

Abstract
Breast cancers are classified into five intrinsic subtypes and 10 integrative clusters based on gene expression patterns and genomic aberrations, respectively. Although the cell-of-origin, adaptive plasticity, and genomic aberrations shape dynamic transcriptomic landscape during cancer progression, how interplay between these three core elements governs obligatory steps for a productive cancer progression is unknown. Here, we used genetic ancestry-mapped immortalized breast epithelial cell lines generated from breast biopsies of healthy women that share gene expression profiles of luminal A, normal-like, and basal-like intrinsic subtypes of breast cancers and breast cancer relevant oncogenes to develop breast cancer progression model. Using flow cytometry, mammosphere growth, signaling pathway, DNA damage response, and in vivo tumorigenicity assays, we provide evidence that establishes cell-context dependent effects of oncogenes in conferring plasticity, self-renewal/differentiation, intratumor heterogeneity, and metastatic properties. By contrast, oncogenic aberrations, independent of cell-context, shaped response to DNA damage-inducing agents. Collectively, this study reveals how the same set of genomic aberration can have distinct effects on tumor characteristics based on cell-of-origin of tumor and highlights the need to utilize multiple “normal” epithelial cell types to decipher oncogenic properties of a gene of interest. Additionally, by creating multiple isogenic cell lines ranging from primary cells to metastatic variants, we provide resources to elucidate cell-intrinsic properties and cell-oncogene interactions at various stages of cancer progression.
Graphical Abstract

Visual Overview: A schematic diagram representing the flow of the study.
Introduction
Molecular profiling of breast tumors has revealed different intrinsic subtypes (1), which include estrogen receptor alpha (ERα)-positive luminal A and luminal B subtypes, HER2+, basal-like and normal-like subtypes. These subtypes of breast cancers have heterogeneous pathologies and different clinical outcome. Similarly, based on genomic aberrations, breast cancers are classified into 10 integrative clusters with distinct outcomes (2). Molecular basis for differing outcomes from different subtypes/clusters is unclear but could be related to interplay between cell-of-origin of tumors and genomic aberrations (3). The normal breast contains different subpopulations of cells, such as stem cells, luminal-progenitor cells, and luminal-differentiated cells. It is proposed that luminal progenitors or bipotent-progenitors are the cell-of-origin of basal breast cancers (4–6). HER2+ cancers may originate from late luminal-progenitors, whereas luminal A and luminal B breast cancers may arise from luminal-differentiated cells (3). Through integration of single cell sequencing data of healthy breast with publicly available breast cancer gene expression datasets, we recently proposed that the majority of breast cancers originate from mature luminal cells (7). However, experimental validation of these possibilities is still challenging because most of the prior culturing methods favored the outgrowth of basal-like breast epithelial cells (8–10). Therefore, developing a model system that utilizes breast epithelial cell lines with luminal characteristics derived from multiple healthy donors would aid in establishing relationship between cell-of-origin, genomic aberrations, and obligatory steps in breast cancer progression.
The normal breast cells progress to cancer due to acquisition of genetic or epigenetic alterations (5). Several breast cancer subtype specific mutations have been reported including PIK3CA mutations in luminal A/B breast cancers, loss of retinoblastoma gene in luminal B breast cancers, and PIK3CA amplifications and TP53 mutations in basal-like breast cancers (11). HER2 amplification is observed in 15% of breast cancers. To date, none of these oncogenic mutations could reproducibly transform breast epithelial cells in vitro and therefore, mutated RAS oncogene in combination with SV40 T/t antigens still remain oncogenes of choice for transformation of breast epithelial cells (12,13). Although initially considered not a relevant oncogene in breast cancer, recent studies have clearly shown the role of RAS oncogene in resistance to endocrine and CDK inhibitor therapies and metastasis of luminal breast cancer (14,15). Simian Virus 40-T/t (SV40-T/t) antigens overexpression results in inactivation of tumor suppressor genes retinoblastoma and p53 by the large-T antigen and Protein Phosphatase 2A (PP2A) by the small-t- antigen (13,16). All three of these signaling pathways are frequently aberrant in breast cancers, as evident from high frequency p53 mutations and PP2A inactivation in integrative cluster 9 subtype of breast cancer (2,10,17), thus justifying the use of the SV40-T/t antigens in mechanistic studies relevant to breast cancer.
We took advantage of our recently developed model system of immortalized breast epithelial cell lines derived from healthy breast tissue of women to address interplay between cell-context and oncogenic aberrations on individual steps of breast cancer progression. Model and results presented in this study differ significantly from previous studies related to cell-of-origin of breast cancer (9,12), as cells were derived from multiple healthy donors of different genetic ancestry. At the time of transformation, these cell lines were diploid in nature (18). Although these cell lines contained heterogenous population cells, RNA-seq followed by PAM50 classification categorized the cell lines into “normal” counterparts of intrinsic subtypes. This diversity in cell characteristics allowed us to discern a strategy to systematically determine how cell-of-origin impacts phenotype of cancer cells with similar genomic aberrations. We demonstrate that interplay between cell-of-origin and genomic aberrations determine stem/progenitor/mature cell hierarchy, self-renewal/differentiation, and metastasis patterns of resulting tumors. Surprisingly, oncogenic aberrations, irrespective of the cell-of-origin of transformed cells, have a direct influence on response to chemotherapeutic drugs. Overall, these findings advance our understanding of interplay between susceptible epithelial cell population and genomic aberrations.
Materials and Methods
Cell culture, Cell line generation and Lentiviral transduction
Immortalized breast epithelial cell lines were cultured as described previously (18). Cells were transformed with oncogenes H-RasG12V, PIK3CAH1047R, and SV40-T/t antigens expressing lentiviruses using vectors pLenti CMV-RasV12-Neo (w108-1) (HRAS G12V, #22259, Addgene), pLenti MNDU3-PGK–PIK3CAH1047R-YFP (10), and pLenti-CMV/TO-SV40 small + Large T (w612-1) (#22298, Addgene), respectively. Cell lines in the laboratory are usually tested for Mycoplasma once in 6 months (Lonza mycoplasma testing kit, last testing was done on January 20, 2021) and cell line authentication/cross contamination yearly using marker short tandem repeat DNA sequencing method (IDEXX BioAnalytics, last testing was done on July 30, 2020). Additional details of cell culture are provided in supplementary materials and methods.
Antibodies and Western blotting
Cell lysates were prepared in radioimmunoassay buffer and analyzed by western blotting as described previously (19). Additional details are provided in Supplementary materials and methods.
Flow cytometry analysis and cell sorting
Flow cytometry was performed as described previously (18). Samples were analyzed and sorted using BD FACSAria and SORPAria. Additional details are provided in Supplementary materials and methods.
Cell proliferation assay and Mammosphere formation assay
For cell proliferation assay, cells were seeded in 96-well plates and grown for 3 days. The mammosphere formation assay was carried out to evaluate the stemness/self-renewal/differentiation properties of cells as described previously (18). Phase contrast images were captured, counted and processed for staining at day 5. Additional details are provided in Supplementary materials and methods
Xenograft study
The Indiana University Animal Care and Use Committee approved the use of animals in this study and all procedures were performed as per NIH guidelines. Transformed cells with 50% basement membrane matrix (BME) type 3 (3632-005-02, Trevigen) were implanted into the mammary fat pad of 5 to 6 week old female NSG (NOD/SCID/IL2Rgnull) mice. Tumor growth was measured weekly and tumor volume was calculated as described previously (20). Additional details are provided in Supplementary materials and methods.
