

Abstract
DDX5 (p68) is a well-known multifunctional DEAD-box RNA helicase and a transcription cofactor. Since its initial discovery more than three decades ago, DDX5 is gradually recognized as a potential biomarker and target for the treatment of various cancer types. Studies over the years significantly expanded our understanding of the functional diversity of DDX5 in various cancer types and extended our knowledge of its Mechanism of Action (MOA). This provides a rationale for the development of novel cancer therapeutics by using DDX5 as a biomarker and a therapeutic target. However, while most of the published studies have found DDX5 to be an oncogenic target and a cancer treatment-resistant biomarker, a few studies have reported that in certain scenarios, DDX5 may act as a tumor suppressor. After careful review of all the available relevant studies in the literature, we found that the multiple functions of DDX5 make it both a superior independent oncogenic biomarker and target for targeted cancer therapy. In this article, we will summarize the relevant studies on DDX5 in literature with a careful analysis and discussion of any inconsistencies encountered, and then provide our conclusions with respect to understanding the MOA of FL118, a novel small molecule. We hope that such a review will stimulate further discussion on this topic and assist in developing better strategies to treat cancer by using DDX5 as both an oncogenic biomarker and therapeutic target.
Keywords: DEAD-box RNA helicase, DDX5, p68, transcription cofactor, DNA damage, DNA repair, cancer therapeutics, cancer biomarker, cancer target
Overview of the DDX5 research area
DDX5 belongs to the DEAD box RNA helicase protein superfamily [1]. The DDX5 protein was initially named as p68 because its molecular weight (MW) is about 68 kD. However, the name p68 is not an exclusive name for DDX5, because several other proteins with MW around 68 kD also use the term “p68”. This can lead to erroneous citations as we noted during our literature review (see below), and this may be the reason that more recent years of publications used DDX5 instead of p68. However, in this article, we will use both terms interchangeably based on the term used in the corresponding published research article. DDX5/p68 was identified by its specific immunological cross reaction with the SV40 large T antigen [2]. The homology between the two small proteins is only in a small region that resembles the epitope of cross-reaction with an antibody named PAb204, suggesting the functional importance of this region. Complementary DNA analysis of DDX5/p68 revealed extensive amino-acid sequence homology to many proteins including the eukaryotic translation initiation factor, elF4A, which has RNA binding and unwinding activity [2]. Consistently, it was found that the purified p68 protein exhibits RNA-dependent ATPase activity and functions as an RNA helicase [2].
Over the past 3 decades, it has been found that p68/DDX5 is a multifunctional protein and is involved in many processes to maintain cancer cell proliferation, tumor development and malignancy through (i) binding, regulation and processing of various types of RNA [3], and (ii) acting as a coactivator to interact with various types of transcription factors (TFs) to control gene transcription [4-6]. The function and regulatory mechanisms of DDX5 have continuously expanded over time. We have summarized the current and latest discoveries on the DDX5 molecule in Tables 1 and 2 without citing specific references related to these findings. However, the specific references can be found from the GeneGo website at https://portal.genego.com/ under the DDX5 molecule interactions. The corresponding references can be found by clicking on the “icon” marker under the “Interaction info” column.
Table 1.
DDX5 incoming signal network*: Molecules that regulate/act on DDX5
| Mechanism and Effects | Classified Molecules |
|---|---|
| 1. Molecules that acetylate DDX5 for activation | Generic enzyme: p300. |
| 2. Molecules that bind on DDX5 for activation | Generic binding protein: ATAD5, RBM4, SUMO-1. Generic RNA: CCAT1, LINC01116. Transcription factor: FOXC1, NANOG, p53. Protein kinase: LMTK3. |
| 3. Molecules that phosphorylate DDX5 for activation | Protein kinase: c-Abl, MAPKAPK2, p38 MAPK. |
| 4. Molecules that transcriptionally regulate DDX5 for activation | Transcription factor: TCF7L2 (TCF4), VDR. |
| 5. Molecules that co-regulates DDX5 for transcription activation | Generic binding protein: Beta-catenin, TDRD3. |
| 6. Molecules that influence DDX5 expression for activation | Compound: Androstanolone extracellular region. Transcription factor: HIF3A, N-Myc. Receptor ligand: Lutropin. |
| 7. Molecules that bind on DDX5 for inhibition | Generic binding protein: Calmodulin, p14ARF, SUMO-2, SUMO-3. Transcription factor: ESR1 (nuclear). Generic RNA: Hm629797, SLC26A4-AS1. |
| 8. Molecules that phosphorylate DDX5 for inhibition | Protein kinase: PKC. |
| 9. Molecules that SUMOylate DDX5 for inhibition | Generic enzyme: PIAS1. |
| 10. Molecules that influence DDX5 expression for inhibition | Receptor ligand: FGF1, MIF. Generic binding protein: ZMF331. |
| 11. miRNA binds on DDX5 for inhibition | Generic RNA: microRNA 134, microRNA 154, microRNA 302a, miR-1-3P, miR-106b-5p, miR-147-3p, miR-17-5p, miR-200a-3p, miR-205-5p, miR-206-3p, miR-218-5p, miR-33a-5p, miR-5590-3p. |
| 12. Molecules that methylate DDX5 with unspecified effects | Generic enzyme: PRMT5. |
| 13. Molecules that bind on DDX5 with unspecified effects | Generic binding protein: 14-3-3 beta/alpha, 14-3-3 gamma, 14-3-3 sigma, 14-3-3 zeta/delta, ACTB, ADA2, AKAP8, Alpha-1 catenin, Alpha-actinin 4, ALY, AOF1, ARVCF, ASCC2, BAF155, BAF60A, BBLF2-BBLF3 (HHV4), Beta-arrestin2, Brca1, BRD3, BRD4, BRD6, BRD7, c-IAP2, Cab45, Casein kinase II, beta chain (Phosvitin), CBX4, CENP-A, ChAF1 subunit A, CIP29, COPS6, CPEB1, Cullin 1, Cullin 2, Cullin 3, Cullin 4A, Cullin 4B, Cullin 5, DGCR8, DLC1 (Dynein LC8a), DnaJB9, DNAJC11, E4/orf4 (HAdV5), EED, EKN1, EWS, FAT10, FMIP, FMR1, FUS, Gamma-synuclein, GBL, GRB2, Histone H1.0, hnRNP A1, hnRNP H1, HOXB-AS3, HZwint-1, IQGAP1, KBTBD4, KIN17, Ku70, Lamin A/C, LEDGF/p52, LSm4, MAML1, MAP1LC3A, MAP1LC3B, Matrin-3, MEX3C, Mi-2 beta, MKI67IP, MMS19L, MTA2, NDRG1, NEDD8, Nesprin 1, Nsp1 (SARS-CoV-2), Nucleolin, Nucleolysin TIAR, Nucleophosmin, p160, p21, PC4, PHLDA3, PQBP-1, Protein p8, Prp8, PTBP1, Rab5ip, RBM16, RBM46, RBM5, RBM7, RP42 (Scro), RPS6, RUFY3, SAM68, SF3A2, SHISA9, Sin3A, SKIV2L, SLM-1, SMARCAD1, SMN, SNRPA, XFm300, SRRF, SRRM1, TFIIS, TIP120A, TNNT1, TNRC6A, TNRC6B, Tob1, TRAF1, U1-70K, U2AF-65, Ubiquitin, UBR4, VHL, ZC3HAV1, ZFP385, ZFYVE21. Compound: 4-Aminophenyl arsonous acid intracellular, Phenylarsonous acid analogs. Transcription factor: AIRE, CREB4, CTCR, DPF2, ERG, EWSR1-FLI1 fusion protein, FOXK2, FUS/DDIT3 fusion protein, HMGB1, HMGI/Y, hnRNP K, MTA1, NF-X1, NK31, NP220, NR2F1, NRB54, PU.1, RARalpha, SMAD3, SMAD5, SPI-B, TARDBP (TDP43), TBX3, TFCP2, X protein (HBV-C), YB-1, ZNF335, ZNF768. Receptor ligand: Amphiregulin, HDGF. Protein kinase: APEG1, Aurora-A, BRD2, c-Raf-1, CDK1 (p34), FAK1, GSK3 beta, IKK-epsilon, MST3, NIK (MAP3K14), p70 S6 kinase2, p90RSK1, PINK1, PKX-alpha, RIPK3. Generic enzyme: APEX2, BCDIN3, BRG1, DDX17, DDX3X, Dyskerin (NAP57), eIF2C2 (Argonaute-2), eIF4A2, ERCC6, GSTK1, HDAC1, HUTH, JAB1, JMJD6, LSD1, MBNL1, MGMT, PARG, PB1 polymerase 1 (Influenza A virus), PHF10, Pin1, PM/SCL-100, RBBP4 (RbAp48), RG9MTD1, RING2, RNF20, SDOS, Sirtuin6, Sirtuin7, STARING, TOP1, TRIM25, WWP2. RAS superfamily: ARF6. Generic protein: Bles03, BRD9, EBNA-LP (HHV4), espF (E. coli), FBXW4, ICP8 (HSV1), NIF3L1BP1, NS3 (DENV2), NS5 protien (DENV2), p15 (PAF), PA (Influenza A virus), PAF1, PB2 (Influenza A virus), Seipin, UL54 (HSV1), WTAP. |
| Voltage-gated ion-channel: CACNB2. Generic receptor: Caspr1, CD4, GABARAP, GABARAPL1, LIMR, Peripherin, SEC62. Generic RNA: CCAT2, LINC00920, LOC100499467, MEG3, MIR222HG, VTRNA1-1. Receptor with enzyme activity: DR5, TGF-beta receptor type I, TrkA. Generic phosphatase: F16P. Generic protease: FAM111A, Otubain1, OTUD3, PSMA3. Ligand-gated ion-channel: GRID1. Transporter: Karyopherin alpha 2. Lipid kinase: PI3K cat class IA (p110-alpha). Protein phosphatase: PP1-cat, PP2A cat (alpha). Lipid phosphatase: PTEN. Generic channel: TRPM7. | |
| 14. Molecules that AMPylate DDX5 with unspecified effects | Generic binding protein: HYPE. |
| 15. Molecules that phosphorylate DDX5 with unspecified effects | Protein kinase: p38alpha (MAPK14), PKC-delta, TLK1. |
| 16. Molecules that dephosphorylate DDX5 with unspecified effects | Protein phosphatase: PP2A catalytic. |
| 17. Molecules that cleave DDX5 with unspecified effects | Generic protease: Granzyme A, PR (HIV-1), Thrombin. |
| 18. Molecules that transport DDX5 with unspecified effects | Transporter: CRM1. |
| 19. Molecules that transcriptionally regulate DDX5 with unspecified effects | Transcription factor: AML1 (RUNX1), Androgen receptor, AP-2, ATF-2, Bcl-6, c-Myc, CREB1, E2A, E2F1, E2F4, ELF2, ETS1, FOXM1, GATA-2, GCR, HES1, HNF4-alpha, KLF4, LBP9, Lef-1, LMO2, Max, Oct-3/4, OTX2, PLZF/RARalpha fusion protein, REV-ERBalpha, REX1, RXR, SOX17, SOX2, SP1, SP3, STAT3, STAT5, YY1, ZFZ, ZNF143, ZNF263, ZNF281. |
| 20. Molecules that transcriptionally co-regulate DDX5 with unspecified effects | Generic binding protein: AEBP1, HEXIM1, SNAP43. Generic enzyme: CARM1, EZH2, PAD4, PLU-1, PRMT1. Protein kinase: CDK9. Transcription factor: MLL1 (HRX), NOTCH1 (NICD), Tat (HIV-1), TIF1-beta. Generic protein: STRA8, ZGLP1. |
| 21. miRNAs that bind to DDX5 transcripts (i.e., DDX5 mRNA) with unspecified effects | Generic RNA: hsa-miR-3169 mature, hsa-miR-3655 mature, hsa-miR-4297 mature, msyutr miT-4317, microRNA 708, miR-1270, miR-142-5p, miR-185-5p, miR-188-5p, miR-199a-5p, miR-199b-5p, miR-205-3p, miR-2054, miR-20a-5p, miR-21-5p, miR-2113, miR-23b-3p, miR-23c, miR-3123, miR-3128, miR-3140-3p, miR-3163, miR-3180-5p, miR-3202, miR-325-5p, miR-338-5p, miR-340-5p, miR-3652, miR-3663-3p, miR-3668, miR-3671, miR-3925-5p, miR-422a, miR-4262, miR-4306, miR-4311, miR-4423-5p, miR-485-5p, miR-509-3-5p, miR-509-5p, miR-511-5p, miR-519b-3p, miR-519c-3p, miR-548a-5p, miR-548b-5p, miR-548c-3p, miR-548c-5p, miR-548d-5p, miR-548i, miR-548k, miR-548I, miR-548w, miR-548y, miR-559, miR-583, miR-586, miR-607, miR-610, miR-613, miR-620, miR-622, miR-628-3p, miR-636, miR-641, miR-645, miR-875-3p, miR-885-5p, miR-92a-3p, miR-93-5p, miR-944, mmu-miR-466q mature. |
Information was retrieved from the GeneGo database at https://portal.genego.com/and organized by the authors.
Relevant references for individual findings can be found in the database from the corresponding molecule by clicking the con under the “Interaction info” section.
Table 2.
