

Abstract
Ischemia-reperfusion (I/R) is a pathological process that occurs in many organs and diseases. Reperfusion, recovery of blood flow, and reoxygenation often lead to reperfusion injury. Drug therapy and early reperfusion therapy can reduce tissue injury and cell necrosis caused by ischemia, leading to irreversible I/R injury. Ferroptosis was clearly defined in 2012 as a newly discovered iron-dependent, peroxide-driven, nonapoptotic form of regulated cell death. Ferroptosis is considered the cause of reperfusion injury. This discovery provides new avenues for the recognition and treatment of diseases. Ferroptosis is a key factor that leads to I/R injury and organ failure. Given the important role of ferroptosis in I/R injury, there is considerable interest in the potential role of ferroptosis as a targeted treatment for a wide range of I/R injury-related diseases. Recently, substantial progress has been made in applying ferroptosis to I/R injury in various organs and diseases. The development of ferroptosis regulators is expected to provide new opportunities for the treatment of I/R injury. Herein, we analytically review the pathological mechanism and targeted treatment of ferroptosis in I/R and related diseases from the perspectives of myocardial I/R injury, cerebral I/R injury, and ischemic renal injury.
1. Introduction
Ischemia-reperfusion (I/R) is a pathological process that occurs in numerous organs and diseases and can lead to cellular damage and death. Ischemia occurs when the blood supply to organs is restricted as a result of an embolus that blocks arterial blood supply. Ischemic events are related to a serious imbalance in cell metabolism, resulting in tissue hypoxia. In the affected ischemic area, reperfusion and restoration of blood flow and reoxygenation are frequently related to “reperfusion injury,” including excessive tissue injury and destructive inflammatory responses [1]. I/R injury is a major pathological factor in many diseases, especially after cardiac trauma. Additionally, I/R injury delays the recovery of transplanted organs and patients undergoing treatment. Pharmacological therapy and early reperfusion therapy can reduce tissue damage and cell necrosis induced by ischemia. However, these treatments can also cause irreversible I/R injuries. Cell death is the major common I/R injury characteristic in all tissues, and consequently, it is a stable pathological index of I/R injury. Ferroptosis is a newly recognized form of programmed cell death that relies on iron and reactive oxygen species (ROS) [2] and is believed to be the cause of reperfusion injury [3]. Thus, the new manner of cell death, ferroptosis, provides new avenues for understanding and treating many diseases. Research on I/R injury has been conducted in numerous organs, including the heart [4], brain [5], kidney [6], and liver [7]. Recent studies have revealed the links between ferroptosis and I/R injury (Figure 1), and ferroptosis inhibitors have successfully prevented or reduced I/R injury in various organs. Ferroptosis is a key driver of I/R injury and organ failure. Inhibiting ferroptosis may become an effective treatment strategy for related organ diseases and will help to reduce cell death during reperfusion injury. Therefore, it is worth emphasizing the importance of exploring the pathological mechanism of ferroptosis in I/R and targeted therapies. In this article, we summarize the available evidence regarding the pathological mechanism of ferroptosis, I/R injury in organs and diseases, and research progress on related therapeutic targets.
Figure 1.
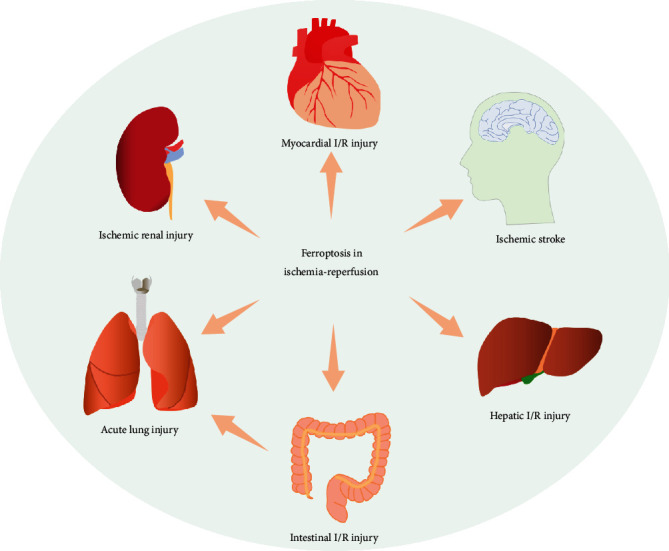
Ferroptosis plays important roles in ischemia-reperfusion (I/R) injury, including myocardial I/R injury, cerebral I/R injury, ischemic renal injury, hepatic I/R injury, intestinal I/R injury, and acute lung injury induced by intestinal I/R.
2. Regulation Mechanism of Ferroptosis
Ferroptosis is an iron-dependent, peroxidation-driven, nonapoptotic form of regulated cell death [3, 8, 9]. It is biochemically, morphologically, and genetically different from apoptosis, necrosis, and other forms of cell death [2, 3, 10, 11]. Ferroptosis was clearly defined in 2012 when it was demonstrated that erastin could inhibit the antioxidant glutathione (GSH) synthesis and trigger iron-dependent cell death, which was also nonapoptotic [2]. Multiple biological regulatory pathways work simultaneously during ferroptosis. GSH deficiency or glutathione peroxidase 4 (GPX4) inactivation causes ferroptosis [2, 12–14]. Furthermore, ferroptosis is characterized by iron-mediated excessive peroxidation of polyunsaturated fatty acids (PUFAs) [15]. The immoderate accumulation of iron-dependent lipid hydroperoxides leads to ferroptosis. The mechanisms and major regulatory factors involved in ferroptosis are shown in Figure 2.
Figure 2.
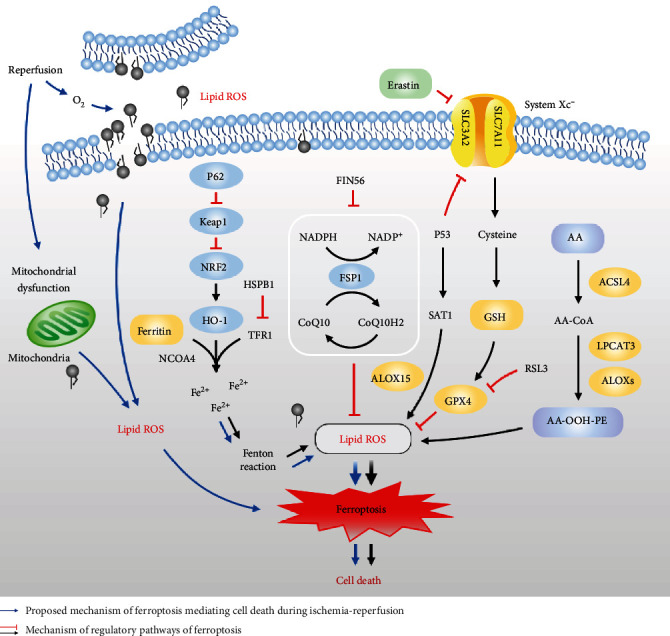
Schematic representation of the proposed mechanism of ferroptosis mediating cell death during ischemia-reperfusion and the mechanism of regulatory pathways of ferroptosis. SLC3A2: solute carrier family 3 member 2; SLC7A11: solute carrier family 7 member 11; Keap1: Kelch-like ECH-associated protein 1; NRF2: nuclear factor erythroid 2-related factor; HO-1: heme oxygenase-1; NCOA4: nuclear receptor coactivator 4; HSPB1: heat shock factor-binding protein 1; TFR1: transferrin receptor 1; FIN56: ferroptosis inducing 56; FSP1: ferroptosis suppressor protein 1; CoQ10: coenzyme Q10; RSL3: (1S, 3R)-RSL; GSH: glutathione; GPX4: glutathione peroxidase 4; ROS: reactive oxygen species; SAT1: spermidine/spermine N1-acetyltransferase 1; ALOX-15: arachidonate lipoxygenase 15; AA: arachidonic acid; ACSL4: acyl-CoA synthetase long-chain family 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; ALOXs: arachidonate lipoxygenases; PE: phosphatidylethanolamine.
