

Abstract
Angiotensin-II (Ang-II), a major target for treatment of cardiovascular disease, promotes cardiovascular dysfunction by directly modulating structure and function of vascular cells. Inflammasome components are expressed in the vasculature and are activated by specific stimuli. However, whether Ang-II activates the inflammasome in vascular cells or inflammasome activation contributes to Ang-II-induced vascular damage is still not fully elucidated. We tested the hypothesis that Ang-II induces endothelial dysfunction, vascular remodeling, and high blood pressure via inflammasome activation. C57BL6/J wild type (WT) and Caspase-1 knockout (Casp1−/−) mice were infused with vehicle or Ang-II for two weeks (490 ng/Kg/day) to determine whether the inflammasome contributes to vascular damage induced by Ang-II. Rat Aortic Vascular Smooth Muscle cells (RASMC) were used to determine if the interaction between Ang-II and inflammasomes causes migration and proliferation of vascular smooth muscle cells. Ex vivo studies revealed that Ang-II infusion induced vascular oxidative stress, endothelial dysfunction and vascular remodeling in WT mice. Casp1−/− mice were protected against Ang-II-induced vascular injury. In vitro experiments, Ang-II activated the NLRP3 inflammasome in RASMC, i.e. Ang-II increased Caspase-1 (Casp1) activity and cleavage of pro-interleukin (IL)-1β. MCC950 (NLRP3 receptor antagonist) prevented Ang-II-induced vascular migration and proliferation, but failed to reduce reactive oxygen species production. In conclusion, Ang-II leads to inflammasome activation in the vasculature contributing to endothelial dysfunction and vascular remodeling. Taken together, we place inflammasomes as a possible therapeutic target in conditions associated with increased Ang-II levels.
Keywords: Angiotensin-II, Inflammasome, vascular damage
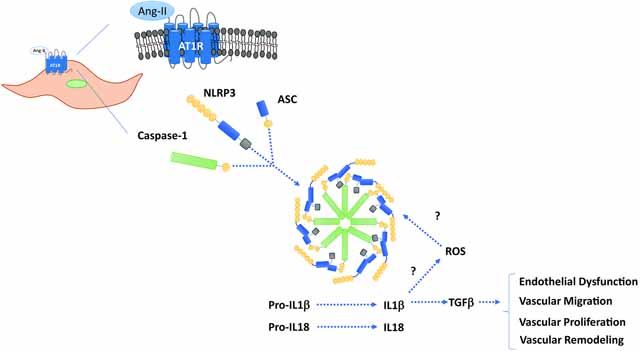
Graphical Abstract
INTRODUCTION
Innate and adaptive immune responses orchestrate cardiovascular health by modulating cellular responses to injury, generation of cytokines, activation and recruitment of immune cells, and cardiovascular structure and function1–3. The first evidence emerged in the sixties, when White and Grollman4 described the effects of two immunosuppressant drugs on blood pressure levels in rats with kidney infarction. Since then, the crosstalk between the cardiovascular and immune systems has been extensively studied5–7. Although the link between overactivation of the immune system and cardiovascular disease is well defined, several pieces of this puzzle are still missing including signals and mechanisms that generate the immune responses. In this context, members of the Nod-like receptor (NLR) family have emerged as major players in the genesis of cardiovascular diseases3, 8–13.
The NLRP3 inflammasome is well expressed in immune cells and is considered a pivotal immune sensor that recognizes endogenous danger signals and triggers sterile inflammation. The activation of NLRP3, well-characterized member of the NLR family, leads to the assembly of the inflammasome platform, a multiprotein oligomer, in which contains Caspase1, a proteolytic protein that processes and increases interleukin (IL)-1β and IL-18 secretion8, 14–19. Compelling evidence shows that NLRP3 inflammasome components are expressed in different cell types including the cardiovascular system and they may participate in the control and organization of cardiovascular function and structure. However, whether Angiotensin-II (Ang-II) activates the NLRP3 inflammasome leading to cardiovascular damage is still poorly understood.
A disequilibrium in Ang-II levels negatively impacts cardiovascular function20–23 by triggering vascular dysfunction, inflammation and remodeling via directly affecting the vasculature, heart and kidneys21 or indirectly regulating immune responses1, 21. Innate immune receptors, such as Toll Like Receptor 4 (TLR4)24, 25 and NLRP310, 26, 27, are expressed across multiple cells including endothelial and vascular smooth muscle cells (VSMC) which, perhaps, are activated Ang-II. Ang-II induces most of its cardiovascular deleterious effects via angiotensin receptor type 1 (AT1R)24, 25. It has been previously demonstrated that Ang-II-induced vascular migration and proliferation depends on a link between AT1R and immune receptors expressed in the vasculature, e.g: TLR424, 25. For these reasons, Ang-II signaling is one of the major therapeutic targets for reverting or minimizing cardiovascular diseases such as arterial hypertension, aneurism, atherosclerosis and heart failure22, 28, 29.
In the present study, we sought to determine whether Ang-II activates the NLRP3 inflammasome and whether NLRP3 activation triggers endothelial dysfunction, vascular remodeling, and high blood pressure. Considering that Casp1 is an essential protein for NLRP3 inflammasome assembly and for the generation of the pro-inflammatory cytokines IL-1β and IL-18, Ang-II was administered to control wild type and Casp1/11 deficient (Casp1−/−) mice. Furthermore, MCC950 (NLRP3 receptor antagonist) was used in isolated VSMC to elucidate the role of inflammasomes on vascular oxidative stress, migration, and proliferation.
MATERIAL AND METHODS
Animals
Eight to ten week–old male C57BL6/J wild type (WT) mice and global knockout mice for Caspase1/11 (Casp1−/−) were kept in the animal facility of the Department of Pharmacology, Ribeirao Preto Medical School, University of Sao Paulo, under controlled temperature (22–24 °C) and humidity, 12-hours (h) light/dark cycles. Mice were fed standard diet and water ad libitum. All experimental procedures were approved by the Ethics Committee on Animal Research of the Ribeirao Preto Medical School, University of Sao Paulo (protocol n° 012/2013–1) and on the University of Pittsburgh (IACUC protocol # 19065333). All experiments were performed in accordance with the Guide Laboratory Animals for The Care and Use of Laboratory Animals.
Ang-II infusion
Eight to ten-week-old male WT and Casp1−/− mice were infused with vehicle or Ang-II (490 ng/min/kg) for 14 days with ALZET osmotic minipumps (Alzet Model 1002; Alzet Corp Durect, Cupertino, CA). Thoracic aortae were used for detection of intracellular reactive oxygen species (ROS) by dihydroethidium fluorescence (DHE, Thermo Fisher, Waltham, MA, USA), for hematoxylin and eosin staining and for Casp1 activity via FLICA 660 Casp1 assay (ImmunoChemistry Technologies, LLC Bloomington, MN, USA). Mesentery beds were used for vascular reactivity studies and for structural studies using a pressure myography (Danish MyoTechnology, Hinnerup). Methods are described in the following sections.
Blood pressure measurement
Systolic blood pressure was measured via CODA® (Kent Scientific corporation, Torrington, CT, USA). After one week of acclimation, blood pressure was recorded at time 0 (before Ang-II treatment), 1 week and 2 weeks post- Ang-II treatment. Mice were placed for 5 minutes in a warm cage to dilate the tail arteries before measuring their blood pressure.
Casp1 activity - FLICA 660 Casp1 Assay
The fluorescent dye FLICA 660 Casp1 Assay (Catalogue# 9122, ImmunoChemistry Technologies, Bloomington, MN, USA) was used to determine caspase1 activity. Aortic rings were frozen in Tissue-Tek® OCT compound (Maumee, OH, USA) embedding medium, cut in histological sections (10 μm), fixed in acetone and incubated for 1 h protected from light at 37 °C, washed, mounted, visualized and analyzed with a Leica DMI 4000B inverted microscope (×40 objective). The images were processed using LAS AF software (Leica Microsystems) and analyzed by the ImageJ analysis software (NIH, Bethesda, MD, USA).
Morphometric analysis of the vascular wall
At 14 days after Ang-II infusion, thoracic aortae were harvested, cleaned of connective tissue, immediately fixed in 4% phosphate-buffered paraformaldehyde at pH 7.4 and embedded in paraffin blocks. Four micrometer-thick slices were stained with hematoxylin and eosin. Cross-Sectional Area (CSA) was calculated by subtracting the lumen internal area (Ai) from the external area (Ae), which was measured in each tissue section. The external diameter (ED) and the internal diameter (ID) were calculated as the square root of 4Ae/p and 4Ai/p, respectively. Media thickness (M) was calculated as (ED - ID)/2. Stained sections were examined with a light microscopy (DMLB; Leica, Bensheim, Germany) and the images were captured at x400. These structural analyses in the media were evaluated by using ImageJ Program.
