

SUMMARY
Cytokines are typically single gene products, except for the heterodimeric interleukin (IL)-12 family. The two subunits (IL-12p40 and IL-12p35) of the prototype IL-12 are known to be simultaneously co-expressed in activated myeloid cells, which secrete the fully active heterodimer to promote interferon (IFN)γ production in innate and adaptive cells. We find that chimeric mice containing mixtures of cells that can only express either IL-12p40 or IL-12p35, but not both together, generate functional IL-12. This alternate two-cell pathway requires IL-12p40 from hematopoietic cells to extracellularly associate with IL-12p35 from radiation-resistant cells. The two-cell mechanism is sufficient to propel local T cell differentiation in sites distal to the initial infection and helps control systemic dissemination of a pathogen, although not parasite burden, at the site of infection. Broadly, this suggests that early secretion of IL-12p40 monomers by sentinel cells at the infection site may help prepare distal host tissues for potential pathogen arrival.
In brief
The innate cytokine IL-12 is a critical enhancer of IFNγ production. Gerber et al. show that IL-12 activity can result from two distinct cells contributing different subunits of IL-12 independently, in vivo. This collaborative, two-cell version of IL-12 could limit pathogen dissemination to tissues distant from the primary infection site.
Graphical Abstract
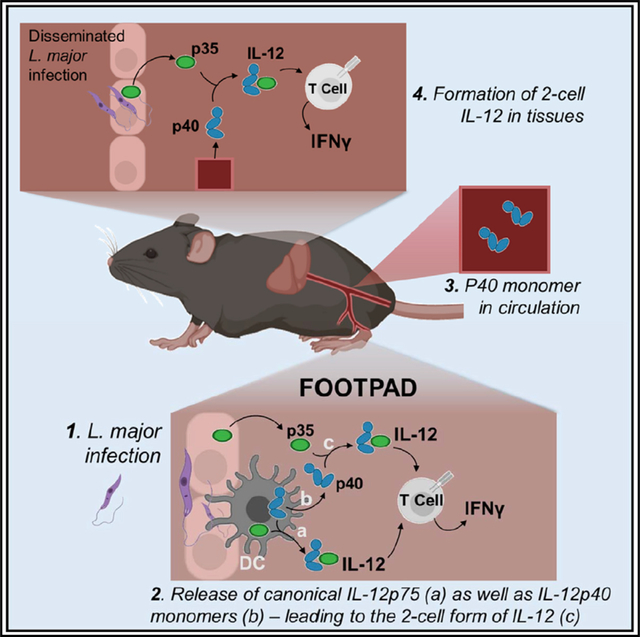
INTRODUCTION
Interleukin (IL)-12 and IL-23 are members of a family of heterodimeric cytokines that play critical roles in regulating the differentiation of the immune response toward distinct effector pathways. IL-12 promotes interferon (IFN)γ secretion by natural killer (NK) cells and T cells, while IL-23 enhances IL-17 production (Tait Wojno et al., 2019). They are also unique among cytokines, as each of these is a heterodimer assembled from proteins encoded by two distinct genetic loci (Gubler et al., 1991; Sieburth et al., 1992). The formation of functional IL-12 or IL-23 requires a common IL-12p40 (p40) subunit that covalently associates with either the IL-13p35 (p35) or IL-23p19 (p19) subunit, respectively (D’Andrea et al., 1992; Oppmann et al., 2000). However, the secretion of IL-12 is canonically thought to rely on p40 and p35 being expressed and assembled within a single myeloid cell (typically dendritic cells [DCs] or macrophages). For instance, innate activation of DCs by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) leads to the upregulation of transcripts for both subunits (Hayes et al., 1995). The subsequent assembly and secretion of the heterodimers are controlled by post-translational checkpoints (Carra et al., 2000; Jalah et al., 2013; Reitberger et al., 2017; Yoon et al., 2000). Importantly, p35 is not secreted alone and requires binding to p40 for correct folding and secretion (Reitberger et al., 2017). The teleological significance of two distinct genetic loci contributing to a tightly regulated single-cell product is not clear.
Importantly, despite this co-dependence on p40 for functional cytokine generation, p35 is widely expressed, while p40 expression is typically limited to DCs and macrophages (Carra et al., 2000; D’Andrea et al., 1992). p40 can also be released from these hematopoietic cells as a monomer and is routinely found in great excess over IL-12 in serum and culture supernatants (Abdi et al., 2006, 2014; Trinchieri, 2003). The precise biological significance of secreted p40 monomer is not yet clear, although recent studies indicate immunomodulatory properties. The p40-p40 homodimer can inhibit the IFNγ-promoting activity of IL-12 (Cooper and Khader, 2007), but the amounts of p40 monomer far exceeds this form, too. Intriguingly, at least in vitro, we previously reported that p40 and p35 can assemble to form functionally active IL-12 (Abdi et al., 2014). If this mechanism operates in vivo, it will add a layer to current models for bridging the innate and adaptive systems. Significantly, this model would suggest that two cells, one hematopoietic (expressing p40) and the other potentially non-hematopoietic (expressing p35), functionally cooperate to determine the trajectory of T cell differentiation in contrast to the single-cell dictation currently predicted by the intracellular assembly model.
To evaluate the different mechanisms by which IL-12 can be generated physiologically, we used a mixed bone marrow chimera strategy where the production of p40 or p35 is separated to different cellular compartments. In this model, cells in the chimeras contain IL-12p40−/− (p40−/−) or IL-12p35−/− (p35−/−) cells such that any cell can either make p40 or p35, but not both. Despite this separation, we found that active IL-12 is available for T cell differentiation, validating this method for IL-12 formation in vivo. We also found that hematopoietic cells are the necessary source of p40 secretion to synergize with p35, expressed by radiation-resistant cells from the non-hematopoietic compartment, for the formation of two-cell IL-12. This two-cell method of IL-12 formation amplified effector IFNγ responses in antigen-specific T cells during immunization and in response to a Leishmania major infection in a polyclonal T cell population. Furthermore, we showed that the two-cell IL-12 was physiologically distinct from the canonical form and plays a major role in controlling parasite dissemination to distal sites but not the primary infection site. Taken together, these data highlight an alternate mechanism by which DCs and stromal cells collaboratively drive T cell differentiation and effector function.
RESULTS
T cell differentiation to IFNγ production is disrupted in p40−/− and p35−/− mice
The biological function of IL-12 in vivo is in promoting the differentiation of IFNγ-producing innate and adaptive cells. While there is considerable literature showing how cytokines such as IL-18 and IL-15 as well as T cell receptor (TCR) signaling pathways themselves influence the initial differentiation of IFNγ-producing cells (Denton et al., 2007; Matson and Singh, 2020; Müller et al., 2001; Tubo and Jenkins, 2014), the role of IL-12 in setting up a feedback loop amplifying this effector mechanism is critical in many responses (Trinchieri, 2003). Immune responses to parasites such as Leishmania are particularly relevant (Park et al., 2000). The differentiation of T cells during Leishmania infection of mice was historically used to outline the original Th1/Th2 paradigm (Heinzel et al., 1989; Scott et al., 1988). In a C57BL/6 (B6) background, the strong differentiation of T cells to an IFNγ-secretion phenotype is associated with better clearance of the subcutaneous infection by L. major while BALB/c animals make an IL-4-dominant, non-resolving response (Fowell and Locksley, 1999). The production of IFNγ by T, NK, or NKT cells in this context was largely dependent on IL-12 from innate sources (Afonso et al., 1994; Sypek et al., 1993). Thus, subcutaneous L. major infections are not cleared in IL-12p40 knockout (KO) (B6.129S1-Il12btm1Jm/J, referred to as p40−/−) animals, where, even in a B6 background, the response to Leishmania proceeds similarly to that in the BALB/c mice (Park et al., 2000). Therefore, we first validated requirements for p40 and p35 in clearing dsRed L. major infections using respective knockout mice on a B6 background (Figure 1). Wild-type B6 (WT) mice began to clear lesions (marked by reduced swelling of the footpads) by 25 days, while both p40−/− and p35−/− (the IL-12p35−/− strain [B6.129S1-Il12atm1Jm/J], referred to as p35−/− here) animals continued to swell (Figures 1A and 1B). L. major infection of both WT and p35−/− mice resulted in systemic p40 release (Figure 1C), and similar to our previous studies with lipopolysaccharide (LPS) challenge (Abdi et al., 2006), only a fraction of this amount was part of the heterodimeric IL-12 cytokine (Figure 1D). The spleen and draining popliteal lymph node (dLN) of p35−/− and WT animals also secreted p40 in ex vivo cultures, without additional pattern-recognition receptor (PRR) stimulation (Figures S1D and S1E). Taken together, these data indicate that, as we previously reported with endotoxin challenge (Abdi et al., 2006), a footpad infection with Leishmania also results in the systemic release of p40.
Figure 1. T cell differentiation is disrupted in p40−/− and p35−/− mice.

(A–H) p40−/− (open circles, blue), p35−/− (open squares, green), and WT (open triangles, black) mice were challenged with 106 dsRed L. major parasites by intradermal injection in the footpad.
(A) Biweekly measurement of footpad thickness following appearance of lesions for p40−/− (open circles, blue), p35−/− (open squares, green), and WT (open triangles, black) through the duration of dsRed L. major infection (n = 3).
(B) Footpad thickness of p40−/− (open circles, blue), p35−/− (open squares, green), and WT (open triangles, black) mice 28 days after dsRed L. major infection (unpaired t test, n = 3).
(C and D) Infected animals were bled 4 h and at days 1, 2, 3, 4, 7, 14, and 21 after L. major infection. Serum (C) p40 and (D) IL-12 levels for p40−/− (open circles, blue), p35−/− (open squares, green), and WT (open triangles, black) animals were measured by ELISA (n = 3).
(E–H) Splenocytes harvested 28 days after L. major infection were restimulated and analyzed via flow cytometry. Gating strategy used in these panels is shown in Figure S1A.
(E) Percentage of IFNγ-producing CD4+ T cells within the CD44hi subset in the spleen measured 28 days after dsRed L. major infection (n = 3).
