-DOPA treatment (one-tailed paired t-test). (C) Spontaneous firing rates of STN neurons in the PD state and during l-DOPA treatment in Monkeys A (red lines) and P (green lines).
Cortically evoked responses of 9 GPe neurons (Monkey A, 4 and Monkey P, 5) were also examined in the PD monkeys before and during STN blockade (Fig. 4E–G). A typical example is shown in Figure 4E. In the PD state, cortical stimulation induced a triphasic response with elongated late excitation (Fig. 4E, upper). During STN blockade, cortical stimulation induced long inhibition with no early excitation and greatly diminished late excitation in the same neuron (Fig. 4E, lower). Other GPe neurons showed similar changes as observed in population PSTHs (Fig. 4F). In the PD state, most GPe neurons exhibited a triphasic response with elongated late excitation (Monkey A, 2/4, red line in Fig. 4F, upper and Monkey P, 4/5, green line in Fig. 4F, lower). STN blockade typically abolished both early and late excitation, and induced long inhibition in both monkeys (purple lines in Fig. 4F, upper and lower). Quantitative analysis (Fig. 4G) revealed that STN blockade diminished both early and late excitation (amplitude).
Spontaneous firing rates were also examined in the same GPi (Monkey A, 4 and Monkey P, 5) and GPe (Monkey A, 4 and Monkey P, 5) neurons before and during STN blockade in the PD monkeys (Fig. 4D,H). In the PD state, the mean firing rates of these GPi and GPe neurons were 58.6 ± 14.6 Hz and 70.3 ± 29.6 Hz, respectively, and were significantly decreased during STN blockade (GPi, 33.3 ± 22.9 Hz, P = 0.0079, and GPe, 17.5 ± 24.6 Hz, P < 0.0001; one-tailed paired t-test).
Effects of l-DOPA Treatment on Cortically Evoked Responses of STN Neurons
To examine possible contribution of neuronal activity changes in the STN to those in the GPi and GPe in the PD state, we recorded cortically evoked responses in one and the same STN neurons in the PD monkeys (17 neurons; Monkey A, 10 and Monkey P, 7) before and during l-DOPA treatment (Fig. 5). Both in the PD state (Monkey A, red line in Fig. 5A, left and Monkey P, green line in Fig. 5A, right) and during l-DOPA treatment (purple lines in Fig. 5A left and right), STN neurons typically exhibited a biphasic response composed of early and late excitation, which was similar to those observed in normal monkeys (Nambu et al. 2000; Iwamuro et al. 2017; Polyakova et al. 2020). Quantitative analysis (Fig. 5B) unveiled no changes in cortically evoked early excitation and inconsistent changes in late excitation during l-DOPA treatment: Late excitation was increased in Monkey A (duration and amplitude), whereas decreased in Monkey P (duration). The mean firing rates of these STN neurons (Fig. 5C) were 44.1 ± 17.6 Hz (Monkey A) and 31.9 ± 15.9 Hz (Monkey P) in the PD state and remained unchanged during l-DOPA treatment (Monkey A, 46.6 ± 17.6 Hz, P = 0.31, and Monkey P, 26.2 ± 12.2 Hz, P = 0.24; one-tailed paired t-test).
Location of Recorded GPi/GPe and STN Neurons
We mapped recording sites of GPi and GPe neurons in the normal and PD states, some of which responded and some of which did not respond to electrical stimulation of the forelimb regions of the M1/SMA (Fig. 6A). Responding neurons (filled circles) were located in the middle of the dorsal-ventral direction in the posterior GPi and GPe, corresponding to the forelimb M1/SMA regions of the GPi and GPe (Nambu et al. 2000; Iwamuro et al. 2017). The border between responding and non-responding (open circles) neurons in the normal state seems to be preserved in the PD state, suggesting that responding areas of the GPe and GPi did not change between the normal and PD states. We also mapped recording and muscimol injection sites in the STN (Fig. 6B), and found that they were located in the dorsolateral STN, corresponding to the forelimb M1/SMA regions of the STN (Nambu et al. 2000; Iwamuro et al. 2017; Polyakova et al. 2020).
Figure 6 .
