

Abstract
Transgene-enhanced oncolytic adenoviruses represent a promising novel therapeutic option for the treatment of cancer. A Phase 1 clinical trial featuring AdAPT-001 is ongoing (NCT04673942). AdAPT-001, a type 5 adenovirus, which carries a TGF-β trap transgene that neutralizes the immunosuppressive cytokine, TGF-β, has been shown in an immunocompetent mouse model to eradicate both locally injected and non-injected tumors. Single dose biodistribution of the TGF-β trap transgene was also evaluated in tumor bearing mice, providing an explanation for systemic activity. The biodistribution and toxicity of a single administration of mouse AdAPT-001 (mAdAPT-001) in 129S1 immunocompetent mice bearing ADS-12 tumors (mouse lung carcinoma) were assessed. mAdAPT-001 was injected intratumorally and intravenously in groups of 25 mice each at varying dose levels. Soluble TGF-β trap was detected in the serum using ELISA. A single AdAPT-001 injection resulted in non-negligible long-term TGF-β trap persistence in the serum over the 14-day study after intravenous and intratumoral administration. No TGF-β-related toxicity was observed. At clinically relevant doses, AdAPT-001 was safe and well tolerated. Systemic levels of the TGF-β trap transgene were observed from both local and intravenous dosing.
Keywords: Adenoviral vector, oncolytic virus, TGF-beta
Introduction
A substantial and unprecedented paradigm shift is underway in medical oncology with the supplementation and even supplantation of indiscriminately cytotoxic chemotherapies by more promising and precise immunotherapies [1], whose function is to harness the cytotoxic potential of the immune system in the severely immune-suppressed and metabolically and structurally hostile tumor microenvironment (TME).
Immune-suppression within the tumor microenvironment accounts, in part, for the absence of benefit in a majority of cancer patients with checkpoint inhibitor (CI) therapies. Hence, it is not enough for T-cells to infiltrate tumors; they must also recognize tumor-specific antigens and overcome the plethora of immunosuppressive and regulatory mechanisms elaborated by the tumor, stromal, lymphatic, vascular and extracellular matrix ensemble, in short, the TME, to mediate cytotoxicity [2].
Whereas CIs appear to function preferentially in T-cell inflamed or immunologically “hot” tumors, the high occurrence of immunosuppressed or immunologically “cold” tumors, which represents the most frequent immune phenotype, imposes a “ceiling” on response rates, generally in the range of 20-40% [3]. These response rates may be augmented with combinatorial checkpoint therapy but only at the expense of unacceptably high rates of Grade 3 and 4 autoimmune-like toxicities [4,5].
A complement to checkpoint inhibitor therapy is oncolytic viruses (OVs).
The advantage of oncolytic viruses is in their replicative capacity, which leads to amplification of expression of the therapeutic transgene that they carry for enhancement of anti-tumor or immunogenic effects; the majority of transgenes used in an oncolytic virotherapy context are directly immunostimulatory [6]; common examples include GM-CSF, IL-18 and IL-12. However, transgenes may also serve to enhance and focus the immunomodulatory functions through their effect on cytokine levels, for example.
AdAPT-001 is a type 5 conditionally replicative adenovirus that is enhanced with an immunomodulatory TGF-β trap. This trap is a fusion protein of soluble TGF-β receptor II and the Fc portion of human IgG1, which “traps” or neutralizes the activity of TGF-β. A pleiotropic cytokine, TGF-β acts in the capacity of a master regulator that controls immune tolerance, differentiation, proliferation and survival [7]. The viral vector, called TAV-255, that carries the TGF-β trap has been engineered with a small 50 bp deletion in the E1A promoter region, which renders it unable to fully replicate and produce progeny in non-transformed cells, although early genes are transcribed. AdAPT-001 is preferentially injected intratumorally, although it may be delivered subcutaneously or intravenously. One potential criticism of intratumoral injection is tumor-specific transgene expression, which theoretically may serve to limit TGF-β-neutralization in distant lesions.