(F) Number of CD44hiIFNγ+CD4+ T cells in the spleen of p40−/− (open circles, blue), p35−/− (open squares, green), and WT (open triangles, black) animals measured 28 days after dsRed L. major infection (unpaired t test, n = 3).
(G) Similar to (D), except CD8+ T cells (n = 3).
(H) Similar to (E), except CD8+ T cells (unpaired t test, n = 3).
(I) Cells from the spleen, draining popliteal lymph nodes (dLNs), and footpad were harvested 28 days after dsRed L. major infection and restimulated with soluble Leishmania antigen (SLA). Concentration of IFNγ in supernatant was measured by ELISA collected from p40−/− (blue), p35−/− (green), and WT (black, shaded) after restimulation for 48 h (unpaired t test, n = 3).
(J and K) SMARTA T cells from CD45.1+ donor animals were sorted and transferred into CD45.2+ p40−/− or CD45.1+CD45.2+ WT recipient animals and challenged 24 h later with GP61–80 and LPS. 4 days after challenge, harvested cells from the spleen and LNs were restimulated with GP61–80 in vitro for 24 or 48 h. Concentrations of IFNγ in the supernatant of (J) splenocyte or (K) LN SMARTA T cells transferred into p40−/− (open bars, blue gradient) or WT (open bars, gray gradient) animals 24 or 48 h after restimulation (n = 3) are shown.
Data in this figure are representative of two independent experiments where n refers to the number of biological replicates in each experiment. (A)–(D), (F), and (H)–(K) are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Colored asterisks indicate relative significance to different groups (blue is significance to p40−/− and green is significance to p35−/−). Anything unmarked is considered not significant.
In this model, IL-12 is typically required for the induction of IFNγ from NK, NKT, and T cells. Accordingly, about 5.2% (±0.76%) of the previously activated (marked by high levels of CD44) CD4 T cells and 13% (±2.1%) of the CD44hi CD8 T cells from WT animals produced IFNγ while those were reduced to 0.37% (±0.06%) CD4 and 0.23% (±0.03%) CD8 in p40−/− animals and 0.8% (±0.02%) CD4 and 1.06% (±0.05%) CD8 in p35−/− mice (Figures 1E, 1H, S1B, and S1C). This also represented an antigen-specific deficit since lymphocytes from various tissues restimulated with soluble Leishmania antigen (SLA) in vitro for 48 h also showed lower IFNγ secretion in p40−/− or p35−/− animals (Figure 1I). Notably, the loss of p40 or p35 does not completely abrogate all of the IFNγ production by T cells. Yet, this is to be expected, as the initial upregulation of IFNγ can be stimulated by other cytokines as well, mainly IL-18 (Koutoulaki et al., 2010). However, IL-12 is more important for the amplification of the IFNγ response, as IFNγ has a positive feedback loop on the expression of both p40 and p35 (Abdi et al., 2006; Liu et al., 2003).
Finally, we validated that the role of p40 and p35 in the generation of an IFNγ response was an antigen-specific phenomenon, rather than L. major specific. This was done using a defined antigen-specific T cell population, lymphocytic choriomeningitis virus (LCMV)-specific TCR-transgenic SMARTA CD4 T cells, transferred into either p40−/− or WT mice and challenged with peptide/LPS. Ex vivo restimulation of these T cells confirmed that IL-12 is critical for the differentiation of SMARTA T cells to produce IFNγ (Figures 1I and 1J), similar to the behavior of polyclonal T cells during Leishmania infection (Figures 1A–1G).
Administration of recombinant p40 allows for antigen-specific T cell differentiation
While knockout mice illustrate the need for p40 and p35 to generate biologically active IL-12-driven IFNγ production by T cells, they cannot be used to determine whether both subunits must be expressed by the same DC or macrophage (canonical IL-12) or each can come from two different cells, hereafter referred to as the “two-cell model.” A prerequisite of the two-cell model is that p40 secreted by DCs should be able to interact with p35 outside the DC and combine to generate active IL-12. There is so far no in vivo evidence for such a functional association of these two subunits. Therefore, we evaluated this possibility using the SMARTA T cell model described above. The expectation was that the quantitative deficiency toward IFNγ production in SMARTA T cells primed in a p40−/− host could be complemented by the administration of exogenous recombinant p40 monomers only if the two-cell model were valid. Accordingly, we primed adoptively transferred SMARTA T cells with peptide mixed with LPS, combined with intraperitoneal (i.p.) administration of recombinant p40 (or PBS as a control) to the p40−/− animals (Figure 2A). The mice were treated daily for 5 days after antigen stimulation, upon which point the T cells were harvested and restimulated in vitro to analyze T cell differentiation.
Figure 2. Administration of recombinant p40 allows for antigen-specific T cell differentiation.

(A) Schematic of experimental design. SMARTA CD4+ T cells from CD45.1+ donors were sorted and transferred into 45.2+ p40−/− animals or WT animals and challenged 24 h later with GP61–80 and LPS. p40−/− recipients were divided into two groups, one that received 1 mg of recombinant p40 and the other that received sterile PBS from days 1 to 5 after transfer. SMARTA T cells were harvested from these animals at day 5 and restimulated in vitro to analyze for T cell differentiation by cytokine production.
(B–D) Splenocytes harvested 5 days after challenge were normalized to SMARTA T cell number and restimulated in vitro with GP61–80 for 48 h. Restimulation doses are in mg. Concentration of (B) IFNγ, (C) IL-4, and (D) IL-17A in supernatant from splenocytes of SMARTA T cells transferred into p40−/− animals treated with PBS (left, blue open bars), p40−/− animals given 1 μg of recombinant p40 (middle, blue filled bars), or WT animals (right, gray filled bars) (n = 4).
Data in this figure are representative of one independent experiment where n refers to the number of biological replicates. (B)–(D) are displayed as mean ± SEM. *,#p < 0.05, **,##p < 0.01, ***,###p < 0.001, ****,####p < 0.0001: *significance relative to the p40−/− PBS-treated animals; #significance relative to the p40−/− animals, which received recombinant p40. Anything unmarked is considered not significant.
In response to increasing doses of GP61–80, as seen in Figures 1J and 1K, T cells transferred into the p40−/− animals that received only PBS did not induce IFNγ production while the ones transferred into WT animals saw an increase of IFNγ production relative to the restimulation dose. Intriguingly, the T cells that were transferred into p40−/− animals that received administration of recombinant p40 saw an intermediate increase in the IFNγ production compared to the T cells in the WT animals (Figure 2B). Given previous reports about p40−/− animals supporting a predisposition toward a T helper (Th)2 phenotype during Leishmania infections, we assessed IL-4 levels in the restimulation as well. IL-4 levels were increased with a restimulation dose in the p40−/− animals treated with PBS but not in the WT animals, as expected. In the p40−/− animals that received recombinant p40, the induction of IFNγ by these T cells also resulted in a decrease in IL-4 production (Figure 2C). Levels of IL-17 were decreased in the T cells that were transferred to WT animals compared to both p40−/− groups (Figure 2D), and levels of IL-2 and IL-10 were unchanged between groups (Figures S2C and S2D). These data indicate that p40 and p35 do not need to be expressed in the same cell for IL-12-induced IFNγ activity to occur.
Chimeric mixing of cells singly producing either p40 or p35, but not both, allows for T cell differentiation in response to antigenic stimulation
Treatment with recombinant p40 showed that the assembly of IL-12 was not limited to a single cell in which both p40 and p35 subunits are expressed. However, this strategy did not answer whether in vivo, in the context of a physiological stimulation, two different cells can contribute to the subunits to make up a functional IL-12 response. To examine the potential of the two-cell model in vivo, we designed a chimeric strategy eliminating the ability of any given cell to make both p40 and p35 on its own (Figure 3A). In chimeric p40−/− animals, irradiated and reconstituted with bone marrow from p35−/− mice (p35−/−→p40−/−), hematopoietic cells from the donor (p35−/−) can make p40 but not p35. The residual host (p40−/−) tissues, alternatively, can express p35 but not p40. This chimera would generate IL-12 only if the collaborative assembly of functional IL-12 envisioned in the two-cell model operated in vivo.
Figure 3. Chimeric mixing of cells producing either p40 or p35, but not both, still allows for T cell differentiation in response to antigenic stimulation.

(A) Schematic of experimental design. Bone marrow chimeras were generated by reconstituting irradiated CD45.1+CD45.2+ p40−/− mice with bone marrow cells from CD45.2+ p35−/− donor mice or irradiated CD45.2+ p40−/− mice with bone marrow cells from CD45.1+CD45.2+ p40−/− or CD45.1+CD45.2+ WT donor mice. SMARTA CD4+ T cells from CD45.1+ donors were sorted and transferred into chimeric animals after reconstitution and challenged 24 h later with GP61–80 and LPS.
(B) Identification of transferred SMARTA T cells in spleens of recipient animals 5 days after challenge (n = 3). Gating strategy for this panel is shown in Figure S2A.
(C–E) Splenocytes harvested 5 days after challenge were normalized to SMARTA T cell number and restimulated in vitro with GP61–80 for 48 h. Restimulation doses are in mg. Concentrations are shown of (C) IFNγ, (D) IL-17A, and (E) IL-4 in supernatant from splenocytes of SMARTA T cells transferred into p40−/− host chimeras reconstituted with p40−/− (left, blue gradient), p35−/− (middle, green gradient), or WT (right, gray gradient) animals after restimulation for 48 h (n = 3).
Data in this figure are representative of one independent experiments where n refers to the number of biological replicates. (C)-(E) are displayed as mean ± SEM. *,#p < 0.05, **,##p < 0.01, ***,###p < 0.001, ****,####p < 0.0001: *significance relative to the p40−/−→p40−/− animals; #relative to the p35−/−→p40−/− animals. Anything unmarked is considered not significant.
