

Abstract
Breast cancer is the most prevalent type of cancer among women. Several types of drugs, targeting the specific proteins expressed on the breast cancer cell surface (such as receptor tyrosine kinases and immune checkpoint regulators) and proteins involved in cell cycle and motility (including cyclin-dependent kinases, DNA stabilisers, and cytoskeleton modulators) are approved for different subtypes of breast cancer. However, breast cancer also has a poor response to conventional chemotherapy due to intrinsic and acquired resistance, and an Akt fingerprint is detectable in most drug-resistant cases. Overactivation of Akt and its upstream and downstream regulators in resistant breast cancer cells is considered a major potential target for novel anti-cancer therapies, suggesting that Akt signalling acts as a cellular mechanism against chemotherapy. The present review has shown that sustained activation of Akt results in resistance to different types of chemotherapy. Akt signalling plays a cellular defence role against chemotherapy and (1) enhances multi-drug resistance, (2) increases reactive oxygen species at breast tumor microenvironment, (3) enhances anaerobic metabolism, (4) inhibits the tricarboxylic cycle, (5) promotes PD-L1 upregulation, (6) inhibits apoptosis, (7) increases glucose uptake, and more importantly (8) recruits and interconnects the plasma membrane, nucleus, endoplasmic reticulum, and mitochondria to hijack breast cancer cells and rescue these cells from chemotherapy. Therefore, Akt signalling is considered a cellular defence mechanism employed against chemotherapeutic effects. In addition, interfering roles of PI3K/Akt signalling on the current cytotoxic and molecularly targeted therapy as well as immunotherapy of breast cancer are discussed with a clinical approach. Although, alpelisib, a PIK3CA inhibitor, is the only PI3K/Akt pathway inhibitor approved for breast cancer, we also highlight well-evaluated inhibitors of PI3K/Akt signalling based on different subtypes of breast cancer, which are under clinical trials whether as monotherapy or in combination with other types of chemotherapy.
Keywords: PI3K/Akt/mTOR inhibitor, breast cancer, drug resistance, targeted therapy, stress conditions
Introduction
Breast cancer is the most prevalent type of cancer among women and the second most prevalent type of cancer worldwide [1]. Endocrine therapy is the first-line therapeutic strategy for oestrogen receptor-positive (ER+) breast cancer patients and uses one of two main drug classes: aromatase inhibitors (AIs) and ER antagonists. Since ER signalling has crosstalk with growth factor signalling, resistance to endocrine therapy is the main barrier for effective treatment of ER+ breast cancer patients [2]. On the other hand, approximately 20% of invasive breast cancer cases are diagnosed as human epidermal growth factor receptor (EGFR) 2-positive (Her2+), making anti-Her2 therapy a second therapeutic strategy against breast cancer [3]. As successful treatment of Her2+ breast cancer is mostly dependent on targeting the specific proteins expressed on the cell surface, several reports have shown that Her2+ breast cancer cells can transform into metastatic and/or breast cancer stem cells (BCSCs) with a poor response to Her2 antagonists [4-6]. However, the triple-negative breast cancer (TNBC) has wider range of chemotherapy is mostly dependent on targeting the specific proteins expressed on the cell surface, such as receptor tyrosine kinases (RTKs) and immune checkpoint regulators, as well as targeting proteins mediating cell cycle, including cyclin-dependent kinases, DNA repair, and cytoskeleton modellers [7].
The phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) signalling in cancer cells mediates chemoresistance in the tumour microenvironment by shielding the immune responses and activating survival signalling pathways in a multitude of human cancers. Overexpression of the PI3K/Akt signalling pathway has active crosstalk with several pathways in different molecular subtypes of breast cancer, including mitogen-activated protein kinase (MAPK), Notch, wnt/β-catenin, and nuclear factor-κB, which leads to chemotherapeutic resistance [8,9]. Multidrug resistance (MDR) proteins are a crucial barrier against chemotherapy by which drugs that have entered cancer cells are pumped out through an ATP-dependent process. The expression of MDR proteins, especially p-glycoprotein (P-gp), and breast cancer resistance protein (BCRP), as well as the phosphorylated Akt, have been detected in paclitaxel- and adriamycin-resistant MDA-MB-231 and MCF-7 breast cancer cell lines [10,11]. In addition, resistance to doxorubicin as a first-line drug is also associated with the expression of P-gp and BCRP. Akt leads to an increase in the cellular level of calcium and a decrease in mitochondrial tricarboxylic acid (TCA) cycle, which eventually increases the level of reactive oxygen species (ROS) as end products of breast cancer cell metabolism [12]; however, ROS activate ROS-dependent apoptosis in breast cancer [1,13]. On the other hand, Akt activates antioxidant signalling and inhibits apoptosis by activating nuclear factor erythroid 2-related factor 2 (Nrf2) [14]. Nrf2 activated by Akt is a cellular response against chemotherapy through the expression of several genes related to antioxidant signalling, MDR, and cell survival [15]. On the other hand, hypoxia-inducible factor-1α (HIF-1α) is a transcription factor whose activation is regulated by both PI3K/Akt and MAPK pathways. HIF-1α upregulates several genes related to glucose metabolism and cancer stemness [16]. Akt enhances hypoxic conditions and ROS levels by activating the main regulators of chemoresistance, HIF-1α and Nrf2 (Figure 1). Therefore, HIF-1α activation by Akt is another cellular response against chemotherapy [17,18]. This releases small molecules, such as calcium, pyruvate, ATP, ADP, and citrate, into the cytoplasm, enhancing the stress condition of cancer cells [19]. Furthermore, HIF-1α leads to cell survival and cancer cell migration as well as ROS creation for ROS scavenger molecules, like glutathione (GSH) [20]. On the other hand, inhibition of ROS generation and autophagy, as well as direct inhibition of Akt, are effective therapeutic strategies for overcoming Akt-promoted drug resistance [21].
Figure 1.
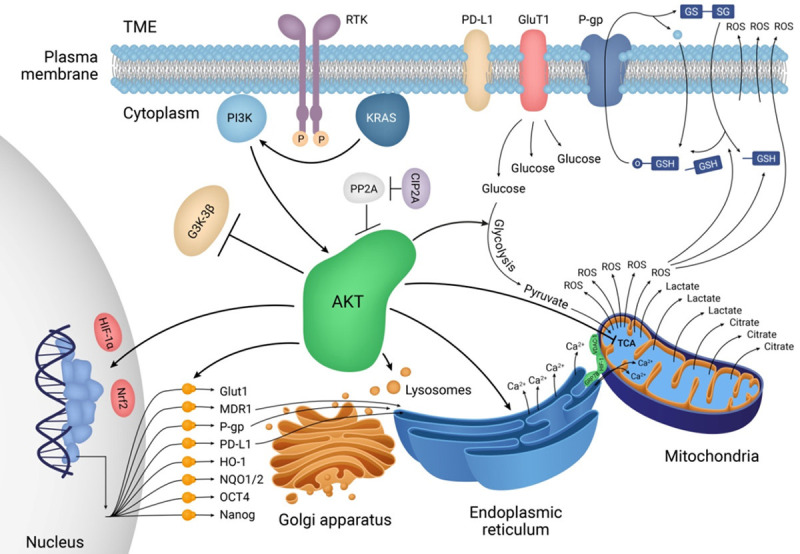
Akt as the master regulator of chemoresistance in breast cancer cells. Akt and its metabolic roles in breast cancer. PI3K activates Akt through Akt recruitment at the plasma membrane. In the case of RAS overactivation, KRAS alternatively activates Akt signalling in an RTK-independent manner. KRAS-dependent activation of Akt upregulates HIF-1α and Nrf2 transcription factors by which numerous genes related to glucose metabolism (e.g., glucose transporter 1), MDRs (e.g., BCRP, MDR1, and P-gp), antioxidants (e.g., HO-1 and NAD(P)H dehydrogenase 1/2), cancer stemness (e.g., OCT4 and Nanog), and immune checkpoints (e.g., PD-L1) are upregulated.
Previously, we reviewed the molecular structure of Akt as well as the detailed mechanisms of Akt signalling and its effects on breast cancer cell metabolism [15]. In the present review, we focus on targeted therapy of breast cancer and the clinical roles of the whole PI3K/Akt signalling pathway inhibitors compared with the current available chemotherapies. Furthermore, we seek to explore the evidence related to the role of three categories of compounds targeting the PI3K/Akt signalling pathway (PIK3CA inhibitors, Akt inhibitors, and mTOR inhibitors) on the future of chemotherapy and immunotherapy in breast cancer.
How resistance to the current chemotherapy is connected to PI3K/Akt signalling?
PI3K/Akt pathway and cytotoxic drugs
CDK4/6 inhibitors
Approximately 20% of breast cancer patients possess impaired p53 tumour suppressors, which in turn have impaired expression of p21, another tumour suppressor; therefore, these cells are unable to check the accuracy of the cell cycle [22]. These cancer patients suffer from uncontrolled cell division due to the overactivation of CDKs. The use of CDK inhibitors, such as palbociclib, ribociclib, and abemaciclib, is critical for inhibition of the cell cycle in ER+ breast cancer cells. As the co-alteration of cell cycle and PI3K pathways have been reported in ER+ breast cancer, the use of CDK inhibitors combined with PI3K inhibitors was rationally recommended for ER+ breast cancer patients [23]. However, Akt activation and expression of MDR genes have been shown in palbociclib-resistant MCF-7 ER+ cells. Everolimus, an mTOR inhibitor, reversed resistance to palbociclib in MCF-7-P cells [24]. The use of palbociclib for TNBC cells with downregulated retinoblastoma tumour suppressor protein (Rb) is beneficial, but Akt signalling was also induced in palbociclib-treated TNBC cells [25]. Therefore, combined chemotherapy of CDK inhibitors and PI3K/Akt/mTOR pathway inhibitors are recommended for TNBC and ER+ breast cancer patients.
Microtubule inhibitors
Microtubule inhibitors are an important category of anti-cancer drugs whose resistance to chemotherapy critically decreases the efficacy of current chemotherapy. For patients suffering from Her2+ and ER+ breast cancers showing resistance to endocrine and anti-Her2 therapies, and for patients suffering from Her2- and ER- breast cancers, microtubule inhibitors, such as paclitaxel and Vinca alkaloids, are considered first-line chemotherapeutics. The actin-binding protein CapG has been reported to induce resistance to paclitaxel in breast cancer by activating Akt signalling through interaction with p300/cAMP-response element binding protein binding protein at the PIK3 regulatory subunit 1 promoter [26]. Moreover, inhibition of CapG and p300/cAMP-response element binding protein binding protein has been shown to attenuate PI3K/Akt signalling, resulting in a higher sensitivity to paclitaxel. In addition, suppression of PI3K/Akt signalling reverses resistance to paclitaxel when breast cancer cells are co-cultured with mesenchymal stem cells [27]. Another study also showed that transgelin-2, another actin-binding protein in breast cancer cells, promotes resistance to paclitaxel through Akt activation [26]. The Akt inhibitor MK-2206 has been shown to reverse resistance to paclitaxel in paclitaxel-resistant ER+ cell [28]. It seems actin-binding proteins are necessary for migration and metastasis of cancer cells, thus the activation of actin polymerization as well as enhancement of Akt-induced chemoresistance [29]. Moreover, paclitaxel-resistant ER+, Her2+, and TNBC cells have upregulated PI3K/Akt signalling and microfilament formation, which leads to metastasis and invasiveness. Since Akt is involved in Nrf2 activation, MDR proteins are also upregulated in paclitaxel-resistant breast cancer cells [30,31].
Since TNBC cells usually have impaired PTEN, they also have overactivated PI3K/Akt signalling. Eribulin is a non-taxane microtubule inhibitor approved for metastatic breast cancer in 2010 [32]. Resistance to eribulin in breast sarcomas has been reported and might be due to increased levels of phoshpo-Akt. PTEN-deficient TNBC cells show higher activity of the PI3K/Akt/mTOR pathway. Accordingly, a combination of an mTOR inhibitor, TAK228, with eribulin was evaluated in such cells. TAK228 is a dual mTORC1/2 inhibitor [33], and mTORC2 has positive feedback on Akt; therefore, TAK228 can inhibit both Akt (indirectly) and mTOR (directly) activation in different TNBC cells, which shows therapeutic effects similar to those of Akt inhibitors. In addition, TAK228 has been shown to synergistically sensitize TNBC cells to eribulin [34,35].
Nucleoside analogues
Antimetabolites are a category of anti-cancer agents that interfere with DNA/RNA metabolism of cancer cells by suppressing DNA replication, transcription, and protein synthesis. The pyrimidine analogue 5-Fluorouracil (5-FU) inhibits thymidylate synthetase. Akt overactivation has been reported in 5-FU-resistant MDA-MB-453 Her2+ breast cancer cells, and the combination of apigenin with 5-FU reverses 5-FU resistance through downregulation of Her2 and Akt [36]. It seems that overexpression of thymidylate synthetase is the main reason for resistance to 5-FU; however, the overactivation of Akt by 5-FU has been shown to lead to 5-FU-related chemotherapy resistance [37]. Furthermore, cellular stress enhanced by activation of chaperones, such as GRP78, located at/in the endoplasmic reticulum recruits Akt to the endoplasmic reticulum and mitochondrial membranes, triggering hypoxia and metabolic reprogramming in TNBC breast cancer cells. As 5-FU induces endoplasmic reticulum stress, GRP78 is upregulated, which then upregulates Akt expression and OCT4. Therefore, targeting Akt and OCT4 as well as inhibition of GRP78 may sensitize breast cancer cells to 5-FU [38].
Gemcitabine is another antimetabolite that inhibits thymidylate synthetase. To decrease the risk of recurrence in breast cancer, anthracyclines are topoisomerase inhibitors used as adjuvant therapy [39]. Patients who do not respond to anthracyclines can receive gemcitabine combined with paclitaxel [40]. Resistance to gemcitabine has also been reported in breast cancer cells with elevated Akt and Src activities. In addition, Akt regulates BCSC migration and resistance of breast cancer cells. Accordingly, Akt and Src inhibition sensitize breast cancer cells to gemcitabine and downregulate BCSC markers, such as CD44 and OCT4 [41]. The stemness of breast cancer cells can be reversed with everolimus [24].
PI3K/Akt pathway and molecularly-targeted therapy
ER antagonists
More than 80% of breast cancer patients have an ER+ subtype, among which 65% are also progestrone receptore-positive (PR+). Howlader et al. (2018) found that hormone-receptor-positive breast cancer subtypes have higher survival compared with other subtypes of breast cancer based on grade [42]. Although ER+ breast cancer cases are treatable with ER antagonists and AIs, poor pathological complete response rates may be due to a change in the histological subtype. Molecular changes in breast cancer subtypes (up to 43%) may lead to a conversion from ER+ or Her2+ to TNBC, thus leading to an increase in the resistance to tamoxifen or trastuzumab after several years of molecularly-targeted therapy. Therefore, a poor pathological complete response does not mean poor prognosis, but can be due to acquired resistance to chemotherapy. In addition, a pathological complete response is useful in accelerating the approval of drugs (Table 1) [43,44]. Downregulation of ER-α is the main reason for resistant ER+ breast cancer cells to tamoxifen. Spalt-like transcription factor 2 (SALL2) is a transcription factor that directly targets ESR1, which upregulates ER and PTEN by targeting ER-α and PTEN promoters followed by downregulation of PI3K/Akt signalling. Hypermethylation of the SALL2 promoter has also been observed in tamoxifen-resistant breast cancer cells [45]. Tumour-associated macrophages, which release CC-chemokine ligand 2, can also increase resistance to tamoxifen through activation of PI3K/Akt signalling [46]. Besides, Ras and other EGFR-associated proteins, like Nogo-B receptor, which recruits Ras to the plasma membrane, also enhance endocrine therapy resistance. Nogo-B receptor regulates oestrogen-induced activation of Akt signalling. Activated ER crosstalks with Akt, and Akt activation is the leading cause of resistance to endocrine therapy [47,48].