A single dose toxicology and biodistribution study was performed to determine the safety profile and level of TGF-β trap expression in the serum after intratumoral and IV injection in an immunocompetent ADS-12 mouse model prior to a Phase 1 study in humans.
Materials and methods
Mice
129S1/SvImJ mice, approximately 6-8 weeks old, were purchased from Jackson Laboratory. The study was approved and conducted in accordance with an Institutional Animal Care and Use Committee.
Test article
The test article was a modified adenovirus mAdAPT-001 (also known as Ad-TAV-mTGFbR-IgG). The virus is a replication-competent adenovirus 5 whose genome has 1) a 50 nucleotide TAV-255 deletion to reduce replication potential in non-cancerous cells, and 2) a inserted murine fusion gene (in place of E1B-19k gene) encoding a secretory fusion protein mTGFbR-IgG. The mTGFbR-IgG fusion protein contains the ligand-binding domain of the mouse TGFß type II receptor (TGFbR) and the Fc domain of mouse IgG1. The mAIM-001 viruses were produced in human A549 cells and pre-formulated as ready to use suspensions in the Vehicle.
The test article was supplied in the following four types of vials, 57 vials per type: 5E9 VP/mL (0.12 mL per vial), 5E10 VP/mL (0.12 mL per vial), 5E11 VP/mL (0.12 mL per vial) and 5E11 VP/mL (0.24 mL per vial). The vials were stored frozen at -80°C.
Control article
The Control Article was the Vehicle of the test article: a solution of 20 mM HEPES, pH7.8, 150 mM NaCl and 10% glycerol. The Vehicle was provided in ready to dose form in the following two types of vials, 57 vials per type: 0.12 mL per vial and 0.24 mL per vial. The vials were stored frozen at -80°C.
Tumor implantation
ADS-12 cell (murine K-ras mutant lung adenocarcinoma) line was provided by EpicentRx. Tumors were established by subcutaneous injection of 1E6 cells in 50 µL in the right flank.
Dose groups, dose concentrations and dose levels
Animals were divided into multiple dosing cohorts as shown in Table 1.
Table 1.
Dose concentrations and dosages
| Group | Dosed Article | Route | Term | Dose Concentration | Dose Level (per animal) | Supply Vials |
|---|---|---|---|---|---|---|
| Group 1 | None | - | Untreated | - | - | - |
| Group 2 | Vehicle | IT | IT Vehicle | 0 (Vehicle) | 0.1 mL | Vehicle 0.12 mL/vial |
| Group 3 | Vehicle | IV | IT Vehicle (high volume) | 0 (Vehicle) | 0.2 mL | Vehicle 0.24 mL/vial |
| Group 4 | ADAPT-001 | IT | IT Low Dose | 5E9 VP/mL | 5E8 VP/0.1 mL | 5E9 VP/mL 0.12 mL/vial |
| Group 5 | ADAPT-001 | IT | IT Medium Dose | 5E10 VP/mL | 5E9 VP/0.1 mL | 5E10 VP/mL 0.12 mL/vial |
| Group 6 | ADAPT-001 | IT | IT High Dose | 5E11 VP/mL | 5E10 VP/0.1 mL | 5E11 VP/mL 0.12 mL/vial |
| Group 7 | ADAPT-001 | IV | IV High Dose | 5E11 VP/mL | 1E11 VP/0.2 mL | 5E11 VP/mL 0.24 mL/vial |
| Group 8 | Vehicle | IV | IV Vehicle | 0 (Vehicle) | 0.1 mL | Vehicle 0.24 mL/vial |
| Group 9 | ADAPT-001 | IV | IV Low Dose | 5E10 VP/mL | 5E9 VP/0.1 mL | 5E11 VP/mL 0.24 mL/vial |
IT: Intratumoral injection. IV: Intravenous injection. VP: Viral particles.