We first wanted to examine this using the same SMARTA T cell model. After flow cytometric analysis of chimeric mice confirming the depletion of host-derived major histocompatibility complex class II (MHC class II)+ cells (Figures S3B and S3C) and subsequent reconstitution with donor cells, we used SMARTA T cells to interrogate the differentiation of naive CD4 T cells driven by IL-12 formed via two-cell assembly. SMARTA T cells were adoptively transferred to p35−/−→p40−/− chimeric animals and challenged with LCMV GP61–80 peptide (with LPS adjuvant). 5 days later, splenocytes were analyzed (Figure 3). SMARTA T cells seeded and expanded to comparable levels in all chimeric hosts following antigen challenge (Figures 3B and S3F). Antigen-specific restimulation of these T cells ex vivo showed that WT→p40 chimeras supported strong IFNγ differentiation (Figure 3C, gray). Conversely, p40−/−→p40−/− chimeras were unable to promote skewing toward IFNγ production (Figure 3C, blue). Interestingly, in p35−/−→p40−/− chimeras, where IL-12 activity is only available through the proposed two-cell model, SMARTA cells were able to produce significant IFNγ compared to p40−/−→p40−/− animals (Figure 3C, green). The p35−/−→p40−/− chimeras also showed a higher production of IL-17A (Figure 3D, green), which can be influenced by IL-23. This suggests that in the loss of canonical IL-12 production, p40 produced by hematopoietic cells will be more available to bind p19 to form IL-23, since in this setup, both subunits can be made by the same DC. Levels of IL-4 in SMARTA T cells from WT→p40−/− and p35−/−→p40−/− chimeras were negligible, while p40−/−→p40−/− chimeras made significantly more (Figure 3E), confirming that IL-4 production is promoted in the absence of both canonical and two-cell IL-12 formation. These data indicate that the alternate pathway for IL-12 assembly involving two separate cells is sufficient for the differentiation of antigen-specific T cells toward IFNγ production in vivo after antigen-specific stimulation.
The two-cell model of IL-12 assembly allows for T cell differentiation to IFNγ during L. major infection
Given that the tenets of Th1/Th2 skewing and the role of IL-12 were first elucidated using L. major infection in mice (Afonso et al., 1994; Heinzel et al., 1989), we chose to use the same infection to evaluate the role of the two-cell model for IL-12 generation during infection response. Using the strategy outlined in the previous section, p40−/− chimeric mice were infected with L. major (Figure 4A). As expected, among T cells in L. major-infected WT→p40−/− chimeras, 9.6% (±0.93%) of the CD4 T cells produced IFNγ, which was significantly higher than that in CD4 T cells from p40−/−→p40−/− chimeras (1.6% [±0.43%]) (Figures 4B and S4B). Strikingly, in the p35−/−→p40−/− chimeras, where individual DCs cannot assemble the IL-12 heterodimer on their own, frequencies of IFNγ-producing CD4 T cells were comparable to those in WT→p40−/− animals, averaging 8.9% (±1.03%) (Figures 3B and S3B). The total number of IFNγ-producing CD44hi CD4 T cells mirrored the pattern seen in the frequencies (Figure 4C). Similarly, for CD8s, the frequency and number of CD44hi T cells producing IFNγ in WT→p40−/− chimeras (13.23% [±2.23%]) were higher than those in p40−/−→p40−/− (2.6% [±0.55%]) but similar to those in p35−/−→p40−/− animals (9.3% [±1.02%]) (Figures 4D, 4E, and S4C). These data suggest that two-cell formation of IL-12 is comparable to canonical IL-12 in driving differentiation of T cells toward an IFNγ response during L. major infection.
Figure 4. Two-cell formation of IL-12 triggers downstream IFNγ in response to L. major infection.

(A) Schematic of experimental design. Bone marrow chimeras were generated by reconstituting irradiated CD45.1+ p40−/− with bone marrow cells from CD45.2+ p40−/−, CD45.2+ p35−/−, or CD45.1+CD45.2+ WT donor mice. Chimeric mice were infected in the footpad dermis with 106 dsRed L. major parasites following reconstitution. 35 days after infection, tissues were harvested and restimulated for intracellular cytokine staining and flow cytometric analysis. Gating strategy used for these panels is shown in Figure S3A.
(B) Percentage and of CD44hiIFNγ+CD4+ T cells in the spleen of p40−/− host chimeras measured 35 days after infection (p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(C) Number of CD44hiIFNγ+CD4+ T cells in the spleen of p40−/− host chimeras reconstituted with p40−/− (circles, blue), p35−/− (squares, green), or WT (triangles, black) donor bone marrow 35 days after infection (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(D) Same as in (B), except CD8+ T cells (p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(E) Same as in (C), except CD8+ T cells (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(F) Percentage of CD44hiIFNγ+CD4+ T cells in lymphocytes harvested from the footpad lesion tissue of p40−/− host chimeras 35 days after infection (p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(G) Number of CD44hiIFNγ+CD4+ T cells in lymphocytes harvested from the footpad lesion of p40−/− host chimeras reconstituted with p40−/− (circles, blue), p35−/− (squares, green), or WT (triangles, black) donor bone marrow 35 days after infection (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−,n= 6; WT→p40−/−, n = 3).
(H) Same as in (F), except CD8+ T cells (p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(I) Same as in (G), except CD8+ T cells (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(J and K) Cells from each tissue were harvested 35 days after infection and restimulated with soluble Leishmania antigen (SLA). Concentrations are shown of IFNγ in supernatant by tissues collected from p40−/−→p40−/− (blue), p35−/−→p40−/− (green), and WT→p40−/− (black) after restimulation for (J) 24 and (K) 48h (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
Data in this figure are representative of two independent experiments where n refers to the number of biological replicates. (C), (E), (G), and (I)–(K) are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Anything not marked is considered not statistically significant.
Similar to the data seen in the SMARTA model in Figure 3, the IL-4 responses were also affected in the chimeric mice. The p40−/−→p40−/− chimeras had increased CD4 T cell production of IL-4 while the p35−/−→p40−/− animals had similar proportions and numbers of CD44hi IL-4-producing T cells to those in the WT→p40−/− animals (Figures S4F and S4G). Interestingly, unlike the data in the SMARTA model, the p35−/−→p40−/− animals saw no significant difference in the proportion and number of CD4 T cells producing IL-17 compared to the p40−/−→p40−/− animals (Figures S4H and S4I). This indicates that the upregulation of p19 versus p35 is context-dependent, lending further evidence for the two-cell model being a specific way for tissues to collaborate and carry out IL-12-specific activities when necessary.
In the L. major model, the primary site of infection is the dermis of the mouse footpad. While T cells recovered from this tissue site in p35−/−→p40−/− chimeras had increased frequencies (6.1% [±1.7%] for CD4s and 5.4% [±3.4%)] in CD8s) of IFNγ production relative to p40−/−→p40−/− chimeras (1.7% [±0.5%] for CD4s and 0.5% [±0.7%] in CD8s), they did not reach the levels seen in WT→p40−/− chimeras (26.2% [±7.1%] for CD4s and 13.4% [±3.1%] in CD8s) (Figures 4F, 4I, S4D, and S4E). Cells taken from various tissue sites and restimulated with SLA for 24 or 48 h confirmed the disparity in IFNγ production between p40−/−→p40−/− and WT→p40−/− animals in an antigen-specific manner. In the antigen-specific restimulation, the IFNγ production in p35−/−→p40−/− chimeras maintained an intermediate phenotype between p40−/−→p40−/− and WT→p40−/− chimeras in all tissues analyzed (Figures 4J and 4K). Taken together, these data suggest that two-cell IL-12 can still elicit IFNγ from CD4 T cells at the primary site of infection but to a lower degree than for canonical IL-12.
Hematopoietic expression of p40 is critical for the two-cell IL-12 activity
The separable contribution by two different cells of subunits toward heterodimeric IL-12 assembly raises several key questions, not the least of which is the category of cells involved. Typically, DCs are considered the primary source of both monomeric p40 and heterodimeric IL-12. While p40 is primarily expressed by DCs and macrophages (typically hematopoietic cells), there is evidence that p35 can be sourced from other cells (Figures S5A and S5B). Thus, we examined a chimeric situation where p35−/− hosts (capable of making p40) were irradiated and reconstituted with p35−/−, p40−/−, or WT bone marrow (Figure 5A).
Figure 5. Hematopoietic cell expression of p40 is critical for two-cell IL-12 formation.

(A) Schematic of experimental design. Bone marrow chimeras were generated by reconstituting irradiated CD45.2+ p35−/− mice with bone marrow cells from CD45.1+ p40−/−, CD45.2+ p35−/−, or CD45.1+CD45.2+ WT donor mice. Chimeric mice were infected in the footpad dermis with 106 dsRed L. major parasites following reconstitution. 35 days after infection, tissues were harvested and restimulated for intracellular cytokine staining and flow cytometric analysis. Gating strategy used for these panels is shown in Figure S3A.
(B) Percentage of CD44hiIFNγ+CD4+ T cells in the spleen of p35−/− host chimeras measured 35 days after infection (p35−/−→p35−/−, n = 3; p40−/−→p35−/−, n = 3; WT→p35−/−, n = 2).
(C) Number of CD44hiIFNγ+CD4+ T cells in the spleen of p35−/− host chimeras reconstituted with p35−/− (squares, green), p40−/− (circles, blue), or WT (triangles, black) donor bone marrow 35 days after infection (unpaired t test, p35−/−→p35−/−, n = 3; p40−/−→p35−/−, n = 3; WT→p35−/−,n= 2 due to animal death).
(D) Same as in (B), except CD8+ T cells (p35−/−→p35−/−, n = 3; p40−/−→p35−/−, n = 3; WT→p35−/−, n = 2).
(E) Same as in (C), except CD8+ T cells (unpaired t test, p35−/−→p35−/−, n = 3; p40−/−→p35−/−, n = 3; WT→p35−/−, n = 2).
Data in this figure are representative of two independent experiments where n refers to the number of biological replicates. The WT groups in these experiments are limited to n = 2 due to technical reasons; however, combining the 600r chimeras (Figure S5) with the WT groups here allows us to have four groups of WT mice, and they were not significantly different in the two experiments. (C) and (E) are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Anything not marked is considered not statistically significant.