2.1. Antioxidant Mechanism of Ferroptosis
System xc− is a cystine-glutamate exchanger, which consists of heterodimers of the solute carrier family 3 member 2 (SLC3A2) and the catalytic subunit solute carrier family 7 member 11 (SLC7A11) [16, 17]. System xc− promotes the exchange process across the plasma membrane of cystine and glutamate. Cystine is necessary for cell survival and is the predominant form of cysteine in extracellular space [18]. System xc− imports extracellular cystine for GSH synthesis and transfers glutamate out of cells [19]. GPX4, an antioxidant enzyme that maintains cellular redox homeostasis, primarily acts as a crucial endogenous antioxidant against phospholipid peroxide, mainly using GSH as a cofactor [20]. The inhibition of system xc− induces GSH depletion and causes the inactivation of GPX4, resulting in the accumulation of toxic lipid ROS and triggering ferroptosis [2, 14]. To maintain the function and activity of GPX4, GSH and selenium (Se) are indispensable [3, 21]. GSH also acts as a major endogenous antioxidant [22] and participates in the regeneration of GPX4 [23]. GSH is synthesized in two steps from glutamate, cysteine, and glycine under the catalysis of enzymes [17]. Erastin and (1S,3R)-RSL (RSL3) were identified as compounds that induce RAS-mutated tumor cell death [9, 24], and some proteins associated with GSH metabolism are related to ferroptosis [8]. GPX4 can act directly or indirectly by ferroptosis agonists, thereby reducing its activity [25]. RSL3 is a ferroptosis inducer that directly inhibits GPX4 by covalently targeting selenocysteine. It inhibits GPX4 activity, thereby causing intracellular accumulation of lipid peroxides and subsequent cell death [14, 26]. Erastin, another representative ferroptosis inducer, depletes cellular cysteine and reduces the biosynthesis of GSH by directly inhibiting system xc− [13]. This leads to the loss of GPX4 activity, leading to the accumulation of lipid peroxides and eventually ferroptosis. The main target of erastin is the exchange of cystine/cystathionine with glutamate [2]. Recent evidence has shown that the ferroptosis suppressor protein 1- (FSP1-) coenzyme Q10- (CoQ10-) (nicotinamide adenine dinucleotide phosphate) NAD(P)H pathway synergistically inhibits the proliferation of lipid peroxides and ferroptosis with GPX4 and GSH, as an independent system [27, 28]. FSP1 functions as an oxidoreductase that reduces CoQ10 using NAD(P)H and produces a lipophilic radical-capture antioxidant (RTA) to prevent the production of lipid peroxides [27, 28]. Ferroptosis inducing 56 (FIN56) has been shown to cause ferroptosis by blocking this pathway [29]. The tumor suppressor protein P53 plays a role in apoptosis, necrosis, and autophagy. P53 can repress SLC7A11 gene expression, affecting the activity of GPX4 and leading to the accumulation of lipid peroxidation and an increase in ferroptosis [30, 31]. P53-SAT1- (spermidine/spermine N1-acetyltransferase 1-) arachidonate lipoxygenase 15 (ALOX-15) pathway also participates in the regulation of ferroptosis [32]. SAT1 acts as a transcriptional target of P53, and its activation can induce ROS-induced lipid peroxidation and ferroptosis.
2.2. Oxidation Mechanisms of Ferroptosis
PUFAs are oxidized to form lipid peroxides [33]. The important factors that cause ferroptosis include the accumulation of lipid peroxides [2, 3]. Oxidized arachidonic acid-containing phosphatidylethanolamine (AA-PE) acts as a signal of ferroptosis. A recent study showed that AA-OOH-PE, instead of other types of phospholipid-OOH, is the key phospholipid that induces ferroptosis [34]. Three enzymes are required for the generation of AA-OOH-PE: arachidonate lipoxygenases (ALOXs), acyl-CoA synthetase long-chain family 4 (ACSL4), and lysophosphatidylcholine acyltransferase 3 (LPCAT3) [35]. ACSL4 catalyzes PUFA (especially AA) to yield CoA-forms [36], and LPCAT3 regulates the esterification of AA-CoA into AA-PE [15]. Under the catalysis of lipoxygenase (LOXs), AA-PE can be oxidized to AA-OOH-PE and play further oxidative roles [26, 34, 37]. When the AA-OOH-PE level exceeds the limit of the reduction system, ferroptosis is induced [17, 36, 38]. In addition to lipid peroxidation, iron, a redox-active metal, is necessary for ferroptosis [39]. As the iron source of the Fenton reaction, in the labile iron pool (LIP), small amounts of iron are free and chelated. Iron released from the LIP can subsequently promote ROS accumulation through the Fenton reaction [40–42]. During ferroptosis, ROS mainly originate from the Fenton reaction. In addition, iron is a prooxidant in ferroptosis and is vital for the function of enzymes, such as lipoxygenases (LOXs) [43, 44]. Ferroptosis is also regulated by proteins involved in iron homeostasis. ShRNA-mediated iron response element binding protein 2 (IREB2) silencing has been demonstrated to reduce sensitivity to ferroptosis by modifying iron uptake, metabolism, and storage [2]. Cysteine desulfurase (NFS1) has also been shown to reduce ferroptosis in lung cancer [45]. Heat shock factor-binding protein 1 (HSPB1), a negative regulator of ferroptosis, reduces iron concentrations by inhibiting the expression of TRF1, thereby inhibiting ferroptosis [46]. Ferritin autophagy can increase the content of Fe2+ [47], thus promoting ferroptosis. At the same time, the ferritin receptor nuclear receptor coactivator 4 (NCOA4) mediates the autophagic degradation of ferritin, increasing free iron [48, 49]. Heme oxygenase-1 (HO-1) decomposes heme to release iron, thereby accelerating ferroptosis [50]. P62 is an autophagy receptor that can inhibit Kelch-like ECH-associated protein 1 (Keap1) directly and promotes nuclear factor erythroid 2-related factor (NRF2) activation simultaneously. An early study showed that oxidative stress induces the expression of HO-1 by activating the p62-Keap1-NRF2 pathway, thus antagonizing ferroptosis [51]. Nrf2 activates the expression of various target genes necessary to regulate ferroptosis by regulating the metabolism of GSH, lipid, iron, and mitochondrial function [51].
3. Pathological Mechanisms and Potential Targeted Therapy of Ferroptosis in Ischemia-Reperfusion Injury
3.1. Role of Ferroptosis in Ischemia-Reperfusion-Related Cell Death
Previous studies have shown that ferroptosis occurs during the reperfusion period and not during the ischemic period in I/R injury models [52]. After I/R, the endogenous mechanisms of scavenging ROS are seriously negated, and ROS cannot be effectively scavenged. Reperfusion of ischemic tissue causes excessive ROS, which mediate I/R injury [53, 54]. There is evidence that I/R injury is accompanied by several cellular events, such as reperfusion-related excessive ROS accompanied by lipid peroxidation [55] and an increase in intracellular iron concentration [56, 57]. Lipid peroxidation and ROS lead to cell injury and death. These cellular events are consistent with the manifestation of iron-dependent ferroptosis, which could be prevented by iron chelators [58]. Figure 2 shows the proposed mechanism of ferroptosis, which mediates cell death during ischemia-reperfusion. Current studies have shown that phospholipid oxidation products exist in myocardial I/R injury [59–62]. In addition to lipid peroxidation, free iron is a necessary condition for ferroptosis. Nonenzymatic production of ROS occurs when metal ions are present. There is a LIP in the cytosol, mitochondria, and lysosomes of cells [41]. The free iron in the LIP could accelerate the lipid peroxidation of saturated fatty acids in humans via the Fenton reaction [63], and iron is involved in producing ROS in mitochondria. Mitochondria of a cell are the main sites for generating ROS [64, 65], and after the injury, they undergo alterations of structure and function. During ischemia, mitochondrial dysfunction leads to excessive ROS production. Therefore, current evidence supports the involvement of ferroptosis in the progression of organ cell injury after I/R. Ferroptosis is an important mechanism that mediates cellular damage to organs and cell death during I/R.
