

Abstract
Background:
Recognition of viral nucleic acids is one of the primary triggers for a type I interferon–mediated antiviral immune response. Inborn errors of type I interferon immunity can be associated with increased inflammation and/or increased susceptibility to viral infections as a result of dysbalanced interferon production. NFX1-type zinc finger–containing 1 (ZNFX1) is an interferon-stimulated double-stranded RNA sensor that restricts the replication of RNA viruses in mice. The role of ZNFX1 in the human immune response is not known.
Objective:
We studied 15 patients from 8 families with an autosomal recessive immunodeficiency characterized by severe infections by both RNA and DNA viruses and virally triggered inflammatory episodes with hemophagocytic lymphohistiocytosis-like disease, early-onset seizures, and renal and lung disease.
Methods:
Whole exome sequencing was performed on 13 patients from 8 families. We investigated the transcriptome, posttranscriptional regulation of interferon-stimulated genes (ISGs) and predisposition to viral infections in primary cells from patients and controls stimulated with synthetic double-stranded nucleic acids.
Results:
Deleterious homozygous and compound heterozygous ZNFX1 variants were identified in all 13 patients. Stimulation of patient-derived primary cells with synthetic double-stranded nucleic acids was associated with a deregulated pattern of expression of ISGs and alterations in the half-life of the mRNA of ISGs and also associated with poorer clearance of viral infections by monocytes.
Conclusion:
ZNFX1 is an important regulator of the response to double-stranded nucleic acids stimuli following viral infections. ZNFX1 deficiency predisposes to severe viral infections and a multisystem inflammatory disease.
Keywords: ZNFX1, type I interferon, susceptibility to viral infections, HLH-like disease, virally induced hepatitis, thrombotic microangiopathy, leukoencephalopathy, brain calcification, interstitial lung disease
Studies of patients showing susceptibility to specific viral infections have helped to elucidate critical pathways in innate and adaptive immunity. Pathogenic variants of genes that disrupt type I and III interferon immune responses (eg, TLR3, UNC93B, IRF7, IRF9) have been found in patients with severe herpes simplex virus type 1 encephalitis, influenza A, and SARS-CoV2 infections.1–5
NFX1-type zinc finger–containing 1 (ZNFX1) is a highly conserved interferon-stimulated double-stranded RNA (dsRNA) sensor that restricts the replication of RNA viruses in mice6 and contributes to transgeneration inheritance in Caenorhabditis elegans by binding to mRNA complexed with short, noncoding RNAs.7 ZNFX1 expression is low in uninfected cells but is rapidly upregulated in response to viral infections and exposure to type I interferons.8 ZNFX1 binds to viral RNA and interacts with the mitochondrial antiviral signaling (MAVS) protein, promoting the expression of interferon-stimulated genes (ISGs). Signaling downstream of ZNFX1 does not depend on 2 other MAVS-associated cytosolic viral sensors (retinoic acid-inducible gene I [RIG-I] and melanoma differentiation–associated protein 5 [MDA5]).6 Furthermore, although studies of ZNFX1-deficient mice and cell lines identified a role for the protein in sensing dsRNA, the protein’s putative role in the human immune response was undefined.
Here, we describe the clinical and molecular features of biallelic ZNFX1 deficiency in 13 patients and 2 clinically affected (but not genotyped) siblings from 8 unrelated kindreds. This early-onset disease is characterized by susceptibility to viral infections, multiorgan dysfunction, and a high mortality rate, indicating the critical role of ZNFX1 in human immunity. Our experimental data demonstrate that ZNFX1 is required for the balanced induction of ISGs downstream of double-stranded nucleic acid sensing in human primary cells.
METHODS
Study participants
Written, informed consent was provided on behalf of all study participants by their parents. The 15 patients described in this article came from Iraq (n = 3), Syria (n = 2), Turkey (n = 4), Germany (n = 2), Australia (n = 2), Egypt (n = 1), and Canada (n = 1). For details on individual patients, please refer to the Patient Clinical History section and the accompanying tables in this article’s Online Repository (at www.jacionline.org).
WES
Whole exome sequencing (WES) was performed on 13 of the affected individuals and on their parents and siblings, as specified. DNA was extracted from blood samples collected in EDTA tubes. Standard methods were used to generate the WES library as well as to filter and prioritize nuclear single-nucleotide variants and indel variants (see the Methods section of the Online Repository at www.jacionline.org).
Functional assays
Quantitative polymerase chain reaction (qPCR) assays, Western blots, immunofluorescence imaging, viral infection, flow cytometry, and transcriptomic analysis were performed according to the standard protocols detailed in the Methods section of the Online Repository.
RESULTS
Severe inflammatory disease and increased susceptibility to viral infections
We investigated 15 patients from 8 families. The patients were abnormally susceptible to viral infections and presented with early-onset, systemic, severe, acute inflammatory disease associated with major dysfunctions of the liver, brain, kidneys, and lungs (Fig 1, A and B, Table I, and see Table E1 in the Online Repository at www.jacionline.org). The severe infections were caused by RNA viruses (influenza A virus [negative single-stranded RNA (ssRNA) (n = 5)], influenza B virus [negative ssRNA (n = 1)], parainfluenza virus [negative ssRNA (n = 1)], respiratory syncytial virus [negative ssRNA (n = 2)], norovirus [positive ssRNA] (n = 2)], and rotavirus [dsRNA (n = 1)]) or DNA viruses (human herpes virus type 6 [n = 3], adenovirus [n = 2], and cytomegalovirus [n = 1]) (Table I). Hence, most of these pathogens were negative ssRNA or DNA viruses. It is noteworthy that live virus vaccines also caused severe vaccine strain infections in 2 patients (measles and varicella zoster virus, respectively) (see Table E2 in the Online Repository at www.jacionline.org). A rotavirus infection recurred in patient 2.2 (P2.2) within a few weeks , and human herpes virus type 6 (with a variable copy number) was detectable in P5.2 for 5 months. Although ongoing disease manifestations in these 2 patients can be attributed to persistent or relapsing viral infections, other patients showed progressive disease even after the virus had been cleared (P8.1) in the apparent absence of infectious agents (P1.2 and P6.1).
FIG 1.