With lethal split dose irradiation, the hematopoietic compartment is ablated in these mice (Figures S3B and S3C). Therefore, in the p40−/−→p35−/− setup, the host cells would be unable to produce p40 for two-cell IL-12 production seen with the p40−/− host chimeras in Figure 3. Thus, only the WT→p35−/− animals were able to induce a CD44hi CD4 population of IFNγ-producing cells, similar to levels seen in WT→p40−/− chimeras, at 10.9% (±1.8%) (Figures 5B and S5C). Both p35−/−→p35−/− and p40−/−→p35−/− chimeras had comparable minimal frequencies (Figures 5B and S5C) as well as numbers of CD44hi IFNγ-producing CD4 T cells in the spleen (Figure 5C). Similar trends were also seen in CD8 cells (Figures 5D, 5E, and S5D). Therefore, a hematopoietic cell is the necessary source of p40 production for two cell-derived IL-12 to be made in vivo.
Unlike the lethal split dose radiation used above, sublethal irradiation at 600 rad allows for the retention of some hematopoietic cells within host animals (Figures S3B and S3C). Under these conditions, p35−/− chimeras would still retain host-derived cells capable of producing p40. Importantly, T cells in these p40−/−→p35−/− chimeric mice produced IFNγ in a comparable proportion to WT→p35−/− animals (Figures S5E and S5G). The total number of IFNγ-producing CD4 (Figure S5F) and CD8 (Figure S5H) T cells in the spleen followed the same trends. Comparing these results with the p40−/−→p35−/− animals that were subjected to split dose lethal irradiation confirms that the hematopoietic compartment is responsible for the production of p40 in the two-cell model. The cellular source of p35 is not as limiting, as suggested by the expression data (Figure S5B) and previous studies that have found p35 expression in keratinocytes and fibroblast-like cells (Aragane et al., 1994; Carrión et al., 2013). As such, when comparing the single-dose 600r chimeras made with p40−/− hosts, where p35 can be produced by the hematopoietic compartment as well as stromal cells, to the lethal split dose in Figure 4, where it is limited to solely stromal cell production, we find that the induction of IFNγ in the p35−/−→p40−/− animals is comparable in both CD4 and CD8 T cells (Figures S5I and S5J). This indicates that p35 is not restricted to the hematopoietic compartment as for p40. Taken together, we infer that bone marrow-derived cells are critical as the source of p40, while p35 can be derived from non-hematopoietic sources for two-cell IL-12 to drive functional IFNγ responses. This agrees with expression data previously published and suggests a localized, tissue expression of p35 while p40, expressed and released by antigen-presenting cells, circulates to form IL-12 wherever p35 is expressed.
The functional significance of two cell- versus one cell-derived IL-12 activity
The chimeric model offered an opportunity to evaluate whether the IFNγ-promoting activity derived from canonical IL-12 versus two-cell IL-12 assembly has differing impacts on the course of the infection. Interestingly, despite the IFNγ production seen from two-cell IL-12 (Figure 4), L. major infection, as monitored by swelling of the footpad midpoint, revealed no difference between p40−/−→p40−/− and p35−/−→p40−/− animals, with both developing non-healing lesions while WT→p40−/− animal lesions healed over time, returning to near baseline levels of footpad thickness (Figures 6A and 6B). Consistent with this, initial analysis of parasite numbers at the footpad showed total parasite titers comparably high, near 1015 for both p40−/−→p40−/− and p35−/−→p40−/− animals, while WT→p40−/− total parasite levels were reduced, near 108 by the experimental endpoint (Figure 6C).
Figure 6. Two cell-derived IL-12 activity is functionally different than one-cell derived IL-12 activity.

Bone marrow chimeras were generated by reconstituting irradiated CD45.1+ p40−/− mice with bone marrow cells from CD45.2+ p40−/−, CD45.2+ p35−/−, or CD45.1+CD45.2+ WT donor mice. Chimeric mice were infected in the footpad dermis with 106 dsRed L. major parasites following reconstitution.
(A) Biweekly measurement of footpad thickness at the midline following establishment of visible lesion for p40−/− host chimeras reconstituted with p40−/− (circles, blue), p35−/− (squares, green), or WT (triangles, black) through the duration of infection (p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(B) Footpad thickness at the midline of p40−/− host chimeras reconstituted with p40−/− (circles, blue), p35−/− (squares, green), or WT (triangles, black) 35 days after L. major infection (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
(C–H) dsRed L. major parasite quantified by limiting dilution from (C) footpad lesion, (D) dLN, (E) non-dLN (ndLN), (F) spleen, (G) liver, and (H) lung tissue of p40−/− host chimeras reconstituted with p40−/− (circles, blue), p35−/− (squares, green), or WT (triangles, black) 35 days after infection (unpaired t test, p40−/−→p40−/−, n = 4; p35−/−→p40−/−, n = 6; WT→p40−/−, n = 3).
Data in this figure are representative of two independent experiments where n refers to the number of biological replicates. All data are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Anything not marked is considered not statistically significant.
Although L. major is traditionally studied as a localized infection (especially in resistant B6 mice), it is also known to disseminate fairly systemically (Kamala and Nanda, 2009; Kurey et al., 2009; Nicolas et al., 2000; Rossi and Fasel, 2018; Sacks and Noben-Trauth, 2002; Wege et al., 2012). Of particular note, the liver is a known site affected in severe L. major infections (Hu et al., 2018). We therefore examined representative tissue sites in the chimeric mice for parasitemia to determine the impact of two-cell IL-12 on parasite control at these sites. Importantly, we used a limiting dilution assay, which only evaluates live replicating parasites rather than genetic material from dead parasites. We validated that disseminating parasites are viable by propidium iodide staining (Figures S6J–S6N).
The data from these assays allowed us to unravel a striking tissue specificity to the biological activity the two-cell IL-12, toward limiting parasite burden in the organs we examined. Unlike the footpad (where the p35−/−→p40−/− chimera failed to control parasite burden any better than did the p40−/− mice; Figure 6C), we noticed that the parasite loads in the dLN were already different (Figure 6D). Compared to the WT mice, p40−/−→p40−/− had an approximately 2-log higher parasite load. However, unlike the footpad samples, p35−/−→p40−/− animals had parasite numbers reduced about 3-fold from those in the p40−/−→p40−/− group, although they were still quite higher than those in the WT→p40−/− group. In striking contrast, when we enumerated parasite count at a distal non-dLN (ndLN) (Figure 6E), the parasite load between the p35−/−→p40−/− chimeras and the WT→p40−/− was not statistically distinguishable, suggesting that both groups control dissemination to this distant site, much better than the p40−/−→p40−/− chimeric mice. Furthermore, evaluation of additional tissue sites (spleen, liver, and lung in Figures 6F–6H) showed a range of differences, with the p35−/−→p40−/− chimeras consistently controlling parasite burden better that the p40−/−→p40−/− chimeras in each case. As shown in Figure 5, chimeras set up with p35−/− host animals are unable to form two-cell IL-12, as the hematopoietic source of p40 is ablated with irradiation in these animals. Thus, both the p35−/−→p35−/− and p40−/−→p35−/− animals have similar parasite titers in all tissues analyzed, confirming the absence of two-cell IL-12 and its role in limiting the dissemination of parasites (Figures S6A–S6H).
Taken together, these data demonstrate a gradation of the impact of two-cell IL-12 activity depending on the tissue type. The primary site of infection and the nearby dLN sites need the canonical one-cell production of IL-12 to clear the parasite. Distant sites, such as the spleen, liver, lung, and ndLNs, can impart significant infection control with solely two-cell production of IL-12.
DISCUSSION
Data presented herein demonstrate using multiple experimental approaches that functional IL-12 can be formed through the collaborative production of individual subunits by two separate cells: p40 from hematopoietic cells and p35 from other cells. Importantly, the chimeric approach also allows us to evaluate the functional significance of this form of IL-12 during protective immune responses to a pathogen, as modeled by L. major. The structural ability of p40 to bind p35 has been extensively characterized, although focusing on assembly within a single cell (Glassman et al., 2021; Lupardus and Garcia, 2008; Yoon et al., 2000). In the present study, we extend previous work (mostly using in vitro models) suggesting that the two proteins can also functionally assemble outside the cell. This functional assembly is consistent with previous literature and gene expression datasets showing that p40 expression is limited to DCs and macrophages (D’Andrea et al., 1992) while p35 expression is more widespread (Dixon et al., 2015; Sha et al., 2015; Shen et al., 2014). In the past decade, the contribution of non-hematopoietic cells toward modulating the trajectory of an immune response has been extensively discussed (Matzinger and Kamala, 2011). The mechanism of IL-12 assembly we demonstrate herein adds an important dimension to this emerging picture, suggesting that heterodimeric cytokines may possess an intrinsic advantage by allowing their subunits to not only reassort to form multiple cytokines, but also source the subunits from different tissues. This has the potential to molecularly encode immunological context, with both sentinel cells (e.g., DCs, macrophages) and stromal cells collaborating to jointly shape the trajectory of T cell differentiation (Abdi and Singh, 2015). Importantly, note that while the data demonstrate that two separate cells can contribute parts of IL-12 in vivo, the significance of the specific source of p35 needs to be resolved further. Importantly, in chimeras where we used WT bone marrow to reconstitute either p40−/− (WT groups in Figures 4B and 4D) or p35−/− (WT groups in Figures 5B and 5D), there was no significant differences in T cell differentiation, suggesting that at least for that parameter, the availability of p40 and p35 from hematopoietic cells was redundant with stromal cells. Intriguingly, we found that the levels of IL-10 secretion by WT T cells developing in a p35−/− stromal environment were significantly (~3.5-fold) greater than those in the p35-sufficient but p40−/− stroma (Figure S5L). Certainly, IL-10 is part of the negative feedback loop elicited by IFNγ and IL-12 (Abdi et al., 2012; D’Andrea et al., 1993); however, how stromal p35 selectively impacts that is not immediately clear and will require additional studies.
If such studies are also extended to additional cytokines such as IL-23, it is possible that a major driver of IFNγ versus IL-17 differentiation in some cases may be the expression of p35 versus p19 by specific stromal cells, rather than DCs themselves. In the chimeric models, although we did notice a trend toward higher IL-17 production in the p35−/− chimeric mice (Figures S4F and S4G), this was not statistically significant. Importantly, the IL-4 production that is associated with IL-12 deficiency was also corrected in the chimeric mice (Figures S4H and S4I). This suggests that the two-cell form of IL-12 can recapitulate most known activities of canonical IL-12.
