

Abstract
Objective:
Hyperuricemia is closely associated with insulin resistance syndrome (and its many cardiometabolic sequalae); however, whether they are causally related has long been debated. We used bidirectional Mendelian randomization (MR) to investigate the potential causal nature and direction between insulin resistance and hyperuricemia, along with gout.
Methods:
We used genome-wide association data (N=288,649 for SU, N=763,813 for gout, N=153,525 for fasting insulin) to select genetic instruments for two-sample MR analyses, using multiple MR methods to address potential pleiotropic associations. We then used individual-level, electronic-medical-record-linked data from UK Biobank (N=360,453 persons of European ancestry) to replicate our analyses via single-sample MR.
Results:
Genetically determined SU, whether inferred from a polygenic score or strong individual loci, was not associated with fasting insulin concentrations. In contrast, genetically determined fasting insulin concentrations were positively associated with SU (0.37 mg/dL per log-unit increase in fasting insulin [95% CI, 0.15 to 0.58], P=0.001). This persisted in outlier-corrected (0.56 mg/dL [0.45 to 0.67]) and multivariable MR analyses conditioned on BMI (0.69 mg/dL [0.53 to 0.85]); all P<0.001. Polygenic scores for fasting insulin were also positively associated with SU among individuals in UK Biobank (P<0.001). Findings for gout were consistent with those for SU bidirectionally.
Conclusions:
These findings provide evidence to clarify core questions about the close association between hyperuricemia and insulin resistance syndrome: hyperinsulinemia leads to hyperuricemia, but not the other way around. Reducing insulin resistance could lower SU and gout risk, whereas lowering SU (e.g., allopurinol) is unlikely to mitigate insulin resistance and its cardiovascular-metabolic sequalae.
INTRODUCTION
The incidence, prevalence, and disability burden of gout have risen substantially over the past decades, especially in the United States. (1) Gout and hyperuricemia, its causal precursor, frequently coexist with metabolic syndrome (2) and are associated with an elevated burden of cardiovascular disease and type 2 diabetes. (3) But despite the close association between hyperuricemia and the insulin resistance syndrome, (4,5) the nature and direction of any causal relations are unclear. Observational studies have identified hyperuricemia as an independent risk factor for insulin resistance and prediabetes, (6) but these findings may represent a case of reverse causality or residual confounding. Conversely, some human physiologic experiments (4,7) suggest that induced hyperinsulinemia can raise serum urate concentrations (SU), but its casual impact at the population level remains unknown.
Clarifying the reason and direction behind the close association between insulin resistance and hyperuricemia could inform the treatment and prevention of these often-overlapping problems. This endeavor can be accomplished using Mendelian randomization (MR), which employs genetic variants as instrumental variables for exposures, allowing one to obtain unconfounded estimates of potential causal effects. Leveraging newly released genome-wide association studies (GWAS), which identified substantially more variants associated with SU and fasting insulin than their predecessors, we performed a bidirectional Mendelian randomization analysis to investigate potential causal relationships between insulin resistance and hyperuricemia, with gout itself as a secondary outcome.
MATERIALS AND METHODS
Study Design
We performed both one- and two-sample MR analyses. First, we conducted a series of univariable two-sample analyses to examine the relationship between SU and fasting insulin, a surrogate measure of insulin resistance. (8) We looked for causal relationships in both directions. We also examined gout in place of SU to ensure consistency of our findings. Given the known correlations between fasting insulin, SU, and body mass index (BMI), we then performed multivariable analyses (9) using BMI-associated genetic variants to partition the total effects of fasting insulin on SU from its direct effects, independent of BMI. Finally, we replicated our findings in a single-sample context, using individual-level data from the UK Biobank resource (UKBB) to assess the relationship between a polygenic score for fasting insulin concentrations and our two outcomes: SU and gout. The UK Biobank obtained ethical approval from the North West - Haydock Research Ethics Committee (16/NW/0274); all participants provided written informed consent.
Data Source and Study Population
Two-Sample MR
For our two-sample analyses, we used summary-level data from the largest available GWAS. For urate and gout, we used summary statistics from the Chronic Kidney Disease Genetics (CKDGen) consortium, consisting of many European-ancestry cohorts. (10) The summary statistics were derived from 288,649 participants for SU and from 13,179 cases and 750,634 controls for gout. For fasting insulin, we used the Meta-Analysis of Glucose- and Insulin-related traits Consortium (MAGIC), (11) which provided summary statistics for fasting insulin, adjusted for measured BMI, based on 153,525 European-ancestry participants without diabetes, as insulin concentrations are affected by diabetes or anti-diabetes medications. Finally, for our multivariable two-sample MR analysis conditioning on BMI, we used BMI association statistics from a recent meta-analysis of the Genetic Investigation of ANthropometric Traits (GIANT) consortium and the UKBB, which featured 681,275 European-ancestry participants in total. (12)
One-Sample MR
For our one-sample analysis, we used individual-level data from the UKBB (application 27892), a prospective cohort of approximately 500,000 individuals aged 40 to 69 years recruited across the United Kingdom. Genotypic and phenotypic data are available as well as biomarker measurements such as SU. Genotyping in this cohort was performed using either the Affymetrix UK BiLEVE Axiom array or the Affymetrix UK Biobank Axiom array. Quality control and imputation were performed centrally by researchers affiliated with the UKBB itself. We limited our analyses to people of European ancestry due to the fact our polygenic score for fasting insulin was based on data from European populations. To do so, we first used principal component analysis to identify genetically European individuals, and then, from this population, removed individuals who did not self-report as White or “do not know/prefer not to answer”, following prior UKBB analyses. (13) We also excluded related individuals. Relatedness was determined as per Bycroft et al. (14) wherein individuals were considered related if they were third-degree relatives or closer (kinship coefficient greater than or equal to 1/2(9/2)), using kinship coefficients provided by the UK Biobank. In total, our final one-sample analysis involved 378,065 individuals.
Outcomes
Two-Sample MR
The primary outcomes for the bi-directional two-sample analyses were age- and sex-adjusted concentrations of SU (mg/dL), as defined by the CKDGen Consortium, (10) and fasting insulin (log pmol/L), as defined by MAGIC. (11,15) SU concentrations averaged 5.1 mg/dL (± standard deviation (SD) 1.5) among European-ancestry members of the Atherosclerosis Risk in Communities (ARIC) cohort, one of the largest European-ancestry population-based cohorts in the CKDGen Consortium. Mean (±SD) fasting insulin concentrations were 1.90±1.7 and 1.82±1.7 log pmol/L, in men and women, respectively, in one of the largest European-ancestry cohorts in MAGIC.