Immunohistochemistry (IHC)
H&E, ERα, GATA3, FOXA1, CK5/6, CK8, CK14, CK19, and EGFR immunostaining was performed at the CLIA-certified Indiana University Health Pathology Laboratory and the whole-slide digital imaging system of Aperio (ScanScope CS) was used for imaging according to protocol described previously (21). Additional details are provided in Supplementary materials and methods
Soft agar colony formation assay and Annexin V/Dead cell apoptosis assay
20,000 cells were resuspended in 0.3% low-melting point agarose (214220, BD Biosciences) containing DMEM/F-12 and 10% FBS and plated on top of 0.6% agarose bottom layer containing DMEM/F-12 and 10% FBS in six-well plates. For annexin V/Dead cell apoptosis assay, after the indicated drug treatment, cells were collected by trypsinization and stained with annexin V-FITC and propidium iodide (PI) using FITC annexin V/dead cell apoptosis kit (V13242; Invitrogen molecular probes) according to the manufacturer’s instructions. Additional details are provided in Supplementary materials and methods.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) with whole cell extracts from immortalized and transformed cell lines was performed as described previously (19). Additional details are provided in Supplementary materials and methods.
Immunofluorescence
Immunofluorescence was carried out as described previously (18). Images were acquired using an Olympus FV10000 MPE inverted confocal microscope and analyzed using Olympus software. Additional details are provided in Supplementary materials and methods
Drug sensitivity colony assay
1000 cells were seeded in 6-well plate, treated with indicated drug for 48 hours, replaced with regular media and allowed to grow for 7 days. Colonies were stained with Coomassie brilliant blue, imaged under microscope and counted by ImageJ software.
Statistical analysis
Statistical analyses were conducted using Prism software program (version 6.0). Data were analyzed using one-way ANOVA. P values below 0.05 were considered statistically significant.
Results
Transformation of immortalized normal breast epithelial cell lines corresponding to intrinsic subtypes
Luminal A (KTB34), luminal-like (KTB6), normal-like (KTB39), and basal-like (KTB22) intrinsic subtypes of immortalized normal breast epithelial cell lines derived from breast biopsies of healthy women described in our previous study (18) were transformed with H-RasG12V, SV40-T/t antigens or combination of both oncogenes using lentivirus-mediated gene transfer. Fig. 1A provides schematic view of the experimental design. H-RasG12V overexpression initiated senescence program at first in all cell lines but eventually transformed clones emerged. Expression levels of oncogenes were similar across cell lines (Fig. 1B–1D) and phase contrast images of the transformed cell lines did not reveal cell line-specific variations in phenotype as all cell lines maintained epithelial morphology (Supplementary Fig. S1A). To determine the effect of H-RasG12V, SV40-T/t antigens and combinations of both on cell proliferation, BrDU-incorporation-ELISA was performed. SV40-T/t antigens promoted cell proliferation at variable levels in all cell subtypes, while H-RasG12V had modest effect on proliferation of luminal A and luminal-like cell lines (Fig. 1E).
Figure 1: Experimental design and phenotypic characterization of transformed cells.

(A) Schematic view of the experimental design. The design involved transformation of immortalized normal breast epithelial cell lines with gene expression patterns overlapping luminal A (KTB34), luminal-like (KTB6), normal-like (KTB39), and basal-like (KTB22) intrinsic subtypes with breast cancer relevant genomic aberrations. Transformed cells were examined for phenotypic plasticity, cancer stem cell (CSC) phenotype, differentiation, signaling pathways alterations, tumor heterogeneity, metastasis and response to chemotherapy.
(B) Levels of mutant H-RasG12V in transduced cell lines.
(C) Levels of SV40-T antigen in the above cell lines.
(D) Levels of both H-RasG12V and SV40-T antigen in double transduced cells. We first generated H-RasG12V cell lines and then introduced SV40-T antigen in most cases.
(E) SV40-T/t antigens but not H-RasG12V increase cell proliferation rates in all cell lines. Cell proliferation rates were determined using BrDU-incorporation-ELISA cell proliferation assay.
(F) CD49f and EpCAM staining patterns of immortalized and transformed cell lines. CD49f+/EpCAM−, CD49f+/EpCAM+ and CD49f-/EpCAM+ cells correspond to stem/basal, progenitor and differentiated cells, respectively. Red arrows indicate transformation-induced changes in phenotype of cells.
(G) CD271 and EpCAM staining patterns of immortalized and transformed cell lines.
Cell type-specific effects of signaling pathways downstream of SV40-T/t antigens on stem/progenitor/mature luminal cell hierarchy
CD49f+/EpCAM−, CD49f+/EpCAM+ and CD49f−/EpCAM+ cells are considered as basal/stem, luminal progenitor and mature luminal cells of the breast (22). PROCR (CD201)+ cells have been described as multipotent stem cells of the mouse mammary gland, whereas CD271+ and CD44+/CD24− cells have been described as minor population of highly invasive cells in luminal cancers and breast cancer stem cells, respectively (23–25). To determine whether H-RasG12V or SV40-T/t antigens alter the phenotype of transformed cells, oncogene overexpressing cells were analyzed by flow cytometry using the above described markers. SV40-T/t antigens overexpression had cell line-specific effects on subpopulation of cells. For example, SV40-T/t antigens altered CD49f/EpCAM staining pattern by increasing intensity of EpCAM staining, which created a subpopulation of cells that are CD49f+/moderate/EpCAMhigh and CD49f+/EpCAMlow (Fig. 1F). These changes were clearly evident in luminal A and basal-like cell lines. Similar cell line-specific changes in stem/progenitor/mature cell hierarchy upon SV40-T/t antigens overexpression were observed when cells were analyzed for CD271/EpCAM, CD201/EpCAM and CD44/CD24 expression patterns (Fig. 1G; Supplementary Fig. S1B–S1D). SV40-T/t antigens created a distinct CD201+/EpCAMlow subpopulation of transformed luminal A and basal-like cell lines. Thus, SV40-T/t antigens influence stem/progenitor/differentiation hierarchy of transformed cells in a cell-context dependent manner. Unlike SV40-T/t antigens, H-RasG12V overexpression had modest effects on phenotype of cell lines (Fig. 1F and 1G; Supplementary Fig. S1C and S1D). Notably, basal-like subtype showed a modest increase in levels of CD49f+/EpCAM−, CD44+/CD24−, CD201+/EpCAM− and CD271+/EpCAM− subpopulations (Fig. 1F and 1G; Supplementary Fig. S1C and S1D). The phenotype of double transformed cells closely resembled that of cells transformed by SV40-T/t antigens with an increase in EpCAM staining intensity and cell type-specific effects on CD271+ subpopulation of cells (Fig. 1F and 1G; Supplementary Fig. S1C and S1D). Taken together, these results indicate that both cell-of-origin and oncogenic mutations determine the stem/progenitor/mature luminal cell hierarchy of transformed cells.
Interplay between cell-of-origin and oncogenes influences self-renewal/differentiation of transformed cells
Mammosphere assay is routinely used as a surrogate assay to measure self-renewal and differentiation capacity of normal and transformed cells. To determine whether transformed luminal A, luminal-like, normal-like or basal-like subtypes show variability in stemness and differentiation properties, we performed serial dilution mammosphere assay. All transformed cells formed variable size mammospheres. SV40-T/t antigens transformed cells formed larger spheres compared to H-RasG12V and immortalized pLKO control cells, which is consistent with the effects of SV40-T/t on cell proliferation (Fig. 2A and 2B). SV40-T/t antigens transformed basal-like subtype displayed higher mammosphere-forming ability than other cell types (Fig. 2A and 2B). In serial dilution mammosphere assay, only SV40-T/t antigens± H-RasG12V transformed cells generated tertiary mammospheres. These results suggest that SV40-T/t antigens confer enhanced self-renewal capacity to transformed cells.
Figure 2: Interplay between cell-of-origin and oncogenes influences self-renewal/differentiation phenotype and signaling pathway activation of transformed cells.

(A) Self-renewal capacity of luminal A, luminal-like, normal-like or basal-like transformed cell lines were measured by mammosphere assay. Primary mammospheres of SV40-T/t antigens ± H-RasG12V transformed basal-like cell line were more efficient than other cells in generating tertiary mammospheres.