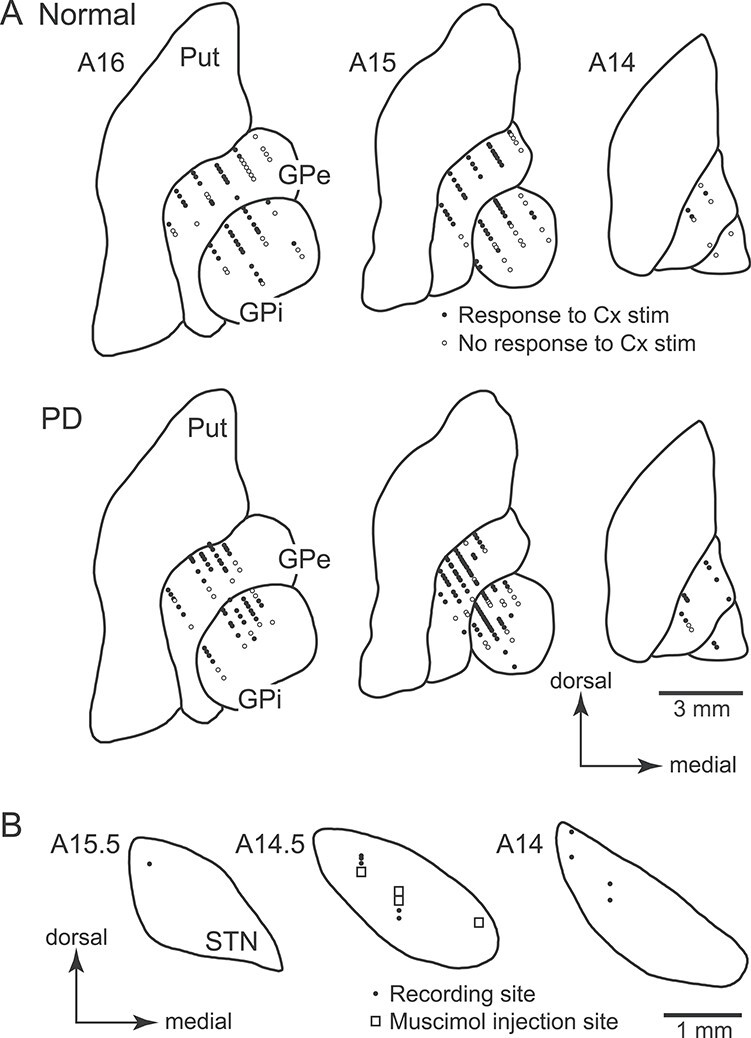
Distribution of recorded GPi, GPe, and STN neurons in Monkey A shown in frontal sections. (A) Locations of GPi and GPe neurons recorded in the normal (upper) and PD (lower) states. Filled circles represent neurons that responded to cortical (Cx) stimulation, whereas open circles represent neurons that showed no responses to Cx stimulation. (B) Locations of recorded STN neurons (filled circles) and muscimol injection sites for STN blockade (open squares). The distance of the sections anterior to the auditory meatus is shown in millimeters in the upper left of each section.
Discussion
In the present study, we analyzed activity of GPi and GPe neurons in the awake state of 2 PD Japanese monkeys that exhibited akinesia/bradykinesia and rigidity, but not tremor. The following results were obtained: 1) In the PD state, cortically evoked inhibition in the GPi mediated by the cortico-striato-GPi direct pathway was drastically diminished, whereas cortically evoked late excitation in the GPe was elongated; 2) l-DOPA treatment ameliorated PD signs and normalized abnormal cortically evoked responses in both the GPi and GPe, that is, clear inhibition appeared in the GPi, and elongated late excitation was diminished in the GPe; 3) STN blockade by muscimol injection again ameliorated PD signs, largely diminished cortically evoked early and late excitation in the GPi and GPe, and unmasked cortically evoked inhibition in the GPi; 4) In the PD state, the mean firing rate remained unchanged in the GPi, whereas that in the GPe was reduced; and 5) Oscillatory neuronal activity was rarely observed in both the GPi and GPe in the PD state. These results suggest that alteration in signal transmission through the cortico-basal ganglia pathways, particularly profoundly reduced information flow through the cortico-striato-GPi direct pathway that is necessary for initiation of voluntary movements (Nambu et al. 2002b, 2015; Nambu 2008; Chiken et al. 2015), is responsible for the manifestation of major PD symptoms (Fig. 7). The contribution of changes in spontaneous firing rates and/or patterns in the GPi appears to be minor.
Figure 7 .
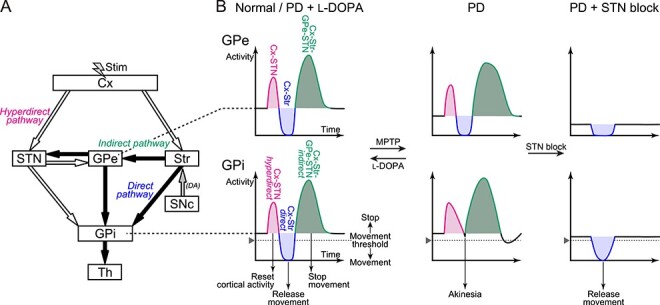
Schematic diagrams showing the basal ganglia circuitry and cortically evoked responses in the GPe and GPi. (A) Basal ganglia circuitry including the cortico (Cx)-STN-GPi hyperdirect, Cx-striato (Str)-GPi direct, and Cx-Str-GPe-STN-GPi indirect pathways. White and black arrows represent excitatory glutamatergic and inhibitory GABAergic projections, respectively. Electrical stimulation (Stim) was applied in the Cx, and responses in the GPe and GPi were recorded. DA, dopamine. (B) Cortically evoked responses in the GPe (upper) and GPi (lower) in the normal or PD state during l-DOPA treatment (left), PD state (middle), and PD state during STN blockade (right). Cortically evoked early excitation (magenta) and following inhibition (blue) and late excitation (green) in the GPe are mediated by the Cx-STN-GPe, Cx-Str-GPe, and Cx-Str-GPe-STN-GPe pathways, respectively, and those in the GPi are mediated by the hyperdirect, direct, and indirect pathways, respectively.
