

Abstract
A new series of pyrimidine and pyridine diamines was designed as dual binding site inhibitors of cholinesterases (ChEs), characterized by two small aromatic moieties separated by a diaminoalkyl flexible linker. Many compounds are mixed or uncompetitive acetylcholinesterase (AChE) and/or butyrylcholinesterase (BChE) nanomolar inhibitors, with compound 9 being the most active on Electrophorus electricus AChE (EeAChE) (Ki = 0.312 μM) and compound 22 on equine BChE (eqBChE) (Ki = 0.099 μM). Molecular docking and molecular dynamic studies confirmed the interaction mode of our compounds with the enzymatic active site. UV–vis spectroscopic studies showed that these compounds can form complexes with Cu2+ and Fe3+ and that compounds 18, 20, and 30 have antioxidant properties. Interestingly, some compounds were also able to reduce Aβ42 and tau aggregation, with compound 28 being the most potent (22.3 and 17.0% inhibition at 100 μM on Aβ42 and tau, respectively). Moreover, the most active compounds showed low cytotoxicity on a human brain cell line and they were predicted as BBB-permeable.
Keywords: acetylcholinesterase inhibitors, butyrylcholinesterase inhibitors, multifunctional compounds, amyloid aggregation, tau aggregation, metal chelation, antioxidant
Introduction
Alzheimer’s disease (AD) is one of the most serious and prevalent neurodegenerative diseases; it accounts for 50–75% of dementia cases in humans, currently affecting 44 million people worldwide,1 and causes huge social economic losses.2 In 2015, the World Alzheimer Report estimated over 9.9 million new cases of dementia each year (approximately one new case every 3 s), which allows one to predict a population of 70 million by 2030 that could rise to 130 million by 2050.3 AD begins with short-term memory loss followed by a progressive decline in memory and cognitive ability associated with severe behavioral abnormalities, such as irritability, anxiety, and depression, and the outcome is always fatal.
Although the precise pathogenetic mechanisms of AD remain to be elucidated, several factors appear to play key roles in the onset and progression of the AD, and they comprise the following: the cholinergic deficit, with reduced levels of acetylcholine (ACh) and many cholinergic markers; the β-amyloid peptide (Aβ) deposition; the hyperphosphorylation and the deposition of the tau protein; the dysregulation of energy metabolism and the increase in oxidative stress; the dyshomeostasis of biometals; and neuroinflammation.4 The cholinergic hypothesis, the first approach developed to describe the pathophysiology of AD, is based on three main evidences observed in AD patients: (i) a severe neurodegeneration of the nucleus basalis of Meynert, the main source of cortical cholinergic innervation; (ii) a severe depletion of the presynaptic cholinergic markers, such as choline acetyl transferase (ChAT); and (iii) the cholinergic antagonist drugs induce memory impairment and cognitive deficits that are alleviated by cholinergic agonists.5−8
These evidences have led to the therapeutic use in AD of cholinesterase inhibitors (ChEIs), able to restore cholinergic tone by blocking the cholinesterase (ChE) activity and consequently increasing the availability of ACh at the synaptic cleft.
To date, the ChEIs remain the primary therapy for AD, with donepezil, galantamine, and rivastigmine, currently approved by the Food and Drug Administration (FDA), along with the NMDA receptor antagonist memantine.9 Unfortunately, these drugs have shown limited clinical results, with short-lasting positive effects and the lack of ability to stop the progression of the disease. The absence of decisive and long-term effects stimulates researchers to develop new therapeutic strategies with the objective of restoring cerebral cholinergic activity, as the degeneration of the cortical cholinergic projection represents a key irreversible event in the progression of AD.
