

Abstract
The expression of Dickkopf-1 (DKK1), a negative regulator of the Wnt/β-catenin signaling pathway, is upregulated in hepatocellular carcinoma (HCC). Here, we investigated the tumorigenic and angiogenic potential of DKK1 in HCC. Stable cell lines were established using the clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease 9 (CRISPR/Cas9)-based DKK1 knock-out system in Hep3B cells and the tetracycline-based DKK1 inducible system in Huh7 cells. Multicellular tumor spheroids (MCTSs) were cultured using Hep3B stable cells. We also employed xenografts generated using Hep3B stable cells and transgenic mouse models established using hydrodynamic tail vein injection. The angiogenic potential increased in HUVECs treated with CM from Huh7 stable cells with high DKK1 expression and Hep3B wild-type cells. DKK1 accelerated the downstream molecules of vascular endothelial growth factor receptor 2 (VEGFR2)-mediated mTOR/p70 S6 kinase (p70S6K) signaling. MCTSs generated using Hep3B wild-type cells promoted compact spheroid formation and increased the expression of CD31 and epithelial-mesenchymal transition (EMT) markers, and increased the VEGFR2-mediated mTOR/p70S6K signaling, compared to the controls (all P<0.01). Xenograft tumors generated using Hep3B cells with DKK1 knock-out (n=10) exhibited slower growth than, the controls (n=10) and the expression of Ki-67, VEGFR2, CD31 and EMT markers decreased (all P<0.05). In addition, forced DKK1 expression with HRAS in transgenic mouse livers (n=5) resulted in the formation of more tumors and increased expression of downstream molecules of VEGFR2-mediated mTOR/p70S6K signaling pathway as well as Ki67, CD31 and EMT markers (P<0.05), compared to that of the controls (n=5). Our findings indicate that DKK1 facilitates angiogenesis and tumorigenesis by upregulating VEGFR2-mediated mTOR/p70S6K signaling in HCC.
Keywords: Dickkopf-1, vascular endothelial growth factor receptor 2, hepatocellular carcinoma, angiogenesis, epithelial-mesenchymal transition
Introduction
Hepatocellular carcinoma (HCC), the primary cancer of the liver, is derived from hepatocytes and occurs in more than approximately 80% of cases of liver cancer [1]. HCC is a hypervascular tumor in which angiogenesis plays an important role in development, invasion and metastasis [2]. Tumor growth relies on angiogenesis, the formation of new blood vessels from pre-existing vasculature, to receive an adequate supply of oxygen and nutrients into the tumors [3,4]. Accordingly, anti-HCC agents, such as sorafenib and lenvatinib, which target vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) receptors have shown survival benefits in HCC treatment [5].
Pro-angiogenic factors such as VEGF, FGF and PDGF are known to activate tyrosine kinases and subsequent downstream intracellular signaling through mitogen-activated protein kinase [6]. Both VEGF and VEGF receptors (VEGFRs), the most prominent and widely researched regulators of angiogenesis, are critical for HCC growth and development [7]. The binding of VEGF to VEGFR leads to endothelial proliferation and migration as well as formation and branching of new tumor blood vessels [8]. In addition, VEGFR1 activation upregulates the expression of epithelial-mesenchymal transition (EMT)-associated factors, such as Snail, Twist and Slug in HCC [9].
Dickkopf-1 (DKK1), which binds to the low-density lipoprotein receptor-related protein-5/6 Wnt co-receptor, is a secreted protein that functions as a negative regulator of Wnt signaling [10-15]. Wnt signaling regulates diverse cellular and biological processes such as proliferation, survival, migration and liver development [16-18]. In addition, the DKK protein family has been found to regulate angiogenesis, and it has been reported that the Wnt signaling pathway and DKK1 modulate tumorigenesis during vasculogenesis and angiogenesis [19].
Several studies have reported that DKK1 expression is upregulated in HCC cell lines [20-22] and the tumor tissues and serum samples of patients with HCC [20,23,24]. Ectopic DKK1 expression promotes HCC cell migration and invasion through β-catenin/matrix metalloproteinase (MMP7) signaling [25]. Moreover, the downregulation of DKK1 expression using siRNA has been shown to inhibit invasion and metastasis in HCCLM3 cells. In contrast, DKK1 overexpression in the HepG2 cell line has been shown to significantly promote its migration and invasiveness [26]. These results indicate that DKK1 regulation can be a promising target for angiogenesis in HCC.
In a previous study, we showed that DKK1 induces angiogenesis by regulating VEGF receptor 2 (VEGFR2) [27], but the correlation between DKK1 and angiogenesis in HCC remains poorly understood. In the present study, we investigated the angiogenic and tumorigenic role of DKK1 in HCC using diverse models of regulated DKK1 expression and found that DKK1 promotes angiogenesis by upregulating VEGFR2-mediated mammalian target of rapamycin (mTOR)/p70 S6 kinase (p70S6K) signaling in HCC.
Material and methods
Cell lines
Huh7 (Korean Cell Line Bank, Seoul, Korea), GP2-293 (Clontech, California, USA), Hep3B, LX2 [Dr. Seo [28]] and WI38 cells were cultured at 37°C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Gibco, Carlsbad, CA, USA), Modified Eagle’s Medium (Gibco) or RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1x penicillin-streptomycin (Welgene, Daegu, Korea). Human umbilical vein endothelial cells (HUVECs) were cultured in Medium 200 (Gibco) supplemented with low serum growth supplement (Gibco) at 37°C with 5% CO2.
Generation of stable cell lines
Stable cell lines were generated using the clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease 9 (CRISPR/Cas9)-based DKK1 knockout system in Hep3B cells and the tetracycline-based DKK1 inducible system in Huh7 cells. The Hep3B wild-type cells used in this study meant that Hep3B cells were untreated, and previous studies showed that Hep3B was originally isolated from a liver biopsy of an 8-year-old black man with primary HCC [29,30]. Alt® CRISPR-Cas9 system, CRISPR RNA (crRNA): Guide RNA (gRNA) targeting the human DKK1 locus along with transactivating crRNA (tracrRNA), were obtained from Integrated DNA Technologies (San Diego, CA, USA) for Hep3B stable cell line. crRNA was used to select target region within the first exon of the human DKK1 gene. Tracr and crRNA were combined according to the manufacturer’s protocol and then crRNA: tracrRNA duplexes were with Cas9 protein. Subsequently, the Alt® CRISPR-Cas9 system was transfected into Hep3B cells using LipofectamineTM RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA). Off-target effects in the two selected stable cell line clones were identified using whole genome sequencing (Promega, Madison, WA, USA) and the knock-out sequence of the human DKK1 exon-1 gene was confirmed by Sanger sequencing (Macrogen, Seoul, Korea).
GP2-293 was transfected to make regulator (Retro-X Tet-On Advanced Inducible Expression System and pVSV-G) and response (pRetroX-tight Pur-DKK1/pVSV-G or pRetroX-tight Pur-Mock/pVSV-G) plasmid (Clontech, California, USA) using CalPhos Mammalian Transfection Kit (Clontech). The regulator plasmid was added to the culture medium of Huh7 cells. Transfected Huh7 cells were selected by on the basis of resistance to the antibiotic, G418 (Takara Bio Inc, Seoul, Korea), followed by the addition of the response plasmid to the culture medium of the selected Huh7 cells. Twenty-four hours after infection, the cells were subjected to puromycin (Takara Bio Inc) selection.
