

Abstract
Background
Ginsenoside Rg1 (Rg1) has been well documented to be effective against various cardiovascular disease. The aim of this study is to evaluate the effect of Rg1 on mechanical stress-induced cardiac injury and its possible mechanism with a focus on the calcium sensing receptor (CaSR) signaling pathway.
Methods
Mechanical stress was implemented on rats through abdominal aortic constriction (AAC) procedure and on cardiomyocytes and cardiac fibroblasts by mechanical stretching with Bioflex Collagen I plates. The effects of Rg1 on cell hypertrophy, fibrosis, cardiac function, [Ca2+]i, and the expression of CaSR and calcineurin (CaN) were assayed both on rat and cellular level.
Results
Rg1 alleviated cardiac hypertrophy and fibrosis, and improved cardiac decompensation induced by AAC in rat myocardial tissue and cultured cardiomyocytes and cardiac fibroblasts. Importantly, Rg1 treatment inhibited CaSR expression and increase of [Ca2+]i, which similar to the CaSR inhibitor NPS2143. In addition, Rg1 treatment inhibited CaN and TGF-β1 pathways activation. Mechanistic analysis showed that the CaSR agonist GdCl3 could not further increase the [Ca2+]i and CaN pathway related protein expression induced by mechanical stretching in cultured cardiomyocytes. CsA, an inhibitor of CaN, inhibited cardiac hypertrophy, cardiac fibrosis, [Ca2+]i and CaN signaling but had no effect on CaSR expression.
Conclusion
The activation of CaN pathway and the increase of [Ca2+]i mediated by CaSR are involved in cardiac hypertrophy and fibrosis, that may be the target of cardioprotection of Rg1 against myocardial injury.
Keywords: Ginsenoside Rg1, calcineurin, CaSR, myocardial remodeling
Graphical abstract
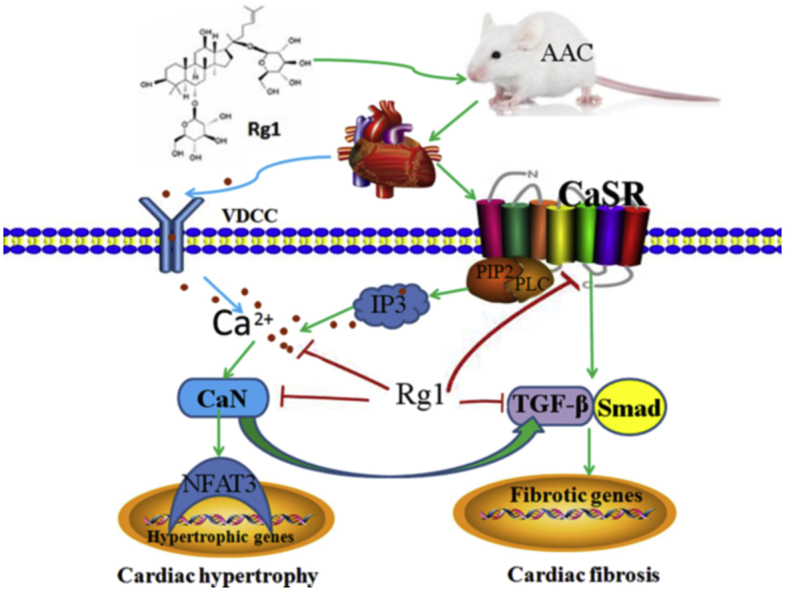
The mechanism of cardioprotection by Ginsenoside Rg1 via downregulation of CaSR. AAC induces overload of [Ca2+]i through CaSR/IP3R pathway. The increased [Ca2+]i leads to activation of CaN signaling pathway, resulting in cardiac hypertrophy and fibrosis. Ginsenoside Rg1 inhibits cardiac hypertrophy and fibrosis through inhibiting [Ca2+]i and downregulation of CaSR, CaN and TGF-β1 signaling pathways.
1. Introduction
Myocardial hypertrophy is the adaptive response of the heart to pressure overload and neurohumoral stimuli and could cause cardiac decompensation and heart failure. Myocardial remodeling is one of the main pathological features of hypertensive heart disease, which including cardiomyocyte hypertrophy and myocardial fibrosis, increasing myocardial stiffness, and eventually leading to systolic and diastolic dysfunction [1,2]. Immune regulation, inflammatory responses, oxidative stress, and especially intracellular Ca2+ overload play crucial roles in the initiation and progression of myocardial remodeling [3,4].
Calcium sensing receptor (CaSR) belongs to G protein coupled receptor family and is expressed in the hearts and neonatal rat cardiomyocytes and cardiac fibroblasts [[5], [6], [7]] and is involved in internal steady state of of calcium and metal ions [8]. According to previous studies, CaSR participates in myocardial ischemia reperfusion(MI/R) injury through activating mitogen-activated protein kinase (MAPK) pathway and endo(sarco)plasmic reticulum pathway, and promoting phospho-protein kinase Cδ translocation on mitochondria and calcium overload [[9], [10], [11]]. CaSR gets involved in myocardial hypertrophy and apoptosis through activation of calcium/calmodulin-dependent protein kinase II (CaMKII) and calcineurin (CaN) signaling pathways in an isoproterenol induced cardiac injury model [12]. CaSR causes cardiac hypertrophy through activating autophagy and promoting the release of Ca2+ from sarcoplasmic reticulum to mitochondria in the rat heart failure model, and aggravates cardiac apoptosis through activating mitochondrial dynamics-mediated apoptotic pathway in rat hypertensive hearts model [13,14]. CaN is one of serine/threonine protein phosphatases, which is activated mainly by the continuous increase of Ca2+. Activated CaN combines with the NFAT-3 transcription factor and promotes dephosphorylation of NFAT-3 in the cytoplasm. After dephosphorylation, NFAT3 was transferred to the nucleus where it regulates the activation of numerous hypertrophy-related genes. Accumulating evidence has illustrated that the CaN/NFAT3 pathway plays a pivotal role in myocardial hypertrophy and fibrosis induced by lipopolysaccharide, isoproterenol and phenylephrine accompanied by increased [Ca2+]i [[15], [16], [17]]. Furthermore, in vivo experiments with aortic constriction-induced pressure overload models in rats and in vitro experiments with mechanical stretch in rat cultured cardiomyocytes demonstrated a prominent role for CaN/NFAT in stretch-induced hypertrophy [[18], [19], [20]]. However, the mechanism by which CaN was activated in the mechanical stress model was unclear.