Nevertheless, two-cell IL-12 does not appear to be sufficient to control L. major at the primary infection site. This is an intriguing observation and illustrates key aspects by which localized immunity in secondary sites can differ from the primary site of infection. For one, the failure to control parasites at the primary site while having robust control at specific distal sites was not correlated with differences in T cell differentiation toward IFNγ production. While there is an extensive literature suggesting that the IL-12/IFNγ axis is critical for the control of Leishmania, the direct evidence in our study is limited to showing that two-cell IL-12 regulates IFNγ and also dissemination. Future experiments will have to evaluate (using IFNγ double knockouts with p40 and p35) whether the IFNγ is causal for limiting tissue dissemination. If it is, one possible explanation for the dichotomy in the functional roles of two-cell versus one-cell IL-12 is that they reflect kinetic variations in the amount of IL-12: while both early and late IL-12 production could be sufficient to drive T cell differentiation, perhaps sustained production is required for control at each site. This is challenging to measure dynamically. For one, the amount of two-cell IL-12 formed in vivo was not detectable using commercial ELISAs (Figure S4J). Yet, clearly, by multiple readouts there is sufficient in vivo availability to drive T cell differentiation. An alternate possibility is that the two-cell form is nucleated by cell-specific process, akin to the trans-presentation of IL-15 (and therefore difficult to detect in circulation).
Canonical IL-12 is formed when p40 disulfide bonds with p35, allowing for both subunits to be secreted. Yet, this covalent association has been shown to be dispensable for secretion of the functional IL-12 cytokine (Reitberger et al., 2017; Yoon et al., 2000). Furthermore, this is not the only route to p35 release, as p35 binds to another known partner (Ebi3) to form IL-35, after these subunits are reportedly released independently (Aparicio-Siegmund et al., 2014; Sawant et al., 2015). We had previously shown that functional IL-12 can be generated in vitro through the collaborative combination of either p40 monomer with p35 released from necrotic cells or the reverse combination of p35 with p40 from serum (Abdi et al., 2014). Thus, mechanistically, the release of p35 in vivo can also be through necrotic cell death upon tissue damage by a pathogen. This would infer that the innate immune system is integrating two signals in informing T cells about the desired route of differentiation (Abdi and Singh, 2015). First, engagement of PRRs on DCs would lead to p40 release, which disseminates systemically albeit in an inert form (does not drive T cell differentiation). Second, the arrival of pathogen in specific tissue sites allows for local and appropriate release of p35. According to this model, distinct tissue sites can control the quantity of the effector response by regulating p35 release and perhaps even the quality by releasing p19 versus p35. A third possibility is in tissue-specific variations in the induction of IFNγ itself. Multiple cytokines (including IL-18 and IL-15) as well as T cell-derived signals (via CD40) can influence the ability of cells to make IFNγ (Abdi et al., 2006; Abdi and Singh, 2010) (Denton et al., 2007; Müller et al., 2001). In many cases, IL-12 is more associated with a feedback loop for amplification of IFNγ production rather than the initial triggering. Tissue levels of such triggers or other negative regulators (transforming growth factor β [TGF]-β, IL-10) can potentially contribute to these differences as well.
L. major infects multiple cell types although often studied as a localized infection. Previous reports, especially in susceptible strains of mice, have shown that the parasite can travel via infected cells to multiple tissues (Kurey et al., 2009; Nicolas et al., 2000; Rossi and Fasel, 2018; Sacks and Noben-Trauth, 2002; Wege et al., 2012). Dissemination of the L. major parasite has been shown to be aided by inflammatory DCs infected with the parasite (Hurdayal et al., 2013, 2020). In addition, L. major is known to downregulate transcription of both p40 and p35 subunits in macrophages and monocytes (Belkaid et al., 1998; Sartori et al., 1997). Taking this into account, production of p35 at disseminated tissue sites would contribute to a greater IL-12 response necessary for local parasite control. The presence of elevated p40 levels detected in the serum allow for the circulation among secondary infection sites and subsequent binding to p35 for the formation of two-cell IL-12. A reviewer of this manuscript suggested that this could perhaps relate to vascularization of each organ and the kinetics of p40 “arrival” or accumulation there. This intriguing hypothesis remains to be tested. In this context, the experiments with recombinant administration of p40 (Figure 2) can be a useful approach.
Collectively, the data we have shown herein establishes that IL-12 activity can come from the collaboration between two distinct cells and impact T cell differentiation to IFNγ production. The functional, in vivo demonstration of the two-cell mechanism of IL-12 assembly allows for consideration of both mechanisms in other disease states as well as potentially understanding clinical conditions where distinct pools of systemic and local IL-12 can have different consequences.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Nevil J. Singh (nsingh@som.umaryland.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data from the GTEx Portal. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 07/15/2020. Accession for these datasets are listed in the Key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Brilliant Violet 605 anti-mouse CD4 clone GK1.5 | Biolegend | Cat# 300555; RRID:AB_2564390 |
| eFluor 450 anti-mouse CD4 clone GK1.5 | eBioscience | Cat# 48–0041-82; RRID:AB_10718983 |
| Brilliant Violet 650 anti-mouse CD8b clone H35–17.2 | BD Biosciences | Cat# 740552; RRID:AB_2740253 |
| FITC anti mouse CD8b clone H35–17.2 | eBioscience | Cat# 11–0080-85; RRID:AB_657775 |
| PE/Cyanine7 anti-mouse/human CD11b clone M1/70 | Biolegend | Cat# 100215; RRID:AB_312798 |
| eFluor 450 anti-mouse CD11c clone N418 | eBioscience | Cat# 48–0114-80; RRID:AB_1548665 |
| PE anti-mouse/human CD44 clone IM7 | BD PharMingen | Cat# 553134; RRID:AB_394649 |
| PE-Cy7 anti-mouse/hman CD44 clone IM7 | Biolegend | Cat# 103030; RRID:AB_830787 |
| PerCP/Cyanine5.5 anti-mouse/human CD44 clone IM7 | Biolegend | Cat# 103032; RRID:AB_2076204 |
| Alexa Fluor 488 anti-mouse CD45.1 clone A20 | Biolegend | Cat# 110718; RRID:AB_492862 |
| APC anti-mouse CD45.2 clone 104 | eBioscience | Cat# 17–0454-82; RRID:AB_469400 |
| Alexa Fluor 700 anti-mouse CD62L clone MEL-14 | Biolegend | Cat# 104426; RRID:AB_493719 |
| PE/Cyanine7 anti-mouse CD62L clone MEL-14 | Biolegend | Cat# 104417; RRID:AB_313102 |
| APC anti-mouse IFNγ clone XMG1.2 | eBioscience | Cat# 17–7311-82; RRID:AB_469504 |
| FITC anti-mouse IFNγ clone XMG1.2 | BD PharMingen | Cat# 554411; RRID:AB_395375 |
| PE anti-mouse IL-4 clone 11B11 | Biolegend | Cat# 504104; RRID:AB_315318 |
| PerCP/Cyanine5.5 anti-mouse IL-10 clone JES5–16E3 | Biolegend | Cat# 505028; RRID:AB_2561523 |
| APC anti-mouse IL-17A clone TC11–18H10.1 | Biolegend | Cat# 506916; RRID:AB_536018 |
| PE anti-mouse MHC Class II (IA/IE) clone M5/114.15.2 | eBioscience | Cat# 12–5321-82; RRID:AB_465928 |
| Brilliant Violet 421 anti-mouse T-bet clone 4B10 | Biolegend | Cat# 644815; RRID:AB_10896427 |
| PE-Cy7 anti-mouse T-bet clone 4B10 | eBioscience | Cat# 12–5825-82; RRID:AB_925761 |
| Brilliant Violet510 anti-mouse TCRb clone H57–597 | Biolegend | Cat# 109233; RRID:AB_2562349 |
| CyChrome anti-mouse TCRb clone H57–597 | BD PharMingen | Cat# 553173; RRID:AB_394685 |
| PE anti-mouse TCR-vb8.3 clone 1B3.3 | BD PharMingen | Cat# 553664; RRID:AB_394980 |
| Biotin anti-mouse NK1.1 clone PK136 | eBioscience | Cat# 13–5941-82; RRID:AB_466804 |
| Biotin anti-mouse CD11c clone N418 | eBioscience | Cat# 13–0114-82; RRID:AB_466363 |
| Biotin anti-mouse CD8a clone 53–6.7 | eBioscience | Cat# 13–0081-82; RRID:AB_466346 |
| Biotin anti-mouse MHC class II clone M5/114.15.2 | eBioscience | Cat# 13–5321-82; RRID:AB_466662 |
| Purified anti-mouse CD16/CD32 clone 2.4G2 | BD PharMingen | Cat# 553141; RRID:AB_394656 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LCMV GP61–80 | AnaSpec | Cat# AS-64851 |
| Collagenase D | Roche | Cat# 11 088 858 011 |
| Propidium Iodide Solution | Sigma-Aldrich | Cat# P4864 |
| Lipopolysaccharides from E. coli | Sigma-Aldrich | Cat# L2880 |
| Medium 199 | GIBCO | Cat# 12350039 |
| RPMI 1640 | GIBCO | Cat# 11875119 |
| Foundation Fetal Bovine Serum | Gemini Bio | Cat# 900–108 |
| HEPES | GIBCO | Cat# 15–630–080 |
| Adenine | Sigma-Aldrich | Cat# A2786 |
| Biotin | Sigma-Aldrich | Cat# B4501 |
| Hemin | Sigma-Aldrich | Cat# 51280 |
| PBS | Quality Biological | Cat# 119–069-101 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17–1440-02 |
| Lectin from Arachis hypogaea (peanut) | Sigma-Aldrich | Cat# L6135 |
| Sodium Pyruvate | GIBCO | Cat# 11360070 |
| Antibiotic-antimycotic | GIBCO | Cat# 15240062 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Brefeldin A | eBioscience | Cat# 00–4506-51 |