One-Sample MR
Since fasting insulin was not measured in the UKBB cohort, the one-sample, individual-level MR analysis was unidirectional, with SU (mg/dL) serving as the primary outcome. Gout was examined as a secondary outcome in both the one- and two-sample analyses. For the one-sample analysis in the UKBB, gout was defined based on either patient report (20% of cases) or diagnoses recorded during primary care encounters (39%) or inpatient hospitalizations (12%), or a combination thereof (30%). This case definition builds upon one concluded to have high precision for detecting association in genetic epidemiological studies of gout (16) and employed in other published studies of gout in the UKBB. (17,18)
Genetic Instruments (Two-Sample and One-Sample MR)
We identified 123 single nucleotide polymorphism (SNPs) for SU (total R2 of 7.2%), (10) and 55 SNPs for gout (total R2 of 1.4%), (10) combining these to produce polygenic instruments for each exposure. We also separately examined the effects of SNPs from two highly-influential SU genes, SLC2A9 (R2=2.4% alone) and ABCG2 (R2=0.7% alone), which are strongly associated with SU concentrations (β=0.33 mg/dL and β=0.25 mg/dL, respectively) and gout risk (OR=1.51 [1.47 to 1.56] and OR=2.04 [1.96 to 2.12], respectively), with little to no evidence of pleiotropy (e.g., associations with related cardiometabolic traits). (19) For fasting insulin, we identified 95 SNPs (total R2 of 1%), (11) and for BMI, we identified 941 (total R2 of 6%). (12) We subsequently pruned these SNPs for linkage disequilibrium at a threshold of R2 < 0.001, leaving 121 independent SNPs for SU (F-statistic=182), 54 for gout (F-statistic=198), 83 for fasting insulin (F-statistic=25), and 925 for BMI (F-statistic=47) (Table S1). The F-statistic is a measure of the strength of association between these genetic instruments and the exposure; (20) values > 10 indicate the instrument is sufficiently strong with low potential for weak instrument bias, (21) which would otherwise drive a two-sample MR estimate toward the null.
Statistical Analysis
Primary Two-Sample MR
We first assessed the associations between genetically determined SU concentrations/gout risk and concentrations of fasting insulin using multiplicative random effects inverse variance weighted (IVW) meta-analysis methods; Wald ratios were generated for the single-SNP estimates. (22) In the opposite direction, we assessed the association between genetically determined fasting insulin concentrations on changes in SU and the odds of gout. Our primary analysis used BMI-adjusted betas as exclusively reported in the latest MAGIC GWAS (n=95 SNPs), (11) whereas our secondary analysis used unadjusted betas from an earlier MAGIC GWAS (n=12 SNPs), (15) where both BMI-unadjusted and BMI-adjusted summary statistics were available (Table S2).
Multivariable Two-Sample MR
We performed multivariable MR analyses to isolate the direct effects of fasting insulin, independent of (or conditional on) BMI, which is known to impact both fasting insulin concentrations (15) and SU. (23,24) Following the procedures for two-sample multivariable MR published by Sanderson et al., (9) these models included variants significantly associated with either fasting insulin or BMI.
Sensitivity MR Analysis for Pleiotropy
We assessed the presence of horizontal pleiotropy using the MR-Egger intercept test, (25) wherein the intercept represents the average pleiotropic effect. An intercept term that is significantly different from zero indicates the presence of unbalanced (directional) pleiotropy, which can bias the IVW estimate. (25) Along with our main (IVW) effect estimates, we generated additional estimates shown to be robust to the presence of horizontal pleiotropy. These included univariable (25) and multivariable (26) MR Egger and univariable weighted median- (27) and mode-based estimates. (28) We conducted leave-one-out analyses, systematically re-calculating the main IVW estimate after removing one variant at a time to visually inspect for influential variants, and re-generated all estimates after removing outliers identified by the MR-PRESSO (Pleiotropy RESidual Sum and Outlier) test. (29)
Two-Sample MR Analysis: Power Calculations
Post-hoc power calculations for the two-sample analysis were performed using the mRnd power calculator (30) based on the proportion of variance explained by the instruments, the numbers of participants in the CKDGen and MAGIC studies, and observed epidemiologic associations and their 95% confidence intervals (31) (Table S3).
One-Sample MR
Polygenic scores were constructed for the UKBB participants from the same fasting insulin SNPs used in the two-sample analysis. To calculate the scores, we used PLINK version 1.9 (www.cog-genomics.org/plink/1.9). Each SNP was weighted according to its effect size for fasting insulin in the latest MAGIC GWAS, as fasting insulin values were not measured in the UKBB cohort. The polygenic scores were normalised, setting the mean to zero and the standard deviation to one. Participants with urate concentrations four or more standard deviations from the mean were excluded. For the analysis of urate concentration as a function of the fasting insulin score, linear regression models were used. For gout, a binary outcome, we used logistic regression. Models were adjusted for age, sex, ten principal components to control for population stratification, and the genotyping platform used. We also controlled for BMI in some models.
Software
The one- and two-sample analyses were conducted using R software (R Project for Statistical Computing, Vienna, Austria, http://www.R-project.org); the R-packages TwoSampleMR, MendelianRandomization, and MVMR; and the MR-Base portal. (32)
RESULTS
Effects of genetically determined serum urate concentration and gout liability on fasting insulin: two-sample MR
In the main IVW analysis, neither genetically determined concentrations of SU (β: 0.0038 log pmol/L fasting insulin per 1 mg/dL increase in SU [95% confidence interval (CI): −0.0390 to 0.0466], p=0.86), nor genetically determined gout liability (β: −0.0026 log pmol/L [95% CI: −0.0235 to 0.0183], p=0.81) had significant effects on fasting insulin (Figure 1a and 1b). These findings were consistent across all MR estimates (Figure 1a and 1b) and did not materially change after removal of the outliers identified by the MR-PRESSO test (Table S4). Furthermore, while the SNPs mapping to the SLC2A9 and ABCG2 genes were strongly associated with SU and odds of gout, neither was associated with changes in fasting insulin (Figure 1a and 1b). Estimates were similar when using the BMI-unadjusted summary statistics for fasting insulin (Table S5). Furthermore, genetically determined SU was not associated with BMI, and thus, no multivariable MR analysis was performed.