At the neuronal level, there are two types of ChEs: acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). AChE activity is dominant in the healthy brain (80%), while BChE exerts a supportive role. However, with the progression of the disease, brain levels of AChE decline to approximately 60%, while those of BChE increase to 120% of normal levels, thus suggesting for the latter a major role in the severe forms of the disease and the possibility of considering it an adequate therapeutic target of AD.10,11
AChE and BChE have 65% amino acid sequence identity; they possess a catalytic active site (CAS), located at the bottom of a 20 Å gorge, constituted by a catalytic triad: Ser203, His447, and Glu202 in human AChE (hAChE) and Ser198, His438, and Glu197 in human BChE (hBChE). An anionic subsite is also present that is constituted by Trp86 in hAChE and Trp82 in hBChE, which bind the quaternary ammonium of the ACh moiety by means of cation−π interaction.12,13 These enzymes exhibit an allosteric modulation site, the peripheral anionic site (PAS), located at the entrance of the active site gorge, which in hAChE consists of five residues, Tyr72, Asp74, Tyr124, Trp286, and Tyr341, while in hBChE is constituted by Tyr332 and Asp70.14,15 Besides its role in the modulation of the catalytic activity, PAS has been reported to be involved in the proaggregating action of AChE toward the Aβ peptide, another important actor in the onset and progression of AD.16,17
The growing scientific evidences highlighting the close correlation between the severe cholinergic depletion and the other pathophysiological events observed in AD, particularly with the abnormal Aβ and tau cascade, give a new interest to the development of new ChEIs for the treatment of AD and, in particular, toward multitarget compounds able to act simultaneously at more levels on the different mechanisms involved in the pathogenesis of AD.18 Many reviews available in the scientific literature summarize the main research lines followed in the development of multitarget compounds for the therapy of AD in both the academic and industrial fields. The majority of articles and patents report studies on dual binding site ChEIs that are able to interact with both CAS and PAS of these enzymes and are endowed with the activity on one or more additional targets, such as a second neurotransmitter system (i.e., serotonergic or monoaminergic), or Aβ and tau production and deposition, the reduction of oxidative stress, or biometal dyshomeostasis.19−21
In our previous studies, we synthesized and in vitro evaluated new series of dual binding site ChEIs, characterized by two small aromatic moieties separated by various functionalized linkers, some of which resulted in nanomolar mixed or uncompetitive inhibitors of both AChE and BChE, able to bind CAS and PAS, able to reduce Aβ aggregation, or endowed with metal chelating properties.22,23
Based on these considerations and as a development of previous works, we decided to study a new series of pyrimidine and pyridine diamine derivatives and evaluate them both in vitro and in cellulo as potential ChEIs endowed with chelating and antioxidant activities as well as direct anti-amyloid aggregation properties, with the aim to develop new multifunctional compounds for AD. These molecules were designed as dual binding site inhibitors, based on the structure of the ChE enzymatic pocket, inserting two small aromatic groups separated by an aliphatic linker (Figure 1). The aromatic moieties are potentially able to interact with the aromatic amino acids located adjacent to the CAS and in the PAS of the enzymes by means of π–π interactions, while the aliphatic linker covers the distance between CAS and PAS and gives flexibility to the compounds. Due to the difficulty to predict the conformational behavior of linear alkyl chains, different length linkers were used, in particular with five or six methylene units. Within the chain, a protonable amine group, both acceptor and donor of hydrogen bond and able to establish cationic−π interactions with the aromatic residues of the enzymes, was inserted to promote interactions with the amino acids of the enzyme gorge. A 2-amino-pyrimidinic or 2-amino-pyridinic moiety was introduced as one of the two aromatic groups since the presence of the two adjacent nitrogen atoms could confer chelating activity to the compounds. On the other side of the aliphatic chain, different aromatic groups were chosen, with the purpose of acquiring further information about the structural requirements for targeting ChEs. Among these groups, phenolic or catecholic rings were also selected to enhance chelating properties and to confer direct antioxidant activity.
Figure 1.

Rational design of the 2-amino-pyrimidine or 2-amino-pyridine derivatives.
Results and Discussion
Chemistry
General Procedure for the Synthesis of Pyrimidine Diamine Derivatives 9–22
The pyrimidine diamine derivatives 9–22 were synthetized following the procedures described in Scheme 1. Initially, the intermediates 3 and 4 were synthesized through two reaction steps (first method, GP-A and procedure 1 or 2 of GP-B in the Experimental Section); subsequently, intermediate 4 was also obtained directly through a single reaction (second method, procedure 3 of GP-B in the Experimental Section). Following the first synthetic method, equimolar amounts of 2-chloropyrimidine, N-Boc-1,5-diaminopentane, or N-Boc-1,6-diaminohexane and triethylamine (TEA) were reacted in methanol at reflux overnight to give compounds 1 or 2, which were purified by column chromatography on a silica gel. Then, intermediates 1 and 2 were deprotected from Boc by treatment with a high excess of trifluoroacetic acid (TFA) in anhydrous dichloromethane to obtain compounds 3 and 4, which were used either as trifluoroacetic salts or as free amines. According to the second synthetic method, intermediate 4 was synthetized directly by the reaction between 2-chloropyrimidine and an excess of 1,6-diaminohexane in the presence of TEA in methanol at reflux overnight. The excess of diamine allowed one to obtain mainly the product of interest, in which only one amino group reacted with 2-chloropyrimidine. The reaction was monitored by ESI-MS, and the crude residue was purified by column chromatography on a silica gel. The second method proved to be more advantageous than the first, as through a single step it was possible to synthetize the desired pyrimidine intermediate with a higher yield.