Table 1.
Common chemotherapy and combination/adjuvant therapies clinically under investigation for breast cancer patients
| Category | Sub-Category | First-line | 1st Target | CID/SIDa | Combination/Replacement | Clinical trial No.b | Clinical phase |
|---|---|---|---|---|---|---|---|
| (The 2nd target) | |||||||
| Cytotoxic agents | CDK inhibitor | Palbociclibl | CDK4/6 | 5330286 | Capecitabine (TS) | NCT03322215 | II |
| Anastrozole (Aromatase) | NCT01723774 | II | |||||
| Exemestane (Aromatase) | NCT02549430 | II | |||||
| Letrozole (Aromatase) | NCT04318223 | II | |||||
| Fulvestrant (ER) | NCT01684215c | I/II | |||||
| Tamoxifen (ER) | NCT03220178 | IV | |||||
| Trastuzumab (Her2) | NCT02384239 | II | |||||
| NCT03304080 | I/II | ||||||
| Anti-microtubule | Paclitaxel | Microtubule | 36314 | Dasatinib (Src) | NCT01306942c | I/II | |
| Trastuzumab (Her2) | NCT03179904 | II | |||||
| Laniquidar (P-gp) | NCT00028873 | II | |||||
| PSC 833 (P-gp) | NCT00001383 | I | |||||
| Ipatasertib (Akt) | NCT03853707 | I/II | |||||
| Atezolizumab (PD-L1) | NCT03515798 | II | |||||
| Pembrolizumab (PD-1) | NCT02365805 | II | |||||
| Bevacizumab (VEGF-A) | NCT01095003 | III | |||||
| 5-FU (TS) | |||||||
| Cyclophosphamide (DNA) | |||||||
| Eribulin | Microtubule | 11354606 | Atezolizumab (PD-L1) | NCT03202316 | II | ||
| Anti-metabolite | 5-FU | TSd | 3385 | Paclitaxel (Microtubule) | NCT03515798 | II | |
| Pembrolizumab (PD-1) | |||||||
| Gemcitabine | TS | 60750 | GB1275 (CD11b) | NCT04060342 | I/II | ||
| Molecularly-targeted agents | Endocrine therapy | Tamoxifen | ER | 2733526 | Ralimetinibe (MAPK) | NCT02322853 | II |
| Palbociclib (CDK4/6) | NCT02384239 | II | |||||
| Everolimus (mTOR) | NCT01298713 | II | |||||
| Fulvestrant | ER | 104741 | Anastrozole (Aromatase) | NCT00738777 | II | ||
| Palbociclib (CDK4/6) | NCT04318223 | II | |||||
| Vandetanib (VEGFR) | NCT02384239 | II | |||||
| Apitolisibf (PI3K) | NCT02530411 | II | |||||
| Everolimus (mTOR) | NCT01437566 | II | |||||
| NCT01797120c | II | ||||||
| Exemestane | Aromatase | 60198 | Bevacizumab (VEGF-A) | NCT00240071c | II | ||
| Fulvestrant (ER) | NCT03575260 | - | |||||
| Everolimus (mTOR) | NCT03695341 | - | |||||
| Anastrozole | Aromatase | 2187 | Fulvestrant (ER) | NCT00738777 | II | ||
| Bevacizumab (VEGF-A) | NCT00240071c | II | |||||
| Palbociclib (CDK4/6) | NCT01723774 | II | |||||
| Ribociclib (CDK4/6) | NCT04256941 | II | |||||
| Abemaciclib (CDK4/6) | NCT01791985 | I/II | |||||
| AZD4547 (FGFR) | |||||||
| Letrozole | Aromatase | 3902 | Bevacizumab (VEGF-A) | NCT00240071c | II | ||
| Palbociclib (CDK4/6) | NCT04256941 | II | |||||
| Ribociclib (CDK4/6) | NCT01791985 | I/II | |||||
| Abemaciclib (CDK4/6) | NCT01082068 | I/II | |||||
| AZD4547 (FGFR) | NCT01231659 | IV | |||||
| Pilaralisibg (PI3K) | NCT01275859 | II | |||||
| Voxtalisibh (PI3K) | |||||||
| Everolimus (mTOR) | |||||||
| Lapatinib (EGFR/Her2) | |||||||
| RTK-targeted therapy | Lapatinib | EGFR/Her2 | 208908 | Epirubicin (DNA) | NCT00753207 | I | |
| Trastuzumab (Her2) | NCT01875666 | I | |||||
| Letrozole (Aromatase) | NCT01275859 | II | |||||
| Trastuzumab | Her2 | 135301230 | Pertuzumab (Her2) | NCT01875666 | I | ||
| Lapatinib (EGFR/Her2) | NCT02073487 | II | |||||
| Paclitaxel (Microtubule) | NCT01265927 | I | |||||
| Imetelstati (Telomerase) | NCT04001621 | II | |||||
| Capecitabine (TS) | NCT00317720c | I/II | |||||
| Pyrotinib (EGFR/Her2) | NCT01306942c | I/II | |||||
| Everolimusj (mTOR) | NCT02129556c | I/II | |||||
| Dasatinib (Src) | NCT00411788c | II | |||||
| Pembrolizumabk (PD-1) | NCT00736970 | II | |||||
| Rapamycin (mTOR) | NCT01007942c | III | |||||
| Deforolimus (mTOR) | |||||||
| Vinorelbine (Tubulin) | |||||||
| T-DM1 | Her2/Tubulin | 135353969 | Lapatinib (EGFR/Her2) | NCT02073487 | II | ||
| Abraxane (Microtubule) | |||||||
| Agents targeting immuno-signalling | Immune checkpoint inhibitor | Atezolizumab | PD-L1 | 249565671 | Ipatasertib (Akt) | NCT03853707 | I/II |
| Paclitaxel (Microtubule) | NCT03202316 | II | |||||
| Eribulin (Microtubule) | |||||||
| Cytokine pathway inhibitors | Ruxolitinib | JAK1/2 | 25126798 | Capecitabine (TS) | NCT01562873c | II | |
| NCT02120417c | II |
CID/SID: PubChem Compound ID/Substance ID;
This table prepared based on the data collected from www.clinicaltrial.gov;
Clinical trial with results;
Thymidylate Synthetase;
Ralimetinib is also known as LY2228820;
Apitolisib is also known as GDC-0980;
Pilaralisib is also known as SAR245408 or XL147;
Voxtalisib is also known as SAR245409 or XL765;
Imetelstat is also known as GRN163L;
Everolimus was formerly known as RAD001;
Pembrolizumab is also known as MK-3475;
Palbociclib is also known as PD-0332991.
Besides, other proteins are also associated with resistance to tamoxifen through activation of PI3K/Akt signalling, such as peptidyl-prolyl isomerase 1, which stabilizes Akt and is overexpressed in breast cancer. Inhibition of peptidyl-prolyl isomerase 1 leads to the downregulation of MAPK/extracellular signal-regulated kinase 1/2 and PI3K/Akt pathways, thereby reducing the proliferation and viability of tamoxifen-resistant breast cancer cells [49]. On the other hand, the eukaryotic regulator 14-3-3ζ has been reported as a prognostic marker in ER+ breast cancer patients; 14-3-3ζ regulates autophagy by interacting with beclin-1 in hepatocellular carcinoma [50,51]. Moreover, sustained activation of Akt upregulates 14-3-3ζ through expression of nicotinamide phosphoribosyl transferase, resulting in resistance to tamoxifen in ER+ breast cancer cells [52]. Akt activates HIF-1α-dependent autophagy in breast cancer, which leads to expression and activation of autophagy-related proteins, such as LC3-I/II and beclin-1 [53,54]. Therefore, 14-3-3ζ may serve as another player, hand-in-hand with Akt signalling to promote hypoxia, autophagy, and resistance in ER+ breast cancer.
The inhibitory effects of oestrogen on paclitaxel-induced apoptosis have been shown in ovarian cancer, which expresses the oestrogen receptor. The PI3K inhibitor LY294002 decreased the negative effect of oestrogen on paclitaxel-related apoptosis in Caov-3 human ovarian cancer cell line; however, transfection of this cell line with ASK1S83A, a mutated Akt with the attenuated ability of phosphorylation, reduced the inhibitory effect of oestrogen [55]. Therefore, as oestrogen promotes breast cancer progression through Akt activation [48], the combinatorial usage of Akt inhibitors would have great potential for treating ER+ breast cancer patients [56,57]. Finally, alpelisib, which sensitizes ER+ breast cancer cells to tamoxifen and fulvestrant [58,59], is the first PI3K inhibitor approved by the US FDA to treat Her2- breast cancer cells possessing mutated PI3K [60].
Aromatase inhibitors
Aromatase is an adrenal enzyme involved in the conversion of androstenedione and estrone to oestrogen. Acquired resistance to AIs, such as anastrozole and letrozole, has been observed in ER+ breast cancer patients. Crosstalk between PI3K/Akt and ER-α signalling has a crucial role in acquired resistance to letrozole. Letrozole-resistant cells have elevated PI3K/Akt signalling levels, which might be due to elevated levels of the catalytic subunit of PIK3CA (P110α) and not Akt [61]. Resistance to AIs is not only dependent on ER, but a higher level of activated Akt has also been detected in AI-resistant patients, possibly due to dysregulation of Her2 [62]. Interestingly, treatment with MK-2206, rapamycin, Akt, and mTORC1 inhibitors has been shown to sensitize ER+ breast cancer cells to AIs [63]. Furthermore, inhibition of Akt signalling has a beneficial role in reversing resistance to Her2 and ER antagonists in Her2+ and ER+ breast cancer cells. Metastatic breast cancer patients who are Her2+ and ER+ and carry a PIK3CA mutation are resistant to AIs and anti-HER2 therapy. However, the PI3K mutation in resistant Her2+ and ER+ breast cancer cells triggers aberrant activation of upstream proteins involved in Akt activation [64].
In addition, exemestane (a steroidal AI) induces autophagy as a cellular defence against chemotherapy, which is associated with acquired resistance in ER+ breast cancer cells [65]. As autophagy is regulated by Akt signalling, Akt and mTOR inhibitors combined with exemestane reverses exemestane-dependent acquired resistance in Her2-ER+ breast cancer [62]. Moreover, autophagy induces resistance to AIs, which has been shown to enable anti-mTOR therapy combined with AIs to sensitize AI-resistant cells to tamoxifen and AIs [66]. However, mTORC2, which has a positive feedback on Akt, does not respond to some mTORC inhibitors, such as rapamycin, which is more specific to mTORC1. Citi et al. (2018) showed that cells with mutated PIK3CA and defective PTEN have different responses to anti-mTOR therapy. Everolimus effectively decreased the proliferation of tumorigenic cell lines with different potency and efficacy. ZR-75-1 was the most sensitive, and T-47D was the most resistant cell line to everolimus. Additionally, no significant changes in phospho-Akt levels have been found in sensitive breast cancer cell lines, MCF-7 and HCC1500, while the resistant cell line, T-47D, had higher levels of phospho-Akt [67]. Petrossian et al. (2018) also showed Akt is overactivated in HR+/Her2- cells, which were resistant to letrozole and everolimus [68]. Therefore, three factors affect the response to anti-mTOR therapy as a replacement for anti-Akt therapy: (1) mutations inducing PIK3CA, (2) mutations inactivating PTEN, and more importantly (3) cellular levels of phospho-Akt (S473/T308) and phospho-extracellular signal-regulated kinase (ERK) 1/2 (T202/T204).
Her2/EGFR antagonists
Lapatinib is a dual TKI against both Her2 and EGF receptors. Lapatinib activates Akt phosphorylation in Her2+ breast cancer and TNBC, resulting in resistance to EGFR therapy [8,69]. Conclusively, overexpression of HIF-1α alters the TME, making it hypoxic, and reprograms the metabolism of breast cancer cells from aerobic to anaerobic [70]. These stressful conditions managed by Akt enable breast cancer cells to be more aggressive and resistant to chemotherapy. A decrease in lapatinib efficacy was also observed in EGFR expressing MDA-MB-231 TNBC cells with induced hypoxia, and downregulation of HIF-1α reversed the response to EGFR-targeted therapy in such a condition [71] (Figure 2).
Figure 2.
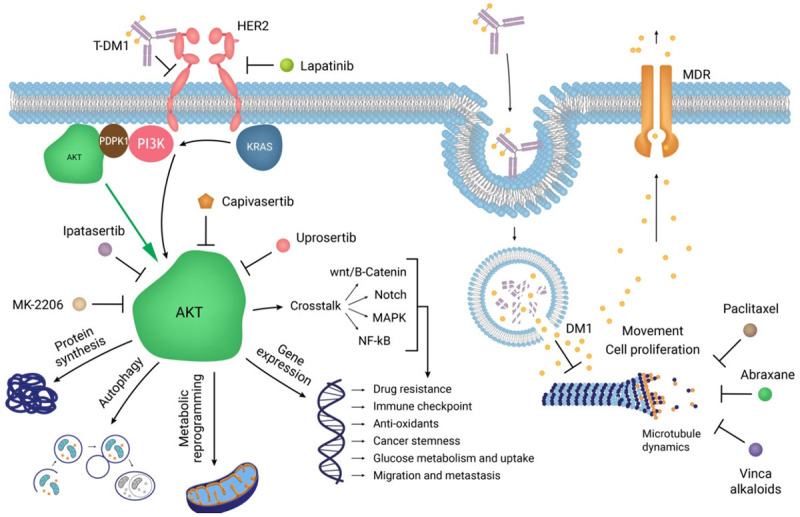
Akt inhibition compared with Her2/EGFR-targeted therapy in breast cancer cells. The mechanisms of actions of different drugs targeting Her2+ breast cancer cells. Akt interconnects different cellular organelles to hijack cellar metabolism, forcefully alters the TME, and promotes breast cancer progression and metastasis. T-DM1, a monoclonal antibody (trastuzumab) conjugated with a small molecule (DM1), is an innovative drug against Her2+ breast cancer, which targets both Her2 signalling and tubulin polymerization. However, internalized DM1 can still be targeted by MDRs and GSH/ROS-mediated recycling systems. Direct targeting of Akt in combination with common chemotherapy reverses resistance to chemotherapy. MK-2206, ipatasertib, capivasertib, and uprosertib are well-studied Akt inhibitors.