| EDTA | Quality Biological | Cat# 351–027-101 |
| Sodium azide | Sigma-Aldrich | Cat# S2002 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat# P8139 |
| Ionomycin calcium salt | Sigma-Aldrich | Cat# I0634 |
| Triethanolamine | Sigma-Aldrich | Cat# 90278 Critical commercial assays |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse IL-17A (homodimer) ELISA Ready-SET-Go! | eBioscience | Cat# 88–7371 RRID:AB_2575104 |
| Mouse IFN gamma ELISA Ready-SET-Go! | eBioscience | Cat# 88–7314 RRID:AB_2575069 |
| Mouse IL-2 Quantikine ELISA Kit | R&D Systems | Cat# M2000 |
| Mouse IL-4 Quantikine ELISA Kit | R&D Systems | Cat# M4000B |
| Mouse IL-12 p40 Quantikine ELISA Kit | R&D Systems | Cat# M1240 |
| Mouse IL-12 p70 Quantikine ELISA Kit | R&D systems | Cat# M1270 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat# 11205D |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554722 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00–5523-00 |
| LIVE/DEAD Fixable Near-IR | Invitrogen | Cat# L10119 |
| Ghost Dye Red 780 | Tonbo Biosciences | Cat# 13–0865 |
| BCA Protein Assay Kit | Pierce | Cat# 23225 |
|
Deposited data | ||
| GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2) | GTEx | https://www.gtexportal.org/home/gene/IL12B |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| L. major-RFP | Kimblin et al., 2008 | Kimblin et al., 2008 |
| Mouse: B6.129S1-Il12btm1Jm/J | The Jackson Laboratory | Cat# 002693 |
| Mouse: IL-12p40−/− CD45.1+ | Bred in-house | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse B6.SJL-PtprcaPepcb/BoyJ | The Jackson Laboratory | Cat# 002014 |
| Mouse: B6NTac | Taconic Biosciences | Cat# B6 |
| Mouse: B6.SJL-Ptprca/BoyAiTac | Taconic Biosciences | Cat# 4007 |
| Mouse: B6 1x2 | Bred in-house | N/A |
| Mouse: B6.129S1-Il12atm1Jm/J | The Jackson Laboratory | Cat# 002692 |
| Mouse: B6.Cg-Ptprca Pepcb Tg(TcrLCMV)1Aox/PpmJ | The Jackson Laboratory | Cat# 030450 |
|
Software and algorithms | ||
| FlowJo (v10.6.2) | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism 7.05 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| GTEx Portal | Broad Institute of MIT and Harvard | http://www.gtexportal.org/home/index.html |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type mice were obtained from both the Jackson Laboratory (C57BL/6J and B6.SJL-PtprcaPepcb/BoyJ) and Taconic Biosciences (B6NTac and B6.SJL-Ptprca/BoyAiTac). B6 mice were bred for dual congenic expression using a dam or sire from opposing place of purchase to eliminate any strain drift between the two wild-type strains. IL-12p40−/− (B6.129S1-Il12btm1Jm/J) and IL-12p35−/− (B6.129S1-Il12atm1Jm/J) were sourced from the Jackson Laboratory. IL-12p40−/− mice were backcrossed to wild-type mice (B6.SJL-PtprcaPepcb/BoyJ) to generate CD45.1 congenic IL-12p40−/− mice. SMARTA TCR transgenic mice were obtained from Jackson Laboratories (B6.Cg-Ptprca Pepcb Tg(TcrLCMV)1Aox/PpmJ). For all experiments, both female and male mice between 4 and 35 weeks of age were used. Animals were bred and maintained under specific pathogen free (SPF) conditions at the University of Maryland, Baltimore. Experiments were performed with animals at least four weeks of age for bone marrow isolation and six weeks of age for all other experiments and approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee.
Parasite lines
A stable transfected line of the Leishmania major FV1 strain expressing a red fluorescent protein was generated as described previously (Kimblin et al., 2008). Briefly, the DsRed gene was amplified by PCR using the pCMV-DsRed-Express plasmid (BD Biosciences/Clontech) as a template and cloned into the SpeI site of the pKSNEO Leishmania expression plasmid. Promastigotes were transfected with the resulting expression plasmid construct [pKSNEO-DsRed] and selected for growth in the presence of 50 μg/ml Geneticin (G418) (Sigma). The resulting parasites are referred to as dsRed L. major. dsRed L. major fluoresces brightly and grows and infects flies and mice normally. L. major-DsRed promastigotes were grown at 25°C in medium 199 (GIBCO) supplemented with 20% heat-inactivated FCS, 15 mM HEPES, 0.1 mM adenine (in 50 mM HEPES), 5 μg/ml hemin (in 50% triethanolamine), and 0.6 μg/ml biotin (in 95% ethanol) (Leish media) and were split into new media every 2–3 days depending on amount of promastigotes in culture.
METHOD DETAILS
Generation of bone marrow chimeras
Femurs and tibias were taken from CD45.2+ IL-12p40−/−, CD45.1+ IL-12p40−/−, CD45.2+ IL-12p35−/− and CD45.1+CD45.2+ B6 mice. The bone marrow was flushed out with a syringe and passed through a 100mm nylon mesh to generate a single cell suspension in 1X PBS supplemented with 5% FCS. Red blood cells were removed from suspension by Ficoll Paque Plus density gradient. Cells were washed twice and resuspended at 50 × 106 cells/ml. Recipient IL-12p40−/− and IL-12p35−/− mice were irradiated with two doses of 600 rad before 5 ×10 bone marrow cells were transferred i.v. The mice rested for 6 weeks for cellular reconstitution.
Preparation of soluble Leishmania antigen
Soluble Leishmania antigen (SLA) was prepared from late-log-phase promastigotes of L. major after several passages in liquid culture. Promastigotes were harvested and counted and adjusted to 2×108 promastigotes/mL. The promastigote suspension was washed three times in 5 mL sterile, cold phosphate-buffered saline (PBS). The suspension underwent five cycles of flash freezing in LN2 and thawing followed by a centrifugation at 8,000 × g for 20 minutes at 4°C. Supernatant containing SLA was collected and protein concentration was determined by BCA assay (Pierce).
Leishmania infection of animals
Infective-stage promastigotes (metacyclics) were isolated from stationary cultures using peanut lectin agglutinin. Mice were infected in the footpad dermis with 106 metacyclic promastigotes in 20 μL using a 27-gauge 1/2 needle. Infection was monitored every 3–4 days by caliper measurement of lesion swelling. To characterize infection site leukocytes, footpad dermal tissue was digested in 1mL 2.5 mg/mL Collagenase D solution in complete RPMI at 37°C for 2 hours and filtered through a 70μm filter. For in vitro re-stimulation, cells were incubated in T cell media alone or separately with 50μg SLA for 48hr.
Adoptive transfer and antigen challenge
Lymph nodes were isolated from SMARTA TCR-tg and mechanically dissociated by mashing tissues through 70 μM nylon mesh. Red blood cells were removed from suspension using Ficoll Paque Plus density gradient. Cell suspension was further enriched for CD4+ T cells using Dynabeads Negative Selection. 100,000 cells were transferred into recipient animals and 24 hours later were challenged with 25μg LCMV GP61–80 peptide (Anaspec) and 5μg LPS or PBS as a control. Spleen tissue was isolated 5d post challenge and analyzed via flow cytometry for SMARTA cell numbers. Cells were set up for in vitro re-challenge in RPMI-1640 (GIBCO) supplemented with 10% FCS (Gemini), 1% sodium pyruvate (GIBCO), 1% antibiotic-antimycotic (GIBCO) and 0.000014% 2-mercaptoethanol (T cell media) alone or doses of LCMV GP61–80 peptide (1μg, 3μg, 10μg), normalized to SMARTA cell numbers, and analyzed 48hr later for cytokine production.
Tissue preparation
Single cell suspensions were prepared from the draining popliteal lymph node, non-draining popliteal lymph node, and spleen by dissociation and passage through 40mm nylon mesh. Lymphocytes were isolated for characterization from the liver, lung and footpad lesions through digestion in 1mL 2.5 mg/mL Collagenase D solution in T cell media at 37°C for 2hr followed by crushing through a 40μm cell strainer.
Intracellular stain and flow cytometry analysis
Single cell suspensions from tissue preparation were re-stimulated with 100ng/ml PMA and 1μg/ml Ionomycin in T cell media for 2 hours at 37°C and treated with 10μg/ml Brefeldin A (eBioscience) for an additional 4 hours at 37°C. For staining, Fc receptor blocking was performed for 15 minutes at 4°C in 1X PBS supplemented with 2% FCS, 0.01% azide, and 1% of each mouse, hamster and rat serums (Fc Block). Surface staining was performed for 30 minutes at 4°C in PBS supplemented with 2% FCS, 0.01% azide and 0.02% EDTA (FACS Buffer) using the antibodies listed. Cells were washed once with FACS buffer. Cells were fixed and permeabilized in BD Cytofix/Cytoperm solution for 20 minutes at 4°C, followed by an overnight incubation at 4°C in 1X eBioscience Fixation/Permeabilization Solution. Intracellular staining was performed using the antibodies described above in 1X eBioscience Permeabilization Buffer for 1 hour at 4°C. Stained cells were washed twice with 1X eBioscience Permeabilization Buffer followed by an additional two washes with FACS buffer. Cells were analyzed on the BD LSR-II cytometer and all data was analyzed using FlowJo (BD Biosciences). Antibodies used in these experiments are listed in the Key resources table.
Parasite quantification
Parasite load was quantified through limiting dilution. Tissues were processed as described above and homogenate was plated in triplicate in serial 10-fold dilutions in 96 well round bottom plates (Corning) containing Leish media. Plates were incubated at 25°C for 10 days, at which point the number of viable parasites was determined microscopically. Total parasite burden was calculated from the highest dilution at which promastigotes could be detected.