DDX5 outgoing signal network*: DDX5 downstream targets
| Mechanism and Effects | Classified Molecules |
|---|---|
| 1. Molecules that DDX5 binds on for activation | Transcription factor: Androgen receptor, c-Fos, CTCF, ESR1 (nuclear), GATA-6, MYOD, NF-AT5, NF-kB1 (p50), NOTCH1 (NICD), p53, RBP-J kappa (CBF1), RUNX2, SRF, STAT3, c-Myc, HES1. Generic binding protein: Beclin 1, b-catenin, Cyclin A2, Cyclin B2, Cyclin D2, hnRNP A2, Rev (HIV1), SUZ12, Bax, Bcl-XL, CDC20, Cyclin D1. Generic enzyme: CBP, DROSHA, G9a, RAD18, Rad50, XRN2. Generic RNA: microRNA let-7a-1. Generic protein: NSSB (HCV). |
| 2. DDX5 co-regulated molecules for transcription activation | Protein kinase: AKT1. Generic binding protein: Bax, Bcl-XL, CDC20, Cyclin D1, Mcl-1, <TS1 (S100A4), ORC6L, p21, PUMA, Stanniocalcin 1. Transcription factor: c-Myc, HES1, MEF2C, MYOG, NFAT-90, SLUG, SNAIL1. Receptor ligand: IL-1 beta, IL-20, VEGF-A, WNT5A. Generic receptor: IL-2R alpha chain. Generic protease: Kallikrein 3 (PSA). Generic RNA: microRNA 181b-1, microRNA 21. Metalloprotease: MMP-2, MMP-9. |
| Transporter: NGAL. Protein Kinase: PLK1. Generic protein: TMEM26. Generic enzyme: TMEM55B. | |
| 3. Molecules that DDX5 binds on for inhibition | RAS superfamily: H-Ras. Generic RNA: microRNA 182, microRNA 196a-2, microRNA 34a. Generic binding protein: Myelin basic protein, Sequestosome 1 (p62). |
| 4. DDX5 co-regulated molecules for transcription inhibition | Transcription factor: C/EBPdelta, KLF4. Voltage-gated ion-channel: CLCA2. Generic binding protein: Ep-CAM, p15. |
| 5. Molecules that DDX5 competes with for inhibition | Generic binding protein: Axin1. |
| 6. Molecules that DDX5 binds on with unspecified effects | Generic enzyme: AARS, ARSL, ADAR1, AMP deaminase 1, ATP8B3, C1TM, C6orf150, CGAT1, CHD5, CYP2A13, CYP4B1, DDX1, DDX17, DDX21, DDX23, DDX24, DDX9, DHX8, DNA ligase IV, eIF2C2 (Argonaute-2), Fibrillarin, FTHC, FTSJ3, HDAC1, HDC, KAR, Lactoperoxidase, METTL3, MOZ, MPG, NS5 (JAEV), PAM, Parvulin 14, PB1 polymerase 1 (Influenza A virus), PDE1C, PDE3B, PDE8B, PISD, POLR2A, PON3, RNA polymerase II, RNMT, RPP38, SATL1, SIA10, SMURF1, TPH1, UGT2A1, XPB, XYLT1. Generic binding protein: ACTL7A, Amyloid beta 42, ANKS1, Ankyrin-B, APC protein, ARC, ARID4A, ASB1, BAT2, C2orf63, CacyBP (SIP), CAMSAP1, CAPZ beta, CAS-L, CBP80, CCAR1, CENP-F, CMTR1, CNOT1, CNTN5, CRFG (NGB), Cyclin A1, Cyclin D3, Cyclin E, Cyclin T1, D52, DAZ2, DEDD, DOM3Z, DZIP, E6 protein (HPV16), ECM29, EED, EEF1E1, eIF2C1 (Argonaute-1), ELAVL1 (HuR), Epiplakin, FAM136A, FAT1, FAT2, FLJ43093, FRYL, GAS11, Gemin5, Hampin, HC II, Hdj-2, Histone H2AX, hnRNP C, HYOU1, IGF2BP3, IRF2BP1, KBTBD5, L-Ficolin, Lba1, LMAN1L, MBD3, MCM10, Mi-2, Mi-2 alpha, Midasin, MKLP1, MYH9, MYL6, Myoglobin, Myosin VIIA, NANOS3, NIPBL, NLRC5, Nop56, Nop58, NP (Influenza A virus), Nucleolin, Oviductin, PABPC1, PES1, Plectin 1, PLEKHA4, PML, RENT1, RENT2, RENT3B, RHOBTB1, RPL10A (CsA-19), RPL13, RPL17, RPL21, RPL26, RPL4, RPL6, RPL7, RPL7A, RPLP0, RPS19, RPS3, RPS3A, RPS9, SAMD13, SEC23A, Sec24C, Septin 4, Serpin B12, SETBP1, SFRS3, SHRM, SMG7, SNX11, SON, SSX5, TANC, Tangerin A, Tau (MAPT), TCP1-eta, TPX2, TRAF5, TSPEAR, ZBTB12, ZNF294, ZNF462, ZNF699, ZNF77, ZNF804B, ZSWIM3. Metalloprotease: ADAM12, SUPT16H. Protein Kinase: AKT2, AKT3, ATM, ATR, Aurora-C, CaMK II alpha, CDKL5, HIPK3, JNK1 (MAPK8), JNK3 (MAPK10), MEK4 (MAP2K4), MYLK1, p90RSK2 (RPS6KA3), RIPK2, SIK, SLK, TAK1 (MAP3K7), Titin, TTK. Ligand-gated ion-channel: ANO7. Generic channel: ATP8B4. Generic protein: C (HCV), C13orf39, C14orf104, C21orf54, C5orf35, XXSX62, Core protein (JAEV), CSPP1, DKFZP434B061, FAM33A, FBF1, FBXW10, FLJ10986, FLJ13231, FLJ14803, KIAA0774, KIAA1109, KIAA1211, KIAA1704, KIAA1731, LL5beta, LSMD1, LZTS2, MLLT11, MNS1, MUM1, NS4B (HCV1b), OSBPL7, SNM1, SPECC1, ST20, SYCP2L, TMEM98, VN1R17P, WDR48. Transcription factor: c-Jun, EBF2, ELYS, FALZ, Fra-1, HEY2, MEIS1, NF45 (ILF2), PXR, RUNX3, SMAD1, SMAD2, SMAD4, SMAD7, STAT1, TBP, TIEG1, TLE4, UNR, ZNF145, ZNF384. Lipid kinase: DGK-zeta. |
| eneric DNA: DNA. Receptor ligand: Galanin, IFN-omega, LAMA2, PACAP, TNF-alpha. GPCR: Gpr123. Generic RNA: HOTAIR, Hotair (mouse), microRNA 145, microRNA 15-1, microRNA 214, microRNA517B, microRNA let-7g, miR-483-5p, mRNA intracellular, RNA. Generic receptor: IL7RA, RELT. | |
| Transporter: IPO11, Karyopherin alpha 6, Rabphilin-3A, SLC21A2, SLC4A2. Regulators (GDI, GAP, GEF, etc.): IQSEC2, LARG, RGS5, SOS2. Voltage-gated ion-channel: Nav1.6. Generic protease: NS3 (JAEV), S1P, SFRS2 (SC-35), Usp24, USP6, USP7. | |
| Generic kinase: PKM2. Protein phosphatase: PP2A cat (beta), SHP-1. RAS superfamily: Rab-32, Rab12, RAB2B. | |
| 7. DDX5 transcriptionally co-regulated molecules with unspecified effects | Metalloprotease: ADAM-TS20. Generic enzyme: AMDHD2, CL6S, HEXA, HHIPL2, Spam1, TERT, ZNRF3. Generic channel: ATP6V1H. Transcription factor: BACH2, MESP2, RARgamma, RLF, Scieraxis, SOX8, TSC-22. Generic binding protein: Cadherin 9, CDC4L, MAEL, MRLPL32, Prickle-1, SERPINB8, SP3A3, SLAP-2, SPAG16, SSX2IP, TFF1, Thrombospondin 4. |
| Ligand-gated ion-channel: GABA-A receptor gamma-2 subunit, GluR6. Receptor ligand: IGF-2, Vitronectin. Voltage-gated ion-channel: KCNH7, KCNQ5. Protein kinase: KSR1. Receptor ligand: LAMB3. Generic protein: LAMP1, MAGEB16, ODZ4, Paralemmin, STOX2. Generic receptor: Neurexin 1-alpha, Neurexin 1-beta, PTCH2. Generic protease: PRSS11 (HtrA1). RAS superfamily: RAB40B. |
Information was retrieved from the GeneGo database at https://portal.genego.com/and organized by the authors.
Relevant references for each finding can be found in the database from the corresponding molecule under the “Interaction info”.
Based on the information summarized in Table 1, DDX5 can be regulated by a lot of different molecules including compound, generic binding protein, generic channel, generic enzyme, generic phosphatase, generic protease, generic protein, generic receptor, generic RNA, ligand-gated ion channel, lipid kinase, lipid phosphatase, protein kinase, protein phosphatase, RAS superfamily, receptor ligand, receptor with enzyme activity, transcription factor, transporter, and voltage-gated ion-channel.
On the other hand, DDX5 also has a lot of downstream targets. As summarized in Table 2, this includes G protein-coupled receptor (GPCR), generic binding protein, generic channel, generic enzyme, generic kinase, generic protease, generic protein, generic receptor, generic RNA, ligand-gated ion-channel, lipid kinase, metalloprotease, protein kinase, protein phosphatase, RAS superfamily, receptor ligand, regulators (GDI, GAP, GEF, etc.), transcription factor, transporter, and voltage-gated ion-channel.
Importantly, most of these findings summarized in Tables 1 and 2 for the DDX5/p68 upstream and downstream specific effects of DDX5/p68 have not been functionally defined. Further elucidating these effects and characterizing the detailed mechanism of action (MOA) will fill a major knowledge gap.
Based on these findings, DDX5/p68 has been identified as a potential oncogenic biomarker and target for the treatment of various cancer types. However, a few studies reported that DDX5 may also possess a tumor suppressor property in certain conditions acting as a favorable cancer biomarker. After carefully reviewing recent and up-to-date studies in the literature, we found that the multiple functions of DDX5 make DDX5 a superior oncogenic biomarker and target for cancer therapy. In this review, we will first begin by discussing the new role of DDX5 in facilitating genome DNA repair, a key role to unify DDX5 as a superior cancer biomarker and target. Then, we will update the studies relevant to individual cancer types to further support our conclusion drawn from literature review. Finally, we will summarize the relationship of DDX5 with anticancer drugs and cancer treatment resistance. We hope that this review will stimulate further discussion to create better strategies for treating cancer by using DDX5 as an oncogenic biomarker and therapeutic target.
The emerged new role of DDX5 in genome DNA repair
The early evidence for p68/DDX5 involvement in a tumor co-suppressor role comes from the fact that p68 acts as a coactivator of the p53 target gene, p21WAF1 [7]. However, later studies demonstrated that while p68 is critical for p53-mediated transactivation of the cell-cycle arrest gene p21WAF1, p68 is dispensable for induction of several pro-apoptotic genes in response to DNA damage [8]. Instead, p68 depletion strikingly inhibits the recruitment of p53 and RNA Pol II to the p21 promoter but not to the apoptotic Bax or PUMA promoters [8], indicating the selective effect on p21 induction (for cell arrest) versus on Bax and PUMA induction (for cell apoptosis). In other words, this finding revealed that p68 depletion may induce apoptosis. Furthermore, using an inducible p68 knockout mouse model, p68 depletion selectively inhibits p21 induction in several tissues, while in the bone marrow, p68 depletion results in an increased sensitivity to γ-irradiation and increased apoptosis [8]. These authors concluded that their studies highlight a novel function of p68 as a modulator to make the decision between p53-mediated growth arrest (e.g., for survival by DNA repair) and apoptosis [8]. Supportively, the early study from Bates, et al. actually observed that cells transfected with p68 siRNA had a somewhat higher background of apoptosis before doxycycline induction of p53 expression [7]. This supports the theory that targeting p68/DDX5 would shift cancer cells from a growth arrest-DNA repair state into an apoptotic state.
The role of DDX5 in the growth arrest-based DNA repair versus apoptosis is emerging. Recent studies have defined a role for DDX5 in damaged DNA repair in addition to its various RNA processing and transcription coactivator roles. Specifically, formation of the aberrant transcription-associated RNA:DNA hybrid (R-loop) formation often causes catastrophic conflicts during replication, resulting in DNA double-strand breaks (DSBs) and genomic instability. Mersaoui et al. demonstrated that DDX5 is a crucial player in R-loop resolution [9]. Specifically, they found that recombinant DDX5 resolves R-loops in an ATP-dependent manner in vitro, leading to R-loop degradation by the XRN2 exoribonuclease. DDX5-deficient cells accumulate R-loops with local reactive oxygen species (ROS)-induced DNA damage and cause spontaneous DNA DSBs (and the unrepaired DNA DSBs will induce cell apoptosis) and hypersensitivity to replication stress [9]. In short, the study revealed that cellular depletion of DDX5 will cause DNA damage accumulation and excessive genomic instability and as a result, cells will go into apoptosis. The follow-up studies from this group further demonstrated that DDX5 was excluded from DSBs in a transcription- and ATM activation-dependent manner; DDX5 deficiency led to delayed exonuclease 1 and replication protein A (RPA) recruitment to laser irradiation-induced DNA damage sites, resulting in homologous recombination repair defects [10]. Thus, these authors defined a role for DDX5 in facilitating the clearance of RNA transcripts overlapping DSBs to ensure proper DNA repair.
The involvement of DDX5 in DNA repair can employ distinct mechanisms. The BRCA2 tumor suppressor is a DNA DSB repair factor essential for maintaining genome integrity and inhibiting cancer development. BRCA2-deficient cells spontaneously accumulate DNA-RNA hybrids, increasing genome instability and cell malignancy. Sessa et al. demonstrated that DDX5 is a BRCA2-interacting protein and associates with DNA-RNA hybrids at the vicinity of DSBs, which is enhanced by BRCA2-mediated stimulation of DDX5 DNA-RNA hybrid-unwinding activity [11]. DDX5-BRCA2 interaction favors DSB repair by homologous recombination, and DDX5 depletion leads to a genome-wide accumulation of DNA-RNA hybrids particularly enriched at DSBs [11]. The studies indicated that DDX5 can help BRCA2 repair DNA DSBs.
However, if a substantially damaged DNA will not be effectively repaired, for example due to DDX5 depletion, cancer cells will have to go into apoptosis. In our view, increasing cancer cell malignant potential or cancer cell death (apoptosis) are the two possible outcomes of a single process, with the decision made by key regulatory proteins including DDX5/p68 which is dependent on the amount of DNA damage and genomic instability intensity (i.e., increased malignant potential with limited DNA repair versus death if the genomic damage cannot be repaired).
Does the DDX5’s DNA repair function play a role in pancreatic ductal adenocarcinoma?
Pancreatic cancer, especially the most common type, pancreatic ductal adenocarcinoma (PDAC, >85%), is known to be a highly malignant and extremely difficult-to-treat cancer and represents ~8% of all cancer-related deaths [12]. Pancreatic cancer has the lowest 5-year survival rate (10%) among all cancers, with the survival in advanced and metastatic disease being only ~3% [12]. Unfortunately, both pancreatic cancer incidence and mortality continues to increase annually [12]. Furthermore, pancreatic cancer is expected to become the second largest cause of cancer-related death by 2030 [13]. These facts indicate that this highly malignant disease should have high genomic instability. The question is: what could the effect of DDX5’s DNA repair function be on the pancreatic cancer malignant behavior?