Assessment of DKK1 using ELISA
The concentration of DKK1 in HCC cell lines was measured using enzyme-linked immunosorbent assay (ELISA) kits (R&D systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. Subsequently, all the groups of supernatants were collected and DKK1 levels were assayed in them.
Cell invasion, tube formation and wound healing assay
The invasiveness of HUVECs was assessed using a transwell chamber (Corning Costar, Cambridge, MA, USA). Next, 3 × 104 HUVECs/well were treated with VEGF or recombinant DKK1 (rDKK1) in each transwell chamber. Following incubation for 24 h, the invading cells were stained with hematoxylin and eosin (H&E). The total number of invaded cells on the lower side of the filter was determined using a light microscope (Olympus, Tokyo, Japan) at 40 × magnification. Invading cells on the lower membrane surface were measured by imageJ software.
Tube formation by HUVECs was measured using Matrigel® (Corning). A 48 well plate (BD Falcon®, Bedford, MA, USA) was coated with 150 μL Matrigel® and seeded with HUVECs (3 × 104/well), followed by the addition of conditioned medium (CM) from Hep3B and Huh7 stable cell lines. The plates were incubated for 4 h, following which tube formation was observed under a light microscope.
Wound healing assays were performed by creating identical wound areas into the cell monolayer using culture-inserts (Ibidi GmbH, Munich, Germany). A 24 well plate was coated with 2% gelatin (BD Biosciences) and seeded with HUVECs (2 × 104/well) on each side of the culture-insert. After attachment, the cells were treated with CM from the HCC stable cell line, followed by detachment of the culture-insert. The wound images were captured at 0 and 6 h using a light microscope.
mRNA isolation and RT-PCR
For PCR, total RNA was extracted using TRIzolTM reagent (Invitrogen, California, USA) and synthesis of first strand cDNA (SuperscriptTM III First-Strand Synthesis System, Invitrogen) was performed according to the manufacturer’s recommended protocols. A PCR master mix (Power SYBRTM Green PCR Master Mix; Applied Biosystems, Warrington, UK) was used for quantitative PCR performed on the StepOnePlus™ PCR System (Applied Biosystems).
Western blot analysis
Cells and tissues were washed and lysed with RIPA buffer containing a protease inhibitor cocktail (Thermo Fisher Scientific) and a phosphatase inhibitor cocktail (GenDEPOT, Katy, TX, USA). Cell and tumor lysates were cleared using centrifugation, separated using SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membranes were incubated with antibodies against DKK1 (R&D Systems), VEGFR2, phosphorylated VEGFR2 (p-VEGFR2), mTOR, phospho-p70S6K, GAPDH, phosphoinositide 3-kinase (PI3K) and p-Akt [at Ser473] (Cell Signaling Technology, Danvers, MA, USA) at the recommended concentrations. The blots were developed using the enhanced chemiluminescence technique (PerkinElmer, Waltham, MA, USA) according to the manufacturer’s instructions.
Spheroid fabrication
Hep3B-only or hybrid spheroids were fabricated in a low attachment multiple well plate (Corning). To model tumor complexity and heterogeneity, we formed multicellular tumor spheroids (MCTSs) with Hep3B stable cell lines and stromal cells, such as HUVECs (human endothelial cells), LX2 (human hepatic stellate cells) and WI38 (human fibroblasts) and counted them prior to mixing them in the desired ratio. A 200 μL cell mixture of these four cell types was prepared in the following ratio: Hep3B stable cells: HUVECs: LX2: WI38=4: 2: 1: 1. This mixture was subsequently pipetted onto the plate. Images of MCTS were acquired using a microscope (Olympus) and imported into ImageJ software. The average radius (r) was then calculated using ImageJ software and used to obtain the volume value (V) with the following formula: V=4/3πr3.
Animals
All experiments involving live mice were performed according to the Guidelines and Regulations for the Care and Use of Laboratory Animals in AAALAC-accredited facilities, and were approved by the Animal Policy and Welfare Committee of the Yonsei University College of Medicine (Permit number: 2018-0088). We purchased 4-5-week-old C57BL/6 male mice from Orientbio (Seongnam, Korea), while 4-week-old BALB/c nude male mice were purchased from Central Lab. Animal Inc. (Seoul, Korea).
Xenograft mouse model
The Hep3B stable cell line (5 × 106 or 7 × 106 cells) in HBSS was mixed with Matrigel® [2:1] (Corning) and then inoculated subcutaneously into the left and right flanks of each BALB/c nude mouse. Tumors were monitored once a week and the tumor volume was calculated with the help of Vernier calipers. The volumes of the tumors were calculated using a standard formula (length × width2 × 0.5) and growth curves were drawn. Nine weeks later, animals in all the groups were sacrificed, following which their tumors were harvested and fixed in 10% formalin.
Histology analysis
The specimens embedded in paraffin blocks were sectioned into 4-μm slices. Specimens for histological analysis were processed using conventional H&E staining for the visualization of general tissue morphology. Stained specimens were inspected using a microscope (Olympus).
Immunohistochemistry and immunofluorescence
Paraffin sections were deparaffinized in xylene and rehydrated through a gradual decrease in ethanol concentration. The antigen epitopes were then unmasked using sodium citrate buffer (pH=6.0). Subsequently, the sections were incubated overnight at 4°C using the following primary antibodies: anti-Ki-67 (Cell Signaling Technology), anti- anti-cluster of differentiation 31 (CD31) (Invitrogen), anti-HRAS (Santa Cruz Biotechnology, California, USA), anti-p-VEGFR2 (Cell signaling) and anti-p-Akt (Cell signaling). After primary incubation, the sections were incubated with the appropriate biotinylated secondary antibodies followed by treatment with freshly prepared 3,3’-diaminobenzidine substrates (Vector Laboratories, Burlingame, CA, USA). Sections were lightly counter-stained with hematoxylin and mounted. For immunofluorescence (IF), the sections were probed with anti-DKK1 (R&D systems), CD31 (Invitrogen), anti-VEGFR2, anti-vimentin, anti-fibronectin and anti-E-cadherin antibodies (Cell Signaling Technology). Next, after washing with phosphate-buffered saline, the sections were incubated with secondary antibodies, stained with 4’,6-diamidino-2-phenylindole and then embedded Fluoromount-GTM (Invitrogen). Fluorescence images were obtained using a Zeiss LSM 700 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Hydrodynamic tail-vein injection
The plasmids pT2/HRASG12V, pT2/shp53, and PT2/C-Luc/PGK-SB13 were prepared using endotoxin-free EndoFree® Plasmid Maxi Kit (Qiagen, Hilden, Germany). DNA mixtures of transposons (pT2 plasmids) and transposase-encoding vector (pPGKSB13) were suspended in Lactated Ringer’s solution and subsequently injected into the lateral tail veins of 5- to 6-week old male mice (0.1 mL/g body weight) in less than 7 s. The livers were harvested 5 weeks following the hydrodynamic transfection, unless specified otherwise.
Statistical analysis
Statistical analyses were conducted using an unpaired parametric Student’s t-test or Fisher’s exact test, as appropriate. A value of P<0.05 was chosen to indicate statistical significance.