Detection of intracellular ROS by dihydroethidium fluorescence
The fluorescent dye DHE was used to determine superoxide anion (O2−) production. Aortic rings were frozen in Tissue-Tek® OCT compound embedding medium, cut in histological sections (10 μm) and incubated for 10 min at 37 °C in Krebs Henseleit solution containing 5 μM DHE. Fluorescent intensity was captured with a Leica DMI 4000B inverted microscope (×40 objective). The images were processed using LAS AF software (Leica Microsystems) and analyzed by the ImageJ analysis software.
Vascular Reactivity Studies
Rings from second-order mesenteric resistance arteries were mounted in a wire myograph (Danysh MyoTechnology) for isometric tension recordings with PowerLab software (AD Instruments). Rings (2 mm) were placed in tissue baths containing warmed (37 °C), aerated (95% O2, 5% CO2) Krebs Henseleit Solution: (in mM: 130 NaCl, 4.7 KCl, 1.17 MgSO4, 0.03 EDTA, 1.6 CaCl2, 14.9 NaHCO3, 1.18 KH2PO4, and 5.5 glucose) and after 30 min of stabilization, curves of tension were performed to adjust the ideal tension for each segment3. Phenylephrine (10 μM) was used to test arterial viability, and the presence of intact endothelium was verified by acetylcholine (ACh) (1 μM)-induced relaxation in vessels contracted with phenylephrine (1 μM). Concentration-response curves for acetylcholine, and sodium nitroprusside (SNP) were performed.
Pressure myography
Structural properties of mesenteric resistance arteries were determined with a pressure myography (Danish Myo Tech, Model 110P and 111P). Vessel segments were placed between two-glass microcannulas and tied with surgical nylon suture. The segment was set to a pressure of 70 mmHg under no-flow conditions and allowed to equilibrate at 37 °C, for 30 min before recording. Experiments were performed in calcium-free (0Ca2+) Krebs Henseleit Solution containing ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) 10 mM and gassed with a mixture of 95% O2 and 5% CO2. Intraluminal pressure was then reduced to 3 mmHg and a pressure–diameter curve was obtained by progressively increasing intraluminal pressure from 3 to 140 mmHg. Wall thickness and CSA were determined30.
Vascular Smooth Muscle Cells
Rat Aortic Smooth Muscle cells (RASMC) were purchased from Lonza (Walkersville, MD, U.S.A) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco from Thermo Fisher Scientific, Waltham, MA, U.S.A) containing 10% fetal bovine serum (FBS, HyClone Logan UT, U.S.A), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco from Thermo Fisher Scientific, Waltham, MA, U.S.A). Cells between passages 3 and 6 were used in all the experiments. Complete medium was replaced by DMEM containing of FBS 0.5% one before the treatments.
RASMC treatments
RASMC were stimulated with Ang-II (0.1 μM Sigma-Aldrich, MO, USA) for 15–30 min (reactive oxygen species measurement) and for 8–24 h (the other experiments). To analyze the impact of NLRP3 on Ang-II effects, RASMC were pre-incubated with a NLRP3 antagonist (MCC950, 1μM, Tocris Minneapolis, MN, U.S.A) 30 min prior Ang-II. To confirm that Ang-II triggers inflammasome activation in isolated vascular cells via AT1R some experiments were performed in presence of losartan (AT1R antagonist, 10μM, Tocris Minneapolis, MN, U.S.A). The involvement of IL-1β was accessed by incubating RASMC with IL-1 β recombinant protein (R&D Systems, Minneapolis, MN, USA). (Lipopolysaccharide (LPS, 500 ng.ml−1 for 6 h, Adipogen Corporation, San Diego, CA, U.S.A) followed by nigericin (20 μM for 40 min, Tocris Minneapolis, MN, U.S.A) was used as a positive control for NLRP3 inflammasome activation.
Western Blot analysis
Total protein was extracted from RASMC. Samples were homogenized in 50 mM Tris/HCl (pH 7.4) lysis buffer [containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 150 mM sodium chloride (NaCl), 1 mM ethylenediamine tetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate (SDS), 2 mM sodium orthovanadate (Na3VO4), 1 mM phenylmethylsulphonyl fluoride (PMSF), 1 μg/ml pepstatin A, 1 μg/ml leupeptin and 1 μg/ml aprotinin]. Total protein was cleared by centrifugation at 12,000 g for 10 min and the pellet was discarded. Proteins (25 μg) were separated by electrophoresis on a polyacrylamide gel and transferred to Immobilon-P poly (vinylidene fluoride) membranes. Non-specific binding sites were blocked with 5% skim milk or 1% bovine serum albumin (BSA) in Tris-buffered saline solution with Tween for 1 h at 24 °C. Membranes were then incubated with specific antibodies overnight at 4 °C. Antibodies were as follows: anti-NLRP3 (1:1000, Novus Biologicals, Centennial, CO, USA, Cat. # MAB7578), anti-Casp1 (1:500, Imgenex, part of Novus Biologicals, Centennial, CO, USA, Cat. # NB100–56565), anti-IL-1β (1:500, Novus Biologicals, Centennial, CO, U.S.A, Cat. # NB600–633), anti-Proliferating cell nuclear antigen (PCNA, 1:1000, Cell Signaling, Danvers, MA, U.S.A Cat. # D3H8P) and anti-β-actin (1:10000 Sigma, St. Louis, MO, USA, Cat. # A3854) which was used as an internal housekeeping control for the samples. After incubation with secondary antibodies, the enhanced chemiluminescence luminol reagent (SuperSignal™ West Femto Maximum Sensitivity Substrate, Thermo Fisher Waltham, MA, U.S.A) was used for antibody detection.
Real time RT (reverse transcription)-PCR
Total mRNA RASMC was extracted (Trizol Plus, Thermo Fisher, Waltham, MA USA), purified with chloroform method, and eluted in 20 μl of diethyl pyrocarbonate (DEPC)-treated water. Complementary DNA was generated by reverse transcription polymerase chain reaction (RT-PCR) with SuperScript III (Thermo Fisher Waltham, MA USA). Reverse transcription was performed at 58 °C for 50 min; the enzyme was heat inactivated at 85 °C for 5 min, and real-time quantitative RT-PCR was performed with the PowerTrack™ SYBR Green Master Mix (Thermo Fisher, Waltham, MA USA). The genes analyzed were: Transforming growth factor β (TGF- β): FW: 5’-GGCACCATCCATGACATGAA-3’ and RV: 5’-CAGGTGTTGAGCCCTTTCCA-3’, Ki67: FW: 5’-CTTTGCGCCATGCTGAAACT-3’ and RV: 5’-ATGACGACCTGGAACATCGG-3’ and collagen III: FW: 5’-AGATGCTGGTGCTGAGAAG-3’ and RV: 5’-TGGAAAGAAGTCTGAGGAAGG-3’. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) was used as house-keeping gene: FW: 5’-ACCCAGAAGACTGTGGATGG-3’ and RV: 5’-CACATTGGGGGTAAGGAACAC-3’. Experiments were performed in a QuantStudio™ 5 Real-Time PCR System, 384-well (Thermo Fisher, Waltham, MA USA). Data were quantified by 2ΔΔ Ct and are presented by fold changes.
ROS measurement in RASMC
ROS was measured by 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA)- Cellular ROS Assay Kit (Abcam, Cambridge, MA USA). Cells were seeded in black 96-well plates and incubated overnight in DMEM with FBS 0.5%. Cells were exposed to different stimuli for short (30–60 min) and long (8–24 h) periods. After stimulation, the medium was replaced by Hanks’ Balanced Salt solution (HBSS Modified medium) and stained with DCFDA for 45 min. Cells were then washed with HBSS Modified medium and the fluorescence was analyzed with excitation / emission at 485 nm / 535 nm in a fluorimeter (FLUOstar Omega, BMG Labtech, Cary, NC USA)
Cell migration assay
Cell migration was analyzed by scratch wounding assay. RASMC were counted using a cell viability analyzer (Vi-Cell BLO, Beckman Coulter, Brea, California USA) and 1×105 cells per well were seeded in 12-well plates. Scratches were performed using a sterile 200 μl pipette tip in 80–90% confluent cells, the medium was replaced to remove any debris, and the attached cells were incubated with different stimuli. FBS 10% was used as a positive control. Images and measurements were obtained before and 12 h after pharmacological treatment using a microscope (Revolve, Echo, San Diego, California USA).
Cell proliferation assay
Cell proliferation was measured by the cell counting kit-8 (CCK-8).100 μl containing 5,000 cells was seeded in a 96-well plate per well. After overnight incubation with DMEM containing FBS 0.5%, cells were stimulated with different drugs in a volume of 10 μl for 12 h. FBS 10% was used as a positive control. CCK-8 solution (10 μl) was added in each well and incubated for 6 h. Before reading, the plate was placed on an orbital shaker for homogenization and absorbance was measured at 450 nm using a microplate reader (SpectraMax M Series Multi-Mode, Orleans Drive Sunnyvale, California USA).