(B) Mammosphere forming efficiency of luminal A, luminal-like, normal-like or basal-like transformed cell lines. SV40-T/t antigens but not H-RasG12V increased the number of mammospheres. Asterisks denote significant differences compared to immortalized cell line.
(C) Cell-of-origin as well as oncogenes have an influence on the levels of stem/basal, progenitor and differentiated cells in the mammospheres based on CD49f/EpCAM staining pattern. Red arrows indicate transformation-induced changes in phenotype of cells in mammospheres.
(D) Cell-of-origin as well as oncogenes have an influence on the levels of cancer stem-like cells (CD44+/CD24−) in mammospheres based on CD44/CD24 staining pattern. Red arrows indicate transformation-induced changes in phenotype of cells in mammospheres.
(E) pAKT (Ser473) and pSTAT3 (Tyr705) but not pERK (Thr202/Tyr204) and PTEN levels showed cell type and/or oncogene-dependent variability. SV40-T/t antigens reduced pAKT in luminal A and luminal-like but not in normal-like or basal cell line. Also note differences in basal pSTAT3 levels in control pLKO cells.
(F) BRD4, involved in epigenetic gene regulation and a drug target, shows cell type as well as oncogene-dependent changes in expression. Arrows show long and short isoforms.
Characterization of cells in primary mammospheres by flow cytometry revealed that oncogenes had cell-of-origin specific effects on subpopulation of differentiated (CD49f−/EpCAM+ and CD44−/CD24+) and stem/basal (CD49f+/EpCAM− and CD44+/CD24−) cells. While immortalized and transformed counterparts of luminal A cell line displayed predominantly luminal progenitor phenotype under mammosphere growth condition, SV40-T/t antigens reduced the number of differentiated cells with the other luminal cell line as well as normal-like cell line (Fig. 2C and 2D). The basal-like cell line underwent dramatic changes in phenotype upon transformation with both oncogenes increasing the proportion of cells with cancer stem cell phenotype (Fig. 2C and 2D). Thus, cell-of-origin has a major influence on whether transformed cells maintain or acquire cancer stem cell phenotype upon transformation. Note that staining patterns of isotype controls for flow cytometry of mammosphere-derived cells are shown in Supplementary Fig. S1E, which were used for gating and to determine quadrants.
Cell-context influences oncogene-induced signaling pathway activation
To extend our observations on cell-of-origin dependent variability in basal and transformation-mediated signaling changes, we measured phospho-AKT (Ser473), phospho-STAT3 (Tyr705), phospho-ERK (Thr202/Tyr204), PTEN, and BRD4 protein levels and DNA binding activity of transcription factors NF-κB, OCT-1, and AP-1 in immortalized and transformed cell lines. SV40-T/t antigen reduced phospho-AKT levels in luminal A and luminal-like cell lines but not in normal-like and basal-like cell lines (Fig. 2E; Supplementary Fig. S2A). SV40-T/t antigens increased phospho-STAT3 in all cell lines, although there was immortalized cell line-specific variability in basal phospho-STAT3 levels (Fig. 2E; Supplementary Fig. S2A). Despite previous studies demonstrating inactivation or loss of PTEN in breast cancers (26), in our model system, transformation with either H-RasG12V or SV40-T/t antigens did not alter PTEN levels (Fig. 2E; Supplementary Fig. S2A). Thus, transformation in the model system used in this study is less likely reliant on PI3K-PTEN-AKT signaling axis. However, we cannot rule out possible differences in post-translational modification of PTEN between immortalized and transformed cells.
Overexpression of H-RasG12V had minimum effect in NF-κB DNA binding activity. However, SV40-T/t antigens increased NF-κB with cell type specific variability in the level of induction (Supplementary Fig. S2B). We did not observe an effect of oncogenes on OCT-1 and AP-1 binding activity (Supplementary Fig. S2C and S2D), which served as controls. These results suggest that while oncogenes primarily determine signaling pathway activation in transformed cells, cell-of-origin has an impact on degree of signaling pathway activation.
BET bromodomain (BRD) proteins have recently been identified as major regulators of oncogenic transcription factors and BRD4 among them has been targeted therapeutically (27). Two isoforms of BRD4 with opposing functions in cancer progression have been described: a long isoform with tumor suppressor activity and a short isoform with pro-metastatic functions (28). While SV40-T/t antigens transformed cells showed increased levels of both long and short forms of BRD4 at variable levels compared to their immortalized counterparts, H-RasG12V increased BRD4 only in basal-like cell line (Fig. 2F; Supplementary Fig. S2E). Since transformed cells expressed higher levels of BRD4 compared to immortalized cells, we examined whether immortalized and H-RasG12V + SV40-T/t antigens transformed luminal A and basal-like cell lines show differences in sensitivity to BET bromodomain inhibitor JQ1 (29). Transformed cells showed lower sensitivity to JQ1 compared to their immortalized cell counterparts, suggesting that BRD4 levels determine sensitivity to JQ1 (Supplementary Fig. S2F).
Tumors originating from luminal A and luminal-like but not normal-like or basal-like cell lines express luminal markers GATA3 and FOXA1.
To determine whether luminal A, luminal-like, normal-like and basal-like subtypes expressing H-RasG12V and SV40-T/t antigens form tumors, we injected 5 × 106 transformed cells with 50% matrigel in 100 microliter HBSS into the mammary fat pad of 6–7 week old female NSG (NOD/SCID/IL2Rgnull) mice. Tumors were analyzed by H&E staining and expression of estrogen receptor alpha (ERα), GATA3, FOXA1, CK5/6, CK8, CK14, CK19, and EGFR using immunohistochemistry. We also created cell lines from half of tumors to characterize the phenotypic cellular heterogeneity using various cell surface markers. Human specific antibody against Na+/K+ ATPase CD298 (ATP1B3) cell surface marker was used to sort the CD298-enriched human tumor cell populations from mouse stromal cells (30).
Double transformed luminal A (13/13), luminal-like (9/13), normal-like (10/18) and basal-like (13/14) cell lines developed tumors in mice at variable frequency and growth rates (Fig. 3A). Tumor incidence rate was statistically significantly higher with transformed luminal A (p=0.009) and basal-like cell line (p=0.04) compared to transformed normal-like cell line. Tumors displayed variable growth rates as well with transformed luminal A cell line-derived tumors displaying highest growth rate (Fig. 3B), demonstrating the influence of cell-of-origin on tumor progression rate. Among H-RasG12V alone transformed cell lines, only basal-like subtype developed tumors, that too at very low frequency, suggesting the influence of cell type on H-RasG12V induced transformation (Supplementary Fig. S3A). Interestingly, despite demonstrating maximum effects on stem/luminal progenitor/mature luminal cell hierarchy, proliferation rate, self-renewal capacity in mammosphere assay, and signaling pathways in vitro, cells overexpressing SV40-T/t antigens failed to generate tumors (Supplementary Fig. S3B). These results highlight how aggressive characteristics displayed by transformed cells in vitro do not translate into similar phenotypes in vivo.
Figure 3: Frequency and characteristics of tumors developed from H-RasG12V+ SV40-T/t transformed luminal A, luminal-like, normal-like and basal-like cell lines.

(A) Tumor incidence by individual cell type transformed with H-RasG12V+ SV40-T/t. N corresponds to number of animals injected.
(B) Growth rate of tumors.
(C) Expression levels of luminal markers ERα, GATA3 and FOXA1 in tumors. H&E staining classified tumors as adenocarcinomas (Ad), squamous carcinoma (Sq), undifferentiated carcinomas (Uc), Adeno-squamous carcinoma (Ad-sq).
(D) Expression levels of cytokeratin CK5/6, CK14, CK19 and EGFR in tumors. Tumors derived from normal-like and basal-like expressed CK5/6.