AdAPT-001 and Control were administered approximately 14 days (14 ± 2 days) after tumor cell injection when the tumor size reached 150 ± 50 mm3. Mice of Groups 2-9 were dosed with either Vehicle or AdAPT-001, IV or IT, on Day 0 as indicated in Table 1. Group-1 mice were not dosed. The animals were euthanized for signs of distress or when the total tumor volume (largest diameter × smallest diameter × depth/2) reached 1000 mm3.
Statistics
Data analysis employed a two-sided Fisher’s exact test for count data to compare the proportion of subjects with presence/absence of TGF-β between IV and IT route of administration and dose level at two timepoints, Day 2 and Day 14. Proportions 95% confidence intervals were estimated via Clopper-Pearson formula. The analysis was stratified by sacrifice day (Day 2 and 14), and the results were summarized in tabular and graphical format. AdAPT-001 TGF-β circulating serum levels (ng/mL, log (10) scale) were plotted by route and dose level and segregated by day of sacrifice as displayed in Figures 1, 2). Descriptive statistics (mean, median, standard deviation, standard error, min, max and 95% confidence interval) were produced for TGF-β concentrations by day, route of administration and dose level.
Figure 1.
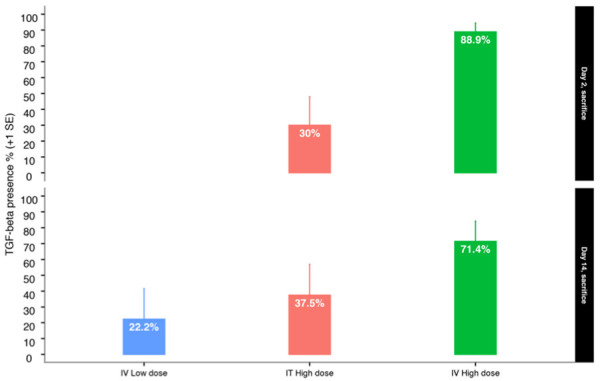
TGF-β serum presence/absence by sacrifice day, route of administration and dose level.
Figure 2.
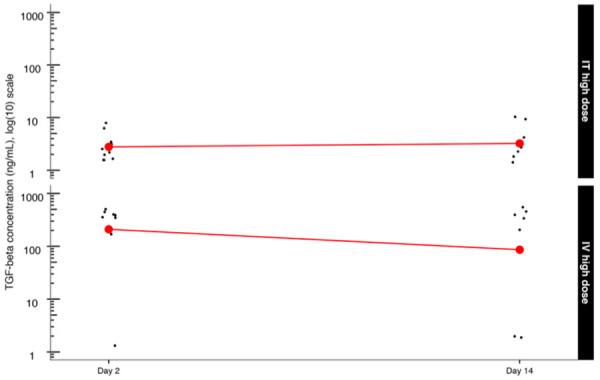
TGF-β Concentration (ng/mL) (log (10) scale) by route of administration and dose level.
Biodistribution samples
After an overnight fast, animals were deeply anesthetized by isoflurane/O2. Terminal blood was collected and processed to obtain serum for ELISA testing. Soluble TGF-β trap was detected in the serum using ELISA [8].
Results
Clinical findings and body weights
5 mice in the high dose IV group developed AdAPT-001-related hepatitis. There were no statistically significant changes in body weight in the AdAPT-001 treated groups compared to their respective control groups (Groups 4, 5, 6 versus Group 2; Group 7 versus Group 3; Group 9 versus Group 8) in either males or females.
Biodistribution of the TGF-β trap
The proportion of TGF-β presence in the serum was compared between AdAPT-001 IT high dose and IV high dose. The difference in the proportion of mice with TGF-β present at Day 2 was statistically significant according to Pearson’s chi-squared two-sided test with Yates’ continuity correction (P=0.0312). Fisher’s exact two-sided test was also statistically significant (P=0.0197).