Furthermore, inhibition of serine/threonine protein phosphatase 2 A, a negative regulator of Akt, by overactivation of cellular inhibitor of serine/threonine protein phosphatase 2 A, which stabilizes Myc oncoprotein, has been observed in lapatinib-resistant Her2+ breast cancer cells and is overcome by combined treatment with Akt inhibitors [72]. However, the inhibitory effects of lapatinib on Akt phosphorylation and MDR expression in doxorubicin-resistant MDA-MB-231 TNBC cells has also been reported [73]. Overall, mutations in ERBB2 and Ras have been shown to affect the response to TKIs through overactivation of Akt signalling. Apart from normal Ras, which directs the MAPK pathway, studies have shown that KRAS causes inhibition of EGFR and alternatively overactivates Akt signalling, leading to EGFR inhibitor resistance [74]. Additionally, ERBB2 mutations at P780-Y781 enhance Akt upregulation and induce lapatinib resistance in breast cancer cells [75].
Trastuzumab, a monoclonal antibody that targets the Her2 receptor, is another first-line medicine for Her2+ breast cancer. Overactivation of Akt has also been observed in trastuzumab-resistant patients with Her2+ metastatic breast cancer [76]. Overexpression of carboxyl-terminal modulator protein, a positive regulator of Akt, is another reason for Akt activation [77]. The insulin receptor, which phosphorylates insulin receptor substrate 1, can activate PI3K/Akt signalling and induce resistance to trastuzumab [78]. Ankyrin repeat domain 44 is a protein-coding gene expressing a subunit of protein phosphatase 6 that is another negative regulator of cell cycle and is involved in several critical cellular processes, such as metabolism, transcription, and apoptosis. The suppression of ankyrin repeat domain 44 increases S473 phosphorylation on Akt in Her2+ER-PR- breast cancer cells. The level of glycolysis and lactate dehydrogenase B activity is also increased in these cells, demonstrating the role of Akt in metabolic reprogramming as mentioned earlier [79]. Therefore, the downregulation of PP6 is associated with partial resistance to trastuzumab.
Finally, in the case of intrinsic and acquired resistance to trastuzumab, trastuzumab emtansine (T-DM1; an antibody-drug conjugate) can be used as a replacement for trastuzumab [80]. T-DM1 not only binds to the Her2 receptor and inhibits downstream MAPK and PI3K/Akt pathways, but is also internalized through receptor-mediated endocytosis. T-DM1 is released in the cytoplasm and inhibits tubulin polymerization by the same mechanism as Vinca alkaloid suppression of the cell cycle [81] (Figure 2). However, resistance to trastuzumab, lapatinib, and T-DM1 has also been observed during multi-Her2 targeted therapy. Recently, a study reported that two Her2+ patients who did not respond to several Her2 antagonists successfully responded to a combination of pembrolizumab (PD-1 inhibitor) and albumin-bound paclitaxel. Another study also reported the use of pembrolizumab combined with trastuzumab has positive effects on the treatment of trastuzumab-resistance [82,83]; however, the sample size of these studies was small, and further elucidation with a higher number of cases is required.
PI3K/Akt pathway and immunotherapy
Cytokine (JAK/STAT) pathway inhibitors
Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signalling plays a crucial role in the development of malignant tumours as well as inflammation and immune responses through interactions with cytokine receptors. Transphosphorylation of JAK kinases leads to phosphorylation of STATs, which translocates into the nucleus after dimerizing and subsequently induces their target genes. JAK/STAT signalling responds to several cytokines and growth factors, including interleukin (IL)-6 and interferon (IFN)-γ, and activates various genes related to cell proliferation, lipid metabolism, differentiation, and anti-apoptosis. However, it also crosstalks with the PI3K/Akt pathway, which promotes metabolic reprogramming, the cell cycle, cell survival, and metastasis in breast cancer cells [84].
In addition, JAK/STAT signalling is a key pathway in response to cytokines secreted via the anti-cancer immune responses, which leads to expression of PD-L1 in breast cancer cells. Research conducted on BT-549, MDA-MB231, and MCF-7 breast cancer cells has indicated that the level of PD-L1 expression in response to IFN-γ is highly elevated whereas 1 µM Akt inhibitor (wortmannin) and EGFR/Her2 dual inhibitor (lapatinib), as well as a higher concentration (50 µM) of MAPK kinase (MEK) inhibitor (PD98059), decreased PD-L1 expression showing engagement of both Akt and MAPK pathways in PD-L1 expression. This study also showed that treatment with IFN-γ also increased the level of STAT1 and JAK2, but not JAK1 and JAK3, indicating the close association of JAK2/STAT signalling with PD-L1 expression in breast cancer cells [85]. In contrast, another study recently reported that increased MAPK/ERK signalling in MMTV-Neu cells lowers activation of STATs, and inhibition of MAPK signalling with MEK inhibitors activated STAT1, STAT3, and STAT5, thus leading to expression of PD-L1 in a murine model and TNBC cells [86].
Several JAK inhibitors, such as ruxolitinib, itacinib, and BSK805, have been tested in pre-clinical and clinical studies to treat breast cancer, among which ruxolitinib as a dual JAK inhibitor targeting both JAK1 and JAK2 is of greater interest. However, a study conducted on several TNBC cell lines showed that JAK-expressing cells that are resistant to MEK inhibitors, including MDA-MB468, do not respond to JAK inhibitors, indicating that an alternative mechanism of JAK activation may be involved in such cells [87]. Consistent with these results, other researchers have observed that MDA-MB468 TNBC cells that acquire resistance to ruxolitinib overexpress Akt. They also showed that Akt knockdown decreased resistance to ruxolitinib [88]. In cases where TNBC cells develop resistance to ruxolitinib, Akt has a compensatory role in decreasing this resistance and supporting the survival of breast cancer cells. Mutated JAK2 can activate Akt and MAPK pathways as well as STAT5 signalling [89]. Crosstalk between STAT5 and Akt has also been reported [90]. However, TNBC cells containing mutated JAK2 with higher Akt and MEK activations may have dual resistance to MEK and JAK2 inhibitors and needs further elucidation (Figure 3).
Figure 3.
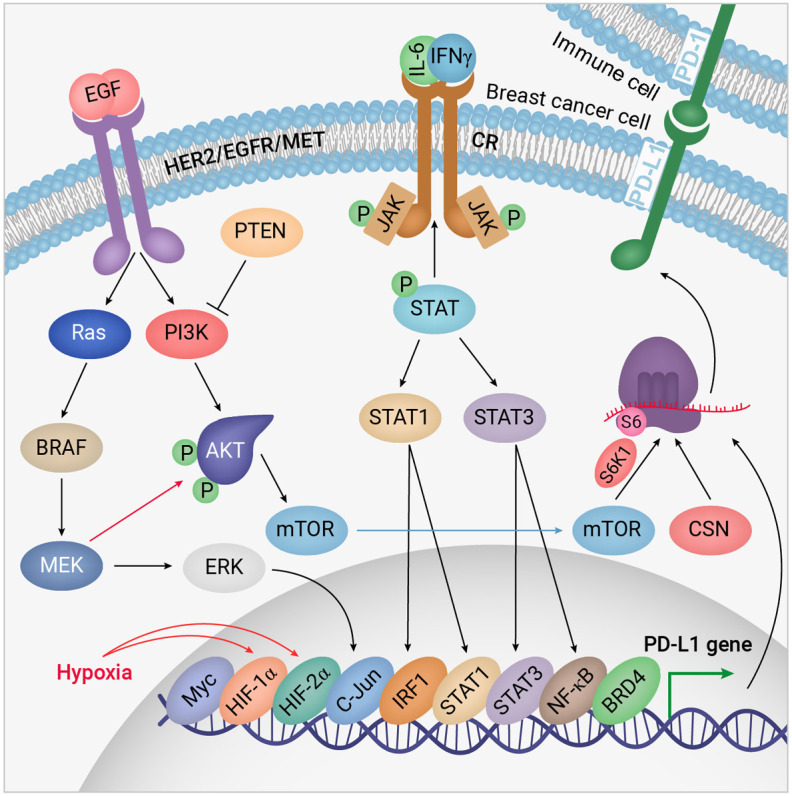
Collaborations of Akt, MAPK, and JAK/STAT pathways in suppressing the anti-cancer immune system. Under hypoxic conditions provided by Akt signalling, PD-L1 is expressed by breast cancer cells to inhibit T-cells. Three main signalling pathways are involved. RTKs triggers PI3K/Akt and MAPK pathways, whereas cytokine receptors (CR) are activated by cytokines released into the TME. JAK/STAT signalling is then activated to recruit STAT1/3 and other transcription factors on some specific gene promoters including the PD-L1 promoter. Simultaneously, proliferation and metastasis are activated and anti-cancer immunity inactivated. S6 and CSN are ribosomal protein S6 and components of the eukaryotic translation factors 3 (eIF-3) complex, respectively, involved in protein synthesis. “Redesigned from Pharmacological Research, Volume 156, Jabbarzadeh Kaboli et al., Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer-A comprehensive review from chemotherapy to immunotherapy, 104806, Copyright (June 2020), with permission from Elsevier”.
In order to evaluate the effects of JAK inhibitors on women who have breast cancer, several clinical trials have initiated, among which only two phase II studies have published the results. One study was conducted on TNBC patients [NCT01562873], while the other was on ER+Her2- breast cancer patients [NCT02120417]. Both of these studies were discontinued because they resulted in a poor improvement in survival rate. In the first study, the safety and efficacy of ruxolitinib were evaluated [NCT01562873] in patients with refractory, metastatic TNBC. Researchers first evaluated the activation of JAK/STAT signalling by immunostaining of phosphorylated STAT3 (N = 171). In 39.2% (67/171) of cases, availability of JAK/STAT signalling was confirmed, among which 21 patients received ruxolitinib. After treatment with 40 cycles of ruxolitinib (1-5 cycles per patient), a different level of toxicity was observed, including fatigue, anaemia, and reduced number of different blood cells such as neutrophils and platelets. As the disease worsened, the treatment was terminated [91]. Lin et al. (2017) showed that ruxolitinib could effectively decrease JAK2/STAT3 signalling, but concluded this compound might be cytostatic, thus failing to reduce tumour burden; however, the number of CD8+ T cells in the TME was decreased, showing diminished anti-cancer immunity of the host during treatment with ruxolitinib [NCT01562873]. The small number of TNBC patients was a limitation of the study. JAK/STAT signalling has a close association with various cellular and extracellular biomarkers. The study did not evaluate the combined administration of ruxolitinib with checkpoint inhibitors and/or other drugs targeting cytokines (e.g., IL-6, IFN-γ, etc.), their receptors, or Akt pathway inhibitors.
Another randomized, double-blind, phase II clinical trial conducted on ruxolitinib as a single agent and in combination with capecitabine in ER+Her2- breast cancer patients (N = 149) showed that while the combination of ruxolitinib and capecitabine was tolerable and showed a greater objective response rate, overall survival rate and PFS were not improved compared with capecitabine alone [92]. The strength of this study was the lower risk of mortality in 40% of cases receiving the combination therapy. Thirty-nine patients (51.3%) who received combination ruxolitinib and capecitabine and 38 (52.1%) who received a combination placebo and capecitabine had died by the final data cutoff time [NCT02120417]. These two phase II studies mentioned here did not support the positive results obtained from preclinical studies. Moreover, both clinical studies ignored the associations of JAK/STAT-linked pathways.
Immune checkpoint (PD-L1) inhibitors
Atezolizumab (a PD-L1 inhibitor) has recently approved for use in combination with paclitaxel for treating TNBC. Atezolizumab affects PD-L1 expression and protein synthesis. Apart from its role in CTL inhibition, atezolizumab downregulates epithelial-mesenchymal transition and cell proliferation and negatively affects hypoxia. Atezolizumab also downregulates Akt signalling in MDA-MB-231 cells [93]. In the phase III clinical trial IMpassion132, the combination of atezolizumab with a first-line chemotherapy, such as gemcitabine, capecitabine, carboplatine, or nab-paclitaxel, in TNBC patients was studied [NCT03371017]. The IMpassion130 clinical trial was designed only for atezolizumab combined with nab-paclitaxel in TNBC patients [NCT02425891]. In IMpassion130, atezolizumab (840 mg) was given for 15 d (from day 1 to 15 in a 28-d cycle) and nab-paclitaxel (100 mg/m2) was given on days 1, 8, and 15 to advanced or metastatic TNBC patients until progression. Significant effects of combined treatment of atezolizumab and nab-paclitaxel were observed on progression-free survival (PFS) and overall survival of TNBC patients, which ultimately led to final approval of this combination by the US FDA [94,95].
Activation of MAPK signalling is associated with a lower level of tumour-infiltrating lymphocytes and leads to evasion of breast tumour cells from the immune system. Hence, MEK inhibitors combined with PD-L1 antagonists enhance the anti-cancer immune response in TNBC mouse models [96]. Accordingly, MAPK signalling has two opposing effects on JAK/STAT signalling. The mechanism by which MEK inhibition activates JAK/STAT has not been elucidated yet. The use of the JAK1/2 inhibitor (ruxolitinib), JAK1 inhibitor (itacinib), and JAK2 inhibitor (BSK805) did not affect JAK/STAT activity induced by MEK inhibition, showing that MEK inhibition uses a different mechanism for JAK/STAT activation that cannot be reversed by JAK inhibitors. On the other hand, combined treatment of EGFR inhibitors, erlotinib and lapatinib, could partially or fully reverse JAK/STAT activity induced by a MEK inhibitor (trametinib) in Her2+ breast cancer cells [86].
One subset of TNBC cells expresses PD-L1. Therefore, if TNBC cells have lower expression of PD-L1, then treatment with MEK inhibitors combined with a PD-L1 inhibitor may be helpful since MEK inhibition promotes PD-L1 expression and makes it a potential target for adjuvant therapy. Measurements of PD-L1, JAK2, STAT1, STAT3, p-STAT1, and p-STAT3 in 11 TNBC cell lines show that cells possessing somatic amplification of the 9p24.1 locus express PD-L1 in response to IFN-γ. Thus, inducible PD-L1 expression was reported, which can be reversed by JAK2 knockdown and a JAK1/2 inhibitor (ruxolitinib) [97]. Furthermore, regarding MEK and Akt inhibition, a study has divided TNBC cells into three categories: (1) MEK inhibition resistant (MDA-MB468, HCC70, and BT20), (2) Akt inhibition resistant (MDA-MB231, MDA-MB435s, and HCC1806), and (3) MEK and Akt inhibition dual resistant (MDA-MB436, SKBR7, and SUM-159PT) [87]. TNBC cells resistant to Akt inhibitors have lower phosphorylated Akt levels; however, metastatic TNBC cells require activation of integrin β1, myosin light chain kinase, and myosin IIA as well as activation of Akt after MEK inhibition. Reports have also shown that higher levels of KRAS might be associated with resistance to tyrosine kinase inhibitors (TKIs) and Akt overactivation [98].