Ginseng, the root of Panax ginseng Meyer, has been used as a traditional medicine for more than a thousand years. In the purified components of ginseng, ginsenoside Rg1 (Rg1) is an abundant and active saponin, which has numerous potential therapeutic effects on metabolic disease. For example, ginsenoside Rg1 improves insulin resistance through decreasing the level of serum inflammatory factors and suppressing glucose output, attenuates the injury of non-alcoholic fatty liver disease through regulating lipid peroxidation, inflammation activation and endoplasmic reticulum stress [21,22]. In addition, recent studies showed ginsenoside Rg1 potential therapeutic effects on cardiovascular diseases. Ginsenoside Rg1 could ameliorate cardiac injury by inhibiting endoplasmic reticulum stress and autophagy in a doxorubicin-induced mouse model [23], improve cardiac function, and reduce cardiac hypertrophy and hypertension in a streptozotocin-induced diabetic rat model [24]. Ginsenoside Rg1 also has protective potential against myocardial ischemia and reperfusion-induced myocardial injury, which may be related to modulating energy metabolism and alleviating myocardial apoptosis [25]. In addition, we recently reported that the cardioprotective role of ginsenoside Rg1 on pressure overload-induced cardiac hypertrophy was partly attributed to the inhibition of the TNF-α/NF-κB signaling pathway [26]. However, the effect of ginsenoside Rg1 on the CaSR- and Ca2+-dependent pathways in mechanical stress-induced hypertrophy and fibrosis is still unclear. Therefore, we conducted the present study on rats through an abdominal aortic constriction (AAC) procedure and on cardiomyocytes and cardiac fibroblasts by mechanical stretching(MS) with Bioflex Collagen I plates to test whether CaSR- and Ca2+-dependent pathways contribute to cardiac injury, and whether this pathway is the target of cardioprotection of Rg1 against mechanical stress-induced myocardial remodeling.
2. Methods
2.1. Chemicals and reagents
Ginsenoside Rg1 (Rg1) was purchased from the Nanjin Jingzhu Biotechnology Company. Dimethyl sulfoxide (DMSO), verapamil, 2-APB and GdCl3 were obtained from Sigma-Aldrich (St. Louis, MO, USA). NPS2143(CaSR inhibitor) was obtained from Selleck Chemicals (Houston, TX, USA). IP3R antibody was purchased from AbSci (Baltimore, MD, USA). NFAT-3 antibody, TGF-β1 antibody and Smad2 antibody were purchased from Abcam (Cambridge, MA, USA). CaSR antibody, CaN antibody, type I collagen antibody, type III collagen antibody and β-actin antibody were obtained from Proteintech Biotechnology.
2.2. Abdominal aortic constriction procedure and drug treatment
Mechanical stress was implemented through abdominal aortic constriction (AAC) procedure on SD rats, and the procedure was followed the Guide for the Care and Use of Laboratory Animals. Forty male SD rats weighing 210 to 240 g were obtained from the Animal Center of Jinzhou Medical University. After three days of pre-adaptation, all the rats were randomly divided into 4 groups (n = 10): the Sham group; AAC group; 12 mg/kg of ginsenoside Rg1 group; and 1 mg/kg of NPS2143 group. A cardiac injury model was established by constriction of the abdominal aorta. Rats were anaesthetized with 20% urethane (0.5 ml/100g, i.p.). Then, a laparotomy was performed, and the aorta was exposed at the level of the renal arteries. The exposed abdominal aorta was ligated with a 0.8 mm silver clip. For the age-matched sham operation, an identical procedure was performed without ligation. Ginsenoside Rg1 was suspended in 0.5% Sodium Carboxymethyl Cellulose (CMC-Na) one day before the surgical procedure and continued for 30 days post-surgery. Rats in the sham and AAC groups were given with an equal volume of CMC-Na. The criteria for the selection of the doses of ginsenoside Rg1 and NPS2143 were based on our previous studies [12,26].
2.3. Echocardiography
Before the rats were sacrificed, echocardiography was used to evaluate the cardiac function of rats. The rats were anaesthetized with inhaled isoflurane, and the left ventricle internal diastolic diameter (LVIDd), the left ventricle ejection fraction (LVEF), and left ventricle fractional shortening (LVFS) were measured and analyzed using the M-mode.
2.4. Heart weight index measurement
After ultrasonic examination, the rats were sacrificed, and the hearts were immediately collected, washed in PBS solution. Then, the left ventricles were separated and weighed. The weight of total body, the weight of heart and the weight of left ventricle were weighed and recorded. Then, heart-weight index(HW/BW) and the left ventricle-weight index(LVW/BW) were calculated according to these data. After weighing, the heart tissues were immediately placed into 4% formaldehyde or Ultra low temperature freezer for the next experiments.
2.5. Morphological staining
After fixed in 4% paraformaldehyde overnight, the heart tissues were embedded in paraffin, cut into 5 μm sections. Then the paraffin sections were stained with hematoxylin-eosin (HE) or Masson's trichrome. The cardiomyocyte cross-sectional diameter and collagen volume fraction (CVF) were determined according to HE staining and Masson's trichrome staining, respectively. For immunohistochemical analyses of CaSR, collagen type I and collagen type III, the paraffin was removed, followed by 10% rabbit serum to block nonspecific binding sites. Then, the sections were treated with CaSR, collagen I and collagen III antibody overnight. After being rinsed in PBS, treated with secondary antibody, and incubated at 37 °C for another 1 h, the sections were stained with diaminobenzidine (DAB) and observed with microscope.
2.6. Isolation of the cardiomyocytes
At the end of the experiment, the rats were anesthetized, and the hearts of rats were collected quickly, installed on the perfusion system, and perfused through the aorta with Tyrode's solution for 5 min and Ca2+-free Tyrode's solution for another 8 min. Then, the heart was perfused with type II collagenase which was dissolved in 50 ml of Ca2+-free Tyrode solution. After 10 min, the type II collagenase was washed out by 5 min of perfusion with Ca2+-free Tyrode's solution. All solutions were inflated with 95% O2 and 5% CO2. After the left and right ventricles are separated, the left ventricle parts were dispersed mechanically. After the cardiomyocyte solutions were adjusted to the same cell density, the cardiomyocytes gradually recovered to normal Ca2+ concentrations.