Results
Establishment and characterization of stable cell lines
Treatment with rDKK1 resulted in a concentration-dependent enhancement of the invasive potential of HUVECs (Figure 1A). Upon confirmation of the expression of DKK1 in various human HCC cell lines, it was found that the levels of secreted DKK1 were high in Hep3B cells, but low in Huh7, SNU475, and SNU449 cells (Figure 1Ba). Accordingly, Huh7 cells with low DKK1 expression and Hep3B cells with high DKK1 expression were selected for the regulation of DKK1 expression. Stable cell lines were established using the CRISPR-Cas9-based DKK1 knockout system in Hep3B cells and the tetracycline-based DKK1 inducible system in Huh7 cells. We used the CRISPR-Cas9 system to select the target region within exon-1 of the human DKK1 gene (Figure 2A, 2B). Subsequently, several colony cell lines were established by transfection, and it was found that the sizes of the DKK1 exon-1 in colonies 73 (Hep3B CRISPR#73 cells) and 131 (Hep3B CRISPR#131 cells) were smaller than those of the positive control (Hep3B wild-type cells) (Figure 1Bb, confirming DKK1 knock-out by sequencing Figure 3A, 3B). We also observed that the expression of DKK1 and the protein levels of secreted DKK1 in Hep3B CRISPR#73 cells were significantly lower than those in Hep3B wild-type and CRISPR#131 cells (all P<0.001) (Figure 1C and 1D). Consequently, Hep3B CRISPR#73 cells were used in this study because DKK1 protein levels in Hep3B CRISPR#73 cells were significantly lower than those in Hep3B CRISPR#131 cells. In addition, the expression of DKK1 and the protein levels of secreted DKK1 were significantly higher in doxycycline-treated pRetroX-tight Pur-DKK1 transfected Huh7 cells (Huh7 Pur-DKK1 cells) than in Huh7 wild-type cells and pRetroX-tight Pur-Mock transfected Huh7 cells (Huh7 Pur-Mock cells) (all P<0.001) (Figure 1E). In addition, we confirmed that the protein levels of secreted DKK1 in CM were higher in Huh7 Pur-DKK1 cells than in Huh7 Pur-Mock cells, when the Huh7 stable cells were treated with doxycycline for 24 h (Figure 1F).
Figure 1.
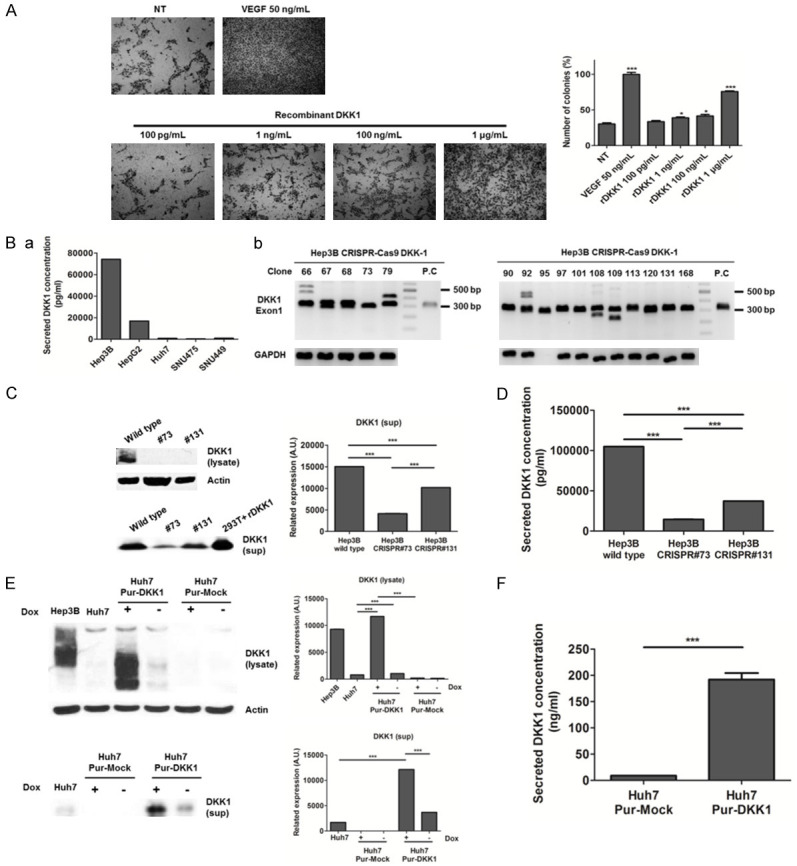
Regulation of DKK1 gene expression in Hep3B and Huh7 cells. A. Effects of DKK1 on invasion assay in HUVECs. NT: no treatment. HUVECs were treated with the indicated concentration of rDKK1 and 50 ng/mL VEGF (as a positive control). B. DKK1 expression levels of human hepatocellular carcinoma cell lines were measured using ELISA (a) and CRISPR-Cas9 based DKK1 knockout was confirmed by PCR (b). C. Western blot analysis of aggregated proteins, collected using centrifugation from Hep3B wild-type, Hep3B CRISPR#73 (#73) and Hep3B CRISPR#131 (#131) cells. DKK1-transfected HEK 293T cells were used as a positive control. D. Protein levels of secreted DKK1 from Hep3B wild type, #73 and #131 cells were measured using ELISA. E. Western blot analysis of aggregated proteins from Hep3B, Huh7, Huh7 Pur-DKK1, Huh7 Pur-Mock cells. Huh7 Pur-DKK1 and Huh7 Pur-Mock cells were cultured in the absence or presence of doxycycline (100 ng/mL) at 37°C for 48 h. Hep3B cells were used as a positive control for DKK1. Lysate: the aggregated fraction after centrifugation; sup: the supernatant (soluble fraction). F. Secreted DKK1 protein levels of CM of Huh7 Pur-Mock and HuH7 Pur-DKK1 cells were measured using ELISA after incubation with doxycycline (100 ng/mL) for 24 h.
Figure 2.

Generation of DKK1 knock-out alleles using CRISPR/Cas9. A. Targeting strategy of the DKK1 exon-1 gene. Blue: primer binding site, Green: sequence of DKK1 exon-1, Yellow: single-guide RNA binding site. B. Sequence of PCR primers for DKK1 exon-1.
Figure 3.

Confirmation of DKK1 exon-1 knock-out in Hep3B cells. A. Sanger sequencing results of gene knock-out in Hep3B CRISPR#73. B. Sanger sequencing results of gene knock-out in Hep3B CRISPR#131.