3.2. Myocardial I/R Injury
3.2.1. Ferroptosis in Myocardial I/R Injury
Coronary artery disease has been suggested to be the most common cardiovascular disease [66], and atherosclerosis is the main cause. Plaque rupture results in acute ischemia and myocardial infarction. A large amount of ROS is produced after reperfusion [67, 68], leading to further damage [69]. During ischemia and early reperfusion, ferritin degrades to release iron and promotes iron-mediated Fenton reaction, leading to oxidative damage and loss of cardiac function associated with I/R injury. In experimental models, inhibition of glutaminolysis, an important part of ferroptosis, was shown to reduce I/R-induced heart injury [70]. Studies have confirmed that ROS accumulation and iron content increase during I/R injury [71, 72], and iron overload is an important cause of myocardial cell injury [73]. A study by Li et al. showed that iron deposition and ROS overproduction occurred during diabetic myocardial injury, and ferroptosis was involved in the I/R injury of diabetic myocardium through the endoplasmic reticulum stress pathway [31]. These studies indicate that ferroptosis plays a key role in myocardial I/R injury. Transferrin receptor 1 (TfR1) is involved in cellular iron uptake. A novel pathway of ubiquitin-specific protease 7 (USP7)/p53/TfR1 is found in rat hearts after I/R treatment, and the upregulation of USP7 accelerates ferroptosis by activating the p53/TfR1 pathway [74]. The study has shown that ferroptosis occurs during reperfusion in rat hearts subjected to I/R [52]. With the prolongation of reperfusion time, the levels of ACSL4 protein in the cardiac tissue gradually increased, accompanied by a decrease in GPX4 levels. The exact role of ferroptosis in myocardial I/R injury remains unclear. Other studies have shown that inhibition of ferroptosis has a protective effect on myocardial I/R-induced rats [75].
3.2.2. Target and Targeted Therapy of Ferroptosis in Myocardial I/R Injury
Targeting ferroptosis is considered a feasible treatment for I/R injury. The exposure of rat cardiomyocytes to simulated I/R injury can cause nonapoptotic cell death, and the addition of ferrostatin-1 during reperfusion can attenuate this effect [76]. Fang et al. [77] found that the in vivo application of ferristatin-1 and an iron chelator plays a role in reducing heart failure caused by I/R. It has been suggested that ferroptosis is related to myocardial I/R injury, and ferristatin-1 may be a feasible treatment to alleviate reperfusion injury. In addition, many studies have demonstrated that iron chelators can block ferroptosis in vitro and in vivo. Deferoxamine (DFO) is the most widely used nontoxic iron chelator that inhibits ferroptosis mediated by lipid peroxidation under various conditions [2]. Chan et al. showed that DFO administered before reperfusion by primary percutaneous coronary intervention could rapidly ameliorate oxidative stress but failed to limit infarct size [78]. In a study by Paraskevaidis et al., intravenous DFO infusion protected the myocardium from reperfusion injury and reduced lipid peroxidation during coronary artery bypass grafting surgery [79]. DFO, which binds iron, has been shown to have a protective effect on myocardial I/R [70]. Shan et al. demonstrated that Cyanidin-3-glucoside (C3G) treatment reduced oxidative stress and Fe2+ content, confirming that C3G effectively alleviated myocardial I/R injury by inhibiting ferroptosis both in vivo and in vitro [72]. C3G treatment could reduce myocardial infarction area, inhibit pathological damage, and inhibit ST-segment elevation. Baicalein (also termed 5,6,7-trihydroxyflavone) is a flavonoid that limits iron accumulation induced by erastin and lipid peroxidation by inhibiting GPX4 degradation and GSH consumption [80]. In addition, baicalein significantly reduced iron-induced lipid peroxidation and inhibited the expression of ALOX-15 in HT22 cells [81]. A recent study has shown that baicalin protects against myocardial I/R injury via inhibiting ACSL4-controlled ferroptosis in a rat model [82]. Ma et al. found that ubiquitin-specific peptidase 22 (USP22) inhibits cardiomyocyte death induced by ferroptosis in myocardial I/R injury via the SIRT1/p53/SLC7A11 axis, providing a novel therapeutic target for the treatment [83]. Chen et al. found that embryonic lethal-abnormal vision like protein 1 (ELAVL1) increased substantially during myocardial I/R injury. ELAVL1 knockout decreased ferroptosis, thus ameliorating I/R injury [84]. FOXC1-activated transcription mediates the increase in ELAVL1 induced by myocardial I/R, and FOXC1 plays a key role in myocardial I/R injury [84]. Inhibition of ELAVL1-mediated ferroptosis might be a new approach for the treatment of myocardial I/R injury. Oxidized phosphatidylcholine (OxPC) production increases during myocardial I/R injury and causes extensive cell death through ferroptosis [85]. Interventions targeting OxPCs may help reduce ferroptosis during I/R injury. Determining appropriate therapeutic targets is vital. Further studies are needed to establish more effective anti-ferroptosis-targeted therapies for patients with myocardial ischemia.
3.3. Cerebral I/R Injury
3.3.1. Ferroptosis in Cerebral I/R Injury
Ischemic stroke is the main cause of destructive cerebrovascular disease and has high mortality and morbidity rates [86]. Restoring blood supply to the ischemic area as soon as possible is crucial for ischemic cerebrovascular disease treatment [87]. Early reperfusion is the most effective therapy for acute cerebral ischemia; however, the increase in ROS and inflammation, and other phenomena in tissues after reperfusion aggravates brain tissue damage [88]. Several studies have indicated that iron chelators attenuate brain injury induced by ischemic stroke in animal models [89, 90] and play an antioxidant, neuroprotective role in stroke patients [91]. Recent studies have suggested that ferroptosis plays a significant role in ischemic stroke [92–94]. Ferroptosis inhibitors, ferrostatin-1 or liprostatin-1, have a protective effect on neuronal injury in middle cerebral artery occlusion (MCAO) mouse models [95]. In brain I/R mice models, the expression levels of proteins related to ferroptosis are abnormal [96]. A significant increase was demonstrated in free iron content and ROS accumulation in brain tissue, whereas the levels of GPX4 and GSH significantly decreased [97]. These studies show that ferroptosis plays a key role in brain I/R injury, and inhibition of ferroptosis could protect brain tissue damage induced by I/R.