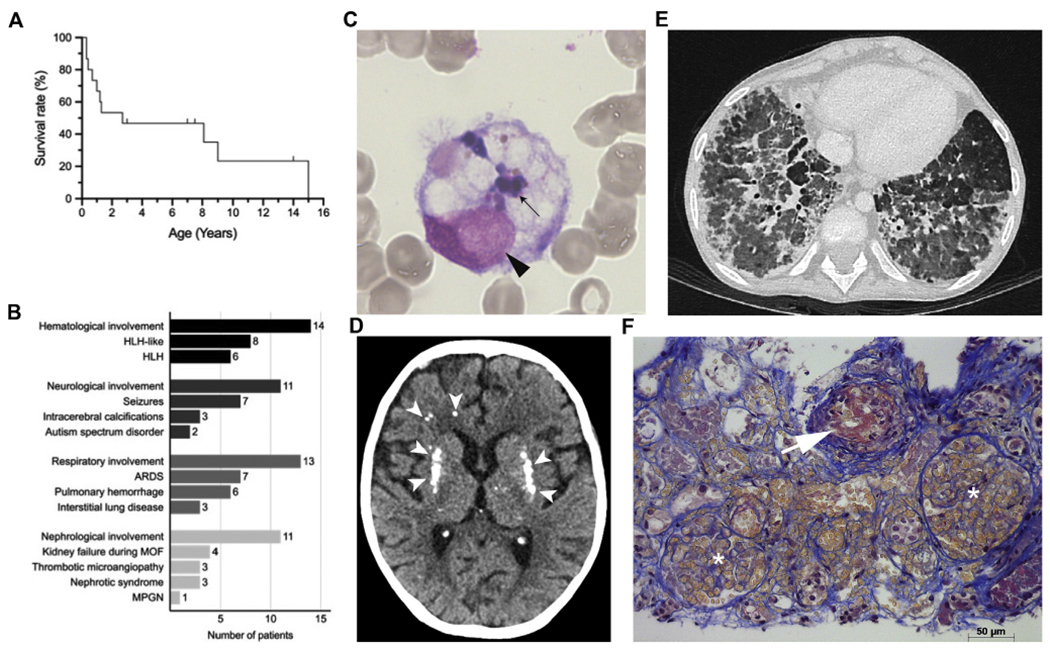
Severe viral infections and inflammatory disease in patients with ZNFX1 deficiency. A, Kaplan-Meier survival curve for patients; dashes indicate ages of patients who are alive. B, Overall inflammatory organ involvement with or without a proven link to infections; number of patients affected. C, May-Gruenwald-Giemsa staining (light microscopy; magnification, ×1000) of a bone marrow aspirate from P5.2. A macrophage with engulfed leukocytes is shown: its nucleus is indicated by an ar, and the engulfed leukocytes are indicated by an arrow. D, Computed tomography image of the brain of P1.2 at the age of 15 years showing calcification of the basal ganglia and white matter abnormalities (white ars). E, A high-resolution computed tomography image of the lungs P1.2 at the age of 9 years and 11 months, showing bilateral diffuse ground glass attenuation, subpleural thickening, and septal thickening. F, Jones staining of a kidney biopsy specimen, highlighting TMA lesions in P5.2. The arrow indicates a small arteriole with endothelial cell swelling and a fibrin/red blood microthrombus obliterating the lumen. Two glomeruli with capillary lumen dilatation and red blood cell stasis are indicated by asterisks. Acute tubular lesions with epithelial cell necrosis, lumen debris, and interstitial hemorrhage are observed (scale bar = 50 μm). ARDS, Acute respiratory distress syndrome; MOF, multiorgan failure; MPGN, membranoproliferative glomerulonephritis.
TABLE I.
Clinical characteristics
| ID | Sex | Viruses eliciting severe disease | HLH or HLH-like disease | Organs affected | Age at last FU | Outcome | |||
|---|---|---|---|---|---|---|---|---|---|
| Liver | CNS | Kidney | Lung | ||||||
| P1.1 | F | — | − | + | − | − | + | 2 y 8 mo | Dead |
| P1.2 | F | Influenza A | − | + | + | + | + | 15 y | Dead |
| P1.3 | M | Influenza A | + | + | + | − | + | 1 y 2 mo | Dead |
| P2.1 | F | — | − | + | − | + | + | 14 y | Alive |
| P2.2 | M | RSV Influenza A | + | + | (+) | + | + | 3 mo | Dead |
| P3.1 | F | NA | NA | NA | + | NA | NA | 5 mo | Dead |
| P3.2 | M | Norovirus HHV6 | + | + | + | + | + | 1 y | Dead |
| P4.1 | F | ADV | + | + | + | + | + | 8 mo | Dead |
| P4.2 | M | ADV Parainfluenza | + | + | + | + | − | 8 y | Alive |
| P5.1 | M | HHV6 (+CMV) | + | + | + | + | + | 3 mo | Dead |
| P5.2 | F | HHV6 (+Sapovirus, rhinovirus) | + | + | − | + | + | 1 y 4 mo | Dead |
| P6.1 | M | Sepsis, germ not identified | − | + | − | + | + | 9 y | Dead |
| P7.1 | F | Vaccine strain measles (+EBV) Influenza B | + | + | + | + | + | 3 y | Alive |
| P8.1 | F | CMV | + | + | + | + | + | 8y | Dead |
| P8.2 | M | Vaccine strain VZV Influenza A | + | + | + | − | + | 7 y | Alive |
| Summary | F/M, 8:7 | Negative ssRNA viruses: 10 Positive ssRNA viruses: 2 dsRNA viruses: 1 dsDNA viruses: 7 |
n = 10 | n = 14 | n = 11 | n = 11 | n = 13 | Alive/dead 4:11 |
|
ADV, Adenovirus; CMV, cytomegalovirus; CNS, central nervous system; dsDNA, double-stranded DNA; F, female; FU, follow-up; HHV6, human herpes virus type 6; HLH, hemophagocytic lymphohistiocytosis; ID, identifier; M, male; NA, not available; RSV, respiratory syncytial virus; SAP, SLAM-associated protein; VZV, varicella zoster virus.
The mortality rate was high: 11 of the 15 patients died during childhood, with 7 deaths occurring before patients reached the age of 3 months (Fig 1, A, and see Table E1). The mean age at death was 3.6 years (median = 1.1 years; range = 3 months to 15 years). Inflammatory episodes with hepatitis and cytopenia were fatal in 7 cases (age at death = 0.3-8 years). Sepsis was reportedly the cause of death in P6.1 (at the age of 9 years). P1.2 died of necrotizing pulmonary aspergillosis 5 years after lung transplantation (ie, at the age of 15 years) . The cause of death was unknown for P1.1 and P3.1 (for their clinical histories, see the Patient Clinical History section in the Online Repository at www.jacionline.org).
Infections leading to severe inflammatory diseases were the initial presentation in 9 of the 15 patients and were present in 12 of the 15 at some point in the course of disease. The systemic inflammatory disease was characterized by episodes of cytopenia and hepatitis. The cytopenia was characterized by anemia in 1 individual and anemia with thrombocytopenia in 12 individuals. In 8 individuals, anemia and anemia with thrombocytopenia were combined with a high leukocyte count (23.8-50.0 × 109/L), neutrophilia, and lymphocytosis. Other initial presentations were seizures (in 3 patients), renal disease (in 2 patients), and lung disease (in 1 patient).
A total of 14 patients had hepatic disease, as evidenced by elevated serum liver enzyme levels (n = 12 patients), hepatomegaly (n = 13), elevated serum lactate dehydrogenase levels (n = 10), coagulopathy (n = 7), and hepatic encephalopathy (n = 1) (see Table E3 in the Online Repository at www.jacionline.org). In 11 patients, hepatic disease was associated with systemic inflammatory disease. A total of 3 patients met the criteria for acute liver failure (see Table E3). Histologic assessment of the liver showed heterogeneous, nonspecific changes, such as necrosis and extramedullary hematopoiesis (in P1.2), necrosis, and lymphocytic infiltration (in P2.1), necrosis and nodular regenerative hyperplasia (in P4.2), and centrilobular necrosis (in P5.2) (see Fig E1 in the Online Repository at www.jacionline.org).
Of the 12 patients with systemic inflammatory disease, 6 met the diagnostic criteria for hemophagocytic lymphohistiocytosis (HLH), including hemophagocytosis in bone marrow aspirates (Table II9 and Fig 1, C). Some patients experienced more than 1 HLH or HLH-like episode associated with hepatitis and leukocytosis, with the latter being much less common in classical HLH. Levels of natural killer cell degranulation and/or cytotoxicity were normal in all patients with HLH. The level of perforin expression was in the lower normal range in P5.1 and P5.2, which was probably due to a heterozygous p.Ala91Val variant in the PRF1 gene that was also carried by their healthy father (data not shown). Spontaneous remission of systemic inflammation was observed in some patients, whereas immunosuppressants were administered to others—with varying degrees of success (see Table E1). The Janus kinase inhibitor ruxolitinib was administered to 1 patient (P5.2); it had a beneficial but transient effect.
TABLE II.