DKK1 enhances the angiogenic potential of HUVECs
We investigated whether DKK1 regulates endothelial cell tube formation and migration, which are two important features of angiogenesis [31]. Tube formation was enhanced in HUVECs treated with concentrated CM from doxycycline-treated in Huh7 Pur-DKK1 cells, whereas it was not enhanced in HUVECs treated with concentrated CM from Hep3B CRISPR#73 cells, compared to the controls (Figure 4A). In addition, HUVECs treated with CM from doxycycline-treated in Huh7 Pur-DKK1 and Hep3B wild-type displayed higher migration ability than the controls (Figure 4Ba). In addition, HUVECs treated with CM from Hep3B CRISPR#73 and #131 cells showed lower migration ability compared to growth medium and CM from Hep3B wild type (P<0.001). Whereas HUVECs treated with CM from CRISPR#73 and CRISPR#131 displayed no differences (Figure 4Bb). The effects of VEGFR2 downstream signaling were examined to understand the mechanism underlying DKK1 induced angiogenesis in HUVECs. We found that DKK1 increased the expression of p-VEGFR2. In addition, DKK1 significantly enhanced the expression of VEGFR2, p110α, p-Akt, p-mTOR and p-p70S6K in HUVECs (all P<0.001) (Figure 4C). Moreover, we demonstrated that HUVECs treated with rDKK1 showed increased Ki67 expression, compared to untreated HUVECs (P<0.01) (Figure 4D). In addition, rDKK1 treatment significantly increased VEGFR2-mediated Akt/mTOR/p70S6K signaling, whereas these protein levels were decreased by the VEGFR2-mediated Akt/mTOR/p70S6K signaling inhibitor, quercetin treatment compared to the control (all P<0.001). However, rDKK1 combined with quercetin treatment ameliorated VEGFR2-mediated Akt/mTOR/p70S6K signaling compared to rDKK1 treatment alone (P<0.001) (Figure 4E). Moreover, quercetin treatment showed no differences in the migration ability of HUVECs, whereas rDKK1 treatment significantly increased it, compared to controls (all P<0.01). However, rDKK1 combined with Quercetin treatment ameliorated the migration ability of HUVECs, compared to rDKK1 treatment (P<0.05) (Figure 4F). These data demonstrate that DKK1 might enhance the angiogenic potential and proliferation of HUVECs through VEGFR2-mediated mTOR/p70S6K signaling in vitro.
Figure 4.

DKK1 increases the angiogenic effects of HUVECs. A. Effects of DKK1 exposure on tube formation in HUVECs. HUVECs were cultured in serum free media (negative control), growth media (positive control) and concentrated CM of Huh7 Pur-Mock, Huh7 Pur-DKK1, Hep3B wild-type and Hep3B CRISPR#73 cells (scale bar: 500 μm). Concentrated CM was harvested from Huh7 Pur-Mock and Pur-DKK1 cells 24 h after doxycycline treatment (100 ng/mL). B. Effects of DKK1 exposure on migration. HUVECs were cultured in concentrated CM from Huh7 Pur-Mock, Huh7 Pur-DKK1, Hep3B wild-type and Hep3B CRISPR#73 cells. Concentrated CM from Huh7 Pur-DKK1 and Huh7 Pur-Mock cells was harvested 24 h after doxycycline treatment (100 ng/mL), Hep3B wild type and Hep3B CRISPR73 (a) and HUVECs were cultured in growth medium and concentrated CM from Hep3B wild type, CRISPR#73 and #131 cells (b). Bottom panel: Graphs show wound size. C. HUVECs lysates were probed with the indicated antibodies. HUVECs were stimulated with 100 ng/mL rDKK1 at each time point. GAPDH was used as loading control. D. HUVECs treated with 100 ng/mL rDKK1 were stained with anti-DKK1 (red) and DAPI (blue) (P<0.01). Ctrl: control (no treatment). E. HUVECs were treated with rDKK1 (100 ng/mL), quercetin (40 μM) and Quercetin (40 μM) + rDKK1 (100 ng/mL) (scale bar: 200 μM). Migration cells were quantified using ImageJ software. F. After starvation in serum-free medium for 6 h, HUVECs were pretreated with or without quercetin (40 μM) and 100 ng/mL rDKK1 for 1 h.
Co-culture with Hep3B stable cells and stromal cells
To investigate whether DKK1 induces angiogenesis in HCC spheroids, we generated an MCTS. We confirmed a profound enhancement of spheroid compactness in MCTS generated using Hep3B wild-type cells, compared to the control (Figure 5A). IF staining of MCTS showed that DKK1 expression levels in MCTS generated using Hep3B wild-type cells were significantly higher than in those generated using Hep3B CRISPR#73 cells (Figure 5B). Expression of the endothelial cell marker, CD31, was enhanced in MCTS generated using Hep3B wild-type, compared to the control. In addition, expression levels of the mesenchymal cell markers, vimentin and Slug, were increased (all P<0.01), whereas expression of the epithelial cell marker, E-cadherin, was attenuated (P<0.001) in MCTS generated using Hep3B wild-type cells, compared to MCTS generated using Hep3B CRISPR#73 cells (Figure 5C). In addition, MCTS generated using Hep3B wild-type cells increased the expression of VEGFR2-mediated mTOR/p70S6K signaling downstream proteins, compared to that generated using Hep3B CRISPR#73 (Figure 5D).
Figure 5.

DKK1 enhanced the compactness and angiogenic effects of HCC-MCTS. A. Hep3B wild-type and CRISPR#73 cells were co-cultured with or without stromal cells (HUVECs: LX2: WI38) for 3 days (scale bar: 200 μm). To calculate the volume of the spheroids, the radius of spheroids of the Hep3B wild-type and CRISPR#73 cells co-cultured with stromal cells were measured (P<0.001) (right panel). B. H&E (scale bar: 50 μm) and IF staining (magnification, 200 ×) for DKK1 on the paraffin sections of HCC-MCTS. C. IHC and IF staining for CD31, vimentin, Slug, and E-cadherin on serial paraffin sections of HCC-MCTS (magnification, 200 ×). D. Lysates of MCTS generated using Hep3B wild-type and CRISPR#73 cells were probed with the indicated antibodies. GAPDH was used as a loading control.
DKK1 promotes tumorigenesis in the xenograft mouse model
The xenograft mouse model was produced using Hep3B wild-type and CRISPR#73 cells to investigate whether DKK1 promotes tumorigenesis. Xenograft tumors generated using Hep3B CRISPR#73 cells (n=10) exhibited slower growth and smaller tumor volume and weight, than the controls (n=10) (Figure 6A-C). Subsequently, the xenograft mouse model using Hep3B CRISPR#73 showed better survival relative to the Hep3B wild-type (Figure 6D). Serum DKK1 levels were significantly higher in the model generated Hep3B wild-type cells than that generated using Hep3B CRISPR#73 cells (P<0.001) (Figure 6E). Immunohistochemistry (IHC) staining showed that Ki-67 expression levels in xenograft tumors generated using Hep3B wild-type cells were significantly higher than those in xenograft tumors generated using Hep3B CRISPR#73 cells (Figure 6F). These results indicate that DKK1 promotes HCC tumorigenesis by affecting tumor cell proliferation.
Figure 6.

DKK1 increased tumorigenesis in the xenograft mouse model. (A) Xenograft tumors generated using Hep3B CRISPR#73 cells exhibited slower growth, (B) smaller tumor volume and (C) smaller tumor weight (P<0.01) (n=10) than, the controls. (D) Kaplan-Meier survival curve of Hep3B wild-type and Hep3B CRISPR#73 tumor-bearing mice (each group, n=5). (E) DKK1 expression levels were detected using ELISA in the serum of the xenograft mouse models. (F) H&E (magnification, 200 ×) and IHC staining (magnification, 400 ×) for Ki-67 on serial paraffin sections of Hep3B wild-type and CRISPR#73 tumors. Graph of Ki-67 positive Hep3B stable cells (P<0.001).