Statistical Analysis
For comparisons of multiple groups, one-way or two-way analysis of variance (ANOVA), followed by the Tukey post-test was used. Differences between two groups were determined using Student’s t test. The relaxation in response to ACh and SNP is expressed as a percentage of the contraction induced by phenylephrine. The concentration-response curves were fitted by nonlinear regression analysis. pD2 (defined as the negative logarithm of the EC50 values) and maximal response (Emax) were determined. Analyses were performed using Prism 8.0 software (GraphPad). A difference was considered statistically significant when P ≤ 0.05.
RESULTS
Ang-II activates inflammasome in the vasculature and in vascular smooth muscle cells
To determine whether Ang-II activates inflammasomes in the vasculature, WT mice (C57BL/6J) were infused with Ang-II via osmotic mini-pumps. Ang-II treatment for 14 days increased the Casp1 activity aorta, measured by FLICA 660 Casp1 assay (Figures 1A and B). In order to determine whether Ang-II directly activates inflammasomes in the vasculature, RASMCs were incubated with Ang-II for 8 and 24 h. Ang-II significantly increased Casp1 (p20) levels and the mature IL-1β form (Figures 2B–D). Interestingly, Ang-II followed by nigericin (inflammasome inducer) or LPS (TLR4 agonist) followed by Ang-II also activated inflammasome. No difference was observed in NLRP3 protein expression (Figure 2A). Moreover, blocking AT1R, by losartan incubation, blunted inflammasome activation triggered by Ang-II (Figure 3A–C).
Figure 1. Ang-II treatment activates vascular inflammasome.
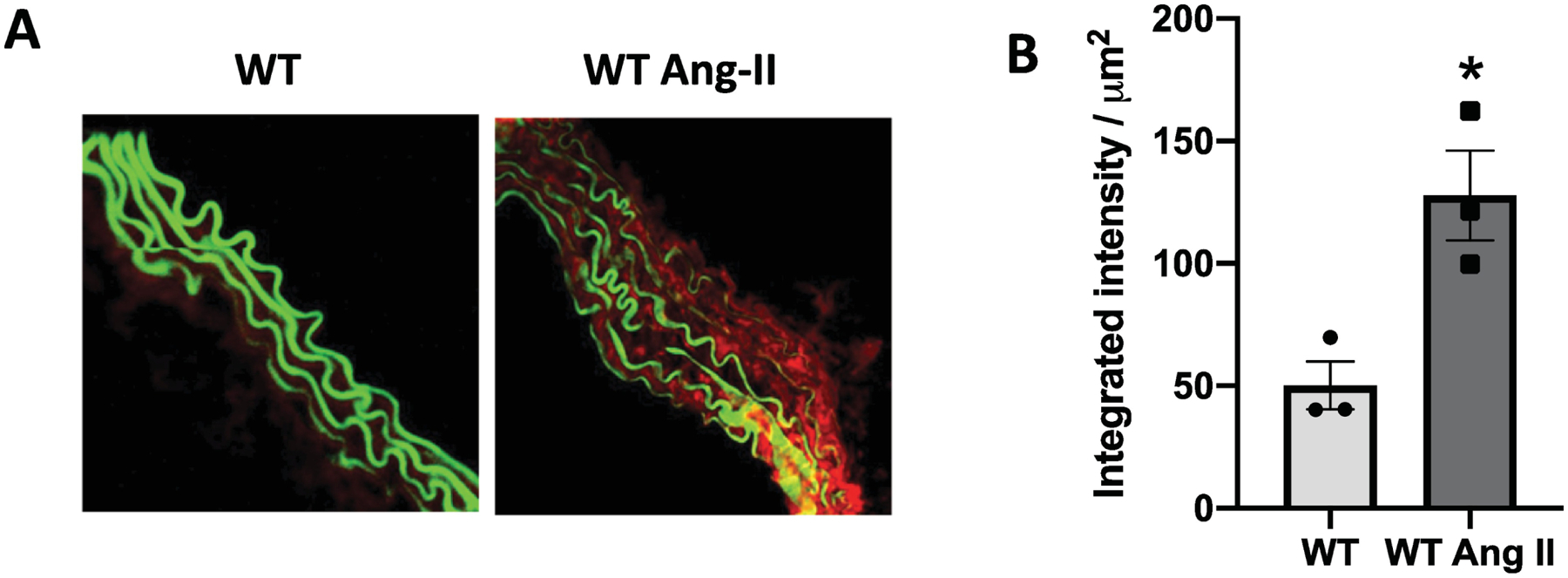
Casp1- mediated specific fluorescent-labeled substrate (FLICA® 660) degradation (in red) is overlaid to the elastin autofluorescence (in green) in thoracic aortae (A) and FLICA® 660 quantification (B) from Ctrl (WT) and Ang-II treated mice (490 ng/min/kg for 14 days with ALZET osmotic minipumps). N= 3. Values are reported as mean ± s.e.m. *P <0.05 vs. WT.
Figure 2. Ang-II activates inflammasome in vascular smooth muscle cells.
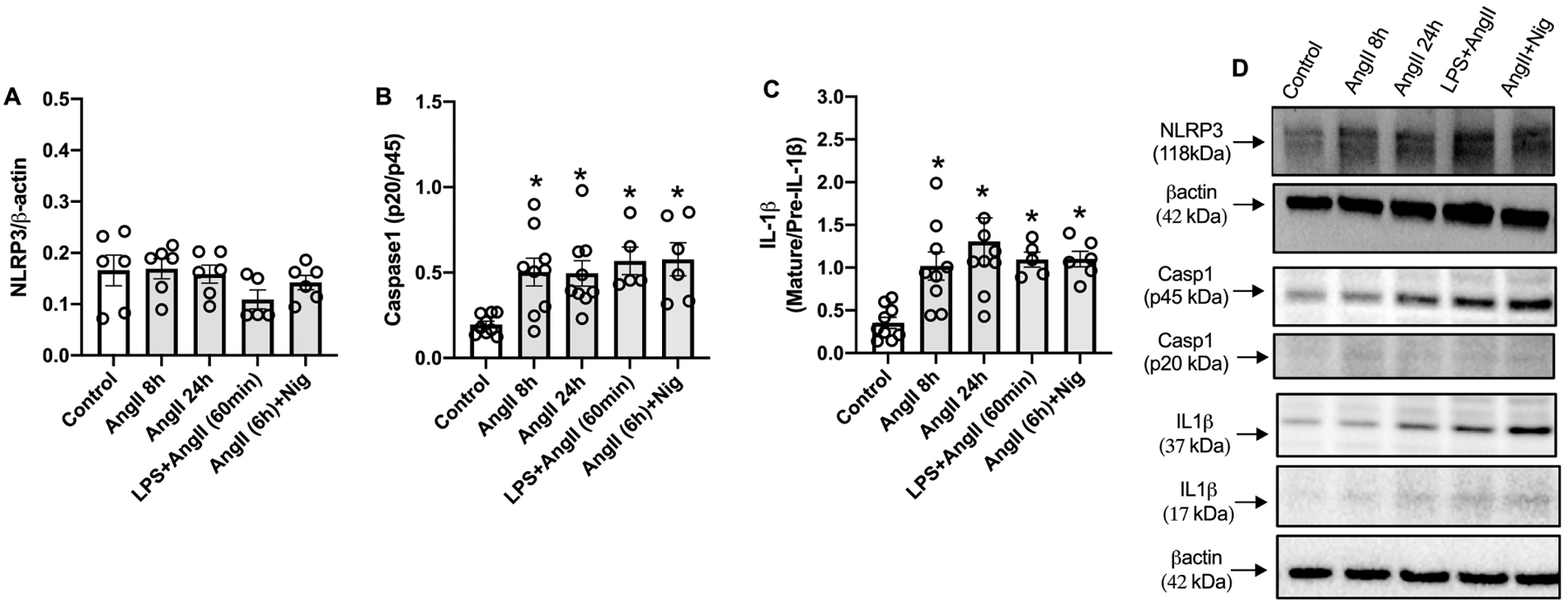
Immunoblots of NLRP3 (A), pro-caspase 1 (p40) and active Casp1 (p20) (B) and pro and mature IL-1β (C) and immunoblot representatives (D) in rat aortic vascular smooth muscle cells (RASMC) treated with Ang-II (0,1 μM) for 8 h or 24. h, or LPS+Ang-II (500 ng/mL for 6 h followed by Ang-II 0,1 μM for 60 min) or Ang-II+Nigericin (Ang-II 0,1 μM for 6 h followed by Ang-II 0,1 μM for 60 min). N= 5 to 9. Values are reported as mean ± s.e.m. *P<0.05 vs. vehicle.
Figure 3.