The estimated odds ratio was 15.4 (presence of TGF-β in serum, IV to IT route, high dose, Day 2). The difference in the proportion of mice with TGF-β present at Day 14 was not statistically significant (P=0.4264, Pearson’s chi-squared test, P=0.3147, Fisher’s exact test). The estimated odds ratio was 26.6 (presence of TGF-β in serum, IV to IT route, high dose, Day 14). Figures 1 and 2 and Tables 2 and 3 show the statistical analysis results.
Table 2.
Statistical summary of TGF-β serum presence/absence by sacrifice day, route of administration and dose level
| Sacrifice day | Dosed article | Group # | term | TGF-beta status | n | % | N | Lower_ci | Upper_ci | se |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | ADAPT-001 | 4 | IT Low Dose | negative | 10 | 100.0 | 10 | 69.2 | 100.0 | < 0.01 |
| 2 | ADAPT-001 | 5 | IT Medium Dose | negative | 10 | 100.0 | 10 | 69.2 | 100.0 | < 0.01 |
| 2 | ADAPT-001 | 6 | IT High Dose | negative | 7 | 70.0 | 10 | 34.8 | 93.3 | 11.9 |
| 2 | ADAPT-001 | 6 | IT High Dose | positive | 3 | 30.0 | 10 | 6.7 | 65.2 | 18.0 |
| 2 | ADAPT-001 | 7 | IT High Dose | negative | 1 | 11.1 | 9 | 0.3 | 48.2 | 18.9 |
| 2 | ADAPT-001 | 7 | IT High Dose | positive | 8 | 88.9 | 9 | 51.8 | 99.7 | 5.5 |
| 2 | Vehicle | 2 | IT Vehicle | negative | 8 | 100.0 | 8 | 63.1 | 100.0 | < 0.01 |
| 2 | Vehicle | 3 | IV Vehicle High Vol | negative | 9 | 100.0 | 9 | 66.4 | 100.0 | < 0.01 |
| 14 | ADAPT-001 | 4 | IT Low Dose | negative | 9 | 100.0 | 9 | 66.4 | 100.0 | < 0.01 |
| 14 | ADAPT-001 | 5 | IT Medium Dose | negative | 9 | 100.0 | 9 | 66.4 | 100.0 | < 0.01 |
| 14 | ADAPT-001 | 6 | IT High Dose | negative | 5 | 62.5 | 8 | 24.5 | 91.5 | 14.8 |
| 14 | ADAPT-001 | 6 | IT High Dose | positive | 3 | 37.5 | 8 | 8.5 | 75.5 | 19.4 |
| 14 | ADAPT-001 | 7 | IV High Dose | negative | 2 | 28.6 | 7 | 3.7 | 71.0 | 21.6 |
| 14 | ADAPT-001 | 7 | IV High Dose | positive | 5 | 71.4 | 7 | 29.0 | 96.3 | 12.7 |
| 14 | ADAPT-001 | 9 | IV Low Dose | negative | 7 | 77.8 | 9 | 40.0 | 97.2 | 9.9 |
| 14 | ADAPT-001 | 9 | IV Low Dose | positive | 2 | 22.2 | 9 | 2.8 | 60.0 | 19.3 |
| 14 | Vehicle | 2 | IT Vehicle | negative | 3 | 100.0 | 3 | 29.2 | 100.0 | < 0.01 |
| 14 | Vehicle | 3 | IV Vehicle High Vol | negative | 8 | 100.0 | 8 | 63.1 | 100.0 | < 0.01 |
| 14 | Vehicle | 8 | IV Vehicle Low Dose | negative | 9 | 100.0 | 9 | 66.4 | 100.0 | < 0.01 |
Table 3.