Figure 1. Causal effect estimates for genetically determined concentration of serum urate (per 1 mg/dL) (A) and odds of gout (B) on BMI-adjusted concentrations of fasting insulin (log pmol/L), ascertained in individuals without diabetes, two-sample analysis.
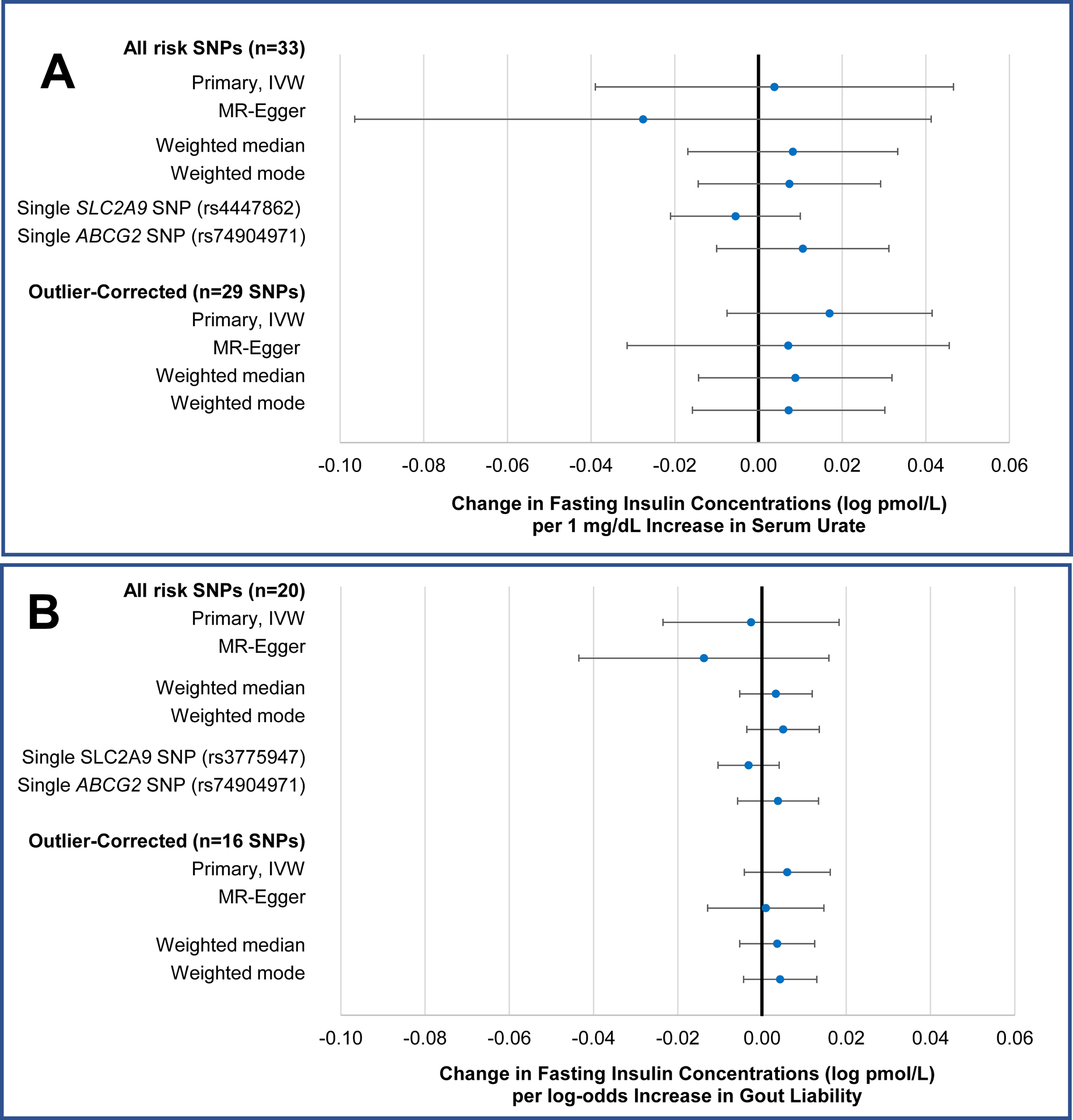
IVW=inverse probability weighted, SNP=single nucleotide polymorphism.
Effects of genetically determined fasting insulin concentrations on serum urate: two-sample MR
In the opposite direction, genetically determined concentrations of fasting insulin (adjusted for BMI) were positively associated with SU (Figure 2a). In the main IVW analysis, a one-unit (one log pmol/L) increase in fasting insulin was associated with a 0.37 mg/dL increase in SU ([95% CI: 0.15 to 0.58], p=0.001). This translates to a 0.63 mg/dL increase in SU per one SD (1.7 log pmol/L) increase in fasting insulin. No pleiotropy was detected (MR-Egger intercept=0.008, p=0.09). All other MR estimates were significant and were numerically larger than the main IVW estimate except for the MR-Egger regression estimate (Figure 2a).
Figure 2. Causal effect estimates for genetically determined concentration of fasting insulin (per log pmol/L) on concentrations of serum urate (mg/dL) (A) and odds of gout (B), ascertained in individuals without diabetes, two-sample analysis.