Several cytokines secreted in breast TME also affect the response of breast tumour cells to immunotherapy. Interleukin (IL)-22 is secreted by immune cells, such as T-helper 17, γδ T, and natural killer cells, which are involved in anti-cancer immunity and enhance Janus kinase/signal transducer and activator of transcription (JAK/STAT)-3/MAPK/Akt signalling pathways, thus promoting PD-L1 expression and inducing resistance to paclitaxel in TNBC [15,99,100]. Stress causes chemokine release into the TME and leads to the accumulation of immune cells. Stimulants of tumour necrosis factor-α and IL-6 induce secretion of IL-8 from mesenchymal cells and phagocytes whose interactions with C-X-C chemokine receptors type 1/2 enhance PI3K/Akt signalling, resulting in the upregulation of PD-L1 [101]. Furthermore, interferon-α, which induces paired-like homeodomain transcription factor 2, is also involved in breast cancer progression. Interferon-α induction of paired-like homeodomain transcription factor 2 activates Akt in letrozole-resistant breast cancer cells [102]. As mentioned above, stress conditions are closely related to resistance to chemotherapy. Therefore, paclitaxel resistance should be taken into account when combined with atezolizumab, and overall, targeting TME stress conditions may be beneficial for reversing resistance to chemotherapy.
The use of JAK/STAT inhibitors is like a double-edged sword. These compounds not only suppress the proliferation of breast cancer cells and inhibit PD-L1 expression, but they may also activate alternative pathways such as Akt signalling, which accelerates the survival of cancer cells. The preclinical studies mentioned in the current article also demonstrate negative results and show resistance to ruxolitinib [88]. However, the above mentioned clinical trials ignored the negative results of preclinical studies and only focused on the positive results. In cases where alternative pathways that have crosstalk with JAK/STAT signalling are taken into account in clinical studies, even negative results increase our knowledge about the behaviour of JAK inhibitors, which will help us better design clinical trials.
PI3K/Akt pathway inhibitors and breast cancer subtypes
The present review has shown that sustained activation of Akt results in resistance to different types of chemotherapy as Akt signalling plays a cellular defence role against chemotherapy that: (1) enhances MDR activation, (2) increases TME ROS levels, (3) enhances anaerobic metabolism, (4) inhibits the TCA cycle, (5) promotes PD-L1 upregulation, (6) inhibits apoptosis, (7) increases glucose uptake, and more importantly (8) recruits and interconnects the plasma membrane, nucleus, endoplasmic reticulum, and mitochondria to hijack breast cancer cells and rescue them from chemotherapy. Therefore, Akt signalling is considered a cellular defence mechanism employed against chemotherapeutic effects. Overall, PI3K/Akt signalling can be inhibited at three main points: (1) PI3K, located upstream, (2) Akt, centrally located, and (3) mTOR, located downstream. However, future comprehensive molecular and cellular investigations need to focus on the roles of Akt signalling in breast cancer. Thus, future research paths on Akt signalling can be divided and summarized in the following categories (Table 2).
Table 2.
Inhibitors of PI3K/Akt/mTOR signalling clinically started or completed as monotherapy or combination therapy for breast cancer
| Compound | 1st Target | CID/SIDa | Combination | Condition | Clinical trial Nob | Clinical phase |
|---|---|---|---|---|---|---|
| (The 2nd target) | Breast Cancer (BC) | |||||
| MK-2206 | Akt | 24964624 | Paclitaxel (Microtubule) | Locally advanced | NCT01263145 | I |
| Anastrozole (Aromatase) | Metastatic solid tumours Metastatic BC | NCT01277757 | II | |||
| Fulvestrant (ER) | Postmenopausal women with metastatic BC | NCT01344031 | I | |||
| Lapatinib (EGFR/Her2) | NCT01245205 | I | ||||
| Trastuzumab (Her2) | NCT01042379 | I | ||||
| Ridaforolimus (mTOR) | NCT01295632 | I | ||||
| Capivasertibc | Akt | 25227436 | Fulvestrant (ER) | Locally advanced (inoperable) | NCT04305496 | III |
| Paclitaxel (Microtubule) | metastatic HR+/HER2- BC Metastatic TNBC | NCT03997123 | III | |||
| Durvalumab (PD-L1) | Refractory Solid tumours | NCT03742102 | I/II | |||
| ER+ BC | NCT02465060 | II | ||||
| Advanced BC | NCT02077569 | II | ||||
| ER+ BC, previously treated with fulvestrant | NCT03182634 | II | ||||
| NCT03310541 | I | |||||
| Uprosertibd | Akt | 51042438 | Trametinibe (MEK1/2) | Metastatic TNBC | NCT01964924 | II |
| NCT00920257 | I | |||||
| NCT01138085 | I | |||||
| M2698 | Akt | 89808643 | Trastuzumab (Her2) | Advanced malignancies | NCT01971515 | I |
| Tamoxifen (ER) | ||||||
| Alpelisibf | PI3K | 56649450 | Fulvestrant (ER) | Prior endocrine therapy, in PIK3CAmutant BC | NCT03056755 | II |
| Letrozole (Aromatase) | HR+ locally-advanced Unresectable or metastatic BC | NCT01923168 | II | |||
| Goserelin (GnRH) | Premenopausal Patients | NCT01870505 | I | |||
| Leuprolide (GnRH) | AR+ PTEN+ BC | NCT02058381 | I | |||
| Exemestane (Aromatase) | PIK3CAmutant PTENmutant | NCT02051751 | I | |||
| Tamoxifen (ER) | anthracycline refractory | NCT02088684 | I | |||
| Paclitaxel (Microtubule) | TNBC | NCT02038010 | I | |||
| Ribociclibg (CDK4/6) | NCT03207529 | I | ||||
| T-DM1 (Her2/Microtubule) | NCT04216472 | II | ||||
| Enzalutamide (Androgen Receptor) | NCT01300962 | I | ||||
| Abraxaneh (Microtubule) | NCT02077933 | I | ||||
| Capecitabine (TS) | ||||||
| Everolimus (mTOR) | ||||||
| Alpelisib | PI3K | 56649450 | Fulvestrant (ER) | Postmenopausal | FDA approval | |
| women, HR+ Her2- PIK3CAmutant advanced or metastatic BC | ||||||
| Apitolisibi | PI3K | 25254071 | Paclitaxel (Microtubule) | Locally recurrent or metastatic BC | NCT01254526 | I |
| Bevacizumab (VEGF-A) | Advanced BC | NCT01437566 | II | |||
| Fulvestrant (ER) | ||||||
| Pilaralisibj | PI3K | 56599306 | Paclitaxel (Microtubule) | Metastatic BC previously treated with trastuzumab | NCT01042925 | I/II |
| Trastuzumab (Her2) | NCT01082068 | I/II | ||||
| Letrozole (Aromatase) | ||||||
| Voxtalisibk | PI3K | 16123056 | Letrozole (Aromatase) | Locally advanced/metastatic solid tumours | NCT01082068 | I/II |
| Pimasertib (MEK1/2) | NCT01390818 | I | ||||
| Rapamycinl | mTORC1 | 5284616 | Trastuzumab (Her2) | Her2+ metastatic BC | NCT00411788 | II |
| Everolimusm | mTOR | 6442177 | Cisplatin (DNA) | Metastatic BC | NCT01031446 | I/II |
| Paclitaxel (Microtubule) | Her2+ metastatic BC | NCT00411788 | II | |||
| Trastuzumab (Her2) | postmenopausal women with metastatic or advanced BC | NCT01007942 | III | |||
| Vinorelbine (Tubulin) | HR+ Her2- BC | NCT01231659 | IV | |||
| Letrozole (Aromatase) | NCT00317720 | I/II | ||||
| Anastrozole (Aromatase) | NCT02291913 | II | ||||
| Exemestane (Aromatase) | NCT01298713 | II | ||||
| Fulvestrant (ER) | NCT01797120 | II | ||||
| Tamoxifen (ER) | ||||||
| Deforolimus | mTOR | 11520894 | MK-2206 (Akt) | Advanced BC | NCT01295632 | I |
| Exemestane (Aromatase) | ER+ BC | NCT01605396 | II | |||
| Dalotuzumab (IGF1R) | Her2+ trastuzumab-refractory metastatic BC | NCT01234857 | II | |||
| Trastuzumab (Her2) | NCT01220570 | I | ||||
| NCT00736970 | II |
CID/SID: PubChem Compound ID/Substance ID;
This table prepared based on the data collected from www.clinicaltrial.gov;
Capivasertib is also known as AZD5363;
Uprosertib is also known as GSK2141795;
Trametinib is also known as GSK1120212;
Alpelisib was formerly known as BYL719;
Ribociclib is also known as LEE011;
Abraxane is also known as nab-paclitaxel or albumin-bound paclitaxel;
Apitolisib is also known as GDC-0980;
Pilaralisib is also known as SAR245408 or XL147;
Voxtalisib is also known as SAR245409 or XL765;
Rapamycin is also known as sirolimus;
Everolimus is also known as RAD001.
PI3K inhibitors
Almost 40% of patients with HR+Her2- breast cancer have mutations in PIK3CA. In a phase III trial, alpelisib combined with fulvestrant was studied in HR+Her2- breast cancer with mutated PIK3CA [NCT02437318]. Two cohorts were designed based on whether patients are PIK3CA-mutants or PIK3CA-wild-type, and then each cohort was sub-grouped into alpelisib plus fulvestrant or placebo plus fulvestrant. As the primary endpoint, PFS was evaluated in both cohorts. The study showed that the PFS doubled (11 months) with combination alpelisib and fulvestrant compared to fulvestrant monotherapy (5.7 months), and overall response rate (ORR) was also higher in the PIK3CA-mutant cohort that received combination alpelisib and fulvestrant. In this study, the most frequent adverse reactions were hyperglycaemia, rash, and gastrointestinal disorders, including diarrhoea, which were also reported with combination therapy in PIK3CA-mutant patients [103].
Consistent with the above trial, a phase II trial was conducted on 131 HR+Her2- breast cancer patients with PIK3CA mutations receiving letrozole combined with alpelisib [NCT01923168]. In contrast with the previous trial, ORR and pathologic complete response were not significantly improved in the alpelisib-treated PIK3CA-mutant cohort of this study. Similarly, two cohorts were studied, PIK3CA-mutant and PIK3CA-wild-type, but with a larger sample size (N = 164 and N = 176, respectively), and two treatment groups in each cohort were created similar to the previous trial: one sub-group received combination therapy (letrozole plus alpelisib) and the other received letrozole monotherapy (letrozole plus placebo). The number of patients in each of the four groups was approximately 60. Comparison of ORRs showed a lower ORR for both PIK3CA-mutant treatment groups. Moreover, 52% of PIK3CA-mutated patients completed the full combination therapy in 24 weeks, and almost half of the patients stopped treatment because of adverse reactions or doctor/patient decision. Therefore, the results were affected by the continuation/discontinuation of therapy. Gastrointestinal problems, hyperglycaemia, fatigue, and skin rashes were the most common adverse events of HR+Her2- breast cancer patients treated with letrozole and alpelisib [104].
Several PI3K inhibitors have been studied for breast cancer so far, including alpelisib, apitolisib, pilaralisib, and voxtalisib. Among them, alpelisib has been approved by the US FDA for the treatment of ER+ breast cancer patients. PI3K inhibitors are effective for patients diagnosed with mutated PI3K and PTEN. Otherwise, Akt can also be activated in an RTK-independent manner. Therefore, RTK signalling (e.g., Her2/EGFR signalling) can be bypassed by RTK-independent activation, and the use of PI3K inhibitors might be less effective than Akt and mTOR inhibitors.
Akt inhibitors
Akt has crosstalk with several oncogenic pathways, such as MAPK, Notch, and wnt/β-catenin, resulting in the structural reformation of cellular metabolism (e.g., mitochondria), cellular migration, BCSC maintenance, and cancer progression. Furthermore, most drugs work against ER+ and Her2+ breast cancer subtypes, and higher mortality rates are also due to ER+ and Her2+ breast cancer patients. In particular, the use of Akt inhibitors is more effective against Her2+ and Her2- breast cancers with overactivated KRAS, and against ER+ breast cancer in which there is ER crosstalk with Akt signalling. A few Akt inhibitors are undergoing clinical trials for breast cancer, including MK-2206, capivasertib, and uprosertib. MK2206 and uprosertib are being investigated for ER+ breast cancer and TNBC subtypes, respectively, whereas capivasertib is being studied for both ER+ breast cancer and TNBC subtypes. However, there is a lack of clinical trials studying the effects of Akt inhibitors for Her2+ breast cancer patients. Treatment of Her2+ breast cancer is strongly dependent on the trastuzumab and lapatinib and other Her2 inhibitors. Since activated PI3K/Akt signalling also causes resistance to anti-Her2 therapy, Akt inhibitors are recommended for Her2+ breast cancer, at least in combination with Her2 inhibitors (Figure 4).
Figure 4.

Interactions of Akt inhibitors with Akt kinase domain. The Akt kinase domain has a consensus D292F293G294 sequence known as the DFG gate, by which Akt kinase is activated/inactivated. While the side chain of the D (Asp) residue faces toward Lys179 (DFGin), the kinase is activated. In contrast, when the side chain of D is located outside of the pocket, and the F (Phe) residue faces Lys179, the enzyme is inactive [105]. To show the interactions of Akt inhibitors, molecular modelling was performed on three well-known Akt inhibitors against the crystal structure of the Akt kinase domain in the active conformation. Structures of the Akt kinase domain (PDB ID: 3OCB) were obtained from the protein databank (www.rcsb.org), and Akt inhibitors were from the PubChem database. After modelling the Akt structure with modeller 9.25 [106] target-template alignment, model building, and model evaluation. This unit describes how to calculate comparative models using the program MODELLER and how to use the ModBase database of such models, and discusses all four steps of comparative modeling, frequently observed errors, and some applications. Modeling lactate dehydrogenase from Trichomonas vaginalis (TvLDH, molecular docking was performed using Autodock 4.2 with 30 repeats for each compound [107,108]. The best interactions of (A-D) capivasertib (AZD5363, CID: 25227436), (E-H) MK-2206 (CID: 24964624), and (I-L) uprosertib (GSK2141795, CID: 51042438) are shown here with binding energies of -10.98, -9.81, and -7.49 kcal/mol, respectively. Capivasertib reached phase I/II/III clinical trials in different subtypes of breast cancer; however, MK-2206 and uprosertib are in phases I and II for metastatic breast cancer and TNBC, respectively.
In a phase I trial, the effects of MK-2206 were studied in combination with trastuzumab in Her2+ breast cancer patients. The maximum tolerated dose of MK-2206 was used combined with trastuzumab four times per day (45-60 mg), (Cohort 1, N = 15) and once a week (135-200 mg), (Cohort 2, N = 19) [NCT00963547]. The authors observed that the pharmacokinetics of MK-2206 were not affected by trastuzumab treatment. This study also indicated that MK-2206 combined with trastuzumab has therapeutic efficacy in Her2+ patients; however, treatment was stopped because adverse effects and disease progression were observed. Researchers also found that PIK3CA mutations were not detected in circulating DNA of patients who responded well to treatment, and three patients who had PIK3CA mutations did not respond to treatment with MK-2206 [105].