Because the studies on DDX5’s DNA repair function mainly employed cancer cells (U2OS osteosarcoma, HeLa, K562), this raises an intriguing possibility that at least in certain types of cancer, if the cancer DNA instability intensity is high (i.e., having high potential malignancy), DDX5 may play a role in stabilizing cancer DNA instability in some degree (i.e., inhibit malignant transformation) before treatment. This notion is consistent with a recent (and also the first) study on DDX5 with PDAC [14]. The study was to determine the DDX5 expression using immunohistochemistry (IHC) on a tissue microarray (TMA) that was constructed with surgery-obtained tissues from 230 PDAC patients who have no preoperative treatment (e.g., no chemotherapy and/or radiotherapy). The IHC data presented in Figure 2 indicated that “DDX5 was stained positive in the nuclei of carcinoma cells (A) and negative in carcinoma cells (B)” [14]. Based on their defined criterion that “DDX5 expression was considered high when more than 50% of the cells were stained and low when less than 50% of the cells were stained”. DDX5 expression was low in 148 cases (64.3%) and high in 82 cases (35.7%) [14]. After analysis of the data with the corresponding clinical information, these authors concluded that low DDX5 expression was typically associated with an advanced pT factor (pT2-pT3: tumor size, >20 mm), lymphatic involvement, advanced tumor-node-metastasis (TNM) stage (stages IIB, III, and IV), and venous involvement [14]. The median overall survival time of patients in the low versus high DDX5 expression groups was 24 versus 38 months, respectively [14].
Figure 2.
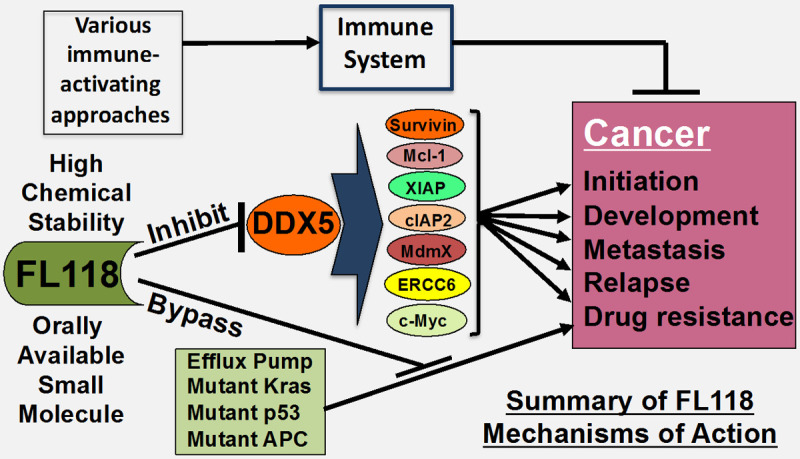
The experiment and observation-based diagram of the FL118’s mechanisms of action (MOA). (1) Use of FL118 affinity purification approach, followed by alternative confirmation, DDX5 was identified as the FL118 direct biochemical target, which was demonstrated being very important for human PDAC and CRC tumors. Our studies relevant to PDAC and CRC revealed that the previously identified FL118 downstream targets (e.g., survivin, Mcl-1, XIAP, cIAP2, etc.) are the downstream targets of DDX5. Additionally, FL118 could bypass additional treatment resistant factors (efflux pump proteins ABCG2 and Pgp, mutated Kras, mutated p53 and mutated APC). (2) Our collaboration studies also revealed that the bone marrow stromal cells-rendered MM cell resistance to CAR T cells or NK cells can be abrogated by FL118 treatment; therefore, it would be attractive for FL118 in combination with immunotherapy for treatment of resistant cancers.
Given that the majority of publications in the past defined a role of DDX5/p68 in promoting cancer development, metastasis and treatment resistance [3-6], this is a surprising result. However, the additional role of DDX5/p68 in DNA repair working together with either p53 [8], BCRA2 [11] or other factors (e.g., ATM) [9,10], may at least partially explain such a surprising result (see below for more discussion). When a cancer type (e.g., PDAC) has a high malignant potential (i.e., higher DNA instability intensity) before treatment, DDX5 may delay the malignant transformation and tumor progression of PDAC by stabilizing the cancer cell genome through facilitation of DNA repair. However, because DDX5/p68 can also control and regulate various RNA processing (e.g., miRNA, lncRNA), ribosome biogenesis and oncogene transcription, downregulation of DDX5 expression and/or inhibition of DDX5 function will not only induce massive DNA damage (beyond the repair limit) but also induce strong apoptotic signaling. Consistent with this notion, previous studies demonstrated that after non-DDX5-targeting drug treatment, either with camptothecin or with heat shock protein 90 (HSP90) inhibitors, the surviving cancer cells always have high DDX5, while the apoptotic cells always have low DDX5 (refer to the “DDX5, anticancer drug and cancer treatment resistance” section below for more information) [15,16]. This significant finding suggests that DDX5 is a treatment resistant factor. Additionally, it is worthy to note that the above-discussed PDAC study excluded the cancer samples from the PDAC patients who have preoperative treatment [14]. This would abrogate a full evaluation of DDX5 as a non-DDX5 targeting drug resistant factor. Furthermore, these authors used percentage cell positivity or negativity as a criterion to separate DDX5 high or low in their IHC study without considering DDX5 expression intensity in the PDAC cells [14]. This may be another weakness in their study, which could make the study incomplete and/or inaccurate, which left a gap for further studies to get a clear conclusion.
Additionally, as we pointed out previously, p68 is not an exclusive name for DDX5, because several other proteins with the MW around 68 kD also use the “p68” term. We should be cautious to avoid confusing citations for DDX5. For example, the double-stranded RNA-dependent protein kinase (p68), an interferon-inducible protein kinase, was cited by the authors of their DDX5 PDAC paper to support their study results (Reference 10 in their paper) [14].
There are examples to support that high expression of an oncogenic and drug target protein can act as a favorable patient survival factor, although in most cases the mechanism is not fully understood. For example, analysis of TCGA data indicated that high expression of HIF2α in clear cell renal cell carcinoma (ccRCC) is associated with favorable patient survival. However, studies have demonstrated that targeting HIF2α for ccRCC treatment is highly effective and HIF2α is a good target for drug development to treat ccRCC tumors [17].
DDX5 in breast, cervical and endometrial cancer
In contrast to the PDAC’s incomplete and controversial study discussed above, review of DDX5/p68-relevant studies from breast, cervical and endometrial cancer in the literature indicated a consistent conclusion that p68/DDX5 is a cancer biomarker and target for cancer prognosis and treatment. We briefly summarize the observations below from the most relevant publications.
The p68/DDX5 signaling and function in breast cancer were recently reviewed [18]. Here, we focus on DDX5 as a breast cancer biomarker and target. Using an integrative biology study (quantitative proteomics, global microRNA profiling, and biochemical characterization of cell lines and tissues) of DDX5 in breast cancer, Wang et al. found that (1) the expression of DDX5 protein increased progressively from luminal to basal breast cancer cells, and correlated positively with that of CD44 in the basal subtypes; and (2) IHC analyses of tissue microarrays containing over 200 invasive human ductal carcinomas indicated that DDX5 was upregulated in the majority of malignant tissues, and its expression is strongly correlated with those of Ki67 and EGFR in the triple-negative breast cancer (TNBC) tumors [19]. These authors further showed that (3) DDX5 regulated a subset of microRNAs including miR-21 and miR-182 in basal breast cancer cells; (4) knockdown of DDX5 resulted in reorganization of actin cytoskeleton and reduction of cellular proliferation in parallel with miR-182 downregulation; and (5) treatment with miR-182 inhibitors resulted in morphological phenotypes resembling those induced by DDX5 knockdown [19]. After pathway and network analyses with bioinformatics tools, these authors concluded that their results reveal a new functional role of DDX5 in breast cancer via the DDX5-->miR-182-->actin cytoskeleton pathway, and suggest the potential clinical utility of DDX5 or miR-182 in the theranostics of breast cancer [19].
Using breast cancer cells as studying models, Mazurek et al. found that (1) DDX5 plays an important role in G1-S-phase transition by directly regulating DNA replication factor expression through promoting the recruitment of RNA polymerase II to E2F-regulated gene promoters; (2) the DDX5 locus is frequently amplified in breast cancer; and (3) breast cancer cells with DDX5 amplification show much more sensitivity to DDX5 depletion for treatment (e.g., trastuzumab) than cells without DDX5 amplification [20], indicating a role of DDX5 in cell resistance and proliferation. These authors proposed that given the high frequency of DDX5 amplification in breast cancer, DDX5 is a promising candidate for targeted therapy of breast tumors. Here, we should point out that the finding of DDX5 helping to correct DNA replication [20] is consistent with the role of DDX5 in DNA repair as shown in recent studies [9,10].
Lyer et al. showed that (1) elevated p68 expression in a cohort of human breast cancers is strongly associated with elevated levels of the oncogenic Polo-like kinase-1 (PLK1); (2) patients expressing detectable levels of both p68 and PLK1 have a poor prognosis; and (3) in the absence of a functional p53, p68 stimulates the expression of PLK1 at basal levels and also in response to the clinically relevant drug, etoposide, while silencing of p68 expression downregulates PLK1 gene expression [21]. These authors also found a competitive/opposing regulation of PLK1 by p68 versus by p53: While the association of p68 with the PLK1 promoter is irrespective of the p53 status, p68 recruitment to the PLK1 promoter is stimulated by etoposide in cells lacking p53 [21].
Given that nuclear accumulation of β-catenin is important for cancer development, it was found that p68 can be transcriptionally upregulated by Wnt/β-catenin signaling in breast cancer cells [22]. This finding further strengthens the involvement of DDX5/p68 in breast cancer progression. Furthermore, a recent study reported that using both proteomic and genomic approaches, DDX5 was found to be the most enriched Fra-1-interacting protein and involved in the Fra1 transcriptional network [23]. This is important because it is known that Fra-1 (a member of the AP-1 family) is overexpressed in TNBC and plays a crucial role in breast cancer growth. These authors demonstrated that (1) DDX5 expression enhances Fra-1 transcriptional activity and potentiates Fra1-driven cell proliferation; (2) the DDX5-targeting gene signature predicts poor clinical outcome in breast cancer patients; and (3) the DDX5 protein level was higher in triple-negative basal-like tumors than in non-basal-like tumors, including luminal A, luminal B, and Her2-enriched subtypes [23]. Collectively, the authors stated that they revealed a role for DDX5 as a regulatory protein of Fra-1 signaling and found that DDX5 is a potential therapeutic target for TNBC.
In cervical cancer, it was shown that (1) p68 mRNA and protein levels are significantly enhanced in cervical cancer cell lines (CaSki, HPV18-positive HeLa, HPV16-positive SiHa and HPV-negative C-33A) compared with the human keratinocyte HaCaT cell line; (2) overexpression of p68 induced an elongated and spindle-shaped morphology in CaSki cells; (3) upregulation of p68 increased the expression of α-smooth muscle actin, vimentin and fibronectin, while epithelial marker E-cadherin was significantly decreased; (4) overexpression of p68 markedly enhanced CaSki cell migration capacity; and (5) knockdown of p68 partially reversed TGF-β1-induced changes in epithelial-mesenchymal transition (EMT) markers and cell morphological changes [24]. Based on these observations, the authors concluded that p68 promotes EMT in cervical cancer cells.
In endometrial cancer (EC), it was reported that (1) the expression of hepatoma-derived growth factor (HDGF) and DDX5 was positively correlated in EC tissues; (2) high expression of DDX5 constituted an unfavorable factor with respect to the clinicopathological characteristics of EC tissues, and the high expression of HDGF and DDX5 led to a worse prognosis for patients with EC; (3) knockdown of HDGF expression significantly decreased EC cellular proliferation, migration, invasion in vitro and tumorigenesis and metastasis in vivo, while overexpression of DDX5 reversed the suppression of shHDGF; and (4) mechanistically, the authors found that HDGF interacts with DDX5 and DDX5 interacts with β-catenin, while HDGF does not interact with β-catenin [25]. The authors proposed that HDGF interacts with DDX5 and promotes the progression of EC through the induction of β-catenin.
In summary, all of the relevant studies in breast, cervical and endometrial cancers indicated that DDX5 is an oncogenic biomarker and target for these cancers’ prognosis and treatment.
DDX5 in colorectal and intestinal cancer
Consistent observations for DDX5/p68 as an oncogenic biomarker and target were also obtained from colorectal cancer (CRC) in the literature. A brief review of these studies is provided below.
Early studies using 50 colorectal adenocarcinoma specimens via IHC and Western blot analyses revealed that (1) p68 protein but not mRNA, is consistently overexpressed in tumors when compared with matched normal tissue; (2) the accumulated p68 showed poly-ubiquitination; and (3) the overexpressed, ubiquitylated p68 is observed in both pre-invasive and invasive lesions, suggesting that the dysregulation of p68 expression occurs early during tumor development [26]. It was also found that (1) the expression of p68/DDX5 and the functionally associated homolog p72/DDX17 strongly increases during the transition of polyp to adenoma to adenocarcinoma in the colon; (2) p68/p72 form complexes with β-catenin and promote β-catenin to activate gene transcription of multiple oncogenes (c-Myc, cyclin D1, c-Jun, and Fra-1); and (3) knockdown of p68/p72 leads to reduced expression of the p68/p72-targeting genes in parallel with inhibiting colon cancer cell proliferation and tumor formation [27]. The authors proposed that p68/p72 overexpression is not only a potential marker of colon cancer but is also causally linked to this disease. Therefore, p68 and p72 may be novel targets in the combat against colon cancer [27].
Another study showed that IHC analyses of the expression of fructose-bisphosphate aldolase A (ALDOA) and DDX5 on a tissue microarray (TMA, containing 141 specimens from 105 CRC patients) revealed that (1) ALDOA and DDX5 were highly expressed in CRC tissues and liver metastatic CRC tissues compared with normal glandular epithelium tissues; and (2) primary CRC tissues highly expressing ALDOA or DDX5 had poor outcome compared with patients who had low expression of those proteins [28]. The authors concluded that high levels of ALDOA and DDX5 contribute to the aggressiveness and poor prognosis of CRC and ALDOA/DDX5 expression could be biomarkers for the prognosis of CRC.
It was also reported that (1) the lncRNA NEAT1 expression is significantly upregulated in CRC tissues compared with its expression in normal tissues; (2) NEAT1 directly bound to DDX5 and sequentially activated the Wnt/β-catenin signaling pathway via DDX5, and fulfilled its oncogenic functions in a DDX5-mediated manner; and (3) analysis of 71 CRC clinical samples with patient outcomes indicated that concomitant NEAT1 and DDX5 protein levels negatively correlated with the overall survival and disease-free survival of CRC patients [29]. The authors concluded that NEAT1 activated Wnt signaling to promote CRC progression and metastasis through the NEAT1/DDX5/Wnt/β-catenin axis, which could be a potential therapeutic target.