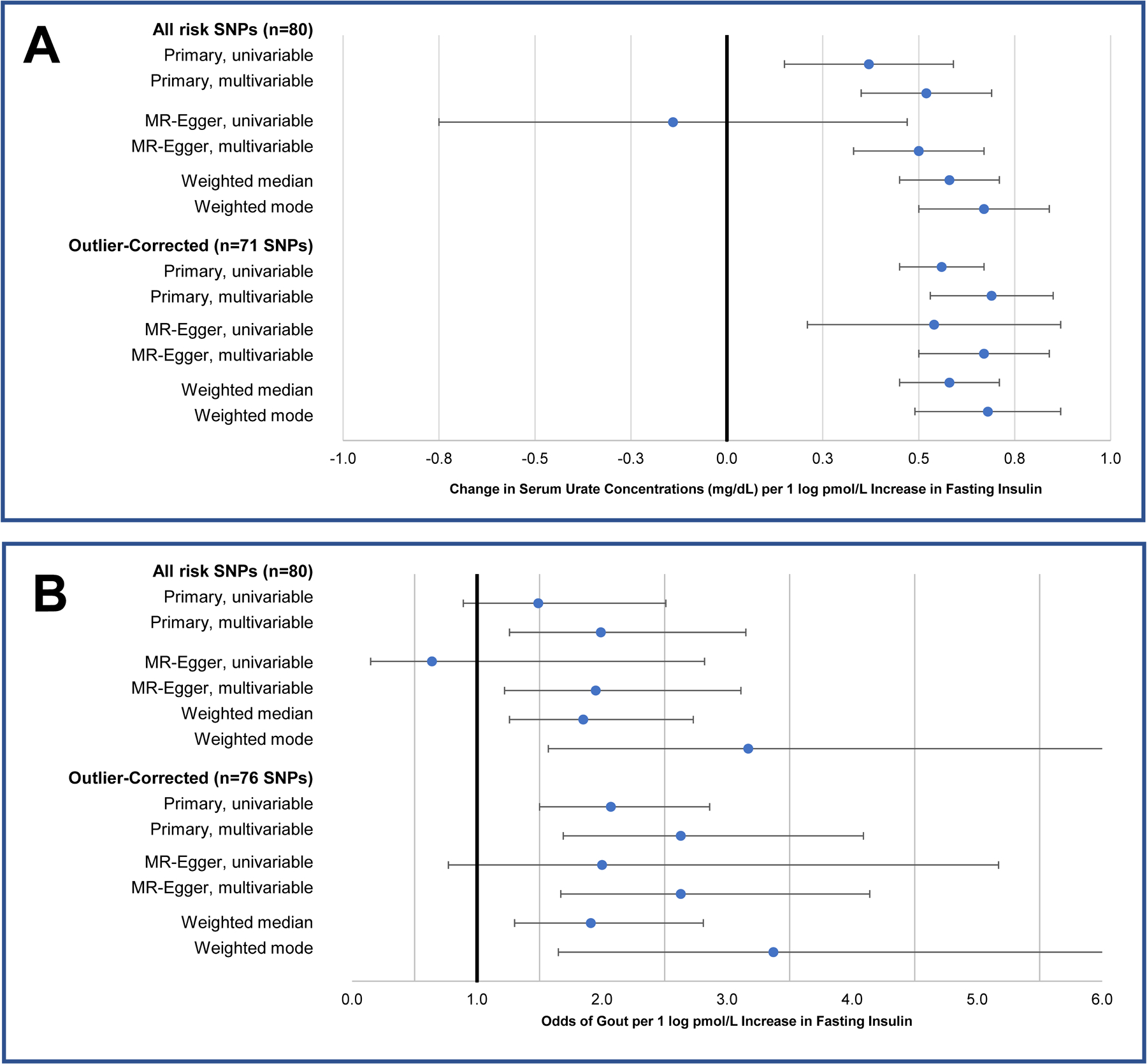
SNP=single nucleotide polymorphism. One of the candidate risk SNPs for fasting insulin was removed during harmonisation due to ambiguity in the strand direction, leaving 80 in the final analysis.
Nine outlier SNPs were identified by the MR-PRESSO test (Table S4). As shown in the leave-one-out plots in Figure S2a and S2b and scatter plots in Figure S4a and S4b, the most influential SNP was rs1260326, mapped to the GCKR gene. Upon the removal of all nine outliers, the IVW estimate strengthened (β: 0.56 mg/dL, [0.45 to 0.67], p<0.001), including the MR-Egger regression estimate (β: 0.54 mg/dL, [0.22 to 0.87], p=0.002) (Figure 2a) and there remained little evidence of pleiotropy (MR-Egger intercept <0.001, p=0.90).
As displayed in Figure 2a, the multivariable IVW estimates for fasting insulin, representing its direct effect on SU conditioned on genetically determined BMI, were larger than their univariable counterparts, reaching 0.52 mg/dL ([0.35 to 0.69]) per log pmol/L of fasting insulin, which translates to 0.88 mg/dL per SD increase in fasting insulin, including outliers, and 0.69 mg/dL ([0.53 to 0.85]), or 1.18 mg/dL per SD of fasting insulin, excluding outliers (both p<0.001). The same pattern was observed for the univariable and multivariable MR-Egger estimates. The univariable and multivariable estimates of the effect of genetically determined BMI on SU were virtually identical, with SU increases of 0.32 and 0.31 mg/dL per one SD increase in BMI, respectively, (both p<0.001).
Effects of genetically determined fasting insulin concentrations on serum urate: one-sample MR
These findings were replicated at the individual level in the UKBB using polygenic risk scores for fasting insulin (Figure 3). SU concentrations among all eligible UKBB participants (n=360,453) averaged 5.19 mg/dL with standard deviation 1.34; 26% had hyperuricemia (SU ≥ 6 mg/dL) (Table S6). Consistent with the two-sample MR, we found a one SD increase in the polygenic risk score corresponded to a significant increase in SU (p=6.3×10−33, Table 1). The effect strengthened after removing the outliers identified in the two-sample MR and grew even larger with additional adjustment for measured BMI. When we excluded ULT users, our effect estimates remained the same to two decimal places.
Figure 3. Serum urate concentration (mean) by decile of the polygenic score for fasting insulin in the UK Biobank.