Diagnostic criteria for hemophagocytic lymphohistiocytosis
| Patient ID | Fever | Splenomegaly | Hemoglobin (g/dL) minimal | Platelet count, minimal | Leukocytes, maximal | Hemophagocytosis | Hyperferritinemia (≥500 mg/L) | Hypertriglyceridemia (fasting level: ≥3.0 mmol/L) or hypofibrinogenemia (≤1.5 g/L) | Elevated soluble CD25 level (≥2400 U/mL) | Low NK and/or T-cell cytotoxicity | NK cell degranulation | Perforin/SAP/XIAP expression | HLH criteria fulfilled | HLH according to an assessment by the attending physicians assessment (age at onset [y]) | HLH trigger |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1.1 | NA | NA | low | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | No HLH | Not applicable |
| P1.2 | Yes | No | 6.8 | 245-425 (age 8-14 y) | 9.2 | NA | Yes (553, age 14 y) | No Fib: 1.99-6.47, age 8-14 y TG 1.2 age 9 y |
NA | NA | NA | NA | NA (2/5; NA: 3)) | No HLH | Not applicable |
| P1.3 | Yes | Yes | 7.1 | 29 | 17.4 | Not in bone marrow (age 11 mo) | Yes (4,511) | Yes TG: 3,32, coagulation defect |
Yes; 3,957 | Normal | Normal CD107 expression | NA | Yes (6/8) | HLH (6 mo) | Rotavirus and norovirus |
| P2.1 | NA | NA | 14.7 | 213 | 9 | NA | No (68) | Yes TG: 4.9 Fib level normal |
NA | Not done | Not done | NA | NA (1/3; NA: 5) | No HLH | Not applicable |
| P2.2 | Yes | Yes | 6.0 | 6 | 25.58 | NA | Yes (34,616) | No Fib: 1.9 TG 0.8 |
NA | Not done | Not done | NA | NA (4/5; NA: 3) | HLH-like (3 mo) | Influenza A |
| P3.1 | NA | NA | NA | low | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Not applicable |
| P3.2 | Yes | No | 8.8 | 34 | 9.12 | NA | No (220) | No Fib 1.55 |
No; 2,122 | Not done | Not done | NA | No (2/6; NA: 2) | HLH-like (1 y) | HHV6 |
| P4.1 | No | Yes | 6.7 | 15 | 33 | In liver (autopsy) | Yes (4,890) | Yes TG: 9.8 Fib not decreased: 5.4 |
No | Normal | Normal CD107 expression | Normal | Yes (5/8) | HLH (7 mo) | ADV |
| P4.2 | Yes | Yes | 7.5 | 62 | 23.8 | No | No (141) | Yes TG: 5.8 Fib not decreased: 4.9 |
NA | Normal | Normal CD107 expression | Normal | NA (4/7; NA: 1) | HLH-like (22 mo) | ADV, Parainfluenza |
| P5.1 | Yes | No | 7.5 | 54 | 36.6 | No (CSF analyzed) | Yes (82,148) | Yes Fib: 0.78 TG not increased: 1.2 |
Yes; 3,185 | Not done | Normal CD107 expression | 13% in NK cells (ref >5%)* | Yes (5/8) | HLH (2 mo) | HHV6, low-level CMV viremia |
| P5.2 | Yes | Yes | 6.8 | 14 | 36.7 | Yes (in BM) | Yes (37,474) | Yes Fib: 1.3 TG 5.5 |
Yes; 12,000 | Not done | Normal CD107 expression | 20% in NK cells (ref >5%)* | Yes (7/8) | HLH (9 mo) | HHV6 |
| P6.1 | Yes | Yes | 9.1 | 63 | 17.3 | NA | NA | NA | NA | NA | NA | NA | NA (3/3; NA:5) | No HLH | Not applicable |
| P7.1 | Yes | Yes | low | 42 | 50 | NA | Yes (12,000) | No | No | Normal NK cytotoxicity | Normal CD107a expression | NA (4/7; NA:1) | HLH-like (5 mo) | ||
| Yes | Yes | 7.5 | 20 | >50 | Yes (BM) | Yes (23,000) | No | NA | Yes (5/7; NA:1) | HLH (1 y) | Vaccine strain measles and low-level EBV viremia | ||||
| Yes | Yes | low | low | high | No (CSF) | Yes (2,546) | No | NA | † | NA (4/7; NA:1) | HLH-like (2 y 9 mo) | Influenza B and Staphylococcus aureus | |||
| P8.1 | Yes | Yes | 5.5 | 25 | Low (0.2) | No | Yes (6,000) | Yes TG 11.82 mmol/L |
Yes; 14,682 | Normal | NA | ND | Yes 6/8 | HLH (8 y) | No infectious agent found |
| P8.2 | No | Yes | 5.2 | 18 | No (BM) | Yes (2,805) | No | Yes (2,534) | Reduced NK cytotoxicity but tested with low NK cell number | NA | NA | NA (4/6, NA:2) | HLH-like (2 mo) | No infectious agent found | |
| Yes | Yes | 6.5 | 95 | NA | Yes (582) | No | No (1,164) | NA | NA | No (4/8) | HLH-like (6 y) | Influenza A |
ADV, Adenovirus; BM, bone marrow; CMV, cytomegalovirus; CSF, cerebrospinal fluid; Fib, Fibrinogen level; HHV6, human herpes virus type 6; HLH, hemophagocytic lymphohistiocytosis; ID, identifier; NA, not available; NK, natural killer; ref, reference; SAP, SLAM-associated protein; TG, triglycerides (fasting level).
Diagnostic criteria for hemophagocytic lymphohistiocytosis; according to the HLH-2004 study group’s criteria (Henter et al9).
Perforin expression was in the lower normal range in P5.1 and P5.2; this was probably due to the concomitant presence of a heterozygous p.Ala91Val variant in PRF1, which was also carried by the healthy father (data not shown). In the (male) patient P5.1, SAP and XIAP expression was measured by flow cytometry and was normal.
An abnormally low percentage of perforin-expressing NK cells, although this might reflect the relative expansion of a CD56 bright NK cell population. The sample was collected during a period of acute illness, when the NK cell count was low (in a context of viral infection/HLH). It is noteworthy that the NK cell count subsequently normalized but perforin release was not retested.
Neurologic involvement was observed in 10 patients, 7 of whom experienced recurrent seizures. In 3 cases, the seizures occurred during an episode of HLH (P4.1, P7.1, and P8.1). A total of 3 patients showed developmental regression (P4.2, P7.1, and P8.1). Neuroimaging showed evidence of multiple focal calcifications in 3 patients (P1.2, P1.3, and P8.1), ischemic lesions (diffusion restriction on magnetic resonance imaging) in 4 patients (P3.2, P4.1, P5.1, and P7.1), and T2 hyperintense lesions in 5 patients (P1.2, P3.2, P4.2, P 5.2, and P7.1) (Fig 1, D and see Fig E2 in the Online Repository at www.jacionline.org). Leptomeningeal enhancement was observed in patients P3.2 and P4.1 during an episode of HLH. Autism spectrum disorder was diagnosed in 2 patients (P4.2 and P8.1).
Lung disease was present in 13 of the 15 patients. Acute respiratory distress syndrome occurred in 7 patients and was mostly associated with viral infections. Recurrent lower respiratory tract infections were observed in 6 patients (P1.1, P1.2, P1.3, P2.1, P2.2, and P8.2). One patient (P2.2) experienced 2 episodes of respiratory syncytial virus bronchiolitis with respiratory failure within the space of a few weeks. A total of 6 patients had pulmonary hemorrhage. Chest computed tomography and a histopathologic assessment of lung biopsy specimens from P1.2 and P1.3 showed interstitial pneumonitis and cholesterol pneumonitis, respectively (Fig 1, E and see Fig E3 in the Online Repository at www.jacionline.org).
There was evidence of renal involvement in 12 patients, including histologically proven thrombotic microangiopathy (TMA) in P2.1, P5.2, and P8.1 (Table I, Fig 1, F, and see Fig E4 in the Online Repository at www.jacionline.org). We variously observed hemolytic uremic syndrome (P2.1), membranoproliferative glomerulonephritis (P6.1), nephrotic syndrome (P4.2, P6.1, and P8.1), mild proteinuria (P2.2), and transiently elevated creatinine levels with glomerulosclerosis, tubular atrophy, and interstitial fibrosis at autopsy (P1.2). Renal failure (in the context of multiorgan failure) occurred in another 4 patients. Taken together, the data indicate that although peripheral destruction might have contributed to the bicytopenia (eg, in cases with TMA, we think that HLH outweighs as the driving force for the bicytopenia).