One mechanism for DDX5 to promote colorectal oncogenesis is through DDX5 in cooperation with β-catenin and NF-κB to upregulate AKT transcription and inhibit the tumor suppressor FOXO3a protein level in an AKT-dependent manner [30]. Another mechanism is that DDX5 can alternatively cooperate and activate Stat3 to increase Stat transcriptional expression, and subsequently, DDX5/Stats cooperatively activate the Stat3 target gene Mcl-1 [31]. An additional mechanism could be through DDX5 involvement of various lncRNAs to promote tumorigenesis [32]. Additionally, use of a mouse model found that knockout of DDX5 in epithelial cells protected mice from intestinal tumorigenesis and dextran sodium sulfate (DSS)-induced colitis [33].
Based on all of the available observations derived from these studies on DDX5/p68 in the literature, it is safe to conclude that DDX5/p68 is an oncogenic biomarker and target in CRC and intestine cancer.
DDX5 in gastric cancer
DDX5 as an oncogenic biomarker and target was also proposed in the studies on gastric cancer. A brief review of the most relevant studies is presented below.
Using proteomics combined with boronate-affinity pull-down, it was found that (1) epigallocatechin-3-gallate (EGCG, Figure 1A), the most abundant polyphenol in green tea, directly binds to p68 in AZ521 human gastric cancer cells; and (2) exposure of AZ521 cells to EGCG lowered the p68 level through proteasomal degradation and inhibits AZ521 cell proliferation [34]. Similarly, another study showed that (1) DDX5 is significantly upregulated in gastric cancer tissues compared with the paired adjacent normal tissues; (2) knockdown of DDX5 inhibited gastric cancer cell proliferation, colony formation and xenografts growth, whereas ectopic expression of DDX5 promoted these cellular functions; and (3) mechanically, DDX5 induced gastric cancer cell growth by activating mTOR/S6K1, and inhibition of mTOR signaling by the mTOR inhibitor everolimus (Figure 1B) significantly attenuated DDX5-mediated cell proliferation [35]. The authors indicated that DDX5 may serve as a therapeutic target in gastric cancer. Additionally, it was also shown that lncRNA MIAT was highly expressed in gastric cancer tissues and cell lines and regulates miR-141/DDX5 pathway; knockdown of MIAT impaired gastric cancer cell proliferation and metastasis by inhibiting DDX5 expression [36].
Figure 1.
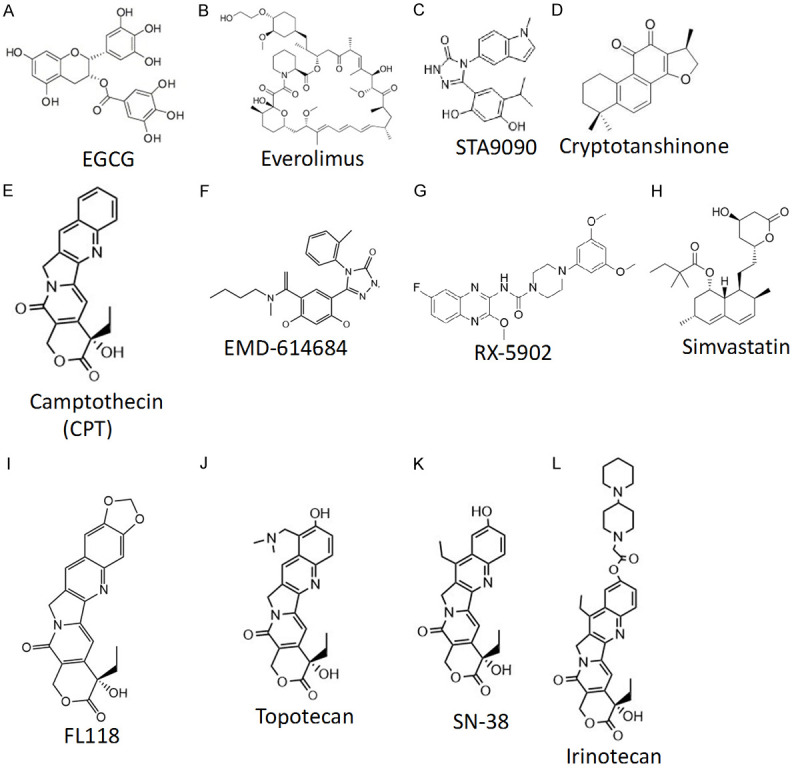
Chemical structures of the compounds that have been discussed in this review article. Each compound chemical structure was either drawn using the ChemDraw Prime 16 software (Perkin Elmer) or downloaded from the compound image collection of online sources.
DDX5 in gliomas
Review of the studies relevant to human gliomas also supports the notion that DDX5/p68 is a glioma prognostic/diagnostic biomarker and target. We briefly review these studies below.
It was reported that (1) p68 protein levels were significantly elevated in high-grade gliomas compared to low-grade gliomas and normal adjacent brain tissues; (2) the expression of p68 was significantly associated with poorer overall survival and enhanced resistance to treatment with radiotherapy plus temozolomide for glioma patients; (3) ectopic expression of p68 enhanced glioma cell proliferation both in vitro and in vivo, but knockdown of p68 prevented glioma cell proliferation [37]. Furthermore, through a tandem affinity purification assay, they found that (4) p68 binds on NF-kB p50 and induces p50 accumulation in the nucleus through release of NF-κB p50 from IκBα and increased NF-κB p50 transcription activity on target gene; and (5) knockdown of NF-κB p50 rescued the phenotypes induced by p68 both in vitro and in vivo [37]. These authors concluded that p68 induces glioma tumor growth through binding with NF-κB p50, regulating p50 nucleus accumulation and transcription activity [37]. Their follow-up studies through microarray analysis further revealed that (1) dual specificity phosphatase 5 (DUSP5) is a downstream target of p68, and negatively regulated by p68; and (2) downregulation of p68 or upregulation of DUSP5 in U87 and LN-229 glioma cells suppressed proliferation, invasion, and migration in parallel with the inhibition of ERK phosphorylation, whereas downregulating DUSP5 rescued the level of ERK phosphorylation [38]. Additionally, it was found that the lncRNA, LINC01116, directly binds to DDX5 in glioma cells and is significantly upregulated in glioma, and positively correlated with clinical malignancy and survival prognosis [39]. LINC01116 overexpression enhanced IL-1β transcription by recruiting DDX5 to the IL-1β promoter in glioma cells to enhance IL-1β expression to attract tumor-associated neutrophils (TANs) into tumor microenvironment to promote tumor cell proliferation [39].
It was also reported that (1) DDX5 and DDX17 expression is upregulated in glioma tissues compared to normal brain tissues; (2) the increased expression of DDX5 and DDX17 is found to be strongly associated with worse overall glioma patient survival; and (3) higher expression of both DDX5 and DDX17 predicted shorter clinical survival time for high-grade glioma patients with radiotherapy or chemotherapy [40]. The authors concluded that overexpressed DDX5 and DDX17 are involved in the clinical progression and poor prognosis of glioma patients, and their upregulation can be used as a reliable clinical predictor for tumor diagnosis and glioma patient survival [40].
DDX5 in leukemia
Data collected from the studies on various types of human leukemia indicated that DDX5/p68 is also a biomarker and target in leukemia. A brief review of the relevant studies is provided below.
It is known that aberrant Notch signaling represents an important oncogenic mechanism for T cell acute lymphoblastic leukemia (T-ALL), an aggressive subset of the most common malignant childhood ALL. In this regard, MAML1 is a major transcription coactivator that regulates Notch oncogenic activities in leukemic cells, although the underlying mechanism is unclear. Lin et al. found that (1) DDX5 interacts with MAML1 in the MAML1 protein complex, and is associated with the endogenous Notch 1 transcription activation complex in human T-ALL leukemic cells; (2) knockdown of DDX5 resulted in the decreased expression of Notch target genes, reduced cell proliferation and increased apoptosis in leukemic cells, while DDX5 depletion inhibited the growth of human leukemia xenograft in nude mice; and (3) DDX5 is highly expressed in primary human T-ALL leukemic cells [41]. The authors proposed that DDX5 may be critical for Notch 1-mediated T-ALL pathogenesis and a potential new target for modulating the Notch signaling in leukemia [41]. Similarly, Mazurek et al. found that acute myeloid leukemia (AML) cells, but not normal bone marrow cells, are dependent on DDX5 [42]. This conclusion is based on the fact that inhibition of DDX5 expression (1) slows AML cell proliferation in vitro and AML progression in vivo but is not toxic to normal bone marrow cells; (2) induces apoptosis in AML cells via induction of reactive oxygen species (ROS); either Bcl2 overexpression or treatment with the ROS scavenger N-acetyl-L-cysteine can block DDX5 inhibition-induced apoptosis; and finally (3) DDX5 is dispensable for normal hematopoiesis and tissue homeostasis [42]. In our view, if these observations can be repeated, we believe that these findings make DDX5 an ideal target in AML without inducing normal hematopoiesis toxicity.
Consistent with the role of DDX5 in T-ALL and AML, using a DDX5-targeting human mAb (2F5), Wu et al. demonstrated that (1) 2F5 selectively inhibits acute promyelocytic leukemia (APL) cell proliferation without toxicity to normal neutrophil and tissues; (2) 2F5 induces G0/G1 phase arrest in APL cells, and promotes APL cell differentiation in parallel with decreased DDX5 expression and increased ROS production; and (3) knockdown of DDX5 inhibits APL cell proliferation, promoted cell differentiation and enhanced ROS production in APL cells, which can be reversed by treatment with ROS inhibitors [43]. Thus, these authors concluded that 2F5 showed the therapeutic value and may provide a novel and valid approach for treatment of relapse/refractory APL [43].
Combining the observations derived from the studies on various types of leukemia (T-ALL, AML, APL), we can conclude that DDX5 is a biomarker and target for multiple leukemia diseases.
DDX5 in liver cancer
Review of the available studies relevant to DDX5 in human hepatocellular carcinoma (HCC) in the literature indicated some inconsistent studies. This may be partially attributed to the uncertainties of the study models/systems used, including HCC cell/tumor models with or without virus infection (e.g., hepatitis B virus/HBV or hepatitis C virus/HCV), as well as other uncertainties such as whether there is an involvement of the double sword property of autophagy. However, after reviewing all of the relevant studies, we conclude that DDX5 can still act as a valid biomarker and target in HCC cancer. Our review and discussion are presented below.
An early study indicated that the expression of seven genes (Dgcr8, p68, p72, Dicer, Ago3, Ago4 and Piwil4) was significantly decreased in primary HCC, especially in non-viral HCC subtypes, compared to the non-cancerous liver; the reduction of two of these genes (Dicer and p68) in HCC was associated with poor prognosis [44]. This could be simply explained as the loss of DDX5 genome repair function would increase HCC malignancy. However, although the mechanism is not fully understood, chronic HBV infection is a major factor contributing to HCC pathogenesis. In this regard, knockdown of DDX5 enhanced transcription from the chronic HBV minichromosome, and paradoxically, chronically infected HBV patients with HCC expressed reduced DDX5 and exhibited poor prognosis after tumor resection [45]. The authors stated that they identify DDX5 as an important player in poor prognosis HCC [45]. Their follow-up studies indicated that DDX5 promotes autophagy and suppresses tumorigenesis; patients with low DDX5 expression showed poor prognosis after tumor resection; and miR-17-5p downregulated DDX5 and impaired autophagy [46]. In our view, this could be well explained by a possible role of DDX5 in HCC genome DNA repair to control HCC over malignancy in the defined conditions.
Another study indicated that DDX5 expression was upregulated in HCC tumors, and the ectopic expression of DDX5 abrogated the tumor-suppressive effect of the knockdown of TINCR (a newly identified lncRNA) [47]. Furthermore, si-TINCR-induced inactivation of AKT signaling was rescued by overexpression of DDX5 [47]. Consistently, knockdown of DDX5 decreased Akt as well as p-Akt (S473) expressions [48]. These findings suggested that DDX5 facilitated HCC cell growth via the Akt signaling pathway. These authors concluded that DDX5 played a crucial role in HCC proliferation and tumorigenesis and may be a novel prognostic marker and potential therapeutic target for HCC [48].
A very recent study has further strengthened the biomarker and target role of DDX5 in HCC. Zhang et al. found that HSP90 interacts directly with DDX5 and inhibits DDX5 protein degradation in the AMPK/ULK1-regulated autophagy pathway, and the subsequent accumulation of DDX5 protein induces the malignant phenotype of HCC through activating the β-catenin signaling pathway [49]. Furthermore, silencing of DDX5 or treatment with HSP90 inhibitor (STA9090, Figure 1C) blocked in vivo tumor growth in a murine HCC xenograft model; high levels of HSP90 and DDX5 protein were associated with poor prognosis [49]. These authors concluded that (1) HSP90 interacted with DDX5 protein and subsequently protected DDX5 protein from AMPK/ULK1-regulated autophagic degradation, and (2) DDX5 and HSP90 are potential therapeutic targets for HCC [49].
In summary, these studies indicate that DDX5 appears to be a cancer biomarker and target for HCC therapeutics. However, it also begs the question of why DDX5 can delay or inhibit the development of malignancy before treatment in some cases, when otherwise it clearly acts as an oncogene and is an oncogenic biomarker and target. As we discussed before, it is likely that if cancer has a high malignancy (i.e., high genome instability), such as PDAC, it is likely that DDX5’s DNA repair role can decrease the malignant potential before treatment. However, when DDX5 expression and/or its multiple functions are abrogated, the malignant cancer cells will be turned into massive apoptosis regardless of the degree of cancer malignancy levels.
DDX5 in lung cancer
It was shown that (1) DDX5 was significantly overexpressed in non-small-cell lung cancer (NSCLC) tissues compared with the matched normal adjacent tissues; (2) overexpression of DDX5 was associated with advanced clinical stage, higher Ki67 index, and shorter overall survival in NSCLC patients; (3) upregulation of DDX5 promoted proliferation of NSCLC cells in vitro and tumor growth of NSCLC xenografts in vivo, whereas downregulation of DDX5 showed the opposite effects; (4) DDX5 directly interacted with β-catenin, promoted its nuclear translocation, and co-activated the expression of cyclin D1 and c-Myc, while silencing β-catenin significantly abrogated DDX5-induced cyclin D1 and c-Myc expression and proliferation in NSCLC cells [50]. This suggests that β-catenin functions as a critical mediator for signaling DDX5 function. These authors proposed that DDX5 may serve as a novel prognostic marker and potential therapeutic target in the treatment of NSCLC [50]. Similarly, Fu et al. demonstrated that preventing lung adenocarcinoma (LADC) cell growth, metastasis, and chemoresistance by the tumor suppressor miR-296-3p can be suppressed by hepatoma-derived growth factor (HDGF) direct interaction with DDX5 to activate the β-catenin/c-Myc pathway [51]. Consistently, reduced miR-296-3p in clinical samples associated with HDGF/DDX5 signaling increase and is an unfavorable factor [51]. Another study revealed that the tumor suppressor function of miR-431 is through miR-431 directly binding on the 3’-UTR of DDX5 to degrade DDX5 [52]; accordingly, the authors showed that miR-431 expression was significantly lower in 122 lung cancer tissue samples compared to the adjacent normal tissues [52]. In contrast, the tumor-promoting function of LncRNA FGD5 antisense RNA1 (FGD5-AS1) in NSCLC cells is through positively regulating the expression of DDX5 via suppressing miR-493-5p, which is associated with enlarged tumor size and lymph node metastasis of the patients [53].