Furthermore, a phase II trial also studied the impact of PIK3CA mutations on treatment with the Akt inhibitor MK-2206 [NCT01277757]. This trial included PIK3CA-mutant and Akt1-mutant breast cancer patients with different subtypes. Experiments were not only performed on pre-treatment and on-treatment biopsies, but also on peripheral blood mononuclear cells and platelet-rich plasma. Significant reduction in phosphorylation was observed in both residues S473 and T308 of Akt in blood samples, but such a decrease was not observed in tumour biopsies. The researchers categorised patients into two cohorts: PIK3CA/Akt1-mutant and PTEN-loss. Overall, they recruited 27 patients; 18 were included in the first cohort and nine in the second. Most patients were HR+ (N = 15), nine had TNBC, and three were Her2+. One PIK3CA-mutant patient (out of 13) with the ER+PR-Her2- phenotype and PIK3CA and KRAS mutations partially responded to MK-2206. Researchers concluded that Her2- patients with KRAS mutations have a better response to Akt monotherapy [106]. However, the number of cases (only one case with a partial response) was too small to convincingly make such an important conclusion. Nonetheless, the conclusion is consistent with other research showing that KRAS-mutant TNBC cells have the potential to alternatively activate Akt signalling [74]. The clinical limitation of this study was due to the small number of cases (N = 27), tumour heterogeneity, and the different breast cancer subtypes included.
The researchers of the above-mentioned trial [NCT00963547] also completely ignored any purposefully targeted therapy of breast cancer patients using MK-2206 treatment. In our opinion, the combination of MK-2206 with other therapeutics in certain subtypes of breast cancer, in which previous treatments with other drugs have failed, is more reasonable than MK-2206 monotherapy in such a diverse range of breast cancer subtypes. Nevertheless, considering the original function of these drugs, drugs that block PI3K/Akt signalling proteins warrant further investigation in the clinic to reach final approval.
Although most studies focused on Akt1, other Akt isoforms (Akt2 and Akt3) have shown opposing activities in different physiological and pathological conditions. Akt1 has crucial initiating effects on breast cancer, while Akt2 promotes breast cancer progression and cell migration [107]. Regarding the differences observed in the activities of other Akt isoforms, knockdown studies have indicated that Akt1 and Akt3 are responsible for clonogenic and DNA double-strand break repair activities, which lead to radio resistance in KRAS-mutated breast cancer cells; however, Akt2 does not have stimulating effects on the repair of double-strand break [108]. On the other hand, Akt3 is upregulated in breast cancer cell models showing resistance to Akt inhibitors such as MK2206. Furthermore, it has been shown that resistance to MK2206 can be reversed in acquired resistant breast cancer cells via Akt3 downregulation [109]. Higher level of Akt1 and Akt3 were detected in lymph node-positive and -negative luminal B breast cancer patients, respectively [110]. In addition, a higher Akt1 level is associated with unfavorable survival, whereas higher Akt2 expression showed a lower risk of progression in Her2+ breast cancer patients [111].
These results indicated Akt2 and Akt3 as attractive targets to develop new inhibitors against PI3K/Akt signaling. Pan-Akt inhibitors or Akt2/Akt3 inhibitors may effectively sensitize breast cancer cells to chemotherapy. A study showed 2, 4, 6-trisubstituted pyridine plays as a scaffold for developing pan-Akt inhibitors [112]. Therefore, developing pan-Akt inhibitors, such as LY294002, reduces the invasive activity of breast carcinoma [113]; however, clinical roles of Akt2 and Akt3 inhibitors should be taken into account in future clinical trials in order to overcome chemoresistance in breast cancer.
mTOR inhibitors
A third option for targeting PI3K/Akt signalling is mTORC1/2. Although there are currently no active clinical trials for Akt inhibitors in Her2+ breast cancer patients, mTOR inhibitors are under clinical investigation for HR+ and Her2+ subtypes. PI3K/Akt is a crucial signalling pathway in resistance to endocrine therapy. The effect of the dual mTORC inhibitor everolimus against HR+ breast cancer has been studied more than other mTOR inhibitors. In a phase II trial, everolimus was used in combination with fulvestrant to postpone resistance to fulvestrant alone in patients who had previously failed therapy with AIs [NCT00570921]. Time to progression as well as ORR and clinical benefit rate were estimated in 31 ER+ breast cancer patients who had a history of unsuccessful treatment with AIs. In this study, combinatorial therapy with fulvestrant and everolimus was effective. The median time to progression, ORR, and clinical benefit rate were estimated as 7.4 months (95% CI, 9-12.1), 13%, and 49% respectively; however, manageable stage 1 and 2 adverse events were observed, which were reverseable [114].
Furthermore, in a phase III trial (BOLERO-2, NCT00863655), everolimus combined with exemestane dramatically improved PFS in patients with HR+Her2- breast cancer who did not respond to AIs perfectly. Median PFS was doubled (7.8 months) in patients treated with combination everolimus and exemestane treatment compared with placebo-controlled cases (3.2 months), and a hazard ratio of 0.45 (95% CI, 0.38-0.54) was estimated for everolimus (N = 485). This study also compared PIK3CA-mutant versus PIK3CA-wild type patients to identify any impact of mutated PIK3CA on everolimus treatment. This study indicated that mTOR inhibitors are also effective with the PIK3CA-wild-type phenotype, and PI3K inhibitors, such as alpelisib, are recommended for PIK3CA-mutant breast cancer patients [115].
To date, rapamycin is being investigated for Her2+ breast cancer patients, and everolimus and deforolimus are being studied against ER+/Her2+ and ER+/Her2- breast cancer subtypes. As mentioned earlier, mTORC2 activates Akt through a positive feedback loop. Therefore, dual mTOR inhibitors (e.g., everolimus and deforolimus), which target both mTORC1 and mTORC2, are greatly effective against Akt signalling; otherwise, mTORC1 inhibitors (e.g., rapamycin) might not be beneficial for the suppression of overall Akt pathway activity. As mTORC1 inhibitors cannot completely inhibit PI3K/Akt signalling, partial activation of Akt may cause the recurrence of breast cancer through the development of BCSC and hypoxia in the TME.
Simultaneous pharmacological targeting of androgen receptor (AR) and PIK3CAH1047R decreases the viability of AR+ TNBC cells and would benefit from the current standard-of-care chemotherapy regimens for TNBC [116,117]. A recent study showed that PIK3CAH1047R/H1047L-mutated metaplastic breast cancers respond better to a combination of PIK3CA and MEK inhibitors BYL-719 and selumetinib [118]. Furthermore, in vivo combination therapy of PI3K and MAPK inhibitors displayed marked antitumour activity in metastatic breast cancers with genomic alterations of PIK3CA, Akt1, BRAF, and PTEN [118,119]. Recent findings have suggested that mutation of PTEN and PIK3CA trigger the inactivation of GSK3 by phosphorylation, thereby mediating response to mTOR inhibitors and mTORC1 inhibition-induced upregulation of PD-L1 in TNBC cancer cells [117,119,120]. Coussy et al. showed that mTOR and PI3K inhibitors have marked antitumour activity in vivo in TNBC harbouring genomic alterations of PIK3CA and Akt1 genes. Interestingly, TNBC cells resistant to enzalutamide show that PIK3CA and Akt1 are potential therapeutic targets [119,121].
Prospective clinical and pre-clinical directions
Constitutively activated Akt has been found in various breast tumour types. Emerging findings of Akt inhibitors suggest that Akt is a critical mediator of oncogenic signalling and participates in the formation of a chemoresistant phenotype in different subtypes of breast cancers. In the last 20 years, many preclinical trials have aimed to improve the effectiveness of antitumour therapy by inhibiting the oncogenic function of PI3K/Akt/mTOR signalling. Furthermore, resistance to CDK inhibitors has also been observed in breast cancer and more specifically, in TNBC patients with MDR proteins [122,123]. As endocrine and anti-Her2 therapies do not have a good effect against TNBC, a combination of cell cycle inhibitors (e.g., CDK and microtubule inhibitors) and immune checkpoint inhibitors have been suggested as a novel therapeutic strategy for TNBC.
In our research over the past few years, we have focused on the role of the Akt pathway in resistance to chemotherapy. Previously, we found that MDA-MB-231 TNBC cells have overactivated Akt in response to lapatinib treatment [8,18]. Having observed increased phosphorylation levels of Akt in lapatinib-treated MDA-MB-231 cells, we sought to observe whether MDA-MB231 cells respond to Akt inhibitors or not. Therefore, we treated TNBC cells with capivasertib, as the most frequent Akt inhibitor in clinical studies. Surprisingly, we observed that capivasertib did not inhibit Akt phosphorylation, and instead, significantly increased the level of Akt phosphorylation at S473. Accordingly, we postulated that both TKIs and Akt inhibitors might trigger Akt activation, which leads to chemoresistance of TNBC cells. These results are consistent with previous studies in which activation of the Akt pathway has been reported in TNBC cells as a response to Akt inhibitors [87]. For further elucidation, we analysed the expressions of several proteins related to Akt pathways, including Akt, PIK3CA, GSK-3β, KRAS, and c-Myc in 10 TNBC cell lines and patients with TNBC. We found that most TNBC cell lines had lower Akt levels, except HCC1143 and HCC1187, which may be related to the resistance to chemotherapy. In contrast, KRAS and c-Myc are strongly expressed in TNBC cell lines [124]. Furthermore, through comparison of data obtained from TNBC and non-TNBC patients, we found that the average expressions of EGFR and tumour suppressors p21 and RB-1 are lower in TNBC patients compared with non-TNBC patients (breast cancer, N = 113; TNBC, N = 115) (unpublished data). Lower RB-1 and p21 levels are associated with downregulation of GLUT1, lower glycolysis, and consumption of alternative compounds like glutamine as a replacement energy source when glucose availability is restricted. Of note, Akt not only affects regulation of genes in the RB-1/E2F/Myc pathway, but also reprograms the metabolism of TNBC cells to provide TNBC-specific TME, which finally leads to resistance of TNBC cells to chemotherapy [125]. Using clinical data, we also found that expression of epigenetic regulators, such as DNA methyltransferases DNMT3A, DNMT3B, and DNMT1, but not histone deacetylases (HDACs) such as HDAC3A and SIRT1, is significantly increased in TNBC compared with non-TNBC patients (unpublished data). We also recently highlighted the effects of JAK/STAT and Akt pathways on resistance to radiotherapy in exploring the possible roles of the IFN/STAT1/Akt pathway in radioresistance. We found that IFN-related DNA damage resistance, STAT1-dependent, and Akt-related genes derived from a primary breast cancer gene expression profile were more broadly associated with resistance to X-ray-induced DNA damage and doxorubicin in breast cancer. Taken together, we suggest that STAT1 and Akt are critical mediators of oncogenic signalling, which participate in the formation of the radioresistant phenotype in breast cancer [126].
Moreover, it should be noted that TNBC patients with MDR have basal-like breast cancer with high expression of tumour-infiltrating lymphocytes and PD-L1. Hence, PD-L1 inhibitors, such as atezolizumab, in combination with routine chemotherapy, has been suggested to be a novel treatment approach for TNBC [127,128]. In this regard, and supporting the investigation of IMpassion130 clinical trials, atezolizumab plus nab-paclitaxel prolonged PFS among patients with TNBC in both the intention-to-treat population and the PD-L1-positive subgroup. Combined PD-L1 inhibitor and nab-paclitaxel treatment in both PD-L1-positive patients and the overall population of the phase III trial significantly improved PFS [129-131]. Immunotherapy with other checkpoint inhibitors that target CTLA-4 and PD-1 is another therapeutic approach that has been successfully applied to the treatment of breast cancer. Growth factor receptors, such as Her2 and EGFR, and immune checkpoints, such as CTLA-4 and PD-1, induce signalling pathways thus leading to cell proliferation and induction of alternative PI3K/Akt signalling [132,133]. Therefor, inhibition of Akt signalling represents an attractive targeted therapy of breast cancer in the modern era of personalized medicine [134,135].
Apart from the Akt upstream RTK, Akt signalling can be activated by crosstalk with other signalling proteins. Therefore, mutations in other signalling proteins affect the Akt pathway and its response to Akt pathway inhibitors. As mentioned earlier, mutations of other signalling proteins, such as KRAS, PIK3CA, EGFR, and mTORC1/2, affect the efficacy of PI3K/Akt pathway inhibitors. In case KRAS is highly mutated, the chance of breast cancer cells acquiring resistance against EGFR/Her2 therapy is increased, and Akt signalling is highly activated in an EGFR-independent manner. On the other hand, according to BOLERO-2 clinical trials, PIK3CA mutations decrease the efficacy of other PI3K/Akt pathway inhibitors, such as everolimus [115]. Other clinical studies mentioned in the present article also highlighted that breast cancer cases possessing PIK3CA mutations, especially patients with ER+ and Her2- phenotypes, have a low response to Akt and mTOR inhibitors. However, the number of studied patients needs to be higher to confirm this finding. A crucial direction for future studies on Akt pathway inhibitors should be the clinical investigation of the negative impact of other signalling proteins on the efficacy of Akt pathway inhibitors.
According to the comprehensive assumptions of the present article, the following challenges of Akt inhibitors should be solved to offer the best treatment and overcome the drug resistance in breast cancer: (i) High heterogeneity and mutagenesis of breast tumours are the main challenges for overcoming drug resistance, incomplete responders, and relapse in individualized treatment of breast cancer. (ii) Inhibition of PI3K/Akt/mTOR may be tolerable because this pathway has crosstalk with immuno-oncologic pathways, such as RAS/MAPK and NF-κB. Certainly, combinatorial approaches to target other pathways and related genes in breast cancer cells have significant benefits in cancer patients undergoing chemotherapy and/or immunotherapy. The development of dose-limiting toxicities has often prevented the use of optimal therapeutic concentrations. (iii) The phenotype of breast cancer individuals and their subtype, as well as the mutations of signalling proteins of other pathways crucially affect the response to Akt pathway inhibitors. (iv) Biotransformation, bioavailability, affinity, and selectivity of Akt pathway inhibitors and biosafety are crucial challenges, and more clinical trials are required to confirm the pharmacokinetics and safety of Akt pathway inhibitors. (v) Epigenetic regulation of the Akt pathway affects the response of breast cancer patients to Akt pathway inhibitors. The activities of proteins, such as DNMTs, SIRTs, and HDACs, and activities of tumour suppressor and oncogenic non-coding microRNAs, should be taken into account while the therapeutic potential of PI3K/Akt signalling is under investigation. Of note, the ratio of microRNAs and other non-coding RNA expression profiles constitutes important mechanisms in controlling their response to Akt inhibitors. Thus, the balance of genetic and epigenetic mutations, as well as corresponding toxicity and adverse effects, should be taken into full account to achieve more stable efficacy during Akt pathway targeting. Based on recent advances reviewed in the present article, our research direction is to determine the mechanism of Akt-dependent chemoresistance and the roles of Akt modifications under the regulation of epigenetic factors and in response to different therapeutic strategies.
Acknowledgements
Parham Jabbarzadeh Kaboli and King-Hwa Ling equally conceived the ideas and organised the study. Parham Jabbarzadeh Kaboli and Saber Imani equally contributed in drafting the manuscript. The authors thank Ella Marushchenko’s studio (scientific-illustrations.com) for the illustration of Figures 1, 2 and 3. Figure 3, previously published in Pharmacological Research, redesigned and reprinted for the American Journal of Cancer Research with permission from Elsevier. The work is funded by the Ministry of Higher Education, Malaysia, through the Fundamental Research Grant Scheme (FRGS/1/2016/SKK10/UPM/01/1) awarded to PJK and KHL.
Disclosure of conflict of interest
None.