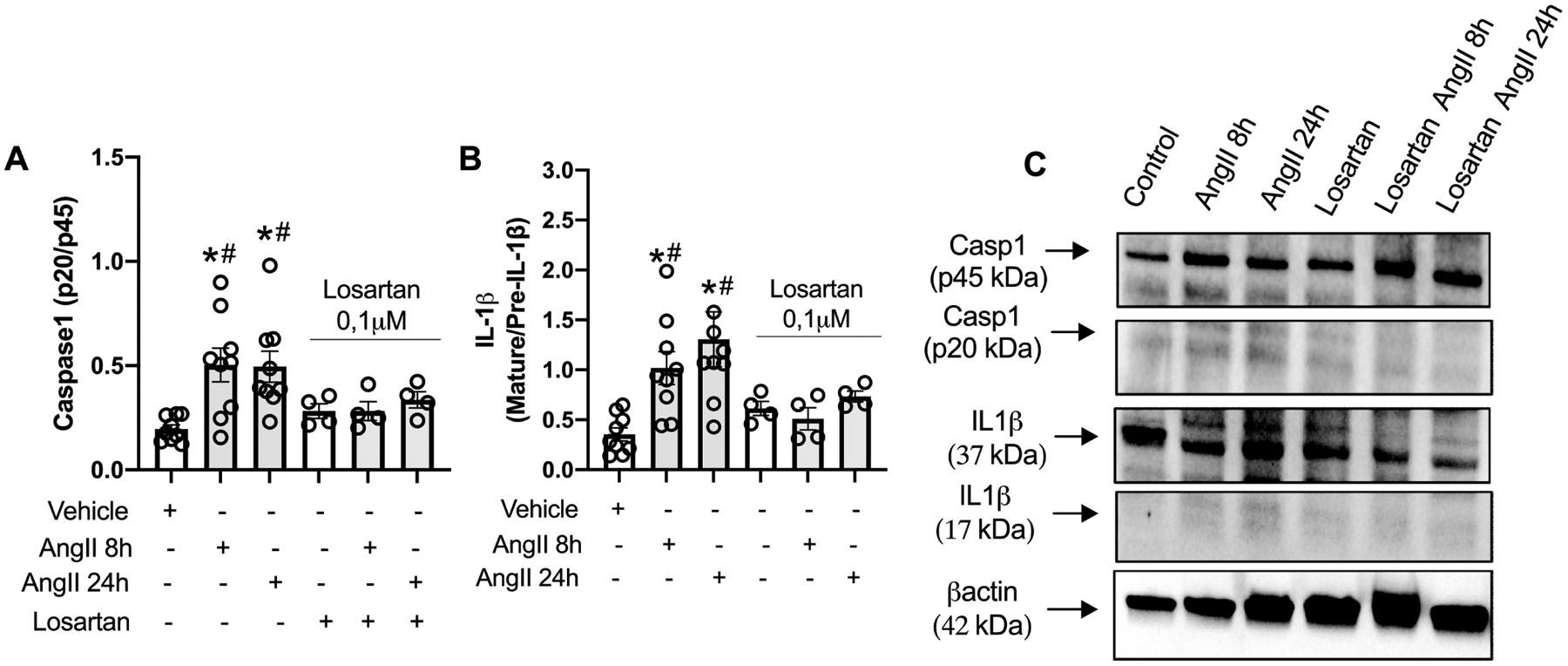
Ang-II activates inflammasome in vascular smooth muscle cells via AT1R. Immunoblots of pro-caspase 1 (p40) and active Casp1 (p20) (A) and pro and mature IL-1β (B) and immunoblot representatives (C) in rat aortic vascular smooth muscle cells (RASMC) treated with Ang-II (0,1 μM) for 8 h or 24h with or without losartan (AT1R antagonist, 10μM). N= 4 to 9. Values are reported as mean ± s.e.m. *P<0.05 vs. vehicle; #P<0.05 vs. in presence of losartan.
Vascular inflammasome contribute to Ang-II-induced vascular remodeling, endothelial dysfunction, and high blood pressure.
To analyze whether inflammasomes contribute to Ang-II-induced vascular remodeling, structural changes in thoracic aorta and in mesenteric arteries were determined in slide sections stained with hematoxylin and eosin and with a myograph for pressurized arteries, respectively. Ang-II increased media thickness (Figures 4A and B) and CSA (Figures 4A and C) in thoracic aorta. Moreover, Casp1−/− mice were protected from Ang-II-induced aortic remodeling (Figures 4A–C). Similarly, Ang-II treatment increased wall thickness (Figure 4D) and CSA (Figure 4E) in mesenteric resistance arteries from WT mice, but not in mesenteric resistance arteries from Casp1−/− mice. In addition, Ang-II triggered endothelial dysfunction, characterized by impaired relaxation to ACh, and Casp1−/− mice were partially protected from endothelial dysfunction (Figure 5A). No difference was found for SNP-induced vascular relaxation (Figure 5B). Furthermore, Ang-II treatment increased the systolic blood pressure in WT mice, whereas Casp1−/− mice were protected from Ang-II-induced high blood pressure (Fig 5C).
Figure 4. Ang-II treatment induces vascular remodeling via inflammasome.
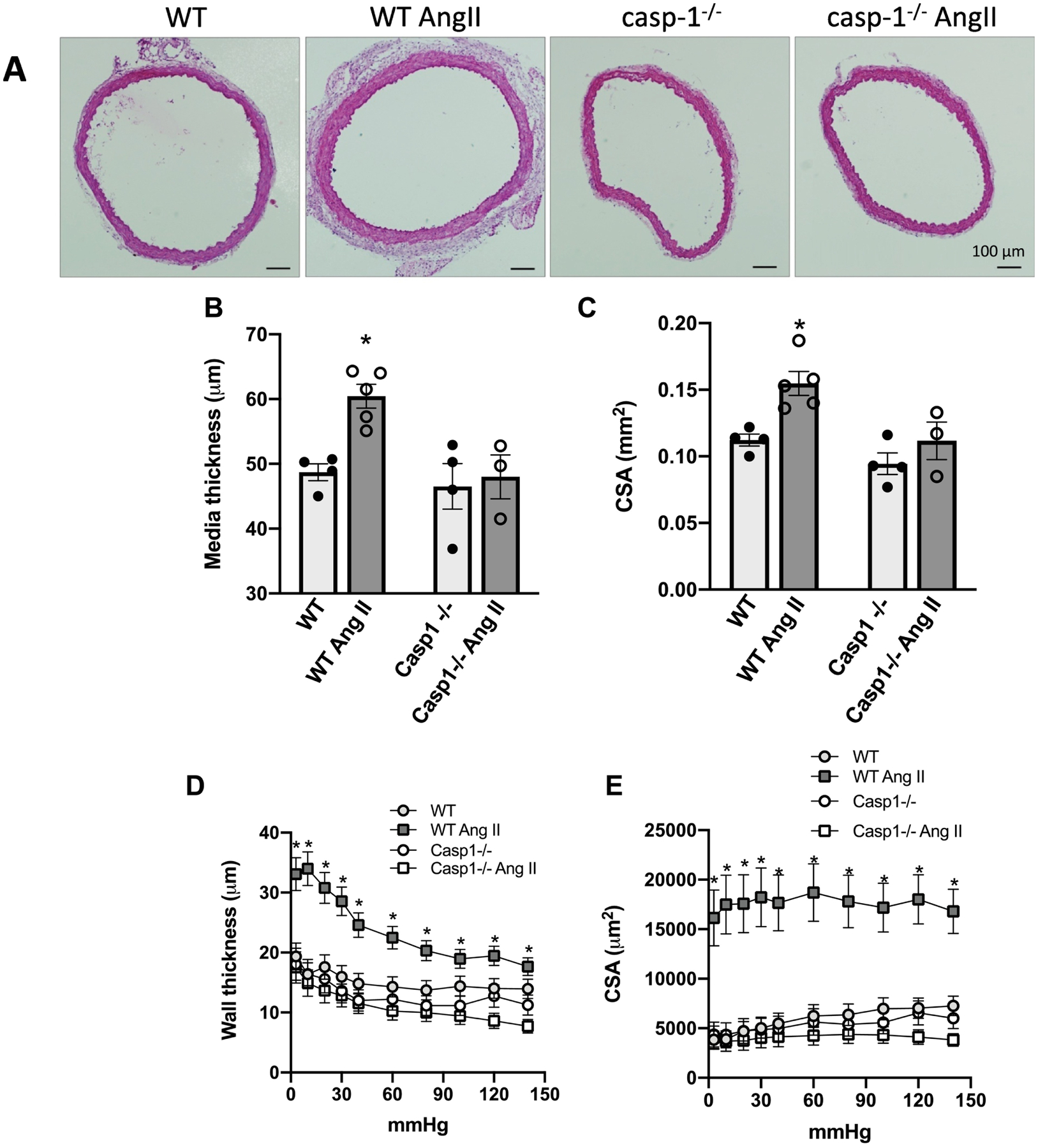
Representative (A) and quantitative histomorphometric analysis of aorta media thickness (B) and aorta cross-sectional area (C). Wall thickness (D) and cross-sectional area (E), determined in a myograph for pressurized arteries, in mesenteric resistance arteries. Aorta and mesenteric arteries from Ctrl (WT) and Casp1−/− mice treated with Ang-II (490 ng/min/kg for 14 days with ALZET osmotic minipumps). Mesenteric arteries were gradually pressurized in passive conditions (0Ca2+ Krebs Henseleit Buffer). Values are reported as mean ± s.e.m. Black lines on the representative images from aortic CSA represent 100 μm. N = 3 to 5. Values are reported as mean ± s.e.m. *P <0.05, vs. WT
Figure 5. Angiotensin-II induces endothelial dysfunction and high blood pressure dependent on inflammasome.
