

Abstract
Lymphangioleiomyomatosis (LAM) is a rare, low-grade metastasizing disease characterized by cystic lung destruction. LAM can exhibit extensive heterogeneity at the molecular, cellular, and tissue levels. However, the molecular similarities and differences among LAM cells and tissue, and their connection to cancer features are not fully understood. By integrating complementary gene and protein LAM signatures, and single-cell and bulk tissue transcriptome profiles, we show sources of disease heterogeneity, and how they correspond to cancer molecular portraits. Subsets of LAM diseased cells differ with respect to gene expression profiles related to hormones, metabolism, proliferation, and stemness. Phenotypic diseased cell differences are identified by evaluating LUM proteoglycan and YB1 transcription factor expression in LAM lung lesions. The RUNX1 and IRF1 transcription factors are predicted to regulate LAM cell signatures, and both regulators are expressed in LAM lung lesions, with differences between spindle-like and epithelioid LAM cells. The cancer single-cell transcriptome profiles most similar to those of LAM cells include a breast cancer mesenchymal cell model and lines derived from pleural mesotheliomas. Heterogeneity is also found in LAM lung tissue, where it is mainly determined by immune system factors. Variable expression of the multifunctional innate immunity protein LCN2 is linked to disease heterogeneity. This protein is found to be more abundant in blood plasma from LAM patients than from healthy women.
Introduction
Lymphangioleiomyomatosis (LAM), a rare multisystem disease that almost exclusively affects women, is characterized by cystic lung destruction, which can lead to respiratory failure in severe cases (1,2). Damage to the lung parenchyma is caused by proliferation of spindle-shaped and epithelioid LAM cells, which express alpha-smooth muscle actin (αSMA). Extrapulmonary LAM manifestations frequently include renal angiomyolipomas (AMLs) and lymphatic abnormalities (1,2). Empirically, LAM is defined as a low-grade metastasizing neoplasm whose cell (or cells) of origin is uncertain (3–5). Recent analyses of single-cell gene expression profiles indicate that LAM cells may originate in the uterus (6) and/or lung mesenchyme (7). However, the similarity with cancer molecular portraits and the extent of disease heterogeneity are not yet fully understood.
LAM can occur sporadically or in the presence of Tuberous Sclerosis Complex (TSC), an autosomal-dominant multisystem disorder caused by mutations in the tumor suppressor genes TSC1 and TSC2 (8). In sporadic LAM (S-LAM), somatic inactivation of TSC2 or much less commonly TSC1 in an unknown cell type(s) appears to be sufficient for disease development (9). Mutations of TSC1 or TSC2 cause abnormal activation of the mechanistic target of rapamycin (mTOR), which is the basis for the current, U.S. Food and Drug Administration-approved, standard of care use of an mTOR inhibitor for lung and kidney disease (10). Rapamycin has greatly improved outcomes for women with LAM, but some patients continue to lose lung function, albeit at a lower rate, whilst receiving treatment (11,12). Disease heterogeneity might contribute to the differences in clinical benefit of rapamycin (13).
mTOR kinase activity is abnormally enhanced in many cancer types, and generally linked to stem cell-like features, which are also present in LAM cells (14–17). Indeed, LAM presents several other fundamental hallmarks of cancer, including continued cell proliferation and resistance to cell death, expression of factors promoting tissue invasion and metastasis, and immune evasion (4,18). In addition, LAM presents features of steroid-sensitive cancers: LAM lung lesions commonly appear heterogeneous, but diseased cells are frequently positive for the expression of estrogen and progesterone receptors (ER and PR, respectively) (19,20); LAM disease is generally more progressive in premenopausal women and may be exacerbated by pregnancy or estrogen-based medications (1,2). Estrogens have been shown to boost the metastatic potential in a LAM cell model (21), and progesterone and estradiol synergistically interact in this process (22). The similarities between LAM and cancer might extend to shared disease susceptibility (23,24) and, overall, suggest that comparison with cancer molecular portraits can provide further insight into LAM biology. Using expression of gene and protein LAM signatures, and by analyzing LAM single-cell and tissue gene expression profiles, we investigated biological features of LAM and their similarities with cancer. This integrative study proposes causes and markers of LAM cell and tissue heterogeneity, reveals potential transcriptional regulators, and defines molecular and cellular relationships with cancer.
Materials and Methods
LAM signatures
Proteins expressed in LAM lung lesions were compiled from the literature using PubMed searches (up to July 2020) with the MeSH terms “lymphangioleiomyomatosis” and “lung”, “tissue”, “protein”, “expression” and/or “immunohistochemistry”, and through curation of the relevant publications. Table 1 lists the compiled proteins making up the “LAM protein” (LAMp) signature; the publication sources are detailed in Supplementary Table S1A. The study of LAMp was accompanied by parallel analyses of a LAMcore signature, which emerged from the analysis of LAM-diseased cells using single-cell RNA-seq (scRNA-seq) assays in four LAM lung tissue samples (6). The signature expression levels or scores were derived from the combined expression analysis of the corresponding gene constituents using the single-sample Gene Set Expression Analysis (ssGSEA) algorithm (25), calculated with the Gene Set Variation Analysis (GSVA) application (https://github.com/rcastelo/GSVA) (26). The same methodology was applied to other gene sets analyzed in this study. A negative signature score for a given cell or tissue means that the gene set has a lower level of expression than the same gene set with a positive value in a different sample, or a different gene set with a positive value (25). The threshold used to define high and low scores of LAMcore and LAMp in LAM cells was based on the corresponding average signature scores. The GSEA (27) tool was applied using standard parameters and curated C2 gene sets.
Table 1.
Compiled LAMp signature.
| Gene name | Gene title | Author(s), year (references listed in Supplementary Table S1A) | LAMcore |
|---|---|---|---|
| ACSS2 | Acyl-CoA synthetase short-chain family member 2 | Krencz et al., 2018 | |
| ACTA2 | Actin alpha 2, smooth muscle | Matthews et al., 1993 | Yes |
| AGTR1 | Angiotensin II receptor type 1 | Valencia et al., 2006 | |
| AGTR2 | Angiotensin II receptor type 2 | Valencia et al., 2006 | |
| ALDH1A1 | Aldehyde dehydrogenase 1 family member A1 | Ruiz de Garibay et al., 2015; Pacheco-Rodriguez et al., 2019 | |
| CD44 | CD44 antigen, homing cell adhesion molecule | Pacheco-Rodriguez et al., 2007 | |
| CD63 | CD63 antigen, granulophysin | Zhe and Schuger, 2004 | |
| CD117 | KIT proto-oncogene | Watz et al., 2007 | |
| CDH1 | E-cadherin | Grzegorek et al., 2015 | |
| CTNNB1 | β-catenin | Flavin et al., 2011 | |
| CTSK | Cathepsin K | Dongre et al., 2017 | Yes |
| CCR2 | C-C motif chemokine receptor 2 | Pacheco-Rodriguez et al., 2009 | |
| CCR10 | C-C motif chemokine receptor 10 | Pacheco-Rodriguez et al., 2009 | |
| CXCR1 | C-X-C motif chemokine receptor 1 | Pacheco-Rodriguez et al., 2009 | |
| CXCR2 | C-X-C motif chemokine receptor 2 | Pacheco-Rodriguez et al., 2009 | |
| CXCR4 | C-X-C motif chemokine receptor 4 | Pacheco-Rodriguez et al., 2009 | |
| CPT1A | Carnitine palmitoyltransferase 1A | Krencz et al., 2018 | |
| DCT | Tyrosinase-related protein 2 | Klarquist et al., 2009 | |
| EGFR | Epidermal growth factor receptor | Grzegorek et al., 2015 | |
| EPOR | Erythropoietin receptor | Ikeda et al., 2011 | |
| ERBB3 | Epidermal growth factor receptor 3 | Kobayashi et al., 2018 | |
| ERBB4 | Epidermal growth factor receptor 4 | Kobayashi et al., 2018 | |
| ESR1 | Estrogen receptor α | Berger et al., 1990; Colley et al., 1989; Kinoshita et al., 1995; Ohori et al., 1991 | Yes |
| FGFR2 | Fibroblast growth factor receptor 2 | Inoue et al., 2002 | |
| FLT4 | Vascular endothelial growth factor receptor 3 | Kumasaka et al., 2004 | |
| FSCN1 | Fascin 1 | Ruiz de Garibay et al., 2015 | |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | Krencz et al., 2018 | |
| GLS | Glutaminase | Krencz et al., 2018 | |
| HMGA2 | High-mobility group AT-hook 2 | D’Armiento et al., 2007 | |
| ID1 | Inhibitor of DNA binding 1 | Ruiz de Garibay et al., 2015 | |
| IGF2 | Insulin-like growth factor 2 | Valencia et al., 2001 | |
| IGFBP2 | Insulin-like growth factor-binding protein 2 | Valencia et al., 2001 | |
| IGFBP4 | Insulin-like growth factor-binding protein 4 | Valencia et al., 2001 | Yes |
| IGFBP5 | Insulin-like growth factor-binding protein 5 | Valencia et al., 2001 | |
| IGFBP6 | Insulin-like growth factor-binding protein 6 | Valencia et al., 2001 | Yes |
| ITGB3 | Integrin subunit β 3 | Ruiz de Garibay et al., 2015 | |
| LGALS3 | Galectin 3 | Klover et al., 2017 | |
| LYVE1 | Lymphatic vessel endothelial hyaluronan receptor 1 | Kumasaka et al., 2008; Davis et al., 2013 | |
| MLANA | Melan-A, melanoma antigen | Matsumoto et al., 1999 | Yes |
| MMP2 | Matrix metallopeptidase 2 | Matsui et al., 2000 | Yes |
| MMP9 | Matrix metallopeptidase 9 | Matsui et al., 2000 | |
| MMP14 | Membrane type 1 (MT1)-MMP | Matsui et al., 2000 | Yes |
| MUC16 | Mucin 16 | Glasgow et al., 2018 | |
| NR2F2 | Nuclear receptor subfamily 2 group F member 2 | Kim et al., 2019 | Yes |
| PDGFRA | Platelet-derived growth factor receptor A | Watz et al., 2007 | Yes |
| PDPN | Podoplanin | Davis et al., 2013 | Yes |
| PGR | Progesterone receptor | Berger et al., 1990; Colley et al., 1989; Kinoshita et al., 1995; Ohori et al., 1991 | Yes |
| PMEL | Premelanosome protein | Bonetti et al., 1993; Hoon et al., 1994 | Yes |
| PRLR | Prolactin receptor | Terasaki et al., 2010 | Yes |
| PROX1 | Prospero homeobox 1 | Davis et al., 2013 | |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | Li et al., 2014 | |
| REN | Renin | Valencia et al., 2006 | |
| RICTOR | RPTOR independent companion of mTOR complex 2 | Krencz et al., 2018 | |
| S100A4 | Fibroblast specific protein 1, metastasin | Clements et al., 2015 | |
| SDC1 | Syndecan 1 | Steagall et al., 2013 | |
| SDC2 | Syndecan 2 | Steagall et al., 2013 | |
| SLC16A1 | Monocarboxylic acid transporter 1 | Krencz et al., 2018 | |
| SOX9 | SRY-box transcription factor 9 | Ruiz de Garibay et al., 2015 | |
| SRC | V-Src avian sarcoma viral oncogene homolog | Tyryshkin et al., 2014 | |
| SYK | Spleen-associated tyrosine kinase | Cui et al., 2017 | |
| THY1 | CD90 antigen | Ando et al., 2016 | |
| TNFRSF11B | Osteoprotegerin | Steagall et al., 2013 | |
| TNFSF11 | Tumor necrosis factor ligand superfamily, member 11 | Steagall et al., 2013 | |
| TRAIL | Tumor necrosis factor ligand superfamily, member 10 | Steagall et al., 2013 | |
| TYRP1 | Tyrosinase related protein 1 | Klarquist et al., 2009 | |
| VEGFC | Vascular endothelial growth factor C | Kumasaka et al., 2004 | |
| VEGFD | Vascular endothelial growth factor D | Seyama et al., 2006 |
Bulk LAM lung and cancer data
Gene expression data from 14 LAM lung bulk tissue samples and control-defined cell lines were obtained from the Gene Expression Omnibus (GEO) reference GSE12027 (28). This report used two cell lines, melanoma (Malme-3M) and pulmonary-artery smooth muscle (PASM), as controls or references because LAM cells are phenotypically similar to smooth muscle cells, and contain the premelanosomal structures found in melanocytes and melanoma cells (29). The CIBERSORTx algorithm (30) was applied to bulk gene expression profiles to infer the proportions of immune cell subsets. The Cancer Genome Atlas (TCGA) normal and primary tumor bulk tissue RNA-seq data (RNA-seq v2 with expectation maximization quantification, RSEM) were obtained from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov). ‘Normal’ refers to histologically normal tissue and tissue adjacent to surgically removed tumors, as defined by TCGA. Metastases and recurrent tumors were excluded from the study. The cancer types are named in accordance with TCGA study abbreviations. For all TCGA studies, gene expression values were log-transformed, and genes with less than 75% representation in a sample were filtered out.