Abbreviations
- Ais
aromatase inhibitors
- BCSCs
breast cancer stem cells
- BCRP
breast cancer resistance protein
- CDK
cyclin-dependent kinase
- DNMT
DNA methyltransferase
- EGFR
epidermal growth factor receptor
- ER
oestrogen receptor
- FDA
food and drug administration
- 5-FU
5-Fluorouracil
- GSH
glutathione
- GSK
glycogen synthase kinase
- GRP78
glucose-regulated protein 78
- HER2
human epidermal growth factor receptor 2
- HIF-1α
hypoxia-inducible factor-1α
- HDAC
histone deacetylase
- IFN-γ
interferon-gamma
- IL-6
Interleukin-6
- JAK
janus kinase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MDR
multidrug resistance
- mTOR
mammalian target of rapamycin
- Nrf2
nuclear factor erythroid 2-related factor 2
- OCT4
octamer-binding transcription factor
- ORR
overall response rate
- PD-L1
programmed death-ligand 1
- P-gp
p-glycoprotein
- PI3K
phosphatidylinositol-3-kinase
- PIK3CA
PI3K catalytic subunit α
- PFS
progression-free survival
- PTEN
phosphatase and tensin homolog
- RTKs
receptor tyrosine kinases
- ROS
reactive oxygen species
- STAT
signal transducers and activators of transcription
- T-DM1
ado-trastuzumab emtansine
- TKI
tyrosine kinase inhibitor
- TME
tumour microenvironment
- TNBC
triple-negative breast cancer
References
- 1.Jabbarzadeh Kaboli P, Rahmat A, Ismail P, Ling KH. Targets and mechanisms of berberine, a natural drug with potential to treat cancer with special focus on breast cancer. Eur J Pharmacol. 2014;740:584–595. doi: 10.1016/j.ejphar.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 2.Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol. 2019;10:245. doi: 10.3389/fendo.2019.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Xu R, Yuan H, Zhang Y, Cheng S. Single-cell RNA sequencing reveals novel gene expression signatures of trastuzumab treatment in HER2+ breast cancer: a pilot study. Medicine. 2019;98:e15872. doi: 10.1097/MD.0000000000015872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodríguez CE, Berardi DE, Abrigo M, Todaro LB, Bal de Kier Joffé ED, Fiszman GL. Breast cancer stem cells are involved in Trastuzumab resistance through the HER2 modulation in 3D culture. J Cell Biochem. 2018;119:1381–1391. doi: 10.1002/jcb.26298. [DOI] [PubMed] [Google Scholar]
- 5.Shah D, Osipo C. Cancer stem cells and HER2 positive breast cancer: the story so far. Genes Dis. 2016;3:114–123. doi: 10.1016/j.gendis.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuyàs E, Gumuzio J, Verdura S, Brunet J, Bosch-Barrera J, Martin-Castillo B, Alarcón T, Encinar JA, Martin ÁG, Menendez JA. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: a potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging. 2020;12:4794–4814. doi: 10.18632/aging.102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vagia E, Mahalingam D, Cristofanilli M. The landscape of targeted therapies in TNBC. Cancers. 2020;12:916. doi: 10.3390/cancers12040916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jabbarzadeh Kaboli P, Ling KH. Lapatinib as a dual tyrosine kinase inhibitor unexpectedly activates Akt in MDA-MB-231 triple-negative breast cancer cells. Lett Drug Des Discov. 2020;17:179292. [Google Scholar]
- 9.Kaboli PJ, Rahmat A, Ismail P, Ling KH. MicroRNA-based therapy and breast cancer: a comprehensive review of novel therapeutic strategies from diagnosis to treatment. Pharmacol Res. 2015;97:104–121. doi: 10.1016/j.phrs.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Zhai Z, Li H, Wang X, Huang Y, Su X. Guajadial reverses multidrug resistance by inhibiting ABC transporter expression and suppressing the PI3K/Akt pathway in drug-resistant breast cancer cells. Chem Biol Interact. 2019;305:98–104. doi: 10.1016/j.cbi.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 11.Qian J, Xia M, Liu W, Li L, Yang J, Mei Y, Meng Q, Xie Y. Glabridin resensitizes p-glycoprotein-overexpressing multidrug-resistant cancer cells to conventional chemotherapeutic agents. Eur J Pharmacol. 2019;852:231–243. doi: 10.1016/j.ejphar.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Tang K, Yu Y, Zhu L, Xu P, Chen J, Ma J, Zhang H, Fang H, Sun W, Zhou L, Wei K, Li F, Lv J, Xie J, Liu Y, Huang B. Hypoxia-reprogrammed tricarboxylic acid cycle promotes the growth of human breast tumorigenic cells. Oncogene. 2019;38:6970–6984. doi: 10.1038/s41388-019-0932-1. [DOI] [PubMed] [Google Scholar]
- 13.Kaushik S, Shyam H, Agarwal S, Sharma R, Nag TC, Dwivedi AK, Balapure AK. Genistein potentiates Centchroman induced antineoplasticity in breast cancer via PI3K/Akt deactivation and ROS dependent induction of apoptosis. Life Sci. 2019;239:117073. doi: 10.1016/j.lfs.2019.117073. [DOI] [PubMed] [Google Scholar]
- 14.De Blasio A, Di Fiore R, Pratelli G, Drago-Ferrante R, Saliba C, Baldacchino S, Grech G, Scerri C, Vento R, Tesoriere G. A loop involving NRF2, miR-29b-1-5p and AKT, regulates cell fate of MDA-MB-231 triple-negative breast cancer cells. J Cell Physiol. 2019;235:1–9. doi: 10.1002/jcp.29062. [DOI] [PubMed] [Google Scholar]
- 15.Jabbarzadeh Kaboli P, Salimian F, Aghapour S, Xiang S, Zhao Q, Li M, Wu X, Du F, Zhao Y, Shen J, Cho CH, Xiao Z. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer - A comprehensive review from chemotherapy to immunotherapy. Pharmacol Res. 2020;156:104806. doi: 10.1016/j.phrs.2020.104806. [DOI] [PubMed] [Google Scholar]
- 16.Chen YH, Su CC, Deng W, Lock LF, Donovan PJ, Kayala MA, Baldi P, Lee HC, Chen Y, Wang PH. Mitochondrial akt signaling modulated reprogramming of somatic cells. Sci Rep. 2019;9:9919. doi: 10.1038/s41598-019-46359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Y, Shao D, Zhao Y, Zhang F, Zheng X, Tan Y, He K, Li J, Chen L. Berberine reverses hypoxia-induced chemoresistance in breast cancer through the inhibition of AMPK-HIF-1α. Int J Biol Sci. 2017;13:794–803. doi: 10.7150/ijbs.18969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jabbarzadeh Kaboli P, Leong MP, Ismail P, Ling KH. Antitumor effects of berberine against EGFR, ERK1/2, P38 and AKT in MDA-MB231 and MCF-7 breast cancer cells using molecular modelling and in vitro study. Pharmacol Rep. 2019;71:13–23. doi: 10.1016/j.pharep.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Wenner CE. Targeting mitochondria as a therapeutic target in cancer. J Cell Physiol. 2012;227:450–456. doi: 10.1002/jcp.22788. [DOI] [PubMed] [Google Scholar]
- 20.Dong X, Yang Y, Zhou Y, Bi X, Zhao N, Zhang Z, Li L, Hang Q, Zhang R, Chen D, Cao P, Yin Z, Luo L. Glutathione S-transferases P1 protects breast cancer cell from adriamycin-induced cell death through promoting autophagy. Cell Death Differ. 2019;26:2086–2099. doi: 10.1038/s41418-019-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou Y, Sarem M, Xiang S, Hu H, Xu W, Shastri VP. Autophagy inhibition enhances Matrine derivative MASM induced apoptosis in cancer cells via a mechanism involving reactive oxygen species-mediated PI3K/Akt/mTOR and Erk/p38 signaling. BMC Cancer. 2019;19:949. doi: 10.1186/s12885-019-6199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger CE, Qian Y, Liu G, Chen H, Chen X. p53, a target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J Biol Chem. 2012;287:30117–30127. doi: 10.1074/jbc.M112.367326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gul A, Leyland-Jones B, Dey N, De P. A combination of the PI3K pathway inhibitor plus cell cycle pathway inhibitor to combat endocrine resistance in hormone receptor-positive breast cancer: a genomic algorithm-based treatment approach. Am J Cancer Res. 2018;8:2359–2376. [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Yang G, Dong H. Everolimus reverses palbociclib resistance in er+ human breast cancer cells by inhibiting phosphatidylinositol 3-kinase(PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway. Med Sci Monit. 2019;25:77–86. doi: 10.12659/MSM.912929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cretella D, Ravelli A, Fumarola C, La Monica S, Digiacomo G, Cavazzoni A, Alfieri R, Biondi A, Generali D, Bonelli M, Petronini PG. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J Exp Clin cancer Res. 2018;37:72. doi: 10.1186/s13046-018-0741-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chi Y, Xue J, Huang S, Xiu B, Su Y, Wang W, Guo R, Wang L, Li L, Shao Z, Jin W, Wu Z, Wu J. Cap G promotes resistance to paclitaxel in breast cancer through transactivation of PIK3R1/P50. Theranostics. 2019;9:6840–6855. doi: 10.7150/thno.36338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Ren Q, Cai Y, Lin T, Zuo W, Wang J, Lin R, Zhu L, Wang P, Dong H, Zhao H, Huang L, Fu Y, Yang S, Tan J, Lan X, Wang S. Mesenchymal stem cells drive paclitaxel resistance in ErbB2/ErbB3-coexpressing breast cancer cells via paracrine of neuregulin 1. Biochem Biophys Res Commun. 2018;501:212–219. doi: 10.1016/j.bbrc.2018.04.218. [DOI] [PubMed] [Google Scholar]
- 28.Lin YH, Chen BY, Lai WT, Wu SF, Guh JH, Cheng AL, Hsu LC. The Akt inhibitor MK-2206 enhances the cytotoxicity of paclitaxel (Taxol) and cisplatin in ovarian cancer cells. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:19–31. doi: 10.1007/s00210-014-1032-y. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Meng T, Zheng X, Liu Y, Hao R, Yan Y, Chen S, You H, Xing J, Dong Y. Transgelin 2 promotes paclitaxel resistance, migration, and invasion of breast cancer by directly interacting with PTEN and activating PI3K/Akt/GSK-3β pathway. Mol Cancer Ther. 2019;18:2457–2468. doi: 10.1158/1535-7163.MCT-19-0261. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Huang X, Lee CK, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010;29:4225–4236. doi: 10.1038/onc.2010.180. [DOI] [PubMed] [Google Scholar]
- 31.Cai J, Chen S, Zhang W, Zheng X, Hu S, Pang C, Lu J, Xing J, Dong Y. Salvianolic acid A reverses paclitaxel resistance in human breast cancer MCF-7 cells via targeting the expression of transgelin 2 and attenuating PI3 K/Akt pathway. Phytomedicine. 2014;21:1725–1732. doi: 10.1016/j.phymed.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Hayasaka N, Takada K, Nakamura H, Arihara Y, Kawano Y, Osuga T, Murase K, Kikuchi S, Iyama S, Emori M, Sugita S, Hasegawa T, Takasawa A, Miyanishi K, Kobune M, Kato J. Combination of eribulin plus AKT inhibitor evokes synergistic cytotoxicity in soft tissue sarcoma cells. Sci Rep. 2019;9:5759. doi: 10.1038/s41598-019-42300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, Patel CG, Zohren F, Wolf JL. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am J Hematol. 2016;91:400–405. doi: 10.1002/ajh.24300. [DOI] [PubMed] [Google Scholar]
- 34.Owusu-Brackett N, Evans KW, Akcakanat A, Yuca E, Tapia C, Rizvi YQ, Dumbrava EI, Janku F, Meric-Bernstam F. TAK228 enhances antitumor activity of eribulin in triple negative breast cancer. Oncotarget. 2019;10:5011–5019. doi: 10.18632/oncotarget.27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen W, Marcinkowski E, Luyimbazi D, Luu T, Xing Q, Yan J, Wang Y, Wu J, Guo Y, Tully D, Han ES, Yost SE, Yuan Y, Yim JH. Eribulin synergistically increases anti-tumor activity of an mTOR inhibitor by inhibiting pAKT/pS6K/pS6 in triple negative breast cancer. Cells. 2019;8:1010. doi: 10.3390/cells8091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi EJ, Kim GH. 5-Fluorouracil combined with apigenin enhances anticancer activity through induction of apoptosis in human breast cancer MDA-MB-453 cells. Oncol Rep. 2009;22:1533–1537. doi: 10.3892/or_00000598. [DOI] [PubMed] [Google Scholar]
- 37.Vinod BS, Antony J, Nair HH, Puliyappadamba VT, Saikia M, Narayanan SS, Bevin A, Anto RJ. Mechanistic evaluation of the signaling events regulating curcumin-mediated chemosensitization of breast cancer cells to 5-fluorouracil. Cell Death Dis. 2013;4:e505. doi: 10.1038/cddis.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao X, Tu Y, Xu Y, Guo Y, Yao F, Zhang X. Endoplasmic reticulum stress confers 5-fluorouracil resistance in breast cancer cell via the GRP78/OCT4/lncRNA MIAT/AKT pathway. Am J Cancer Res. 2020;10:838–855. [PMC free article] [PubMed] [Google Scholar]
- 39.Shah AN, Gradishar WJ. Adjuvant anthracyclines in breast cancer: what is their role? Oncologist. 2018;23:1153–1161. doi: 10.1634/theoncologist.2017-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer KB, Martino Andrade AJ, Venturelli AC, Kita DH, Machado DLB, Adams Philipsen R, do Nascimento Silva AA, Cantão I, Moreira DL, da Silva Junior VA, Stumpp T, Morais RN. Identification of a critical window for ganciclovir-induced disruption of testicular development in rats. Toxicol Sci. 2018;162:488–498. doi: 10.1093/toxsci/kfx276. [DOI] [PubMed] [Google Scholar]
- 41.Wu ZH, Lin C, Liu MM, Zhang J, Tao ZH, Hu XC. Src inhibition can synergize with gemcitabine and reverse resistance in triple negative breast cancer cells via the AKT/c-Jun pathway. PLoS One. 2016;11:e0169230. doi: 10.1371/journal.pone.0169230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howlader N, Cronin KA, Kurian AW, Andridge R. Differences in breast cancer survival by molecular subtypes in the united states. Cancer Epidemiol Biomarkers Prev. 2018;27:619–626. doi: 10.1158/1055-9965.EPI-17-0627. [DOI] [PubMed] [Google Scholar]
- 43.Gahlaut R, Bennett A, Fatayer H, Dall BJ, Sharma N, Velikova G, Perren T, Dodwell D, Lansdown M, Shaaban AM. Effect of neoadjuvant chemotherapy on breast cancer phenotype, ER/PR and HER2 expression - implications for the practising oncologist. Eur J Cancer. 2016;60:40–48. doi: 10.1016/j.ejca.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 44.Yao JH, Zhang WQ, Shao Y, Zhu Y, Liu J, Jin GS, Yang Y. Molecular phenotype discordance between primary and synchronous metastatic lesions of breast cancer and its determinant role in guiding treatment decisions: a case report. Am J Transl Res. 2020;12:5866–5873. [PMC free article] [PubMed] [Google Scholar]