(E) Cell lines derived from tumors of luminal A, luminal-like, normal-like and basal-like double transformed cells were characterized by flow cytometry using CD49f/EpCAM and CD44/CD24 markers to determine stem/luminal progenitor/mature luminal cell hierarchy (CD49f/EpCAM) and cancer stem cell (CD44+/CD24−) properties.
Representative H&E staining patterns of tumors are shown in Fig. 3C. Consistent with phenotypic heterogeneity within cell lines observed in vitro, transformed luminal A cell line generated poorly differentiated carcinoma, moderately differentiated squamous cell carcinoma, well-differentiated squamous cell carcinoma and adenocarcinoma. Luminal-like-double transformed cells derived tumors showed undifferentiated carcinoma, poorly differentiated squamous carcinoma, moderately differentiated squamous cell carcinoma, pleomorphic carcinoma with areas of multi-nucleated cells and adenocarcinoma. Normal-like -double transformed cells derived tumors were anaplastic carcinoma with squamous cell features, undifferentiated carcinoma, poorly differentiated squamous cell carcinoma, and anaplastic squamous sarcoma. Basal-like -double transformed cells derived tumors showed undifferentiated squamous cell carcinoma, squamous cell carcinoma, anaplastic, adenosquamous and adenoid cystic carcinomas. Tumor derived from H-RasG12V transformed basal-like cells showed undifferentiated carcinoma (Supplementary Fig. S3C).
Transformed luminal A cells-derived tumors showed ERα-/GATA3-/FOXA1+, ERα-/GATA3+/FOXA1- and ERα-/GATA3+/FOXA1+ patterns (Fig. 3C). Luminal-like cells-derived tumors were ERα-/GATA3+/FOXA1- and ERα-/GATA3-/FOXA1- (Fig. 3C). In general, adenocarcinomas were GATA3+. None of the normal-like cells-derived tumors expressed luminal markers, whereas tumors derived from basal-like cell line expressed very low levels of GATA3 and FOXA1 (Fig. 3C). Tumor derived from H-RasG12V transformed basal-like cells did not express any luminal markers (Supplementary Fig. S3C). Thus, cell-of-origin rather than oncogenic aberrations determine the expression patterns of luminal cell markers in tumors.
CK5/6 but not CK14 expression patterns enabled us to distinguish luminal-like cells derived tumors from normal-like and basal-like cell derived tumors. The number of CK5/6+ tumor cells was much higher with normal-like and basal-like cell derived tumors compared to luminal A and luminal-like cell derived tumors (Fig. 3D). Since few of the luminal A and luminal-like tumors were squamous histotypes, CK14 expression was much more common, although intensity was stronger with basal like cells-derived tumors (Fig. 3D). CK19 expression was uniformly low across tumors and EGFR expression was variable and did not show any recognizable patterns. However, EGFR-positivity of many of these tumors, despite expressing luminal markers such as GATA3 and FOXA1, suggests that tumors represent recently modified intrinsic subtypes of breast cancers- myoluminal and myobasal subtypes (31,32). Tumor derived from H-RasG12V transformed basal-like cells was CK5/6-/CK14+/CK19- (Supplementary Fig. S3D). Tumors derived from each cell subtype were negative for CK8 expression (Supplementary Fig. S3D and S3E). Taken together, immunohistochemical analysis of ERα, GATA3, FOXA1, CK5/6, CK14 and CK19 again confirmed the inter-tumor heterogeneity, which could be due to differences in cell-of-origin of tumors.
Tumors derived from Luminal A and luminal-like transformed cells display phenotypic heterogeneity
Since each cell lines generated distinct subtypes of tumors, we further characterized cell lines established from tumors for phenotypic heterogeneity. Cell lines were established after sorting human cells from mouse xenografts by flow cytometry using CD298 marker (Supplementary Fig. S3F). Cell lines established from tumors of luminal A-H-RasG12V+SV40-T/t transformed cells or luminal-like–H-RasG12V+SV40-T/t transformed cells contained morphologically distinct subpopulation of cells, whereas those derived from normal-like and basal-like cell lines were similar to parental cell lines (Supplementary Fig. S4A). Tumor-derived cell lines retained the expression of both mutant Ras and SV40-T/t antigens (Supplementary Fig. S4B). Tumor-derived cell lines of luminal A-H-RasG12V+SV40-T/t transformed cells showed two distinct subpopulations: CD49f+/EpCAM− and CD49f+/EpCAM+; CD44+/CD24− and CD44+/CD24+; CD201+/EpCAM− and CD201+/EpCAM+; and CD271+/low/EpCAM− and CD271+/low/EpCAM+ cells (Fig. 3E; Supplementary Fig. S4C). These results indicate that there is acquired plasticity or clonal selection of transformed cells in vivo as the majority of transformed cells gained CD201 expression or CD201+ transformed cells were selected in vivo. Note that transformed luminal A cell line in vitro contained very low number of CD201+/EpCAM− cells (Supplementary Fig. S1C). In contrast to luminal A and luminal-like tumor derived cell lines, normal-like and basal-like tumor-derived cells displayed one dominant population of cells, which were CD49f+/EpCAM+, CD44+/CD24+, CD201+/EpCAM+ or CD271low/EpCAM+ (Fig. 3E; Supplementary Fig. S4C). Thus, despite the same oncogenic mutations, cell-of-origin of transformed cells determine cancer stem cell and/or differentiation properties in vivo.
Since luminal A and luminal-like cell tumor-derived cell lines contained phenotypically distinct population of cells and to further rule out the possibility of any contaminating mouse cells, we used JAM-A and EpCAM to sort JAM-A+/EpCAM+ and JAM-A−/EpCAM− subpopulation of cells and propagated these cells for further characterization. JAM-A has previously been shown to be expressed in glioma stem but not in normal brain cells and we had demonstrated its expression in breast epithelial cells (33,34). JAM-A+/EpCAM+ cells displayed cuboidal morphology, whereas JAM-A−/EpCAM− cells displayed spindle morphology (Supplementary Fig. S4D). Both population of cells expressed SV40-T antigen and H-RasG12V confirming that these cells are derived from original transplanted breast epithelial cells (Supplementary Fig. S4E). JAM-A+/EpCAM+ cells were CD49f+/EpCAM+, CD44+/CD24+, CD201-/EpCAM+, CD201+/EpCAM+ and CD271low/EpCAM+ (Supplementary Fig. S4F). By contrast, JAM-A−/EpCAM− cells were CD49f+/EpCAM−, CD44+/CD24−, CD201+/EpCAM− and CD271low/EpCAM−. While the phenotype of JAM-A+/EpCAM+ cells were similar to that of in vitro transformed cells, JAM-A−/EpCAM− phenotype was acquired by transformed cells in vivo. Note that oncogene-activated signaling pathways in tumor-derived cells and their in vitro counterparts were similar from all four cell types (Supplementary Fig. S4G – S4J).
Single cell-derived luminal A and luminal-like transformed cells display phenotypic heterogeneity.
Phenotypic heterogeneity noted above in tumor-derived cell lines could be due to heterogeneity in immortalized cell lines from which transformed cells originated or due to acquired plasticity of transformed cells in vivo. To distinguish between these possibilities, we used soft agar assay to isolate single cell-derived tumorigenic clones. Tumor-derived cells from luminal A–H-RasG12V+SV40T/t, luminal-like–H-RasG12V+SV40T/t, and basal-like–H-RasG12V+SV40T/t transformed cells generated soft agar clones (Fig. 4A). Cells from tumor of normal-like–H-RasG12V+SV40T/t transformed cells formed smaller colonies. We next established 2D cultures of these soft agar clones and confirmed SV40-T/t antigens and mutant H-RasG12V expression (Fig. 4B). Tumor cell lines derived from soft agar clones showed morphological changes with aggressive phenotype such as limited cell-cell adhesion and spindle shape compared to parental cells (Supplementary Fig. S5A). These results suggest that anchorage-independent growth enforced by soft agar assay selects for transformed cells that have limited cell-cell adhesion properties.