LAM scRNA-seq data
The scRNA-seq LAM lung data were analyzed by implementing the process described in the original study (6), including preprocessing of LAM1–4 samples, and using the Seurat v3 package (31). Data were downloaded from the GEO reference GSE135851. Briefly, only cells with ≥ 500 expressed (unique molecular identifier (UMI) > 0) genes and < 10% UMIs mapping to mitochondrial genes were considered, except for the analysis of tissue sample LAM4, which included cells with ≥ 300 expressed (UMI > 0) genes and < 10% UMIs mapping to mitochondrial genes. Expression of each gene in a given cell was determined by computing the natural logarithm of (10,000 UMIgene/UMIcell +1). Highly variable genes (n = 2,000) were used for dimension reduction, applying runPCA (PCA n = 200) and the Uniform Manifold Approximation and Projection (UMAP) method (32). The previously defined LAM cell cluster was identified based on the expression of specific genes (6). We identified LAM cells (n = 133) in this dataset by applying the same analytical procedure as originally reported, excluding lung mesenchymal cells, and by confirming the expression of defined LAM genes in a precise cell cluster (Supplementary Figure S1). This analytical process did not enable us to rule out the possible existence of additional diseased cells, but we limited our analyses to the defined cluster for consistency with the original analysis (6).
Integration of LAM and cancer scRNA-seq data
Cancer cell line data, from a recent study of cell heterogeneity (33), were downloaded from the Broad Institute’s single-cell portal (https://singlecell.broadinstitute.org/single_cell). The cancer cell lines of breast, head and neck, kidney, lung, ovarian, pancreatic, skin, stomach, and uterine tissue were selected (n = 23,546 cells; Supplementary Table S1B), and genes that did not pass the original quality control were excluded from subsequent analyses. For each cell line, a Seurat object was created and only genes detected in ≥ 2 cells were considered, with ≥ 500 UMIs and < 5% UMIs mapping to mitochondrial genes. After merging objects, gene expression was determined by computing the natural logarithm of (10,000 UMIgene/UMIcell +1). The LAM and cancer cell profiles were integrated using the FindIntegrationAnchors and IntegrateData functions. Highly variable genes (n = 2,000) were also used for dimensional reduction by applying runPCA and UMAP (PCA n = 200).
Functional annotations
Enrichment at transcription factor-binding sites was determined with oPOSSUM v3.0 (http://opossum.cisreg.ca), using default parameters and JASPAR core profiles, and promoter genomic sequences set to −2 kb from the transcription start site. GO and KEGG pathway enrichment analyses were performed in g:Profiler in R (https://biit.cs.ut.ee/gprofiler/page/r) using the default parameters for an unranked gene list.
LAM lung tissue
Formalin-fixed paraffin-embedded LAM lung tissue from seven patients was analyzed in this study by immunohistochemical assays. These samples corresponded to lung biopsies or organ transplants of individuals with a clinical LAM diagnosis based on outlined guidelines (1), and collected by Spanish LAM reference hospitals (University Hospital Vall d’Hebron; University Hospital La Princesa; University Hospital Clínica Puerta del Hierro; and University Hospital of Bellvitge), coordinated by the Spanish LAM Association (AELAM). LAM diagnosis included in all cases transbronchial lung tissue biopsy and subsequent pathological evaluation, high-resolution computed tomography scanning, and pulmonary function tests. All patients provided written, informed consent and the biomarker study was approved by the ethics committee of IDIBELL. The studies conformed to the principles set out in the World Medical Association Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Immunohistochemistry
Assays were performed on serial paraffin sections using an EnVision kit (Dako). Antigens were retrieved using citrate pH 6.0 (anti-LUM, anti-LCN2, and anti-RUNX1) or EDTA pH 9.0 (anti-IRF1 and anti-YB1) buffer. Endogenous peroxidase was blocked by pre-incubation in a solution of 3% H2O2, performed in 1x phosphate-buffered saline with 10% goat serum. Slides were incubated overnight at 4°C with primary antibody diluted in blocking solution. The antibodies were anti-IRF1 (C-20, Santa Cruz Biotechnology; dilution 1:100), anti-LCN2 (MAB1757, R&D Systems; dilution 1:50), anti-LUM (sc-166871, Santa Cruz Biotechnology; dilution 1:50), anti-RUNX1 (ab23980, Abcam; dilution 1:200), and anti-YB1 (ab76149, Abcam; dilution 1:50). Secondary peroxidase-conjugated antibodies (Envision+ system-HRP, Dako) were used. Positive controls are shown in Supplementary Figure S2. Sections were counterstained with hematoxylin and examined under a Nikon Eclipse 80i microscope.
Plasma samples
LAM blood samples were collected during the 2017 and 2018 annual AELAM patient conferences, so the period between undertaking pulmonary function tests and sample acquisition varied, making it unfeasible to assess LCN2 levels by pulmonary function. The data collected consisted of age at diagnosis, age at sample extraction, diagnosis of AML, pneumothorax, TSC, and therapy used at the time of sample extraction. All patients provided written informed consent, and the study was approved by the ethics committees of IDIBELL and the Instituto de Investigación Sanitaria La Princesa, Hospital de Henares, Spain. Control samples were obtained from healthy women. VEGF-D levels were measured using a commercially available ELISA kit according to the manufacturer’s protocol (R&D Systems). Emphysema plasma samples were collected from patients attending the Interstitial Lung Disease Unit of the University Hospital of Bellvitge. These were adult individuals (31–59 years old) with a smoking history and dyspnea on exertion, but whose FEV1/FVC ratio was less than 0.70, and who had not been diagnosed with chronic obstructive pulmonary disease. The plasma samples of patients with Langerhans cell histiocytosis and Sjögren syndrome were collected by the ILD Center of Excellence, St. Antonius Hospital Biobank, Nieuwegein, The Netherlands. The study was approved by the St. Antonius Hospital ethics committee (reference R05–08A), and all participants provided written informed consent. Detailed information about the individuals included in the plasma study is provided in Supplementary Table S1C. The studies conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
LCN2 quantification
LCN2 was quantified using the human LCN2/NGAL Quantikine Enzyme-linked Immunosorbent Assay (ELISA) Kit (R&D Systems) in accordance with the manufacturer’s instructions. The samples were diluted 1:100. Plasma samples of patients with cerebrospinal fluid leak were used as positive controls (34). The limits of quantification and detection were 52 pg/ml and 23 pg/ml, respectively.
Statistical analyses and data availability
The correlation between signature scores was computed using Spearman’s rank (rs) correlation coefficient. The two-tailed Mann–Whitney U test was used to assess signature scores and gene expression differences between pairs of groups. The log-fold change of expression differences between two defined cell groups was used to rank genes for GSEA (27). Genes differentially expressed between cell subsets were identified using the FindMarkers function of the Seurat v3 package with default parameters. The bimodality of the LAMcore score distribution in LAM cells was determined using the bimodality coefficient and Hartigan’s dip statistic (35). All data and reagents in this study are available from the authors upon request.
Results
LAM protein and gene expression signatures
LAM lung lesions often display heterogeneity at the molecular and cellular levels (36). Even diagnostic markers —typically αSMA and the premelanosomal protein gp100, detected using the HMB-45 monoclonal antibody— may be heterogeneous among diseased cells (1,2,37,38). Since the activities of genes and proteins are precisely coordinated to execute cellular functions (39), analysis of a set of biomarkers corresponding to proteins expressed in LAM lung lesions could provide further insight into disease biology (40). To define this LAM-associated protein set, we compiled evidence from the literature preceding single-cell LAM studies (6,7); this curation provided a list of 67 proteins shown to be expressed in lung LAM lesions (Table 1), but whose levels relative to other conditions, tissue and/or cells are generally unclear. This 67-protein set defined the LAMp signature used in subsequent analyses. Using gene expression data of bulk LAM lung tissue nodules (28), 24 (35%) of the LAMp genes showed significant overexpression relative to two control cell lines (Supplementary Figure S3). This percentage of overexpressed LAMp genes in bulk LAM tissue was found to be higher than would be expected at random considering all the genes examined (one-tailed proportion test, P = 0.029).
To complement the study of LAMp, we used a transcriptional signature of LAM single cells, named LAMcore, which was obtained by the landmark analysis of single-cell RNA-seq (scRNA-seq) profiles in four LAM lung tissues (6). Of the 777 genes in LAMcore, 14 were identified in LAMp (hypergeometric test for overlap P < 10−6; Table 1). Then, using the scRNA-seq data from the LAMcore study (6), we identified 21% of LAMp genes (14 of 66 available for analysis) as being significantly (false-discovery rate (FDR) < 5%) overexpressed in LAM cells relative to non-LAM cells (Supplementary Table S2A). This proportion was again found to be higher than would be expected at random (hypergeometric test for overlap P = 5 × 10−15). Therefore, as expected, the literature-curated set of proteins expressed in LAM lung lesions (i.e., LAMp) and the single-cell LAM cell transcriptome signature (i.e., LAMcore) significantly overlap with respect to their gene identities and differences in expression relative to other lung cell types.