- 45.Ye L, Lin C, Wang X, Li Q, Li Y, Wang M, Zhao Z, Wu X, Shi D, Xiao Y, Ren L, Jian Y, Yang M, Ou R, Deng G, Ouyang Y, Chen X, Li J, Song L. Epigenetic silencing of SALL2 confers tamoxifen resistance in breast cancer. EMBO Mol Med. 2019;11:e10638–e10638. doi: 10.15252/emmm.201910638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, Wang J, Zhou X, Zhang H, Zhang Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020;111:47–58. doi: 10.1111/cas.14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Y, Hu W, Liu T, Rana U, Aguilera-Barrantes I, Kong A, Kumar SN, Wang B, Gao P, Wang X, Duan Y, Shi A, Song D, Yang M, Li S, Han B, Zhao G, Fan Z, Miao QR. Nogo-B receptor increases the resistance of estrogen receptor positive breast cancer to paclitaxel. Cancer Lett. 2018;419:233–244. doi: 10.1016/j.canlet.2018.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han R, Gu S, Zhang Y, Luo A, Jing X, Zhao L, Zhao X, Zhang L. Estrogen promotes progression of hormone-dependent breast cancer through CCL2-CCR2 axis by upregulation of Twist via PI3K/AKT/NF-κB signaling. Sci Rep. 2018;8:9575. doi: 10.1038/s41598-018-27810-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang S, Chen Y, Liang ZM, Li NN, Liu Y, Zhu Y, Liao D, Zhou XZ, Lu KP, Yao Y, Luo ML. Targeting pin1 by all-trans retinoic acid (ATRA) overcomes tamoxifen resistance in breast cancer via multifactorial mechanisms. Front cell Dev Biol. 2019;7:322. doi: 10.3389/fcell.2019.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, Danes CG, Guo H, Lan KH, Ensor J, Hittelman W, Hung MC, Yu D. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009;69:3425–3432. doi: 10.1158/0008-5472.CAN-08-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang Y, Zhang Y, Liu S, Sun Z, Wang C, Li L, Zhou W, Cheng S. 14-3-3ζ binds to and stabilizes phospho-beclin 1(S295) and induces autophagy in hepatocellular carcinoma cells. J Cell Mol Med. 2020;24:954–964. doi: 10.1111/jcmm.14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ge X, Zhao Y, Dong L, Seng J, Zhang X, Dou D. NAMPT regulates PKM2 nuclear location through 14-3-3ζ: Conferring resistance to tamoxifen in breast cancer. J Cell Physiol. 2019;234:23409–23420. doi: 10.1002/jcp.28910. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Gao L, Li B, Liu C, Hong S, Min J, Hong L. Knockdown of hypoxia-inducible factor 1α (HIF-1α) promotes autophagy and inhibits phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway in ovarian cancer cells. Med Sci Monit. 2019;25:4250–4263. doi: 10.12659/MSM.915730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng M, Zhang W, Lingling Y, Tan J, Chen Z. HIF-1a regulates hypoxia-induced autophagy via translocation of ANKRD37 in colon cancer. Exp Cell Res. 2020;395:112175. doi: 10.1016/j.yexcr.2020.112175. [DOI] [PubMed] [Google Scholar]
- 55.Mabuchi S, Ohmichi M, Kimura A, Nishio Y, Arimoto-Ishida E, Yada-Hashimoto N, Tasaka K, Murata Y. Estrogen inhibits paclitaxel-induced apoptosis via the phosphorylation of apoptosis signal-regulating kinase 1 in human ovarian cancer cell lines. Endocrinology. 2004;145:49–58. doi: 10.1210/en.2003-0792. [DOI] [PubMed] [Google Scholar]
- 56.Turner NC, Alarcón E, Armstrong AC, Philco M, López Chuken YA, Sablin MP, Tamura K, Gómez Villanueva A, Pérez-Fidalgo JA, Cheung SYA, Corcoran C, Cullberg M, Davies BR, deBruin EC, Foxley A, Lindemann JPO, Maudsley R, Moschetta M, Outhwaite E, Pass M, Rugman P, Schiavon G, Oliveira M. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA. Ann Oncol. 2019;30:774–780. doi: 10.1093/annonc/mdz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, Simigdala N, Polanska UM, Campbell H, Rani A, Liccardi G, Johnston S, Davies BR, Dowsett M, Martin LA. AKT antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther. 2015;14:2035–2048. doi: 10.1158/1535-7163.MCT-15-0143. [DOI] [PubMed] [Google Scholar]
- 58.Chen IC, Hsiao LP, Huang IW, Yu HC, Yeh LC, Lin CH, Wei-Wu Chen T, Cheng AL, Lu YS. Phosphatidylinositol-3 kinase inhibitors, buparlisib and alpelisib, sensitize estrogen receptor-positive breast cancer cells to tamoxifen. Sci Rep. 2017;7:9842. doi: 10.1038/s41598-017-10555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vernieri C, Corti F, Nichetti F, Ligorio F, Manglaviti S, Zattarin E, Rea CG, Capri G, Bianchi GV, de Braud F. Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications. Breast Cancer Res. 2020;22:33. doi: 10.1186/s13058-020-01271-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rozeboom B, Dey N, De P. ER+ metastatic breast cancer: past, present, and a prescription for an apoptosis-targeted future. Am J Cancer Res. 2019;9:2821–2831. [PMC free article] [PubMed] [Google Scholar]
- 61.Hoeflich KP, Guan J, Edgar KA, O’Brien C, Savage H, Wilson TR, Neve RM, Friedman LS, Wallin JJ. The PI3K inhibitor taselisib overcomes letrozole resistance in a breast cancer model expressing aromatase. Genes Cancer. 2016;7:73–85. doi: 10.18632/genesandcancer.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gilabert M, Launay S, Gonçalves A. Exemestane-everolimus in HER2-negative, hormonal receptor-positive, post-menopausal metastatic breast cancer with resistance to non-steroidal aromatase inhibitor: a new option. Bull Cancer. 2014;101:325–333. doi: 10.1684/bdc.2014.1910. [DOI] [PubMed] [Google Scholar]
- 63.Vilquin P, Villedieu M, Grisard E, Ben Larbi S, Ghayad SE, Heudel PE, Bachelot T, Corbo L, Treilleux I, Vendrell JA, Cohen PA. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer. 2013;133:1589–1602. doi: 10.1002/ijc.28182. [DOI] [PubMed] [Google Scholar]
- 64.Sun B, Ding L, Wu S, Meng X, Song S. Combined treatment with everolimus and fulvestrant reversed anti-HER2 resistance in a patient with refractory advanced breast cancer: a case report. Onco Targets Ther. 2016;9:3997–4003. doi: 10.2147/OTT.S104398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amaral C, Augusto TV, Tavares-da-Silva E, Roleira FMF, Correia-da-Silva G, Teixeira N. Hormone-dependent breast cancer: targeting autophagy and PI3K overcomes exemestane-acquired resistance. J Steroid Biochem Mol Biol. 2018;183:51–61. doi: 10.1016/j.jsbmb.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 66.Lui A, New J, Ogony J, Thomas S, Lewis-Wambi J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer. 2016;16:487. doi: 10.1186/s12885-016-2490-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Citi V, Del Re M, Martelli A, Calderone V, Breschi MC, Danesi R. Phosphorylation of AKT and ERK1/2 and mutations of PIK3CA and PTEN are predictive of breast cancer cell sensitivity to everolimus in vitro. Cancer Chemother Pharmacol. 2018;81:745–754. doi: 10.1007/s00280-018-3543-6. [DOI] [PubMed] [Google Scholar]
- 68.Petrossian K, Nguyen D, Lo C, Kanaya N, Somlo G, Cui YX, Huang CS, Chen S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res Treat. 2018;170:499–506. doi: 10.1007/s10549-018-4779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Shea J, Cremona M, Morgan C, Milewska M, Holmes F, Espina V, Liotta L, O’Shaughnessy J, Toomey S, Madden SF, Carr A, Elster N, Hennessy BT, Eustace AJ. A preclinical evaluation of the MEK inhibitor refametinib in HER2-positive breast cancer cell lines including those with acquired resistance to trastuzumab or lapatinib. Oncotarget. 2017;8:85120–85135. doi: 10.18632/oncotarget.19461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu KC, Cheng KS, Wang YW, Chen YF, Wong KL, Su TH, Chan P, Leung YM. Perturbation of akt signaling, mitochondrial potential, and ADP/ATP ratio in acidosis-challenged rat cortical astrocytes. J Cell Biochem. 2017;118:1108–1117. doi: 10.1002/jcb.25725. [DOI] [PubMed] [Google Scholar]
- 71.El Guerrab A, Zegrour R, Nemlin CC, Vigier F, Cayre A, Penault-Llorca F, Rossignol F, Bignon YJ. Differential impact of EGFR-targeted therapies on hypoxia responses: implications for treatment sensitivity in triple-negative metastatic breast cancer. PLoS One. 2011;6:e25080. doi: 10.1371/journal.pone.0025080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao M, Howard EW, Parris AB, Guo Z, Zhao Q, Ma Z, Xing Y, Liu B, Edgerton SM, Thor AD, Yang X. Activation of cancerous inhibitor of PP2A (CIP2A) contributes to lapatinib resistance through induction of CIP2A-Akt feedback loop in ErbB2-positive breast cancer cells. Oncotarget. 2017;8:58847–58864. doi: 10.18632/oncotarget.19375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwon YS, Chun SY, Nam KS, Kim S. Lapatinib sensitizes quiescent MDA-MB-231 breast cancer cells to doxorubicin by inhibiting the expression of multidrug resistance-associated protein-1. Oncol Rep. 2015;34:884–890. doi: 10.3892/or.2015.4047. [DOI] [PubMed] [Google Scholar]
- 74.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3:112–123. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 75.Yang ZY, Yang L, Xu CW, Wang XJ, Lei L. An insertion mutation of ERBB2 enhances breast cancer cell growth and confers resistance to lapatinib through AKT signaling pathway. Biol Open. 2020;9:bio047662. doi: 10.1242/bio.047662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim JW, Kim DK, Min A, Lee KH, Nam HJ, Kim JH, Kim JS, Kim TY, Im SA, Park IA. Amphiregulin confers trastuzumab resistance via AKT and ERK activation in HER2-positive breast cancer. J Cancer Res Clin Oncol. 2016;142:157–165. doi: 10.1007/s00432-015-2012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen YC, Li HY, Liang JL, Ger LP, Chang HT, Hsiao M, Calkins MJ, Cheng HC, Chuang JH, Lu PJ. CTMP, a predictive biomarker for trastuzumab resistance in HER2-enriched breast cancer patient. Oncotarget. 2017;8:29699–29710. doi: 10.18632/oncotarget.10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiu N, He YF, Zhang SM, Zhan YT, Han GD, Jiang M, He WX, Zhou J, Liang HL, Ao X, Xia HM, Li J, Yang YY, He ZM, Zou ZZ, Li HS. Cullin7 enhances resistance to trastuzumab therapy in Her2 positive breast cancer via degrading IRS-1 and downregulating IGFBP-3 to activate the PI3K/AKT pathway. Cancer Lett. 2019;464:25–36. doi: 10.1016/j.canlet.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 79.La Ferla M, Lessi F, Aretini P, Pellegrini D, Franceschi S, Tantillo E, Menicagli M, Marchetti I, Scopelliti C, Civita P, DeAngelis C, Diodati L, Bertolini I, Roncella M, McDonnell LA, Hochman J, Del Re M, Scatena C, Naccarato AG, Fontana A, Mazzanti CM. ANKRD44 gene silencing: a putative role in trastuzumab resistance in Her2-like breast cancer. Front Oncol. 2019;9:547. doi: 10.3389/fonc.2019.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peddi PF, Hurvitz SA. Ado-trastuzumab emtansine (T-DM1) in human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2014;6:202–209. doi: 10.1177/1758834014539183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hunter FW, Barker HR, Lipert B, Rothé F, Gebhart G, Piccart-Gebhart MJ, Sotiriou C, Jamieson SMF. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2020;122:603–612. doi: 10.1038/s41416-019-0635-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Loi S, Giobbie-Hurder A, Gombos A, Bachelot T, Hui R, Curigliano G, Campone M, Biganzoli L, Bonnefoi H, Jerusalem G, Bartsch R, Rabaglio-Poretti M, Kammler R, Maibach R, Smyth MJ, Di Leo A, Colleoni M, Viale G, Regan MM, André F. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019;20:371–382. doi: 10.1016/S1470-2045(18)30812-X. [DOI] [PubMed] [Google Scholar]
- 83.Li B, Tao W, Shao-Hua Z, Ze-Rui Q, Fu-Quan J, Fan L, Ze-Fei J. Remarkable response with pembrolizumab plus albumin-bound paclitaxel in 2 cases of HER2-positive metastatic breast cancer who have failed to multi-anti-HER2 targeted therapy. Cancer Biol Ther. 2018;19:292–295. doi: 10.1080/15384047.2017.1414761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang M, Chen H, Zhou L, Huang X, Su F, Wang P. Identification of SOCS family members with prognostic values in human ovarian cancer. Am J Transl Res. 2020;12:1824–1838. [PMC free article] [PubMed] [Google Scholar]
- 85.Nakayama Y, Mimura K, Tamaki T, Shiraishi K, Kua LF, Koh V, Ohmori M, Kimura A, Inoue S, Okayama H, Suzuki Y, Nakazawa T, Ichikawa D, Kono K. Phospho-STAT1 expression as a potential biomarker for anti-PD-1/anti-PD-L1 immunotherapy for breast cancer. Int J Oncol. 2019;54:2030–2038. doi: 10.3892/ijo.2019.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Franklin DA, James JL, Axelrod ML, Balko JM. MEK inhibition activates STAT signaling to increase breast cancer immunogenicity via MHC-I expression. Cancer Drug Resist. 2020;3:603–612. doi: 10.20517/cdr.2019.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van derNoord VE, McLaughlin RP, Smid M, Foekens JA, Martens JWM, Zhang Y, van de Water B. An increased cell cycle gene network determines MEK and Akt inhibitor double resistance in triple-negative breast cancer. Sci Rep. 2019;9:13308. doi: 10.1038/s41598-019-49809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bousoik E, Nabiee R, Amirrad F, Nichols A, Witt R, Mahdipoor P, Montazeri Aliabadi H. Heterogeneity and plasticity of human breast cancer cells in response to molecularly-targeted drugs. Front Oncol. 2019;9:1070. doi: 10.3389/fonc.2019.01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ginzburg YZ, Feola M, Zimran E, Varkonyi J, Ganz T, Hoffman R. Dysregulated iron metabolism in polycythemia vera: etiology and consequences. Leukemia. 2018;32:2105–2116. doi: 10.1038/s41375-018-0207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rädler PD, Wehde BL, Wagner KU. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol Cell Endocrinol. 2017;451:31–39. doi: 10.1016/j.mce.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stover DG, Gil Del Alcazar CR, Brock J, Guo H, Overmoyer B, Balko J, Xu Q, Bardia A, Tolaney SM, Gelman R, Lloyd M, Wang Y, Xu Y, Michor F, Wang V, Winer EP, Polyak K, Lin NU. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer. 2018;4:10. doi: 10.1038/s41523-018-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Shaughnessy J, De Michele A, Ma CX, Richards P, Yardley DA, Wright GS, Kalinsky K, Steis R, Diab S, Kennealey G, Geschwindt R, Jiang W, Rugo HS. A randomized, double-blind, phase 2 study of ruxolitinib or placebo in combination with capecitabine in patients with advanced HER2-negative breast cancer and elevated C-reactive protein, a marker of systemic inflammation. Breast Cancer Res Treat. 2018;170:547–557. doi: 10.1007/s10549-018-4770-6. [DOI] [PubMed] [Google Scholar]