Figure 4: Single cell-derived tumor cells generate metastatic tumors.

(A) Isolation of individual tumor cell derived cell lines using soft agar assay. Soft agar colonies from various tumor-derived cell lines are shown.
(B) SV40-T antigen and H-RasG12V expression in soft agar colony-derived cell lines. β–actin was used as an internal control.
(C) The majority of cells in soft agar clones-derived cell lines are enriched for CD49f+/EpCAM− stem cell markers.
(D) Tumor developed from the soft agar clones of luminal A tumor cells (cell lines with prominent CD49f+/EpCAM− cells, left, and CD49f+/EpCAM+ cells, right) metastasized to lungs. The expressions of GATA3 and FOXA1 were analyzed in tumors and lungs metastasis by immunohistochemistry. Representative immunohistochemistry data are shown.
To further characterize these clones for stem/luminal progenitor/mature luminal cell and cancer stem cell phenotypes, we performed flow cytometry with CD49f, EpCAM, CD44, and CD24 surface markers. The majority of clones contained cells that were CD49f+/EpCAM− and CD44+/CD24−, suggesting preferential growth of tumor cells with cancer stem cell properties in soft agar assay (Fig. 4C; Supplementary Fig. S5B). However, clone 6 of tumor 1, clones 3 and 6 of tumor 2 and clones 2, 4, and 5 of tumor 3 (luminal A-derived) contained a subpopulation of cells that were CD49f+/EpCAM+ and CD44+/CD24+ suggesting clonal differences in plasticity (Fig. 4C; Supplementary Fig. S5B). Cell surface staining patterns with isotype control antibodies are shown in Supplementary Fig. S5C, which were used for gating and to determine quadrants.
Interestingly, those clones with two different population of cells were also morphologically heterogeneous in 2D culture with both spindle and cuboidal cells (Supplementary Fig. S5A). Thus, it is likely that cell-of-origin determines plasticity of transformed cells.
To determine whether soft agar clones derived from luminal A–H-RasG12V+SV40T/t tumor cells form tumors of specific histotype, we injected 5×106 cells from tumor 1 clone 6 (prominent CD49f+/EpCAM− subpopulations) and tumor 2 clone 6 (predominant CD49f+/EpCAM+ subpopulation). CD49f+/EpCAM− cell-derived tumors were undifferentiated carcinomas, whereas tumors from CD49f+/EpCAM+ cells were carcinomas (Fig. 4D). Tumors in both cases metastasized to lungs (Fig. 4D). Tumor developed from prominent CD49f+/EpCAM− subpopulation contained a small fraction of cells that were GATA3+, whereas tumor and metastasis developed from CD49f+/EpCAM+ cells showed strong GATA3-positivity (Fig. 4D).
Metastatic properties are governed by both cell-context and oncogenes
We next examined the relationship between cell context and oncogenes in metastatic progression. All luminal A–H-RasG12V+SV40-T/t, luminal-like–H-RasG12V+SV40-T/t and two out of three basal-like–H-RasG12V+SV40-T/t cells-derived tumors showed metastasis to lungs, whereas none of the normal-like–H-RasG12V+SV40-T/t cells-derived tumors showed lung metastasis (Fig. 5A and 5B). Lung metastasis expressed GATA3 but not FOXA1 in luminal A and luminal-like cells-derived tumors. Interestingly, lung metastasis of basal-like-derived tumors expressed luminal markers FOXA1 and contained few GATA3+ cells, suggesting that tumor and lung metastasis in basal-like cell lines originated from a small fraction of luminal-like cells within the basal-like cell line (Fig. 5C).
Figure 5: Bidirectional crosstalk between cell-context and oncogenes determine metastatic properties.

(A) H&E staining shows tumors developed from the luminal A, luminal-like and basal-like but not normal-like double transformed cell lines metastasize to lungs.
(B) Frequency of metastasis of luminal A, luminal-like, normal-like and basal-like cell derived tumors.
(C) Expression levels of GATA3 and FOXA1 in tumors and lung metastasis.
(D) Cells derived from tumors and lung metastasis of luminal A and luminal-like double transformed cells were characterized by flow cytometry using CD49f/EpCAM and CD44/CD24 markers to determine stem/luminal progenitor/mature luminal properties and cancer stem cell phenotype.
(E) Histotype and expression levels of luminal markers ERα, GATA3 and FOXA1 in tumors developed from luminal A cells double transformed with SV40-T/t and PIK3CA (luminal A-SV40-T/t + PIK3CA cells)
(F) Expression levels of cytokeratin CK5/6, CK14, CK19 and EGFR in tumors developed from luminal A-SV40-T/t + PIK3CA cells.
(G) Unlike cells from SV40-T/t + H-RasG12V-derived tumors, cells derived from tumors of luminal A-SV40-T/t + PIK3CA were not enriched for CD49f+/EpCAM− stem cells or CD44+/CD24− cancer stem cells.
(H) Tumors derived from luminal A-SV40-T/t + PIK3CA transformed cells fail to metastasize to lungs.
Cell lines were created from matched tumors and metastasis to determine whether metastatic cells acquire additional phenotype compared to primary tumor cells (Fig. 5D; Supplementary Fig. S6A and S6B). Luminal A–H-RasG12V+SV40T/t tumor and metastatic cells showed similar staining patterns i.e. CD49f+/low/EpCAM−, CD44+/CD24−, CD201+/EpCAM−, and CD271low/EpCAM− (Fig. 5D; Supplementary Fig. S6B). Similar results were obtained with luminal-like–H-RasG12V+SV40T/t tumor and metastatic cells i.e. CD49f+/EpCAM+, CD44+/CD24−, CD44+/CD24+, CD201+/EpCAM−, CD201+/EpCAM+, CD271−/EpCAM+ and CD271+/EpCAM+ (Fig. 5D; Supplementary Fig. S6B). Thus, at least phenotypically, primary and metastatic cells are similar.
To determine the role of oncogenes in metastatic phenotype, we created a new series of cell lines that overexpressed breast cancer relevant mutant PIK3CA and SV40-T/t antigens. Luminal A and normal-like cells were transformed with either mutant PIK3CA (PIK3CAH1047R) alone or a combination of SV40-T/t antigens and PIK3CAH1047R (Supplementary Fig. S6C). The normal-like cells transformed with PIK3CAH1047R alone or in combination with SV40-T/t antigens did not form tumor. Luminal A cells transformed with PIK3CAH1047R alone did not form tumor, but in combination with SV40-T/t antigens formed tumors. Luminal A-SV40T/t+PIK3CAH1047R transformed cells derived tumors were adenocarcinoma and poorly differentiated squamous cell carcinoma (Fig. 5E). Staining of tumors developed from luminal A-SV40T/t+PIK3CAH1047R transformed cells showed both ERα−/GATA3+/FOXA1+ and ERα−/GATA3−/FOXA1− subpopulations (Fig. 5E). Keratins expression profiles of tumors were CK5/6−/CK14+ with a small population of CK19+ cells and all the tumors were EGFR+ (Fig. 5F).
Cell lines were established after sorting human cells from mouse xenografts of luminal A-SV40T/t+PIK3CAH1047R transformed cells using CD298 marker (Supplementary Fig. S6D). Tumor-derived cell lines of luminal A-SV40T/t+PIK3CAH1047R transformed cells displayed CD49f+/EpCAM+ and CD49f−/EpCAM+; CD44+/CD24+; CD201+/EpCAM+ and CD201−/EpCAM+; and CD271−/EpCAM+ and CD271+/EpCAM+ phenotypes and minimally enriched for cells with cancer stem cell phenotype (Fig. 5G; supplementary Fig. S6E) compared to luminal A-H-RasG12V+SV40T/t transformed cells (See Fig. 3E; Supplementary Fig. S4C). None of the luminal A-SV40T/t+PIK3CAH1047R cells-derived tumors metastasized to lungs (Fig. 5H), whereas luminal A–H-RasG12V+SV40T/t cells-derived tumors showed metastasis to lung (see Fig. 5A and 5B). These results suggest that a bidirectional regulatory relationship between cell-context and oncogenes determine acquisition of cancer stem cell phenotype and metastasis properties in vivo.