Complementarity of LAM signatures
To further assess the LAMp and LAMcore signature scores relative to the disease, we compared their expression values (excluding shared genes; Table 1) between LAM and non-LAM scRNA-seq profiles (6). We carried out non-parametric, single-specimen gene set enrichment analysis (ssGSEA) (25) to compute signature values in individual cell transcriptomes. This yielded an enrichment score denoting the degree to which the genes in a given set were upregulated or downregulated in a coordinated manner within a cell. The LAMp score was computed using 56 (84%) of the literature-curated genes: 11 were not measured or were entirely unexpressed in the dataset (AGTR2, CCR2, CXCR1, DCT, FLT4, ITGB3, MMP9, REN, TNFRSF11B, TNFSF11, and VEGFD). The LAMcore was scored on the basis of all the originally defined genes in this signature (n = 777) (6). As expected, both signatures showed a significantly higher level of expression in LAM than in non-LAM cells (Figure 1A). Unexpectedly, however, the signatures were found to be negatively correlated in LAM cells (Spearman’s correlation coefficient (rs) = −0.35, P = 5 × 10−5; Figure 1B, left panel), but strongly positively correlated in non-LAM cells (rs = 0.42, P < 2 × 10−16; Figure 1B, right panel). It is of particular note that LAMcore was positively correlated (rs = 0.81, P < 2 × 10−16) with a gene set characteristic of LAM cells identified in an independent scRNA-seq study (7).
Figure 1. LAM signatures and potential diseased-cell heterogeneity.
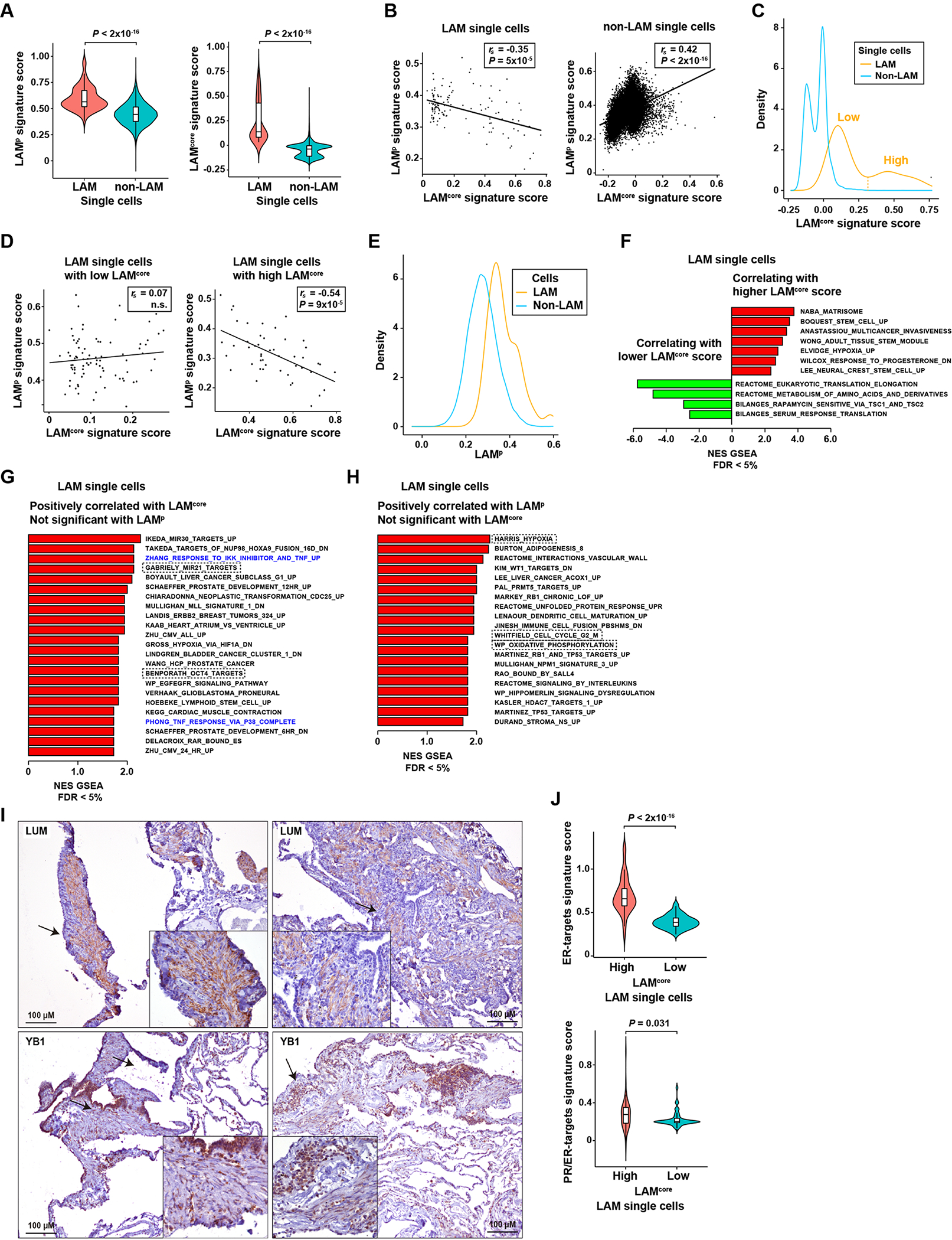
A, Violin plots showing higher LAMp and LAMcore signature scores in LAM cells than in non-LAM cells (scRNA-seq data from four LAM lung tissue (6)). Significance is indicated by Mann–Whitney test P values. The Y-axis scores show the combined expression value of the genes included in the corresponding signatures, computed using the ssGSEA algorithm (Methods). B, Scatter plots showing negative and positive LAMp-LAMcore score correlations in LAM (left panel) and non-LAM (right panel) cells, respectively. Spearman correlation coefficients (rs) and P values are indicated. The Y- and X-axis scores show the combined expression value of the genes included in the corresponding signatures, computed using the ssGSEA algorithm (Methods). C, Density plot showing the bimodal distribution of LAMcore in LAM and non-LAM cells. The threshold distinguishing high and low LAMcore in LAM cells (n = 48 and n = 85, respectively) is indicated. D, Scatter plots showing the LAMp-LAMcore correlation in LAM cells with defined low (left panel) or high (right panel) LAMcore, using the threshold depicted in panel C. E, Density plot showing the distribution LAMp scores in LAM and non-LAM cells. F, Histogram depicting Gene Set Expression Analysis (GSEA) C2-curated gene sets positively (red) or negatively (green) correlated (FDR < 5%) with LAMcore (excluding LAMp) in LAM cells. The X-axis depicts the normalized enrichment score (NES) of the GSEA. The complete lists of correlated gene sets are provided in Supplementary Table S2C (positive correlations) and 2D (negative correlations). G, Histogram depicting GSEA C2-curated gene sets positively correlated (FDR < 5%) with the LAMcore score, but not the LAMp score, in LAM cells. The gene sets reported in the Results are indicated by dashed rectangles; TNF-target gene sets are denoted in blue. H, Histogram depicting GSEA C2-curated gene sets positively correlated (FDR5 < 5%) with the LAMp score, but not the LAMcore score, in LAM cells. The gene sets reported in the Results are indicated by dashed rectangles. I, Top panels, LUM cytoplasmic expression in spindle-like cells in LAM lung. The arrows indicate the areas magnified in the insets. Bottom panels, YB1 nuclear expression in LAM epithelioid and spindle-like cells. LAM patients n = 7. The positive controls for the assays are shown in Supplementary Figure S2. J, Violin plots showing higher levels of expression of hormone-related signatures (ER-positively regulated genes (43) and progesterone-induced genes in ER-positive breast cancer (44)) in LAM cells with high LAMcore scores. Significance is indicated by Mann–Whitney test P values.
The differential correlation of LAMp and LAMcore signatures might indicate that although the two sets partially overlap with respect to their constituents, and are co-expressed in non-LAM cells, they recognize partially different diseased-cell phenotypes. Following up on this idea, the LAMcore signature score was found to be bimodally distributed in LAM cells (Figure 1C). The bimodality allowed us to define two cell subsets (Figure 1C), which might be indicative of disease heterogeneity. Evaluation of the LAMp-LAMcore relationship in these subsets revealed a negative correlation only in LAM cells with high LAMcore values (Figure 1D). The LAMcore was also bimodally distributed in non-LAM cells (Figure 1C). All the lung mesenchymal cells, which we expected to be similar to LAM cells (6), were located within the highest 10% of LAMcore values in this analysis. In turn, the LAMp score distribution did not show a significant difference from unimodality in LAM cells and non-LAM cells (Figure 1E).
Following on from the observation of two potential LAMcore-based subsets of diseased cells, for each measured gene expression, we computed the log-fold change between the LAM cells with high and low LAMcore values (Supplementary Table S2B), and used these differences to evaluate associations with a curated collection of gene sets representing defined biological states in health and disease (27). This analysis demonstrated the phenotypic variability among LAM cells: the LAMcore score was found to be positively correlated (FDR < 5%) with sets linked to the extracellular matrix organization (“NABA matrisome”), hypoxia under certain conditions (“Elvidge MCF7 hypoxia upregulated”), cell stemness (“Boquest CD31-negative stromal stem cell upregulated”, “Wong adult tissue cell stem module”, and “Lee neural stem cell upregulated”), and cancer cell invasion (“Anastassiou multicancer invasiveness signature”), amongst other similar associations (Figure 1F and Supplementary Table S2C). In turn, the LAMcore score in LAM cells was found to be negatively correlated with sets linked to amino acid metabolism (“Reactome metabolism of amino acids and derivatives pathway”), translation (“Reactome eukaryotic translation elongation pathway”), and sensitivity to rapamycin (“Bilanges rapamycin sensitive via TSC1/2”) (41), amongst other similar associations (Figure 1F and Supplementary Table S2D). An analogous analysis of LAMp scores in LAM cells did not reveal any correlations that were the opposite of those detected with LAMcore with an FDR < 5%, but several sets were uniquely and significantly correlated with LAMcore or LAMp (Supplementary Table S2C–F): positive correlations with LAMcore that were not identified (nominal P > 0.10) with LAMp included sets linked to the onco-miR-21 (“Gabriely miR-21 targets in glioma”) and OCT4 (“Benporath OCT4 targets in human embryonic stem cells”) targets, among others (Figure 1G). On the other hand, uniquely positive correlations with LAMp included sets linked to the cell cycle (“Whitfield genes expressed in G2/M phase”), hypoxia (“Harris hypoxia induced”), and oxidative phosphorylation (“WikiPathway oxidative phosphorylation”), among others (Figure 1H). Therefore, LAM cells with high or low values of LAMcore and LAMp signatures are differentially associated with biological processes and functions.