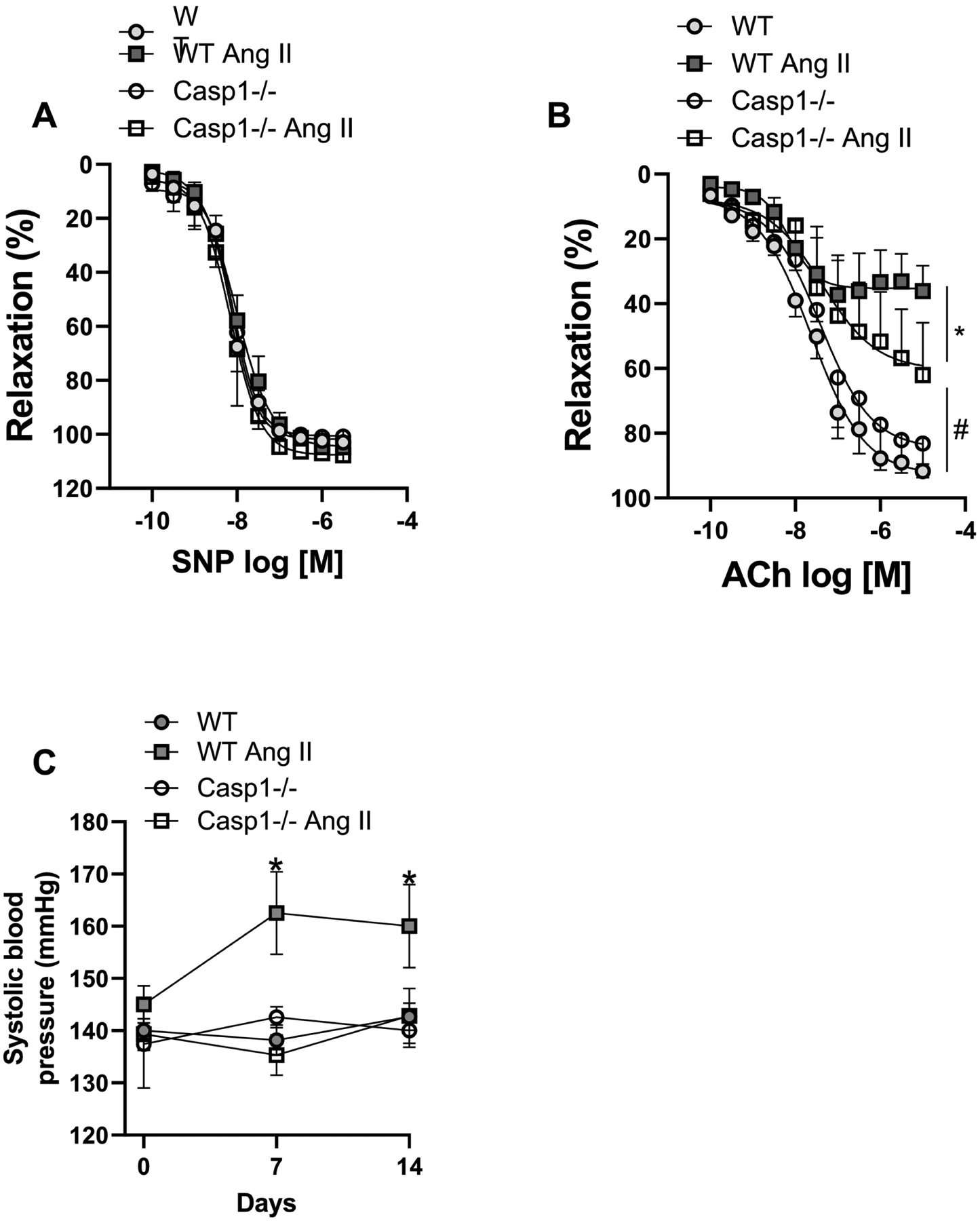
Concentration-response curves were performed to acetylcholine (ACh) (A) and to sodium nitroprusside (SNP) (B) in segments of mesenteric arteries (2nd order). (C) Systolic blood pressure measured by tail plestimography.from Ctrl (WT) and Casp1−/− mice treated with Ang-II (490 ng/min/kg for 14 days with ALZET osmotic minipumps). N = 4 to 7. *P<0.05 vs. WT; #P<0.05 vs. Ang-II and Casp-1 groups.
Inflammasome blockade reduces Ang-II-induced vascular oxidative stress ex vivo, but not in vitro
DHE staining confirmed that Ang-II treatment increases ROS production in WT mice aortas and the lack of Casp1 prevents Ang-II-induced vascular oxidative stress (Figures 6A and B). ROS production is considered a rapid event, unless whether changes in expression of oxidant or antioxidants proteins are involved. Thus, to check whether vascular inflammasome is a downstream platform in Ang-II induced oxidative stress signaling, RASMC were treated for short and long periods with Ang-II with vehicle or MCC950. Unexpectedly, inflammasome blockade failed to prevent ROS production induced by Ang-II in vitro, both at short and long-term incubations (Figure 6C), suggesting that inflammasome indirectly stimulate ROS production in vivo.
Figure 6. Inflammasome modulates Ang-II-induced vascular oxidative stress in vivo, but not in vitro.
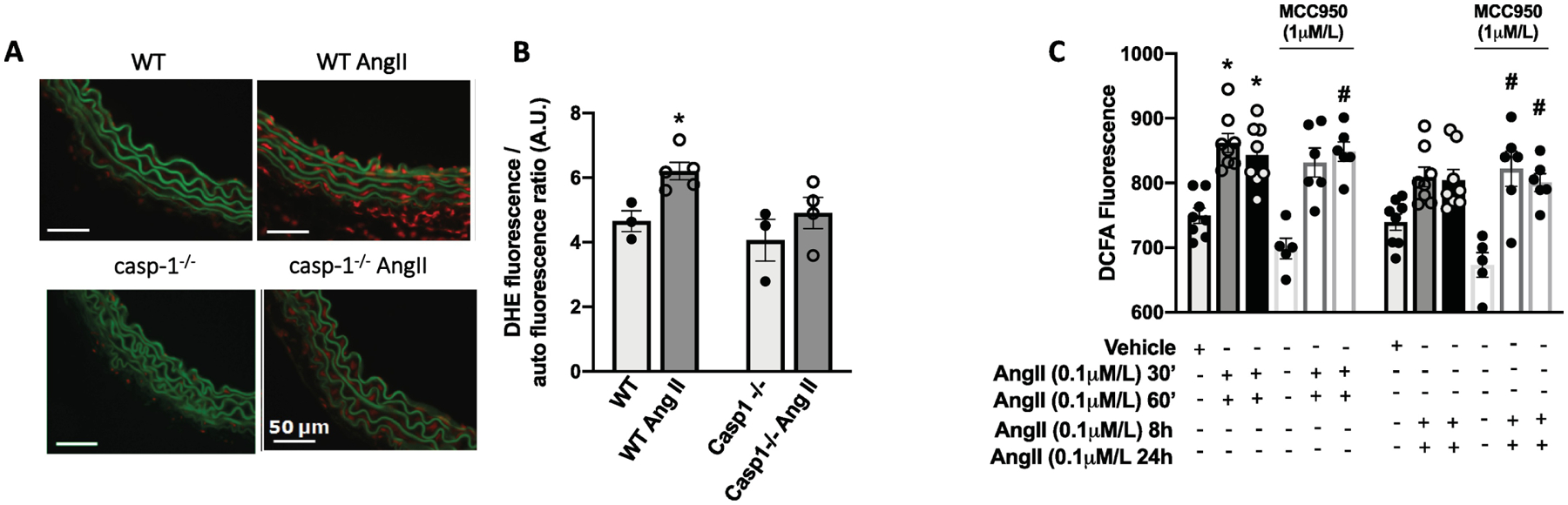
Representative dihydroethidium (DHE) staining (in red) of transverse aortic sections (×400) (A) and DHE fluorescence quantification of aortic sections (B) from Ctrl (WT) and Casp1−/− mice treated with Ang-II (490 ng/min/kg for 14 days with ALZET osmotic minipumps). ROS measurement in Rat Aortic Vascular Smooth Muscle Cells (RASMC) by 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) incubated with Ang-II (0,1 μM) for 30 or 60 min and 8 h or 24 h. Some experiments were performed in the presence of a selective NLRP3 antagonist (MMC950, 1 μM, 30 min). N = 3–5 in vascular tissues and N=5–8 in RASMC. Values are reported as mean ± s.e.m. *P<0.05 vs. WT; #P<0.05 vs. vehicle; **P<0.05 vs MCC950 alone.