Oncogenes, irrespective of cell context, determine susceptibility to DNA damaging agents
To understand whether bidirectional relationship between cell-context and oncogenes extend to response to chemotherapy, we determined double stranded DNA break (DSB) response of immortalized and transformed cells upon treatment with chemotherapeutic agents. Flow cytometry was used to identify induction of an established marker for unrepaired DSBs γ-H2AX in response to 48 hours of treatment with doxorubicin, paclitaxel and cisplatin (35). The type of oncogene instead of cell-context of transformed cells influenced DSB response, as doxorubicin increased γ-H2AX levels more efficiently in SV40-T/t antigens overexpressing cells compared to H-RasG12V overexpressing cells (Fig. 6A). Similar effects were observed with paclitaxel (Fig. 6B) and cisplatin treatment (Fig. 6C). However, H-RasG12V dominantly reduced DSB response because γ-H2AX levels were lower in double transformed cells compared to SV40-T/t transformed cells.
Figure 6: Oncogenes determine susceptibility to DNA damaging agents independent of cell-of-origin of transformation.

(A) Double stranded DNA break (DSB) response in luminal A, luminal-like, normal-like and basal-like immortalized, transformed and tumor cells was examined after treatment with doxorubicin (Dox, 250 nM for 48 hours) by flow cytometry using pγ-H2AX as a marker.
(B) DSB response in luminal A, luminal-like, normal-like and basal-like immortalized, transformed and tumor cells was examined after treatment with paclitaxel (Pac, 100 nM for 48 hours) by flow cytometry using pγ-H2AX.
(C) Same assay as in A and B except that cells were treated with 5 μM cisplatin for 48 hours.
(D) Immunofluorescence staining with pγ-H2AX and RAD51 antibodies was used to measure the response to doxorubicin. Recruitment of RAD51 to pγ-H2AX-positive foci indicating repair of damaged DNA is evident in pLKO and H-RasG12V transformed cells but not in SV40-T/t antigen expressing cells. H-RasG12V restores repair process as higher levels of pγ-H2AX and RAD51 co-localization was observed in double transformed cells compared to SV40-T/t transformed cells.
To further investigate oncogene-dependent changes in DSB response and potential resolution of DSB, we performed co-localization studies of γ-H2AX and RAD51. Co-localization of these two molecules indicates active repair of DSBs (36). Doxorubicin-treated pLKO and H-RasG12V transformed cells showed strong γ-H2AX and RAD51 co-localization and distinct foci, whereas signals in SV40-T/t antigens transformed cells were much more diffuse (Fig. 6D). Taken together, these results indicate that while DNA repair pathways are relatively unaffected in pLKO and H-RasG12V transformed cells, SV40-T/t antigens induced signals disrupt DNA repair.
Therapy-induced apoptosis is oncogene-dependent
To elucidate the role of oncogenes in chemotherapy-induced apoptosis, annexin V staining was performed to measure response to doxorubicin, paclitaxel, and cisplatin. SV40-T/t antigens transformed cells compared to immortalized pLKO or H-RasG12V transformed cells were more sensitive to doxorubicin, paclitaxel and cisplatin-induced apoptosis (Fig. 7A–7D). Colony formation assay was used to further confirm oncogene-dependent susceptibility of transformed cells to chemotherapy. As with other two assays, SV40-T/t antigens transformed cells were more sensitive to doxorubicin and showed reduced number of colonies compared to immortalized or H-RasG12V transformed cells (Supplementary Fig. S7A and S7B).
Figure 7: Therapy-induced apoptosis is oncogene-dependent.
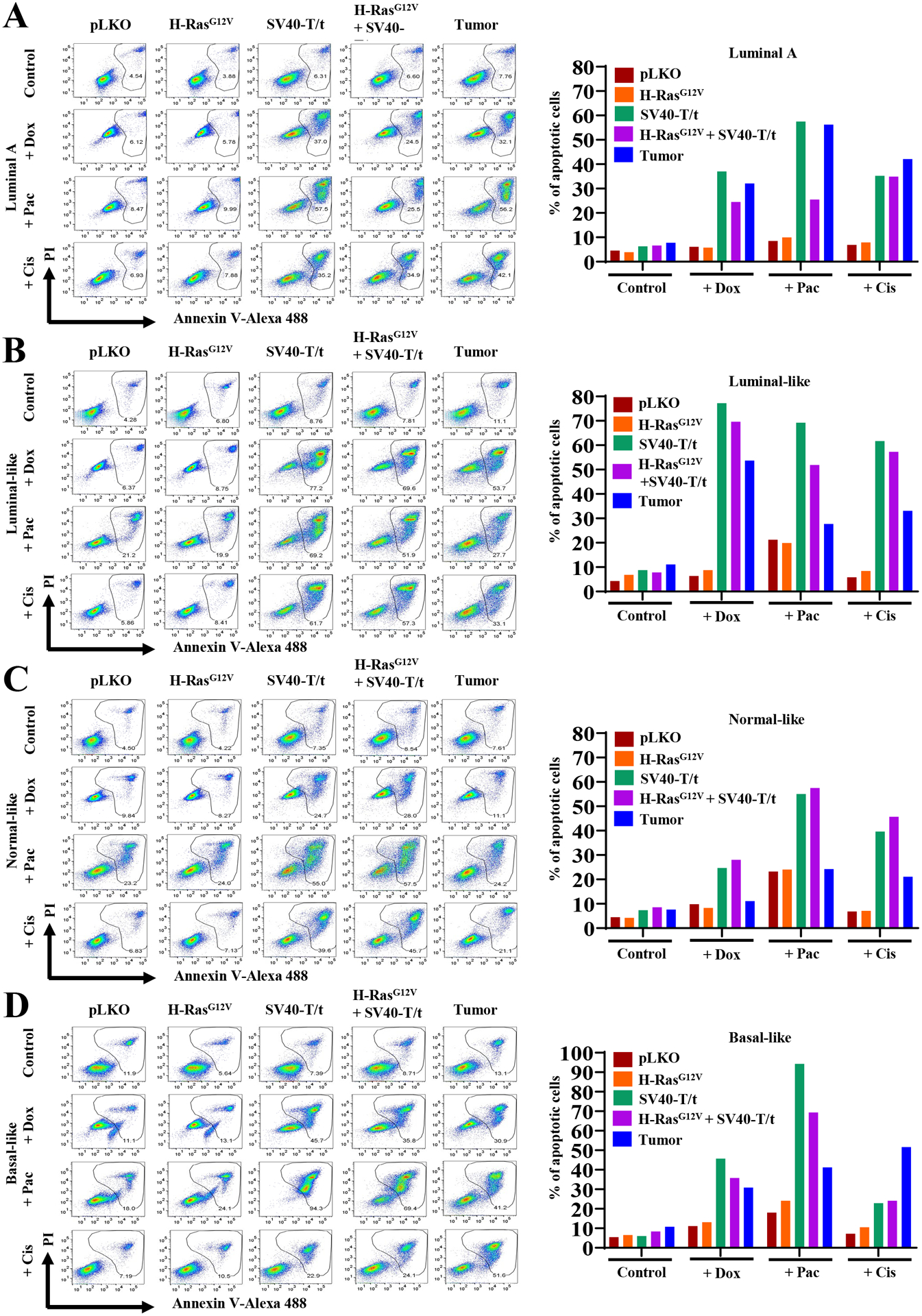
Annexin V staining was used to measure response to chemotherapeutic drugs doxorubicin, paclitaxel and cisplatin. Percentage of Annexin V ± PI positive cells under various conditions are shown on the right.