Scheme 1. Synthetic Route to Pyrimidine Diamine 9–22 Derivatives.

Reagents and conditions: (a) TEA (1 equiv), MeOH, reflux, 20 h; (b) TFA (20 equiv), dry CH2Cl2, room temp, 3.5 h; (c) opportune aldehyde (1 equiv), dry CH2Cl2, dry K2CO3 (for 9–16 and 21) or activated molecular sieves (for 17–20 and 22), room temp, 12 h; and (d) NaBH4 (3 equiv), MeOH, room temp, 2 h.
Equimolar amounts of amine intermediates 3 or 4 and the appropriate aldehyde were dissolved in dry dichloromethane in the presence of potassium carbonate or molecular sieves as desiccant agents. The choice of the suitable drying agent was based on the starting reagents: when compound 3 or 4 was used as trifluoroacetic salt, potassium carbonate was utilized both as a desiccant, either as a base to deprotonate the amino groups of the intermediates; when the reaction was carried out using aldehydes containing acidic groups susceptible to react with potassium carbonate (such as indole −NH or phenol −OH), molecular sieves were selected as desiccant agents. These reactions were monitored by IR spectroscopy to confirm the imine formation, with the appearance of a stretching absorption band around 1640 cm–1 and the disappearance of the aldehyde band at about 1700 cm–1. The obtained imine intermediates were reduced by treatment with sodium borohydride in methanol, and after the reaction, the excess of sodium borohydride was decomposed by the addition of water or 1 M HCl. The addition of hydrochloric acid was necessary to avoid the oxidation of phenolic derivatives at basic pH.
The residues obtained were purified by column chromatography on a silica gel and/or by crystallization. Moreover, the structures of final compounds were confirmed by spectroscopic analysis; as an example, in the 1H NMR spectra, the appearance of the methylene singlet between 3.89 and 3.63 ppm was observed. The detailed synthetic procedures and the analytical and spectroscopic data of synthesized compounds are reported in the Experimental Section and agree with the proposed structures.
General Procedure for the Synthesis of Pyridine Diamine Derivatives 23–33
Pyridine diamine derivatives 23–33 were synthetized following the procedures described in Scheme 2. Intermediates 5 and 6 were obtained by Ullmann coupling catalyzed by CuI and 2-isobutyrylcyclohexanone, as described by Shafir and Buchwald.24 A Schlenk flask was charged with N-Boc-1,5-diaminopentane or N-Boc-1,6-diaminohexane and solid reactants (CuI and Cs2CO3), and then it was evacuated and backfilled with N2. Under a counter flow of N2, 2-iodopyridine, dimethylformamide (DMF), and finally 2-isobutyrylcyclohexanone were added. The reaction was monitored by ESI-MS, and the best yield was obtained stirring the mixture at 40 °C for 21 h. Purification by column chromatography on a silica gel led to the isolation of the products of interest with quantitative yield. Then, intermediate 5 and 6 were deprotected from Boc by treatment with a high excess of TFA in anhydrous dichloromethane to obtain compounds 7 and 8, which were used either as trifluoroacetic salts or as free amines. Differently from what has been said for the pyrimidine intermediates, in this case, it was not possible to synthetize compounds 5 and 6 directly through a single reaction between 2-iodopyridine and an excess of the opportune diamine. In fact, in this way, the product in which both amino groups reacted with 2-iodopyridine was mainly obtained.
Scheme 2. Synthetic Route to Pyridine Diamine 23–33 Derivatives.

Reagents and conditions: (a) CuI (0.05 equiv), Cs2CO3 (2 equiv), 2-isobutyrylcyclohexanone (0.2 equiv), DMF, N2, 40 °C, 21 h; (b) TFA (20 equiv), dry CH2Cl2, room temp, 3.5 h; (c) opportune aldehyde (1 equiv), dry CH2Cl2, dry K2CO3 (for 23–28 and 33) or activated molecular sieves (for 29–32), room temp, 12 h; and (d) NaBH4 (3 equiv), MeOH, room temp, 2 h.
Then, intermediates 7 and 8 were reacted with the appropriate aldehydes, and the imines obtained were reduced with sodium borohydride, as previously described for the pyrimidine derivatives. The residues obtained were purified by column chromatography on a silica gel or by crystallization. The structures of final compounds were confirmed by spectroscopic analysis; as an example, in the 1H NMR spectra, the appearance of the methylene singlet between 3.90 and 3.63 ppm was observed. The detailed synthetic procedures and the analytical and spectroscopic data of synthesized compounds are reported in the Experimental Section and agree with the proposed structures.