Statistical summary of TGF-β concentration (ng/mL) by sacrifice day, route of administration and dose level
| Dosed article | term | Sacrifice day | n | mean | median | sd | min | max | se | lower_ci | upper_ci |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ADAPT-001 | IT high dose | 2 | 10 | 3.2 | 2.0 | 2.0 | 2.0 | 6.9 | 0.6 | 2.0 | 4.5 |
| ADAPT-001 | IT high dose | 14 | 8 | 4.3 | 2.0 | 4.0 | 2.0 | 13.2 | 1.4 | 1.5 | 7.1 |
| ADAPT-001 | IT high dose | 2 | 9 | 341.7 | 396.0 | 153.9 | 2.0 | 546.0 | 51.3 | 241.1 | 442.2 |
| ADAPT-001 | IT high dose | 14 | 7 | 301.7 | 252.0 | 250.6 | 2.0 | 585.0 | 94.7 | 116.1 | 487.3 |
AdAPT-001 TGF-beta circulating serum levels (ng/mL, log (10) scale) were plotted by route and dose level and segregated by day of sacrifice as displayed in Figure 1 and Table 2 (original scale). The comparison between Day 2 IT high dose TGF-beta concentration appears to have been maintained at Day 14, indicative of a long-circulating half-life.
A similar observation also appears to hold for TGF-beta IV high dose between Day 2 (high dose) and Day 14 (high dose) TGF-beta concentrations (Figure 2).
Discussion and conclusion
In general, oncolytic viruses have been administered intratumorally (IT) rather than intravenously (IV) to prevent inactivation by neutralizing antibodies (nAbs) and sequestration by the reticuloendothelial system [9]. However, like the widely cited dictum to “act locally but think globally”, in situ replication of oncolytic virus abscopally activates the immune system, due to 1) the generation of so-called danger signals, which stimulate toll-like receptors (TLR) [10], and 2) the release of tumor-derived antigens, which may prime cytotoxic lymphocytes. Additionally, in the case of AdAPT-001, adenoviral particles are potentially small enough (~90 nm) [11] to extravasate from the leaky, tortuous tumor neovessels, seed the systemic circulation and infect distant metastases. Viremic dissemination may also occur through continuous release of progeny from infected tumor cells.
TAV-255, the oncolytic virus used in this study, which is detargeted from normal cells by virtue of a 50 bp deletion in the E1A promoter, has previously been dosed in two patients who benefited based on serial clinical, radiological and histopathological observations for multiple months under compassionate use protocols [12]. In both patients and on separate occasions, viremia for 72 hours after dosing was observed along with minimal toxicity [13].
In this toxicology and biodistribution study of a single administration of mouse AdAPT-001 in a syngeneic tumor model, viral hepatitis developed in the high dose intravenous group only. Intravenously injected Ad particles are sequestered by the liver, which is well-known to cause an acute inflammatory transaminitis and vascular damage [14]; however, in this case, with AdAPT-001, hepatotoxicity occurred at doses of adenovirus two logs over the planned clinical dose in humans. Additionally, in humans, pre-existing anti-adenovirus neutralizing antibodies, which were presumably not present in the immunologically-naïve mice, would be expected to prevent sequestration of blood-born adenovirus (Ad) in the liver and viral hepatitis.
Biodistribution analysis demonstrated that the soluble mouse TGF-β trap is observed in the serum, at non-negligible levels and for a duration of at least 14 days, when the virus is dosed both intravenously and intratumorally. Laboratory studies have demonstrated that although the TAV-255 [15] base vector is able to penetrate normal, non-transformed cells, infection is abortive (i.e., it fails to produce new virus particles and to induce cell lysis); however, early genes like TGF-β trap transgene are transcribed, which likely explains its appearance in the serum. Moreover, the enhanced persistence of the trap in circulation is attributed to two factors: 1) the presence of the Fc domain, which interacts with the salvage neonatal Fc-receptor [16], FcRn [17], whose function is to prevent degradation of IgG and albumin in the endosome and 2) the slower renal clearance of a larger sized molecule [18]. The potential mechanisms of action of intratumorally-dosed AdAPT-001 are illustrated in Figure 3.
Figure 3.