The predicted subsets of LAM-diseased cells were further evaluated by immunohistochemical studies of the gene products with the largest difference in expression between LAMcore-low and LAMcore-high cells: the keratan sulfate proteoglycan lumican (LUM), predicted to be overexpressed in LAMcore-high cells; and the transcription factor Y-Box binding protein 1 (YB1), predicted to be overexpressed in LAMcore-low cells (Supplementary Table S2B). Both proteins were identified as being expressed in LAM lung lesions, but LUM expression was mostly limited to diseased cells with a spindle cell-like phenotype, while the level of YB1 expression was lower in this type than in epithelioid cells, when detected within the lesions and in the epithelial layer (Figure 1I).
Given the key role of estrogens and progesterone in LAM biology and disease progression (42), we further evaluated differences between LAM cells with high and low LAMcore relative to the expression of signatures of ER-target genes (43) and progesterone-induced genes in ER-positive breast cancer (44)). The LAMcore-high, but not the LAMcore-low LAM cells tended to be positively associated with these two signatures (Supplementary Figure S4). Indeed, among the curated gene sets, a positive association with LAMcore corresponded to genes that were downregulated in primary ovarian cells exposed to progesterone (45) (“Wilcox response to progesterone downregulated”; Figure 1D). Violin plots further illustrate the overexpression of ER/PR-related signatures in LAM cells with high LAMcore scores (Figure 1J). Collectively, these data depict two subsets of LAM cells that differ in gene expression profiles of key biological states, which may in turn be connected to cancer features.
LAM signatures are commonly overexpressed in normal tissue relative to tumor tissue
To evaluate associations between the defined LAMp and LAMcore signatures, and cancer molecular portraits, we examined their values (excluding shared genes) in gene expression data of normal-adjacent and primary tumor tissue of 15 solid cancer types studied in The Cancer Genome Atlas (TCGA) (46). These cancers comprised tissue-of-origin categories proposed for LAM, in particular those of breast (breast invasive carcinoma (BRCA)), kidney (kidney renal clear cell carcinoma (KIRC)), lung (lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC)), and uterus (uterine corpus endometrial carcinoma (UCEC)). In addition to the positive LAMp-LAMcore correlation in non-LAM cells (Figure 1B, right panel), the scores of these signatures also proved to be positively correlated in most normal tissue settings, and in all tumor types (Supplementary Figure S5A). LAMp and LAMcore were mostly underexpressed in tumors relative to normal tissue, with the exception of glioblastoma (GBM), head and neck squamous cell carcinoma (HNSC), pancreatic adenocarcinoma (PAAD), and stomach adenocarcinoma (STAD) (Figure 2). The LAMcore signature did not show any significant differences between normal and tumor tissues in GBM, HNSC, and PAAD, whereas LAMp showed the opposite trend in these types and in STAD, i.e., significantly higher levels of expression in tumors than in normal tissue counterparts (Figure 2). Intriguingly, these cancer types (i.e., GBM, HNSC, PAAD, and STAD) are characterized by high stromal content and/or mesenchymal cell features, which may broaden the concept that the signatures capture some largely overlapping phenotypes. Nonetheless, both signatures generally showed higher levels of expression in normal tissue, which is consistent with LAM being a low-grade neoplasm (5).
Figure 2. LAM signatures commonly show higher levels of expression in normal-adjacent tissue relative to primary-tumor tissue, with exceptions linked to stromal content and/or mesenchymal features.
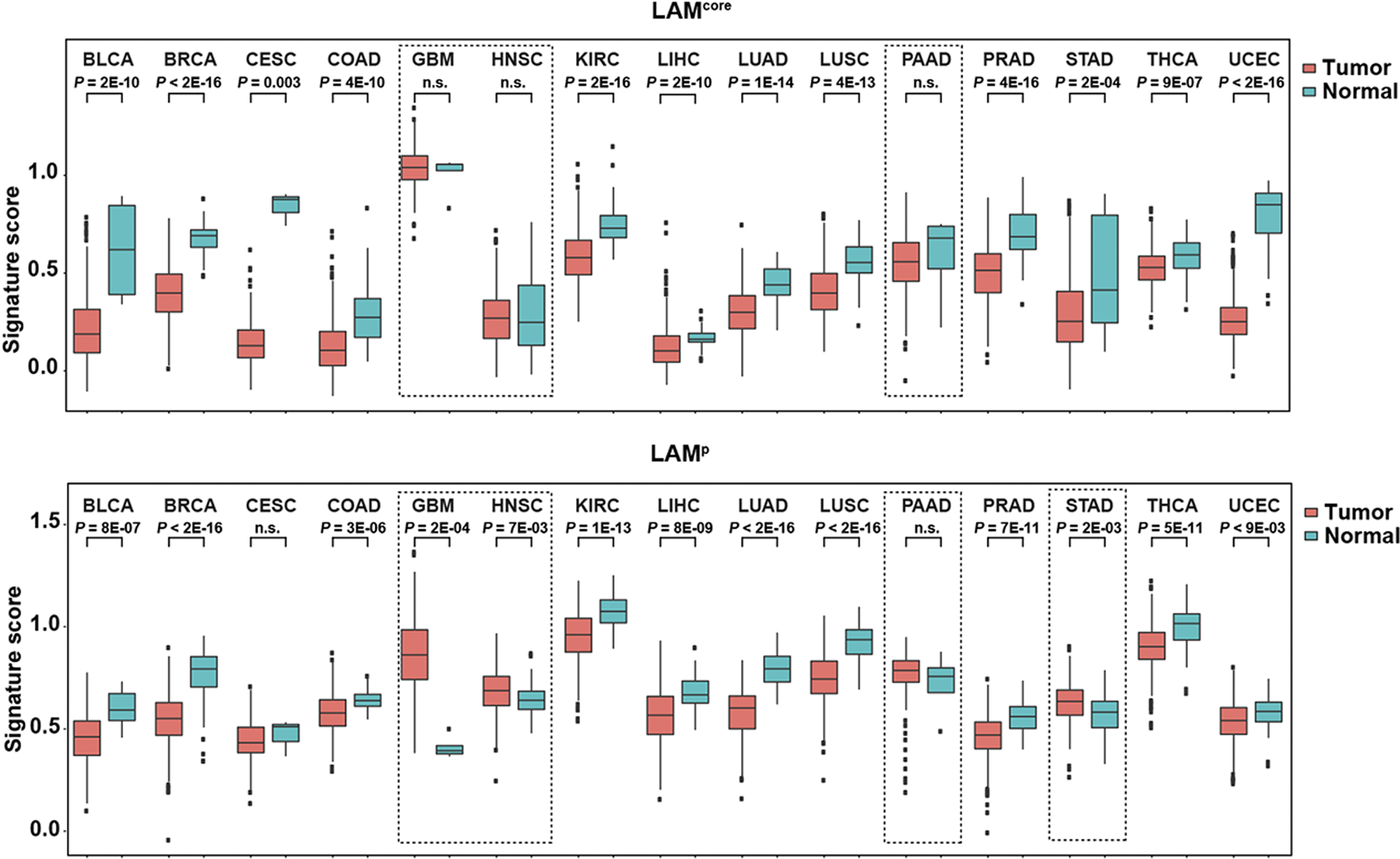
Whisker plots showing signature scores (LAMcore, top panel; and LAMp, bottom panel) in normal-adjacent tissue and primary tumors of 15 solid cancer types analyzed in TCGA (study acronyms are depicted). Score differences between normal and tumor tissue in each setting were assessed with the Mann–Whitney test. The dashed rectangles in the LAMcore panel indicate cancer types with no differences between normal and tumor tissue (GBM, HNSC, and PAAD); levels of expression were significantly higher in normal tissue than in all the other cancer types. The dashed rectangles in the LAMp analysis indicate cancer types with higher levels of expression in tumors relative to normal tissue (GBM, HNSC, and STAD) or with no difference between the two (PAAD).
Transcriptome-based similarities between LAM and cancer cells
Unsupervised hierarchical clustering of TCGA gene expression data using the LAMcore gene list commonly identified relatively high scores in normal-adjacent tissue of urothelial bladder carcinoma (BLCA), HNSC, KIRC, and STAD studies; in tumors, high signature scores were also widely identified in BLCA, KIRC, and STAD (Supplementary Figure S5B). An analogous analysis of LAMp mainly identified high scores in normal-adjacent tissue of BRCA, but tumor analyses yielded similar results to those found in LAMcore, largely identifying high scores in BLCA, HNSC, KIRC, and STAD, as well as in PAAD (Supplementary Figure S5B). These cancer connections broaden the range of observations described above regarding tumor stromal content and cancer cell phenotype.
The availability of single-cell transcriptome profiles of LAM cells from different patients (6), and of cancer cell lines representing a variety of neoplasms (33), offers the possibility of determining disease similarities more precisely. For this purpose, the data from both studies were processed and integrated for cell clustering analysis (Methods). The cancer cell lines that were clustered closer to LAM cells originated from breast, gastric, lung, and urothelial cancer (Figure 3A). These similarities involved six cancer cell lines with at least two cells identified: BT549, corresponding to triple-negative breast cancer with a mesenchymal phenotype (47); LMSU, corresponding to a gastric lymph node metastasis; BFTC909, corresponding to a transitional cell or urothelial carcinoma; and three models of pleural mesothelioma, represented by the ACCMESO1, NCIH226, and NCIH2452 cell lines (Figure 3B). The BT549 cell line mapped most closely to LAM cells and is a model that is sensitive to mTOR inhibition; targeting mTOR with everolimus in this setting reduces VEGF expression (48). The expression of ER in this cell line is not completely null and can be upregulated using chromatin remodelers (49). In addition, we identified three pleural mesothelioma cell models, which also typically show PI3K/mTOR overactivation and sensitivity to rapamycin (50).
Figure 3. Cancer cell lines similar to LAM cells.