DKK1 promotes epithelial-mesenchymal transition makers in xenograft mouse model
To investigate the mechanism of HCC tumorigenesis by DKK1, we observed the angiogenesis and EMT markers of xenograft mouse models using IF study. IF staining of serial sections of xenograft tumors showed that DKK1 expression levels in xenograft tumors generated using Hep3B wild-type cells were significantly higher than in those generated using Hep3B CRISPR#73 (P<0.05). In addition, the expression of angiogenesis markers, VEGFR2 and CD31, in xenograft tumors generated using Hep3B wild-type cells were significantly higher than in those generated using Hep3B CRISPR#73 cells (all P<0.05) (Figure 7A, 7B). The expression levels of mesenchymal markers of vimentin (P<0.05) and fibronectin (P<0.01) were significantly attenuated in xenograft tumors generated using Hep3B CRISPR#73 cells, compared to the controls, whereas the expression of the epithelial marker, E-cadherin, was significantly enhanced (P<0.05) in xenograft tumors generated using Hep3B CRISPR#73 cells, compared to the controls (Figure 7C, 7D). These results suggest that DKK1 increased the EMT markers in HCC.
Figure 7.
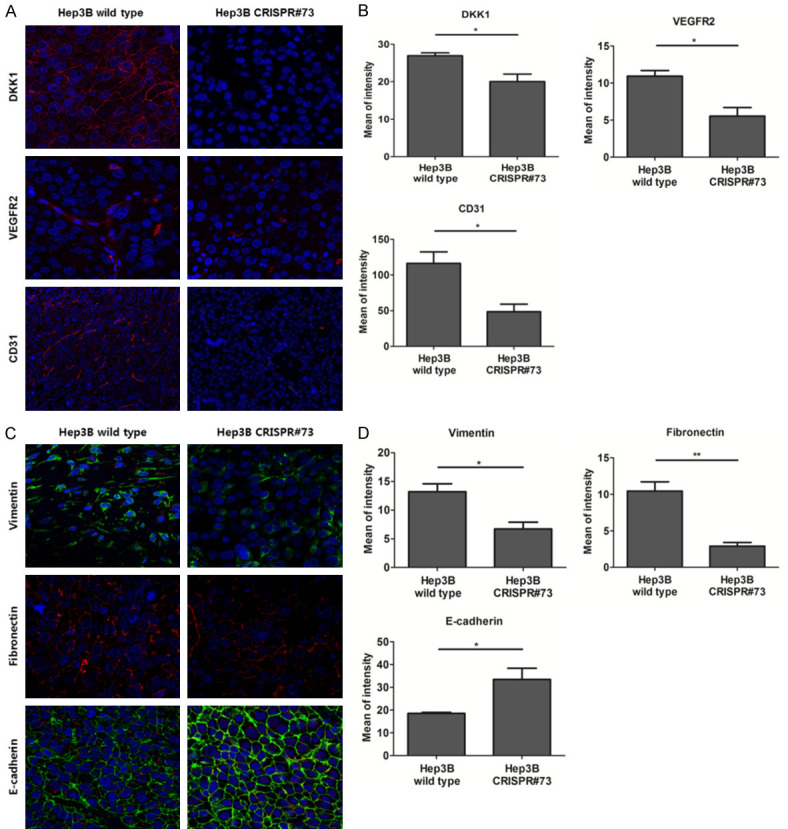
DKK1 increased angiogenesis and EMT markers in the xenograft mouse model. A. Observation of IF staining for DKK1, VEGFR2 and CD31 on serial paraffin sections of xenograft tumors generated using Hep3B wild-type and CRISPR#73 with confocal microscopy. B. Quantification of DKK1 (P<0.05), VEGFR2 (P<0.05) and CD31 (P<0.05) IF, presented as the mean fluorescence intensity. C. Observation of IF staining for vimentin, fibronectin and E-cadherin on serial paraffin sections of xenograft tumors generated using Hep3B wild-type and CRISPR#73 cells with confocal microscopy. D. Quantification of vimentin (P<0.05), fibronectin (P<0.01) and E-cadherin (P<0.05) IF, presented as the mean fluorescence intensity.
DKK1 enhanced tumor growth in the engineered mouse model
Hydrodynamic injection was performed to investigate the tumorigenic potential of DKK1. We previously confirmed that HCC is induced by the co-expression of HRASG12V with a shRNA downregulating p53 in the liver [32]. To investigate the effects of DKK1 in the liver, we expressed DKK1 together with the aforementioned oncogenic combinations. The liver was harvested 5 weeks after hydrodynamic injection (Figure 8A). DKK1-2A-HRASG12V + shp53 mice (HI_DKK1 mice) (n=5) had significantly more tumors with larger size than those of luciferase-2A-HRASG12V + shp53 mice (HI_Luc2A mice) (n=5) (Figure 8B). IHC staining showed that HRAS expression levels were similar between the HI_DKK1 and HI_Luc2A groups and that Ki-67 staining of HI_DKK1 tumors was slightly higher than that of HI_Luc2A tumors (Figure 8C). The expression levels of downstream molecules of VEGFR2-mediated mTOR/p70S6K signaling, were higher in HI_DKK1 tumors than in HI_Luc2A tumors (Figure 8D, 8E). In addition, IF staining showed that the expression levels of DKK1 and angiogenesis markers, VEGFR2 and CD31, were significantly increased in HI_tumors compared to HI_Luc2A tumors (all P<0.001) (Figure 9A, 9B). Moreover, expression levels of the epithelial marker, E-cadherin, was significantly attenuated and the mesenchymal markers of fibronectin were significantly enhanced in HI_DKK1 tumors, compared to the control (all P<0.001) (Figure 9C, 9D). These results indicate that DKK1 promotes tumorigenesis and angiogenesis by upregulating VEGFR2-mediated mTOR/p70S6K signaling and EMT markers in the mouse liver.
Figure 8.

DKK1 increased tumorigensis in the transgenic mouse model. A. DNA mixtures were injected into the lateral tail veins of 4-week-old mice and the livers were harvested at 9 weeks. B. Forced expression of DKK1 with HRAS in transgenic mouse liver resulted in the formation of more tumors (n=5), compared to the controls. C, D. H&E and IHC staining for HRAS, Ki-67, p-Akt and p-VEGFR2 on serial paraffin sections of HI_Luc2A and HI_DKK1 tumors (magnification, 200 ×). E. Lysates of HI_Luc2A and HI_DKK1 tumors were probed with the indicated antibodies. GAPDH was used as a loading control.
Figure 9.