Enzymatic Assays
On the synthesized compounds, enzymatic inhibition studies were carried out toward Electrophorus electricus AChE (EeAChE) and equine BChE (eqBChE) according to Ellman’s spectrophotometric method.25 Initially, for each compound, the percentages of inhibition were determined at the inhibitor concentration equal to 9 μM and, for the most potent compounds, also at 900 and 90 nM in the presence of 0.0833 U/mL of the enzyme and 100 μM of acetylthiocholine.
Based on the results illustrated in Table 1, related to pyrimidine diamine derivatives, there are no significant differences between the percentages of inhibition toward EeAChE of the compounds having an aliphatic linker with five methylene units (9–12) compared to the corresponding derivatives with six methylene units (13, 14, 16, and 21). On the other hand, toward eqBChE, the compounds having a five-methylene chain show a lower inhibitory potency compared to the six methylene derivatives. As a consequence of this observation, for the synthesis of the subsequent compounds, we decided to use an aliphatic linker with six methylene units.
Table 1. Inhibition of EeAChE and eqBChE Activities by Pyrimidine Diamine Derivatives 9–22.


Data are the average of three replicates.
nd stands for not determined.
Among pyrimidine diamine derivatives, the most potent compounds on EeAChE, with approximately 90% of inhibition at 9 μM and between 40 and 60% at 900 nM, are 9 and 13, both with an unsubstituted phenyl ring on one side of the aliphatic chain, 19, with a 3-hydroxyphenyl ring, and 20, having a 2,3-dihydroxyphenyl ring. The insertion of a halogen atom in the para position of the phenyl ring (compounds 10, 14, and 15) as well as the replacement of the phenyl with the 3,4,5-trimethoxyphenyl (17) or 2,6-dichloropyridine ring (12 and 21) causes a drastic reduction of the inhibitory potency on EeAChE; the replacement of the phenyl with the 2-methoxyphenyl (11 and 16), 3-methoxy-4-hydroxyphenyl (18), or indole (22) ring reduces the percentages of inhibition toward EeAChE from 90% to 60–70% at an inhibitor concentration of 9 μM.
Generally, these pyrimidine diamine derivatives have low inhibitory potency toward eqBChE (8–45% at 9 μM), with the exception of compound 18, with the 3-methoxy-4-hydroxyphenyl ring, which shows 84% inhibition at 9 μM and 44% at 900 nM, and 22, with the indole group, having a 94% inhibition at 9 μM and 65% at 900 nM.
Among pyridine diamine derivatives, whose data are reported in Table 2, the compounds having an aliphatic chain with five methylene units (23 and 24) show a lower inhibitory potency toward both enzymes compared to the corresponding derivatives with a six-methylene chain (25 and 26). Generally, pyridine diamine derivatives have similar or lower percentages of inhibition on EeAChE compared to the corresponding pyrimidine derivatives (Tables 1 and 2) with the exception of 29 and 33, having the 3,4,5-trimethoxyphenyl or 2,6-dichloropyridine ring, respectively. On the contrary, pyridine derivatives are more potent on eqBChE than the corresponding pyrimidine compounds. Among pyridine derivatives, the most potent compound on EeAChE is 25, having an unsubstituted phenyl ring on one side of the aliphatic chain, with a 73% inhibition at 9 μM. Among the compounds with six methylene units, the insertion of a halogen atom in the para position on the phenyl ring (26 and 27) and the replacement of the phenyl with the 3,4,5-trimethoxyphenyl group (29) cause a reduction of the percent inhibition at 9 μM on EeAChE from 73% to 24–30%. Furthermore, the replacement of the phenyl with the 2-methoxyphenyl (28), 3-methoxy-4-hydroxyphenyl (30), or 2,3-dihydroxyphenyl (32) ring reduces the inhibition at 9 μM to about 60%, while the replacement with the 3-hydroxyphenyl (31) or 2,6-dichlropyridine (33) ring reduces inhibition to about 40%. On eqBChE, the most potent compound is 30, with the 3-methoxy-4-hydroxyphenyl ring, that shows 91% inhibition at 9 μM and 51% at 900 nM.
Table 2. Inhibition of EeAChE and eqBChE Activities by Pyridine Diamine Derivatives 23–33.


Data are the average of three replicates.
nd stands for not determined.