2.7. Cell culture and mechanical stretching
The cardiomyocytes and cardiac fibroblasts were from neonatal 1- to 3-day-old SD rats were cultured and isolated with differential adherence methods. Cells were cultured in DMEM supplemented with 12% (v/v) fetal bovine serum and 100 U/mL penicillin/streptomycin. For MS, the cardiomyocytes or cardiac fibroblasts were cultured on Bioflex Collagen I plates with serum-free medium, and stretched in a Flexcell FX-5000 tension system by 20% above the initial length at a frequency of 1 Hz for up to 24 h. The control cells were cultured in Bioflex Collagen I plates but did not stretch.
2.8. RT-PCR
The extraction and quantification of total RNA from cardiac tissue, cardiomyocytes and fibroblasts were completed according to the kits. 2 μg of total RNA was used from each sample, and was reverse transcribed using AMV reverse transcriptase with random hexamers for 50 min at 42 °C. Then, the products after amplification of cDAN were used in agarose gel electrophoresis, stained with nucleic acid dye and exposed to UV irradiation. The mRNA expression of type I/III collagen, ANP and BNP was expressed as a ratio to GADPH mRNA according to gray value. The primer sequences are: ANP forward: CCTGGACTGGGGAAGTCAAC, reverse: GTCAATCCTACCCCCGAAGC; BNP forward: CGAGACAAGAGAGAGCAGGAC, reverse: TCTGGAGACTGGCTAGGACT; Collagen I forward: GATGGACTCAACGGTCTCCC, reverse: CGGCCACCATCTTGAGACTT; Collagen III forward: TTCCTGGGAGAAATGGCGAC, reverse: ACCAGCTGGGCCTTTGATAC; GAPDH forward: GTATCGGACGCCTGGTTAC, reverse: CTGTGCCGTTGAACTTGCC.
2.9. Fluo-3/AM staining for intracellular [Ca2+]i
After different treatments, the isolated cardiomyocytes and cultured cardiomyocytes were cultured with 6 μM Fluo-3/AM for 40 min at 37 °C avoid light, washed three times with Ca2+-free PBS and incubated further in complete medium. Data of [Ca2+]i were estimated by the fluorescence intensity determined by Fluo-3 in cultured and isolated cardiomyocytes with excitation and emission at 488 and 530 nm, respectively. The changes in [Ca2+]i are represented as changes in fluorescence intensity analyzed with Image Pro Plus 6.0.
2.10. Immunofluorescence
Cardiomyocytes were fixed, permeabilized, blocked, and incubated with a CaSR antibody (1:150) at 4 °C overnight, followed by incubation with the fluorescent goat anti-rabbit secondary antibody at 37 °C for 1.5 h. Finally, the cytoskeleton was stained by 1 μM of rhodamine-labeled phalloidin, and nucleus was stained by 1 μM of DAPI. Fluorescence images were captured and processed with fluorescence microscope.
2.11. EdU incorporation assay
Cell proliferation was assessed by an EdU-488 cell proliferation kit. Four hours before the end of stretching, cardiac fibroblasts were treated with EdU working solution, and at the end of the experiment, the cardiac fibroblasts were fixed with 4% paraformaldehyde and incubated with click additive solution and Hoechst 33342. Then, images were captured and processed with Leica DMI3000B fluorescence microscope.
2.12. Extraction of proteins and western blot
Nuclear protein fractions from the heart tissues and cells were extracted by a protein extraction kit according to the instructions of manufacturer. BCA method was used to analysis protein concentration. For Western blotting, protein extracts (20 μg) were fractionated by 8%-12% SDS-PAGE (1.5 h, 90V), transferred onto PVDF membranes(GE Healthcare Life Sciences, USA) and blocked with 2% BSA for 1.5 h. After washed three times with TBST, the PVDF membranes were incubated with different primary antibodies of NFAT-3(1:1000), CaSR(1:1000), CaN(1:1000), and β-actin(1:5000) followed by incubating with secondary antibodies conjugated with horseradish peroxidase. Detection was performed with enhanced ECL kit (Future Biotech, China). The results were analyzed with Quantity One software (Bio-Rad Laboratories, Hercules).
2.13. Statistical analysis
All data are expressed as the mean ± standard deviation. SPSS 19.0 software was used to analyze all the data. Differences between the means of groups were determined by one-way ANOVA followed by Bonferroni's test. Significance was defined as P < 0 05.
3. Results
3.1. Ginsenoside Rg1 improved cardiac function and attenuated cardiac hypertrophy in an AAC model
Echocardiographic results showed that LVEF and LVFS decreased, while LVIDd increased in the AAC group rats. It is suggested that hypertrophy is in the decompensated period and cardiac function is decreased (Table 1). The heart volume of AAC group was larger than that of sham group, but not increased after ginsenoside Rg1 and NPS2143 treatment(Fig. 1A). Furthermore, morphological analyses showed that the cardiomyocyte cross-sectional diameter was increased in the AAC group(Fig. 1B and C), along with increased HW/BW and LVW/BW ratios(Fig. 1D and E) and ANP and BNP mRNA expression(Fig. 1F–H). However, ginsenoside Rg1 and NPS2143 treatment significantly improved left ventricle dysfunction in the AAC rat, as shown by the increased LVEF and LVFS and decreased LVIDd. In addition, ginsenoside Rg1 and NPS2143 resulted in significant reductions in the cardiomyocyte cross-sectional diameter, ratio of HW/BW and LVW/BW and mRNA expression of ANP and BNP. These results suggested that ginsenoside Rg1 treatment could mitigate AAC-induced cardiac hypertrophy and improve the impaired cardiac function.
Table 1.
Parameters of Cardiac Function in rats.
| Group | LVEF (%) | LVFS (%) | LVIDd (mm) |
|---|---|---|---|
| Sham | 78.69 ± 2.65 | 39.15 ± 3.31 | 6.88 ± 0.34 |
| AAC | 57.97 ± 6.96∗∗ | 27.30 ± 2.95∗∗ | 8.70 ± 0.55∗∗ |
| AAC + Rg1 | 69.14 ± 4.31# | 33.21 ± 3.67# | 7.62 ± 0.51# |
| AAC + NPS2143 | 66.26 ± 6.25# | 33.69 ± 4.30# | 7.58 ± 0.35# |
LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVIDd, left ventricular internal diastolic diameter. Data were presented as the mean ± SD, n = 6 rats for each group. ∗∗P < 0.01 versus the Sham group; #P < 0.05 versus the AAC group.
Fig. 1.