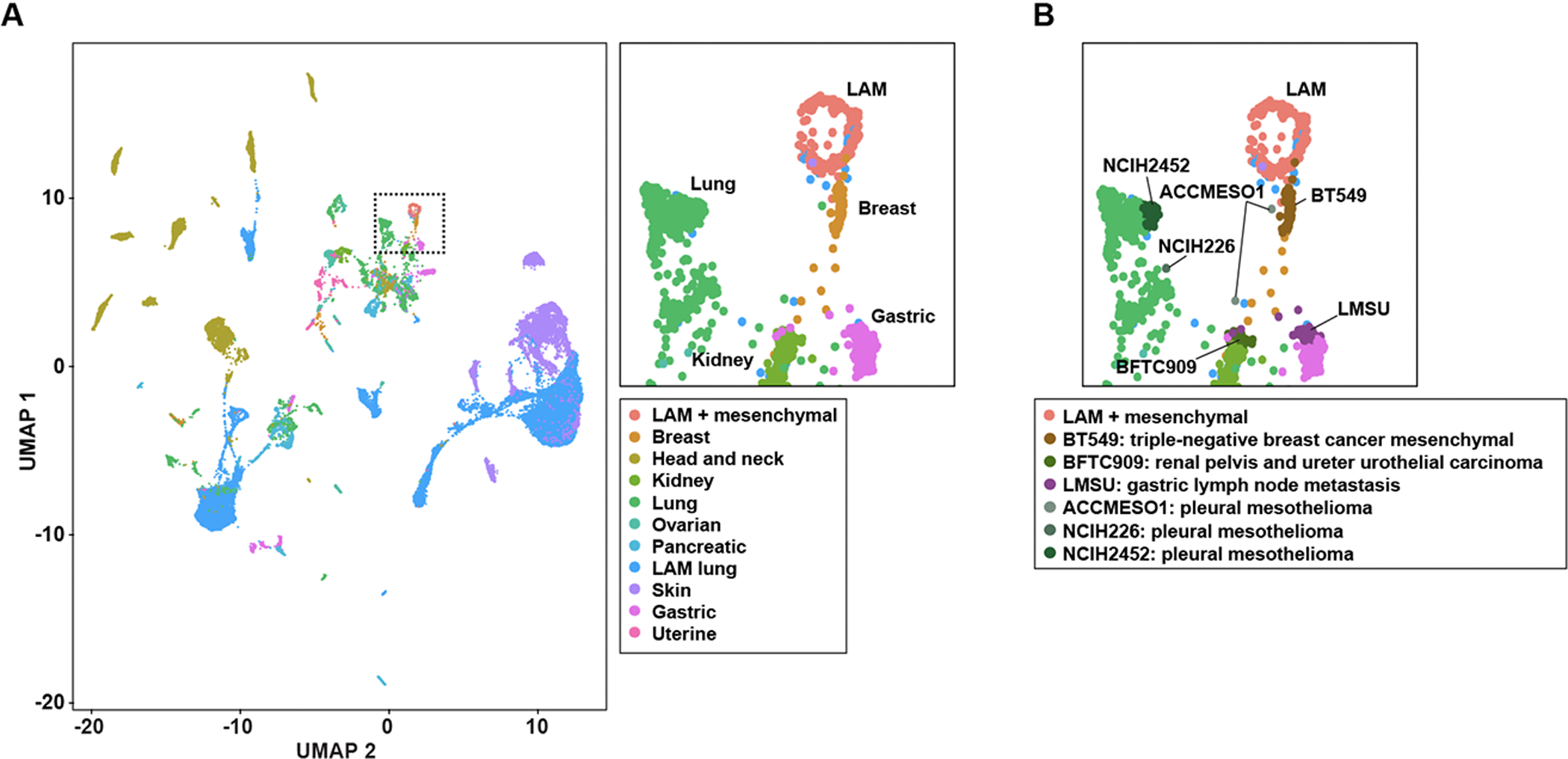
A, UMAP projection of LAM and cancer cell scRNA-seq data: global view, left panel; and zoom-in to LAM cells (plus lung mesenchymal cells, as originally annotated (6)) and closely positioned cancer cell lines, top right panel. The tissue of origin of the cancer cell lines is depicted in the inset. B, UMAP zoom-in projection indicating the six cancer cell lines (inset) positioned close to LAM cells.
To further assess the similarity between the identified cancer cell lines and LAM cells, we determined which genes were differentially expressed between these two groups and all other cancer cells included in the initial clustering analysis. Fifty genes were identified as commonly overexpressed (FDR < 5%; Supplementary Table S2G) in the six cancer cell lines and LAM cells. This number represented a higher proportion than would be expected by chance: 819 and 225 genes were overexpressed in the LAM cells and the six cancer cell lines, respectively; hypergeometric test P = 1 × 10−24. The 50-gene set included two known markers of LAM, ACTA2 and MMP2 (both included in LAMp and LAMcore; Table 1), and the autophagy receptor SQSTM1/p62; of the remaining genes, 18 were also included in LAMcore (Supplementary Table S2G). In addition, analysis of Gene Ontology (GO) terms and pathway annotations in the 50-gene set exposed the strongest associations (FDR < 5%) with extracellular matrix biology (Supplementary Table S2H), which was consistent with the identification of “NABA matrisome” as the highest positive correlation with LAMcore in LAM cells (Figure 1F). Therefore, single-cell transcriptome analyses connect LAM to four cancer settings, including those of breast cancer, gastric lymph node metastasis, mesothelioma, and urothelial carcinoma.
Prediction of LAM master regulators
Having determined the similarities between the LAMcore and LAMp signatures in single-cell and bulk tissue data, the representation of transcriptional factor-binding motifs in the corresponding gene promoters —covering a length 2 kb upstream from each transcription start site— was evaluated using JASPAR core binding profiles (51). Excluding shared genes, 11 transcription factors were identified as being overrepresented (Z-scores > 5, P = 5 × 10−7) in both signatures: CEBPA, ELF5, FOXA1, IRF1, NFATC2, RUNX1, SOX5, SOX9, SPIB, STAT1, and TBP (Supplementary Table S2I). STAT1 is a mediator of LAM cell survival and proliferation (52,53), and ELF5 and FOXA1 are coregulators of ER function (54). SOX9 supports mTOR signaling and lung metastasis in breast cancer (55), and is also expressed in LAM lung lesions (16), which means that it is a component of LAMp (Table 1).
Of the remaining transcription factor predictions, RUNX1 regulates the fate of ER-positive mammary epithelial luminal cells (56) and its expression characterizes circulating tumor cells of endometrial cancers, in which it promotes invasion and metastasis (57). Intriguingly, the previous results identified LAMcore and LAMp positive correlations (FDR < 5%) with a set corresponding to genes regulated by RUNX1-RUNX1T1 (58) (Supplementary Table S2C). The transcription factor predictions also included IRF1, which could provide a link to chemokine expression in LAM lung lesions and fluids (28). Therefore, we evaluated the expression of these two factors in the LAM lung tissue of seven patients using immunohistochemical assays. RUNX1 was strongly positive in LAM epithelioid cells in four of the patients; diseased cells with a spindle cell-like phenotype showed weak positivity for this factor in all cases (Figure 4, top panels). In turn, LAM cells with a spindle cell-like phenotype showed clear IRF1 expression in five of the patients and, as expected, infiltrating immune cells were also found to be strongly positive (Figure 4, bottom panels).
Figure 4. Expression of RUNX1 and IRF1 transcription factor in LAM lung lesions.
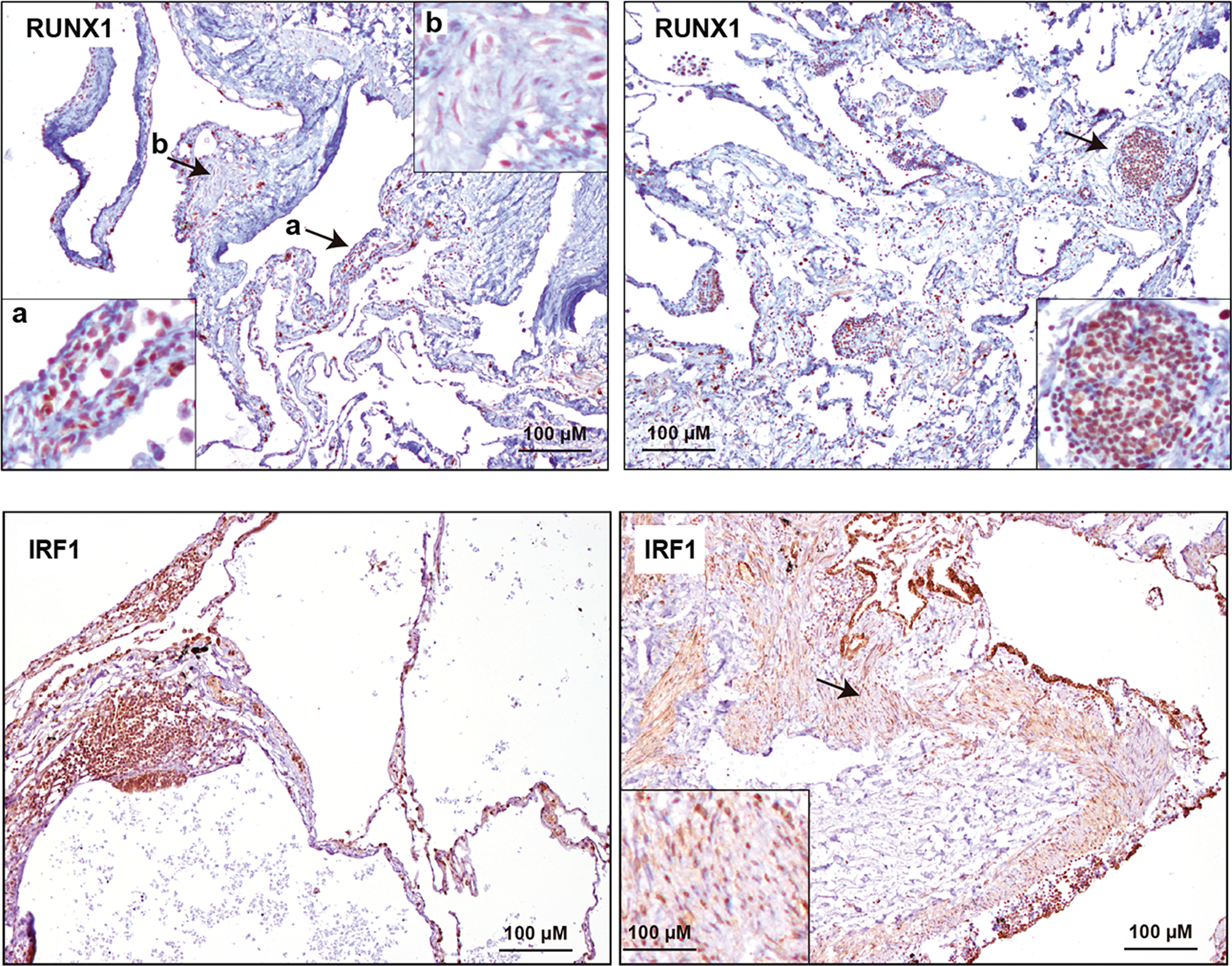
Top panels, positive RUNX1 expression in LAM epithelioid cells (left panel a) and weaker expression in LAM spindle cells (left panel b). The arrows indicate the areas magnified in the insets. Right panel, positive RUNX1 expression in a LAM lung nodule. The alveolar epithelium also appears positive. Bottom panels, positive IRF1 expression in LAM spindle cells (right panel). The arrow indicates the area magnified in the inset. Left panel, positive IRF1 expression in a LAM lung nodule. The alveolar epithelium also appears positive. LAM patients n = 7. The positive controls for the assays are shown in Supplementary Figure S2.