Interestingly, it was also found that (1) DDX5 depletion in small cell lung cancer (SCLC) specifically reduced intracellular succinate (a TCA cycle intermediate serving as a direct electron donor to mitochondrial complex II); (2) DDX5 is overexpressed in SCLC cell lines and its downregulation resulted in various metabolic and cellular alterations in parallel with reduced growth and mitochondrial dysfunction [54]. These authors proposed that the oncogenic role of DDX5, at least in part, manifests as upregulation of respiration to support the energy demands of cancer cells [54].
Based on all of the observations derived from the reviewed studies, we conclude that DDX5 could use multiple ways to express its oncogenic property as a cancer biomarker and target in lung cancer.
DDX5 in osteosarcoma
Osteosarcoma (OS) is a common primary bone malignant tumor, and currently, there are no effective therapeutic regimens for OS treatment. Recent studies indicated that DDX5 is an OS biomarker and target. We briefly summarize these studies below.
It was found that (1) DDX5 interacted with the transcription factor 12 (TCF12, an important molecule in the Wnt signaling pathway) in both OS tissues and MG63 cells; (2) the expression of both DDX5 and TCF12 proteins was significantly higher in OS patients tissues and in the OS MG63 cells than in the corresponding normal tissues and human osteoblast cell hFOB 1.19; (3) overexpression of both DDX5 and TCF12 was associated with clinicopathological features and poor prognosis of OS patients; (4) siRNA knockdown of DDX5 not only inhibited MG63 cell proliferation, migration and invasion, respectively, but also promoted apoptosis of MG63 cells; and (5) DDX5 knockdown also inhibited the expression of TCF12 and decreased the mRNA and protein levels of Cyclin E1 (an important regulator of G1 to S phase progression) [55]. The authors concluded that DDX5 interacts with TCF12 and promotes the progression of OS by stimulating cell cycle progression, and thus, DDX5 and TCF12 could be potential biomarkers for the diagnosis and treatment of OS [55].
Interestingly, another study found that the long non-coding RNA (lncRNA) DLEU1 upregulated DDX5 through the miR-671-5p/DDX5 pathway to realize its oncogenic function in promoting OS cell proliferation, migration, invasion and tumor progression [56]. Supportively, another very recent study found that the circular RNA, Circ-XPR1 was upregulated in OS, which in turn upregulated DDX5 through inhibiting miR-214-5p, and knockdown of Circ-XPR1 significantly reduced DDX5 and OS cell proliferation [57].
DDX5 in prostate cancer
Androgen receptor (AR) is known to be a critical protein molecule extensively involved in prostate cancer development and treatment resistance. Clark et al. found that (1) the interaction of p68 with AR in prostate cancer (PCa) LNCaP cells resulted in the recruitment of AR and p68 to the promoter region of the AR-regulated prostate-specific antigen (PSA) gene; (2) RNAi silencing of p68 reduced AR-regulated PSA expression; (3) tyrosine phosphorylation of p68 by c-Abl kinase enhances the coactivation of ligand-dependent transcription of AR-regulated luciferase reporters, independent of ATP-binding; and (4) increased expression of p68 in PCa compared with benign tissue were revealed in a prostate TMA containing 147 cancer and 44 benign biopsies [58]. Their follow-up study indicated that (1) p68 interacts with β-catenin in PCa cell nuclei [59]. The PCa cell models used in the study were the androgen-dependent LNCaP cells but androgen independent in a hormone refractory derivative of the same cell line being representative of a castration resistant PCa (CRPC) disease type; and (2) p68 interacts with RNA polymerase II (RNAP II) and is recruited to the elongating regions of the AR-mediated PSA gene and possibly facilitating RNAP II transcription of AR-mediated genes [59]. The significance of these studies is the identification of p68 as a novel AR and β-catenin transcription coactivator in PCa and thus p68 possibly plays a role in PCa progression to hormone-refractory disease.
Additionally, using resveratrol-immobilized beads, Taniguchi et al. identified 11 novel resveratrol-binding proteins and one of the 11 proteins is DDX5. They found that (1) treatment with resveratrol induced degradation of DDX5 in PCa cells; (2) depletion of DDX5 caused apoptosis by inhibiting mTORC1 signaling; and (3) knockdown of DDX5 attenuated PCa cell growth and the inhibitory activities of resveratrol against mTORC1 signaling [60].
It was also found that (1) higher level of the oncogenic LncRNA CCAT1 leads to increased mortality in CRPC patients; and (2) the CCAT1 protein promotes PCa cell proliferation and PCa xenograft tumor growth, because CCAT1 acts as a scaffold for DDX5 and AR transcriptional complex to facilitate the expression of AR-regulated genes in nuclei, thus stimulating CRPC progression [61]. Additionally, another study found that DDX5 is fused in frame with the ETs transcription factor ETV4 in PCa cells and led to the expression of a DDX5-ETV4 fusion protein [62].
In summary, these studies support the conclusion that DDX5 is an oncogenic biomarker and target in the PCa disease.
DDX5 in squamous cell carcinoma (SCC)
The studies relevant to SCC also support the notion that DDX5 is a cancer biomarker and target. We briefly review these studies below.
Differential display and Northern blots discovered that p68 was overexpressed in head and neck SCC (HNSCC) cells but not normal upper aerodigestive tract mucosal keratinocytes [63]. The authors concluded that p68 may be involved in the process of malignant transformation or progression of HNSCC [63].
Using 24 human cutaneous SCC tissue specimens and their adjacent tissues plus 6 normal foreskin samples to compare the expression of p68 with that of Ki-67 by double immunofluorescent staining, the study found that p68 protein was overexpressed in all 24 SCC cases (100%), whereas very low expression of p68 was detected in normal foreskin [64]. Moreover, p68 protein expression was higher in cases of cutaneous SCC with metastasis than in cases without metastasis [64]. These authors concluded that the high frequency of p68 expression in cutaneous SCC indicates that p68 might be involved in the development and progression of cutaneous SCC.
Another study has shown that in esophageal SCC (ESCC) cells, knockdown of DDX5 inhibits ESCC cell proliferation and metastasis in parallel with the downregulation of CDK2, Cyclin D1, and Vimentin but the upregulation of E-cadherin [65]. The authors proposed that the downregulation of DDX5 in ESSC correlates to lower malignancy and presents a novel target for the development of new treatment strategies [65].
LncRNA regulates DDX5 in neuroblastoma and thyroid cancer
Long non-coding RNA (lncRNA) is known to play essential roles in tumor progression. However, the functional roles and underlying mechanisms of lncRNAs in pediatric neuroblastoma (NB) remain elusive. Zhao et al. found that (1) the lncRNA NHEG1 bound to and stabilized DDX5 protein through repressing proteasome-mediated degradation, resulting in β-catenin transactivation that altered target gene expression associated with NB progression; (2) NHEG1 harbored oncogenic properties via its interplay with DDX5; (3) RNAi silencing NHEG1 or DDX5 reduced tumor growth and prolonged survival of nude mice bearing xenograft tumors; and (4) high NHEG1 or DDX5 expression was associated with poor survival of NB patients [66]. These authors concluded that lncRNA NHEG1 exhibits oncogenic activity that affects NB progression via stabilizing the DDX5 protein, which may serve as a potential therapeutic target for NB [66]. In contrast, but consistently, Yuan et al. found that one mechanism for the lncRNA SLC26A4-AS1 to play a tumor suppressor role in thyroid cancer is through SLC26A4-AS1 simultaneous interaction with both DDX5 and the E3 ligase, TRIM25, to promote DDX5 degradation through the ubiquitin-proteasome pathway [67].
These studies also support the general notion that DDX5 is a cancer biomarker and target in neuroblastoma and thyroid cancer.
DDX5, anticancer drug and cancer treatment resistance
DDX5 and camptothecin
In an attempt to answer why seemingly identical cells respond differently to a drug, Cohen et al. studied the dynamics and variability of the protein response of human cancer cells to camptothecin (CPT, a well-characterized drug with topoisomerase1 as its therapeutic target) using a dynamic-proteomics approach [15]. They found that the cells differ in the behavior of a subset of proteins corresponding to the outcomes: cell death or survival. Specifically, it was found that DDX5 and the replication factor RFC1 show cell-cell differences that correlated with cell fate. DDX5 increased markedly in surviving cells. In contrast, cells that underwent morphological changes associated with cell death showed decreased DDX5 [15]. Then, the authors asked whether DDX5 plays a functional role in the response to CPT. They knocked down DDX5 by RNAi and found that after adding CPT to the DDX5-silenced cells, the cell death rate doubled when compared with cells treated with nonspecific RNAi or no RNAi [15]. These results indicate that DDX5 rises in cells that survive and decreases in cells that die, and silencing DDX5 causes accelerated cell killing by CPT. Therefore, DDX5 may play a functional role in the escape of cells from the CPT action as the CPT resistant factor.
Relevant to the studies discussed above [15], it was recently reported that in osteosarcoma (OS) cell models, camptothecin induces DDX5 degradation [68]. However, we have identified several serious inconsistencies in this publication. For example, the word “camptothecin (cpt)” was used in the Title and Abstract but in the second paragraph of the Introduction, the authors stated that “Recently, cpt has been shown diverse anticancer properties against various tumors in humans [3-7]”. Interestingly, references 3-7 are about a different small molecule called cryptotanshinone (Figure 1D) not camptothecin (Figure 1E). Because camptothecin has been known to possess antitumor activity against various human cancers for more than 5 decades, we believe that the “cpt” compound used in their study was likely cryptotanshinone but not camptothecin. Additionally, there are inconsistencies in the paper [68]. For example, the Figure 3E result with 2 h and 4 h cpt treatment in their paper is not consistent with the Figure 3E-quantified data shown in Figure 3F with 2 h and 6 h; and the Figure 4A (NC panel) showing significant DNA damage (γH2AX positivity) in the absence of cpt treatment is not consistent with Figure 4C showing no γH2AX positivity without cpt treatment. In summary, the data and conclusion presented in this paper should be reviewed with caution until the inconsistencies can be clarified and additional studies are performed [68].
DDX5 and HSP90 inhibitor
We discussed the finding that DDX5 markedly increased in surviving cells and significantly decreased in dying cells after CPT treatment in the study reported by Cohen et al. [15]. The authors from the same PI group further asked whether bimodality is a special case for CPT, or whether it occurs for other drugs as well. To address this, the authors tested an HSP90 inhibitor (HSP90i, a drug with a different mechanism of action from CPT) [16]. Using single-cell dynamic proteomics to follow 100 proteins in space and time, they found bimodal dynamics (increasing in some cells and decreasing in others) for a quarter of the proteins including DDX5 and RFC1 [16], which were also found in previous studies with CPT [15]. The two drugs (CPT, HSP90i) shared some aspects in their single-cell response. This includes 7 of the bimodal proteins and translocation of oxidative response proteins to the nucleus, but differed in other aspects, with HSP90i (EMD-614684, Figure 1F) showing more bimodal proteins [16]. Interestingly, the cell cycle phase at drug administration impacted the probability of cell death from HSP90i but not from camptothecin [16]. These authors concluded that the bimodal proteins associated with cell fate (cell survival or death) may be potential drug targets to enhance the effects of therapy [16]. Consistent with this study, a very recent study reviewed earlier found that HSP90 interacts directly with DDX5 to protect DDX5 from degradation, and the subsequent accumulation of DDX5 protein induces the malignant phenotype of HCC [49]. High HSP90 and DDX5 were associated with poor prognosis [49].
DDX5 and RX-5902
Previous studies indicated that RX-5902 (Figure 1G) exhibits strong growth inhibition in various human cancer cell lines with IC50 values ranging from 10-20 nM [69]. Using a drug affinity responsive target stability (DARTS) method followed by a confirmation with the direct binding of 3H-labeled RX-5902 to p68 RNA helicase (DDX5), Kost et al. found that Y593 phospho-p68 is a cellular target of RX-5902 [70]. They further showed that RX-5902 inhibited the β-catenin-dependent ATPase activity of p68 RNA helicase in an in vitro system, and that treatment of cancer cells with RX-5902 resulted in the downregulation of the expression of certain genes (e.g., c-Myc, cyclin D1 and p-c-Jun) relevant to β-catenin pathway [70]. Therefore, the authors proposed that RX-5902 binding to Y593 phospho-p68 to inhibit Y593 phospho-p68-β-catenin interactions may contribute to the anticancer activity of this compound [70]. However, the mode of action and conformational changes of this interaction remain elusive. Comparative docking-for-functional analysis of phospho-p68 against RX-5902 and β-catenin uncovered a spectrum of structural linkages associated with the molecular basis of β-catenin-dependent ATPase activity. These authors believe that this may provide a platform for the rational design of specific and potent inhibitors against phospho-p68 with a special emphasis on anticancer activity [71].
The efficacy of RX-5902 was further evaluated in preclinical models of triple-negative breast cancer (TNBC) [72]. A panel of 18 TNBC cell lines was exposed to RX-5902, and changes in proliferation, apoptosis, cellular ploidy, and effector protein expression were assessed. Gene expression profiling was used in sensitive and resistant cell lines with pathway analysis to explore pathways associated with sensitivity to RX-5902 [72]. RX-5902 was active in vivo against both cell line and patient-derived xenograft (PDX) tumor models, and showed potent antiproliferative activity in vitro against TNBC cell lines with an average IC50 of 56 nM in sensitive cell lines [72]. RX-5902 treatment induced apoptosis, G2-M cell-cycle arrest and aneuploidy in a subset of cell lines, and decreased nuclear β-catenin and Mcl-1 [72]. RX-5902 exhibited dose-proportional pharmacokinetics and plasma and tumor tissue in nude mice [72]. Pathway analysis demonstrated an increase in the epithelial-to-mesenchymal transformation (EMT), TGFβ, and Wnt/beta-catenin pathways associated with sensitivity to RX-5902 [72]. Together, the authors believe that this study supports the continued investigation of RX-5902 in TNBC and combinations with immunotherapy.