Abnormal Cortically Evoked Responses in the GPi and GPe and Their Relationship with PD Symptoms
In the normal state, cortical stimulation typically induced a triphasic response composed of early excitation followed by inhibition and late excitation in the GPi (Nambu et al. 2000; Tachibana et al. 2008; Iwamuro et al. 2017), whereas in the PD state, cortically evoked inhibition mediated by the cortico-striato-GPi direct pathway was largely diminished (Fig. 2 and Table 3). The result indicates that information flow through the direct pathway (Fig. 7A) responsible for the initiation of voluntary movements (Nambu et al. 2002b, 2015; Nambu 2008; Chiken et al. 2015) is severely impaired in the PD state (Fig. 7B, GPi, left and middle). Monkey A with more severer PD signs showed more profound loss of cortically evoked inhibition in the GPi compared with Monkey P (Fig. 2 and Table 3), supporting this notion. Inhibitory responses of GPi neurons during passive limb movements, which are presumably transferred through the direct pathway, decreased in PD monkeys (Boraud et al. 2000), suggesting that not only electrically induced information flow but also natural information flow through the direct pathway is impaired. These changes through the direct pathway can be caused by the following intra- and extra-striatal mechanisms. 1) Activity of striatal neurons is controlled by dopaminergic inputs from the SNc. Loss of dopaminergic inputs through D1 receptors (D1Rs) coupled with Gs/olf proteins onto the striato-GPi direct pathway neurons decreases their intrinsic excitability in the PD state (Surmeier et al. 2007; Gerfen and Surmeier 2011), causing diminished cortically evoked inhibition in the GPi. 2) D1Rs are also expressed in the striato-GPi GABAergic axon terminals presynaptically and increase GABA release (Kliem et al. 2007). Loss of dopamine should decrease GABA release from the striato-GPi terminals within the GPi, which would cause diminished cortically evoked inhibition in the GPi. Actually, supplementation of dopamine restored cortically evoked inhibition in the GPi in the present study (Fig. 3), although it may not fully restore basal ganglia functions to the normal state (Heimer et al. 2006). Loss of signal transmission through D1Rs reduced locomotor activity in mice and diminished cortically evoked inhibition in the GPi, supporting both mechanisms discussed above (Chiken et al. 2015).
An increase of early excitation in the GPi was also found in the PD state (Fig. 2 and Table 3). These observations suggest that information flow through the cortico-STN-GPi hyperdirect pathway is enhanced, contradicting the recent studies (Mathai et al. 2015; Chu et al. 2017). Several possible mechanisms for increased early excitation in the GPi can be considered: 1) Decreased spontaneous firing rates of GPe neurons in the PD state (Fig. 2) reduce tonic inhibitory inputs to the STN, resulting in increased excitability of STN neurons; 2) Loss of direct dopaminergic inputs from the SNc to STN may also be involved (Obeso et al. 2004; Rommelfanger and Wichmann 2010: Galvan et al. 2014); and 3) Cortically evoked early excitation in the GPi is relatively enhanced due to decrease of the following inhibition in the PD state (Fig. 2 and Table 3). However, 1) and 2) are less probable because, in the present study, early excitation in the GPe mediated by the cortico-STN-GPe pathway was not enhanced in the PD state (Fig. 2 and Table 3), and early excitation in the STN in the PD state was not diminished by l-DOPA treatment (Fig. 5).
Similarly in the GPe, in the normal state, cortical stimulation induced a triphasic response composed of early excitation followed by inhibition and late excitation (Nambu et al. 2000; Kita et al. 2004; Iwamuro et al. 2017). However, in the PD state, this triphasic response pattern was basically preserved, and cortically evoked late excitation mediated by the cortico-striato-GPe-STN-GPe pathway (Ryan and Clark 1991; Nambu et al. 2000; Kita et al. 2004; Sano et al. 2013) was elongated (Fig. 2 and Table 3). The result indicates that information flow through the cortico-striato-GPe-STN-GPe pathway (Fig. 7A) is enhanced in the PD state (Fig. 7B, GPe, left and middle). l-DOPA administration actually shortened and normalized cortically evoked late excitation (Fig. 3). The following mechanisms could explain these changes through the cortico-striato-GPe-STN-GPe pathway: 1) Loss of dopaminergic inputs through D2 receptors (D2Rs) coupled with Gi/o proteins onto the striato-GPe indirect pathway neurons should increase their intrinsic excitability in the PD state (Surmeier et al. 2007; Gerfen and Surmeier 2011); 2) D2Rs are also expressed in the striato-GPe GABAergic axon terminals presynaptically, and loss of dopaminergic inputs would enhance GABA release from the striato-GPe terminals within the GPe (Hadipour-Niktarash et al. 2012); 3) Signal transmission from the GPe to STN could be enhanced in the PD state (Fan et al. 2012; Chu et al. 2015); and 4) Signal transmission from the STN to GPe would be enhanced in the PD state. Concerning 1) and 2), these effects would lead to an increase in both the cortically evoked inhibition and following late excitation in the GPe. Inhibition in the PD state was actually reduced during l-DOPA treatment (Fig. 3). Although an increase in inhibition was not detected in the PD state (Table 3), enhanced inhibition in the GPe might be masked by decreased spontaneous firing rates (Fig. 2). As for 1), 2), and 3), in the PD state, these effects would lead to an increase in the cortically evoked late excitation in the STN, which is mediated by the cortico-striato-GPe-STN pathway (Maurice et al. 1998; Polyakova et al. 2020); however, it is not plausible because late excitation in the STN in the PD state was diminished only in Monkey P by l-DOPA treatment (Fig. 5). In terms of 4), this possibility has not been examinedyet.