Potential LAM tissue heterogeneity
The above results broaden the concept of heterogeneity among LAM cells. Next, we aimed to assess heterogeneity among LAM lung tissues. For this study, the variance of gene expression measured by microarray assays in 14 LAM lung tissue samples (28) was computed, and the gene decile with the highest variance was defined. This gene subset showed significant overrepresentation of several Gene Ontology (GO) terms corresponding to immune system function, including “Chemokine activity” and “Cytokine activity” (Figure 5A and Supplementary Table S2J). Several other terms overrepresented in the LAM variable gene set were linked to cell differentiation, development, and DNA transcription (Supplementary Table S2J). In parallel, pathway-based analyses identified “Cytokine-cytokine receptor interaction” as the most overrepresented (Figure 5A and Supplementary Table S2J), reinforcing the concept that genes with large expression variation among LAM lung tissue are mainly linked to the immune system.
Figure 5. Molecular features of LAM tissue heterogeneity and identification of a subgroup.
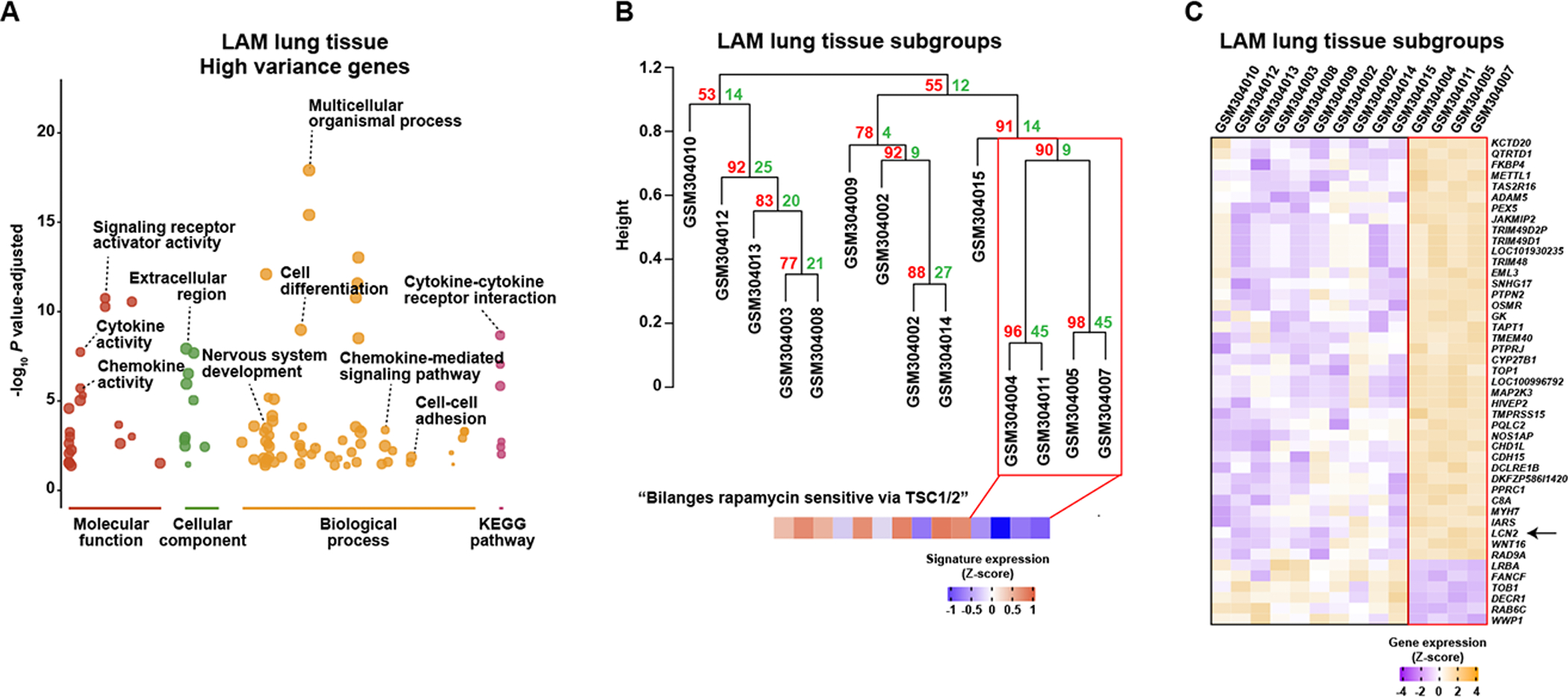
A, Overrepresented GO and KEGG annotations in the gene decile with most variable expression among 14 LAM lung nodules (GEO GSE12027); complete results are presented in Supplementary Table S2J. B, Unsupervised hierarchical clustering of LAM lung nodules based on the nine principal components derived from the variable genes (accounting for 76% of the variation). The numbers in red and green show the approximately unbiased (AU) P value (x100) and the bootstrap probability (BP x100) of the Pvclust algorithm. The immune-related cluster considered significant is marked by a red rectangle. GEO samples are detailed (GSM numbers). The bottom panel shows the expression of the signature of “Bilanges rapamycin sensitive via TSC1/2” gene set, which is significantly underexpressed (Mann–Whitney test P = 0.01) in the depicted LAM subgroup. C, Expression profiles of genes differentially expressed (FDR < 5%) between the two LAM subgroups (panel B). LCN2 is identified by an arrow.
Hierarchical unsupervised clustering with bootstrap resampling using the aforementioned gene decile identified a LAM subgroup comprising four of the 14 lung samples (Figure 5B). The four-sample subgroup showed a lower level of expression of the signature corresponding to “Bilanges rapamycin sensitive via TSC1/2” (41) (Figure 5B). Differential gene expression analysis identified 37 overexpressed and six underexpressed genes (FDR < 5%) in this subgroup, and several of the highlighted genes were linked to immune system functions (LCN2, MAP2K3, OSMR, PTPN2, PTPRJ, and TRIM48; Figure 5C). The total set of 43 genes presented significant overrepresentation of IRF1-binding sites (Z-score = 4.63), but an underrepresentation of RUNX1-binding sites (Z-score = −7.23). We then inferred the immune cell contents (30) in the 14 samples and found that the immune-associated subgroup had significantly higher levels of eosinophil numbers (Mann–Whitney test, P = 0.008; Supplementary Table S2K). Collectively, these data depict two subsets of LAM lung lesions that differ in immune system factors. Therefore, variability of immune cell content and/or of expression of defined factors by LAM and/or tissue microenvironment cells may contribute to disease heterogeneity. It is of particular note that LAMcore in LAM cells showed positive correlations with TNF-induced gene sets (Figure 1G).
LCN2 as tissue and plasma biomarker
A review of the cancer-related literature featuring the overexpressed genes identified in the immune-associated LAM tissue subgroup highlighted lipocalin 2 (LCN2; Figure 5C). LCN2 is overexpressed and associated with poor prognosis in several solid malignancies (59), and mediates breast cancer metastasis to lung (60). LCN2 is a multifunctional, innate-immunity protein that protects from inflammation in epithelial tissue (61). Immunohistochemical analysis of LAM lung tissue (n = 7) revealed strong LCN2 expression in all cases, in epithelioid and spindle-like diseased cells (Figure 6A). The degree of LCN2 positivity varied between lesions (Figure 6A). Unexpectedly, however, the level of LCN2 expression in scRNA-seq LAM cells was found to be significantly lower than in non-LAM cells (log-fold change = −0.32, P = 0.001). This apparent inconsistency with the immunohistochemistry results is reminiscent of the negative correlation between LAMp and LAMcore, and could indicate that the identities of the diseased cells analyzed by immunohistochemistry and scRNA-seq assays are not the same.
Figure 6. LCN2 as a LAM tissue and plasma marker.
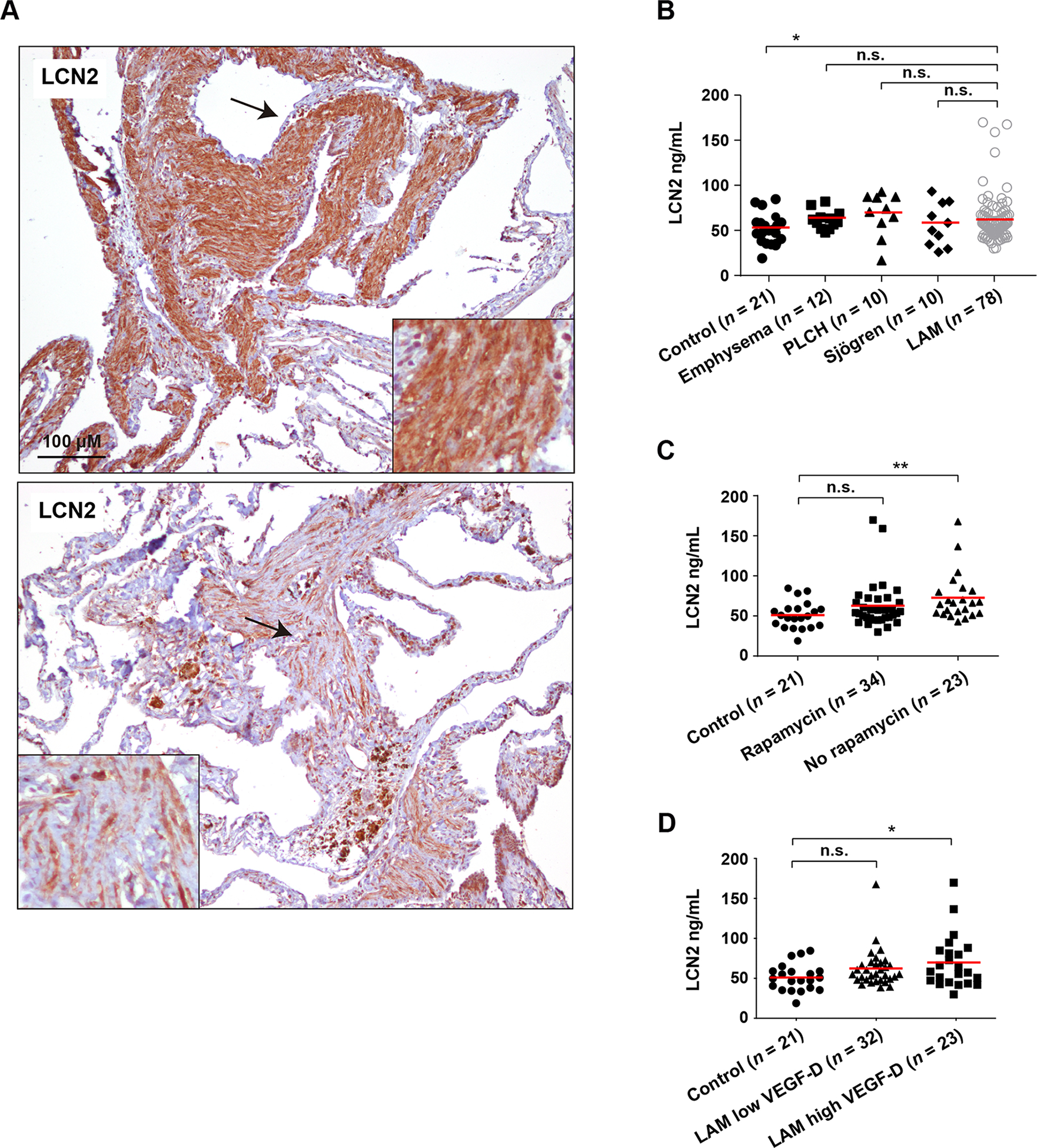
A, Detection of LCN2 expression in LAM lung lesions. Variation of expression is appreciable between the top and bottom lung tissue lesions. The arrows indicate the areas magnified in the insets. LAM patients n = 7. B, LCN2 plasma levels are significantly higher in LAM patients than in healthy women. The asterisk indicates statistical significance using a two-tailed Mann–Whitney test (*P < 0.05). LCN2 plasma levels are not significantly (n.s.) different in LAM patients relative to patients with related pulmonary diseases. The horizontal red lines indicate average values. C, LCN2 plasma levels are higher in LAM patients not treated with rapamycin, relative to healthy women. The asterisks indicate statistical significance using a two-tailed Mann–Whitney test (*P < 0.01). D, LCN2 plasma levels are higher in LAM patients with high VEGF-D levels (> 800 pg/ml), relative to healthy women. The asterisk indicates statistical significance using a two-tailed Mann–Whitney test (*P < 0.05).