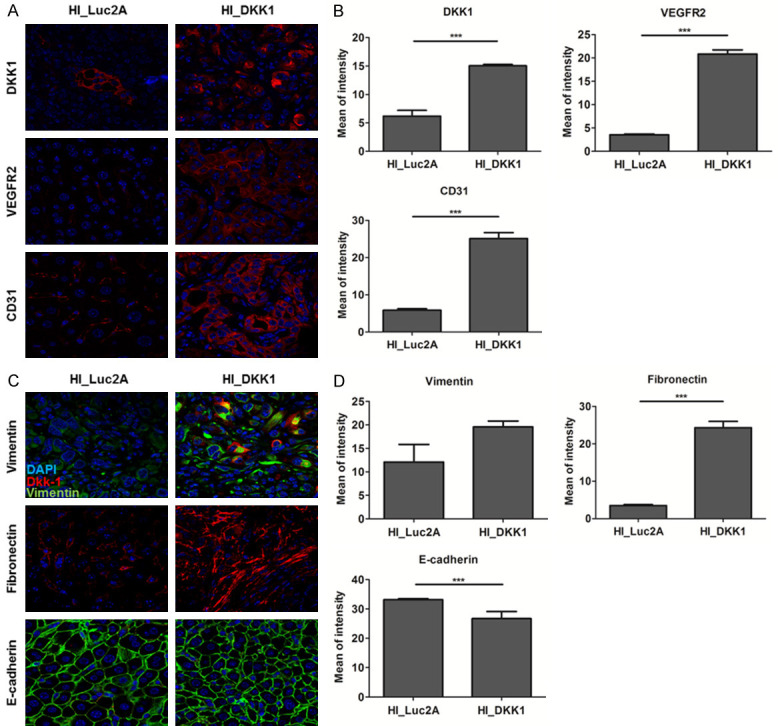
DKK1 increased angiogenesis and EMT markers in the transgenic mouse model. A. Observation of IF staining for DKK1, VEGFR2 and CD31 on serial paraffin sections of HI_Luc2A and HI_DKK1 tumors using confocal microscopy. B. Quantification of DKK1, VEGFR2 and CD31, presented as the mean fluorescence intensity. C. IF staining for vimentin, fibronectin and E-cadherin on serial paraffin sections of HI_Luc2A and HI_DKK1 tumors. D. Quantification of vimentin, fibronectin and E-cadherin, presented as the mean fluorescence intensity.
Discussion
We previously reported that DKK1 promotes angiogenesis via VEGFR2 regulation [27]. However, the specific interaction between DKK1 and VEGFR2 in HCC remains to be ascertained. In this study, we first confirmed that rDKK1 increases the invasion of HUVECs and then established stable cell lines with regulated DKK1 expression. Second, we found that the tube formation and migration abilities of HUVECs were significantly enhanced upon treatment with concentrated CM with high DKK1 concentration. Third, a signaling study showed that DKK1 was correlated with the VEGFR2-mediated mTOR/p70S6K signaling pathway. Fourth, we also found that DKK1 enhanced spheroid compactness and EMT markers in HCC-MCTS models. In addition, we reported that DKK1 promoted angiogenesis and tumorigenesis in xenograft tumor and mouse liver, which was supported by the increased expression of endothelial and EMT markers, such as CD31, vimentin, fibronectin and decreased expression of E-cadherin. Based on these results, we concluded that DKK1 promotes angiogenesis and tumorigenesis via upregulation of VEGFR2-mediated mTOR/p70S6K signaling and EMT markers.
Our study has several unique findings. First, our study provides additional support to the fact that DKK1 is significantly associated with angiogenesis of endothelial cells. To identify the angiogenic role of DKK1 in endothelial cells, we established stable cell lines using CRISPR/Cas9-based DKK1 knockout system in Hep3B and the tetracycline-based DKK1 inducible system in Huh7 cells. In Hep3B stable cell lines, two clones (Hep3B CRISPR#73 and #131) were selected because they were slightly smaller than the size of the upper part of the positive control band. Moreover, the Sanger sequencing results were used for selection. To date, the CRISPR-Cas9 system has shown great potential for genome editing, but several limitations still exist [33]. For instance, homology-directed repair and insertion-deletion mutations in some genome sites have shown low efficiency. In addition, it has been found that some regions of the genome are not cut very well with CRISPR, and some cell types do not easily accept foreign DNA, RNA, or RNA-protein complexes [34]. All these factors might be associated with the minimal detection of DKK1 in our study. Nevertheless, the secreted DKK1 levels in Hep3B CRISPR#73 and #131 were significantly lower than those in Hep3B wild-type cells, which enabled us to investigate the effects of DKK1 inhibition on angiogenesis and tumorigenesis in vitro and in vivo. To date, it has been reported that DKK1 promotes the angiogenic effects of endothelial cells and fibroblasts [35,36]. Smadja et al. [35] showed that DKK1 enhanced the proangiogenic potential of human endothelial colony-forming cells and these angiogenic effects have been attributed to enhancement of VEGFR2. In addition, Jiang et al. [37] showed that DKK1 is correlated with angiogenesis in fibroblasts through increased expression of VEGF and also found that HIF-1α may be associated with DKK1-induced HUVECs migration. Our data similarly found that DKK1 raised the angiogenic potential of endothelial cells, which was supported by the increased effects of HUVEC invasion, tube formation and migration. Our results showed that the angiogenic effects of HUVECs were increased by DKK1 in HCC cells.
Second, because we noticed a close association between DKK1 and angiogenesis, we focused on the mechanism involved. We found that DKK1 promoted HUVECs invasion and migration by upregulating VEGFR2-mediated mTOR/p70S6K pathway. mTOR expression is frequently upregulated in cancer, including HCC and is associated with poor prognosis, poorly differentiated tumors and early recurrence [38]. In addition, Trinh et al. [39] determined that VEGF-A signaling acts on tumor cells as a stimulator of the Akt/mTOR pathway. Our results showed that DKK1 increased p-VEGFR2 and p-p70S6K protein levels in HUVECs. p70S6K, which is activated in signaling pathways that include mTOR, is significantly associated with HCC [40,41]. Li et al. [40] suggested that the expression level of p-p70S6K was increased in HCC compared to that in cirrhotic nodules and normal liver tissue using immunostaining. In addition, Kristine et al. [42] showed that patients with breast tumors having increased expression of p-p70S6K showed increased metastasis and worse disease-free survival. In addition, to identify the effects of DKK1 in VEGFR2 signaling, we used the quercetin. In a previous study, Pratheeshkumar et al. [43] found that quercetin inhibited VEGFR2-mediated Akt/mTOR/p70S6K signaling. We found that DKK1 combination of auercetin ameliorated VEGFR2-mediated Akt/mTOR/p70S6K signaling and angiogenesis effects, compared to DKK1 alone. These results suggest that DKK1 promotes angiogenesis through VEGFR2-mediated Akt/mTOR/p70S6K signaling. Based on these results, our data suggest that the DKK1-induced VEGFR2 downstream pathway is involved in the angiogenesis of endothelial cells. Considering that DKK1 is a major player in VEGFR2-mediated mTOR/p70S6K, DKK1 may be an attractive target for preventing the incidence of angiogenesis.
Third, we showed the influence of DKK1 on spheroid formation using Hep3B, HUVEC, LX2 and WI-38 cells. Our results reveal a striking correlation between the compactness ability of DKK1 and MCTS. In ovarian cancer cells, Katharine et al. [44] first indicated that a cell line possessing myofibroblast-like properties could form compact spheroids and subsequently, Katharine suggested that compact spheroid formation may facilitate ovarian cancer cell invasion. In addition, Cho et al. [45] found that YAP/TAZ levels were significantly different for each type of HCC-MCTS model, while Hep3B MCTS had the highest level of YAP/TAZ expression. YAP and TAZ are known to contribute to cell cycle [46] and Jorgenson et al. [47] reported that TAZ activation is associated with fibroblast spheroid growth. In summary, although Kelm et al. [48] reported an inverse association between tumor cell spheroid cohesiveness and invasive potential, a positive correlation between tumorigenicity and spheroid formation of cancer cells has been suggested [49]. Taken together, our MCTS results show that a DKK1-mediated increase in compactness, in addition to a rise in the expression levels of vimentin and Slug in MCTS, may promote cancer cell invasion. In addition, the relationship between YAP/TAZ and the ability of DKK1 including spheroid compactness should be further studied.