Information flow through the cortico-striato-GPe-STN-GPi indirect pathway (Fig. 7A), which shares several common components with the cortico-striato-GPe-STN-GPe pathway, might also be enhanced in the PD state (Fig. 7B, middle). Late excitation in the GPi mediated by the indirect pathway was significantly reduced during l-DOPA treatment (Fig. 3); however, changes in cortically evoked late excitation were not detected in the PD state (Fig. 2 and Table 3). Instead, in the PD state, cortically evoked long-lasting inhibition was frequently observed after late excitation in the GPi (Figs 2–4). This may be caused by the augmented late excitation in the GPe (Figs 2–4 and Table 3) that could inhibit GPi activity (Fig. 7B, middle) through direct GPe-GPi GABAergic inhibitory projections (Fig. 7A; Smith and Bolam 1989; Bolam and Smith 1992; Shink and Smith 1995; Kita 2001).
Effects of l-DOPA Treatment and STN Blockade on Cortically Evoked Responses in the GPi and GPe
l-DOPA treatment effectively improved PD motor signs, particularly akinesia/bradykinesia, gross motor skills, and rigidity. At the same time, cortically evoked responses in both the GPi and GPe were normalized, that is, cortically evoked inhibition in the GPi was recovered and abnormally augmented late excitation in the GPe was diminished (Fig. 3). In addition, STN blockade again effectively alleviated such PD motor signs, and extensively diminished early and late excitation in both the GPi and GPe, resulting in a clear manifestation of cortically evoked inhibition in the GPi (Fig. 4). Similar effects were observed in normal monkeys with STN blockade as well (Nambu et al. 2000). These results suggest that the proper balance between excitatory inputs through the hyperdierect and indirect pathways and inhibitory inputs through the direct pathway to the GPi is crucial for normal motor control and that a reduction of inhibitory inputs through the direct pathway to the GPi is correlated with appearance of PD signs. The effectiveness of l-DOPA treatment and STN blockade in reducing major PD signs may be caused by the recovery of signal transmission through the direct pathway (Fig. 7B, left and right).
Little Changes in Spontaneous Firings in the GPi and GPe in the PD State
According to the classical model of the basal ganglia, dopaminergic inputs exert excitatory effects on striatal direct pathway neurons through D1Rs and inhibitory effects on striatal indirect pathway neurons through D2Rs, and thus loss of dopaminergic inputs increases and decreases the mean firing rates of GPi and GPe neurons, respectively, and increased GPi firings lead to a decrease in spontaneous firing rates of thalamic and cortical neurons, resulting in akinesia (Albin et al. 1989; DeLong 1990; Gerfen et al. 1990). The model has been supported by a number of subsequent studies (Filion and Tremblay 1991; Boraud et al. 1996; Wichmann et al. 2002; Soares et al. 2004). The present study also showed decreased firing rates of GPe neurons in the PD monkeys (Fig. 2), as reported by recent studies (Raz et al. 2000; Rivlin-Etzion et al. 2008; Tachibana et al. 2011). However, the present study did not detect any firing rate changes in the GPi of the PD monkeys with moderate and severe motor signs (Fig. 2), which is in accordance with several recent studies (Wichmann et al. 1999; Raz et al. 2000; Rivlin-Etzion et al. 2008; Tachibana et al. 2011). In the present study, the paradoxical effects of l-DOPA were observed in the PD monkeys: l-DOPA treatment decreased firing rates of GPi neurons in the PD monkeys that were not different from those in the normal state; and it did not change firing rates of GPe neurons in the PD monkeys that were lower than those in the normal state (Fig. 3). The PD state may accompany morphological changes in the basal ganglia (Villalba et al. 2009; Fan et al. 2012; Chu et al. 2015, 2017; Mathai et al. 2015; Suarez et al. 2016), and l-DOPA treatment may not be able to fully recover neuronal activity to the normal state (Heimer et al. 2006).
In previous studies, firing pattern changes, especially oscillatory activity in the theta and/or beta ranges, were commonly found in the basal ganglia of PD patients (Hurtado et al. 1999; Levy et al. 2000; Magnin et al. 2000) and PD monkeys (Bergman et al. 1994; Nini et al. 1995; Raz et al. 2000; Soares et al. 2004; Tachibana et al. 2011), proposing that oscillatory activity may disturb information flow through the basal ganglia and cause PD symptoms (Bergman et al. 1998a; Brown 2007). In the present study, the burst index was increased in both the GPi and GPe in the PD state (Table 2), which is consistent with previous studies (Wichmann and Soares 2006; Tachibana et al. 2011); however, most neurons in the GPi and GPe of the PD monkeys with mild and severe motor signs did not exhibit oscillatory activity in the PD state (see Supplementary Fig. S2). A previous study reported that abnormal synchronized oscillatory activity emerged later than major motor signs during progressive PD (Leblois et al. 2007).