LCN2 is a plasma biomarker of several diseases linked to tissue inflammation, including acute and chronic kidney disease, chronic obstructive pulmonary disease, and cancers (62). We analyzed LCN2 levels in plasma from 78 LAM patients, 21 healthy age-matched female controls, and 32 patients with LAM-related pulmonary diseases (12 patients with emphysema, 10 with pulmonary Langerhans cell histiocytosis, and 10 with Sjögren syndrome) using an enzyme-linked immunosorbent assay (ELISA). The level of plasma LCN2 was significantly higher in LAM patients than in healthy women (two-tailed Mann-Whitney test P = 0.047), but there were no significant differences between patients with LAM and those with the other pulmonary diseases (Figure 6B). LAM patients who were not taking rapamycin showed higher LCN2 plasma levels than did healthy women (two-tailed Mann–Whitney test P = 0.003; Figure 6C). Four LAM cases appeared to be outliers, with relatively high LCN2 levels (Figure 6B,C); if these cases were ignored, there was no significant difference between LAM patients and healthy women, although the overabundance of LCN2 in untreated LAM relative to healthy women remained significant (two-tailed Mann–Whitney test P = 0.009). Measuring VEGF-D in LAM plasma has been clinically implemented for disease diagnosis and monitoring (11). Patients with a defined high plasma VEGF-D level (> 800 pg/ml) also showed overabundance of LCN2 relative to healthy women (two-tailed Mann–Whitney test P = 0.041; Figure 6D), which further connects LCN2 to the pathology.
Discussion
Using complementary gene/protein signatures, this study indicates sources of LAM cell and tissue heterogeneity, in parallel with the similarities and differences with respect to cancer molecular portraits. A literature-based signature of proteins expressed in LAM lung lesions (LAMp) captures a diseased-cell phenotype that partially differs from that identified by RNA-seq profiling of LAM single diseased cells (LAMcore (6)). These signatures are found to be specifically anti-correlated in LAM cells, in particular in diseased cells with high values of LAMcore. The signature distinctions might be influenced by their different origin (i.e., proteins expressed in LAM lung lesions, LAMp; or genes expressed in LAM lung single cells, LAMcore). However, both signatures significantly overlap in terms of gene identities, are positively coexpressed in most normal and cancer settings, and are overexpressed in LAM single cells relative to non-LAM single cells. As inferred from the curated gene set associations with LAMp and LAMcore scores, LAM cell subsets differ in terms of their hormone-mediated signaling, proliferation, and stemness features, which in turn may be linked to distinct metabolic activities (e.g., metabolism of amino acids and rapamycin-sensitivity genes). These observations are further evidence of the existence of a phenotypic gradient of diseased cells that may be associated with the established spindle-like and epithelioid forms (13). Paracrine interactions in the tissue microenvironment may also contribute to the gradient. However, TSC1 or TSC2 mutations in profiled single cells need to be identified in order to assign diseased-cell features definitively.
When compared with gene expression profiles in cancer studies, the LAMp and LAMcore signatures show frequent associations with normal-adjacent tissue and/or with primary tumors predicted to have high stromal content and/or mesenchymal cell characteristics. These may be sources of bias when trying to assess LAM cell origin using bulk tissue data. Inference of the LAM origin may also be hindered because normal cells are not expected to show abnormal activation of mTOR, even though normal stem cells are frequently characterized by mTOR activation (63). On the other hand, cancer cells commonly harbor many other genetic alterations that cause complex biological changes, despite frequently showing mTOR overactivation (64). In spite of these limitations, we have compared LAM and cancer single-cell transcriptome profiles, the results demonstrating connections with the diseased cell phenotype (e.g., BT549, mesenchymal; LMSU, metastatic) and/or proposed disease origins. The identification of three pleural mesothelioma cell lines with their characteristic overactivation of PI3K/mTOR and sensitivity to mTOR inhibition (50) is particularly intriguing, especially considering the recent demonstration that Tsc2 loss in the lung mesenchymal lineage causes LAM-like disease in mice (7). In parallel, our study exposes RUNX1 as being a regulator of LAM cell signatures. This factor is highly expressed in the mesenchymal and epithelial compartments of the developing and postnatal lung (65), and regulates lung inflammation (66). Moreover, genetic variation in the RUNX1 locus, and in IRF1, has been associated with pulmonary function measures in the general population (67,68) (Supplementary Figure S6). Therefore, LAM cells might originate in a lung cell population that is regulated by these factors during tissue development and/or inflammation-related insults.
By analyzing gene expression profiles in bulk LAM lung tissue, our study also indicates sources of disease heterogeneity. This heterogeneity appears to result mainly from the interplay with the immune system, as is supported by the expression of IRF1 and RUNX1 in LAM lung lesions. The activity of the immune system may influence LAM progression and application of immunotherapies (18,69,70). A 43-gene set is proposed to differentiate two types of lesions, and the expression of the multifunctional innate immunity protein LCN2 may contribute to this distinction, further linking the disease to inflammation (60,61). LCN2 is a lipocalin that stabilizes matrix metalloproteinases, which are known to mediate lung tissue destruction in LAM (71–73). Moreover, RUNX1 negatively regulates LCN2 expression and prevents autophagy in skeletal muscle (74), whereas LCN2 expression is induced by IFNγ and TNFα (75); further, LAMcore positively correlates with TNF-induced gene sets. The depicted diseased tissue, as well as single-cell differences might influence the response to rapamycin, but analyses of relatively large cohorts are required to test this hypothesis.
Supplementary Material
Implications:
This study identifies LAM molecular and cellular features, master regulators, cancer similarities, and potential causes of disease heterogeneity.
Acknowledgments
We thank the individuals who provided samples and the AELAM foundation for its continued support for LAM research. We also wish to thank Oscar M. Tirado for support with LUM immunohistochemistry. Our results are partly based upon data generated by TCGA Research Network (https://www.cancer.gov/tcga), and we express our gratitude to TCGA consortia and coordinators for producing the data and the clinical information used in our study. This research was partially supported by AELAM (ICO-IDIBELL agreement, to M.A. Pujana), The LAM Foundation Seed Grant 2019, to M.A. Pujana, Carlos III Institute of Health grant PI18/01029 to M.A. Pujana and ICI19/00047 to M. Molina-Molina [co-funded by European Regional Development (ERDF), a way to build Europe], Generalitat de Catalunya SGR grant 2017-449, to M. A. Pujana, the CERCA Program for IDIBELL institutional support, and ZonMW- TopZorg grant 842002003, to C.H.M. van Moorsel. M. Plass was supported by a “Ramon y Cajal” contract of the Spanish Ministry of Science and Innovation (RYC2018-024564-I) and J. Moss was supported by the Intramural Research Program of NIH/NHLBI.
Footnotes
Conflicts of Interest Statement: B. Saez reports personal fees from MSD, Janssen, and Chiesi outside the submitted work. C. Valenzuela reports personal fees from Boehringer Ingelheim, F. Hoffmann-La Roche, Ltd., and BMS outside the submitted work. M. Molina- Molina reports grants from Roche and Boehringer Ing, grants and personal fees from Esteve-Teijin, and personal fees from Origo Pharma outside the submitted work. No disclosures were reported by the other authors.
Supplementary material
The manuscript is accompanied by six supplementary figures and two supplementary tables.