One patient (P4.2) underwent allogeneic hematopoietic stem cell transplantation (HSCT) at the age of 3 years. Five years later, he is in good health but still has a significant developmental delay. Brain magnetic resonance imaging of this patient showed that the white matter changes present at the age of 32 months had stabilized at the age of 42 months (ie, 6 months after HSCT) and had even regressed 5 years after HSCT (Fig 2, A–F). Although the patient has made developmental progress since the HSCT, he continues to show developmental delay and has been diagnosed with autism. Another survivor (P2.1, now aged 14 years) has renal disease but never experienced HLH-like disease. The third and fourth survivors, P7.1 (now aged 3) and P8.2 (now aged 7 years), are stable, although both show severe neurologic impairments. P7.1 is receiving immunoglobulin replacement therapy, but P8.2 is not receiving any immunomodulatory treatment at all.
FIG 2.
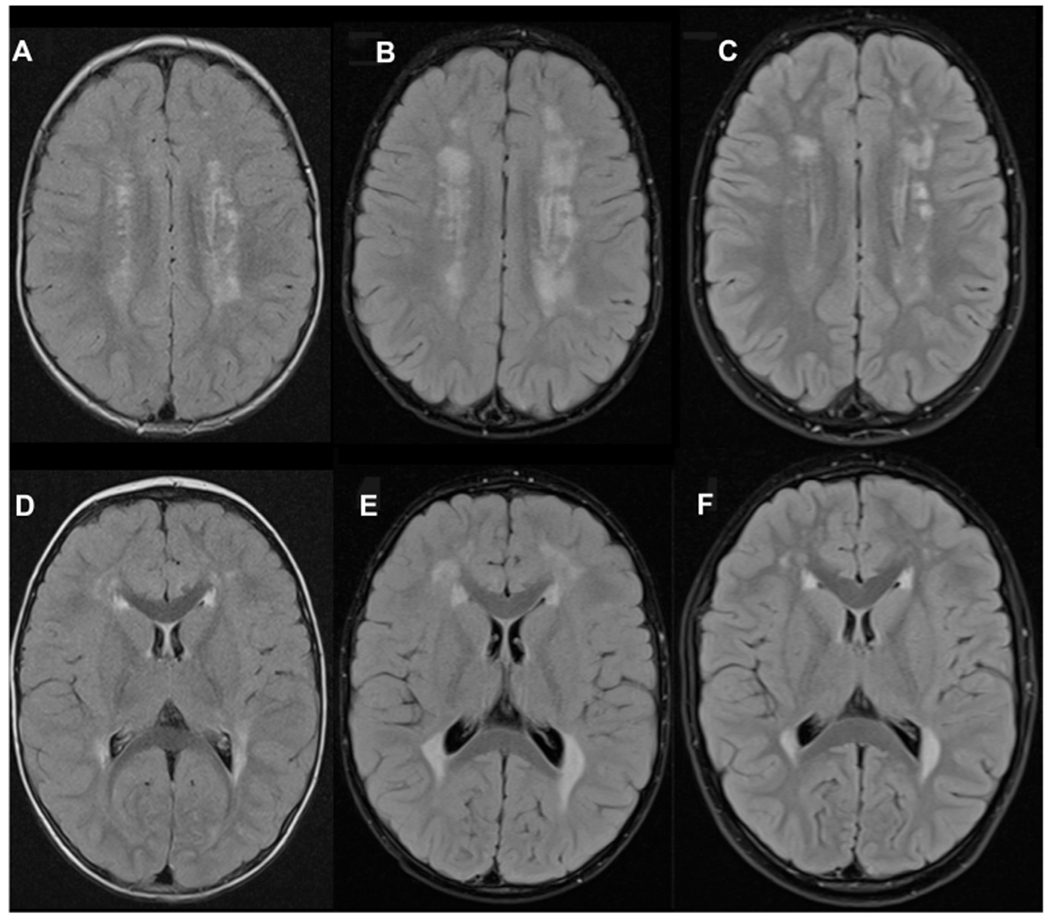
Regression of white matter changes in the brain of patient P4.2 following HSCT. Axial fluid–attenuated inversion recovery. Magnetic resonance images at the ages of 32 months (A and D), 42 months (B and E), and 8 years (C and F) demonstrating an initial increase in periventricular and deep white matter changes 6 months after HSCT (B and E) and then marked regression seen at last follow-up (5 years after HSCT) (C and F).
Biallelic ZNFX1 variants in the patients
By using WES, we identified 11 biallelic ZNFX1 variants in 13 patients (ie, in all 8 families studied; Fig 3, A). There were 5 truncating variants and 6 missense variants. In all of the patients, ZNFX1 was the only candidate gene that segregated with the disease. Only 1 variant (p.C1264S) is listed in the Genome Annotation Database (https://gnomad.broadinstitute.org/) as being heterozygous and occurring at a frequency of 1.22 × 10−5. All missense variants were predicted to be deleterious by several tools, including combined annotation-dependent depletion (CADD), PROVEAN PolyPhen-2, and the algorithm Sorting Intolerant from Tolerant (SIFT) (see Table E4 in the Online Repository at www.jacionline.org).
FIG 3.
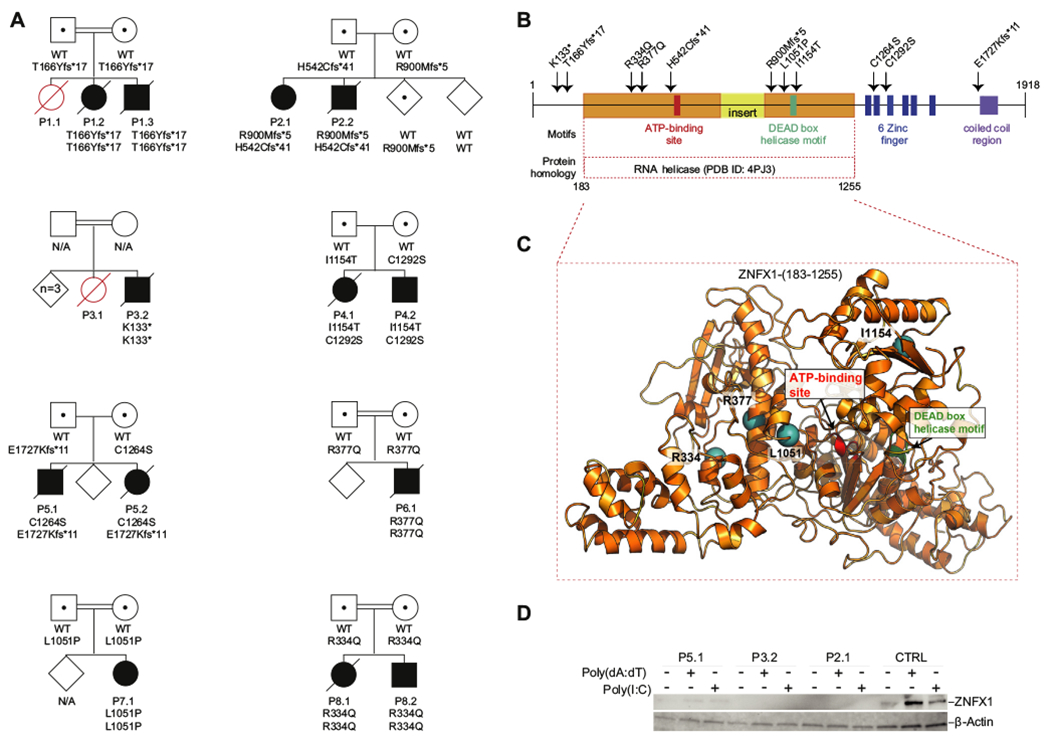
Biallelic ZNFX1 variants lead to the loss of protein expression in response to stimulation by intracellular nucleic acids. A, The pedigrees of the 8 families. Patients carrying homozygous or compound heterozygous deleterious variants in ZNFX1 are indicated by solid symbols. Healthy individuals carrying heterozygous variants are indicated by dotted symbols. Affected persons with an unknown genotype are indicated by open red symbols, whereas unaffected individuals are indicated by open diamonds. Circles indicate females, and squares indicate males. Slashes over symbols indicate deceased patients. N/A (meaning not available) indicates that sequencing was not performed. B, Predicted domains and identified variants in the ZNFX1 amino acid sequence. The 11 deleterious variants identified are indicated by arrows. The domain homologous to the RNA helicase Aquarius (Protein Data Bank identifier 4PJ3) is highlighted in orange, with an insert shown in yellow. C, A ribbon diagram of a homology model of ZNFX1 (183-1255), based on the structural template RNA helicase Aquarius (Protein Data Bank identifier 4PJ3) is shown. Locations of the 4 missense variants within this domain are shown as teal spheres in the present study. D, A protein immunoblot for ZNFX1 in dermal fibroblasts from a healthy donor (control [CTRL]) and from P5.1, P3.2, and P2.1 under resting conditions and 24 hours after transfection with the nucleic acids poly(dA:dT) or poly(I:C). β-Actin was used as a loading control.