3.3.2. Target and Targeted Therapy of Ferroptosis in Cerebral I/R Injury
A deeper understanding of the target and targeted treatment of ferroptosis will be beneficial for treating related diseases. Pharmacological Se drives GPX4 expression to reduce ferroptosis, provide neuroprotection, and have a therapeutic effect on stroke [92]. A previous study found that the tau-mediated iron export pathway prevents injury induced by ferroptosis after ischemic stroke and the therapeutic potential of ferroptotic inhibition in this injury [95]. Guan et al. observed that ROS and iron levels were increased in the brains of ischemic stroke model gerbils [98]. Carvacrol exerts neuroprotective effects in cerebral ischemia by increasing the expression of GPX4 and inhibiting ferroptosis [98]. Carthamin yellow is a flavonoid isolated from safflower. In a recent study, using MCAO model rats, carthamin yellow improved cerebral I/R injury by reducing ferroptosis [96]. Galangin is a flavonoid that is mainly extracted from Chinese medicinal herbs [99]. Galangin upregulates SLC7A11 and indirectly increases the expression of GPX4, thereby inhibiting ferroptosis and attenuating cerebral I/R injury in gerbils. Galangin treatment has been shown to increase the survival rate of neurons in the brain of gerbils and alleviate learning and memory impairment after I/R [100]. It has been hypothesized that PIEZO1 participates in cerebral I/R injury by regulating ferroptosis, which might be a potential therapeutic treatment to protect against neuronal damage [101]. Furthermore, Cui et al. identified that ACSL4 is a novel neuronal death regulator, as knockout of ACSL4 could protect mice from cerebral ischemia, while forced overexpression of ACSL4 aggravated ischemic brain injury. The intervention of ACSL4 expression could be a potential therapeutic target for ischemic stroke [102]. The therapeutic targets of ferroptosis in ischemic stroke still need to be further studied to determine appropriate and more effective anti-ferroptosis-targeted therapies.
3.4. Ischemic Renal Injury
3.4.1. Ferroptosis in Ischemic Renal Injury
Pathophysiological renal I/R injuries include renal tubular injury, inflammation, and vascular dysfunction. The precipitating factors and major early events of acute kidney injury (AKI) are renal tubular epithelial cell injury and subsequent cell death [103]. In recent years, the role of ferroptosis in renal I/R injury has been investigated. It has been suggested that ferroptosis has pathophysiological relevance in AKI [2, 103–105]. In mice with renal I/R injury, administration of ferrostatin 1 or 16–86 15 min before ischemia, renal tissue injury, and serum creatinine and urea were decreased 48 h after ischemia, indicating that ferroptosis plays an important role in the pathogenesis [106]. In mouse models of AKI induced by I/R injury, ferroptosis is directly related to the synchronous necrosis of renal tubules [106]. Ding et al. demonstrated that I/R induced upregulation of miR-182-5p and miR-378a-3p and induced ferroptosis in renal injury by downregulating GPX4 and SLC7A11 [107]. Recent studies have suggested that ferroptosis plays an important role in renal I/R injury; however, the exact mechanism of ferroptosis needs to be further studied.
3.4.2. Target and Targeted Therapy of Ferroptosis in Ischemic Renal Injury
Pannexin1 (PANX1) is an ATP-releasing pathway family protein that can promote apoptosis during kidney injury. A previous study has shown that the deletion of PANX1 prevents renal I/R injury by attenuating ferroptosis activated by mitogen-activated protein kinase/extracellular signal-regulated kinase signaling [108]. Macrophage migration inhibitory factor (MIF) is a stress-regulatory cytokine. It has been reported that MIF can effectively limit necrotic ptosis, restore intracellular GSH, and reduce lipid peroxidation to alleviate oxidative stress [109]. MIF protects renal tubular epithelial cells and plays a role in renal protection during experimental I/R injury. Pachymic acid is a lanostane-type triterpenoid found in Poria cocos. According to research, under the treatment of pachymic acid, the renal function of mice with renal I/R injury can be improved, and renal injury can be alleviated. The protective effect of pachymic acid may be related to the inhibition of renal ferroptosis through directly or indirectly activating Nrf2 and upregulating downstream GPX4, system xc−, and HO-1 [110]. Additionally, XJB-5-131 is a mitochondrial-targeted nitroxide with a high affinity for tubular epithelial cells [111]. XJB-5-131 inhibits lipid peroxidation after I/R injury and inhibits I/R-induced ferroptosis in tubular epithelial cells, thus improving ischemic renal injury [111]. Irisin is a type of exercise-induced hormone that can ameliorate mitochondrial function and reduce ROS production. In renal I/R mice models, irisin treatment can reduce I/R-induced AKI by upregulating GPX4, a pivotal regulator of ferroptosis [112]. Inhibition of ferroptosis protects renal tubular epithelial cells and reduces renal I/R injury. These results indicate that the crucial endogenous antioxidant GPX4 and its cofactor GSH are important targets for protection against renal I/R injury.
3.5. Mechanism and Targeted Therapy of Ferroptosis in I/R Injury in Other Organs
Hepatic I/R, as a surgical complication after liver transplantation, can cause inflammation and immune reactions, lead to rejection, and affect prognosis [113, 114]. Galeano et al. demonstrated that subchronic, low-level iron administration has significant hepatoprotection against I/R injury [115]. A previous study has shown that ferroptosis is a mechanism of liver I/R injury in a mouse model of hepatic injury. Li et al. demonstrated that iron overload is a new risk factor for hepatic I/R injury. Ferroptosis mediates the relationship between iron overload and liver injury, showing that ferroptosis is a prospective target for preventing and treating hepatic I/R injury [116]. A recent study has also implicated that ferroptosis is associated with intestinal I/R injury [117]. Increasing evidence shows that ROS play a key role in the pathogenesis of intestinal I/R. The primary contributors of ferroptosis, including ROS generation and increase in lipid peroxidation, are related to intestinal I/R injury [118]. Deng et al. found that capsiate, a gut microbiota metabolite, can promote the expression of GPX4 by activating TRPV1 and inhibiting ferroptosis induced by intestinal I/R [119]. In addition, intestinal I/R has been shown to induce acute lung injury [120, 121]. An earlier study demonstrated ferroptosis in acute lung injury induced by intestinal I/R [122]. It has been shown that Nrf2 inhibits ferroptosis by promoting the expression of SLC7A11 and HO-1 and plays a protective role in acute lung injury following intestinal I/R [122]. The inhibitor of apoptosis-stimulating protein of p53 (iASPP) is the only inhibitor of apoptosis-stimulating protein in the p53 family. A recent study has suggested that in Nrf2-/- mice with acute lung injury induced by intestinal I/R, iASPP can effectively alleviate acute lung injury and inhibit ferroptosis through the Nrf2/HIF-1α/TF signaling pathway [123].
4. Conclusions
In recent years, great progress has been made in applying ferroptosis in I/R injury in various organs and diseases (see Figure 3). Table 1 summarizes the main findings of studies that investigated the relevant mechanisms and targets of ferroptosis in I/R injury. However, there are several unclear mechanisms of ferroptosis in I/R injury. First, existing research on the pathological effects of ferroptosis has mostly concentrated on the animal and cell levels, and evaluation of its clinical efficacy is rare. Second, there is relatively little research on the target of ferroptosis in I/R injury in various organs and the application of targeted therapy, which requires more in-depth and extensive investigations. With the in-depth exploration of research, suppressing ferroptosis could be an effective strategy for treating I/R-related organ diseases. The development of ferroptosis modulators is expected to provide new opportunities for the treatment of I/R injury and related diseases. Therefore, it is necessary to conduct such studies to screen new targeted ferroptosis therapies with potentially protective effects against I/R injury in the future.
Figure 3.