For a selection of compounds, among the most potent as inhibitors of EeAChE and/or eqBChE, the inhibition constant (Ki) and the corresponding inhibition mechanism were determined according to Dixon’s method,26 reporting in graph the reciprocal of the hydrolysis rate vs the inhibitor concentrations at a fixed concentration of the substrate. The recorded data were analyzed with the enzyme kinetic module of SigmaPlot to find the best fitting model of inhibition using the linear regression analysis. The reference kinetic models used in the regression analysis were competitive, noncompetitive, uncompetitive, and mixed. Each determination was repeated five times, and incorrect values were discarded to reduce the standard deviation within the limit of 5%. In this way, the regression lines obtained have a linear regression coefficient (R2) higher than 0.95. Dixon’s plots of all tested compounds are reported in the Supporting Information (Figures S1–S12).
The tested compounds revealed a mixed or uncompetitive inhibition mechanism toward EeAChE and eqBChE, with Ki according to the order of low micromolar or nanomolar (Table 3). The mixed inhibition mechanism might suggest an interaction of the compound with both CAS and PAS of the enzyme and a possible involvement in the inhibition of Aβ plaque formation induced by AChE. For compounds inhibiting the enzymes with a mixed mechanism, Table 3 also reports the values of α, which indicate whether the inhibitor preferentially binds to the free enzyme (α > 1) or to the enzyme–substrate complex (α < 1).
Table 3. Inhibition Mechanisms Determined on EeAChE and eqBChE, Inhibition Constants (Ki), and α Values.
|
EeAChE |
eqBChE |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cmpd | mechanism | Ki ± SDa (μM) | R2 | α | cmpd | mechanism | Ki ± SDa (μM) | R2 | α | |
| 9 | mixed | 0.312 ± 0.108 | 0.982 | 7.5 | 18 | mixed | 3.034 ± 0.604 | 0.986 | 7.3 | |
| 13 | mixed | 0.426 ± 0.132 | 0.991 | 0.3 | 22 | mixed | 0.099 ± 0.071 | 0.990 | 10 | |
| 19 | uncomp. | 0.509 ± 0.018 | 0.992 | 25 | mixed | 2.373 ± 0.304 | 0.992 | 2.2 | ||
| 23 | mixed | 0.743 ± 0.316 | 0.983 | 2.2 | 26 | mixed | 3.465 ± 1.480 | 0.950 | 0.5 | |
| 25 | mixed | 0.995 ± 0.374 | 0.988 | 0.8 | 28 | mixed | 3.434 ± 0.701 | 0.988 | 0.8 | |
| 28 | mixed | 1.323 ± 0.622 | 0.990 | 1.2 | 30 | mixed | 1.105 ± 0.189 | 0.983 | 1.4 | |
Each data was obtained with three substrate and five inhibitor concentrations; each measurement was carried out in five replicates (see the Experimental Section).
The pyrimidine amine compounds are more potent on EeAChE than the pyridine analogs, as confirmed by Ki values of 9 and 13 (pyrimidine derivatives with the phenyl ring on one side of the aliphatic chain, respectively, with five or six methylene units) compared to Ki values of 23 and 25 (pyridine derivatives with the phenyl ring on one side of the aliphatic chain, respectively, with five or six methylene units). All the compounds tested on EeAChE show a mixed inhibition mechanism, with the exception of 19, which acts with an uncompetitive mechanism. The most potent inhibitors of EeAChE are compounds 9 and 13, with Ki values of 312 ± 108 and 426 ± 132 nM, respectively.
Generally, pyridine amine derivatives are more potent on eqBChE than the corresponding pyrimidine derivatives. All the compounds tested on eqBChE show a mixed inhibition mechanism. Compound 22 (a pyrimidine derivative with the indole group on one side of the aliphatic chain) is the most potent inhibitor of eqBChE, with Ki equal to 99 ± 71 nM.
For the pyrimidine amine derivative 20, with a 2,3-dihydroxyphenyl ring on one side of the aliphatic chain, it was necessary to determine the IC50 on EeAChE (the concentration of inhibitor required to inhibit 50% of the enzyme) instead of the Ki due to the instability of this molecule in the aqueous solution for the time required for Ki determination. The IC50 value was obtained by plotting the percentages of inhibition toward EeAChE vs the concentration of inhibitor expressed in a logarithmic scale at a fixed substrate concentration (100 μM). The recorded data were analyzed with the enzyme kinetic module of SigmaPlot. All measurements were replicated three times, and the IC50 value obtained was confirmed by repeating the experiment twice. For compound 20, the IC50 on EeAChE is 622 ± 30 nM, as shown in Table 4. The IC50 plot of the tested compound is reported in the Supporting Information (Figure S13).
Table 4. IC50 on EeAChE for Compound 20 and on Both ChEs for Tacrine and Donepezil, Used as the Reference Standard.
| cmpd | EeAChE, IC50 ± SE (nM) | eqBChE, IC50 ± SE (nM) |
|---|---|---|
| 20 | 621.9 ± 29.5 | nda |
| tacrine | 41.0 ± 4.9 | 3.7 ± 0.5 |
| donepezil | 16.2 ± 1.8 | 1727 ± 300 |
nd stands for not determined.