Ginsenoside Rg1 inhibited cardiac hypertrophy induced by AAC. A: Visual size of the hearts. B: H&E staining microscope imaging; C: Cardiomyocyte cross-sectional diameter analyzed by H&E staining results. D-E: Data on HW/BW (mg/g) and LVW/BW (mg/g). F-H: mRNA expression of BNP and ANP determined by RT-PCR. Data were presented as the mean ± SD, n = 4 for B-C and F-H; n = 8 for D and E. ∗∗P < 0.01 compared with the Sham group; ##P < 0.01 compared with the AAC group.
3.2. Ginsenoside Rg1 reduced AAC-induced myocardial fibrosis
Mechanical stress induced by AAC led to significant pathological myocardial remodeling, including myocardial fibrosis and collagen deposition. The result from Masson staining and immunohistochemistry showed that the fibrotic area(Fig. 2A and D) and collagen I/III deposition (Fig. 2B–C and E-F) in the AAC group were significantly increased compared with that in the sham operation group. After treatment with ginsenoside Rg1, fibrotic area and collagen I/III deposition were significantly attenuated. In addition, the protein expressions levels of TGF-β1 and Smad2 were enhanced in the AAC group compared with the sham-operated group(Fig. 2G and H), and all of these changes were reversed by ginsenoside Rg1 treatment or NPS2143.
Fig. 2.

Ginsenoside Rg1 attenuated myocardial fibrosis induced by AAC. A and D: Representative pictures of Masson trichrome and data of CVF according to Masson trichrome. B-C and E-F: Representative pictures of collagen I/III immunohistochemistry and statistical data on collagen I/III expression. G-I: TGF-β1 and Smad2 protein expression determined by western blot analysis in heart tissue. All data were presented as the mean ± SD, ∗∗P < 0.01 compared with the Sham group; ##P < 0.01 compared with the AAC group (n = 4).
3.3. Ginsenoside Rg1 treatment inhibited the Ca2+/CaSR/CaN signaling pathway
Up-regulation of CaSR is involved in the cardiac hypertrophy and fibrosis induced by AAC through increased intracellular Ca2+ and Ca2+-depend signaling pathway. In current study, isolated rat cardiomyocytes were used in different groups to investigate the effect of ginsenoside Rg1 on intracellular Ca2+. Consistent with the expression of CaSR and CaN, intracellular Ca2+ was increased in the AAC group compared with the sham group. Treatment with ginsenoside Rg1 inhibited the enhanced intracellular Ca2+ (Fig. 3C and D)as well as the expression of CaSR (Fig. 3A and B), CaN and nuclear NFAT-3 (Fig. 3E–G). In addition, NPS2143 showed the similar effects to ginsenoside Rg1, suggesting that the cardioprotective effect of ginsenoside Rg1 was associated with inhibition of Ca2+/CaSR signaling pathway activation.
Fig. 3.

Ginsenoside Rg1 treatment inhibited the Ca2+/CaSR/CaN signaling pathway. A-B: Representative pictures of CaSR immunohistochemistry and statistical data on CaSR expression. C-D: [Ca2+]i fluorescence assayed by Fluo-3/AM incubation. E-G: CaN and nucleus NFAT-3 expressions determined by western blot analysis in myocardial tissue. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Sham group; ##P < 0.01 versus the AAC group (n = 4).
3.4. Ginsenoside Rg1 treatment attenuated cardiac hypertrophy in cultured cardiomyocytes
In the present study, mechanical stress was imposed on cultured cardiomyocytes by MS with Bioflex Collagen I plates. The results showed that treated cultured cardiomyocytes with 1 Hz stretch for 24 h caused cardiomyocytes hypertrophy, such as the increased cell surface area (Fig. 4A and B) and increased ANP and BNP mRNA expression (Fig. 4C–E). Furthermore, 10 μM ginsenoside Rg1 inhibited stretch-caused cardiac hypertrophy. NPS2143 showed the similar anti-hypertrophic effect with ginsenoside Rg1.
Fig. 4.

Ginsenoside Rg1 treatment attenuated cardiac hypertrophy in cultured cardiomyocytes. A-B: Cultured cardiomyocytes surface area were measured according to rhodamine-labeled phalloidin staining. The bars represent the cell surface area. C-E: Cardiomyocytes mRNA expressions of ANP and BNP determined by RT-PCR. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Con group; ##P < 0.01 compared with the MS group (n = 4).
3.5. Ginsenoside Rg1 inhibited fibrosis in cultured cardiac fibroblasts
Myocardial fibroblasts exist in the interstitial tissue of healthy myocardium, which may be an obvious source of fibrosis after myocardial injury. In the present study, cardiac fibroblasts were used to investigate the effect of Rg1 on cardiac fibrosis. The results showed that Rg1 administration significantly decreased MS-induced collagen synthesis, as determined by measurement of collagen I and collagen III mRNA. In addition, EdU incorporation was used to evaluate fibroblast proliferation. The number of EdU-positive cells was increased in MS-treated cardiac fibroblasts but decreased in the Rg1 treatment group. In addition, Rg1 treatment inhibited the enhanced protein expression of TGF-β1 and Smad2 in cardiac fibroblasts induced by MS (Fig. 5).
Fig. 5.

Ginsenoside Rg1 inhibited fibrosis in cultured cardiac fibroblasts. A: Representative images of cardiac fibroblasts analyzed according to EdU incorporation assay. B-D: Cardiac fibroblasts collagen I and III expressions determined by RT-PCR. E-G: TGF-β1 and Smad2 protein expression determined according to western blot analysis in cardiac fibroblasts. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Con group; ##P < 0.01 versus the MS group (n = 4).
3.6. Ginsenoside Rg1 inhibited CaSR and CaN signaling in cardiomyocytes and fibroblasts
To further investigate the cardioprotection of Rg1 against cardiac injury, we examined the effects of Rg1 on CaSR, CaN and nucleus NFAT3 in cardiomyocytes and cardiac fibroblasts. The results from immunofluorescence and Western blot analyses showed that CaSR and CaN signaling were up-regulated by MS, and Rg1 treatment significantly suppressed CaSR, CaN and NFAT3 expression both in cardiomyocytes (Fig. 6A–C) and cardiac fibroblasts(Fig. 6D–F). The effects of Rg1 on the protein expression of CaSR, CaN and nucleus NFAT3 were similar to CaSR inhibitor NPS2143.
Fig. 6.