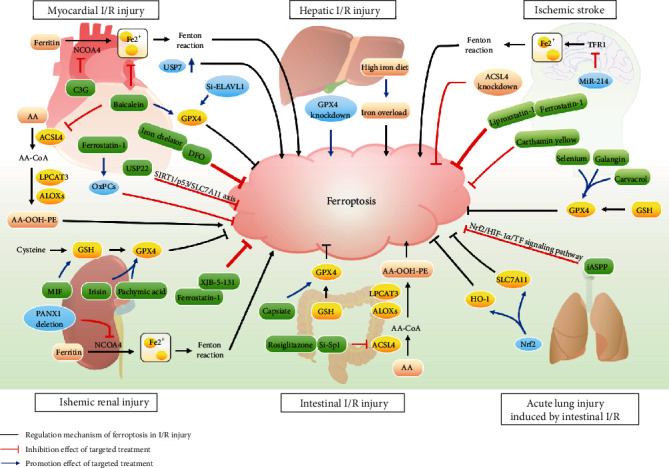
Pathological mechanism and targeted therapies of ferroptosis in ischemia-reperfusion (I/R) injury-related diseases. NCOA4: nuclear receptor coactivator 4; C3G: cyanidin-3-glucoside; USP7: ubiquitin-specific protease 7; USP7 ↑: the upregulation of USP7; ELAVL1: embryonic lethal-abnormal vision like protein 1; GPX4: glutathione peroxidase 4; AA: arachidonic acid; ACSL4: acyl-CoA synthetase long-chain family 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; ALOXs: arachidonate lipoxygenases; PE: phosphatidylethanolamine; OxPCs: oxidized phosphatidylcholines; USP22: ubiquitin-specific peptidase 22; DFO: deferoxamine; TFR1: transferrin receptor 1; GSH: glutathione; MIF: macrophage migration inhibitory factor; Sp1: special protein 1; SLC7A11: solute carrier family 7 member 11; NRF2: nuclear factor erythroid 2-related factor; HO-1: heme oxygenase-1; iASPP: inhibitor of apoptosis-stimulating protein of P53.
Table 1.
Studies reporting the mechanisms and targets of ferroptosis in ischemia-reperfusion injury.
| Ferroptosis in I/R injury | Experiment | Number (n) | Model/study population | Intervention | Target | Reference |
|---|---|---|---|---|---|---|
| Myocardial I/R injury | In vitro | Not applicable | Mouse embryonic fibroblasts | Not available | Glutaminolysis | [70] |
| In vitro | Not applicable | H9c2 cells | C3G | Ferroptosis-related protein, NCOA4, RSL3 | [72] | |
| In vitro | Not applicable | Adult rat cardiomyocytes | Ferrostatin-1 | OxPCs | [76] | |
| In vitro | Not applicable | H9c2 cells | Baicalin | ACSL4 | [82] | |
| In vitro | Not applicable | Human cardiomyocytes | ELAVL1 knockout | Not available | [84] | |
| In vivo | Not applicable | I/R-treated Sprague–Dawley rats | Not available | Upregulation of USP7 | [74] | |
| In vivo | Not applicable | Murine models of doxorubicin- and I/R-induced cardiomyopathy | Ferrostatin-1 and iron chelator | Not available | [77] | |
| In vivo | Not applicable | Sprague–Dawley rats | C3G | Ferroptosis-related protein, NCOA4, RSL3 | [72] | |
| In vivo | Not applicable | Myocardial I/R rat model | Baicalin | ACSL4 | [82] | |
| In vivo | Not applicable | Sprague–Dawley rats | USP22 | Not available | [83] | |
| Clinical finding | 60 | Patients with ST-elevation-myocardial infarct | DFO | Not available | [78] | |
| Clinical finding | 45 | Patients undergoing coronary artery bypass grafting | DFO | Not available | [79] | |
|
| ||||||
| Cerebral I/R injury | In vitro | Not applicable | Primary cortical neurons and hippocampal HT22 cells | Selenium | GPX4 | [92] |
| In vivo | Not applicable | C57BL/6 mice | Selenium | GPX4 | [92] | |
| In vivo | Not applicable | Sprague–Dawley rats | Liproxstatin-1, Ferrostatin-1 | Not available | [95] | |
| In vivo | Not applicable | Brain I/R mice model | Carthamin yellow | Not available | [96] | |
| In vivo | Not applicable | Mice models of cerebral I/R | MiR-214 | PVT1, TFR1, TP53 | [97] | |
| In vivo | Not applicable | Ischemic stroke model gerbils | Carvacrol | GPX4 | [98] | |
| In vivo | Not applicable | Ischemic stroke model gerbils | Galangin | SLC7A11, GPX4 | [100] | |
| In vivo | Not applicable | Mice models | Knockdown of ACSL4 | Not available | [102] | |
|
| ||||||
| Ischemic renal injury | In vivo | Not applicable | C57BL/6 mice | Ferrostatin 1 or 16–86 | Not available | [106] |
| In vivo | Not applicable | I/R-induced renal injury rat models | Not available | GPX4, SLC7A11 | [107] | |
| In vivo | Not applicable | Mice models | PANX1 deletion | Not available | [108] | |
| In vivo | Not applicable | Mif-/- mice | MIF | GSH | [109] | |
| In vivo | Not applicable | Renal I/R injury mice model | Pachymic acid | GPX4, SLC7A11, and HO-1 | [110] | |
| In vivo | Not applicable | Renal I/R mice model | XJB-5-131 | Not available | [111] | |
| In vivo | Not applicable | Renal I/R mice model | Irisin | GPX4 | [112] | |
|
| ||||||
| Hepatic I/R injury | In vivo | Not applicable | Gpx4-/- mice | Knockout of GPX4 | Not available | [6] |
| In vivo | Not applicable | Murine hepatic I/R injury model | Iron overload | Not available | [116] | |
|
| ||||||
| Intestinal I/R injury | In vitro | Not applicable | Caco-2 cells | Rosiglitazone, si-special protein 1 | ACSL4 | [117] |
| In vitro | Not applicable | Ileum organoid H/R model | Capsiate | GPX4, TRPV1 | [119] | |
| In vivo | Not applicable | Murine model of intestinal I/R | Rosiglitazone, si-special protein 1 | ACSL4 | [117] | |
| In vivo | Not applicable | Mouse intestinal I/R model | Capsiate | GPX4, TRPV1 | [119] | |
|
| ||||||
| Acute lung injury induced by intestinal I/R | In vivo | Not applicable | C57BL/6J mice and Nrf2-/- mice | Nrf2 | SLC7A11, HO-1 | [122] |
| In vivo | Not applicable | Nrf2-/- mice with acute lung injury induced by intestinal I/R | Inhibitor of apoptosis-stimulating protein of P53 | Nrf2 | [123] | |
Acknowledgments
This work was supported by the CACMS Innovation Fund (grant number CI2021A00919) and the National Key R&D Program of China (grant numbers 2018YFC1704901 and 2018YFC1704900).
Contributor Information
Lijing Zhang, Email: dzmyyccu@163.com.
Yanwei Xing, Email: xingyanwei12345@163.com.
Conflicts of Interest
The authors declare that no conflict of interest exists.
Authors' Contributions
Xinye Li, Ning Ma, Juping Xu, Yanchi Zhang, and Pan Yang contributed equally to this work.