Subsequently, it was also evaluated whether β-catenin signaling blockade by RX-5902 would enhance the efficacy of anti-CTLA-4 and anti-PD-1 immune checkpoint blockade in immunocompetent, preclinical models of TNBC [73]. Using immune system-humanized mice implanted with MDA-MB-231 xenograft tumors, the authors showed that the addition of RX-5902 to CTLA-4 or PD-1 inhibitors resulted in decreased tumor growth; immunologic analyses demonstrated a significant increase in the number of activated T cells in tumor infiltrating lymphocytes (TILs) with RX-5902 treatment compared to vehicle [73]. In the RX-5902/nivolumab combination group, there was a significant increase in the percentage of CD4+ T cells in TILs and increased systemic granzyme B production [73]. These authors summarized that RX-5902 enhanced the efficacy of Nivolumab (a PD-1 blocking antibody) in a humanized, preclinical model of TNBC. Several changes in immunologic profiles (e.g., an increase in activated TILs and a decrease in human myeloid populations) were noted in mice treated with RX-5902 alone or in combination [73]. RX-5902 could potentiate the effects of checkpoint inhibitors of CTLA4 and the PD-1 inhibitor in the 4 T-1 murine TNBC model [73]. They concluded that RX-5902 may have important immunomodulatory, as well as antitumor activity in TNBC when combined with a checkpoint inhibitor.
RX-5902 monotherapy in advanced solid tumors for a phase 1 study was registered by Rexahn Pharmaceuticals, Inc. in 2013 and the trial was completed at the end of 2019 (https://clinicaltrials.gov/ct2/show/NCT02003092). The major results were communicated in 2017 [74]. As of May 2017, 35 subjects (23 Females, 12 males) were treated with oral RX-5902. The dose limiting toxicities were G4 hyponatremia (n=1) and G3 fatigue (n=1) at 300 mg administered daily for 4 weeks. The maximum tolerated dose of 250 mg was considered as the recommended Phase 2 dose (RP2D). Of the 35 subjects treated, 15 subjects (breast ER+/PR+/Her2-, triple negative breast (n=2), cervical (n=2), neuroendocrine (n=3), paraganglioma, colorectal (n=3), pancreatic, ovarian, head and neck cancers) experienced stable disease; 2 subjects have received treatment for >2.5 years. The most common related adverse events were G1/G2 anorexia, G1/G2 nausea, G1/G2 vomiting, G1/G2 diarrhea, G1 weight loss and G1/G2 fatigue. Oral RX-5902 was bioavailable with median Tmax of 2 hours and median elimination half-life of 12 hours [74]. These investigators concluded that RX-5902 is safe and well-tolerated at the doses and schedules tested. The RP2D of 250 mg of RX-5902 administered daily for 5 consecutive days for 4 weeks is being studied further in metastatic TNBC in phase 2 studies [74].
It is recognized that RX-5902 specifically binds to the Y593 phospho-p68 [70] and controls the phospho-p68 associated downstream signaling function [70,71]. However, many studies suggest that both phosphorylated p68 (a small portion of the total p68) and unphosphorylated p68 (the major portion of the total p68) play important roles in cancer development, metastasis, and treatment resistance. Therefore, it would be ideal for a drug that could bind to the p68/DDX5 protein itself and inhibit the function of both phosphorylated and unphosphorylated p68 downstream signaling as well as the expression of p68/DDX5 in terms of using DDX5/p68 as a target for high effective cancer therapeutics.
DDX5 and simvastatin
Simvastatin is a well-known cholesterol lowering agent, and it belongs to a group of drugs known as Statins. Clinical trials of simvastatin (Figure 1H) with other targeted or chemotherapeutic drugs have been conducted over the past 15 years in various types of cancer with both positive and negative outcomes [75-80]. In preclinical studies, simvastatin has also been found to have antitumor activity in various types of cancer cells and/or tumor models including renal cell carcinoma (RCC), which is known to have a high mortality rate and steadily risen incidence as indicated in the recent review [17]. With this being said, a recent paper reported that simvastatin suppresses RCC cells by inhibiting DDX5 expression and increasing the expression of DUSP5 (a dual specificity protein phosphatase that can de-phosphate and inactivate ERK1/2 MAP kinase) [81]. Using various methods, these authors showed that simvastatin not only inhibited RCC cell viability, migration, and invasion, but also regulated the cell cycle and induced apoptosis [81]. Mechanistically, simvastatin significantly suppressed DDX5 and promoted DUSP5 expression [81]. These authors concluded that simvastatin-induced inhibition of RCC is via regulation of the DDX5/DUSP5 axis [81].
DDX5 and FL118
The FL118 chemical structure is shown in Figure 1I. By using genetically engineered cancer cell models containing the survivin gene promoter-driven luciferase reporter as an assay biomarker [82] through high throughput screening (HTS) of compound libraries; followed by in vitro-&-in vivo analyses of the HTS-resulted hits-to-lead compounds, we discovered FL118 [83]. The chemical structure of FL118 is similar to CPT and its analogues, irinotecan, SN-38 (the active metabolite of irinotecan) and topotecan. However, FL118 has a unique structure of “10,11-methylenedioxy”, and none of the other CPTs has such a structure (Figure 1E, 1J-L). CPTs use topoisomerase 1 (Top1) as their cancer therapeutic target; high expression of Top1 makes CPTs more effective but resistant to other chemotherapeutic drugs [84-86]. In contrast, FL118 does not use Top1 as its major therapeutic target, evidenced by the fact that inhibition of Top1 activity by FL118 is at the µM level, but inhibition of cancer cell growth by FL118 is in the range of pM to low nM levels [83]. FL118 exhibits high efficacy to human tumors that have no or low Top1 expression [87]. This is because FL118 inhibits multiple drug resistance proteins (survivin, MCL-1, XIAP, cIAP2, MDM4) [83,88] and key DNA damage repair regulators, ERCC1 [89] and ERCC6 [90]. Additionally, ABC efflux pump protein transporters are known to be drug resistance factors for CPTs [91-95]. However, FL118 is not a substrate of efflux pump protein transporters such as ABCG2/BCRP and MDR1/Pgp, and can bypass such protein expression-mediated drug resistance [96,97]. Furthermore, it is well known that TP53/p53 or KRAS mutations are challenging treatment resistant factors. However, FL118 exhibited better anticancer efficacy in CRC cells with mutant p53 [88], and our recent studies indicated that FL118 exhibited even better anticancer efficacy either in human bladder cancer cells with KRAS mutation or in human CRC cells with KRAS mutation in comparison with bladder cancer cells or CRC cells with wild type KRAS [98,99].
It is known that cancer stem cells (CSC) play a critical role in treatment resistance and metastasis. In many cases, FL118-induced elimination of human tumor in animal models did not have tumor relapse by the end of the studies. This is consistent with the fact that FL118 inhibits CSC markers/targets (ABCG2, ALDH1A1, Oct4) and reduces the invasive capability of CSC spreading [89]. Our studies also revealed that FL118 targets drug resistant and CSC population cells, eliminates human PDAC tumors and inhibits human PDAC metastasis in animal models [90]. Additional features for FL118 include that FL118 is orally available, highly stable chemically, accumulates and resides in tumors, and is rapidly cleared from the bloodstream (favorable pharmacokinetics-PK) [96]. Studies from the CRO Covance Lab indicate that FL118 exhibits favorable toxicity profiles in dogs [90]. Furthermore, our collaborative studies demonstrated that the bone marrow mesenchymal stromal cells (BMMSC)-rendered multiple myeloma (MM) cell resistance to CAR T cells or NK cells can be abrogated by FL118 treatment through the inhibition of survivin, MCL-1 and XIAP [100,101].
Considering all of these unique characteristics of FL118, one critical question that needs to be answered is why FL118 possesses so many antitumor advantages. We used an FL118 affinity purification approach and identified FL118 direct biochemical targets [102]. Specifically, we conjugated FL118 to agarose beads and made an FL118 affinity purification column in parallel with a control column without conjugated FL118 on the agarose beads, then we passed the whole cancer cell lysates through the FL118 affinity column and the control column in parallel. After stringently washing off non-specific protein binding, we eluted FL118-binding proteins with 8 M urea. After a series of processes of de-urea, concentration of the elution solution with nano-Sep Omega Devices, gradient PAGE gel separation of the concentrated elution samples, isolation of the protein band from the gel, in gel digestion, peptide purification, mass spectrometry analysis, and protein database searching with the identified peptide sequences, we were able to identify DDX5/p68 as the direct biochemical target of FL118 [102]. After alternative confirmation and isothermal titration calorimetry (ITC) analysis, we found that FL118 has a very high affinity with DDX5 protein (KD =34.4 nM). Using our improved state-of-the art proteomics technology [103], we also found that DDX5 is a major FL118 target (unpublished data). Our data indicated that DDX5 is a major target for FL118 in PDAC and CRC. We have done numerous studies on the functional relationship of DDX5 and FL118. For example, our studies indicated that survivin, Mcl-1, XIAP, cIAP2, mutant Kras are all downstream targets of DDX5 and that PDAC patient-derived xenograft (PDX) tumors with mutant p53 exhibit higher mutant Kras expression and greater sensitivity to FL118 than tumors with wild type p53 and wild type Kras [104].
In consideration of all these findings together, we outlined an FL118 mechanism of action model (Figure 2) to explain why FL118 exhibits high antitumor efficacy in PDAC and CRC, and why it is desired for FL118 to enter clinical trials in the near future.
Conclusion
DDX5/p68 appears to be highly expressed in various types of cancer tissues in comparison with the adjacent normal tissues. Although a few studies reported that high DDX5 expression may delay tumor malignancy in certain types of cancer before treatment, the emerging role of DDX5 in genome DNA repair (preventing excessive genomic instability) could explain these seemingly paradoxical observations. Therefore, based on the overwhelming demonstration in the literature as reviewed in this article, we conclude that the DNA repair function of DDX5 plus its RNA helicase and transcription co-activator functions makes DDX5 a superior oncogenic biomarker and target for targeted cancer therapeutics. This conclusion is consistent with the fact that after non-DDX5 targeting drug treatment, the final surviving cancer cells always have high DDX5 expression, and the dying cells have low DDX5 expression, indicating that DDX5 is a cancer drug treatment resistant factor. Thus, targeting DDX5 for cancer therapeutics alone or in combination with immunotherapy (e.g., in the case of FL118 shown in Figure 2) would be innovative approaches to overcome treatment resistance and increase patient response rates, while avoiding the high patient toxicity which often results from multiple chemotherapeutic drug combination treatment.
Acknowledgements
The research work over the 15+ years performed in the Roswell Park Li Lab or in the Canget BioTekpharma LLC (Canget) Research Lab was sponsored in part by the 2020 Pancreatic Cancer Action Network (PanCAN) Translational Research Grant (20-65-FENG), NIH Grants (R43CA221389, R44CA176937, R03CA182552, R21CA180764, R01CA133241, R01CA109481, P30CA016056) and DOD grants (PC110408, BC083321) as well as many public and private foundations (i.e., Roswell Park Alliance Foundation, Concern Foundation, Elsa U. Pardee Foundation, Susan G Komen Breast Cancer Foundation, Wendy Will Case Cancer Fund; Charlotte Geyer Foundation; Mesothelioma Applied Research Foundation). We would like to thank other members in the Li Lab and/or in Canget Research Lab for their cooperation during preparation of this work. The authors also thank Ms. Katherine Plante for her editorial proofreading of the manuscript before submission.
Disclosure of conflict of interest
FL118 and FL118 core structure-based analogs will be further developed in Canget BioTekpharma LLC (www.canget-biotek.com), a spinoff company from Roswell Park Comprehensive Cancer Center (www.roswellpark.org). FL and XL are two of the eighteen initial investors of Canget for development of FL118 and FL118 core structure platform-relevant anticancer agents.