Ginsenoside Rg1 inhibited CaSR and CaN signaling in cardiomyocytes and fibroblasts. A: Representative images of CaSR immunofluorescence staining. Blue is the nucleus of DAPI staining; red is the cytoskeleton of rhodamine-labeled phalloidin staining; green is CaSR expression; the last line is a merge graph of three kinds of coloring. B-C: CaN and nucleus NFAT3 protein expression determined according to western blot analysis in cardiomyocytes. D-F: CaSR, CaN and nucleus NFAT3 protein expression determined according to western blot in cardiac fibroblasts. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Con group; ##P < 0.01 versus the MS group (n = 4).
Up-regulation of CaSR leads to [Ca2+]i increase through the phospholipase C (PLC) -inositol 1, 4, 5, riphosphate (IP3) pathway. To investigate the effects of Rg1 on increase in [Ca2+]i induced by MS, we incubated the cells with Fluo-3/AM to examine the [Ca2+]i via fluorescence changes. In contrast to the above experiments, IP3R inhibitor (2-APB, 20 μM) and L-type Ca2+channel inhibitor (verapamil, 10 μM) were used in this assay. The results showed that [Ca2+]i was significantly increased by MS and all of Rg1, NPS2143, 2-APB and verapamil inhibited the MS-induced enhancement of [Ca2+]i. However, Rg1 and 2-APB showed a more obvious inhibition effect than NPS2143 and verapamil. In addition, MS significantly increased IP3R protein expression, which was abrogated by Rg1, NPS2143 and 2-APB but not verapamil. These results indicated that CaSR/PLC/IP3 pathway is at least partially involved in the increases in [Ca2+]i induced by MS (Fig. 7).
Fig. 7.

The effect of Rg1 on [Ca2+]i and IP3R expression. A: [Ca2+]i fluorescence assayed by Fluo-3/AM incubation. B: Data on [Ca2+]i fluorescence intensity analyzed with Image Pro Plus 6.0. C: The protein expression of IP3R in cardiomyocytes. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Con group; ##P < 0.01 versus the MS group (n = 4).
3.7. CaSR mediated CaN pathway activation induced by MS
Finally, CaSR agonist (GdCl3) and CaN inhibitor (CsA) were used to examine the regulation of CaSR on CaN pathway activation induced by MS. The results showed that although GdCl3 could increase the cardiac size and [Ca2+]i, and upregulate the CaN pathway, it could not further increase the cardiac size, [Ca2+]i and upregulate CaN pathway when combined with MS. CsA, an inhibitor of CaN, could significantly inhibit the CaN signaling pathway, attenuate cardiac hypertrophy, and moderately regulate [Ca2+]i both in the presence and absence of GdCl3. However, CsA had no effect on CaSR expression neither in the presence or in absence of GdCl3. Combined with the above-mentioned results, we confirmed that up-regulation of Ca2+-dependent CaN/NFAT3 pathway is involved in the CaSR-mediated cardiac injury induced by MS(Fig. 8).
Fig. 8.

CaSR mediated CaN pathway activation induced by MS. A: Cultured cardiomyocytes surface area were measured according to rhodamine-labeled phalloidin staining. B: [Ca2+]i fluorescence assayed by Fluo-3/AM incubation. C: Data of cell surface area. D: [Ca2+]i fluorescence intensity analyzed with Image Pro Plus 6.0. E-H: CaSR, CaN and nucleus NFAT3 protein expression determined according to western blot in cardiomyocytes. Data were presented as the mean ± SD, ∗∗P < 0.01 versus the Con group; ##P < 0.01 versus the MS group; ▲▲P < 0.01 versus the GdCl3 group (n = 4).
4. Discussion
As a major ingredient of P. ginseng, ginsenoside Rg1 has beneficial effects on the immune system, the central nervous system and endocrine system, and especially the cardiovascular system [[27], [28], [29]]. Previous studies demonstrated that ginsenoside Rg1 administration improved cardiac function, alleviated cardiac injury, modulated myocardial energy metabolism, and inhibited the cardiac inflammation and oxidative stress induced by ischemia-reperfusion injury and glucose deprivation [25,30]. Recent studies, including reports from our laboratory, have demonstrated that Rg1 could attenuate cardiac hypertrophy and cardiac remodeling and preserve cardiac systolic and diastolic function against pressure overload, and the mechanism was related to inhibiting TNF-α/NF-κB and enhancing angiogenesis by increasing the expression of HIF-1 and VEGF [26,31]. Intracellular Ca2+ overload plays a crucial role in the transition of cardiac hypertrophy to cardiac remodeling and heart failure. Based on the above studies, the present research further investigated the protective effect of Rg1 on pressure overload-induced cardiac remodeling by focusing on CaSR, Ca2+ and its related CaN pathway. In contrast to previous studies, current studies use in vitro models of increased cardiac after-load via MS in cardiomyocytes and cardiac fibroblasts and demonstrated for the first time that up-regulation of Ca2+-dependent CaN pathway mediated by CaSR contribute to the cardiac hypertrophy and fibrosis induced by MS. Furthermore, Rg1 showed a cardiac protective effect induced by MS through inhibiting CaSR/CaN signaling and deceasing [Ca2+]i.