Live parasite analysis by flow cytometry
Animals were infected as described above and the infection allowed to proceed for 22 days. At the endpoint, tissues were harvested and processed as described above in preparation for flow cytometry analysis. Once tissues were in single cell suspension, surface staining was done in 1X PBS at 25°C for 10min, followed by propidium iodide (Sigma) staining at 10ug/mL in 1X PBS for 10min at 25°C in the dark. Cells were analyzed on the BD LSR-II cytometer and all data was analyzed using FlowJo (BD Biosciences). Antibodies used for this experiment are listed in the Key resources table.
Cytokine production quantification
Tissue samples were processed as described above and single cell suspensions were plated in triplicate in 96-well plates (Corning). Levels of IL-17A and IFNγ in supernatants were assessed by sandwich enzyme-linked immunosorbent assay (ELISA) by using the Ready-SET-Go! systems (eBioscience). IL-12p40, IL-12p70, IL-2 and IL-4 were measured using Quantikine ELISA systems (R&D Systems).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data analyzed in this study was analyzed using GraphPad Prism software, version 7.05. Information about specific statistical tests used for each experiment are listed in the figure legends.
Supplementary Material
Highlights.
Both p35 and p40 subunits of IL-12 are required to differentiate IFNγ-producing T cells
p35 and p40 produced by two different cells can collaborate to drive T cell responses
The two-cell IL-12 activity requires hematopoietic p40 but can use stromal p35
Two-cell IL-12 activity is useful for controlling pathogen dissemination in vivo
ACKNOWLEDGMENTS
We thank members of the Nevil laboratory—Courtney Matson, Kenneth Rosenberg, Gideon Wolf, and Zachary Fasana—for assistance with mouse takedowns, discussions, and critiques during the development of this work; Dr. David L. Sacks for the dsRed-L. major parasites; Drs. Yasmine Belkaid and Alain Debrabant for assistance setting up L. major protocols; and the University of Maryland Greenebaum Comprehensive Cancer Center Flow Cytometry Core and Dr. Xiaoxuan Fan for flow cytometry support. This work was supported by NIH grants R01 AI110719, R21 AI149076, and R21 AI166330 (to N.J.S.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109816.
REFERENCES
- Abdi K, and Singh NJ (2010). Antigen-activated T cells induce IL-12p75 production from dendritic cells in an IFN-g-independent manner. Scand. J. Immunol 72, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi K, and Singh NJ (2015). Making many from few: IL-12p40 as a model for the combinatorial assembly of heterodimeric cytokines. Cytokine 76, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi K, Singh N, and Matzinger P (2006). T-cell control of IL-12p75 production. Scand. J. Immunol 64, 83–92. [DOI] [PubMed] [Google Scholar]
- Abdi K, Singh NJ, and Matzinger P (2012). Lipopolysaccharide-activated dendritic cells: “Exhausted” or alert and waiting? J. Immunol 188, 5981–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi K, Singh NJ, Spooner E, Kessler BM, Radaev S, Lantz L, Xiao TS, Matzinger P, Sun PD, and Ploegh HL (2014). Free IL-12p40 monomer is a polyfunctional adapter for generating novel IL-12-like heterodimers extracellularly. J. Immunol 192, 6028–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, and Scott P (1994). The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 263, 235–237. [DOI] [PubMed] [Google Scholar]
- Aparicio-Siegmund S, Moll JM, Lokau J, Grusdat M, Schröder J, Plöhn S, Rose-John S, Grötzinger J, Lang PA, Scheller J, and Garbers C (2014). Recombinant p35 from bacteria can form interleukin (IL-)12, but not IL-35. PLoS ONE 9, e107990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragane Y, Riemann H, Bhardwaj RS, Schwarz A, Sawada Y, Yamada H, Luger TA, Kubin M, Trinchieri G, and Schwarz T (1994). IL-12 is expressed and released by human keratinocytes and epidermoid carcinoma cell lines. J. Immunol 153, 5366–5372. [PubMed] [Google Scholar]
- Belkaid Y, Butcher B, and Sacks DL (1998). Analysis of cytokine production by inflammatory mouse macrophages at the single-cell level: Selective impairment of IL-12 induction in Leishmania-infected cells. Eur. J. Immunol 28, 1389–1400. [DOI] [PubMed] [Google Scholar]
- Carra G, Gerosa F, and Trinchieri G (2000). Biosynthesis and posttranslational regulation of human IL-12. J. Immunol 164, 4752–4761. [DOI] [PubMed] [Google Scholar]
- Carrión M, Pérez-García S, Jimeno R, Juarranz Y, González-Álvaro I, Pablos JL, Gutiérrez-Cañas I, and Gomariz RP (2013). Inflammatory mediators alter interleukin-17 receptor, interleukin-12 and −23 expression in human osteoarthritic and rheumatoid arthritis synovial fibroblasts: Immunomodulation by vasoactive intestinal peptide. Neuroimmunomodulation 20, 274–284. [DOI] [PubMed] [Google Scholar]
- Cooper AM, and Khader SA (2007). IL-12p40: An inherently agonistic cytokine. Trends Immunol. 28, 33–38. [DOI] [PubMed] [Google Scholar]
- D’Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, Chan SH, Kobayashi M, Young D, Nickbarg E, et al. (1992). Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J. Exp. Med 176, 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, and Trinchieri G (1993). Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med 178, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton AE, Doherty PC, Turner SJ, and La Gruta NL (2007). IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur. J. Immunol 37, 368–375. [DOI] [PubMed] [Google Scholar]
- Dixon KO, van der Kooij SW, Vignali DA, and van Kooten C (2015). Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur. J. Immunol 45, 1736–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowell DJ, and Locksley RM (1999). Leishmania major infection of inbred mice: Unmasking genetic determinants of infectious diseases. BioEssays 21, 510–518. [DOI] [PubMed] [Google Scholar]
- Glassman CR, Mathiharan YK, Jude KM, Su L, Panova O, Lupardus PJ, Spangler JB, Ely LK, Thomas C, Skiniotis G, and Garcia KC (2021). Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 184, 983–999.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler U, Chua AO, Schoenhaut DS, Dwyer CM, McComas W, Motyka R, Nabavi N, Wolitzky AG, Quinn PM, Familletti PC, et al. (1991). Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. USA 88, 4143–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MP, Wang J, and Norcross MA (1995). Regulation of interleukin-12 expression in human monocytes: Selective priming by interferon-γ of lipopolysaccharide-inducible p35 and p40 genes. Blood 86, 646–650. [PubMed] [Google Scholar]
- Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, and Locksley RM (1989). Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med 169, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Marshall C, Darby J, Wei W, Lyons AB, and Körner H (2018). Absence of tumor necrosis factor supports alternative activation of macrophages in the liver after infection with Leishmania major. Front. Immunol 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdayal R, Nieuwenhuizen NE, Revaz-Breton M, Smith L, Hoving JC, Parihar SP, Reizis B, and Brombacher F (2013). Deletion of IL-4 receptor alpha on dendritic cells renders BALB/c mice hypersusceptible to Leishmania major infection. PLoS Pathog. 9, e1003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdayal R, Nieuwenhuizen NE, Khutlang R, and Brombacher F (2020). Inflammatory dendritic cells, regulated by IL-4 receptor alpha signaling, control replication, and dissemination of Leishmania major in mice. Front. Cell. Infect. Microbiol 9, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalah R, Rosati M, Ganneru B, Pilkington GR, Valentin A, Kulkarni V, Bergamaschi C, Chowdhury B, Zhang GM, Beach RK, et al. (2013). The p40 subunit of interleukin (IL)-12 promotes stabilization and export of the p35 subunit: Implications for improved IL-12 cytokine production. J. Biol. Chem 288, 6763–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamala T, and Nanda NK (2009). Protective response to Leishmania major in BALB/c mice requires antigen processing in the absence of DM. J. Immunol 182, 4882–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimblin N, Peters N, Debrabant A, Secundino N, Egen J, Lawyer P, Fay MP, Kamhawi S, and Sacks D (2008). Quantification of the infectious dose of Leishmania major transmitted to the skin by single sand flies. Proc. Natl. Acad. Sci. USA 105, 10125–10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutoulaki A, Langley M, Sloan AJ, Aeschlimann D, and Wei XQ (2010). TNFα and TGF-β1 influence IL-18-induced IFNγ production through regulation of IL-18 receptor and T-bet expression. Cytokine 49, 177–184. [DOI] [PubMed] [Google Scholar]
- Kurey I, Kobets T, Havelková H, Slapnicková M, Quan L, Trtková K, Grekov I, Svobodová M, Stassen AP, Hutson A, et al. (2009). Distinct genetic control of parasite elimination, dissemination, and disease after Leishmania major infection. Immunogenetics 61, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao S, Herman LM, and Ma X (2003). Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-γ-primed IL-12 production by IFN regulatory factor 1. J. Exp. Med 198, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus PJ, and Garcia KC (2008). The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J. Mol. Biol 382, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CA, and Singh NJ (2020). Manipulating the TCR signaling network for cellular immunotherapy: Challenges & opportunities. Mol. Immunol 123, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P, and Kamala T (2011). Tissue-based class control: The other side of tolerance. Nat. Rev. Immunol 11, 221–230. [DOI] [PubMed] [Google Scholar]