Abbreviations
- AML
acute myeloid leukemia
- APL
acute promyelocytic leukemia
- AR
androgen receptor
- ccRCC
clear cell renal cell carcinoma
- CPT
camptothecin
- CRC
colorectal cancer
- CRPC
castration resistant prostate cancer
- CSC
cancer stem cells
- DSBs
double-strand breaks
- EC
endometrial cancer
- EGCG
epigallocatechin-3-gallate
- EMT
epithelial-mesenchymal transition
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HDGF
hepatoma-derived growth factor
- HSP90
heat shock protein 90
- HSP90i
HSP90 inhibitor
- HTS
high throughput screening
- IHC
immunohistochemistry
- lncRNA
long non-coding RNA
- mCRPC
metastatic castration resistant prostate cancer
- MOA
mechanism of action
- MW
molecular weight
- NB
neuroblastoma
- OS
osteosarcoma
- PCa
prostate cancer
- PDAC
pancreatic ductal adenocarcinoma
- ROS
reactive oxygen species
- SCC
squamous cell carcinoma
- T-ALL
T cell acute lymphoblastic leukemia
- TNBC
triple-negative breast cancer
- Top1
topoisomerase 1
References
- 1.Linder P, Jankowsky E. From unwinding to clamping-the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 2.Hirling H, Scheffner M, Restle T, Stahl H. RNA helicase activity associated with the human p68 protein. Nature. 1989;339:562–564. doi: 10.1038/339562a0. [DOI] [PubMed] [Google Scholar]
- 3.Xing Z, Ma WK, Tran EJ. The DDX5/Dbp2 subfamily of DEAD-box RNA helicases. Wiley Interdiscip Rev RNA. 2019;10:e1519. doi: 10.1002/wrna.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuller-Pace FV, Moore HC. RNA helicases p68 and p72: multifunctional proteins with important implications for cancer development. Future Oncol. 2011;7:239–251. doi: 10.2217/fon.11.1. [DOI] [PubMed] [Google Scholar]
- 5.Dai TY, Cao L, Yang ZC, Li YS, Tan L, Ran XZ, Shi CM. P68 RNA helicase as a molecular target for cancer therapy. J Exp Clin Cancer Res. 2014;33:64. doi: 10.1186/s13046-014-0064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nyamao RM, Wu J, Yu L, Xiao X, Zhang FM. Roles of DDX5 in the tumorigenesis, proliferation, differentiation, metastasis and pathway regulation of human malignancies. Biochim Biophys Acta Rev Cancer. 2019;1871:85–98. doi: 10.1016/j.bbcan.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, Gregory DJ, Lane DP, Perkins ND, Fuller-Pace FV. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005;24:543–553. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicol SM, Bray SE, Black HD, Lorimore SA, Wright EG, Lane DP, Meek DW, Coates PJ, Fuller-Pace FV. The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell-cycle arrest after DNA damage. Oncogene. 2013;32:3461–3469. doi: 10.1038/onc.2012.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mersaoui SY, Yu Z, Coulombe Y, Karam M, Busatto FF, Masson JY, Richard S. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019;38:e100986. doi: 10.15252/embj.2018100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Z, Mersaoui SY, Guitton-Sert L, Coulombe Y, Song J, Masson JY, Richard S. DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions. NAR Cancer. 2020;2:zcaa028. doi: 10.1093/narcan/zcaa028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sessa G, Gomez-Gonzalez B, Silva S, Perez-Calero C, Beaurepere R, Barroso S, Martineau S, Martin C, Ehlen A, Martinez JS, Lombard B, Loew D, Vagner S, Aguilera A, Carreira A. BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repairdouble dagger. EMBO J. 2021;40:e106018. doi: 10.15252/embj.2020106018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 13.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 14.Morimachi M, Hirabayashi K, Takanashi Y, Kawanishi A, Saika T, Ueyama Y, Nakagohri T, Nakamura N, Suzuki H, Kagawa T. Low expression of DDX5 is associated with poor prognosis in patients with pancreatic ductal adenocarcinoma. J Clin Pathol. 2020;73 doi: 10.1136/jclinpath-2020-207002. jclinpath-2020-207002. [DOI] [PubMed] [Google Scholar]
- 15.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, Cohen L, Danon T, Perzov N, Alon U. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 16.Zimmer A, Amar-Farkash S, Danon T, Alon U. Dynamic proteomics reveals bimodal protein dynamics of cancer cells in response to HSP90 inhibitor. BMC Syst Biol. 2017;11:33. doi: 10.1186/s12918-017-0410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li F, Aljahdali IAM, Zhang R, Nastiuk KL, Krolewski JJ, Ling X. Kidney cancer biomarkers and targets for therapeutics: survivin (BIRC5), XIAP, MCL-1, HIF1alph, HIF2alph, NRF2, MDM2, MDM4, p53, KRAS and AKT in renal cell carcinoma. J Exp Clin Cancer Res. 2021;40:254. doi: 10.1186/s13046-021-02026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashemi V, Masjedi A, Hazhir-Karzar B, Tanomand A, Shotorbani SS, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Anvari E, Baradaran B, Jadidi-Niaragh F. The role of DEAD-box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J Cell Physiol. 2019;234:5478–5487. doi: 10.1002/jcp.26912. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Huang J, Hu Z. RNA helicase DDX5 regulates microRNA expression and contributes to cytoskeletal reorganization in basal breast cancer cells. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.011932. M111.011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazurek A, Luo W, Krasnitz A, Hicks J, Powers RS, Stillman B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2012;2:812–825. doi: 10.1158/2159-8290.CD-12-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer RS, Nicol SM, Quinlan PR, Thompson AM, Meek DW, Fuller-Pace FV. The RNA helicase/transcriptional co-regulator, p68 (DDX5), stimulates expression of oncogenic protein kinase, Polo-like kinase-1 (PLK1), and is associated with elevated PLK1 levels in human breast cancers. Cell Cycle. 2014;13:1413–1423. doi: 10.4161/cc.28415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guturi KK, Sarkar M, Bhowmik A, Das N, Ghosh MK. DEAD-box protein p68 is regulated by beta-catenin/transcription factor 4 to maintain a positive feedback loop in control of breast cancer progression. Breast Cancer Res. 2014;16:496. doi: 10.1186/s13058-014-0496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He H, Song D, Sinha I, Hessling B, Li X, Haldosen LA, Zhao C. Endogenous interaction profiling identifies DDX5 as an oncogenic coactivator of transcription factor Fra-1. Oncogene. 2019;38:5725–5738. doi: 10.1038/s41388-019-0824-4. [DOI] [PubMed] [Google Scholar]
- 24.Li MY, Liu JQ, Chen DP, Li ZY, Qi B, Yin WJ, He L. p68 prompts the epithelial-mesenchymal transition in cervical cancer cells by transcriptionally activating the TGF-beta1 signaling pathway. Oncol Lett. 2018;15:2111–2116. doi: 10.3892/ol.2017.7552. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Liu C, Wang L, Jiang Q, Zhang J, Zhu L, Lin L, Jiang H, Lin D, Xiao Y, Fang W, Guo S. Hepatoma-derived growth factor and DDX5 promote carcinogenesis and progression of endometrial cancer by activating beta-catenin. Front Oncol. 2019;9:211. doi: 10.3389/fonc.2019.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Causevic M, Hislop RG, Kernohan NM, Carey FA, Kay RA, Steele RJ, Fuller-Pace FV. Overexpression and poly-ubiquitylation of the DEAD-box RNA helicase p68 in colorectal tumours. Oncogene. 2001;20:7734–7743. doi: 10.1038/sj.onc.1204976. [DOI] [PubMed] [Google Scholar]
- 27.Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–7578. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
- 28.Dai L, Pan G, Liu X, Huang J, Jiang Z, Zhu X, Gan X, Xu Q, Tan N. High expression of ALDOA and DDX5 are associated with poor prognosis in human colorectal cancer. Cancer Manag Res. 2018;10:1799–1806. doi: 10.2147/CMAR.S157925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, Xu M, Sun H, Liu C, Wei P, Du X. The lncRNA NEAT1 activates Wnt/beta-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. 2018;11:113. doi: 10.1186/s13045-018-0656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar M, Khare V, Guturi KK, Das N, Ghosh MK. The DEAD box protein p68: a crucial regulator of AKT/FOXO3a signaling axis in oncogenesis. Oncogene. 2015;34:5843–5856. doi: 10.1038/onc.2015.42. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar M, Khare V, Ghosh MK. The DEAD box protein p68: a novel coactivator of Stat3 in mediating oncogenesis. Oncogene. 2017;36:3080–3093. doi: 10.1038/onc.2016.449. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Liu X, Cui X, Tan Y, Wang Q, Wang Y, Xu C, Fang C, Kang C. LncRNA PRADX-mediated recruitment of PRC2/DDX5 complex suppresses UBXN1 expression and activates NF-kappaB activity, promoting tumorigenesis. Theranostics. 2021;11:4516–4530. doi: 10.7150/thno.54549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abbasi N, Long T, Li Y, Yee BA, Cho BS, Hernandez JE, Ma E, Patel PR, Sahoo D, Sayed IM, Varki N, Das S, Ghosh P, Yeo GW, Huang WJM. DDX5 promotes oncogene C3 and FABP1 expressions and drives intestinal inflammation and tumorigenesis. Life Sci Alliance. 2020;3:e202000772. doi: 10.26508/lsa.202000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka T, Ishii T, Mizuno D, Mori T, Yamaji R, Nakamura Y, Kumazawa S, Nakayama T, Akagawa M. (-)-Epigallocatechin-3-gallate suppresses growth of AZ521 human gastric cancer cells by targeting the DEAD-box RNA helicase p68. Free Radic Biol Med. 2011;50:1324–1335. doi: 10.1016/j.freeradbiomed.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 35.Du C, Li DQ, Li N, Chen L, Li SS, Yang Y, Hou MX, Xie MJ, Zheng ZD. DDX5 promotes gastric cancer cell proliferation in vitro and in vivo through mTOR signaling pathway. Sci Rep. 2017;7:42876. doi: 10.1038/srep42876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sha M, Lin M, Wang J, Ye J, Xu J, Xu N, Huang J. Long non-coding RNA MIAT promotes gastric cancer growth and metastasis through regulation of miR-141/DDX5 pathway. J Exp Clin Cancer Res. 2018;37:58. doi: 10.1186/s13046-018-0725-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang R, Jiao Z, Li R, Yue H, Chen L. p68 RNA helicase promotes glioma cell proliferation in vitro and in vivo via direct regulation of NF-kappaB transcription factor p50. Neuro Oncol. 2012;14:1116–1124. doi: 10.1093/neuonc/nos131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R, Bao HB, Du WZ, Chen XF, Liu HL, Han DY, Wang LG, Wu JN, Wang CL, Yang MC, Liu ZW, Zhang N, Teng L. P68 RNA helicase promotes invasion of glioma cells through negatively regulating DUSP5. Cancer Sci. 2019;110:107–117. doi: 10.1111/cas.13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang T, Cao L, Dong X, Wu F, De W, Huang L, Wan Q. LINC01116 promotes tumor proliferation and neutrophil recruitment via DDX5-mediated regulation of IL-1beta in glioma cell. Cell Death Dis. 2020;11:302. doi: 10.1038/s41419-020-2506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo Q, Que T, Luo H, Meng Y, Chen X, Huang H, Hu R, Luo K, Zheng C, Yan P, Gong J, Fu H, Liu J, Tang Q, Huang G. Upregulation of DEAD box helicase 5 and 17 are correlated with the progression and poor prognosis in gliomas. Pathol Res Pract. 2020;216:152828. doi: 10.1016/j.prp.2020.152828. [DOI] [PubMed] [Google Scholar]
- 41.Lin S, Tian L, Shen H, Gu Y, Li JL, Chen Z, Sun X, You MJ, Wu L. DDX5 is a positive regulator of oncogenic NOTCH1 signaling in T cell acute lymphoblastic leukemia. Oncogene. 2013;32:4845–4853. doi: 10.1038/onc.2012.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazurek A, Park Y, Miething C, Wilkinson JE, Gillis J, Lowe SW, Vakoc CR, Stillman B. Acquired dependence of acute myeloid leukemia on the DEAD-box RNA helicase DDX5. Cell Rep. 2014;7:1887–1899. doi: 10.1016/j.celrep.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, You YQ, Ma YX, Kang YH, Wu T, Wu XJ, Hu XX, Meng QH, Huang Y, Zhang N, Pan XB. DDX5-targeting fully human monoclonal autoantibody inhibits proliferation and promotes differentiation of acute promyelocytic leukemia cells by increasing ROS production. Cell Death Dis. 2020;11:552. doi: 10.1038/s41419-020-02759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitagawa N, Ojima H, Shirakihara T, Shimizu H, Kokubu A, Urushidate T, Totoki Y, Kosuge T, Miyagawa S, Shibata T. Downregulation of the microRNA biogenesis components and its association with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2013;104:543–551. doi: 10.1111/cas.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Xing Z, Mani SK, Bancel B, Durantel D, Zoulim F, Tran EJ, Merle P, Andrisani O. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology. 2016;64:1033–1048. doi: 10.1002/hep.28698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Zhang Y, Zhu X, Chen C, Zhang C, Xia Y, Zhao Y, Andrisani O, Kong L. DEAD box protein 5 inhibits liver tumorigenesis by stimulating autophagy via interaction with p62/SQSTM1. Hepatology. 2019;69:1046–1063. doi: 10.1002/hep.30300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao H, Xie Z, Tang G, Wei S, Chen G. Knockdown of terminal differentiation induced ncRNA (TINCR) suppresses proliferation and invasion in hepatocellular carcinoma by targeting the miR-218-5p/DEAD-box helicase 5 (DDX5) axis. J Cell Physiol. 2020;235:6990–7002. doi: 10.1002/jcp.29595. [DOI] [PubMed] [Google Scholar]
- 48.Xue Y, Jia X, Li L, Dong X, Ling J, Yuan J, Li Q. DDX5 promotes hepatocellular carcinoma tumorigenesis via Akt signaling pathway. Biochem Biophys Res Commun. 2018;503:2885–2891. doi: 10.1016/j.bbrc.2018.08.063. [DOI] [PubMed] [Google Scholar]
- 49.Zhang T, Yang X, Xu W, Wang J, Wu D, Hong Z, Yuan S, Zeng Z, Jia X, Lu S, Safadi R, Han S, Yang Z, Neckers LM, Liangpunsakul S, Zhou W, Lu Y. Heat shock protein 90 promotes RNA helicase DDX5 accumulation and exacerbates hepatocellular carcinoma by inhibiting autophagy. Cancer Biol Med. 2021;18:693–704. doi: 10.20892/j.issn.2095-3941.2020.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z, Luo Z, Zhou L, Li X, Jiang T, Fu E. DDX5 promotes proliferation and tumorigenesis of non-small-cell lung cancer cells by activating beta-catenin signaling pathway. Cancer Sci. 2015;106:1303–1312. doi: 10.1111/cas.12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu Q, Song X, Liu Z, Deng X, Luo R, Ge C, Li R, Li Z, Zhao M, Chen Y, Lin X, Zhang Q, Fang W. miRomics and proteomics reveal a miR-296-3p/PRKCA/FAK/Ras/c-Myc feedback loop modulated by HDGF/DDX5/beta-catenin complex in lung adenocarcinoma. Clin Cancer Res. 2017;23:6336–6350. doi: 10.1158/1078-0432.CCR-16-2813. [DOI] [PubMed] [Google Scholar]