Hemodynamic overload caused by mechanical stress contributes to the development of cardiac hypertrophy and the transition from compensated hypertrophic state to cardiac remodeling until heart failure [32]. At the cellular level, cardiomyocyte hypertrophy is first dominant response to mechanical stress, while the progress of cardiac remodeling and clinical heart failure is related to myocardial cell degeneration, myocardial fibrosis and loss. In the present study, cardiomyocytes and cardiac fibroblasts were cultured separately to evaluate the effect of Rg1 on cardiac injury. The results showed that Rg1 administration inhibited cardiomyocyte hypertrophy and attenuated the cardiac fibroblast proliferation and fibrosis induced by MS, demonstrating the protective effect of Rg1 on cardiac hypertrophy and fibrosis, which was further shown in an in vivo study, as indicated by the improved cardiac function, decreased cross-sectional diameter and collagen deposition in the AAC group rats. In addition, with the improvement of hypertrophy and fibrosis, Rg1 administration abrogated CaN/NFAT3 signaling activation, which was induced by mechanical stretch. As a hypertrophic signaling pathway, CaN signaling contributes to various cardiac hypertrophy and remodeling models, and the activated mechanism depends on intracellular Ca2+. Under physiological conditions, intracellular Ca2+ for myocardial contractility is provided by Ca2+ entering through the L-type and T-type Ca2+-channels and Na+-Ca2+ exchangers and from the sarcoplasmic reticulum [6]. In hypertrophied myocardium, the relative contribution of these Ca2+-regulating mechanisms changed dramatically, and the CaN activation mechanism was also correspondingly different. Previous studies have indicated that Ca2+ is increased by CaSR and subsequently activates the CaN signaling pathway, thereby contributing to cardiac hypertrophy [12,33]. Increases in intracellular Ca2+ and the resulting activation of Ca2+-dependent signaling pathways in cardiomyocytes have a critical role in the pathogenesis of cardiac hypertrophy. The CaSR inhibitor Calhex 231 ameliorates cardiac hypertrophy and attenuates [Ca2+]i, while the CaSR agonists GdCl3 aggravates cardiac hypertrophy by increasing [Ca2+]i [12,34]. Consistent with previous studies, we showed that, accompanied by cardiac hypertrophy, CaN and [Ca2+]i increased in pressure-overload rat hearts and stretch-treated cardiomyocytes. A previous study showed that MS activated CaSR, which contributed to attenuating vascular calcification in human aortic smooth muscle cells [35]. However, the effect of MS on CaSR in cardiomyocytes and cardiac fibroblasts was not investigated. One novel finding is that CaSR expression was upregulated alone with cardiac hypertrophy induced by MS, which distinguishes our report from previous studies. Moreover, cardiomyocytes from the NPS2143 group subjected to MS showed minimal activation of CaN signaling and [Ca2+]i. Although GdCl3 could increase the cardiac size and [Ca2+]i and upregulate CaN pathways, it could not further increase the cardiac size and [Ca2+]i and upregulate CaN pathways when combined with MS. These results indicate that CaSR-regulated increases in [Ca2+]i and CaN/NFAT3 pathway activation contribute to the cardiac hypertrophy and fibrosis induced by mechanical stress. However, the increase of [Ca2+]i caused by mechanical stretch may be partly due to activation of CaSR since verapamil, an L-type Ca2+ channel inhibitor, also inhibits [Ca2+]i, but has no effect on IP3R protein expression. Indeed, some reports have shown that all L-type Ca2+ channels, capacitive Ca2+ entry, Na+/H+ exchangers, Na+/Ca2+ exchangers and stretch-activated channels contribute to [Ca2+]i and cardiac hypertrophy induced by mechanical stress [20,36,37]. It is not surprising that multiple intracellular mechanisms are responsible for [Ca2+]i overload to orchestrate the hypertrophic response and that these pathways are interdependent. HIMF overexpression increased the cytosolic Ca2+ concentration and activated the CaN, which could be prevented by the L-type Ca2+ channel blocker nifedipine or the CaSR inhibitor Calhex 231 [38], suggesting that a reciprocal yet reinforcing relationship between different Ca2+ activation mechanisms contributes to cardiac injury. Nevertheless, further investigations are still warranted to delineate the mechanisms of the interaction between different calcium channels that are responsible for Ca2+ regulation.
The TGF-β1/Smad signaling pathway is a classical pathway for fibrosis that plays an important role in models of pressure overload-induced cardiac fibrosis [39,40]. CaSR promotes high glucose-induced myocardial fibrosis via Ca2+ activation and the TGF-β1/Smads pathway in cardiac fibroblasts [41]. Accompanied by myocardial fibroblast proliferation and fibrosis induced by phenylephrine, Ca2+/CaN/NFAT signaling was activated, and these effects were abolished by nifedipine (a blocker of Ca2+ influx), BAPTA-AM (an intracellular Ca2+ buffer), and CsA [15]. The current study confirmed previous findings and found that CaSR and CaN signaling contributes to cardiac fibrosis in pressure overloaded rat tissues, and this finding was further confirmed by an in vivo study with a cardiac fibroblast stretch model. Furthermore, Rg1 administration not only inhibited cardiac fibrosis, but also CaN activation, an effect of the CaSR inhibitor NPS2143. These results illustrated that CaSR regulates CaN activation in a mechanical stress-induced cardiac fibrosis model, which is the mechanism underlying the protection of Rg1 on cardiac injury.
We firstly demonstrated that increase of [Ca2+]i and CaN/NFAT3 pathway activation mediated by CaSR contribute to cardiac hypertrophy and fibrosis caused by mechanical stress. The protective effect of Rg1 on mechanical stress-caused cardiac hypertrophy and fibrosis may be partly mediated via inhibiting of CaSR expression, [Ca2+]i elevation and activation of CaN/NFAT3 pathway.
Acknowledgement
The present study was supported by Nation Science Foundation Project (81973553), Guide Planned Project of Liaoning Province (No. 2019-ZD-0617 and JYTJCZR2020077).
References