References
- 1.Eltzschig H. K., Eckle T. Ischemia and reperfusion--from mechanism to translation. Nature Medicine . 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon S., Lemberg K., Lamprecht M., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stockwell B., Friedmann Angeli J., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell . 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ten Hove M., Lygate C. A., Fischer A., et al. Reduced inotropic reserve and increased susceptibility to cardiac ischemia/reperfusion injury in phosphocreatine-deficient guanidinoacetate-N-methyltransferase-knockout mice. Circulation . 2005;111(19):2477–2485. doi: 10.1161/01.CIR.0000165147.99592.01. [DOI] [PubMed] [Google Scholar]
- 5.Hanson L. R., Roeytenberg A., Martinez P. M., et al. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. The Journal of Pharmacology and Experimental Therapeutics . 2009;330(3):679–686. doi: 10.1124/jpet.108.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedmann Angeli J., Schneider M., Proneth B., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature Cell Biology . 2014;16(12):1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montalvo-Jave E. E., Escalante-Tattersfield T., Ortega-Salgado J. A., Piña E., Geller D. A. Factors in the pathophysiology of the liver ischemia-reperfusion injury. The Journal of Surgical Research . 2008;147(1):153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yagoda N., von Rechenberg M., Zaganjor E., et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature . 2007;447(7146):864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W., Stockwell B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & Biology . 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fatokun A., Dawson V. L., Dawson T. M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. British Journal of Pharmacology . 2014;171(8):2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., Tuo Q. Z., Lei P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. Journal of Molecular Neuroscience: MN . 2018;66(2):197–206. doi: 10.1007/s12031-018-1155-6. [DOI] [PubMed] [Google Scholar]
- 12.Bochkov V., Oskolkova O., Birukov K., Levonen A. L., Binder C. J., Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxidants & Redox Signaling . 2010;12(8):1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon S., Patel D., Welsch M., et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife . 2014;3, article e02523 doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W., SriRamaratnam R., Welsch M., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell . 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon S., Winter G., Musavi L., et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chemical Biology . 2015;10(7):1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bröer S., Wagner C. A. Structure-function relationships of heterodimeric amino acid transporters. Cell Biochemistry and Biophysics . 2002;36(2-3):155–168. doi: 10.1385/CBB:36:2-3:155. [DOI] [PubMed] [Google Scholar]
- 17.Liang C., Zhang X., Yang M., Dong X. Recent progress in ferroptosis inducers for cancer therapy. Advanced Materials (Deerfield Beach, Fla.) . 2019;31(51, article e1904197) doi: 10.1002/adma.201904197. [DOI] [PubMed] [Google Scholar]
- 18.Conrad M., Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: cystine supplier and beyond. Amino Acids . 2012;42(1):231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 19.Bridges R., Natale N. R., Patel S. A. System xc− cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. British Journal of Pharmacology . 2012;165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso B. R., Hare D. J., Bush A. I., Roberts B. R. Glutathione peroxidase 4: a new player in neurodegeneration? Molecular Psychiatry . 2017;22(3):328–335. doi: 10.1038/mp.2016.196. [DOI] [PubMed] [Google Scholar]
- 21.Ingold I., Berndt C., Schmitt S., et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell . 2018;172(3):409–422.e21. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 22.Meister A., Anderson M. E. Glutathione. Annual Review of Biochemistry . 1983;52(1):711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 23.Maiorino M., Conrad M., Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxidants & Redox Signaling . 2018;29(1):61–74. doi: 10.1089/ars.2017.7115. [DOI] [PubMed] [Google Scholar]
- 24.Dolma S., Lessnick S., Hahn W., Stockwell B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell . 2003;3(3):285–296. doi: 10.1016/S1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 25.Ke B., Tian M., Li J., Liu B., He G. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Medicinal Research Reviews . 2016;36(6):983–1035. doi: 10.1002/med.21398. [DOI] [PubMed] [Google Scholar]
- 26.Yang W., Kim K., Gaschler M., Patel M., Shchepinov M. S., Stockwell B. R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America . 2016;113(34):E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bersuker K., Hendricks J., Li Z., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature . 2019;575(7784):688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doll S., Freitas F., Shah R., et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature . 2019;575(7784):693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 29.Shimada K., Skouta R., Kaplan A., et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nature Chemical Biology . 2016;12(7):497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang L., Kon N., Li T., et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature . 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W., Li W., Leng Y., Xiong Y., Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA and Cell Biology . 2020;39(2):210–225. doi: 10.1089/dna.2019.5097. [DOI] [PubMed] [Google Scholar]
- 32.Ou Y., Wang S., Li D., Chu B., Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proceedings of the National Academy of Sciences of the United States of America . 2016;113(44):E6806–E6812. doi: 10.1073/pnas.1607152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris I., DeNicola G. M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends in Cell Biology . 2020;30(6):440–451. doi: 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Kagan V., Mao G., Qu F., et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature Chemical Biology . 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei P., Bai T., Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Frontiers in Physiology . 2019;10:p. 139. doi: 10.3389/fphys.2019.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doll S., Proneth B., Tyurina Y., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology . 2017;13(1):91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forcina G., Dixon S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics . 2019;19(18, article e1800311) doi: 10.1002/pmic.201800311. [DOI] [PubMed] [Google Scholar]
- 38.Lagarde M., Calzada C., Jouvène C., et al. Functional fluxolipidomics of polyunsaturated fatty acids and oxygenated metabolites in the blood vessel compartment. Progress in Lipid Research . 2015;60:41–49. doi: 10.1016/j.plipres.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Hassannia B., Vandenabeele P., vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell . 2019;35(6):830–849. doi: 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Galaris D., Barbouti A., Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochimica et Biophysica Acta Molecular Cell Research . 2019;1866(12):p. 118535. doi: 10.1016/j.bbamcr.2019.118535. [DOI] [PubMed] [Google Scholar]
- 41.Kakhlon O., Cabantchik Z. I. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radical Biology & Medicine . 2002;33(8):1037–1046. doi: 10.1016/S0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 42.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutation Research . 2003;531(1-2):81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 43.Grivennikova V., Vinogradov A. D. Generation of superoxide by the mitochondrial complex I. Biochimica et Biophysica Acta . 2006;1757(5-6):553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Halliwell B., Cross C. E. Oxygen-derived species: their relation to human disease and environmental stress. Environmental Health Perspectives . 1994;102(Supplement 10):5–12. doi: 10.1289/ehp.94102s105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez S., Sviderskiy V., Terzi E., et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature . 2017;551(7682):639–643. doi: 10.1038/nature24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X., Ou Z., Xie M., et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene . 2015;34(45):5617–5625. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terman A., Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxidants & Redox Signaling . 2013;18(8):888–898. doi: 10.1089/ars.2012.4885. [DOI] [PubMed] [Google Scholar]