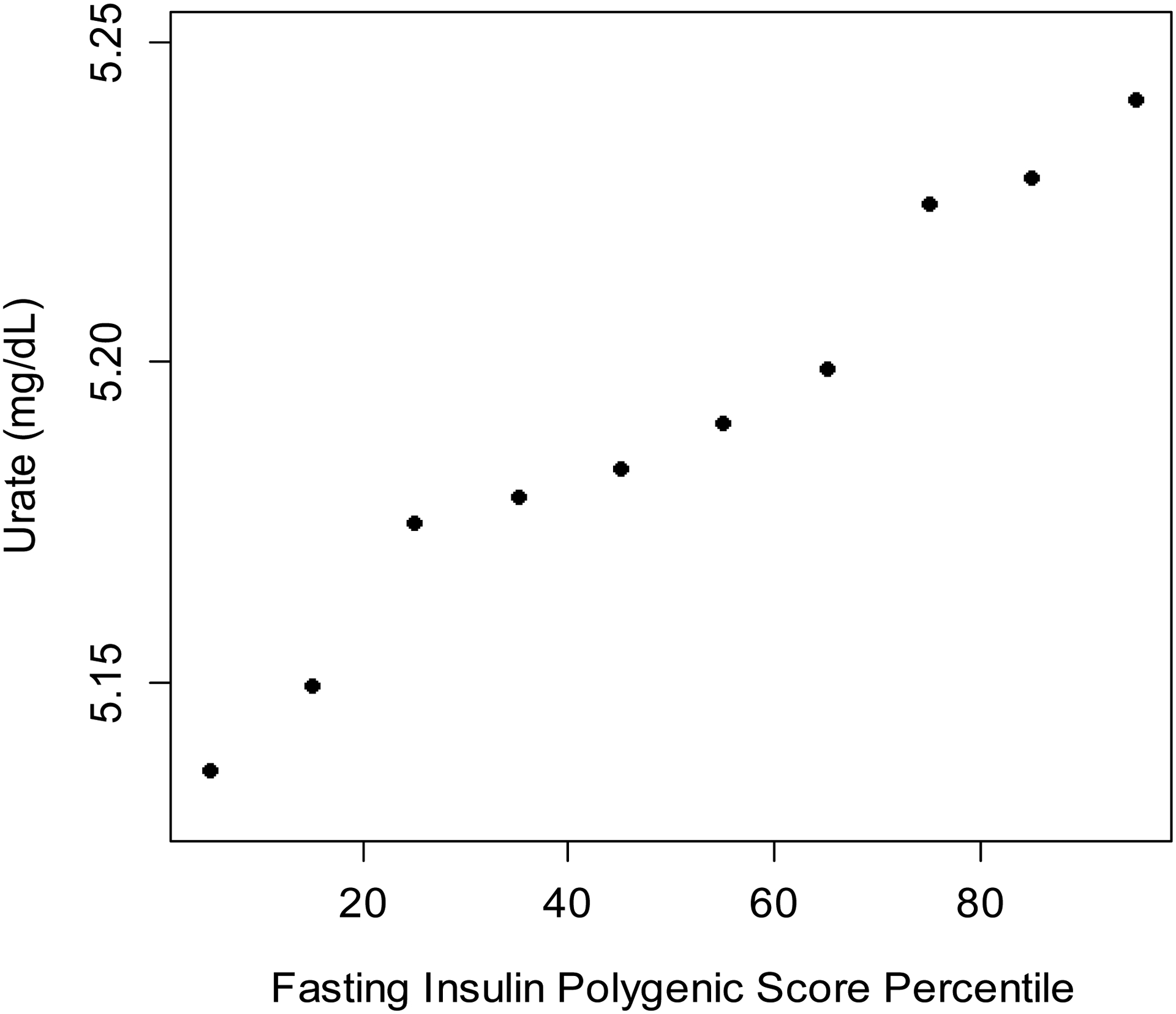
The 71-SNP polygenic score, which excluded outliers, was used to generate this figure.
Table 1. Association of polygenic scores for fasting insulin with serum urate concentrations and odds of gout in the UK Biobank (one-sample analysis).
All models were adjusted for age and sex as well as ten principal components and the genotyping platform used.
| Change in urate (mg/dl) per SD in fasting insulin score (95% CI) | P | Odds ratio for gout per SD in fasting insulin score (95% CI) | P | |
|---|---|---|---|---|
| Unadjusted for BMI | ||||
| All SNPs | 0.023 (0.019 to 0.026) | 6.3×10−33 | 1.03 (1.01 to 1.05) | 9.5×10−4 |
| Excluding outliers | 0.031 (0.027 to 0.035) | 8.2×10−61 | 1.05 (1.03 to 1.07) | 1.0×10−7 |
| Adjusted for BMI | ||||
| All SNPs | 0.031 (0.028 to 0.034) | 1.8×10−69 | 1.05 (1.03 to 1.07) | 8.8×10−7 |
| Excluding outliers | 0.040 (0.036 to 0.043) | 8.6×10−114 | 1.07 (1.05 to 1.09) | 9.8×10−13 |
Effects of genetically determined fasting insulin concentrations on risk of gout: one- and two-sample MR
The estimated effects of genetically determined fasting insulin on gout risk followed a similar pattern to SU. In the two-sample MR, the odds ratio (OR) for gout (per log pmol/L insulin) increased from 1.49 [95% CI: 0.89 to 2.51], p=0.13 in the initial analysis to 2.07 [1.50 to 2.86], p<0.001 when four outliers were excluded (Figure 2b). As with SU, the multivariable OR was larger than the univariable, reaching 2.63 ([1.69 to 4.10], p<0.001). In the one-sample analysis (n=12,920), we observed a similar result, namely that there was a statistically significant positive association between the polygenic score for fasting insulin and odds ratio for gout (p=9.5×10−4, see Table 1) which increased when we adjusted for BMI and excluded outlier SNPs. Our definition of gout captured 93% of participants on urate-lowering therapy. Limiting our definition of gout to self-reported cases only did not change the significance of our results, or the direction of effect.
DISCUSSION
This first bidirectional Mendelian randomization analysis of SU and fasting insulin provides evidence that genetically elevated fasting insulin, a measure of insulin resistance and precursor to cardiometabolic diseases, is causally associated with hyperuricemia, as well as the clinical endpoint of gout. Effects were consistent across summary-level and individual-level analyses and strengthened upon the removal of outliers and controlling for BMI. Conversely, our data do not support a causal effect of SU on fasting insulin concentrations.
Our null findings on the effects of SU are consistent with an earlier, individual-level MR analysis of multiple population-based cohorts (e.g., ARIC, Framingham) within the CHARGE consortium (33) wherein an eight-SNP genetic risk score for SU was not associated with fasting insulin concentrations. These findings also agree with prior MR analyses which similarly found no causal effects between SU and clinical cardiometabolic endpoints, (19) including coronary heart disease (34,35) and type 2 diabetes. (19) Furthermore, the two pivotal individual genes (SLC2A9 and ABCG2) accounting for 34% and 10%, respectively, of the total proportion of variance in SU concentration explained by the polygenic instrument for SU, (10) were not associated with fasting insulin concentrations in this MR analysis, nor the prior CHARGE consortium analysis. (33) Thus, a causal role of SU on insulin resistance seems highly unlikely.