Therefore, firing rate and pattern changes could not fully explain PD signs. Alteration of signal transmission through the hyperdirect, direct, and indirect pathways that causes dynamic activity changes in the GPi/GPe is a fundamental feature of PD, and they may secondarily cause firing rate and pattern changes in the GPi/GPe (Nambu et al. 2015). Depending on the activity balance between the 3 pathways, spontaneous firing rates may be modulated (Leblois et al. 2006), or oscillatory activity may be induced (Terman et al. 2002; Leblois et al. 2006; Tachibana et al. 2011).
The Pathophysiology of Tremor and Rigidity
Oscillatory activity synchronized with tremor was reported in the STN and thalamus, especially cerebellar receiving thalamus, of PD patients, and lesion in the STN or thalamus abolished tremor, suggesting its causal role (Lenz et al. 1988; Fox et al. 1991; Rodriguez et al. 1998; Ohye et al. 2012). African green monkeys developed prolonged episodes of low- (5–7 Hz) and high- (10–16 Hz) frequency tremor and exhibit strong oscillatory and correlated GPe/GPi activity at low- and high-frequencies (Bergman et al. 1994, 1998b; Raz et al. 2000). On the other hand, macaque monkeys usually failed to develop low-frequency tremor and showed various degrees of oscillatory GPe/GPi activity in the PD state: no tremor and almost no oscillatory activity (in the present study), and infrequent short episodes of high-frequency (10–16 Hz) postural/action tremor and oscillatory (8–15 Hz) activity (Tachibana et al. 2011). Taken together, these observations suggest that the level of synchronized oscillatory activity seems to be related to tremor.
In PD patients, stretch reflexes, especially long-latency reflexes, were enhanced and may serve as the basis for rigidity (Berardelli et al. 1983; Rothwell et al. 1983). However, the mechanism by which dopamine deficiency enhances stretch reflexes is not known yet. Descending pathways from the basal ganglia to the spinal cord through the pedunculopontine tegmental nucleus (PPN) may also contribute to rigidity. The GPi projects to the PPN, whose activity decreases muscle tone (Takakusaki et al. 2004), and loss of inhibition in the GPi in the PD state (Fig. 7B, GPi, middle) continuously suppresses PPN activity, resulting in increased muscletone.
Pathophysiology of PD and Therapeutic Mechanisms of l-DOPA Treatment and STN Blockade
Based on the present study and our dynamic model of the cortico-basal ganglia functions explaining the control mechanism of voluntary movements (Nambu et al. 2002b, 2015; Nambu 2008), we would like to propose a new model that explains the pathophysiology of PD symptoms and therapeutic mechanisms of l-DOPA treatment and STN blockade (Fig. 7). In the normal state, signals through the corico-striato-GPi direct pathway play a key role in the initiation of voluntary movements (Fig. 7A): The GPi, the output nucleus of the basal ganglia, is continuously suppressing thalamocortical activity in the basal state, and when cortical neurons are excited, signals through the direct pathway induce cortically derived phasic inhibition in the GPi, which increases thalamocotical activity by a disinhibitory mechanism, leading to a release of motor actions (Fig. 7B, left). On the other hand, signals through the cortico-STN hyperdirect and cortico-striato-GPe-STN-GPi indirect pathways reset on-going cortical activity and stop movements, respectively. In the PD state, dopamine deficiency causes large suppression of signal transmission through the direct pathway, which leads to a failure to induce enough inhibition in the GPi, resulting in akinesia/bradykinesia (Fig. 7B, middle).
l-DOPA treatment restores signal transmission through the direct pathway, and enables a release of motor actions upon cortical excitation (Fig. 7B, left). On the other hand, STN blockade suppresses signal transmission through the cortico-STN-GPi hyperdirect and cortico-GPe-STN-GPi indirect pathways extensively, and thus diminished signal transmission through the direct pathway is unmasked, and enables a release of motor actions (Fig. 7B, right). The same mechanism may also apply to deep brain stimulation (DBS) in the STN in PD patients, because STN-DBS appears to block signal transmission through the STN (Maurice et al. 2003; Chiken and Nambu 2014). The present findings that restoration of signal transmission through the direct pathway can alleviate PD symptoms will provide an important clue to develop novel therapeutic methods and to find more effective manipulating targets for the treatment ofPD.
Authors’ Contributions
M.T. and A.N. designed the research; S.C., M.T., and A.N. performed the research; S.C. analyzed the data; S.C., M.T., and A.N. wrote the paper.
Funding
MEXT KAKENHI (“Non-linear Neuro-oscillology,” 15H05873 to A.N.); JSPS KAKENHI (16K07014 to S.C., and 19KK0193 and 26250009 to A.N.); JST CREST (JPMJCR1853 to S.C.); AMED (JP20dm0307005, JP20dm0207050 to A.N.).
Notes
We thank S. Sato, H. Isogai, K. Awamura, K. Miyamoto, T. Sugiyama, and N. Suzuki for technical assistance, and Y. Yamagata for her critical reading of the manuscript. We also thank the late Dr Hironobu Tokuno for discussion on the early idea of the presentwork.
One Japanese monkey used in the present study was provided through National BioResource Project (NBRP) “Japanese Monkeys” of AMED. Conflict of Interest: The authors declare no conflicts of interest.