Ang-II induces RASMC migration and proliferation through inflammasome activation
To determine whether Ang-II promotes vascular remodeling by directly regulating inflammasome in the vasculature, the effects of Ang-II on the migration and proliferation of vascular cells were determined in the presence of NLRP3 blockade. After 12 h of incubation, Ang-II triggered vascular smooth muscle cells migration, which was prevented by blocking inflammasome activation (Figure 7A and B). Furthermore, Ang-II incubation for 24 h induced cell proliferation (Figure 8A), as well as, PCNA protein levels, Ki67, and TGF- β gene expression (proliferative markers). Interestingly, inflammasome blockade inhibited the expression of Ang-II proliferative markers (Figures 8A–D). In order to analyze whether IL-1β (inflammasome end-product) regulates TGF- β, we stimulated RASMC with IL-β, we observed that IL-β induces TGF- β expression time-dependent manner (Figure 8E)
Figure 7. Angiotensin-II induces vascular migration via inflammasome.
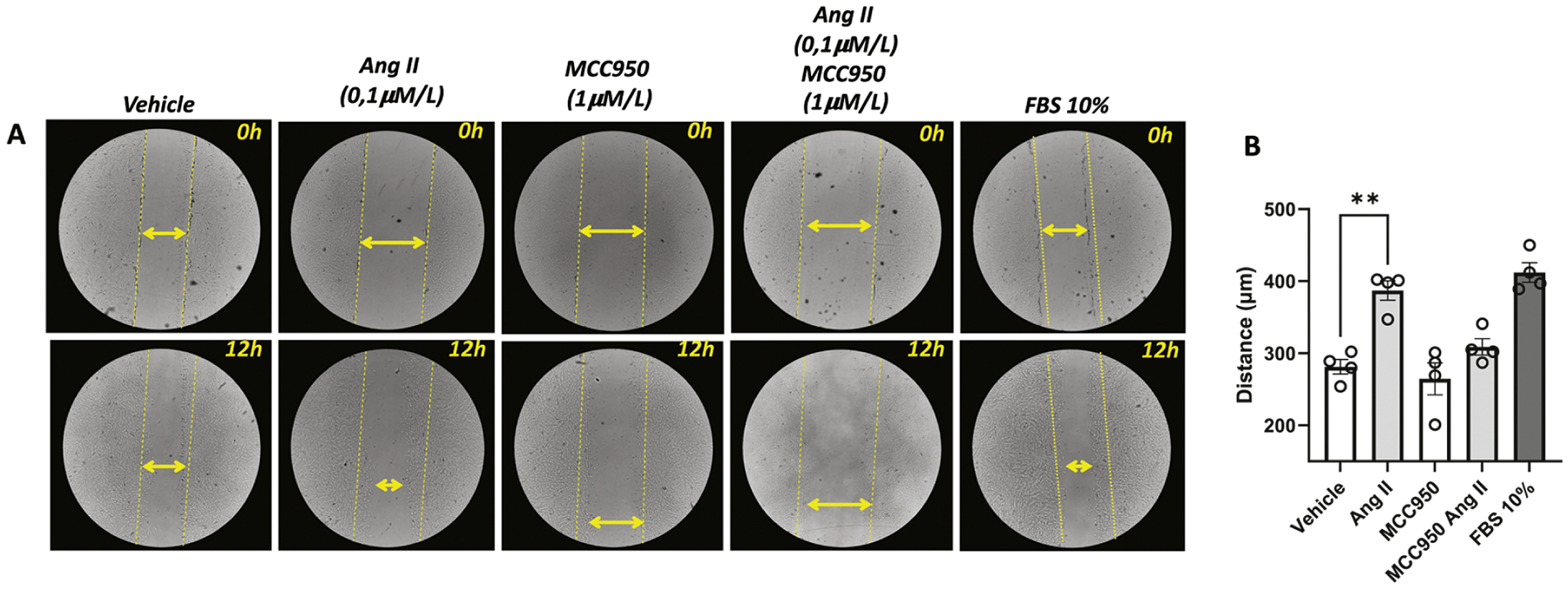
Cell migration was analyzed by scratch wounding assay. RASMC were counted using (1×105 cells per well) and seeded in 12-well dark plates. Photos were obtained right after the scratch and 12 h after incubation with Ang-II (0,1 μM). Some experiments were performed in the presence of a selective NLRP3 antagonist (MMC950, 1 μM, 30 min). Fetal Bovine Serum (FBS, 10%) was used as a positive control. Images represent individual experiments from the same well. Yellow lines represent the distance of the scratches before any stimulus. Yellow double arrows represent the distance of the scratches after 24h of the stimulus. N= 4. Values are reported as mean ± s.e.m. **P<0.05 vs. vehicle.
Figure 8. Angiotensin-II induces vascular proliferation and increases TGF-β via inflammasome.
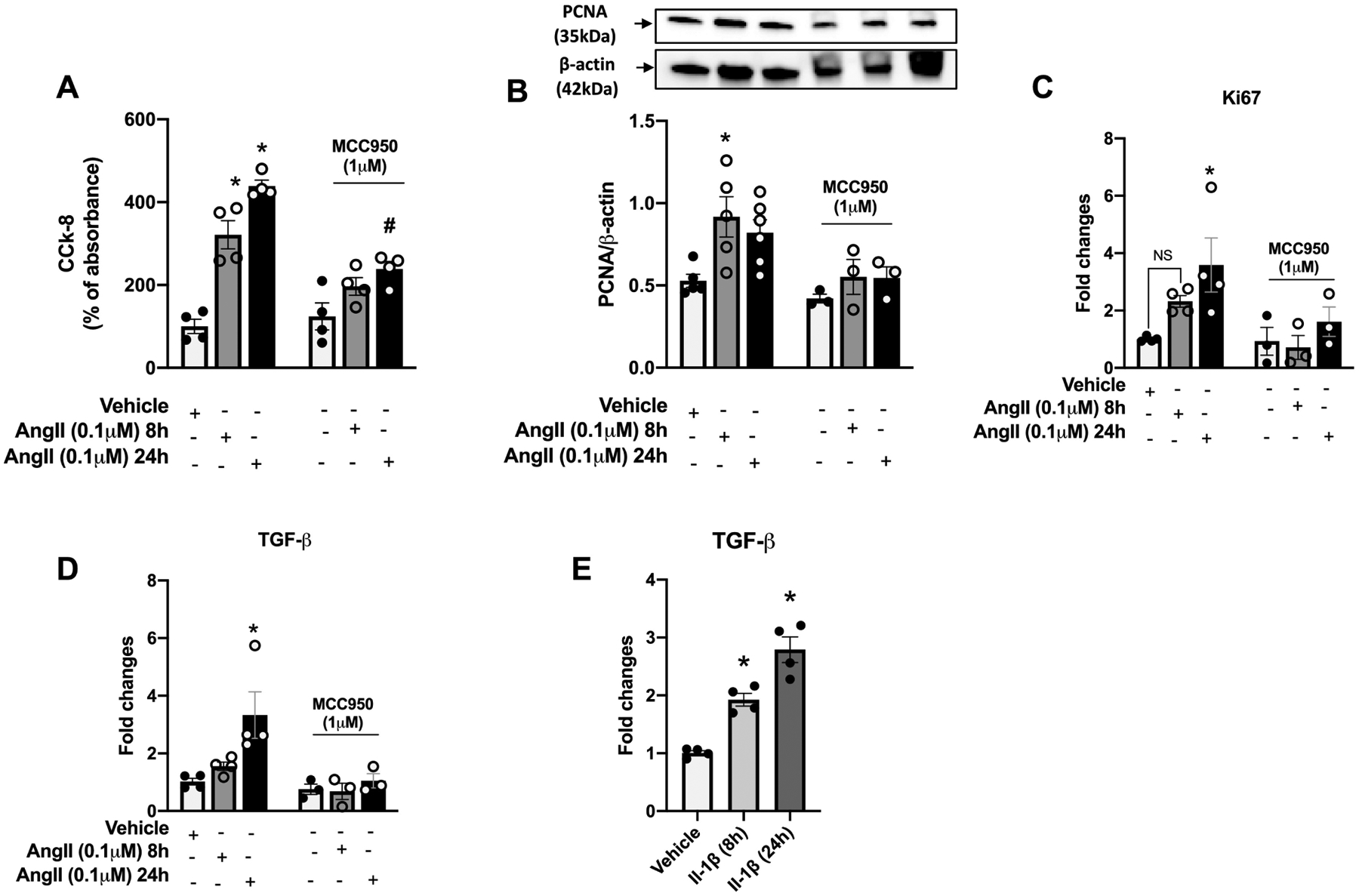
Cell counting kit-8 (CCK-8) (A), proliferating cell nuclear antigen (PCNA) protein expression (B) and Ki67 (C) and TGF-β (D) gene expressions in Rat Aortic Vascular Smooth Muscle Cells (RASMC) incubated with Ang-II (0,1 μM) for 8 h or 24 h. TGF-β (E) gene expressions in RASMC incubated with IL-1β (5ng/mL) for 8 h or 24 h. Some experiments were performed in the presence of a selective NLRP3 antagonist (MMC950, 1 μM, 30 min). N = 4 to 6. Values are reported as mean ± s.e.m. *P<0.05 vs. vehicle.