- 48.Gao M., Monian P., Pan Q., Zhang W., Xiang J., Jiang X. Ferroptosis is an autophagic cell death process. Cell Research . 2016;26(9):1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou W., Xie Y., Song X., et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy . 2016;12(8):1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daher R., Manceau H., Karim Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse Medicale (Paris, France: 1983) . 2017;46(12):e272–e278. doi: 10.1016/j.lpm.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 51.Sun X., Ou Z., Chen R., et al. Activation of the p62-Keap 1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology (Baltimore, Md.) . 2016;63(1):173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang L., Luo X., Tu H., et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn-Schmiedeberg's Archives of Pharmacology . 2021;394(2):401–410. doi: 10.1007/s00210-020-01932-z. [DOI] [PubMed] [Google Scholar]
- 53.Hess M., Manson N. Molecular oxygen: Friend and foe: The role of the oxygen free radical system in the calcium paradox, the oxygen paradox and ischemia/reperfusion injury. Journal of Molecular and Cellular Cardiology . 1984;16(11):969–985. doi: 10.1016/S0022-2828(84)80011-5. [DOI] [PubMed] [Google Scholar]
- 54.Becker L. C., Ambrosio G. Myocardial consequences of reperfusion. Progress in Cardiovascular Diseases . 1987;30(1):23–44. doi: 10.1016/0033-0620(87)90009-0. [DOI] [PubMed] [Google Scholar]
- 55.Farmer E. E., Mueller M. J. ROS-mediated lipid peroxidation and RES-activated signaling. Annual Review of Plant Biology . 2013;64(1):429–450. doi: 10.1146/annurev-arplant-050312-120132. [DOI] [PubMed] [Google Scholar]
- 56.Zhao Y., Xin Z., Li N., et al. Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic Biol Med . 2018;124:1–11. doi: 10.1016/j.freeradbiomed.2018.05.082. [DOI] [PubMed] [Google Scholar]
- 57.Scindia Y., Leeds J., Swaminathan S. Iron homeostasis in healthy kidney and its role in acute kidney injury. Seminars in Nephrology . 2019;39(1):76–84. doi: 10.1016/j.semnephrol.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 58.Galaris D., Barbouti A., Korantzopoulos P. Oxidative stress in hepatic ischemia-reperfusion injury: the role of antioxidants and iron chelating compounds. Current Pharmaceutical Design . 2006;12(23):2875–2890. doi: 10.2174/138161206777947614. [DOI] [PubMed] [Google Scholar]
- 59.White C. W., Ali A., Hasanally D., et al. A cardioprotective preservation strategy employing ex vivo heart perfusion facilitates successful transplant of donor hearts after cardiocirculatory death. The Journal of Heart and Lung Transplantation . 2013;32(7):734–743. doi: 10.1016/j.healun.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 60.White C. W., Hasanally D., Mundt P., et al. A whole blood-based perfusate provides superior preservation of myocardial function during ex vivo heart perfusion. The Journal of Heart and Lung Transplantation . 2015;34(1):113–121. doi: 10.1016/j.healun.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 61.Ganguly R., Hasanally D., Stamenkovic A., et al. Alpha linolenic acid decreases apoptosis and oxidized phospholipids in cardiomyocytes during ischemia/reperfusion. Molecular and Cellular Biochemistry . 2018;437(1-2):163–175. doi: 10.1007/s11010-017-3104-z. [DOI] [PubMed] [Google Scholar]
- 62.Yeang C., Hasanally D., Que X., et al. Reduction of myocardial ischaemia-reperfusion injury by inactivating oxidized phospholipids. Cardiovascular Research . 2019;115(1):179–189. doi: 10.1093/cvr/cvy136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valko M., Jomova K., Rhodes C. J., Kuča K., Musílek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Archives of Toxicology . 2016;90(1):1–37. doi: 10.1007/s00204-015-1579-5. [DOI] [PubMed] [Google Scholar]
- 64.Brand M. D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biology and Medicine . 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 65.Ježek P., Hlavatá L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. The International Journal of Biochemistry & Cell Biology . 2005;37(12):2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 66.Anderson J. L., Morrow D. A. Acute myocardial infarction. The New England Journal of Medicine . 2017;376(21):2053–2064. doi: 10.1056/NEJMra1606915. [DOI] [PubMed] [Google Scholar]
- 67.Granger D., Kvietys P. R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biology . 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.An N., Gao Y., Si Z., et al. Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Frontiers in Immunology . 2019;10:p. 1592. doi: 10.3389/fimmu.2019.01592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang X., An N., Zhong C., et al. Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib- induced atrial fibrillation. Redox Biology . 2020;30:p. 101432. doi: 10.1016/j.redox.2020.101432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Molecular Cell . 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baba Y., Higa J., Shimada B., et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. American Journal of Physiology-Heart and Circulatory Physiology . 2018;314(3):H659–H668. doi: 10.1152/ajpheart.00452.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shan X., Lv Z., Yin M., Chen J., Wang J., Wu Q. N. The Protective Effect of Cyanidin-3-Glucoside on Myocardial Ischemia- Reperfusion Injury through Ferroptosis. Oxidative Medicine and Cellular Longevity . 2021;2021:15. doi: 10.1155/2021/8880141.8880141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park E., Chung S. W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death & Disease . 2019;10(11):p. 822. doi: 10.1038/s41419-019-2064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang L., Zhou Y., Xiong X., et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radical Biology & Medicine . 2021;162:339–352. doi: 10.1016/j.freeradbiomed.2020.10.307. [DOI] [PubMed] [Google Scholar]
- 75.Li J., Cao F., Yin H., et al. Ferroptosis: past, present and future. Cell Death & Disease . 2020;11(2):p. 88. doi: 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stamenkovic a., Pierce G. N., Ravandi A. phospholipid oxidation products in ferroptotic myocardial cell death. American Journal of Physiology-Heart and Circulatory Physiology . 2019;317(1):h156–h163. doi: 10.1152/ajpheart.00076.2019. [DOI] [PubMed] [Google Scholar]
- 77.Fang X., Wang H., Han D., et al. Ferroptosis as a target for protection against cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America . 2019;116(7):2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan W., Taylor A., Ellims A., et al. Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation-myocardial infarction. Circulation: Cardiovascular Interventions . 2012;5(2):270–278. doi: 10.1161/CIRCINTERVENTIONS.111.966226. [DOI] [PubMed] [Google Scholar]
- 79.Paraskevaidis I., Iliodromitis E., Vlahakos D., et al. Deferoxamine infusion during coronary artery bypass grafting ameliorates lipid peroxidation and protects the myocardium against reperfusion injury: immediate and long-term significance. European Heart Journal . 2005;26(3):263–270. doi: 10.1093/eurheartj/ehi028. [DOI] [PubMed] [Google Scholar]
- 80.Xie Y., Song X., Sun X., et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochemical and Biophysical Research Communications . 2016;473(4):775–780. doi: 10.1016/j.bbrc.2016.03.052. [DOI] [PubMed] [Google Scholar]
- 81.Li Q., Li Q., Jia J., et al. Baicalein exerts neuroprotective effects in FeCl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Frontiers in Pharmacology . 2019;10:p. 638. doi: 10.3389/fphar.2019.00638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fan Z., Cai L., Wang S., Wang J., Chen B. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Frontiers in Pharmacology . 2021;12:p. 628988. doi: 10.3389/fphar.2021.628988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma S., Sun L., Wu W., Wu J., Sun Z., Ren J. USP22 protects against myocardial ischemia-reperfusion injury via the SIRT1-p 53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Frontiers in Physiology . 2020;11:p. 551318. doi: 10.3389/fphys.2020.551318. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Chen H., Xiao Z., Ling X., Xu R. N., Zhu P., Zheng S. Y. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Molecular Medicine (Cambridge, Mass.) . 2021;27(1):p. 14. doi: 10.1186/s10020-021-00271-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stamenkovic A., O’Hara K. A., Nelson D. C., et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. American Journal of Physiology-Heart and Circulatory Physiology . 2021;320(3):H1170–h1184. doi: 10.1152/ajpheart.00237.2020. [DOI] [PubMed] [Google Scholar]