Fourth, we found that DKK1 leads to poor survival and there was an increase in the expression of angiogenesis and EMT markers in the mouse model, indicating the enhancement of angiogenesis and EMT markers upon DKK1 stimulation. Similar to the findings of the present study, Yao et al. [50] showed that in vasculogenic mimicry, DKK1 increases the expression level of EMT-related protein in non-small cell lung cancer. In addition, Maha et al. [51] found that since DKK1 facilitates tumor invasion and migration through transforming growth factor beta 1, it may induce EMT in HCC cell lines. Indeed, increasing evidence indicates a central role of EMT, which might occur at the leading edge of tumor cells, under particular factors derived from the tumor microenvironment in HCC [52]. In addition to EMT, DKK1 is significantly associated with endothelial-to-mesenchymal transition (EndMT) [27,53]. Previously, Choi et al. [27] found that DKK1 increases the expression of EndMT-related proteins (N-cadherin, Twist and vimentin) in endothelial cells and Cheng et al. [53] demonstrated that DKK1 enhances EndMT in aortic endothelial cells by augmenting ALK/Smad signaling to modulate the endothelial cell phenotype. In contrast, some studies have shown that EMT is negatively regulated by DKK1 [54-56]. In spite of this controversy regarding the interaction between DKK1 and EMT/EndMT [57,58], based on the findings of our current study, we postulate that DKK1 may help to develop effective therapies against HCC.
There are several limitations to this study. First, our data show that DKK1 activates VEGFR2 downstream signaling, but only at the endothelial cell level. The mechanism of DKK1 should be further investigated in the HCC cell line or HCC mouse model. Second, our data confirmed that DKK1 activates downstream molecules of VEGFR2 signaling, but further studies are required to determine the underlying mechanism. In addition, since DKK1 is known to be a Wnt signaling antagonist, it needs to be a studied whether the activation of VEGFR2 signaling by DKK1 is dependent or independent of Wnt signaling.
In conclusion, our findings demonstrated that DKK1 facilitates endothelial cell angiogenesis through the upregulation of VEGFR2-mediated mTOR/p70S6K signaling and promotes HCC progression through the activation of EMT factors in diverse HCC models. Our results suggest that DKK1 is a potential therapeutic target for HCC.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2019R1A2C4070136) and by a faculty research grant from Yonsei University College of Medicine (6-2018-0130).
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Liu K, Min XL, Peng J, Yang K, Yang L, Zhang XM. The changes of HIF-1α and VEGF expression after TACE in patients with hepatocellular carcinoma. J Clin Med Res. 2016;8:297–302. doi: 10.14740/jocmr2496w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- 5.Daher S, Massarwa M, Benson AA, Khoury T. Current and future treatment of hepatocellular carcinoma: an updated comprehensive review. J Clin Transl Hepatol. 2018;6:69–78. doi: 10.14218/JCTH.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong L, Zhang X, Li C, Zhou L. Potential therapeutic targets and small molecular drugs for pediatric B-precursor acute lymphoblastic leukemia treatment based on microarray data. Oncol Lett. 2017;14:1543–1549. doi: 10.3892/ol.2017.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arciero CA, Sigurdson ER. Liver-directed therapies for hepatocellular carcinoma. J Natl Compr Canc Netw. 2006;4:768–774. doi: 10.6004/jnccn.2006.0067. [DOI] [PubMed] [Google Scholar]
- 8.Amini A, Masoumi Moghaddam S, Morris DL, Pourgholami MH. The critical role of vascular endothelial growth factor in tumor angiogenesis. Curr Cancer Drug Targets. 2012;12:23–43. doi: 10.2174/156800912798888956. [DOI] [PubMed] [Google Scholar]
- 9.Yi ZY, Feng LJ, Xiang Z, Yao H. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in hepatocellular carcinoma cells. J Invest Surg. 2011;24:67–76. doi: 10.3109/08941939.2010.542272. [DOI] [PubMed] [Google Scholar]
- 10.Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- 11.Fedi P, Bafico A, Nieto Soria A, Burgess WH, Miki T, Bottaro DP, Kraus MH, Aaronson SA. Isolation and biochemical characterization of the human Dkk-1 homologue, a novel inhibitor of mammalian Wnt signaling. J Biol Chem. 1999;274:19465–19472. doi: 10.1074/jbc.274.27.19465. [DOI] [PubMed] [Google Scholar]
- 12.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417:664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 13.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 14.Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, Dorward DW, Glinka A, Grinberg A, Huang SP, Niehrs C, Izpisúa Belmonte JC, Westphal H. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell. 2001;1:423–434. doi: 10.1016/s1534-5807(01)00041-7. [DOI] [PubMed] [Google Scholar]
- 15.Zorn AM. Wnt signalling: antagonistic Dickkopfs. Curr Biol. 2001;11:R592–595. doi: 10.1016/s0960-9822(01)00360-8. [DOI] [PubMed] [Google Scholar]
- 16.Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–1305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- 17.Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, Sylvester KG. Wnt/beta-catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology. 2007;133:1579–1591. doi: 10.1053/j.gastro.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 18.Choi HJ, Park H, Lee HW, Kwon YG. The Wnt pathway and the roles for its antagonists, DKKS, in angiogenesis. IUBMB Life. 2012;64:724–731. doi: 10.1002/iub.1062. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Ma JX. Wnt pathway antagonists and angiogenesis. Protein Cell. 2010;1:898–906. doi: 10.1007/s13238-010-0112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tung EK, Mak CK, Fatima S, Lo RC, Zhao H, Zhang C, Dai H, Poon RT, Yuen MF, Lai CL, Li JJ, Luk JM, Ng IO. Clinicopathological and prognostic significance of serum and tissue Dickkopf-1 levels in human hepatocellular carcinoma. Liver Int. 2011;31:1494–1504. doi: 10.1111/j.1478-3231.2011.02597.x. [DOI] [PubMed] [Google Scholar]
- 21.Kwack MH, Hwang SY, Jang IS, Im SU, Kim JO, Kim MK, Kim JC, Sung YK. Analysis of cellular changes resulting from forced expression of Dickkopf-1 in hepatocellular carcinoma cells. Cancer Res Treat. 2007;39:30–36. doi: 10.4143/crt.2007.39.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SU, Park JH, Kim HS, Lee JM, Lee HG, Kim H, Choi SH, Baek S, Kim BK, Park JY, Kim DY, Ahn SH, Lee JD, Han KH. Serum Dickkopf-1 as a biomarker for the diagnosis of hepatocellular carcinoma. Yonsei Med J. 2015;56:1296–1306. doi: 10.3349/ymj.2015.56.5.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Q, Fan J, Yang XR, Tan Y, Zhao W, Xu Y, Wang N, Niu Y, Wu Z, Zhou J, Qiu SJ, Shi YH, Yu B, Tang N, Chu W, Wang M, Wu J, Zhang Z, Yang S, Gu J, Wang H, Qin W. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. Lancet Oncol. 2012;13:817–826. doi: 10.1016/S1470-2045(12)70233-4. [DOI] [PubMed] [Google Scholar]