ZNFX1 is a 1918–amino acid multidomain protein comprising a large helicase domain with an ATP-binding site10 and a DEAD helicase box,11 6 zinc fingers, and a coiled-coil region (Fig 3, B). The large helicase domain is homologous to the human RNA helicase Aquarius that is involved in RNA splicing.12 The spatial distribution of the patients’ 4 missense variants within the RNA helicase motif are shown in the 3-dimensional model of ZNFX1 in Fig 3, C.
ZNFX1 mRNA is ubiquitously expressed in human tissues, albeit predominantly in the hematopoietic system (see Fig E5 in the Online Repository at www.jacionline.org). Low ZNFX1 protein expression was noted in fibroblasts under resting conditions, whereas a rapid upregulation was observed after 24 hours of stimulation with transfected poly(I:C) or poly(dA:dT) (Fig 3, D). ZNFX1 could not be detected in whole cell extracts of fibroblasts from 2 of the patients carrying biallelic stop codons (p.R900Mfs*5/p.H542Cfs*41 in P2.1 and p.K133*/p.K133* in P3.2), whereas low levels of ZNFX1 could be detected in extracts from stimulated dermal fibroblasts isolated from P5.1, who bears 1 missense variant (p.C1264S) and 1 C-terminally truncating variant (E1727Kfs*11). Conceivable lower-molecular-weight forms of ZNFX1 were not detected with this approach.
Impaired viral clearance and skewed ISG expression in ZNFX1 deficiency
Because ZNFX1 deficiency was associated with severe viral infections in the patients, we evaluated the capability of patient cells to initiate an antiviral interferon response leading to elimination of infection with vesicular stomatitis virus (VSV) or influenza virus in vitro. Indeed, after prestimulation through transfection of poly(I:C), (LyoVec poly(I:C)), the monocytes of P2.1 were less efficient in clearing VSV than the control monocytes were (Fig 4, A and B). In notable contrast, the baseline expression of ISGs seen in peripheral blood isolated from the patients (see Fig E6, A in the Online Repository at www.jacionline.org) was higher than in the controls. This difference was biologically relevant because it was associated with a moderate resistance of unstimulated patient monocytes to the VSV and influenza virus infections (see Fig E6, B).
FIG 4.
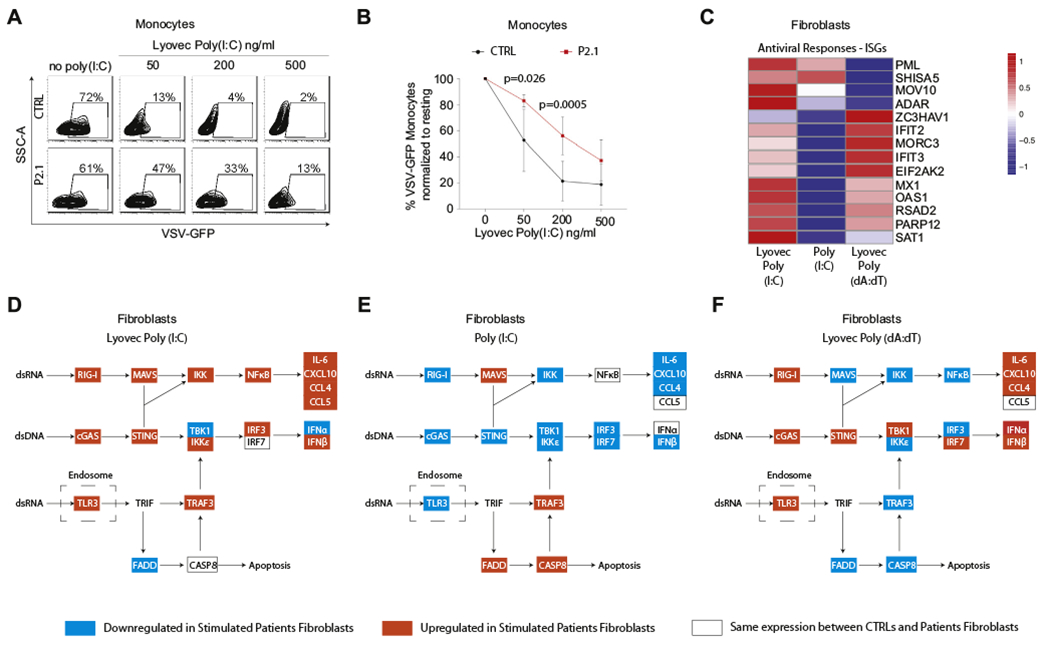
Biallelic defects in ZNFX1 deregulate ISGs’ expression and protection against viral infections in response to treatment with nucleic acids. A and B, Flow cytometry analysis of monocytes from P2.1 and a healthy control (CTRL) pretreated for 12 hours with different concentrations of LyoVec-poly(I:C) and subsequently infected with VSV–green fluorescent protein (GFP) for 5 hours. Representative plots of a single experiment showing VSV-GFP signal versus area of side scattered signal (SSC-A) (A) and mean percentage of VSV-GFP-positive monocytes relative to the unstimulated condition (no LyoVec-poly(I:C)) for 4 repeats (B). Error bars refer to the SD (n = 4). P values were calculated by using 2-way ANOVA and the Sidak multiple comparisons test. C, Transcriptomic analysis results for selected ISGs involved in antiviral responses are summarized in a heat map showing the mean difference in fold induction of ISGs expression from resting conditions in dermal fibroblasts from 4 patients (P1.2, P2.1, P3.2, and P5.2) over that in dermal fibroblasts from 4 different age-matched, healthy controls. Three different stimulations were used: 18 hours of intracellular poly(I:C) (LyoVec Poly (I:C)), 6 hours of soluble poly(I:C) (Poly (I:C)), or 18 hours of transfected poly(dA:dT) (LyoVec Poly (dA:dT)). D-F, The same data were used to study the activity of canonic double-stranded nucleic acids sensing pathways according to the Kyoto Encyclopedia of Genes and Genomes. Colored highlights indicate the rate of fold induction of gene expression in patients over that in the controls: red highlights the indicated increase, blue highlights the indicated decrease, and white boxes indicate no difference. Results from stimulation with LyoVec-poly(I:C) is shown in (D), with soluble poly(I:C) in (E) and LyoVec-poly(dA:dT) in (F). CASP8, Caspase 8.
The defective ability of the patient monocytes to establish a fully competent antiviral defense program in monocytes following stimulation with intracellular poly(I:C) could not be attributed to a generally weak response to intracellular double-stranded nucleic acids. In patient-derived dermal fibroblasts stimulated with intracellular poly(I:C) or poly(dA:dT), we found an enhanced expression of the ISGs IFIT1 and OAS2 (see Fig E7, A and C in the Online Repository at www.jacionline.org). Transfection with poly(dA:dT) also caused an increased rate of expression of IFIT2, whereas transfection with poly(I:C) did not affect the expression pattern of this ISG. On the other hand, patient fibroblasts exposed to poly(I:C) in solution failed to increase the expression of IFIT1 and IFIT2 to the levels observed in the control fibroblasts under the same conditions (Fig E7, B).