- 86.Cao G., Jiang N., Hu Y., et al. Ruscogenin attenuates cerebral ischemia-induced blood-brain barrier dysfunction by suppressing TXNIP/NLRP3 inflammasome activation and the MAPK pathway. International Journal of Molecular Sciences . 2016;17(9):p. 1418. doi: 10.3390/ijms17091418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pamenter M., Hogg D., Gu X., Buck L. T., Haddad G. G. Painted turtle cortex is resistant to an in vitro mimic of the ischemic mammalian penumbra. Journal of Cerebral Blood Flow and Metabolism . 2012;32(11):2033–2043. doi: 10.1038/jcbfm.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li W., Tan C., Liu Y., et al. Resveratrol ameliorates oxidative stress and inhibits aquaporin 4 expression following rat cerebral ischemia-reperfusion injury. Molecular Medicine Reports . 2015;12(5):7756–7762. doi: 10.3892/mmr.2015.4366. [DOI] [PubMed] [Google Scholar]
- 89.Groenendaal F., Shadid M., McGowan J., Mishra O. P., van Bel F. Effects of Deferoxamine, a Chelator of Free Iron, on Na+,K+-ATPase Activity of Cortical Brain Cell Membrane during Early Reperfusion after Hypoxia-Ischemia in Newborn Lambs. Pediatric Research . 2000;48(4):560–564. doi: 10.1203/00006450-200010000-00023. [DOI] [PubMed] [Google Scholar]
- 90.Shadid M., Buonocore G., Groenendaal F., et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neuroscience Letters . 1998;248(1):5–8. doi: 10.1016/S0304-3940(98)00303-6. [DOI] [PubMed] [Google Scholar]
- 91.Selim M. Treatment with the iron chelator, deferoxamine mesylate, alters serum markers of oxidative stress in stroke patients. Translational Stroke Research . 2010;1(1):35–39. doi: 10.1007/s12975-009-0001-0. [DOI] [PubMed] [Google Scholar]
- 92.Alim I., Caulfield J., Chen Y., et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell . 2019;177(5):1262–1279.e25. doi: 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 93.Anrather J., Iadecola C. Inflammation and stroke: an overview. Neurotherapeutics . 2016;13(4):661–670. doi: 10.1007/s13311-016-0483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tuo Q., Zhang S. T., Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Medicinal Research Reviews . 2021 doi: 10.1002/med.21817. [DOI] [PubMed] [Google Scholar]
- 95.Tuo Q., Lei P., Jackman K., et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry . 2017;22(11):1520–1530. doi: 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- 96.Guo H., Zhu L., Tang P., et al. Carthamin yellow improves cerebral ischemia-reperfusion injury by attenuating inflammation and ferroptosis in rats. International Journal of Molecular Medicine . 2021;47(4) doi: 10.3892/ijmm.2021.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu J., Xu F., Lu H. lncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sciences . 2020;260:p. 118305. doi: 10.1016/j.lfs.2020.118305. [DOI] [PubMed] [Google Scholar]
- 98.Guan X., Li X., Yang X., et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sciences . 2019;235:p. 116795. doi: 10.1016/j.lfs.2019.116795. [DOI] [PubMed] [Google Scholar]
- 99.Zhang W., Lan Y., Huang Q., Hua Z. Galangin induces B16F10 melanoma cell apoptosis via mitochondrial pathway and sustained activation of p38 MAPK. Cytotechnology . 2013;65(3):447–455. doi: 10.1007/s10616-012-9499-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guan X., Li Z., Zhu S., et al. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sciences . 2021;264:p. 118660. doi: 10.1016/j.lfs.2020.118660. [DOI] [PubMed] [Google Scholar]
- 101.Guo X., Lu Y., Zhang H., Huang J. Q., Li Y. W. PIEZO1 might be involved in cerebral ischemia-reperfusion injury through ferroptosis regulation: a hypothesis. Medical Hypotheses . 2021;146:p. 110327. doi: 10.1016/j.mehy.2020.110327. [DOI] [PubMed] [Google Scholar]
- 102.Cui Y., Zhang Y., Zhao X., et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain, Behavior, and Immunity . 2021;93:312–321. doi: 10.1016/j.bbi.2021.01.003. [DOI] [PubMed] [Google Scholar]
- 103.Linkermann A., Stockwell B., Krautwald S., Anders H. J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nature Reviews Immunology . 2014;14(11):759–767. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 104.Xie Y., Hou W., Song X., et al. Ferroptosis: process and function. Cell Death and Differentiation . 2016;23(3):369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang W., Stockwell B. R. Ferroptosis: death by lipid peroxidation. Trends in Cell Biology . 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Linkermann A., Skouta R., Himmerkus N., et al. Synchronized renal tubular cell death involves ferroptosis. Proceedings of the National Academy of Sciences of the United States of America . 2014;111(47):16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ding C., Ding X., Zheng J., et al. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death & Disease . 2020;11(10):p. 929. doi: 10.1038/s41419-020-03135-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Su L., Jiang X., Yang C., et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. The Journal of Biological Chemistry . 2019;294(50):19395–19404. doi: 10.1074/jbc.RA119.010949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stoppe C., Averdunk L., Goetzenich A., et al. The protective role of macrophage migration inhibitory factor in acute kidney injury after cardiac surgery. Science Translational Medicine . 2018;10(441, article eaan4886) doi: 10.1126/scitranslmed.aan4886. [DOI] [PubMed] [Google Scholar]
- 110.Jiang G., Liao Y., Huang L., Zeng X. J., Liao X. H. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Molecular Medicine Reports . 2021;23(1) doi: 10.3892/mmr.2020.11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao Z., Wu J., Xu H., et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia−reperfusion injury. Cell Death & Disease . 2020;11(8):p. 629. doi: 10.1038/s41419-020-02871-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang J., Bi J., Ren Y., et al. Involvement of GPX4 in irisin's protection against ischemia reperfusion-induced acute kidney injury. Journal of Cellular Physiology . 2021;236(2):931–945. doi: 10.1002/jcp.29903. [DOI] [PubMed] [Google Scholar]
- 113.Ali J., Davies S., Brais R., et al. Analysis of ischemia/reperfusion injury in time-zero biopsies predicts liver allograft outcomes. Liver Transplantation . 2015;21(4):487–499. doi: 10.1002/lt.24072. [DOI] [PubMed] [Google Scholar]
- 114.Kim W., Lake J., Smith J., et al. OPTN/SRTR 2016 annual data report: liver. American Journal of Transplantation . 2018;18:172–253. doi: 10.1111/ajt.14559. [DOI] [PubMed] [Google Scholar]
- 115.Galleano M., Tapia G., Puntarulo S., Varela P., Videla L. A., Fernández V. Liver preconditioning induced by iron in a rat model of ischemia/reperfusion. Life Sciences . 2011;89(7-8):221–228. doi: 10.1016/j.lfs.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 116.Yamada N., Karasawa T., Wakiya T., et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: potential role of ferroptosis. American Journal of Transplantation . 2020;20(6):1606–1618. doi: 10.1111/ajt.15773. [DOI] [PubMed] [Google Scholar]
- 117.Li Y., Feng D., Wang Z., et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death and Differentiation . 2019;26(11):2284–2299. doi: 10.1038/s41418-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hu Y., Mao Z., Xu L., et al. Protective effect of dioscin against intestinal ischemia/reperfusion injury via adjusting miR-351-5p-mediated oxidative stress. Pharmacological Research . 2018;137:56–63. doi: 10.1016/j.phrs.2018.09.016. [DOI] [PubMed] [Google Scholar]
- 119.Deng F., Zhao B., Yang X., et al. The gut microbiota metabolite capsiate promotes Gpx4 expression by activatingTRPV1to inhibit intestinal ischemia reperfusion-induced ferroptosis. Gut Microbes . 2021;13(1):1902719–1902721. doi: 10.1080/19490976.2021.1902719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mura M., Andrade C., Han B., et al. Intestinal ischemia-reperfusion-induced acute lung injury and oncotic cell death in multiple organs. Shock (Augusta, Ga.) . 2007;28(2):227–238. doi: 10.1097/01.shk.0000278497.47041.e3. [DOI] [PubMed] [Google Scholar]
- 121.Stallion A., Kou T., Latifi S., et al. Ischemia/reperfusion: a clinically relevant model of intestinal injury yielding systemic inflammation. Journal of Pediatric Surgery . 2005;40(3):470–477. doi: 10.1016/j.jpedsurg.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 122.Dong H., Qiang Z., Chai D., et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging . 2020;12(13):12943–12959. doi: 10.18632/aging.103378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li Y., Cao Y., Xiao J., et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death & Differentiation . 2020;27(9):2635–2650. doi: 10.1038/s41418-020-0528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]