- 24.Patil MA, Chua MS, Pan KH, Lin R, Lih CJ, Cheung ST, Ho C, Li R, Fan ST, Cohen SN, Chen X, So S. An integrated data analysis approach to characterize genes highly expressed in hepatocellular carcinoma. Oncogene. 2005;24:3737–3747. doi: 10.1038/sj.onc.1208479. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Li M, Li Q, Wang CJ, Xie SQ. DKK1 promotes hepatocellular carcinoma cell migration and invasion through β-catenin/MMP7 signaling pathway. Mol Cancer. 2013;12:157. doi: 10.1186/1476-4598-12-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao YM, Liu Z, Liu HL. Dickkopf-1 (DKK1) promotes invasion and metastasis of hepatocellular carcinoma. Dig Liver Dis. 2013;45:251–257. doi: 10.1016/j.dld.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 27.Choi SH, Kim H, Lee HG, Kim BK, Park JY, Kim DY, Ahn SH, Han KH, Kim SU. Dickkopf-1 induces angiogenesis via VEGF receptor 2 regulation independent of the Wnt signaling pathway. Oncotarget. 2017;8:58974–58984. doi: 10.18632/oncotarget.19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Y, Kim SH, Kim KM, Choi EK, Kim J, Seo HR. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci Rep. 2016;6:36750. doi: 10.1038/srep36750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 30.Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209:497–499. doi: 10.1126/science.6248960. [DOI] [PubMed] [Google Scholar]
- 31.Tahergorabi Z, Khazaei M. A review on angiogenesis and its assays. Iran J Basic Med Sci. 2012;15:1110–1126. [PMC free article] [PubMed] [Google Scholar]
- 32.Ju HL, Ahn SH, Kim DY, Baek S, Chung SI, Seong J, Han KH, Ro SW. Investigation of oncogenic cooperation in simple liver-specific transgenic mouse models using noninvasive in vivo imaging. PLoS One. 2013;8:e59869. doi: 10.1371/journal.pone.0059869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Xu J, Ge S, Lai L. CRISPR/Cas: advances, limitations, and applications for precision cancer research. Front Med (Lausanne) 2021;8:649896. doi: 10.3389/fmed.2021.649896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alyami MZ, Alsaiari SK, Li Y, Qutub SS, Aleisa FA, Sougrat R, Merzaban JS, Khashab NM. Cell-type-specific CRISPR/Cas9 delivery by biomimetic metal organic frameworks. J Am Chem Soc. 2020;142:1715–1720. doi: 10.1021/jacs.9b11638. [DOI] [PubMed] [Google Scholar]
- 35.Smadja DM, d’Audigier C, Weiswald LB, Badoual C, Dangles-Marie V, Mauge L, Evrard S, Laurendeau I, Lallemand F, Germain S, Grelac F, Dizier B, Vidaud M, Bièche I, Gaussem P. The Wnt antagonist Dickkopf-1 increases endothelial progenitor cell angiogenic potential. Arterioscler Thromb Vasc Biol. 2010;30:2544–2552. doi: 10.1161/ATVBAHA.110.213751. [DOI] [PubMed] [Google Scholar]
- 36.Weng LH, Ko JY, Wang CJ, Sun YC, Wang FS. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012;64:3267–3277. doi: 10.1002/art.34602. [DOI] [PubMed] [Google Scholar]
- 37.Jiang SJ, Li W, Li YJ, Fang W, Long X. Dickkopf-related protein 1 induces angiogenesis by upregulating vascular endothelial growth factor in the synovial fibroblasts of patients with temporomandibular joint disorders. Mol Med Rep. 2015;12:4959–4966. doi: 10.3892/mmr.2015.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855–865. doi: 10.1016/j.jhep.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trinh XB, Tjalma WA, Vermeulen PB, Van den Eynden G, Van der Auwera I, Van Laere SJ, Helleman J, Berns EM, Dirix LY, van Dam PA. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer. 2009;100:971–978. doi: 10.1038/sj.bjc.6604921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Tan D, Zhang Z, Liang JJ, Brown RE. Activation of Akt-mTOR-p70S6K pathway in angiogenesis in hepatocellular carcinoma. Oncol Rep. 2008;20:713–719. [PubMed] [Google Scholar]
- 41.Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, Liu Y, Gui B, Sini M, Smith JW, Dombrowski F, Calvisi DF, Evert M, Chen X. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice. Hepatology. 2015;61:200–213. doi: 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P, Yang W, Yin G, Hittelman WN, Yu D. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;66:2028–2037. doi: 10.1158/0008-5472.CAN-04-4559. [DOI] [PubMed] [Google Scholar]
- 43.Pratheeshkumar P, Budhraja A, Son YO, Wang X, Zhang Z, Ding S, Wang L, Hitron A, Lee JC, Xu M, Chen G, Luo J, Shi X. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR- 2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS One. 2012;7:e47516. doi: 10.1371/journal.pone.0047516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sodek KL, Ringuette MJ, Brown TJ. Compact spheroid formation by ovarian cancer cells is associated with contractile behavior and an invasive phenotype. Int J Cancer. 2009;124:2060–2070. doi: 10.1002/ijc.24188. [DOI] [PubMed] [Google Scholar]
- 45.Cho K, Ro SW, Lee HW, Moon H, Han S, Kim HR, Ahn SH, Park JY, Kim DY. YAP/TAZ suppress drug penetration into hepatocellular carcinoma through stromal activation. Hepatology. 2021 doi: 10.1002/hep.32000. [DOI] [PubMed] [Google Scholar]
- 46.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 47.Jorgenson AJ, Choi KM, Sicard D, Smith KM, Hiemer SE, Varelas X, Tschumperlin DJ. TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression. Am J Physiol Cell Physiol. 2017;312:C277–C285. doi: 10.1152/ajpcell.00205.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol Bioeng. 2003;83:173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 49.Casey RC, Burleson KM, Skubitz KM, Pambuccian SE, Oegema TR Jr, Ruff LE, Skubitz AP. Beta 1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am J Pathol. 2001;159:2071–2080. doi: 10.1016/s0002-9440(10)63058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao L, Zhang D, Zhao X, Sun B, Liu Y, Gu Q, Zhang Y, Zhao X, Che N, Zheng Y, Liu F, Wang Y, Meng J. Dickkopf-1-promoted vasculogenic mimicry in non-small cell lung cancer is associated with EMT and development of a cancer stem-like cell phenotype. J Cell Mol Med. 2016;20:1673–1685. doi: 10.1111/jcmm.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fezza M, Moussa M, Aoun R, Haber R, Hilal G. DKK1 promotes hepatocellular carcinoma inflammation, migration and invasion: Implication of TGF-β1. PLoS One. 2019;14:e0223252. doi: 10.1371/journal.pone.0223252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798–808. doi: 10.1016/j.jhep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Cheng SL, Shao JS, Behrmann A, Krchma K, Towler DA. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1679–1689. doi: 10.1161/ATVBAHA.113.300647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitra A, Menezes ME, Shevde LA, Samant RS. DNAJB6 induces degradation of beta-catenin and causes partial reversal of mesenchymal phenotype. J Biol Chem. 2010;285:24686–24694. doi: 10.1074/jbc.M109.094847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menezes ME, Devine DJ, Shevde LA, Samant RS. Dickkopf1: a tumor suppressor or metastasis promoter? Int J Cancer. 2012;130:1477–1483. doi: 10.1002/ijc.26449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5995. doi: 10.1158/0008-5472.CAN-05-0616. discussion 5995. [DOI] [PubMed] [Google Scholar]