Transcriptomic analysis of dermal fibroblasts derived from 4 patients and 4 controls (treated with intracellular or soluble dsRNA or double-stranded DNA) confirmed the qPCR data showing an increased rate of expression of ISGs in response to intracellular double-stranded nucleic acids (Fig E7, A and C), as evidenced by overexpression of ISGs involved in antiviral responses (Fig 4, C). Although treatment with soluble poly(I:C) (no LyoVec poly(I:C) confirmed qPCR data; see Fig E7, B) showing a marked reduction in the expression of most ISGs involved in antiviral defense (Fig 4, C), it was associated with elevated levels of expression of ISGs known to modulate the p53-dependent apoptosis pathways (promyelocytic leukemia protein [PML] and Shisa family member 5 [SHISA5]). Analysis of pathways belonging to the canonic sensing of intracellular and extracellular double-stranded nucleic acid sensing revealed that intracellular poly(I:C) caused a heightened fold expression in ISGs belonging to the RIG-I-MAVS pathway in patient fibroblasts compared with that in the control fibroblasts (Fig 4, D). Consistent with an upregulation of this pathway, we observed elevated transcript levels of cytokines such as IL-6, C-X-C motif chemokine ligand 10 (CXCL10), C-C motif chemokine ligand 4, C-C motif chemokine ligand 5, and IFN-β. Stimulation with soluble poly(I:C) instead resulted in lower fold induction of type I interferons and other nuclear factor κB–responsive ISGs (Fig 4, E) in patient fibroblasts. Interestingly, the rates of expression of transcripts encoding known apoptosis-inducing proteins (FADD and caspase 8) were upregulated in poly(I:C)-stimulated patient fibroblasts, which is consistent with the known role of some components of the TLR3 signaling pathway in inducing dsRNA-induced cell death through caspase 8. Stimulation of the patient fibroblasts with intracellular poly(dA:dT) resulted in higher expression of ISGs belonging to the STING pathway than in the control fibroblasts, including downstream type I interferons and interferon-responsive cytokines and chemokines (Fig 4, F).
Therefore, absence of ZNFX1 in primary fibroblasts results in hyperresponses to double-stranded nucleic acid stimulation. In the case of intracellular RNA and DNA, this results in enhanced interferon responses, whereas extracellular soluble RNA induces a transcriptome pattern corresponding to apoptosis via caspase 8, thus lowering other interferon responses. Overall, dysregulation of interferon responses prevents acquisition of protection from infections following prestimulation. These results place ZNFX1 as an essential protein in balancing viral sensing.
ZNFX1 is required for a balanced posttranscriptional regulation of ISGs
Because previous work has demonstrated that ZNFX1 in lower eukaryotes binds to endogenous transcripts and regulates their processing by microRNA, we evaluated whether posttranscriptional mechanisms might influence the differential rate of expression of some ISGs detected in the patient fibroblasts. Therefore, to understand the mechanism underlying higher ISG expression rates after extended (18-hour) stimulation with double-stranded nucleic acids (Fig 5, A), we examined whether the absence of ZNFX1 promotes the stability of ISG mRNAs in response to intracellular poly(dA:dT). To this end, we added 6-dichlorobenzimidazole 1-β-D-ribofuranoside, an inhibitor of transcription elongation by RNA polymerase II, to the cultures 18 hours after poly(dA:dT) transfection. The levels of ISG mRNAs at 0, 30, 60, and 90 minutes after initiation of the 6-dichlorobenzimidazole 1-β-D-ribofuranoside treatment were higher in the patients than in the controls, which indicated that ISG mRNAs were more stable in the absence of ZNFX1 (Fig 5, B). Secretion of IFN-β and CXCL10 from fibroblasts in response to stimulation with poly(I:C)LyoVec and poly(dA:dT)LyoVec was elevated in patients when compared with that in healthy controls (Fig 5, C and D). Finally, supplementation of fibroblasts with a ZNFX1 wild-type construct lowered secretion of IFN-β and CXCL10.
FIG 5.
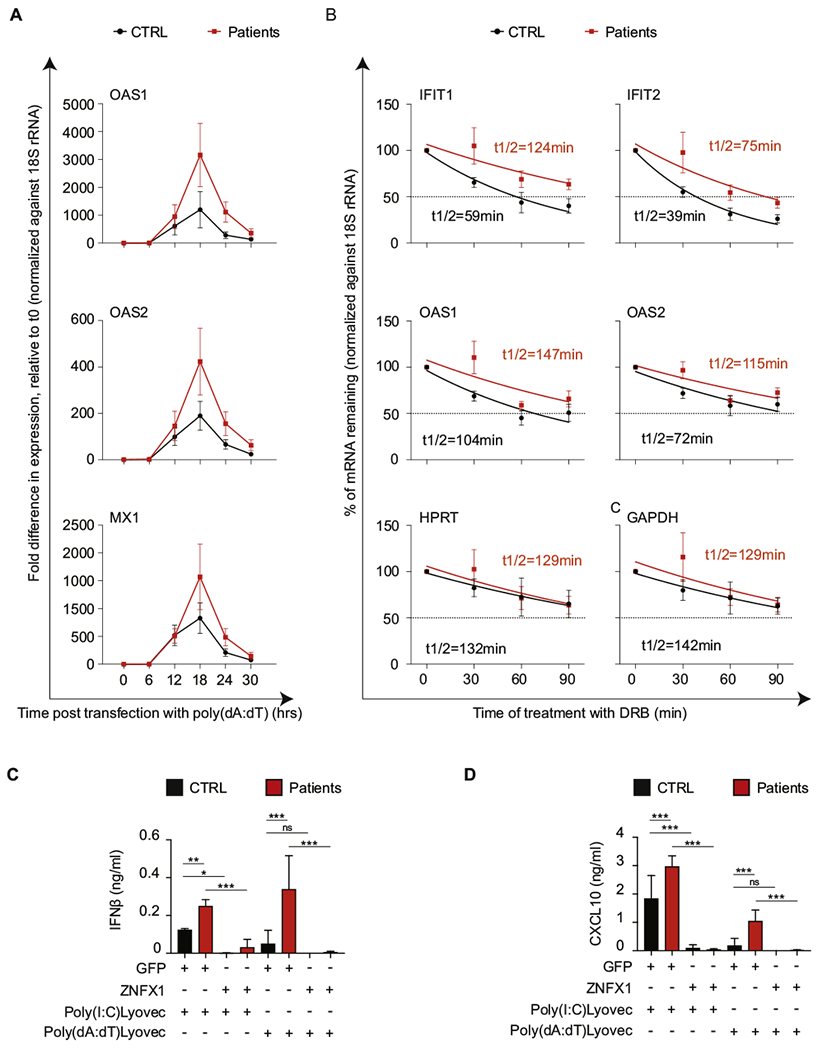
Increased ISG expression in response to transfected poly(dA:dT) in biallelic defects in ZNFX1 is associated with increased mRNA stability. A, The mRNA expression levels of OAS1, OAS2, and MX1 (representative ISGs) by skin fibroblasts from P1.2, P2.1, P3.2, and P5.2 (red squares) and 4 healthy controls (CTRLs) (black circles) at baseline (zero hours) and at different time points (6, 12, 18, 24. and 30 hours) after stimulation with transfection reagent–complexed poly(dA:dT). B, Mean values of mRNA stability of representative ISG mRNAs in fibroblasts from 4 healthy controls and 4 patients (P1.2, P2.1, P3.2, and P5.2). Gene transcription was inhibited by the addition of 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB) 18 hours after transfection with LyoVec-poly(dA:dT).qPCR was performed at the indicated time points after the addition of DRB. The amount of mRNA at each time point was normalized against ribosomal 18S RNA and represented relative to the amount at the time of DRB addition (time zero). The half-life (t1/2) of each mRNA (red for P1.2 and black for CTRL) was calculated by using nonlinear regression analysis. A representative result of 3 independent experiments is shown. Concentrations of IFN-β (C) and CXCL10 (D) in the supernatant of dermal fibroblasts from 3 healthy controls (CTRL [black bars]) and 3 patients (P1.2, P3.2, P5.2 [red bars]) following 18 hours of stimulation with poly(I:C)LyoVec or poly(dA:dT)LyoVec. Fibroblasts were transfected with plasmids expressing ZNFX1 or green fluorescent protein. Shown is the mean of 3 repeats for each of the 3 samples(n = 9), with error bars showing the SD. P values were calculated by using ordinary 1-way ANOVA as follows: ns = 0.12; *P = .033l; **P = .002; and ***P < .001.