Molecular Docking Studies
Molecular modeling analyses were performed to provide structural insights on the binding mode of the investigated compounds into the hAChE and hBChE catalytic pocket. For the current study, the X-ray crystallographic structures of the hAChE in complex to donepezil (PDB code:4EY7)13 and the hBChE in complex to the naphthamide derivative (PDB code:5NN0)27 were used. As there is no crystal structure of eqBChE in the protein data bank (PDB), hBChE was used for the computational study. Indeed, the sequence of eqBChE derived from the Uniprot Database shares 90% sequence identity with that of the adopted hBChE (PDB code:5NN0), above all compared residues of the binding site (Figure S14). Regarding AChE, even if the apo-crystal structures of EeAChE are present in PDB, they cannot be considered as good quality models. After the alignment of EeAChE (PDB code:1C2O) with the adopted hAChE structure (PDB code:4EY7), 94.7% of the identity of the aligned sequences within a range of 7 Å from the binding site is obtained (Figure S15). According to a previous work23 and the criteria for the identification of reliable complexes reported in the literature,28 the selected models were considered as a good starting point for in silico investigation.
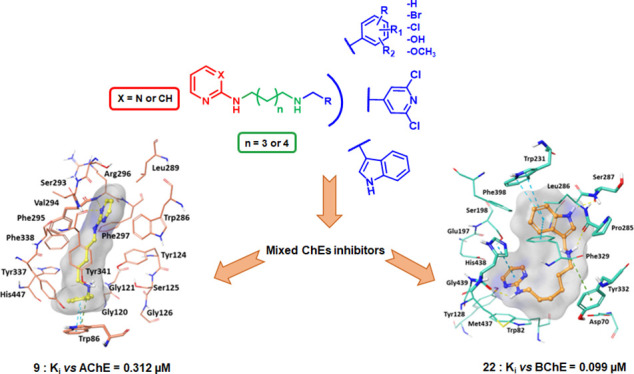
Our docking protocol was validated by docking co-crystallized ligands into the binding site. Root mean square deviation (RMSD) values between the native pose of hAChE and hBChE ligands and the related best redocked conformations were found to be 0.12 and 0.96 Å, respectively, thus revealing the reliability of the docking protocol (Figure S16). According to previously published studies,23,29 no linear correlation between docking score and the experimental data is expected (Table S1). Nevertheless, the obtained docking poses for the analyzed compounds were comparable with the binding mode of donepezil into AChE (Figure S17). It is widely known that donepezil is a dual binding site inhibitor, i.e., engaging simultaneously π–cation and π–π interactions with Trp86 and Trp286 of the CAS and the PAS, respectively. Moreover, the ketone group of the indanone ring of donepezil formed a hydrogen bond with Phe295 of the mid-gorge binding site. In detail, the CAS region is characterized by Trp86, Tyr119, Tyr124, Tyr133, Glu202, Ser203, Trp439, His447, and Tyr449, and the PAS region is composed of Tyr72, Asp74, Thr75, Trp286, Leu289, Tyr341, and Val365 residues. The mid-gorge pocket of hAChE consists of Leu76, Phe295, Arg296, Phe297, Tyr337, and Phe338, and it is about 20 Å deep by an average of 13 Å wide.
Particularly, it is shown that, regardless of the aliphatic chain length, the protonated amine group interacted with Trp86 of the anionic site of CAS for all the docked compounds except for compound 29, which interacted with Tyr341 and Tyr337 of the mid-gorge site.
Both 2-amino-pyrimidinic and 2-amino-pyridinic moieties exhibited hydrogen bonds with Phe295 and Arg296 in the mid-gorge pocket, except for compound 13, whose pyrimidine cycle was more exposed to the solvent. The introduction of the other aromatic ring contributed to the stabilization of the compounds in the CAS region. Molecular recognition studies revealed the major binding interactions between our most potent inhibitors with hAChE (compounds 9, 13, 19, and 20) (Figure 2) and hBChE (compounds 22 and 30) (Figure 3).
Figure 2.

Best docking pose of compounds: (a) 9, (b) 13, (c) 19, and (d) 20 into the hAChE active site shown as wire representation. Most relevant interacting residues are displayed as thin salmon tubes. Compounds 9, 13, 19, and 20 are illustrated in yellow, green, pink, and blue carbon ball and stick representation, respectively. Hydrogen bonds, π–π interactions, and π–cation contacts are, respectively, represented by yellow, blue, and green dotted lines.