- 1.Li X., Chu G., Zhu F., Zheng Z., Wang X., Zhang G., Wang F. Epoxyeicosatrienoic acid prevents maladaptiveremodeling in pressure overload by targeting calcineurin/NFAT and Smad-7. Exp Cell Res. 2019 doi: 10.1016/j.yexcr.2019.111716. [DOI] [PubMed] [Google Scholar]
- 2.Zheng R.H., Bai X.J., Zhang W.W., Wang J., Bai F., Yan C.P., James E.A., Bose H.S., Wang N.P., Zhao Z.Q. Liraglutide attenuates cardiac remodeling and improves heart function after abdominal aortic constriction through blocking angiotensin II type 1 receptor in rats. Drug Des Devel Ther. 2019;13:2745–2757. doi: 10.2147/DDDT.S213910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang W.Y., Huo J.Y., Chen C., Chen R., Ge T.T., Chang Q., Hu J.W., Geng J., Jiang Z.X., Shan Q.J. Renal denervation ameliorates post-infarction cardiac remodeling in rats through dual regulation of oxidative stress in the heart and brain. Biomed Pharmacother. 2019;118 doi: 10.1016/j.biopha.2019.109243. [DOI] [PubMed] [Google Scholar]
- 4.Glasenapp A., Derlin K., Wang Y., Bankstahl M., Meier M., Wollert K.C., Bengel F.M., Thackeray J.T. Multimodality imaging of inflammation and ventricular remodeling in pressure overload heart failure. J Nucl Med. 2019 doi: 10.2967/jnumed.119.232488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Y.H., Liu M.N., Li H., Shi S., Zhao Y.J., Wang R., Xu C.Q. Calcium-sensing receptor induces rat neonatal ventricular cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;350:942–948. doi: 10.1016/j.bbrc.2006.09.142. [DOI] [PubMed] [Google Scholar]
- 6.Tfelt-Hansen J., Hansen J.L., Smajilovic S., Terwilliger E.F., Haunso S., Sheikh S.P. Calcium receptor is functionally expressed in rat neonatal ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1165–H1171. doi: 10.1152/ajpheart.00821.2005. [DOI] [PubMed] [Google Scholar]
- 7.Chi J., Wang L., Zhang X., Fu Y., Liu Y., Chen W., Liu W., Shi Z., Yin X. Activation of calcium-sensing receptor-mediated autophagy in angiotensinII-induced cardiac fibrosis in vitro. Biochem Biophys Res Commun. 2018;497:571–576. doi: 10.1016/j.bbrc.2018.02.098. [DOI] [PubMed] [Google Scholar]
- 8.Spurr N.K. Genetics of calcium-sensing--regulation of calcium levels in the body. Curr Opin Pharmacol. 2003;3:291–294. doi: 10.1016/s1471-4892(03)00034-1. [DOI] [PubMed] [Google Scholar]
- 9.Jiang C.M., Han L.P., Li H.Z., Qu Y.B., Zhang Z.R., Wang R., Xu C.Q., Li W.M. Calcium-sensing receptors induce apoptosis in cultured neonatal rat ventricular cardiomyocytes during simulated ischemia/reperfusion. Cell Biol Int. 2008;32:792–800. doi: 10.1016/j.cellbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Lu F., Tian Z., Zhang W., Zhao Y., Bai S., Ren H., Chen H., Yu X., Wang J., Wang L. Calcium-sensing receptors induce apoptosis in rat cardiomyocytes via the endo(sarco)plasmic reticulum pathway during hypoxia/reoxygenation. Basic Clin Pharmacol Toxicol. 2010;106:396–405. doi: 10.1111/j.1742-7843.2009.00502.x. [DOI] [PubMed] [Google Scholar]
- 11.Zheng H., Liu J., Liu C., Lu F., Zhao Y., Jin Z., Ren H., Leng X., Jia J., Hu G. Calcium-sensing receptor activating phosphorylation of PKCdelta translocation on mitochondria to induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol Cell Biochem. 2011;358:335–343. doi: 10.1007/s11010-011-0984-1. [DOI] [PubMed] [Google Scholar]
- 12.Lu M., Leng B., He X., Zhang Z., Wang H., Tang F. Calcium sensing receptor-related pathway contributes to cardiac injury and the mechanism of astragaloside IV on cardioprotection. Front Pharmacol. 2018;9:1163. doi: 10.3389/fphar.2018.01163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu F.H., Fu S.B., Leng X., Zhang X., Dong S., Zhao Y.J., Ren H., Li H., Zhong X., Xu C.Q. Role of the calcium-sensing receptor in cardiomyocyte apoptosis via the sarcoplasmic reticulum and mitochondrial death pathway in cardiac hypertrophy and heart failure. Cell Physiol Biochem. 2013;31:728–743. doi: 10.1159/000350091. [DOI] [PubMed] [Google Scholar]
- 14.Hong S., Zhang X., Zhang X., Liu W., Fu Y., Liu Y., Shi Z., Chi J., Zhao M., Yin X. Role of the calcium sensing receptor in cardiomyocyte apoptosis via mitochondrial dynamics in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Biochem Biophys Res Commun. 2017;487:728–733. doi: 10.1016/j.bbrc.2017.04.126. [DOI] [PubMed] [Google Scholar]
- 15.Wang J., Wang Y., Zhang W., Zhao X., Chen X., Xiao W., Zhang L., Chen Y., Zhu W. Phenylephrine promotes cardiac fibroblast proliferation through calcineurin-NFAT pathway. Front Biosci (Landmark Ed) 2016;21:502–513. doi: 10.2741/4405. [DOI] [PubMed] [Google Scholar]
- 16.Tsai C.Y., Kuo W.W., Shibu M.A., Lin Y.M., Liu C.N., Chen Y.H., Day C.H., Shen C.Y., Viswanadha V.P., Huang C.Y. E2/ER beta inhibit ISO-induced cardiac cellular hypertrophy by suppressing Ca2+-calcineurin signaling. PLoS One. 2017;12 doi: 10.1371/journal.pone.0184153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C.J., Cheng Y.C., Lee K.W., Hsu H.H., Chu C.H., Tsai F.J., Tsai C.H., Chu C.Y., Liu J.Y., Kuo W.W. Lipopolysaccharide induces cellular hypertrophy through calcineurin/NFAT-3 signaling pathway in H9c2 myocardiac cells. Mol Cell Biochem. 2008;313:167–178. doi: 10.1007/s11010-008-9754-0. [DOI] [PubMed] [Google Scholar]
- 18.Saygili E., Rana O.R., Meyer C., Gemein C., Andrzejewski M.G., Ludwig A., Weber C., Schotten U., Kruttgen A., Weis J. The angiotensin-calcineurin-NFAT pathway mediates stretch-induced up-regulation of matrix metalloproteinases-2/-9 in atrial myocytes. Basic Res Cardiol. 2009;104:435–448. doi: 10.1007/s00395-008-0772-6. [DOI] [PubMed] [Google Scholar]
- 19.Finsen A.V., Lunde I.G., Sjaastad I., Ostli E.K., Lyngra M., Jarstadmarken H.O., Hasic A., Nygard S., Wilcox-Adelman S.A., Goetinck P.F. Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin-NFAT pathway. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou N., Li L., Wu J., Gong H., Niu Y., Sun A., Ge J., Zou Y. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem Biophys Res Commun. 2010;397:263–269. doi: 10.1016/j.bbrc.2010.05.097. [DOI] [PubMed] [Google Scholar]