Collectively, these findings demonstrate that ZNFX1 is important for viral defense and acts as a buffer in keeping a balanced interferon response to double-stranded nucleic acids, via a program of posttranscriptional regulation, toward a less inflammatory but more protective response, placing it as an essential protein in balancing the innate immune response.
DISCUSSION
To the best of our knowledge, this is the first report on human ZNFX1 deficiency. This deleterious deficiency is associated with susceptibility to viral infections as well as subsequent multiorgan dysfunction and inflammation. The consistent clinical phenotype observed among the 15 patients from 8 unrelated families with distinct ethnic backgrounds suggests that ZNFX1 is the causative gene for this disease.
Compared with ZNFX1-deficient mice in prior studies,6 our patients exhibited a broader range of virally induced disease that includes both RNA and DNA viruses, suggesting that ZNFX1 has additional roles beyond sensing cytosolic viral dsRNA in humans.
We have shown that although transfection with synthetic dsRNA and double-stranded DNA (dsDNA) oligos (mimicking infections with DNA and RNA viruses) causes an upregulation of inflammatory pathways, pretreatment with intracellularly delivered dsRNA does not protect patient-derived monocytes from infection. This lack of protection may be due to the complex gene signature seen in the patient fibroblasts following treatment with nucleic acids, which on the one hand promotes interferon-associated inflammation but on the other hand interferes with mechanisms of antiviral response. Previous work has demonstrated that ZNFX1 deficiency does not predispose mice or human cell lines to DNA virus infections.6 Therefore, the damage caused by DNA viruses in 6 of the patients might be directly linked to an insufficient resolution of the interferon response to the infection and not to excessive viral load. Extracellular dsRNA-mimicking oligos also cause a hyperresponse, although in this case, the signature corresponds to apoptosis with increased expression of FADD and caspase 8 and lower expression of inflammatory cytokines.
Consistent with increased susceptibility to viral infections, a respiratory syncytial virus infection recurred in 1 patient within a few weeks and 2 patients experienced vaccine strain infections (measles and varicella zoster virus, respectively). These are extremely rare events in immunocompetent hosts,13 but they are well documented in patients with defective type I and III interferon immune responses.14–18 Amelioration of central nervous system manifestations after HSCT points to an immune-driven disease. Although these observations do not fully exclude a tissue-specific role of ZNFX1 in neurons, liver cells, lung cells, and renal cells, our clinical observations and in vitro data clearly show that ZNFX1 deficiency has an impact on the immune system. In this regard, central nervous system manifestations could be caused by either HLH activity and/or viral infections.
For many viral infections, the severity of clinical disease is thought to be associated with a high viral load.19–21 In addition to cell-autonomous impairment of inflammation control, poor viral control might also contribute to the immune disease observed in patients with ZNFX1 deficiency. Thus, viral infections with RNA viruses (norovirus [positive ssRNA] and influenza A virus, RSV, parainfluenza virus, and influenza B virus [negative ssRNA]) were directly linked to HLH or to HLH-like manifestations in 7 patients. Multiorgan involvement might be suggestive of hyperinflammation caused by viral escape and viremia. However, a persistent viral load was observed in only some of the patients with ZNFX1 deficiency, whereas the others continued to display an immune disease either after viral clearance or in the absence of an identified pathogen.
Occurrence of complement-mediated TMA has recently been reported in a cohort of patients with therapy-refractory HLH.22 TMA has also been described as a dose-dependent adverse reaction to recombinant type I interferons in the treatment of viral hepatitis and multiple sclerosis.23–27 The overexpression of inflammatory genes seen in cells of ZNFX1-deficient patients after exposure to intracellular double-stranded sDNA and dsRNA might therefore be implicated in the pathogenesis of TMA that is observed in these patients.
Our observation that in the absence of ZNFX1 the half-life of ISGs is increased following extensive stimulation (for 24 hours) with intracellular DNA offers an attractive mechanism that is in line with the findings of previous work showing the essential role of ZNFX1 in posttranscriptional regulation of mRNA in lower eukaryotes.7,28 Nevertheless, whether ZNFX1 is directly involved in regulating the half-life of ISGs, or whether regulation of ISG mRNA stability is a result of alternative mechanisms that are secondary to its possible role in sensing nucleic acids, remains unclear. Furthermore, because of limited sample availability, fibroblasts from patients carrying biallelic missense mutations in ZNFX1 were not included in functional studies, and therefore, no conclusions on phenotype to genotype association could be drawn.
Viral infections in patients with ZNFX1 deficiency were associated with HLH-like episodes. HLH is characterized by fever, hepatosplenomegaly, pancytopenia, hyperferritinemia, severe coagulopathy, and hypercytokinemia. Viral infections are known to be major HLH triggers.29 To date, variants in 6 different genes (PRF1, UNC13D, STXBP2, STX11, RAB27A, and LYST) are known to directly affect perforin-mediated cytotoxicity and thereby cause HLH.30 Variants in other genes (SH2D1A, CD48, BIRC4, NLRC4, HAVCR2 [TIM-3], CDC42, RC3H1, HEM1, and AP3B3A) have been linked to HLH and HLH-like disease.31–36 ZNFX1 must now be added to this list.
Clinical observations in patients with ZNFX1 deficiency have revealed an interplay between inflammation and immunodeficiency. At present, there are few treatment options for individuals with ZNFX1 deficiency. In our study, treatment with immunosuppressants (including a Janus kinase inhibitor) led to only transient benefit. In 1 patient, HSCT arrested the HLH-like episodes and was followed by improvements in neurologic development. We recommend that (1) variants in ZNFX1 be included in genomic screens for patients experiencing severe viral infections and HLH and (2) HSCT be evaluated as a treatment for patients with ZNFX1 deficiency.
We have shown that ZNFX1 is important for sensing of viral-derived double-stranded nucleic acids in humans. Our data furthermore implicate a role for ZNFX1 in posttranscriptional regulation of ISGs, as was previously found for other proteins such as ZAP.37 Higher expression of ISGs was seen in the peripheral blood of patients with ZNFX1, together with a lower predisposition to infection, suggesting that a persistent status of hyperinflammation might on the one hand provide some levels of protection from viral infections but might on the other hand contribute to multiorgan damage. The mechanism by which ZNFX1 regulates the stability of mRNA remains elusive, but its ability to bind dsRNA is suggestive of a RNA interference mechanism mediated by small RNAs, as shown in lower eukaryotes.
Supplementary Material
Clinical implications:
ZNFX1 deficiency should be considered in patients with severe viral infections and signs of virally triggered hemophagocytic lymphohistiocytosis-like disease with hepatitis, encephalopathy, interstitial lung disease, and/or microangiopathy.