- Müller U, Köhler G, Mossmann H, Schaub GA, Alber G, Di Santo JP, Brombacher F, and Hölscher C (2001). IL-12-independent IFN-γ production by T cells in experimental Chagas’ disease is mediated by IL-18. J. Immunol 167, 3346–3353. [DOI] [PubMed] [Google Scholar]
- Nicolas L, Sidjanski S, Colle JH, and Milon G (2000). Leishmania major reaches distant cutaneous sites where it persists transiently while persisting durably in the primary dermal site and its draining lymph node: A study with laboratory mice. Infect. Immun 68, 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725. [DOI] [PubMed] [Google Scholar]
- Park AY, Hondowicz BD, and Scott P (2000). IL-12 is required to maintain a Th1 response during Leishmania major infection. J. Immunol 165, 896–902. [DOI] [PubMed] [Google Scholar]
- Reitberger S, Haimerl P, Aschenbrenner I, Esser-von Bieren J, and Feige MJ (2017). Assembly-induced folding regulates interleukin 12 biogenesis and secretion. J. Biol. Chem 292, 8073–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, and Fasel N (2018). How to master the host immune system? Leishmania parasites have the solutions!. Int. Immunol 30, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks D, and Noben-Trauth N (2002). The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol 2, 845–858. [DOI] [PubMed] [Google Scholar]
- Sartori A, Oliveira MA, Scott P, and Trinchieri G (1997). Metacyclogenesis modulates the ability of Leishmania promastigotes to induce IL-12 production in human mononuclear cells. J. Immunol 159, 2849–2857. [PubMed] [Google Scholar]
- Sawant DV, Hamilton K, and Vignali DA (2015). Interleukin-35: Expanding its job profile. J. Interferon Cytokine Res 35, 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P, Natovitz P, Coffman RL, Pearce E, and Sher A (1988). Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J. Exp. Med 168, 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha X, Meng S, Li X, Xi H, Maddaloni M, Pascual DW, Shan H, Jiang X, Wang H, and Yang XF (2015). Interleukin-35 inhibits endothelial cell activation by suppressing MAPK-AP-1 pathway. J. Biol. Chem 290, 19307–19318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, et al. (2014). IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D, Jabs EW, Warrington JA, Li X, Lasota J, LaForgia S, Kelleher K, Huebner K, Wasmuth JJ, and Wolf SF (1992). Assignment of genes encoding a unique cytokine (IL12) composed of two unrelated subunits to chromosomes 3 and 5. Genomics 14, 59–62. [DOI] [PubMed] [Google Scholar]
- Sypek JP, Chung CL, Mayor SE, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, and Schaub RG (1993). Resolution of cutaneous leishmaniasis: Interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med 177, 1797–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait Wojno ED, Hunter CA, and Stumhofer JS (2019). The immunobiology of the interleukin-12 family: Room for discovery. Immunity 50, 851–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Tubo NJ, and Jenkins MK (2014). TCR signal quantity and quality in CD4+ T cell differentiation. Trends Immunol. 35, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wege AK, Florian C, Ernst W, Zimara N, Schleicher U, Hanses F, Schmid M, and Ritter U (2012). Leishmania major infection in humanized mice induces systemic infection and provokes a nonprotective human immune response. PLoS Negl. Trop. Dis 6, e1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon C, Johnston SC, Tang J, Stahl M, Tobin JF, and Somers WS (2000). Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 19, 3530–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data from the GTEx Portal. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 07/15/2020. Accession for these datasets are listed in the Key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Brilliant Violet 605 anti-mouse CD4 clone GK1.5 | Biolegend | Cat# 300555; RRID:AB_2564390 |
| eFluor 450 anti-mouse CD4 clone GK1.5 | eBioscience | Cat# 48–0041-82; RRID:AB_10718983 |
| Brilliant Violet 650 anti-mouse CD8b clone H35–17.2 | BD Biosciences | Cat# 740552; RRID:AB_2740253 |
| FITC anti mouse CD8b clone H35–17.2 | eBioscience | Cat# 11–0080-85; RRID:AB_657775 |
| PE/Cyanine7 anti-mouse/human CD11b clone M1/70 | Biolegend | Cat# 100215; RRID:AB_312798 |
| eFluor 450 anti-mouse CD11c clone N418 | eBioscience | Cat# 48–0114-80; RRID:AB_1548665 |
| PE anti-mouse/human CD44 clone IM7 | BD PharMingen | Cat# 553134; RRID:AB_394649 |
| PE-Cy7 anti-mouse/hman CD44 clone IM7 | Biolegend | Cat# 103030; RRID:AB_830787 |
| PerCP/Cyanine5.5 anti-mouse/human CD44 clone IM7 | Biolegend | Cat# 103032; RRID:AB_2076204 |
| Alexa Fluor 488 anti-mouse CD45.1 clone A20 | Biolegend | Cat# 110718; RRID:AB_492862 |
| APC anti-mouse CD45.2 clone 104 | eBioscience | Cat# 17–0454-82; RRID:AB_469400 |
| Alexa Fluor 700 anti-mouse CD62L clone MEL-14 | Biolegend | Cat# 104426; RRID:AB_493719 |
| PE/Cyanine7 anti-mouse CD62L clone MEL-14 | Biolegend | Cat# 104417; RRID:AB_313102 |
| APC anti-mouse IFNγ clone XMG1.2 | eBioscience | Cat# 17–7311-82; RRID:AB_469504 |
| FITC anti-mouse IFNγ clone XMG1.2 | BD PharMingen | Cat# 554411; RRID:AB_395375 |
| PE anti-mouse IL-4 clone 11B11 | Biolegend | Cat# 504104; RRID:AB_315318 |
| PerCP/Cyanine5.5 anti-mouse IL-10 clone JES5–16E3 | Biolegend | Cat# 505028; RRID:AB_2561523 |
| APC anti-mouse IL-17A clone TC11–18H10.1 | Biolegend | Cat# 506916; RRID:AB_536018 |
| PE anti-mouse MHC Class II (IA/IE) clone M5/114.15.2 | eBioscience | Cat# 12–5321-82; RRID:AB_465928 |
| Brilliant Violet 421 anti-mouse T-bet clone 4B10 | Biolegend | Cat# 644815; RRID:AB_10896427 |
| PE-Cy7 anti-mouse T-bet clone 4B10 | eBioscience | Cat# 12–5825-82; RRID:AB_925761 |
| Brilliant Violet510 anti-mouse TCRb clone H57–597 | Biolegend | Cat# 109233; RRID:AB_2562349 |
| CyChrome anti-mouse TCRb clone H57–597 | BD PharMingen | Cat# 553173; RRID:AB_394685 |
| PE anti-mouse TCR-vb8.3 clone 1B3.3 | BD PharMingen | Cat# 553664; RRID:AB_394980 |
| Biotin anti-mouse NK1.1 clone PK136 | eBioscience | Cat# 13–5941-82; RRID:AB_466804 |
| Biotin anti-mouse CD11c clone N418 | eBioscience | Cat# 13–0114-82; RRID:AB_466363 |
| Biotin anti-mouse CD8a clone 53–6.7 | eBioscience | Cat# 13–0081-82; RRID:AB_466346 |
| Biotin anti-mouse MHC class II clone M5/114.15.2 | eBioscience | Cat# 13–5321-82; RRID:AB_466662 |
| Purified anti-mouse CD16/CD32 clone 2.4G2 | BD PharMingen | Cat# 553141; RRID:AB_394656 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LCMV GP61–80 | AnaSpec | Cat# AS-64851 |
| Collagenase D | Roche | Cat# 11 088 858 011 |
| Propidium Iodide Solution | Sigma-Aldrich | Cat# P4864 |
| Lipopolysaccharides from E. coli | Sigma-Aldrich | Cat# L2880 |
| Medium 199 | GIBCO | Cat# 12350039 |
| RPMI 1640 | GIBCO | Cat# 11875119 |
| Foundation Fetal Bovine Serum | Gemini Bio | Cat# 900–108 |
| HEPES | GIBCO | Cat# 15–630–080 |
| Adenine | Sigma-Aldrich | Cat# A2786 |
| Biotin | Sigma-Aldrich | Cat# B4501 |
| Hemin | Sigma-Aldrich | Cat# 51280 |
| PBS | Quality Biological | Cat# 119–069-101 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17–1440-02 |
| Lectin from Arachis hypogaea (peanut) | Sigma-Aldrich | Cat# L6135 |
| Sodium Pyruvate | GIBCO | Cat# 11360070 |
| Antibiotic-antimycotic | GIBCO | Cat# 15240062 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Brefeldin A | eBioscience | Cat# 00–4506-51 |
| EDTA | Quality Biological | Cat# 351–027-101 |
| Sodium azide | Sigma-Aldrich | Cat# S2002 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat# P8139 |
| Ionomycin calcium salt | Sigma-Aldrich | Cat# I0634 |
| Triethanolamine | Sigma-Aldrich | Cat# 90278 Critical commercial assays |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse IL-17A (homodimer) ELISA Ready-SET-Go! | eBioscience | Cat# 88–7371 RRID:AB_2575104 |
| Mouse IFN gamma ELISA Ready-SET-Go! | eBioscience | Cat# 88–7314 RRID:AB_2575069 |
| Mouse IL-2 Quantikine ELISA Kit | R&D Systems | Cat# M2000 |
| Mouse IL-4 Quantikine ELISA Kit | R&D Systems | Cat# M4000B |
| Mouse IL-12 p40 Quantikine ELISA Kit | R&D Systems | Cat# M1240 |
| Mouse IL-12 p70 Quantikine ELISA Kit | R&D systems | Cat# M1270 |
| Dynabeads M-280 Streptavidin | Invitrogen | Cat# 11205D |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554722 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00–5523-00 |
| LIVE/DEAD Fixable Near-IR | Invitrogen | Cat# L10119 |
| Ghost Dye Red 780 | Tonbo Biosciences | Cat# 13–0865 |
| BCA Protein Assay Kit | Pierce | Cat# 23225 |
|
Deposited data | ||
| GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2) | GTEx | https://www.gtexportal.org/home/gene/IL12B |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| L. major-RFP | Kimblin et al., 2008 | Kimblin et al., 2008 |
| Mouse: B6.129S1-Il12btm1Jm/J | The Jackson Laboratory | Cat# 002693 |
| Mouse: IL-12p40−/− CD45.1+ | Bred in-house | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse B6.SJL-PtprcaPepcb/BoyJ | The Jackson Laboratory | Cat# 002014 |
| Mouse: B6NTac | Taconic Biosciences | Cat# B6 |
| Mouse: B6.SJL-Ptprca/BoyAiTac | Taconic Biosciences | Cat# 4007 |
| Mouse: B6 1x2 | Bred in-house | N/A |
| Mouse: B6.129S1-Il12atm1Jm/J | The Jackson Laboratory | Cat# 002692 |
| Mouse: B6.Cg-Ptprca Pepcb Tg(TcrLCMV)1Aox/PpmJ | The Jackson Laboratory | Cat# 030450 |
|
Software and algorithms | ||
| FlowJo (v10.6.2) | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism 7.05 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| GTEx Portal | Broad Institute of MIT and Harvard | http://www.gtexportal.org/home/index.html |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.