Conversely, in the opposite direction, fasting insulin concentrations were positively associated with SU and this relationship grew larger when outliers were removed. The most prominent outlier was rs1260326, mapped to the GCKR gene, which affects multiple cardiometabolic pathways. (19,33) This SNP was significantly associated with fasting insulin concentrations, but is also a likely causal variant of SU, (10) making a strong case for its removal on the basis of horizontal pleiotropy. The unidirectional causal effects observed for fasting insulin in our study are consistent with another causal indicator reported by the CKDGen consortium (i.e., genetic causal proportion = −0.49, p=2.80×10−2) (10) that suggested fasting insulin is partially genetically causal to urate concentrations. Our findings also corroborate previous physiologic experiments demonstrating insulin’s anti-uricosuric property, with exogenous insulin reducing the renal excretion of urate (7,36) in both healthy and hypertensive individuals. Insulin may increase renal urate reabsorption via stimulation of GLUT9 (encoded by SLC2A9) and other renal urate transporters involved in urate reabsorption. (37) Insulin resistance manifests early in the progression to type 2 diabetes, (38) and the early pathophysiologic changes could raise SU concentrations before dysglycemia becomes clinically evident, a theory supported by the Whitehall II cohort study, (39) wherein participants who eventually developed type 2 diabetes already had lower levels of insulin sensitivity at baseline (up to 13 years prior to diagnosis) than those who did not develop diabetes.
Whilst obesity and insulin resistance are positively correlated, we provide evidence that a portion of fasting insulin’s effects on SU are independent of genetically determined BMI, with multivariable effect estimates that were larger than the univariable. At the same time, our results suggest there is at least some portion of the SU-raising effect of obesity independent of the insulin pathway. Negative confounding by BMI is consistent with prior reports from MAGIC investigators, (15,40) and the phenomenon of lipodystrophic insulin resistance, (41) wherein a lack (or dysfunction) of white adipose tissue, especially subcutaneous gluteofemoral fat, leads to insulin resistance and metabolic syndrome in non-obese or lean individuals. (42) Subtle lipodystrophy is believed to be a major contributor to metabolic syndrome at the population level. (42)
Related to this, a potential caveat of our analysis is that the SNP-insulin association estimates from the MAGIC summary statistics were adjusted for age, sex, and BMI (as measured in study participants who underwent genotyping), while the corresponding estimates for SU were not adjusted for BMI. MAGIC investigators opted to adjust for BMI as this had increased the number of insulin-associated variants detected in their previous GWAS, (15) including some insulin-raising alleles associated with lower BMI. While this adjustment can help isolate SNPs impacting insulin resistance independently of BMI, it can also raise concerns about collider bias (43) (e.g., inducing a spurious association between the SNP and fasting insulin). However, MAGIC investigators evaluated this possibility and found no evidence of collider bias in the vast majority of SNPs tested. (11,41) Moreover, there were no meaningful differences in the effect estimates we generated using the BMI-unadjusted and BMI-adjusted fasting insulin summary statistics from the earlier MAGIC GWAS (Table S2). Our examining the impact of fasting insulin on SU with a multivariable MR model that included BMI-associated SNPs (the potential collider) (9) should further alleviate these concerns.
Our novel findings explain the core reason and direction underpinning the close association between hyperuricemia and insulin resistance syndrome, with implications for the prevention and management of both conditions and their cardiometabolic sequala. Large-scale pharmaceutical trials of drugs that substantially lowered SU have not, to date, reported cardiovascular-metabolic benefits, such as weight change, lipid profile, blood pressure, or renal function. (44,45) Building upon this, our data suggest interventions targeting SU alone (e.g., urate lowering drugs) are unlikely to lower insulin concentrations and, in turn, the risk of insulin resistance or metabolic syndrome and its cardiovascular-metabolic consequences. Instead, our data suggest lifestyle modifications specifically shown to improve insulin concentrations and insulin resistance (e.g., a ‘green’ Mediterranean diet emphasising consumption of plant proteins over red/processed meats and other animal proteins, (46)) would lower SU, in addition to providing other cardiometabolic risk benefits. Indeed, a higher-protein, low-carbohydrate diet was associated with reductions in BMI (median 2.7 kg/m2) and SU (median 1.6 mg/dL), and improvements in dyslipidemia, in a pilot open-label trial of gout patients, (47) while in a recent analysis of the Dietary Intervention Randomized Controlled Trial (DIRECT), three healthy weight-loss diets significantly reduced SU, particularly among those with baseline hyperuricemia (by 1.9 to 2.4 mg/dL over 6 months, the maximum weight-loss phase, and by 1.1 to 1.4 mg/dL over 24 months). (48) This reduction was independently driven by reductions in plasma insulin concentrations in addition to weight reduction. (48)
Our analysis has some limitations. While the genetic association data were sourced from large, multi-national disease consortia, they pertained mainly to European ancestry/white British populations. This served to minimise confounding by differences in population structure, (21) but our findings should be confirmed in other ancestral populations. Since insulin concentrations were not measured in the UKBB cohort, we could not replicate our analysis of the effect of genetically determined SU on fasting insulin in the single-sample setting. However, none of the estimates from the two-sample analysis suggested a causal role for these exposures. With an R2 of 1.3%, the 80-SNP instrument for fasting insulin explained a comparatively low proportion of the phenotypic variance (e.g., overall variance in measured fasting insulin concentrations) than did the instruments for SU (R2=7.1%) and BMI (R2=6%). This was evident in the one-sample MR, wherein the polygenic risk score for fasting insulin was positively correlated with measured SU concentrations (Figure 3), although the absolute difference between the extreme deciles of the risk score was small (~0.10 mg/dL). Of note, since fasting insulin concentrations were not measured in the UKBB cohort, different methods were required for the one- and two-sample analyses, and the resultant effect estimates cannot be directly compared. The estimates generated by the two-sample MR represent the change in concentrations of SU (and odds of gout) associated with a one-unit change in genetically determined concentrations of fasting insulin itself, while the estimates generated by the one-sample MR represent the changes associated with a one-SD change in the polygenic score for fasting insulin, which is less sensitive. As such, the findings of our one-sample analysis served to reinforce the presence of causal effects of fasting insulin observed in our two-sample analysis, rather than quantify the magnitude of these effects. (49) Importantly, the variants are a proxy for the genetic predisposition towards raised fasting insulin concentrations, while environmental factors (50) appear to make a larger contribution to the total phenotypic variance in fasting insulin. BMI, for example, accounted for one-third of the variance in fasting insulin concentrations in one MAGIC cohort. (15)
While weak instrument bias would drive the two-sample MR estimates towards the null (in the absence of substantial overlap between samples), we observed significant causal effects for fasting insulin, but not SU, whose instrument was stronger. Indeed, we had >99% power to detect a causal effect of SU on fasting insulin concentrations matching that observed in a representative sample of US adults (Table S4). (31) We sourced data from recently published, mainly population-based genome-wide association studies, and the findings of the two-sample univariable and multivariable analyses were generally consistent for SU and gout, and robust to sensitivity analyses, especially after outliers were removed. Moreover, the positive associations between genetically instrumented fasting insulin concentrations and SU were replicated at the individual level, before and after adjustment for measured BMI.