Figure 3.

Best docking pose of compounds: (a) 22 and (b) 30 into hBChE active site shown as wire representation. The most relevant interacting residues are displayed as thin light green tubes. Compounds 22 and 30 are illustrated in orange and pink carbon ball and stick representation, respectively. Hydrogen bonds, π–π interactions, and π–cation contacts are, respectively, represented by yellow, blue, and green dotted lines.
The compound 9 pyrimidine group formed two hydrogen bonds with Phe295 and Arg296 (Figure 2a). The phenyl ring also created a π–π stacking interaction with Trp86, while the protonated amine contributed to the accommodation at the CAS and PAS sites by means of a π–cation with Trp86 and a salt bridge with Asp74, respectively. The aliphatic extension of one methylene unit in compound 13 (Figure 2b) induced a destabilization of the hydrogen bonds between the pyrimidine portion and mid-gorge amino acids. Conversely, this orientation allowed a π–cation interaction between the protonated amine group and the indole ring of Trp86, with the loss of the interaction with Asp74 observed for compound 9, still maintaining the π–π interaction between the phenyl group and Trp86.
Concerning compounds 19 and 20 (Figure 2c,d), they showed very similar docking poses orientating the protonated amine group toward Trp86 and interacting with Tyr133 and Glu202 through two and three hydrogen bonds with the phenolic and cathecolic group, respectively.
As regard hBChE, all the investigated compounds were able to accommodate into the binding cavity with a lower binding affinity with respect to that of hAChE due to the presence of more hydrophilic and charged residues in the PAS and mid-gorge site.
In detail, the pyrimidine moiety of compound 22 (Figure 3a) created a π–π interaction with His438, and this residue also engaged a hydrogen bond with the amine group. Furthermore, a π–cation and a hydrogen bond were formed between the ligand protonated amine and Tyr332 and Pro285, respectively. The indole moiety pointed to Trp231 in CAS and Phe329 in mid-gorge regions, providing also a hydrogen bond with Ser287. Compound 30 (Figure 3b) showed a different docking pose orientating the pyridine toward Trp231 and Phe329. It interacted with His438 by means of the hydroxyphenyl at position 4, while the amine group bound to the pyridine. Its 3-methoxy-4-hydroxyphenyl ring was able to engage a π–π stacking interaction with Trp82. With Asp70, belonging to the PAS region, that phenolic moiety established one hydrogen bond and the protonated amine group an additional salt bridge.
Molecular Dynamic (MD) Studies
The best docked poses of compounds 9, 13, 19, and 20 into binding pockets of hAChE and compounds 22 and 30 into that of hBChE were submitted to 250 ns of molecular dynamic (MD) simulations. The results of MDs were investigated in terms of stability of the complexes and conformational flexibility of hChEs in the presence of the promising inhibitors by monitoring the single contributions of hydrophobic, water-bridge, and hydrogen bonding interactions.
The stability of the hChEs complexes trajectories can be monitored by the RMSD of the protein’s backbone atoms from its initial to final conformation over an MD time of 250 ns (Figure 4). As shown, the RMSD plot indicated that all compounds in complex to hAChE (Figure 4a) and hBChE (Figure 4b) maintained overall stability during the whole MD, thus ensuring a good equilibrium for the systems. Particularly, compounds 9, 13, and 20 in complex to hAChE were associated with increased conformational stability with RMSD average values of 1.89, 2.25, and 2.04 Å, respectively, in comparison to compound 19 (2.60 Å). As to compounds 22 and 30 in complex to hBChE, a similar stability trend was observed, resulting in very low fluctuations and RMSD average values of 2.13 and 2.06 Å, respectively.
Figure 4.

(a) RMSD of compounds 9, 13, 19, and 20 and backbone atoms of hAChE. (b) RMSD of compounds 22 and 30 and backbone atoms of hBChE.