References
- 1.Johnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, Harari S, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010;35:14–26. [DOI] [PubMed] [Google Scholar]
- 2.McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice guidelines: Lymphangioleiomyomatosis diagnosis and management. Am J Respir Crit Care Med 2016;194:748–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henske EP, McCormack FX. Lymphangioleiomyomatosis - A wolf in sheep’s clothing. J Clin Invest 2012;122:3807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krymskaya VP, McCormack FX. Lymphangioleiomyomatosis: A monogenic model of malignancy. Annu Rev Med 2017;68:69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCormack FX, Travis WD, Colby TV, Henske EP, Moss J. Lymphangioleiomyomatosis: calling it what it is: a low-grade, destructive, metastasizing neoplasm. Am J Respir Crit Care Med 2012;186:1210–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo M, Yu JJ, Perl AK, Wikenheiser-Brokamp KA, Riccetti M, Zhang EY, et al. Single cell transcriptomic analysis identifies a unique pulmonary lymphangioleiomyomatosis cell. Am J Respir Crit Care Med 2020;202:1373–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obraztsova K, Basil MC, Rue R, Sivakumar A, Lin SM, Mukhitov AR, et al. mTORC1 activation in lung mesenchyme drives sex- and age-dependent pulmonary structure and function decline. Nat Commun 2020;11:5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345–56. [DOI] [PubMed] [Google Scholar]
- 9.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A 2000;97:6085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011;364:1595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young L, Lee H-S, Inoue Y, Moss J, Singer LG, Strange C, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med 2013;1:445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bee J, Fuller S, Miller S, Johnson SR. Lung function response and side effects to rapamycin for lymphangioleiomyomatosis: A prospective national cohort study. Thorax 2018;73:369–75. [DOI] [PubMed] [Google Scholar]
- 13.Miller S, Stewart ID, Clements D, Soomro I, Babaei-Jadidi R, Johnson SR. Evolution of lung pathology in lymphangioleiomyomatosis: associations with disease course and treatment response. J Pathol Clin Res 2020;6:215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krymskaya VP. Smooth muscle-like cells in pulmonary lymphangioleiomyomatosis. Proc Am Thorac Soc 2008;5:119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Julian LM, Delaney SP, Wang Y, Goldberg AA, Doré C, Yockell-Lelièvre J, et al. Human pluripotent stem cell-derived TSC2-haploinsufficient smooth muscle cells recapitulate features of lymphangioleiomyomatosis. Cancer Res 2017;77:5491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz de Garibay G, Herranz C, Llorente A, Boni J, Serra-Musach J, Mateo F, et al. Lymphangioleiomyomatosis biomarkers linked to lung metastatic potential and cell stemness. PLoS One 2015;10:e0132546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacheco-Rodríguez G, Steagall WK, Samsel L, Dagur PK, McCoy JP, Tunc I, et al. Circulating lymphangioleiomyomatosis tumor cells with loss of heterozygosity in the TSC2 gene show increased aldehyde dehydrogenase activity. Chest 2019;156:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H-J, Krymskaya VP, Henske EP. Immunotherapy for lymphangioleiomyomatosis and tuberous sclerosis: Progress and future directions. Chest 2019;156:1062–7. [DOI] [PubMed] [Google Scholar]
- 19.Berger U, Khaghani A, Pomerance A, Yacoub MH, Coombes RC. Pulmonary lymphangioleiomyomatosis and steroid receptors. An immunocytochemical study. Am J Clin Pathol 1990;93:609–14. [DOI] [PubMed] [Google Scholar]
- 20.Colley MH, Geppert E, Franklin WA. Immunohistochemical detection of steroid receptors in a case of pulmonary lymphangioleiomyomatosis. Am J Surg Pathol 1989;13:803–7. [DOI] [PubMed] [Google Scholar]
- 21.Yu JJ, Robb VA, Morrison TA, Ariazi EA, Karbowniczek M, Astrinidis A, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A 2009;106:2635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Zhang E, Lao T, Pereira AM, Li C, Xiong L, et al. Progesterone and estradiol synergistically promote the lung metastasis of tuberin-deficient cells in a preclinical model of lymphangioleiomyomatosis. Horm Cancer 2014;5:284–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nuñez O, Román A, Johnson SR, Inoue Y, Hirose M, Casanova Á, et al. Study of breast cancer incidence in patients of lymphangioleiomyomatosis. Breast Cancer Res Treat 2016;156:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nuñez O, Baldi BG, Radzikowska E, Carvalho CRR, Herranz C, Sobiecka M, et al. Risk of breast cancer in patients with lymphangioleiomyomatosis. Cancer Epidemiol 2019;61:154–6. [DOI] [PubMed] [Google Scholar]
- 25.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hänzelmann S, Castelo R, Guinney J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacheco-Rodriguez G, Kumaki F, Steagall WK, Zhang Y, Ikeda Y, Lin J-P, et al. Chemokine-enhanced chemotaxis of lymphangioleiomyomatosis cells with mutations in the tumor suppressor TSC2 gene. J Immunol1950. 2009;182:1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhe X, Schuger L. Combined smooth muscle and melanocytic differentiation in lymphangioleiomyomatosis. J Histochem Cytochem 2004;52:1537–42. [DOI] [PubMed] [Google Scholar]
- 30.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37:38–44. [DOI] [PubMed] [Google Scholar]
- 33.Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet 2020;52:1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llorens F, Hermann P, Villar-Piqué A, Diaz-Lucena D, Nägga K, Hansson O, et al. Cerebrospinal fluid lipocalin 2 as a novel biomarker for the differential diagnosis of vascular dementia. Nat Commun 2020;11:619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfister R, Schwarz KA, Janczyk M, Dale R, Freeman JB. Good things peak in pairs: A note on the bimodality coefficient. Front Psychol 2013;4:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonetti F, Pea M, Martignoni G, Zamboni G, Iuzzolino P. Cellular heterogeneity in lymphangiomyomatosis of the lung. Hum Pathol 1991;22:727–8. [DOI] [PubMed] [Google Scholar]
- 37.Bonetti F, Chiodera PL, Pea M, Martignoni G, Bosi F, Zamboni G, et al. Transbronchial biopsy in lymphangiomyomatosis of the lung. HMB45 for diagnosis. Am J Surg Pathol 1993;17:1092–102. [DOI] [PubMed] [Google Scholar]
- 38.Hoon V, Thung SN, Kaneko M, Unger PD. HMB-45 reactivity in renal angiomyolipoma and lymphangioleiomyomatosis. Arch Pathol Lab Med 1994;118:732–4. [PubMed] [Google Scholar]
- 39.Vidal M, Cusick ME, Barabási A-L. Interactome networks and human disease. Cell 2011;144:986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nijmeh J, El-Chemaly S, Henske EP. Emerging biomarkers of lymphangioleiomyomatosis. Expert Rev Respir Med 2018;12:95–102. [DOI] [PubMed] [Google Scholar]
- 41.Bilanges B, Argonza-Barrett R, Kolesnichenko M, Skinner C, Nair M, Chen M, et al. Tuberous sclerosis complex proteins 1 and 2 control serum-dependent translation in a TOP-dependent and -independent manner. Mol Cell Biol 2007;27:5746–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prizant H, Hammes SR. Minireview: Lymphangioleiomyomatosis (LAM): The “other” steroid-sensitive cancer. Endocrinology 2016;157:3374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AAM, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009. [DOI] [PubMed] [Google Scholar]
- 44.Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015;523:313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilcox CB, Feddes GO, Willett-Brozick JE, Hsu L-C, DeLoia JA, Baysal BE. Coordinate up-regulation of TMEM97 and cholesterol biosynthesis genes in normal ovarian surface epithelial cells treated with progesterone: implications for pathogenesis of ovarian cancer. BMC Cancer 2007;7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173:400–416. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alshaker H, Wang Q, Böhler T, Mills R, Winkler M, Arafat T, et al. Combination of RAD001 (everolimus) and docetaxel reduces prostate and breast cancer cell VEGF production and tumour vascularisation independently of sphingosine-kinase-1. Sci Rep 2017;7:3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramadan WS, Vazhappilly CG, Saleh EM, Menon V, AlAzawi AM, El-Serafi AT, et al. Interplay between epigenetics, expression of estrogen receptor-α, HER2/ERBB2 and sensitivity of triple negative breast cancer cells to hormonal therapy. Cancers 2018;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altomare DA, You H, Xiao G-H, Ramos-Nino ME, Skele KL, De Rienzo A, et al. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene 2005;24:6080–9. [DOI] [PubMed] [Google Scholar]
- 51.Ho Sui SJ, Fulton DL, Arenillas DJ, Kwon AT, Wasserman WW. oPOSSUM: Integrated tools for analysis of regulatory motif over-representation. Nucleic Acids Res 2007;35:W245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Hashemite N, Kwiatkowski DJ. Interferon-gamma-Jak-Stat signaling in pulmonary lymphangioleiomyomatosis and renal angiomyolipoma: A potential therapeutic target. Am J Respir Cell Mol Biol 2005;33:227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goncharova EA, Goncharov DA, Damera G, Tliba O, Amrani Y, Panettieri RA, et al. Signal transducer and activator of transcription 3 is required for abnormal proliferation and survival of TSC2-deficient cells: Relevance to pulmonary lymphangioleiomyomatosis. Mol Pharmacol 2009;76:766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tarulli GA, Laven-Law G, Shakya R, Tilley WD, Hickey TE. Hormone-sensing mammary epithelial progenitors: Emerging identity and hormonal regulation. J Mammary Gland Biol Neoplasia 2015;20:75–91. [DOI] [PubMed] [Google Scholar]
- 55.Mateo F, Arenas EJ, Aguilar H, Serra-Musach J, de Garibay GR, Boni J, et al. Stem cell-like transcriptional reprogramming mediates metastatic resistance to mTOR inhibition. Oncogene 2017;36:2737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Bragt MPA, Hu X, Xie Y, Li Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. eLife 2014;3:e03881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alonso-Alconada L, Muinelo-Romay L, Madissoo K, Diaz-Lopez A, Krakstad C, Trovik J, et al. Molecular profiling of circulating tumor cells links plasticity to the metastatic process in endometrial cancer. Mol Cancer 2014;13:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tonks A, Pearn L, Musson M, Gilkes A, Mills KI, Burnett AK, et al. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia 2007;21:2495–505. [DOI] [PubMed] [Google Scholar]
- 59.Xu W-X, Zhang J, Hua Y-T, Yang S-J, Wang D-D, Tang J-H. An integrative pan-cancer analysis revealing LCN2 as an oncogenic immune protein in tumor microenvironment. Front Oncol 2020;10:605097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ören B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J Pathol 2016;239:274–85. [DOI] [PubMed] [Google Scholar]
- 61.X X, Bs Y, M V-K. Lipocalin 2: An emerging player in iron homeostasis and inflammation. Annu Rev Nutr 2017;37:103–30. [DOI] [PubMed] [Google Scholar]
- 62.Pitteri SJ, Faca VM, Kelly-Spratt KS, Kasarda AE, Wang H, Zhang Q, et al. Plasma proteome profiling of a mouse model of breast cancer identifies a set of up-regulated proteins in common with human breast cancer cells. J Proteome Res 2008;7:1481–9. [DOI] [PubMed] [Google Scholar]
- 63.Meng D, Frank AR, Jewell JL. mTOR signaling in stem and progenitor cells. Dev 2018;145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011;12:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levanon D, Brenner O, Negreanu V, Bettoun D, Woolf E, Eilam R, et al. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech Dev 2001;109:413–7. [DOI] [PubMed] [Google Scholar]
- 66.Tang X, Sun L, Jin X, Chen Y, Zhu H, Liang Y, et al. Runt-related transcription factor 1 regulates LPS-induced acute lung injury via NF-κB signaling. Am J Respir Cell Mol Biol 2017;57:174–83. [DOI] [PubMed] [Google Scholar]
- 67.Portas L, Pereira M, Shaheen SO, Wyss AB, London SJ, Burney PGJ, et al. Lung development genes and adult lung function. Am J Respir Crit Care Med 2020;202:853–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 2019;51:481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu H-J, Lizotte PH, Du H, Speranza MC, Lam HC, Vaughan S, et al. TSC2-deficient tumors have evidence of T cell exhaustion and respond to anti-PD-1/anti-CTLA-4 immunotherapy. JCI Insight 2018;3:e98674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maisel K, Merrilees MJ, Atochina-Vasserman EN, Lian L, Obraztsova K, Rue R, et al. Immune checkpoint ligand PD-L1 is upregulated in pulmonary lymphangioleiomyomatosis. Am J Respir Cell Mol Biol 2018;59:723–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi T, Fleming MV, Stetler-Stevenson WG, Liotta LA, Moss J, Ferrans VJ, et al. Immunohistochemical study of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in pulmonary lymphangioleiomyomatosis (LAM). Hum Pathol 1997;28:1071–8. [DOI] [PubMed] [Google Scholar]
- 72.Atochina-Vasserman EN, Guo C-J, Abramova E, Golden TN, Sims M, James ML, et al. Surfactant dysfunction and lung inflammation in the female mouse model of lymphangioleiomyomatosis. Am J Respir Cell Mol Biol 2015;53:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nascimento ECT do, Baldi BG, Mariani AW, Annoni R, Kairalla RA, Pimenta SP, et al. Immunohistological features related to functional impairment in lymphangioleiomyomatosis. Respir Res 2018;19:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, et al. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 2005;19:1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao P, Elks CM, Stephens JM. The induction of lipocalin-2 protein expression in vivo and in vitro. J Biol Chem 2014;289:5960–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.