DISCUSSION
This study demonstrates that Ang-II activates inflammasome in the vasculature by acting directly on vascular cells. This interaction regulates endothelial function as well as the migration and proliferation of vascular smooth cells. Ang-II induces vascular dysfunction, inflammation, and remodeling due to its potential to promote oxidative stress and to activate innate and adaptive immune responses1, 22, 23, 29. In addition, Ang-II vascular signaling is considered one of the main therapeutic targets to decrease blood pressure and to treat aneurism, atherosclerosis and heart failure22, 31. Although Ang-II induces well characterized vascular damage, many gaps exist in the mechanisms involved.
In 2007 Guzik et al.1 showed for the first time that a specific immune cell population is involved in the genesis of Ang-II-induced hypertension and vascular damage. The authors showed that Rag1−/− mice (knockout mice for mature B and T lymphocytes) are partially protected from Ang-II-induced hypertension, demonstrating only slight endothelial dysfunction in response to Ang-II. Interestingly, immune receptors and their downstream proteins are broadly expressed in mammalian cells including vascular cells, but the meaning of their presence in non-immune cells is still poorly understood.
Ang-II-induced vascular changes is well established1, 22, 23, 29, 31, but it is unknown whether vascular inflammasomes control the vascular abnormalities. Compelling evidence places inflammasomes as a major component in aneurisms, atherosclerosis, hypertension, and infarctions. Here, we show that Ang-II induces endothelial dysfunction, oxidative stress, and vascular remodeling dependent on inflammasome activation, placing this inflammatory sensor as a relevant clinical target. These findings corroborate our previous studies, showing a similar phenotype in aldosterone-treated mice and db/db mice3, 32. Although Ang-II-induced vascular damage relies on inflammasome activation, it is still not clear whether this response is a direct effect of the platform stimulation in the vasculature, or whether the immune system rules the vascular damage.
We next determined whether Ang-II stimulates inflammasome and regulates vascular remodeling by acting directly on vascular smooth muscle cells. In vitro, Ang-II per se promoted inflammasome activation, which was blunted by antagonizing AT1R. Furthermore, combining Ang-II with LPS or nigericin, which are well characterized inflammasome activators, also induced inflammasome activation, suggesting that Ang-II generates both the primary and secondary signals involved in inflammasome activation via AT1R activation.
Taking advantage of a specific NLRP3 antagonist (MCC950)33, we investigated the role of inflammasomes on vascular oxidative stress, migration, and proliferation in RASMC. Differing from our ex vivo findings, inflammasome inhibition did not prevent the effects of Ang-II on ROS production in RASMC, suggesting that inflammasome (Casp1 −/−) might be regulating an additional or alternative mechanism in vivo. Casp1 is an important regulator of different inflammasomes including the NLRC4 and NLRP115–17, the antioxidant properties conferred by deficiency in casp1 in Ang-II treated mice might not have been confirmed in isolated VSMC because we used a NLRP3 antagonist, then other inflammasomes independent on Casp1 might be controlling the vascular redox signaling. It is worth highlighting that we used a deficient mouse for Casp1 and 11, then we also cannot exclude that the antioxidant profile found in ex vivo studies might have been generated by Casp11 deficiency, which has been also associated with cardiovascular risk by activating different signalings34. Furthermore, it is well-known that Ang-II induces immune cells infiltration into the vasculature, which contributes to local oxidation by immune cells secreting cytokines, such as IL-1β and IL-18 (end products of inflammasome activation)35–37, thus infiltrated immune cell in the vasculature may be leading to vascular oxidation via cytokines release. Furthermore, high levels of Ang-II reduce antioxidant enzyme expression and/or activity38, 39, and increases levels of NADPH oxidases40–42, which may not be regulated by inflammasomes in vascular cells. It also may explain why inflammasome blockade did not prevent ROS generation induced by Ang-II in vitro. Further studies are necessary to solve this such divergence between the ex vivo and in vitro effects of inflammasomes.
An intriguing finding from our study it is that Casp1 deficiency fully abolished Ang-II-induced vascular remodeling in large and small blood vessels, but partially the endothelial dysfunction in small arteries. In other studies, inflammasome activation has been involved on controlling endothelial function and vascular phenotype10, 19, 27, 32, 43. Based on our data, we may suggest that inflammasome seems to be more important for vascular growth than endothelial function, at least in the context of Ang-II-induced hypertension. Further studies are necessary to dissect by which mechanisms inflammasome controls endothelial cells and VSMCs regulation.
Inflammasomes are linked to vascular smooth muscle cells phenotypic transformation, proliferation, and vascular remodeling. In induced26 and genetic rodent models of hypertension27, histone acetylation and NFκB activation mediate these effects and such changes seem to not be exclusive to the vasculature44–46. Ang-II treatment promoted vascular remodeling in the thoracic aorta and mesenteric artery of WT mice and induced vascular migration and proliferation in RASMC. Blockade of inflammasome signaling via genetic manipulation or pharmacological treatment provided protection from vascular changes triggered by Ang-II. TGF-β is a pleiotropic growth factor47 that induces vascular migration and proliferation48, and it is sensitive to Ang-II effects49, 50. In renal cells, inflammasomes seem to control TGF-β expression and activity51. Furthermore, pulmonary and vascular cells with long term IL-1β, the final product of the inflammasome cascade, exposure also exhibit increased TGF-β expression52, 53. Similarly, we observed that IL-1β promoted TGF-β expression in a time-dependent manner in isolated vascular smooth muscle cells. Altogether, it is possible to speculate that Ang-II activates inflammasomes in vascular smooth muscle cells, promoting vascular migration and proliferation via TGF-β signaling.
In conclusion, our study shows that Ang-II generates both signals for inflammasome activation via AT1R activation, leading to structural vascular alterations possibly by regulating TGF-β signaling. In addition, our study also shows that inflammasome plays a major role in Ang-II-induced endothelial dysfunction (Fig. 8). At this point, we may suggest inflammasomes as a therapeutic target to reduce high blood pressure and minimize cardiovascular damage associated with hypertension.