Supplementary Material
References
- Albin RL, Young AB, Penney JB. 1989. The functional anatomy of basal ganglia disorders. Trends Neurosci. 12:366–375. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. 1990. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13:266–271. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. 1986. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 9:357–381. [DOI] [PubMed] [Google Scholar]
- Alvarez L, Macias R, Guridi J, Lopez G, Alvarez E, Maragoto C, Teijeiro J, Torres A, Pavon N, Rodriguez-Oroz MC, et al. 2001. Dorsal subthalamotomy for Parkinson's disease. Mov Disord. 16:72–78. [DOI] [PubMed] [Google Scholar]
- Baron MS, Wichmann T, Ma D, DeLong MR. 2002. Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J Neurosci. 22:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardelli A, Sabra AF, Hallett M. 1983. Physiological mechanisms of rigidity in Parkinson's disease. J Neurol Neurosurg Psychiatry. 46:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. 1990. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 249:1436–1438. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. 1994. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 72:507–520. [DOI] [PubMed] [Google Scholar]
- Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, Vaadia E. 1998a. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci. 21:32–38. [DOI] [PubMed] [Google Scholar]
- Bergman H, Raz A, Feingold A, Nini A, Nelken I, Hansel D, Ben-Pazi H, Reches A. 1998b. Physiology of MPTP tremor. Mov Disord. 13(Suppl 3):29–34. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Smith Y. 1992. The striatum and the globus pallidus send convergent synaptic inputs onto single cells in the entopeduncular nucleus of the rat: a double anterograde labelling study combined with postembedding immunocytochemistry for GABA. J Comp Neurol. 321:456–476. [DOI] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Bioulac B, Gross C. 1996. High frequency stimulation of the internal Globus Pallidus (GPi) simultaneously improves parkinsonian symptoms and reduces the firing frequency of GPi neurons in the MPTP-treated monkey. Neurosci Lett. 215:17–20. [DOI] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Bioulac B, Gross CE. 2000. Ratio of inhibited-to-activated pallidal neurons decreases dramatically during passive limb movement in the MPTP-treated monkey. J Neurophysiol. 83:1760–1763. [DOI] [PubMed] [Google Scholar]
- Brown P. 2007. Abnormal oscillatory synchronisation in the motor system leads to impaired movement. Curr Opin Neurobiol. 17:656–664. [DOI] [PubMed] [Google Scholar]
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. 1983. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 80:4546–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiken S, Nambu A. 2014. Disrupting neuronal transmission: mechanism of DBS? Front Syst Neurosci. 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiken S, Shashidharan P, Nambu A. 2008. Cortically evoked long-lasing inhibition of pallidal neurons in a transgenic mouse model of dystonia. J Neurosci. 28:13967–13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiken S, Sato A, Ohta C, Kurokawa M, Arai S, Maeshima J, Sunayama-Morita T, Sasaoka T, Nambu A. 2015. Dopamine D1 receptor-mediated transmission maintains information flow through the cortico-striato-entopeduncular direct pathway to release movements. Cereb Cortex. 25:4885–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Atherton JF, Wokosin D, Surmeier DJ, Bevan MD. 2015. Heterosynaptic regulation of external globus pallidus inputs to the subthalamic nucleus by the motor cortex. Neuron. 85:364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, McIver EL, Kovaleski RF, Atherton JF, Bevan MD. 2017. Loss of hyperdirect pathway cortico-subthalamic inputs following degeneration of midbrain dopamine neurons. Neuron. 95:1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotzias GC, Papavasiliou PS, Gellene R. 1969. Modification of Parkinsonism--chronic treatment with L-dopa. N Engl J Med. 280:337–345. [DOI] [PubMed] [Google Scholar]
- DeLong MR. 1971. Activity of pallidal neurons during movement. J Neurophysiol. 34:414–427. [DOI] [PubMed] [Google Scholar]
- DeLong MR. 1990. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13:281–285. [DOI] [PubMed] [Google Scholar]
- Fan KY, Baufreton J, Surmeier DJ, Chan CS, Bevan MD. 2012. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. J Neurosci. 32:13718–13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. 2008. The history of dopamine and levodopa in the treatment of Parkinson's disease. Mov Disord. 23(Suppl 3):S497–S508. [DOI] [PubMed] [Google Scholar]
- Fahn S, Jankovic J, Hallett M. 2011. Principles and practice of movement disorders. 2nd ed. Philadelphia: Elsevier Saunders. [Google Scholar]
- Filion M, Tremblay L. 1991. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 547:142–151. [PubMed] [Google Scholar]
- Fox MW, Ahlskog JE, Kelly PJ. 1991. Stereotactic ventrolateralis thalamotomy for medically refractory tremor in post-levodopa era Parkinson's disease patients. J Neurosurg. 75:723–730. [DOI] [PubMed] [Google Scholar]
- Galvan A, Hu X, Rommelfanger KS, Pare JF, Khan ZU, Smith Y, Wichmann T. 2014. Localization and function of dopamine receptors in the subthalamic nucleus of normal and parkinsonian monkeys. J Neurophysiol. 112:467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. 2011. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 34:441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, Sibley DR. 1990. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 250:1429–1432. [DOI] [PubMed] [Google Scholar]