In conclusion, this study provides robust evidence that insulin resistance has a positive causal effect on serum urate concentrations, with this relationship operating only in one direction. Interventions to reduce insulin resistance may lower SU concentrations and gout risk, conferring additional metabolic health benefits.
Supplementary Material
Figure S1: Leave-one-out sensitivity analysis of the effect of genetically determined concentrations of serum urate (per 1 mg/dL) (A) and odds of gout (B) on BMI-adjusted concentrations of fasting insulin (log pmol/L), re-generating the inverse variance weighted estimate after each SNP is removed, two-sample analysis. The SNP at the top (rs1260326) maps to the GCKR gene; rs74904971 (third from the bottom in A and bottom of B) maps to the ABCG2 gene.
Figure S2: Leave-one-out sensitivity analysis of the effect of genetically determined concentrations of fasting insulin (per log pmol/L), ascertained in individuals without diabetes, on concentrations of serum urate (mg/dL) (A) and odds of gout (B), re-generating the inverse variance weighted estimate after each SNP is removed, two-sample analysis. The SNP at the top (rs1260326) maps to the GCKR gene.
Figure S3: Scatterplot of the associations of the serum urate-associated SNPs (A) and gout-associated SNPs (B) with serum urate (or gout) and fasting insulin concentrations, and estimated causal effect, by MR test. The slope of each line corresponds to the causal effect estimated by each method.
Figure S4: Scatterplot of the associations of the fasting insulin-associated SNPs with fasting insulin and serum urate concentrations (A) and odds of gout (B), and estimated causal effect, by MR test. The slope of each line corresponds to the causal effect estimated by each method
Sources of Funding:
This research was supported by grants P50-AR-060772, R01-AR-065944, and R01-AR-056291 from the National Institutes of Health. The funder had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. NM is supported by a Fellowship Award from the Canadian Institutes of Health Research. MJO is supported by the National Institutes of Health Ruth L. Kirschstein Institutional National Research Service Award program [T32-DK-110919]. CY is supported by the National Institutes of Health Ruth L. Kirschstein Institutional National Research Service Award program [T32-AR-007258] and a Scientist Development Award from the Rheumatology Research Foundation.
Disclosures
NM, MJO, CY, TRM, and AL have no conflicts of interest to disclose. DBM reports research support from Astra Zeneca and consulting fees from Horizon and Kowa Pharmaceuticals. HKC reports research support from Ironwood and Horizon, and consulting fees from Ironwood, Selecta, Horizon, Takeda, Kowa, and Vaxart. The authors declare no support from any organisation for the submitted work other than that described above, and no financial relationships in the previous two years with any organisations that might have an interest in the submitted work.
REFERENCES
- 1.Safiri S, Kolahi A-A, Cross M, Carson-Chahhoud K, Hoy D, Almasi-Hashiani A, et al. Prevalence, Incidence, and Years Lived With Disability Due to Gout and Its Attributable Risk Factors for 195 Countries and Territories 1990–2017: A Systematic Analysis of the Global Burden of Disease Study 2017. Arthritis Rheumatol Hoboken NJ 2020;72:1916–1927. [DOI] [PubMed] [Google Scholar]
- 2.Choi HK, Ford ES. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med 2007;120:442–447. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Pandya BJ, Choi HK. Comorbidities of Gout and Hyperuricemia in the US General Population: NHANES 2007–2008. Am J Med 2012;125:679–687.e1. [DOI] [PubMed] [Google Scholar]
- 4.Facchini F, Chen YD, Hollenbeck CB, Reaven GM. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991;266:3008–3011. [PubMed] [Google Scholar]
- 5.Rathmann W, Funkhouser E, Dyer AR, Roseman JM. Relations of hyperuricemia with the various components of the insulin resistance syndrome in young black and white adults: the CARDIA study. Coronary Artery Risk Development in Young Adults. Ann Epidemiol 1998;8:250–261. [DOI] [PubMed] [Google Scholar]
- 6.Krishnan E, Pandya BJ, Chung L, Hariri A, Dabbous O. Hyperuricemia in Young Adults and Risk of Insulin Resistance, Prediabetes, and Diabetes: A 15-Year Follow-up Study. Am J Epidemiol 2012;176:108–116. [DOI] [PubMed] [Google Scholar]
- 7.Ter Maaten JC, Voorburg A, Heine RJ, Ter Wee PM, Donker AJ, Gans RO. Renal handling of urate and sodium during acute physiological hyperinsulinaemia in healthy subjects. Clin Sci Lond Engl 1979 1997;92:51–58. [DOI] [PubMed] [Google Scholar]
- 8.Laakso M How Good a Marker Is Insulin Level for Insulin Resistance? Am J Epidemiol 1993;137:959–965. [DOI] [PubMed] [Google Scholar]
- 9.Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol 2019;48:713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet 2019;51:1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Spracklen CN, Marenne G, Varshney A, Corbin LJ, Luan J, et al. The Trans-Ancestral Genomic Architecture of Glycaemic Traits.; 2020. Available at: http://biorxiv.org/lookup/doi/10.1101/2020.07.23.217646. Accessed August 19, 2020. [DOI] [PMC free article] [PubMed]
- 12.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet 2018;27:3641–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole JB, Florez JC, Hirschhorn JN. Comprehensive genomic analysis of dietary habits in UK Biobank identifies hundreds of genetic associations. Nat Commun 2020;11:1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cadzow M, Merriman TR, Dalbeth N. Performance of gout definitions for genetic epidemiological studies: analysis of UK Biobank. Arthritis Res Ther 2017;19:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tai V, Narang RK, Gamble G, Cadzow M, Stamp LK, Merriman TR, et al. Do Serum Urate–Associated Genetic Variants Differentially Contribute to Gout Risk According to Body Mass Index? Analysis of the UK Biobank. Arthritis Rheumatol 2020;72:1184–1191. [DOI] [PubMed] [Google Scholar]
- 18.Sandoval-Plata G, Nakafero G, Chakravorty M, Morgan K, Abhishek A. Association between serum urate, gout and comorbidities: a case-control study using data from the UK Biobank. Rheumatol Oxf Engl 2020. [DOI] [PubMed] [Google Scholar]