- 52.Xu CM, Chen LX, Gao F, Zhu MF, Dai Y, Xu Y, Qian WX. MiR-431 suppresses proliferation and metastasis of lung cancer via down-regulating DDX5. Eur Rev Med Pharmacol Sci. 2019;23:699–707. doi: 10.26355/eurrev_201901_16883. [DOI] [PubMed] [Google Scholar]
- 53.Cui F, Luo P, Bai Y, Meng J. Silencing of long non-coding RNA FGD5-AS1 inhibits the progression of non-small cell lung cancer by regulating the miR-493-5p/DDX5 axis. Technol Cancer Res Treat. 2021;20:1533033821990007. doi: 10.1177/1533033821990007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Xing Z, Russon MP, Utturkar SM, Tran EJ. The RNA helicase DDX5 supports mitochondrial function in small cell lung cancer. J Biol Chem. 2020;295:8988–8998. doi: 10.1074/jbc.RA120.012600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, Wang Q, Wang Q, Liu J, Jiang X, Zhang Y, Liu Y, Zhou F, Liu H. DEAD-box helicase 5 interacts with transcription factor 12 and promotes the progression of osteosarcoma by stimulating cell cycle progression. Front Pharmacol. 2018;9:1558. doi: 10.3389/fphar.2018.01558. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Chen X, Zhang C, Wang X. Long noncoding RNA DLEU1 aggravates osteosarcoma carcinogenesis via regulating the miR-671-5p/DDX5 axis. Artif Cells Nanomed Biotechnol. 2019;47:3322–3328. doi: 10.1080/21691401.2019.1648285. [DOI] [PubMed] [Google Scholar]
- 57.Mao X, Guo S, Gao L, Li G. Circ-XPR1 promotes osteosarcoma proliferation through regulating the miR-214-5p/DDX5 axis. Hum Cell. 2021;34:122–131. doi: 10.1007/s13577-020-00412-z. [DOI] [PubMed] [Google Scholar]
- 58.Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ, Fuller-Pace FV, Robson CN. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938–7946. doi: 10.1158/0008-5472.CAN-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark EL, Hadjimichael C, Temperley R, Barnard A, Fuller-Pace FV, Robson CN. p68/DdX5 supports beta-catenin & RNAP II during androgen receptor mediated transcription in prostate cancer. PLoS One. 2013;8:e54150. doi: 10.1371/journal.pone.0054150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taniguchi T, Iizumi Y, Watanabe M, Masuda M, Morita M, Aono Y, Toriyama S, Oishi M, Goi W, Sakai T. Resveratrol directly targets DDX5 resulting in suppression of the mTORC1 pathway in prostate cancer. Cell Death Dis. 2016;7:e2211. doi: 10.1038/cddis.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.You Z, Liu C, Wang C, Ling Z, Wang Y, Wang Y, Zhang M, Chen S, Xu B, Guan H, Chen M. LncRNA CCAT1 promotes prostate cancer cell proliferation by interacting with DDX5 and MIR-28-5P. Mol Cancer Ther. 2019;18:2469–2479. doi: 10.1158/1535-7163.MCT-19-0095. [DOI] [PubMed] [Google Scholar]
- 62.Han B, Mehra R, Dhanasekaran SM, Yu J, Menon A, Lonigro RJ, Wang X, Gong Y, Wang L, Shankar S, Laxman B, Shah RB, Varambally S, Palanisamy N, Tomlins SA, Kumar-Sinha C, Chinnaiyan AM. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–7637. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beier UH, Maune S, Meyer JE, Gorogh T. Overexpression of p68 mRNA in head and neck squamous cell carcinoma cells. Anticancer Res. 2006;26:1941–1946. [PubMed] [Google Scholar]
- 64.Wang SJ, Zhang C, You Y, Shi CM. Overexpression of RNA helicase p68 protein in cutaneous squamous cell carcinoma. Clin Exp Dermatol. 2012;37:882–888. doi: 10.1111/j.1365-2230.2012.04365.x. [DOI] [PubMed] [Google Scholar]
- 65.Ma L, Zhao X, Wang S, Zheng Y, Yang S, Hou Y, Zou B, Dong L. Decreased expression of DEAD-Box helicase 5 inhibits esophageal squamous cell carcinomas by regulating endoplasmic reticulum stress and autophagy. Biochem Biophys Res Commun. 2020;533:1449–1456. doi: 10.1016/j.bbrc.2020.10.026. [DOI] [PubMed] [Google Scholar]
- 66.Zhao X, Li D, Yang F, Lian H, Wang J, Wang X, Fang E, Song H, Hu A, Guo Y, Liu Y, Li H, Chen Y, Huang K, Zheng L, Tong Q. Long noncoding RNA NHEG1 drives beta-catenin transactivation and neuroblastoma progression through interacting with DDX5. Mol Ther. 2020;28:946–962. doi: 10.1016/j.ymthe.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan J, Song Y, Pan W, Li Y, Xu Y, Xie M, Shen Y, Zhang N, Liu J, Hua H, Wang B, An C, Yang M. LncRNA SLC26A4-AS1 suppresses the MRN complex-mediated DNA repair signaling and thyroid cancer metastasis by destabilizing DDX5. Oncogene. 2020;39:6664–6676. doi: 10.1038/s41388-020-01460-3. [DOI] [PubMed] [Google Scholar]
- 68.Zhao X, Bao M, Zhang F, Wang W. Camptothecin induced DDX5 degradation increased the camptothecin resistance of osteosarcoma. Exp Cell Res. 2020;394:112148. doi: 10.1016/j.yexcr.2020.112148. [DOI] [PubMed] [Google Scholar]
- 69.Lee YB, Gong YD, Yoon H, Ahn CH, Jeon MK, Kong JY. Synthesis and anticancer activity of new 1-[(5 or 6-substituted 2-alkoxyquinoxalin-3-yl)aminocarbonyl]-4-(hetero)arylpiperazine derivatives. Bioorg Med Chem. 2010;18:7966–7974. doi: 10.1016/j.bmc.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 70.Kost GC, Yang MY, Li L, Zhang Y, Liu CY, Kim DJ, Ahn CH, Lee YB, Liu ZR. A novel anti-cancer agent, 1-(3,5-Dimethoxyphenyl)-4-[(6-Fluoro-2-Methoxyquinoxalin-3-yl)Aminocarbonyl] Piperazine (RX-5902), Interferes with beta-catenin function through Y593 phospho-p68 RNA helicase. J Cell Biochem. 2015;116:1595–1601. doi: 10.1002/jcb.25113. [DOI] [PubMed] [Google Scholar]
- 71.Ali W, Shafique S, Rashid S. Structural characterization of beta-catenin and RX-5902 binding to phospho-p68 RNA helicase by molecular dynamics simulation. Prog Biophys Mol Biol. 2018;140:79–89. doi: 10.1016/j.pbiomolbio.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 72.Capasso A, Bagby SM, Dailey KL, Currimjee N, Yacob BW, Ionkina A, Frank JG, Kim DJ, George C, Lee YB, Benaim E, Gittleman B, Hartman SJ, Tan AC, Kim J, Pitts TM, Eckhardt SG, Tentler JJ, Diamond JR. First-in-class phosphorylated-p68 inhibitor RX-5902 inhibits beta-catenin signaling and demonstrates antitumor activity in triple-negative breast cancer. Mol Cancer Ther. 2019;18:1916–1925. doi: 10.1158/1535-7163.MCT-18-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tentler JJ, Lang J, Capasso A, Kim DJ, Benaim E, Lee YB, Eisen A, Bagby SM, Hartman SJ, Yacob BW, Gittleman B, Pitts TM, Pelanda R, Eckhardt SG, Diamond JR. RX-5902, a novel beta-catenin modulator, potentiates the efficacy of immune checkpoint inhibitors in preclinical models of triple-negative breast cancer. BMC Cancer. 2020;20:1063. doi: 10.1186/s12885-020-07500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diamond J, Eckhardt G, Gluck L, Gutierrez M, Peterson C, Pila R, Benaim E. Phase 1 study of RX-5902, a novel orally bioavailable inhibitor of phosphorylated P68, which prevents β-catenin translocation in advanced solid tumors. Ann Oncol. 2017;28:v83. [Google Scholar]
- 75.Strandberg TE, Pyorala K, Cook TJ, Wilhelmsen L, Faergeman O, Thorgeirsson G, Pedersen TR, Kjekshus J, Group S. Mortality and incidence of cancer during 10-year follow-up of the Scandinavian simvastatin survival study (4S) Lancet. 2004;364:771–777. doi: 10.1016/S0140-6736(04)16936-5. [DOI] [PubMed] [Google Scholar]
- 76.Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, Kim HT, Lee JS. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011;17:1553–1560. doi: 10.1158/1078-0432.CCR-10-2525. [DOI] [PubMed] [Google Scholar]
- 77.Han JY, Lim KY, Yu SY, Yun T, Kim HT, Lee JS. A phase 2 study of irinotecan, cisplatin, and simvastatin for untreated extensive-disease small cell lung cancer. Cancer. 2011;117:2178–2185. doi: 10.1002/cncr.25790. [DOI] [PubMed] [Google Scholar]
- 78.Hong JY, Nam EM, Lee J, Park JO, Lee SC, Song SY, Choi SH, Heo JS, Park SH, Lim HY, Kang WK, Park YS. Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemother Pharmacol. 2014;73:125–130. doi: 10.1007/s00280-013-2328-1. [DOI] [PubMed] [Google Scholar]
- 79.Lee Y, Lee KH, Lee GK, Lee SH, Lim KY, Joo J, Go YJ, Lee JS, Han JY. Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced nonadenocarcinomatous non-small cell lung cancer. Cancer Res Treat. 2017;49:1001–1011. doi: 10.4143/crt.2016.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim Y, Kim TW, Han SW, Ahn JB, Kim ST, Lee J, Park JO, Park YS, Lim HY, Kang WK. A single arm, phase II study of simvastatin plus XELOX and bevacizumab as first-line chemotherapy in metastatic colorectal cancer patients. Cancer Res Treat. 2019;51:1128–1134. doi: 10.4143/crt.2018.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qiu Y, Zhao Y, Wang H, Liu W, Jin C, Xu W. Simvastatin suppresses renal cell carcinoma cells by regulating DDX5/DUSP5. Scand J Urol. 2021;55:337–343. doi: 10.1080/21681805.2021.1876163. [DOI] [PubMed] [Google Scholar]
- 82.Li F. Compositions and methods for identifying agents that alter expression of survivin (Patent US7569221) In: USPTO, editor. USPTO. US 7569221 B2 ed. USA: Health Research Inc., Roswell Park Cancer Institute; 2009. pp. 1–22. [Google Scholar]
- 83.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kotoh S, Naito S, Yokomizo A, Kumazawa J, Asakuno K, Kohno K, Kuwano M. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin-resistant bladder cancer cells. Cancer Res. 1994;54:3248–3252. [PubMed] [Google Scholar]
- 85.Sakai A, Kasahara K, Ohmori T, Kimura H, Sone T, Fujimura M, Nakao S. MET increases the sensitivity of gefitinib-resistant cells to SN-38, an active metabolite of irinotecan, by up-regulating the topoisomerase I activity. J Thorac Oncol. 2012;7:1337–1344. doi: 10.1097/JTO.0b013e31825cca4c. [DOI] [PubMed] [Google Scholar]
- 86.Smith PJ, Makinson TA, Watson JV. Enhanced sensitivity to camptothecin in ataxia-telangiectasia cells and its relationship with the expression of DNA topoisomerase I. Int J Radiat Biol. 1989;55:217–231. doi: 10.1080/09553008914550271. [DOI] [PubMed] [Google Scholar]
- 87.Li F, Ling X, Harris DL, Liao J, Wang Y, Westover D, Jiang G, Xu B, Boland PM, Jin C. Topoisomerase I (Top1): a major target of FL118 for its antitumor efficacy or mainly involved in its side effects of hematopoietic toxicity? Am J Cancer Res. 2017;7:370–382. [PMC free article] [PubMed] [Google Scholar]
- 88.Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 2014;74:7487–7497. doi: 10.1158/0008-5472.CAN-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang J, Liu Z, Zhang D, Liu R, Lin Q, Liu J, Yang Z, Ma Q, Sun D, Zhou X, Jiang G. FL118, a novel survivin inhibitor, wins the battle against drug-resistant and metastatic lung cancers through inhibition of cancer stem cell-like properties. Am J Transl Res. 2017;9:3676–3686. [PMC free article] [PubMed] [Google Scholar]
- 90.Ling X, Wu W, Fan C, Xu C, Liao J, Rich LJ, Huang RY, Repasky EA, Wang X, Li F. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J Exp Clin Cancer Res. 2018;37:240. doi: 10.1186/s13046-018-0899-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, Traxler P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 92.Shishido Y, Ueno S, Yamazaki R, Nagaoka M, Matsuzaki T. ABCG2 inhibitor YHO-13351 sensitizes cancer stem/initiating-like side population cells to irinotecan. Anticancer Res. 2013;33:1379–1386. [PubMed] [Google Scholar]
- 93.Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein (Pgp) inhibitor GF120918. J. Clin. Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 94.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP): two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 95.Filipski E, Berland E, Ozturk N, Guettier C, van der Horst GT, Levi F, Okyar A. Optimization of irinotecan chronotherapy with P-glycoprotein inhibition. Toxicol Appl Pharmacol. 2014;274:471–479. doi: 10.1016/j.taap.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 96.Ling X, Liu XJ, Zhong K, Smith N, Prey J, Li F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am J Transl Res. 2015;7:1765–1781. [PMC free article] [PubMed] [Google Scholar]
- 97.Westover D, Ling X, Lam H, Welch J, Jin C, Gongora C, Del Rio M, Wani M, Li F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol Cancer. 2015;14:92. doi: 10.1186/s12943-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Santha S, Ling X, Aljahdali IAM, Rasam SS, Wang X, Liao J, Wang J, Fountzilas C, Li Q, Qu J, Li F. Mutant kras as a biomarker plays a favorable role in FL118-induced apoptosis, reactive oxygen species (ROS) production and modulation of survivin, Mcl-1 and XIAP in human bladder cancer. Cancers (Basel) 2020;12:3413. doi: 10.3390/cancers12113413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thangaiyan R, Aljahdali IA, Lent-Moore KY, Liao J, Ling X, Li F. Kras mutation subtypes distinctly affect colorectal cancer cell sensitivity to FL118, a novel inhibitor of survivin, Mcl-1, XIAP, cIAP2 and MdmX. Am J Transl Res. 2021;13:7458–7474. [PMC free article] [PubMed] [Google Scholar]
- 100.Holthof LC, van der Schans JJ, Katsarou A, Poels R, Gelderloos AT, Drent E, van Hal-van Veen SE, Li F, Zweegman S, van de Donk NWCJ, Themeli M, Groen RWJ, Mutis T. Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery of CAR T cells through inhibition of apoptosis. Clin Cancer Res. 2021;27:3793–3803. doi: 10.1158/1078-0432.CCR-20-2188. [DOI] [PubMed] [Google Scholar]
- 101.Holthof LC, Stikvoort A, van der Horst HJ, Gelderloos AT, Poels R, Li F, Groen RWJ, Zweegman S, van de Donk N, O’Dwyer M, Mutis T. Bone marrow mesenchymal stromal cell-mediated resistance in multiple myeloma against nk cells can be overcome by introduction of CD38-CAR or TRAIL-variant. Hemasphere. 2021;5:e561. doi: 10.1097/HS9.0000000000000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ling X, Li QY, Li F. Zhejiang University of Technology, China, and Canget, USA; 2019. Matter of composition, synthesis, formulation and application of FL118 platform positions 7 and 9-derived analogues for treatment of human disease (PCT/US2019/051595) [Google Scholar]
- 103.Wang X, Jin L, Hu C, Shen S, Qian S, Ma M, Zhu X, Li F, Wang J, Tian Y, Qu J. Ultra-high-resolution ionstar strategy enhancing accuracy and precision of MS1-based proteomics and an extensive comparison with state-of-the-art SWATH-MS in large-cohort quantification. Anal Chem. 2021;93:4884–4893. doi: 10.1021/acs.analchem.0c05002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ling X, Wu W, Liao J, Santha S, Aljahdali I, Wang J, Fountzilas C, Iyer RV, Boland PM, Li F. Sensitivity of colorectal or pancreatic cancers with high DDX5 and mutations in Kras and p53 genes to FL118 treatment. J. Clin. Oncol. 2020;38(Suppl):e16102. [Google Scholar]