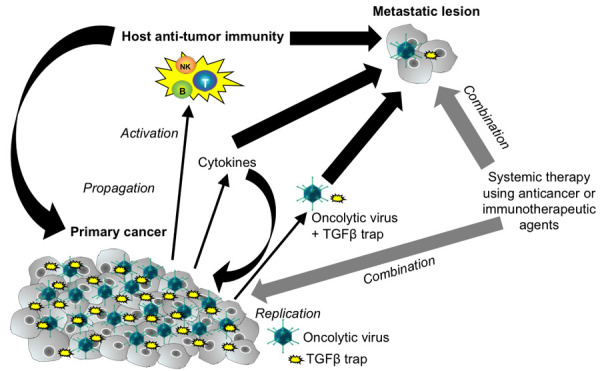
Potential Mechanisms of Action of AdAPT-001 from Intratumoral Administration. Local replication of AdAPT-001 induces specific and abscopal antitumor immunity in the course of its oncolytic activities. The TGF-beta trap is present systemically and virus itself may also enter the systemic circulation due to extravasation from neovessels, continuous release of progeny and infection of circulating cancer cells that are shed from the tumor. A combination with immune checkpoint inhibitors or chemotherapy is expected to enhance the activity of AdAPT-001.
The long circulating half-life of the TGF-β trap in the absence of observable toxicity from prolonged TGF-β inhibition suggests that AdAPT-001 may be dosed intravenously, since higher levels of the TGF-β trap are seen in the serum after IV dosing, although it is presently unknown whether accumulation of the trap occurs in non-injected tumors.
In conclusion, due to the specificity of the virus for neoplastic cells and the systemicity of TGF-β trap transgene it is possible to speculate that AdAPT-001 may be used not only to treat locally injected tumors but also micrometastatic and metastatic disease. A phase 1 clinical trial featuring AdAPT-001 is ongoing (NCT04673942).
Disclosure of conflict of interest
None.
References
- 1.Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6:140. doi: 10.1186/s40425-018-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. 2016;2:286–294. doi: 10.1016/j.trecan.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 5.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loskog A. Immunostimulatory gene therapy using oncolytic viruses as vehicles. Viruses. 2015;7:5780–5791. doi: 10.3390/v7112899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lequin RM. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA) Clin Chem. 2005;51:2415–2418. doi: 10.1373/clinchem.2005.051532. [DOI] [PubMed] [Google Scholar]
- 9.Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8:301–13. [PubMed] [Google Scholar]
- 10.Struzik J, Szulc-Dąbrowska L. NF-κB signaling in targeting tumor cells by oncolytic viruses-therapeutic perspectives. Cancers (Basel) 2018;10:426. doi: 10.3390/cancers10110426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yumul R, Richter M, Lu Z, Saydaminova K, Wang K, Wang CHK, Carter D, Lieber A. Epithelial Junction opener improves oncolytic adenovirus therapy in mouse tumor models. Hum Gene Ther. 2016;27:325–337. doi: 10.1089/hum.2016.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larson C, Oronsky B, Varner G, Caroen S, Burbano E, Insel E, Hedjran F, Carter CA, Reid TR. A practical guide to the handling and administration of personalized transcriptionally attenuated oncolytic adenoviruses (PTAVs) Oncoimmunology. 2018;7:e1478648. doi: 10.1080/2162402X.2018.1478648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouvet M, Larson C, Oronsky B, Reid T, Morris C. Extended treatment with MY-NEOVAX, personalized neoantigen-enhanced oncolytic viruses, for two end-stage cancer patients. Oxford Med Case Rep. 2019;11:461–463. doi: 10.1093/omcr/omz105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worgall S, Wolff G, Falck-Pedersen E, Crystal RG. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum Gene Ther. 1997;8:37–44. doi: 10.1089/hum.1997.8.1-37. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Hedjran F, Larson C, Perez GL, Reid T. A novel immunocompetent murine model for replicating oncolytic adenoviral therapy. Cancer Gene Ther. 2015;22:17–22. doi: 10.1038/cgt.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 17.Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194:4595–4603. doi: 10.4049/jimmunol.1403014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22:868–876. doi: 10.1016/j.copbio.2011.06.012. [DOI] [PubMed] [Google Scholar]