Supplementary Material
FUNDING INFORMATION
This work was supported by grants from São Paulo Research Foundation (2011/12604-2 to SBC and to the Center of Research in Inflammatory Diseases 2013/08216-2), Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (88887.092495/2015-00 to RCT and TBN), and NIH-R00 (4R00HL14013903) to TBN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST
None
REFERENCES
- 1.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thang LV, Demel SL, Crawford R, Kaminski NE, Swain GM, Van Rooijen N, Galligan JJ. Macrophage depletion lowers blood pressure and restores sympathetic nerve alpha2-adrenergic receptor function in mesenteric arteries of doca-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2015;309:H1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho FN, Pequeno IO, Olivon VC, Neves KB, Lopes RA, Campos E, Aguiar Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga TT, Luiz JP, Cau SB, Elias PL, Moreira AC, Camara NO, Zamboni DS, Alves-Filho JC, Tostes RC. Nlrp3 inflammasome mediates aldosterone-induced vascular damage. Circulation. 2016 [DOI] [PubMed] [Google Scholar]
- 4.White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron. 1964;1:93–102 [DOI] [PubMed] [Google Scholar]
- 5.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–264 [PubMed] [Google Scholar]
- 6.Olsen F Transfer of arterial hypertension by splenic cells from doca-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C. 1980;88:1–5 [DOI] [PubMed] [Google Scholar]
- 7.Tousoulis D, Andreou I, Antoniades C, Tentolouris C, Stefanadis C. Role of inflammation and oxidative stress in endothelial progenitor cell function and mobilization: Therapeutic implications for cardiovascular diseases. Atherosclerosis. 2008;201:236–247 [DOI] [PubMed] [Google Scholar]
- 8.Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of nlrp3 inflammasomes: Ros as trigger or effector? Antioxid Redox Signal. 2015;22:1111–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butts B, Gary RA, Dunbar SB, Butler J. The importance of nlrp3 inflammasome in heart failure. J Card Fail. 2015;21:586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL. Endothelial nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 2015;1853:396–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conforti-Andreoni C, Ricciardi-Castagnoli P, Mortellaro A. The inflammasomes in health and disease: From genetics to molecular mechanisms of autoinflammation and beyond. Cell Mol Immunol. 2011;8:135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He J, Yang Y, Peng DQ. Monosodium urate (msu) crystals increase gout associated coronary heart disease (chd) risk through the activation of nlrp3 inflammasome. Int J Cardiol. 2012;160:72–73 [DOI] [PubMed] [Google Scholar]
- 13.Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, Krohg-Sorensen K, Skjelland M, Espevik T, Aukrust P, Lengquist M, Hedin U, Jansson JH, Fransen K, Hansson GK, Eriksson P, Sirsjo A. Nlrp3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. Il-1r1/myd88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022 [DOI] [PubMed] [Google Scholar]
- 17.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the nlrp3 inflammasome during apoptosis. Immunity. 2012;36:401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, Cheng X, Wang J, Qin X, Yu J, Ji Y, Yang X, Wang H. Caspase-1 inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ Res. 2016;118:1525–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakaue T, Suzuki J, Hamaguchi M, Suehiro C, Tanino A, Nagao T, Uetani T, Aono J, Nakaoka H, Kurata M, Sakaue T, Okura T, Yasugi T, Izutani H, Higaki J, Ikeda S. Perivascular adipose tissue angiotensin ii type 1 receptor promotes vascular inflammation and aneurysm formation. Hypertension. 2017;70:780–+ [DOI] [PubMed] [Google Scholar]
- 21.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circulation Research. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daugherty A, Manning MW, Cassis LA. Angiotensin ii promotes atherosclerotic lesions and aneurysms in apolipoprotein e-deficient mice. Journal of Clinical Investigation. 2000;105:1605–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruder-Nascimento T, Chinnasamy P, Riascos-Bernal DF, Cau SB, Callera GE, Touyz RM, Tostes RC, Sibinga NE. Angiotensin ii induces fat1 expression/activation and vascular smooth muscle cell migration via nox1-dependent reactive oxygen species generation. J Mol Cell Cardiol. 2014;66:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Batista PR, Palacios R, Martin A, Hernanz R, Medici CT, Silva MA, Rossi EM, Aguado A, Vassallo DV, Salaices M, Alonso MJ. Toll-like receptor 4 upregulation by angiotensin ii contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One. 2014;9:e104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernanz R, Martinez-Revelles S, Palacios R, Martin A, Cachofeiro V, Aguado A, Garcia-Redondo L, Barrus MT, de Batista PR, Briones AM, Salaices M, Alonso MJ. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin ii-induced hypertension. Br J Pharmacol. 2015;172:3159–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren XS, Tong Y, Ling L, Chen D, Sun HJ, Zhou H, Qi XH, Chen Q, Li YH, Kang YM, Zhu GQ. Nlrp3 gene deletion attenuates angiotensin ii-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol Biochem. 2017;44:2269–2280 [DOI] [PubMed] [Google Scholar]
- 27.Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, Zhou YB, Han Y, Chen Q, Li YH, Kang YM, Zhu GQ. Nlrp3 inflammasome activation contributes to vsmc phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8:e3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritz E Role of renal angiotensin ii type 1 receptors in the genesis of hypertension: Guyton revisited - angiotensin ii causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc natl acad sci USA 103: 17985–17990, 2006 crowley s.D., gurley s.B., herrera m.J., ruiz p., griffiths r., kumar a.P., kim h.-s., smithies o., le t.H., coffman t.M. J Am Soc Nephrol. 2007;18:356–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin ii causes hypertension and cardiac hypertrophy through its receptors in the kidney. P Natl Acad Sci USA. 2006;103:17985–17990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva MA, Cau SB, Lopes RA, Manzato CP, Neves KB, Bruder-Nascimento T, Mestriner FL, Montezano AC, Nguyen Dinh Cat A, Touyz RM, Tostes RC. Mineralocorticoid receptor blockade prevents vascular remodelling in a rodent model of type 2 diabetes mellitus. Clin Sci (Lond). 2015;129:533–545 [DOI] [PubMed] [Google Scholar]
- 31.De Mello WC, Danser AH. Angiotensin ii and the heart : On the intracrine renin-angiotensin system. Hypertension. 2000;35:1183–1188 [DOI] [PubMed] [Google Scholar]
- 32.Ferreira NS, Bruder-Nascimento T, Pereira CA, Zanotto CZ, Prado DS, Silva JF, Rassi DM, Foss-Freitas MC, Alves-Filho JC, Carlos D, Tostes RC. Nlrp3 inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA. A small-molecule inhibitor of the nlrp3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng C, Wang R, Tan H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int J Biol Sci. 2019;15:1345–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabkin SW. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat Clin Pract Cardiovasc Med. 2009;6:192–199 [DOI] [PubMed] [Google Scholar]
- 36.Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, Morris MA, Taylor-Fishwick DA, Nadler JL. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab. 2013;15 Suppl 3:117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang KT, Sullivan JC, Pollock JS. Superoxide dismutase activity in small mesenteric arteries is downregulated by angiotensin ii but not by hypertension. Toxicol Res. 2018;34:363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khaper N, Singal PK. Modulation of oxidative stress by a selective inhibition of angiotensin ii type 1 receptors in mi rats. J Am Coll Cardiol. 2001;37:1461–1466 [DOI] [PubMed] [Google Scholar]
- 40.Li WJ, Liu Y, Wang JJ, Zhang YL, Lai S, Xia YL, Wang HX, Li HH. “Angiotensin ii memory” contributes to the development of hypertension and vascular injury via activation of nadph oxidase. Life Sci. 2016;149:18–24 [DOI] [PubMed] [Google Scholar]
- 41.Nakashima T, Umemoto S, Yoshimura K, Matsuda S, Itoh S, Murata T, Fukai T, Matsuzaki M. Tlr4 is a critical regulator of angiotensin ii-induced vascular remodeling: The roles of extracellular sod and nadph oxidase. Hypertens Res. 2015;38:649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin ii stimulates nadh and nadph oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148 [DOI] [PubMed] [Google Scholar]
- 43.Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, Neves KB, Alves-Lopes R, Campos E, Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga T, Luiz JP, Cau SB, Elias PC, Moreira AC, Camara NO, Zamboni DS, Alves-Filho JC, Tostes RC. Nlrp3 inflammasome mediates aldosterone-induced vascular damage. Circulation. 2016;134:1866–1880 [DOI] [PubMed] [Google Scholar]
- 44.Zhang LL, Huang S, Ma XX, Zhang WY, Wang D, Jin SY, Zhang YP, Li Y, Li X. Angiotensin(1–7) attenuated angiotensin ii-induced hepatocyte emt by inhibiting nox-derived h2o2-activated nlrp3 inflammasome/il-1beta/smad circuit. Free Radic Biol Med. 2016;97:531–543 [DOI] [PubMed] [Google Scholar]
- 45.Wen Y, Liu Y, Tang T, Lv L, Liu H, Ma K, Liu B. Nlrp3 inflammasome activation is involved in ang ii-induced kidney damage via mitochondrial dysfunction. Oncotarget. 2016;7:54290–54302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, Zhang A. Angiotensin ii stimulates the nlrp3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis (Basel). 2018;4:83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haque S, Morris JC. Transforming growth factor-beta: A therapeutic target for cancer. Hum Vaccin Immunother. 2017;13:1741–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu YT, Chen L, Tan ZB, Fan HJ, Xie LP, Zhang WT, Chen HM, Li J, Liu B, Zhou YC. Luteolin inhibits vascular smooth muscle cell proliferation and migration by inhibiting tgfbr1 signaling. Front Pharmacol. 2018;9:1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin ii in renal fibrosis. Hypertension. 1998;31:181–188 [DOI] [PubMed] [Google Scholar]
- 50.Li YS, Ni SY, Meng Y, Shi XL, Zhao XW, Luo HH, Li X. Angiotensin ii facilitates fibrogenic effect of tgf-beta1 through enhancing the down-regulation of bambi caused by lps: A new pro-fibrotic mechanism of angiotensin ii. PLoS One. 2013;8:e76289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, Beck PL, Muruve DA. Inflammasome-independent nlrp3 augments tgf-beta signaling in kidney epithelium. J Immunol. 2013;190:1239–1249 [DOI] [PubMed] [Google Scholar]
- 52.Yue TL, Wang XK, Olson B, Feuerstein G. Interleukin-1 beta (il-1 beta) induces transforming growth factor-beta, (tgf-beta 1) production by rat aortic smooth muscle cells. Biochem Biophys Res Commun. 1994;204:1186–1192 [DOI] [PubMed] [Google Scholar]
- 53.Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.