- Hadipour-Niktarash A, Rommelfanger KS, Masilamoni GJ, Smith Y, Wichmann T. 2012. Extrastriatal D2-like receptors modulate basal ganglia pathways in normal and parkinsonian monkeys. J Neurophysiol. 107:1500–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer G, Rivlin-Etzion M, Bar-Gad I, Goldberg JA, Haber SN, Bergman H. 2006. Dopamine replacement therapy does not restore the full spectrum of normal pallidal activity in the 1-methyl-4-phenyl-1,2,3,6-tetra-hydropyridine primate model of Parkinsonism. J Neurosci. 26:8101–8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado JM, Gray CM, Tamas LB, Sigvardt KA. 1999. Dynamics of tremor-related oscillations in the human globus pallidus: a single case study. Proc Natl Acad Sci U S A. 96:1674–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison WD, Allan RJ, Opitz H, Levy R, Dostrovsky JO, Lang AE, Lozano AM. 1998. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson's disease. Ann Neurol. 44:622–628. [DOI] [PubMed] [Google Scholar]
- Iwamuro H, Tachibana Y, Ugawa Y, Saito N, Nambu A. 2017. Information processing from the motor cortices to the subthalamic nucleus and globus pallidus and their somatotopic organizations revealed electrophysiologically in monkeys. Eur J Neurosci. 46:2684–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H. 2001. Neostriatal and globus pallidus stimulation induced inhibitory postsynaptic potentials in entopeduncular neurons in rat brain slice preparations. Neuroscience. 105:871–879. [DOI] [PubMed] [Google Scholar]
- Kita H, Nambu A, Kaneda K, Tachibana Y, Takada M. 2004. Role of ionotropic glutamatergic and GABAergic inputs on the firing activity of neurons in the external pallidum in awake monkeys. J Neurophysiol. 92:3069–3084. [DOI] [PubMed] [Google Scholar]
- Kliem MA, Maidment NT, Ackerson LC, Chen S, Smith Y, Wichmann T. 2007. Activation of nigral and pallidal dopamine D1-like receptors modulates basal ganglia outflow in monkeys. J Neurophysiol. 98:1489–1500. [DOI] [PubMed] [Google Scholar]
- Leblois A, Boraud T, Meissner W, Bergman H, Hansel D. 2006. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J Neurosci. 26:3567–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois A, Meissner W, Bioulac B, Gross CE, Hansel D, Boraud T. 2007. Late emergence of synchronized oscillatory activity in the pallidum during progressive Parkinsonism. Eur J Neurosci. 26:1701–1713. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Tasker RR, Kwan HC, Schnider S, Kwong R, Murayama Y, Dostrovsky JO, Murphy JT. 1988. Single unit analysis of the human ventral thalamic nuclear group: correlation of thalamic "tremor cells" with the 3-6 Hz component of parkinsonian tremor. J Neurosci. 8:754–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Hutchison WD, Lozano AM, Dostrovsky JO. 2000. High-frequency synchronization of neuronal activity in the subthalamic nucleus of parkinsonian patients with limb tremor. J Neurosci. 20:7766–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Lang AE, Dostrovsky JO, Pahapill P, Romas J, Saint-Cyr J, Hutchison WD, Lozano AM. 2001. Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain. 124:2105–2118. [DOI] [PubMed] [Google Scholar]
- Magnin M, Morel A, Jeanmonod D. 2000. Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neuroscience. 96:549–564. [DOI] [PubMed] [Google Scholar]
- Marsden CD. 1982. Basal ganglia disease. Lancet. 2:1141–1147. [DOI] [PubMed] [Google Scholar]
- Mathai A, Ma Y, Pare JF, Villalba RM, Wichmann T, Smith Y. 2015. Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain. 138:946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Deniau JM, Glowinski J, Thierry AM. 1998. Relationships between the prefrontal cortex and the basal ganglia in the rat: physiology of the corticosubthalamic circuits. J Neurosci. 18:9539–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Thierry AM, Glowinski J, Deniau JM. 2003. Spontaneous and evoked activity of substantia nigra pars reticulata neurons during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 23:9929–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WC, DeLong MR. 1987. Altered tonic activity of neurons in the globus pallidus and subthalamic nucleus in the primate MPTPT model of parkinsonism. In: Carpenter MB, Jayaraman A, editors. The basal ganglia II: structure and functions – current concepts. New York: Plenum, pp. 415–427. [Google Scholar]
- Miller WC, DeLong MR. 1988. Parkinsonian symptomatology. An anatomical and physiological analysis. Ann N Y Acad Sci. 515:287–302. [DOI] [PubMed] [Google Scholar]
- Nambu A. 2008. Seven problems on the basal ganglia. Curr Opin Neurobiol. 18:595–604. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Hamada I, Kita H, Imanishi M, Akazawa T, Ikeuchi Y, Hasegawa N. 2000. Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. J Neurophysiol. 84:289–300. [DOI] [PubMed] [Google Scholar]
- Nambu A, Kaneda K, Tokuno H, Takada M. 2002a. Organization of corticostriatal motor inputs in monkey putamen. J Neurophysiol. 88:1830–1842. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Takada M. 2002b. Functional significance of the cortico-subthalamo-pallidal 'hyperdirect' pathway. Neurosci Res. 43:111–117. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tachibana Y, Chiken S. 2015. Cause of parkinsonian symptoms: firing rate, firing pattern or dynamic activity changes? Basal Ganglia. 5:1–6. [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H. 1995. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J Neurophysiol. 74:1800–1805. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz M, Marin C, Alonso F, Zamarbide I, Lanciego JL, Rodriguez-Diaz M. 2004. The origin of motor fluctuations in Parkinson's disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 62(Suppl 1):S17–S30. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, et al. 2017. Past, present, and future of Parkinson's disease: a special essay on the 200th anniversary of the Shaking Palsy. Mov Disord. 32:1264–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohye C, Higuchi Y, Shibazaki T, Hashimoto T, Koyama T, Hirai T, Matsuda S, Serizawa T, Hori T, Hayashi M, et al. 2012. Gamma knife thalamotomy for Parkinson disease and essential tremor: a prospective multicenter study. Neurosurgery. 70:526–535. [DOI] [PubMed] [Google Scholar]