- 21.Fan X., Zhang C., Niu S., Fan B., Gu D., Jiang K., Li R., Li S. Ginsenoside Rg1 attenuates hepatic insulin resistance induced by high-fat and high-sugar by inhibiting inflammation. Eur J Pharmacol. 2019;854:247–255. doi: 10.1016/j.ejphar.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y., Yang C., Zhang S., Li J., Xiao Q., Huang W. Ginsenoside Rg1 protects against non-alcoholic fatty liver disease by ameliorating lipid peroxidation, endoplasmic reticulum stress, and inflammasome activation. Biol Pharm Bull. 2018;41:1638–1644. doi: 10.1248/bpb.b18-00132. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z.M., Li C.B., Liu Q.L., Li P., Yang H. Ginsenoside Rg1 prevents doxorubicin-induced cardiotoxicity through the inhibition of autophagy and endoplasmic reticulum stress in mice. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19113658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin Q., Lin N., Huang H., Zhang X., Cao X., Wang Y., Li P. Ginsenoside Rg1 ameliorates cardiac oxidative stress and inflammation in streptozotocin-induced diabetic rats. Diabetes Metab Syndr Obes. 2019;12:1091–1103. doi: 10.2147/DMSO.S208989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L., Pan C.S., Yan L., Cui Y.C., Liu Y.Y., Mu H.N., He K., Hu B.H., Chang X., Sun K. Ginsenoside Rg1 ameliorates rat myocardial ischemia-reperfusion injury by modulating energy metabolism pathways. Front Physiol. 2018;9:78. doi: 10.3389/fphys.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang F., Lu M., Yu L., Wang Q., Mei M., Xu C., Han R., Hu J., Wang H., Zhang Y. Inhibition of TNF-alpha-mediated NF-kappaB activation by ginsenoside Rg1 contributes the attenuation of cardiac hypertrophy induced by abdominal aorta coarctation. J Cardiovasc Pharmacol. 2016;68:257–264. doi: 10.1097/FJC.0000000000000410. [DOI] [PubMed] [Google Scholar]
- 27.Xu T.Z., Shen X.Y., Sun L.L., Chen Y.L., Zhang B.Q., Huang D.K., Li W.Z. Ginsenoside Rg1 protects against H2O2induced neuronal damage due to inhibition of the NLRP1 inflammasome signalling pathway in hippocampal neurons in vitro. Int J Mol Med. 2019;43:717–726. doi: 10.3892/ijmm.2018.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park S., Ahn I.S., Kwon D.Y., Ko B.S., Jun W.K. Ginsenosides Rb1 and Rg1 suppress triglyceride accumulation in 3T3-L1 adipocytes and enhance beta-cell insulin secretion and viability in Min6 cells via PKA-dependent pathways. Biosci Biotechnol Biochem. 2008;72:2815–2823. doi: 10.1271/bbb.80205. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y., Zou Y., Lin L., Zheng R. Ginsenoside Rg1 activates dendritic cells and acts as a vaccine adjuvant inducing protective cellular responses against lymphomas. DNA Cell Biol. 2017;36:1168–1177. doi: 10.1089/dna.2017.3923. [DOI] [PubMed] [Google Scholar]
- 30.Xu Z., Li C., Liu Q., Yang H., Li P. Ginsenoside Rg1 protects H9c2 cells against nutritional stress-induced injury via aldolase/AMPK/PINK1 signalling. J Cell Biochem. 2019;120:18388–18397. doi: 10.1002/jcb.29150. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y.J., Zhang X.L., Li M.H., Iqbal J., Bourantas C.V., Li J.J., Su X.Y., Muramatsu T., Tian N.L., Chen S.L. The ginsenoside Rg1 prevents transverse aortic constriction-induced left ventricular hypertrophy and cardiac dysfunction by inhibiting fibrosis and enhancing angiogenesis. J Cardiovasc Pharmacol. 2013;62:50–57. doi: 10.1097/FJC.0b013e31828f8d45. [DOI] [PubMed] [Google Scholar]
- 32.Tornatore T.F., Dalla Costa A.P., Clemente C.F., Judice C., Rocco S.A., Calegari V.C., Cardoso L., Cardoso A.C., Goncalves A., Jr., Franchini K.G. A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. Am J Physiol Heart Circ Physiol. 2011;300:H902–H912. doi: 10.1152/ajpheart.00319.2010. [DOI] [PubMed] [Google Scholar]
- 33.Wang L.N., Wang C., Lin Y., Xi Y.H., Zhang W.H., Zhao Y.J., Li H.Z., Tian Y., Lv Y.J., Yang B.F. Involvement of calcium-sensing receptor in cardiac hypertrophy-induced by angiotensinII through calcineurin pathway in cultured neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2008;369:584–589. doi: 10.1016/j.bbrc.2008.02.053. [DOI] [PubMed] [Google Scholar]
- 34.Liu L., Wang C., Sun D., Jiang S., Li H., Zhang W., Zhao Y., Xi Y., Shi S., Lu F. Calhex(2)(3)(1) ameliorates cardiac hypertrophy by inhibiting cellular autophagy in vivo and in vitro. Cell Physiol Biochem. 2015;36:1597–1612. doi: 10.1159/000430322. [DOI] [PubMed] [Google Scholar]
- 35.Calaghan S.C., White E. The role of calcium in the response of cardiac muscle to stretch. Prog Biophys Mol Biol. 1999;71:59–90. doi: 10.1016/s0079-6107(98)00037-6. [DOI] [PubMed] [Google Scholar]
- 36.Molostvov G., Hiemstra T.F., Fletcher S., Bland R., Zehnder D. Arterial expression of the calcium-sensing receptor is maintained by physiological pulsation and protects against calcification. PLoS One. 2015;10 doi: 10.1371/journal.pone.0138833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruwhof C., van Wamel J.T., Noordzij L.A., Aydin S., Harper J.C., van der Laarse A. Mechanical stress stimulates phospholipase C activity and intracellular calcium ion levels in neonatal rat cardiomyocytes. Cell Calcium. 2001;29:73–83. doi: 10.1054/ceca.2000.0158. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S., Wang G., Liu W., Ding W., Dong M., Zheng N., Ye H., Liu J. Hypoxia-induced mitogenic factor promotes cardiac hypertrophy via calcium-dependent and hypoxia-inducible factor-1alpha mechanisms. Hypertension. 2018;72:331–342. doi: 10.1161/HYPERTENSIONAHA.118.10845. [DOI] [PubMed] [Google Scholar]
- 39.Wei W.Y., Zhang N., Li L.L., Ma Z.G., Xu M., Yuan Y.P., Deng W., Tang Q.Z. Pioglitazone alleviates cardiac fibrosis and inhibits endothelial to mesenchymal transition induced by pressure overload. Cell Physiol Biochem. 2018;45:26–36. doi: 10.1159/000486220. [DOI] [PubMed] [Google Scholar]
- 40.Chen H.H., Zhao P., Zhao W.X., Tian J., Guo W., Xu M., Zhang C., Lu R. Stachydrine ameliorates pressure overload-induced diastolic heart failure by suppressing myocardial fibrosis. Am J Transl Res. 2017;9:4250–4260. [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan H., Fan Y., Wang Y., Gao T., Shao Y., Zhao B., Li H., Xu C., Wei C. Calciumsensing receptor promotes high glucoseinduced myocardial fibrosis via upregulation of the TGFbeta1/Smads pathway in cardiac fibroblasts. Mol Med Rep. 2019;20:1093–1102. doi: 10.3892/mmr.2019.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]