- 19.Keenan T, Zhao W, Rasheed A, Ho WK, Malik R, Felix JF, et al. Causal Assessment of Serum Urate Levels in Cardiometabolic Diseases Through a Mendelian Randomization Study. J Am Coll Cardiol 2016;67:407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wald A The Fitting of Straight Lines if Both Variables are Subject to Error. Ann Math Stat 1940;11:284–300. [Google Scholar]
- 23.Larsson SC, Burgess S, Michaëlsson K. Genetic association between adiposity and gout: a Mendelian randomization study. Rheumatol Oxf Engl 2018;57:2145–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyngdoh T, Vuistiner P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, et al. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. PLoS One 2012;7:e39321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 2017;32:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rees JMB, Wood AM, Burgess S. Extending the MR-Egger method for multivariable Mendelian randomization to correct for both measured and unmeasured pleiotropy. Stat Med 2017;36:4705–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 12 01;46:1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brion M-JA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 2013;42:1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi HK, Ford ES. Haemoglobin A1c, fasting glucose, serum C-peptide and insulin resistance in relation to serum uric acid levels--the Third National Health and Nutrition Examination Survey. Rheumatology 2008;47:713–717. [DOI] [PubMed] [Google Scholar]
- 32.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 2010;3:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White J, Sofat R, Hemani G, Shah T, Engmann J, Dale C, et al. Plasma urate concentration and risk of coronary heart disease: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 2016;4:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efstathiadou A, Gill D, McGrane F, Quinn T, Dawson J. Genetically Determined Uric Acid and the Risk of Cardiovascular and Neurovascular Diseases: A Mendelian Randomization Study of Outcomes Investigated in Randomized Trials. J Am Heart Assoc 2019;8:e012738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muscelli E, Natali A, Bianchi S, Bigazzi R, Galvan AQ, Sironi AM, et al. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am J Hypertens 1996;9:746–752. [DOI] [PubMed] [Google Scholar]
- 37.Mount D, Mandal A. Insulin: Genetic and Physiological Influences on Human Uric Acid Homeostasis [abstract]. Arthritis Rheumatol 70. [Google Scholar]
- 38.Weir GC, Bonner-Weir S. Five Stages of Evolving Beta-Cell Dysfunction During Progression to Diabetes. Diabetes 2004;53:S16–S21. [DOI] [PubMed] [Google Scholar]
- 39.Tabák AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimäki M, Witte DR. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. The Lancet 2009;373:2215–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott RA, Fall T, Pasko D, Barker A, Sharp SJ, Arriola L, et al. Common Genetic Variants Highlight the Role of Insulin Resistance and Body Fat Distribution in Type 2 Diabetes, Independent of Obesity. Diabetes 2014;63:4378–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.EPIC-InterAct Consortium, Cambridge FPLD1 Consortium, Lotta LA, Gulati P, Day FR, Payne F, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 2017;49:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest 2019;129:4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL, Kraft P. Adjusting for Heritable Covariates Can Bias Effect Estimates in Genome-Wide Association Studies. Am J Hum Genet 2015;96:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, et al. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N Engl J Med 2018;378:1200–1210. [DOI] [PubMed] [Google Scholar]
- 45.Doria A, Galecki AT, Spino C, Pop-Busui R, Cherney DZ, Lingvay I, et al. Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. N Engl J Med 2020;382:2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsaban G, Yaskolka Meir A, Rinott E, Zelicha H, Kaplan A, Shalev A, et al. The effect of green Mediterranean diet on cardiometabolic risk; a randomised controlled trial. Heart Br Card Soc 2020. [DOI] [PubMed] [Google Scholar]
- 47.Dessein PH, Shipton EA, Stanwix AE, Joffe BI, Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Ann Rheum Dis 2000;59:539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yokose C, McCormick N, Rai SK, Lu N, Curhan G, Schwarzfuchs D, et al. Effects of Low-Fat, Mediterranean, or Low-Carbohydrate Weight Loss Diets on Serum Urate and Cardiometabolic Risk Factors: A Secondary Analysis of the Dietary Intervention Randomized Controlled Trial (DIRECT). Diabetes Care 2020;43:2812–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological Challenges in Mendelian Randomization: Epidemiology 2014;25:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snieder H, Boomsma DI, Doornen LJ van, Neale MC. Bivariate genetic analysis of fasting insulin and glucose levels. Genet Epidemiol 1999;16:426–446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Leave-one-out sensitivity analysis of the effect of genetically determined concentrations of serum urate (per 1 mg/dL) (A) and odds of gout (B) on BMI-adjusted concentrations of fasting insulin (log pmol/L), re-generating the inverse variance weighted estimate after each SNP is removed, two-sample analysis. The SNP at the top (rs1260326) maps to the GCKR gene; rs74904971 (third from the bottom in A and bottom of B) maps to the ABCG2 gene.
Figure S2: Leave-one-out sensitivity analysis of the effect of genetically determined concentrations of fasting insulin (per log pmol/L), ascertained in individuals without diabetes, on concentrations of serum urate (mg/dL) (A) and odds of gout (B), re-generating the inverse variance weighted estimate after each SNP is removed, two-sample analysis. The SNP at the top (rs1260326) maps to the GCKR gene.
Figure S3: Scatterplot of the associations of the serum urate-associated SNPs (A) and gout-associated SNPs (B) with serum urate (or gout) and fasting insulin concentrations, and estimated causal effect, by MR test. The slope of each line corresponds to the causal effect estimated by each method.
Figure S4: Scatterplot of the associations of the fasting insulin-associated SNPs with fasting insulin and serum urate concentrations (A) and odds of gout (B), and estimated causal effect, by MR test. The slope of each line corresponds to the causal effect estimated by each method