- Polyakova Z, Chiken S, Hatanaka N, Nambu A. 2020. Cortical control of subthalamic neuronal activity through the hyperdirect and indirect pathways in monkeys. J Neurosci. 40:7451–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Vaadia E, Bergman H. 2000. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J Neurosci. 20:8559–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A, Simcox E, Turnbull D. 2014. Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev. 14:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Saban G, Rosin B, Haber SN, Vaadia E, Prut Y, Bergman H. 2008. Low-pass filter properties of basal ganglia cortical muscle loops in the normal and MPTP primate model of parkinsonism. J Neurosci. 28:633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MC, Guridi OJ, Alvarez L, Mewes K, Macias R, Vitek J, DeLong MR, Obeso JA. 1998. The subthalamic nucleus and tremor in Parkinson's disease. Mov Disord. 13(Suppl 3):111–118. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rojas R, Carballo-Barreda M, Alvarez L, Guridi J, Pavon N, Garcia-Maeso I, Macías R, Rodriguez-Oroz MC, Obeso JA. 2018. Subthalamotomy for Parkinson's disease: clinical outcome and topography of lesions. J Neurol Neurosurg Psychiatry. 89:572–578. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Wichmann T. 2010. Extrastriatal dopaminergic circuits of the basal ganglia. Front Neuroanat. 4:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell JC, Obeso JA, Traub MM, Marsden CD. 1983. The behaviour of the long-latency stretch reflex in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry. 46:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan LJ, Clark KB. 1991. The role of the subthalamic nucleus in the response of globus pallidus neurons to stimulation of the prelimbic and agranular frontal cortices in rats. Exp Brain Res. 86:641–651. [DOI] [PubMed] [Google Scholar]
- Sano H, Chiken S, Hikida T, Kobayashi K, Nambu A. 2013. Signals through the striatopallidal indirect pathway stop movements by phasic excitation in the substantia nigra. J Neurosci. 33:7586–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppi K, Weintraub D, Coelho M, Perez-Lloret S, Fox SH, Katzenschlager R, Hametner EM, Poewe W, Rascol O, Goetz CG, et al. 2011. The movement disorder society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord. 26(Suppl 3):S42–S80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shink E, Smith Y. 1995. Differential synaptic innervation of neurons in the internal and external segments of the globus pallidus by the GABA- and glutamate-containing terminals in the squirrel monkey. J Comp Neurol. 358:119–141. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bolam JP. 1989. Neurons of the substantia nigra reticulata receive a dense GABA-containing input from the globus pallidus in the rat. Brain Res. 493:160–167. [DOI] [PubMed] [Google Scholar]
- Smith RD, Zhang Z, Kurlan R, McDermott M, Gash DM. 1993. Developing a stable bilateral model of parkinsonism in rhesus monkeys. Neuroscience. 52:7–16. [DOI] [PubMed] [Google Scholar]
- Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. 2004. Role of external pallidal segment in primate parkinsonism: comparison of the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism and lesions of the external pallidal segment. J Neurosci. 24:6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R. 2016. L-DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb Cortex. 26:4253–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. 2007. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30:228–235. [DOI] [PubMed] [Google Scholar]
- Tachibana Y, Kita H, Chiken S, Takada M, Nambu A. 2008. Motor cortical control of internal pallidal activity through glutamatergic and GABAergic inputs in awake monkeys. Eur J Neurosci. 27:238–253. [DOI] [PubMed] [Google Scholar]
- Tachibana Y, Iwamuro H, Kita H, Takada M, Nambu A. 2011. Subthalamo-pallidal interactions underlying parkinsonian neuronal oscillations in the primate basal ganglia. Eur J Neurosci. 34:1470–1484. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Saitoh K, Harada H, Kashiwayanagi M. 2004. Role of basal ganglia-brainstem pathways in the control of motor behaviors. Neurosci Res. 50:137–151. [DOI] [PubMed] [Google Scholar]
- Terman D, Rubin JE, Yew AC, Wilson CJ. 2002. Activity patterns in a model for the subthalamopallidal network of the basal ganglia. J Neurosci. 22:2963–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Lee H, Smith Y. 2009. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp Neurol. 215:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, Soares J. 2006. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol. 95:2120–2133. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR. 1994. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 72:521–530. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. 1999. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp Brain Res. 125:397–409. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Kliem MA, Soares J. 2002. Slow oscillatory discharge in the primate basal ganglia. J Neurophysiol. 87:1145–1148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.