- 93.Saleh R, Taha ZR, Sasidharan Nair V, Alajez MN, Elkord E. PD-L1 blockade by atezolizumab downregulates signaling pathways associated with tumor growth, metastasis, and hypoxia in human triple negative breast cancer. Cancers. 2019;11:1050. doi: 10.3390/cancers11081050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Henschel V, Molinero L, Chui SY, Maiya V, Husain A, Winer EP, Loi S, Emens LA. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59. doi: 10.1016/S1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 95.Cortés J, André F, Gonçalves A, Kümmel S, Martín M, Schmid P, Schuetz F, Swain SM, Easton V, Pollex E, Deurloo R, Dent R. IMpassion132 Phase III trial: atezolizumab and chemotherapy in early relapsing metastatic triple-negative breast cancer. Futur Oncol. 2019;15:1951–1961. doi: 10.2217/fon-2019-0059. [DOI] [PubMed] [Google Scholar]
- 96.Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV, Sánchez V, Sanders ME, Cook RS, Pilkinton MA, Mallal SA, Wang K, Miller VA, Stephens PJ, Yelensky R, Doimi FD, Gómez H, Ryzhov SV, Darcy PK, Arteaga CL, Balko JM. Correction: RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint Inhibitors. Clin cancer Res. 2019;25:1437. doi: 10.1158/1078-0432.CCR-18-4264. [DOI] [PubMed] [Google Scholar]
- 97.Chen M, Pockaj B, Andreozzi M, Barrett MT, Krishna S, Eaton S, Niu R, Anderson KS. JAK2 and PD-L1 amplification enhance the dynamic expression of PD-L1 in triple-negative breast cancer. Clin Breast Cancer. 2018;18:e1205–e1215. doi: 10.1016/j.clbc.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 98.Choi C, Kwon J, Lim S, Helfman DM. Integrin β1, myosin light chain kinase and myosin IIA are required for activation of PI3K-AKT signaling following MEK inhibition in metastatic triple negative breast cancer. Oncotarget. 2016;7:63466–63487. doi: 10.18632/oncotarget.11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lebeau J, Rainbolt TK, Wiseman RL. Coordinating mitochondrial biology through the stress-responsive regulation of mitochondrial proteases. Int Rev Cell Mol Biol. 2018;340:79–128. doi: 10.1016/bs.ircmb.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jabbarzadeh Kaboli P, Zhang L, Xiang S, Shen J, Li M, Zhao Y, Wu X, Zhao Q, Zhang H, Lin L, Yin J, Wu Y, Wan L, Yi T, Li X, Cho CH, Li J, Xiao Z, Wen Q. Molecular markers of regulatory T cells in cancer immunotherapy with special focus on acute myeloid leukemia (AML) - a systematic review. Curr Med Chem. 2019;26:175370. doi: 10.2174/0929867326666191004164041. [DOI] [PubMed] [Google Scholar]
- 101.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 102.Xu YY, Yu HR, Sun JY, Zhao Z, Li S, Zhang XF, Liao ZX, Cui MK, Li J, Li C, Zhang Q. Upregulation of PITX2 promotes letrozole resistance via transcriptional activation of IFITM1 signaling in breast cancer cells. Cancer Res Treat. 2019;51:576–592. doi: 10.4143/crt.2018.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, Yamashita T, Lu YS, Inoue K, Takahashi M, Pápai Z, Longin AS, Mills D, Wilke C, Hirawat S, Juric D. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380:1929–1940. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 104.Mayer IA, Prat A, Egle D, Blau S, Fidalgo JAP, Gnant M, Fasching PA, Colleoni M, Wolff AC, Winer EP, Singer CF, Hurvitz S, Estévez LG, van Dam PA, Kümmel S, Mundhenke C, Holmes F, Babbar N, Charbonnier L, Diaz-Padilla I, Vogl FD, Sellami D, Arteaga CL. A phase II randomized study of neoadjuvant letrozole plus alpelisib for hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer (NEO-ORB) Clin Cancer Res. 2019;25:2975–2987. doi: 10.1158/1078-0432.CCR-18-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hudis C, Swanton C, Janjigian YY, Lee R, Sutherland S, Lehman R, Chandarlapaty S, Hamilton N, Gajria D, Knowles J, Shah J, Shannon K, Tetteh E, Sullivan DM, Moreno C, Yan L, Han HS. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15:R110. doi: 10.1186/bcr3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xing Y, Lin NU, Maurer MA, Chen H, Mahvash A, Sahin A, Akcakanat A, Li Y, Abramson V, Litton J, Chavez-MacGregor M, Valero V, Piha-Paul SA, Hong D, Do KA, Tarco E, Riall D, Eterovic AK, Wulf GM, Cantley LC, Mills GB, Doyle LA, Winer E, Hortobagyi GN, Gonzalez-Angulo AM, Meric-Bernstam F. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019;21:78. doi: 10.1186/s13058-019-1154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hinz N, Jücker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun Signal. 2019;17:154. doi: 10.1186/s12964-019-0450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Toulany M, Maier J, Iida M, Rebholz S, Holler M, Grottke A, Jüker M, Wheeler DL, Rothbauer U, Rodemann HP. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov. 2017;3:17072. doi: 10.1038/cddiscovery.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stottrup C, Tsang T, Chin YR. Upregulation of AKT3 confers resistance to the AKT inhibitor MK2206 in breast cancer. Mol Cancer Ther. 2016;15:1964–1974. doi: 10.1158/1535-7163.MCT-15-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonin S, Pracella D, Barbazza R, Dotti I, Boffo S, Stanta G. PI3K/AKT signaling in breast cancer molecular subtyping and lymph node involvement. Dis Markers. 2019;2019:7832376. doi: 10.1155/2019/7832376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Christodoulou C, Oikonomopoulos G, Koliou GA, Kostopoulos I, Kotoula V, Bobos M, Pentheroudakis G, Lazaridis G, Skondra M, Chrisafi S, Koutras A, Bafaloukos D, Razis E, Papadopoulou K, Papakostas P, Kalofonos HP, Pectasides D, Skarlos P, Kalogeras KT, Fountzilas G. Evaluation of the insulin-like growth factor receptor pathway in patients with advanced breast cancer treated with trastuzumab. Cancer Genomics Proteomics. 2018;15:461–471. doi: 10.21873/cgp.20105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trejo-Soto PJ, Hernández-Campos A, Romo-Mancillas A, Medina-Franco JL, Castillo R. In search of AKT kinase inhibitors as anticancer agents: structure-based design, docking, and molecular dynamics studies of 2,4,6-trisubstituted pyridines. J Biomol Struct Dyn. 2018;36:423–442. doi: 10.1080/07391102.2017.1285724. [DOI] [PubMed] [Google Scholar]
- 113.Kozlova NI, Morozevich GE, Ushakova NA, Berman AE. Implication of integrin α2β1 in proliferation and invasion of human breast carcinoma and melanoma cells: noncanonical function of akt protein kinase. Biochemistry (Mosc) 2018;83:738–745. doi: 10.1134/S0006297918060111. [DOI] [PubMed] [Google Scholar]
- 114.Massarweh S, Romond E, Black EP, Van Meter E, Shelton B, Kadamyan-Melkumian V, Stevens M, Elledge R. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat. 2014;143:325–332. doi: 10.1007/s10549-013-2810-9. [DOI] [PubMed] [Google Scholar]
- 115.Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, Anderson I, Robinson DM, He W, Garg A, McDonald ER 3rd, Bitter H, Huang A, Taran T, Bachelot T, Lebrun F, Lebwohl D, Baselga J. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J. Clin. Oncol. 2016;34:419–426. doi: 10.1200/JCO.2014.60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, Chen X, Balko JM, Gómez H, Arteaga CL, Mills GB, Sanders ME, Pietenpol JA. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014;16:406. doi: 10.1186/s13058-014-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hatem R, El Botty R, Chateau-Joubert S, Servely JL, Labiod D, de Plater L, Assayag F, Coussy F, Callens C, Vacher S, Reyal F, Cosulich S, Diéras V, Bièche I, Marangoni E. Targeting mTOR pathway inhibits tumor growth in different molecular subtypes of triple-negative breast cancers. Oncotarget. 2016;7:48206–48219. doi: 10.18632/oncotarget.10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Coussy F, ElBotty R, Lavigne M, Gu C, Fuhrmann L, Briaux A, deKoning L, Dahmani A, Montaudon E, Morisset L, Huguet L, Sourd L, Painsec P, Chateau-Joubert S, Larcher T, Vacher S, Melaabi S, Salomon AV, Marangoni E, Bieche I. Combination of PI3K and MEK inhibitors yields durable remission in PDX models of PIK3CA-mutated metaplastic breast cancers. J Hematol Oncol. 2020;13:13. doi: 10.1186/s13045-020-0846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coussy F, Lavigne M, deKoning L, Botty REl, Nemati F, Naguez A, Bataillon G, Ouine B, Dahmani A, Montaudon E, Painsec P, Chateau-Joubert S, Laetitia F, Larcher T, Vacher S, Chemlali W, Briaux A, Melaabi S, Salomon AV, Guinebretiere JM, Bieche I, Marangoni E. Response to mTOR and PI3K inhibitors in enzalutamide-resistant luminal androgen receptor triple-negative breast cancer patient-derived xenografts. Theranostics. 2020;10:1531–1543. doi: 10.7150/thno.36182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun SY. mTOR-targeted cancer therapy: great target but disappointing clinical outcomes, why? Front Med. 2020;15:221–231. doi: 10.1007/s11684-020-0812-7. [DOI] [PubMed] [Google Scholar]
- 121.Gerratana L, Basile D, Buono G, DePlacido S, Giuliano M, Minichillo S, Coinu A, Martorana F, DeSanto I, Del Mastro L, De Laurentiis M, Puglisi F, Arpino G. Androgen receptor in triple negative breast cancer: a potential target for the targetless subtype. Cancer Treat Rev. 2018;68:102–110. doi: 10.1016/j.ctrv.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 122.Němcová-Fürstová V, Kopperová D, Balušíková K, Ehrlichová M, Brynychová V, Václavíková R, Daniel P, Souček P, Kovář J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol Appl Pharmacol. 2016;310:215–228. doi: 10.1016/j.taap.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 123.Gómez-Miragaya J, Palafox M, Paré L, Yoldi G, Ferrer I, Vila S, Galván P, Pellegrini P, Pérez-Montoyo H, Igea A, Muñoz P, Esteller M, Nebreda AR, Urruticoechea A, Morilla I, Pernas S, Climent F, Soler-Monso MT, Petit A, Serra V, Prat A, González-Suárez E. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of a CD49f+ tumor-initiating population. Stem Cell Reports. 2017;8:1392–1407. doi: 10.1016/j.stemcr.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kaboli PJ, Chen Y, Xiang S, Xiao Z. Genetic analysis of triple-negative breast cancer cell lines and the therapeutic role of Akt/Nrf2 crosstalk in resistance to EGFR inhibitors. FASEB J. 2020;34:1. [Google Scholar]
- 125.Wu Q, Ba-Alawi W, Deblois G, Cruickshank J, Duan S, Lima-Fernandes E, Haight J, Tonekaboni SAM, Fortier AM, Kuasne H, McKee TD, Mahmoud H, Kushida M, Cameron S, Dogan-Artun N, Chen W, Nie Y, Zhang LX, Vellanki RN, Zhou S, Prinos P, Wouters BG, Dirks PB, Done SJ, Park M, Cescon DW, Haibe-Kains B, Lupien M, Arrowsmith CH. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat Commun. 2020;11:4205. doi: 10.1038/s41467-020-18020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu S, Imani S, Deng Y, Pathak JL, Wen Q, Chen Y, Wu J. Targeting IFN/STAT1 pathway as a promising strategy to overcome radioresistance. Onco Targets Ther. 2020;13:6037–6050. doi: 10.2147/OTT.S256708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kagihara JA, Andress M, Diamond JR. Nab-paclitaxel and atezolizumab for the treatment of PD-L1-positive, metastatic triple-negative breast cancer: review and future directions. Expert Rev Precis Med drug Dev. 2020;5:59–65. doi: 10.1080/23808993.2020.1730694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Simmons CE, Brezden-Masley C, McCarthy J, McLeod D, Joy AA. Positive progress: current and evolving role of immune checkpoint inhibitors in metastatic triple-negative breast cancer. Ther Adv Med Oncol. 2020;12:1758835920909091. doi: 10.1177/1758835920909091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 130.Reddy SM, Carroll E, Nanda R. Atezolizumab for the treatment of breast cancer. Expert Rev Anticancer Ther. 2020;20:151–158. doi: 10.1080/14737140.2020.1732211. [DOI] [PubMed] [Google Scholar]
- 131.Ahmed FS, Gaule P, McGuire J, Patel K, Blenman K, Pusztai L, Rimm DL. PD-L1 protein expression on both tumor cells and macrophages are associated with response to neoadjuvant durvalumab with chemotherapy in triple-negative breast cancer. Clin Cancer Res. 2020;26:5456–5461. doi: 10.1158/1078-0432.CCR-20-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mezni E, Vicier C, Guerin M, Sabatier R, Bertucci F, Gonçalves A. New therapeutics in HER2-positive advanced breast cancer: towards a change in clinical practices? Cancers. 2020;12:1573. doi: 10.3390/cancers12061573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Akula SM, Abrams SL, Steelman LS, Emma MR, Augello G, Cusimano A, Azzolina A, Montalto G, Cervello M, McCubrey JA. RAS/RAF/MEK/ERK, PI3K/PTEN/AKT/mTORC1 and TP53 pathways and regulatory miRs as therapeutic targets in hepatocellular carcinoma. Expert Opin Ther Targets. 2019;23:915–929. doi: 10.1080/14728222.2019.1685501. [DOI] [PubMed] [Google Scholar]
- 134.Gupta GK, Collier AL, Lee D, Hoefer RA, Zheleva V, Siewertsz van Reesema LL, Tang-Tan AM, Guye ML, Chang DZ, Winston JS, Samli B, Jansen RJ, Petricoin EF, Goetz MP, Bear HD, Tang AH. Perspectives on triple-negative breast cancer: current treatment strategies, unmet needs, and potential targets for future therapies. Cancers. 2020;12:2392. doi: 10.3390/cancers12092392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Giotta Lucifero A, Luzzi S, Brambilla I, Schena L, Mosconi M, Foiadelli T, Savasta S. Potential roads for reaching the summit: an overview on target therapies for high-grade gliomas. Acta Biomed. 2020;91:61–78. doi: 10.23750/abm.v91i7-S.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]