Acknowledgments
Funded by grants from the Schwyzer-Winiker Foundation (Zurich, Switzerland [to S.P. and J.P.S.]), the University of Zurich’s Clinical Research Priority Program CYTIMM-Z (to R.P. and J.P.S.), the Novartis Foundation for Biomedical Research (Basel, Switzerland [to D.K. and J.P.S.]), Promedica, the Béatrice Ederer-Weber Foundation (Zurich, Switzerland), the Helmut Horten Foundation (Zurich, Switzerland), the University of Zurich’s Cancer Research Center Funding program (to S.V. and J.P.S.), National Institutes of Health grants 5K08 AI116979-04 (to J.C.) and 1R01AI139633-01 (to R.S.G.), the Perkin Fund (to R.S.G.), the Deutsche Forschungsgemeinschaft, Germany (grants SFB1160, TPA01 and EXC-2189, as well as grant 390939984 [to S.E.]), the Bundesministerium für Bildung und Forschung, Germany (grant HCQ4Surfdefect/E-Rare-3 [to M.G.]), the Deutsche Forschungsgemeinschaft (grant 970/9-1 [to M.G.]), the German Center for Lung Research (DZL) (grant FKZ 82DZL23A2 [to M.F.]), and the Dietmar Hopp Foundation (St. Leon-Rot, Germany; grant 23011235 [to C.S.]).
We thank the patients and their families for their kind cooperation and the participating health care staff for their support. We thank Dr David Fraser (Biotech Communication SARL, Ploudalmézeau, France) for copyediting assistance, as well as Sonja Brun, Elena Kuzmenko, Dina Pitts, and Stefanie Herter for technical assistance.
Abbreviations used
- CXCL10
C-X-C Motif chemokine ligand 10
- dsRNA
Double-stranded RNA
- HLH
Hemophagocytic lymphohistiocytosis
- HSCT
Hematopoietic stem cell transplantation
- ISG
Interferon-stimulated gene
- MAVS
Mitochondrial antiviral signaling protein
- MDA5
Melanoma differentiation-associated protein 5
- PML
Promyelocytic leukemia protein
- qPCR
Quantitative PCR
- RIG-I
Retinoic acid–inducible gene I
- SAP
SLAM-associated protein
- SHISA5
Shisa family member 5
- ssRNA
Single-stranded RNA
- STING
Stimulator of interferon response cGAMP interactor
- TMA
Thrombotic microangiopathy
- TLR3
Toll-like receptor 3
- VSV
Vesicular stomatitis virus
- WES
Whole exome sequencing
- XIAP
X-linked inhibitor of apoptosis
- ZNFX1
NFX1-type zinc finger–containing 1
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006;314: 308–12. [DOI] [PubMed] [Google Scholar]
- 2.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015;348:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez N, Melki I, Jing H, Habib T, Huang SSY, Danielson J, et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J Exp Med 2018;215:2567–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med 2019; 216:2038–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020;370:eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Yuan S, Jia X, Ge Y, Ling T, Nie M, et al. Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat Cell Biol 2019;21:1346–56. [DOI] [PubMed] [Google Scholar]
- 7.Ishidate T, Ozturk AR, Durning DJ, Sharma R, Shen EZ, Chen H, et al. ZNFX-1 functions within perinuclear nuage to balance epigenetic signals. Mol Cell 2018; 70:639–49.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamada H, Yamamura M, Ohi H, Kobayashi Y, Niwa K, Oyama T, et al. Characterization of the human zinc finger nfx-1-type containing 1 encoding ZNFX1 gene and its response to 12-O-tetradecanoyl-13-acetate in HL-60 cells. Int J Oncol 2019; 55:896–904. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- 10.Saraste M, Sibbald PR, Wittinghofer A. The P-loop–a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci 1990;15:430–4. [DOI] [PubMed] [Google Scholar]
- 11.Linder P, Lasko PF, Ashburner M, Leroy P, Nielsen PJ, Nishi K, et al. Birth of the D-E-A-D box. Nature 1989;337:121–2. [DOI] [PubMed] [Google Scholar]
- 12.De I, Bessonov S, Hofele R, dos Santos K, Will CL, Urlaub H, et al. The RNA helicase Aquarius exhibits structural adaptations mediating its recruitment to spliceosomes. Nat Struct Mol Biol 2015;22:138–44. [DOI] [PubMed] [Google Scholar]
- 13.Sood SB, Suthar K, Martin K, Mather K. Vaccine-associated measles in an immunocompetent child. Clin Case Rep 2017;5:1765–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monafo WJ, Haslam DB, Roberts RL, Zaki SR, Bellini WJ, Coffin CM. Disseminated measles infection after vaccination in a child with a congenital immunodeficiency. J Pediatr 1994;124:273–6. [DOI] [PubMed] [Google Scholar]
- 15.Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci U S A 2013;110:3053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duncan CJ, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med 2015; 7ll:307ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol 2017;139:1995–7.e9. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med 2019;216:2057–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pujadas E, Chaudhry F, McBride R, Richter F, Zhao S, Wajnberg A, et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med 2020;8:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malavige GN, Jones L, Kamaladasa SD, Wijewickrama A, Seneviratne SL, Black AP, et al. Viral load, clinical disease severity and cellular immune responses in primary varicella zoster virus infection in Sri Lanka. PLoS One 2008;3:e3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavreys L, Baeten JM, Overbaugh J, Panteleeff DD, Chohan BH, Richardson BA, et al. Virus load during primary human immunodeficiency virus (HIV) type 1 infection is related to the severity of acute HIV illness in Kenyan women. Clin Infect Dis 2002;35:77–81. [DOI] [PubMed] [Google Scholar]
- 22.Gloude NJ, Dandoy CE, Davies SM, Myers KC, Jordan MB, Marsh RA, et al. Thinking beyond HLH: clinical features of patients with concurrent presentation of hemophagocytic lymphohistiocytosis and thrombotic microangiopathy. J Clin Immunol 2020;40:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kavanagh D, McGlasson S, Jury A, Williams J, Scolding N, Bellamy C,et al. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood 2016;128:2824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt D, Kavanagh D, Drummond I, Weller B, Bellamy C, Overell J, et al. Thrombotic microangiopathy associated with interferon beta. N Engl J Med 2014;370: 1270–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harvey M, Rosenfeld D, Davies D, Hall BM. Recombinant interferon alpha and hemolytic uremic syndrome: cause or coincidence? Am J Hematol 1994;46: 152–3. [DOI] [PubMed] [Google Scholar]
- 26.Beuthien W, Mellinghoff HU, Kempis J. Vasculitic complications of interferon-alpha treatment for chronic hepatitis C virus infection: case report and review of the literature. Clin Rheumatol 2005;24:507–15. [DOI] [PubMed] [Google Scholar]
- 27.Szilasiova J, Gdovinova Z, Jautova J, Baloghova J, Ficova M, Bohus P. Cutaneous vasculitis associated with interferon beta-1b treatment for multiple sclerosis. Clin Neuropharmacol 2009;32:301–3. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 2004;427:645–9. [DOI] [PubMed] [Google Scholar]
- 29.Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol 2011;11:512–6. [DOI] [PubMed] [Google Scholar]
- 30.Pachlopnik Schmid J, Cote M, Menager MM, Burgess A, Nehme N, Menasche G, et al. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev 2010;235: 10–23. [DOI] [PubMed] [Google Scholar]
- 31.Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 2011;117:1522–9. [DOI] [PubMed] [Google Scholar]
- 32.Volkmer B, Planas R, Gossweiler E, Lunemann A, Opitz L, Mauracher A, et al. Recurrent inflammatory disease caused by a heterozygous mutation in CD48. J Allergy Clin Immunol 2019. [DOI] [PubMed] [Google Scholar]
- 33.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014;46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet 2018;50:1650–7. [DOI] [PubMed] [Google Scholar]
- 35.Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med 2019;216:2778–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tavernier SJ, Athanasopoulos V, Verloo P, Behrens G, Staal J, Bogaert DJ, et al. A human immune dysregulation syndrome characterized by severe hyperinflammation with a homozygous nonsense Roquin-1 mutation. Nat Commun 2019;10:4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwerk J, Soveg FW, Ryan AP, Thomas KR, Hatfield LD, Ozarkar S, et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat Immunol 2019;20:1610–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.