(A) Luminal A immortalized, transformed and primary tumor-derived cells.
(B) Luminal-like immortalized, transformed and primary tumor-derived cells.
(C) Normal-like immortalized, transformed and primary tumor-derived cells.
(D) Basal-like immortalized, transformed and primary tumor-derived cells.
Isogenic cell line resource to study the interplay between cell-of-origin and oncogenes on various facets of cancer progression
During the course of this investigation we have created an important resource for research community to further investigate interplay between cell-of-origin and defined oncogenes in cancer progression. These cell lines are suitable to study cell-context dependent oncogene-induced epigenomic changes under the same genetic background. Phenotype and genetic ancestry of these cell lines are described in Supplementary Table S1. Since transformed variants of several of these cell lines spontaneously metastasize to lungs, these cell lines are useful for screening of drugs that not only target primary tumors but also metastasis.
Discussion
Tumor heterogeneity has a serious clinical consequence as it contributes to drug resistance, metastatic spread, and even improper molecular classification of cancer that affect treatment decision making (37). It is believed that tumor heterogeneity has at least three origins; pliancy of initially transformed cell (cell-of-origin), adaptive plasticity of transformed cells, and genomic aberrations (37,38). Although multiple breast cancer subtypes including basal type are suggested to originate from luminal progenitor cells (5,6), the most susceptible population within the heterogenous population of luminal cells are yet to be identified and experimentally analyzed. The majority of transformation assays of breast epithelial cells gave rise to squamous carcinomas (39). The adenocarcinoma phenotype has been hard to recapitulate in in vivo tumor models (40,41), although this tumor histotype comprises the majority of tumors naturally occurring in breast and other cancers (42). Ince et al., have shown that the same set of oncogenes can generate metastatic adenocarcinomas or non-metastatic squamous carcinomas depending on growth media used for initial isolation/propagation of cells, which provided first indication to cell-of-origin determining histotype of tumors (12). Previous studies in this respect had a major limitation as breast epithelial cell lines used were derived from reduction mammoplasty samples or normal tissues adjacent to tumors with aberrant genomes, which we and others have shown them to be molecularly/histologically abnormal (43–45). Here, we used the cell lines derived from biopsies of healthy donors of different genetic ancestry to develop an assay system that closely recapitulates naturally occurring human breast cancer, including their metastatic behavior. As we reported previously, these cell lines remained diploid when we tested them at ~20 passage (18). The use of reduction mammaplasty samples instead of normal breast epithelial cells could be a reason for discrepancy between data presented here and by Nguyen et al., (10). Authors using purified luminal progenitors (CD49f+/EpCAM+) and basal cells (CD49f+/EpCAM−) and activated K-RasG12D oncogene suggested that potent oncogenic role of this oncogene rather than the epithelial cell type of the breast determines histopathology of resulting tumors. Our study, however, suggests the role of cell-of-origin in determining histopathology of tumors.
Cell line models used here allowed us systematic analyses that could address the following questions: 1) Can we achieve transformation of breast epithelial cells derived from healthy donors using a single oncogene or need more than one oncogene?; 2) Instead of one “normal” cell line typically used in the literature to understand the signaling axis downstream of oncogenic activation, do breast epithelial cell lines derived from multiple donors reveal similar downstream signaling by an oncogene?; 3) Will the use of multiple cell lines allow us to dissect the roles of cell-of-origin and oncogenic mutations on various steps of the oncogenic processes including acquiring cancer stem cell phenotype, metastasis patterns, and response to therapy?; and 4) do cells enriched for luminal and basal cell gene expression patterns differ in their susceptibility to transformation? Our results suggest that cell-of-origin determines histology of tumors as only cells enriched for luminal epithelial gene expression patterns gave rise to adenocarcinomas, whereas all tumors originating from cells with basal or normal-like intrinsic breast cancer subtype gene expression rarely generated adenocarcinoma. While H-RasG12V alone was able to transform basal-like cell line at a very low frequency, luminal-like cell lines required two oncogenes (H-RasG12V or PIK3CAH1047R and SV40-T/t). Thus, cell-of-origin also determines requirement of oncogenes for transformation. It is unclear at present which among known downstream targets of SV40-T/t antigens (RB, p53, PP2A or p16 inactivation (46)) is essential or sufficient along with H-RasG12V or PIK3CAH1047R for transformation. Further studies are required in this direction.
Signaling pathway activation in transformed cells is dependent on bidirectional interaction between cell-context and oncogenes. For example, SV40-T/t antigens reduced pAKT in luminal-like cell lines but not in basal or normal-like cell lines. SV40-T/t antigens induced NF-κB DNA binding more robustly in the normal-like cell line compared to luminal-like cell lines. H-RasG12V but not SV40-T/t antigens had cell type specific effects on BRD4 induction. These cell type-specific effects of oncogenes in inducing signaling pathways may be a reason for lack of uniform activity of drugs that target signaling pathways downstream of oncogenes. Also, oncogene-induced increase in BRD4 levels correlated with lower response to JQ1. Thus, an interplay between oncogenic aberrations and cell-of-origin of tumor may determine sensitivity to targeted therapies such as JQ1, which is often difficult to discern from genomic analyses of tumors.
We observed that bidirectional interaction between cell-context and oncogenic signals additionally determine clonal diversity, cancer stem cell phenotype, and metastasis patterns. For example, SV40-T/t antigens overexpression resulted in significant phenotypic diversity only in luminal A and basal-like cell lines but not in normal-like cell line while H-RasG12V did not cause phenotypic diversity in any cell lines. In vivo, luminal A and luminal-derived tumors but not tumors derived from normal or basal-like cell lines gained CD201+/EpCAM− phenotype. While both luminal and basal-like cell lines transformed with H-RasG12V plus SV40-T/t antigens developed metastatic tumors, luminal cell lines transformed with PIK3CAH1047R plus SV40-T/t antigens developed only non-metastatic tumors. Clearly, interactions between genomic aberrations and potentially the epigenome of the cell types are required for cancer cells to acquire metastatic properties. A systematic epigenome, transcriptome, and proteome analysis of isogenic cell line models listed in Table S1 may be essential to reveal complex interplay between cell-context and genomic aberrations.
Only property of tumor cells that is independent of cell-of-origin is the response to chemotherapeutic agents. SV40-T/t antigens overexpressing but not H-RasG12V overexpressing cells were sensitive to all three drugs that we have tested in multiple assays. Mechanisms behind their sensitivity are unknown but further exploration may yield important clues to mechanisms of chemotherapeutic resistance.
In conclusion, using unique set of immortalized luminal A, luminal-like, normal-like and basal-like cell lines generated using breast biopsies of healthy women, we have created a model system to study the effects of cellular pliancy and genomic aberrations on various steps of cancer progression. Visual overview provides a synapsis our findings. Our studies clearly indicate the need to use multiple “normal” cell line resources to understand interplay between cell type and genomic aberrations as well as for identifying universally activated signaling pathway downstream of a genomic aberration, which is critical for developing targeted therapies. These unique cell line models will be highly useful in understanding the mechanisms that contribute to tumor heterogeneity, developmental hierarchy for breast cancer cells, therapy resistance and may help to develop predictive markers of breast cancer metastasis in future.
Supplementary Material
Implications:
Our findings demonstrate that how an interplay between the normal cell type that encountered genomic aberrations and type of genomic aberration influences heterogeneity, self-renewal/differentiation and tumor properties including propensity for metastasis.
Acknowledgments
We thank Dr. Connie J. Eaves (University of British Columbia, Vancouver, Canada) for mutant PIK3CA vector, members of the IUSCC flow cytometry core, confocal microscopy core, animal facility, and tissue procurement cores at the IU Simon Cancer center and Susan G Komen Tissue Bank for various tissues and reagents. We also thank countless number of women for donating their breast tissue for research purpose as well as volunteers who facilitated tissue collection. We also thank Dr. Rakesh Kumar for his suggestions to improve the manuscript. This work is supported by DOD-W81XWH-15-1-0707, DOD-WH1XWH2010577, Susan G. Komen for the Cure (SAC110025) and 100 Voices of Hope to HN. Susan G. Komen for the Cure, Breast Cancer Research Foundation and Vera Bradley Foundation for Breast Cancer Research provide funding support to Komen Normal Tissue Bank.
Footnotes
Disclosure of potential conflicts of interest: Authors have no conflict of interest to declare.
References
- 1.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98(19):10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prat A, Perou CM. Mammary development meets cancer genomics. Nat Med 2009;15(8):842–4 doi nm0809–842 [pii] 10.1038/nm0809-842. [DOI] [PubMed] [Google Scholar]
- 4.Fu NY, Rios AC, Pal B, Law CW, Jamieson P, Liu R, et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat Cell Biol 2017;19(3):164–76 doi 10.1038/ncb3471. [DOI] [PubMed] [Google Scholar]
- 5.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011;8(2):149–63 doi 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009;15(8):907–13 doi nm.2000 [pii] 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 7.Bhat-Nakshatri P, Gao P, Sheng L, McGuire PC, Xuei X, Wan J, Liu Y, Althouse SK, Colter A, Sandusky G, Storniolo A, and Nakshatri H. A single cell atlas of the healthy breast tissues reveals clinically relevant clusters of breast epithelial cells. Cell Reports Medicine 2021;2:100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res 2010;12(5):R87 doi 10.1186/bcr2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci U S A 2012;109(8):2772–7 doi 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen LV, Pellacani D, Lefort S, Kannan N, Osako T, Makarem M, et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature 2015;528(7581):267–71 doi 10.1038/nature15742. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70 doi 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007;12(2):160–70. [DOI] [PubMed] [Google Scholar]
- 13.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001;15(1):50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright KL, Adams JR, Liu JC, Loch AJ, Wong RG, Jo CE, et al. Ras Signaling Is a Key Determinant for Metastatic Dissemination and Poor Survival of Luminal Breast Cancer Patients. Cancer Res 2015;75(22):4960–72 doi 10.1158/0008-5472.CAN-14-2992. [DOI] [PubMed] [Google Scholar]
- 15.Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer discovery 2020;10(8):1174–93 doi 10.1158/2159-8290.CD-19-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 2002;22(7):2111–23 doi 10.1128/mcb.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deeb KK, Michalowska AM, Yoon CY, Krummey SM, Hoenerhoff MJ, Kavanaugh C, et al. Identification of an integrated SV40 T/t-antigen cancer signature in aggressive human breast, prostate, and lung carcinomas with poor prognosis. Cancer Res 2007;67(17):8065–80 doi 10.1158/0008-5472.CAN-07-1515. [DOI] [PubMed] [Google Scholar]
- 18.Kumar B, Prasad MS, Bhat-Nakshatri P, Anjanappa M, Kalra M, Marino N, et al. Normal breast-derived epithelial cells with luminal and intrinsic subtype-enriched gene expression document inter-individual differences in their differentiation cascade. Cancer Res 2018;78(17):5107–23 doi 10.1158/0008-5472.CAN-18-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhat-Nakshatri P, Sweeney CJ, Nakshatri H. Identification of signal transduction pathways involved in constitutive NF-kappaB activation in breast cancer cells. Oncogene 2002;21(13):2066–78. [DOI] [PubMed] [Google Scholar]
- 20.Kumar S, Kishimoto H, Chua HL, Badve S, Miller KD, Bigsby RM, et al. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am J Pathol 2003;163(6):2531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad M, Kumar B, Bhat-Nakshatri P, Anjanappa M, Sandusky G, Miller KD, et al. Dual TGFbeta/BMP Pathway Inhibition Enables Expansion and Characterization of Multiple Epithelial Cell Types of the Normal and Cancerous Breast. Mol Cancer Res 2019;17(7):1556–70 doi 10.1158/1541-7786.MCR-19-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev 2014;28(11):1143–58 doi 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Cai C, Dong X, Yu QC, Zhang XO, Yang L, et al. Identification of multipotent mammary stem cells by protein C receptor expression. Nature 2014;517:81–4 doi 10.1038/nature13851. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Villadsen R, Sorlie T, Fogh L, Gronlund SZ, Fridriksdottir AJ, et al. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci U S A 2012;109(16):6124–9 doi 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100(7):3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbognin L, Miglietta F, Paris I, Dieci MV. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers (Basel) 2019;11(9) doi 10.3390/cancers11091401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shu S, Polyak K. BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb Symp Quant Biol 2016;81:123–9 doi 10.1101/sqb.2016.81.030908. [DOI] [PubMed] [Google Scholar]
- 28.Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res 2011;71(8):3121–31 doi 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahni JM, Keri RA. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol Res 2018;129:156–76 doi 10.1016/j.phrs.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526(7571):131–5 doi 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathews JC, Nadeem S, Levine AJ, Pouryahya M, Deasy JO, Tannenbaum A. Robust and interpretable PAM50 reclassification exhibits survival advantage for myoepithelial and immune phenotypes. NPJ Breast Cancer 2019;5:30 doi 10.1038/s41523-019-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roelands J, Mall R, Almeer H, Thomas R, Mohamed MG, Bedri S, et al. Ancestry-associated transcriptomic profiles of breast cancer in patients of African, Arab, and European ancestry. NPJ Breast Cancer 2021;7(1):10 doi 10.1038/s41523-021-00215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell reports 2014;6(1):117–29 doi 10.1016/j.celrep.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakshatri H, Anjanappa M, Bhat-Nakshatri P. Ethnicity-Dependent and -Independent Heterogeneity in Healthy Normal Breast Hierarchy Impacts Tumor Characterization. Scientific reports 2015;5:13526 doi 10.1038/srep13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010;24(4):679–86 doi 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 36.Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA damage foci: Meaning and significance. Environ Mol Mutagen 2015;56(6):491–504 doi 10.1002/em.21944. [DOI] [PubMed] [Google Scholar]
- 37.Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Science translational medicine 2012;4(127):127ps10 doi 10.1126/scitranslmed.3003854. [DOI] [PubMed] [Google Scholar]
- 38.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019;24(1):65–78 doi 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res 2005;7(4):171–9 doi 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene 2000;19(8):968–88. [DOI] [PubMed] [Google Scholar]
- 41.Liu BY, McDermott SP, Khwaja SS, Alexander CM. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc Natl Acad Sci U S A 2004;101(12):4158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirot F, Chaltiel D, Ben Lakhdar A, Mathieu MC, Rimareix F, Conversano A. Squamous cell carcinoma of the breast, are there two entities with distinct prognosis? A series of 39 patients. Breast Cancer Res Treat 2020;180(1):87–95 doi 10.1007/s10549-020-05525-5. [DOI] [PubMed] [Google Scholar]
- 43.Degnim AC, Visscher DW, Hoskin TL, Frost MH, Vierkant RA, Vachon CM, et al. Histologic findings in normal breast tissues: comparison to reduction mammaplasty and benign breast disease tissues. Breast Cancer Res Treat 2012;133(1):169–77 doi 10.1007/s10549-011-1746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teschendorff AE, Gao Y, Jones A, Ruebner M, Beckmann MW, Wachter DL, et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nature communications 2016;7:10478 doi 10.1038/ncomms10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakshatri H, Kumar B, Burney HN, Cox ML, Jacobsen M, Sandusky GE, et al. Genetic ancestry-dependent differences in breast cancer-induced field defects in the tumor-adjacent normal breast. Clin Cancer Res 2019;25(9):2848–59 doi 10.1158/1078-0432.CCR-18-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005;24(52):7729–45 doi 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.