The compound 9 protonated amine group interacted mainly with the amino acids of both CAS and PAS regions of the hAChE pocket. Indeed, Asp74, Trp86, His447, and Tyr337 for 46, 53, 38, and 46% of the MD time were engaged, respectively. The pyrimidine ring also formed a π–π interaction with Tyr341 (35%) of the mid-gorge site. By carefully looking at compound 13 accommodation into the hAChE pocket during MD, it can be observed that the protonated amine group participated in major interactions with Trp86 (24%), Glu202 (96%), His447 (17%), and Gly448 (97%). Tyr124 also provided a significant hydrogen bond in the overall simulation (96%), while Phe297 and Tyr341 engaged π–π stacking interactions with the pyrimidine moiety and one of its nitrogen atoms displayed two water bridges, with Phe295 (42%) and Arg296 (21%), located at the mid-gorge. Finally, the phenyl ring was surrounded by the aromatic residues of the CAS region through a hydrophobic interaction with Trp86 and Tyr449 for 35 and 41% of the MD time. The hydroxyphenyl in compound 19 and the 2,3-dihydroxyphenyl ring in compound 20 were inserted into the CAS region, interacting with Tyr133 for 84% and with Glu202 for 67% of the MD time, respectively. Also, the 2,3-dihydroxyphenyl ring formed a strong π–π stacking interaction with Trp86 during the whole trajectory. The pyrimidine moiety formed a water bridge with Phe295 for 33 and 58% of MDs for compounds 19 and 20, respectively. The last one was also stabilized by the hydrogen bond between the nitrogen atom of the pyrimidine ring and Tyr124 at the CAS site (62%). The protonated nitrogen of compound 19 exhibited a noticeable 96% π–cation with Trp86, 89% hydrogen bond with Glu202, and 36% water-bridge interaction with His447 residue, while, in compound 20, the positively charged group displayed long persistency in the interaction with Asp74 (69%), Trp86 (66%), and Ser125 (86%).
Analyzing the main interactions of compounds 22 and 30 during MDs, it was possible to observe the effect of the different orientation of the former vs the latter in hBChE.
Indeed, the indole moiety of compound 22 engaged hydrophobic interactions with Trp231 (45%) and Phe329 (28%), a hydrogen bond with Ser287 (44%), and a water bridge with the Pro285 residue in CAS and mid-gorge pockets. The flexibility of the ligand allowed one to orient the protonated amine group toward the catalytic region forming a hydrogen bond with His438, a water bridge with Glu197, and a salt bridge with Trp82 at the CAS site. The insertion of pyrimidine contributed to the stabilization of hBChE into the CAS region due to the interaction with Trp82, Tyr440, and Trp430. Instead, the opposite orientation of compound 30 favored the interaction of the pyridine ring in the π–π stacking interactions with Trp231 and Phe329 for 50 and 69% of the time of the simulation. This also directed the amino group toward Ser198 (20%) of the catalytic region and the 3-methoxy-4-hydroxyphenyl ring toward His438 (37%). Such rearrangement led to the protonated amine group anchoring to Asp70 (10%) and Trp82 (35%) at PAS and CAS sites, respectively.
In summary, MD results reveal the role of the pyrimidine ring, protonated amine group, and substituted aromatic moiety of the pyrimidine derivatives in occupying CAS, PAS, and mid-gorge cavities, thus suggesting their promising role as dual binding site inhibitors.
The protonated amine group inserted into linear alkyl chains of five or six methylene units is one of the crucial pharmacophore features for ChEs inhibition, and it mainly establishes strong interactions with the catalytic site and PAS region.
The pyrimidine diamine scaffold bearing the indole moiety and 3-methoxy-4-hydroxyphenyl ring drives the selectivity of the molecule toward BChE inhibition.
Instead, phenolic and catecholic rings represent high selectivity for AChE inhibition, strongly supported by the interaction with Tyr133 and Glu202 of the AChE binding pocket.
Finally, the phenyl ring of compounds 9 and 13, which differ only in the length of the alkyl chain, is well stabilized into the bottom of the gorge.
Chelation Studies
For a selection of compounds, among the most potent as inhibitors of EeAChE and/or eqBChE, metal chelation studies were performed by using UV–vis spectrophotometric analysis, a method widely used for investigating chelates, to define their possible multitarget profile.30,31
The chelating ability of pyrimidine and pyridine compounds 9, 13, 18, 19, 20, 25, 28, and 30 on bio-metals Fe3+, Cu2+, and Zn2+ was evaluated. Initially, the UV–vis spectrum of the ligand was recorded and compared with the spectra obtained by adding an excess of metal to the ligand solution, maintaining the same concentration of ligand (ligand/metal ratio 1:3, 1:5, or 1:10). The variation of the UV–vis spectrum of the ligand in the presence of metal ions is indicative of the formation of the complex. Based on this, it has been observed that all the tested pyrimidine and pyridine derivatives have the ability to chelate Fe3+ and Cu2+ ions, while only compound 20 shows a chelating capacity also on Zn2+. Based on these results, it can be stated that the chelating activity of these compounds is due to the presence of two adjacent nitrogen atoms in the 2-aminopyrimidine or 2-aminopyridine moiety. For derivatives 18–20 and 30, it is also possible that the phenolic portion participates in the formation of the complex. In particular, the catechol ring in 20 could be responsible for the ability of this molecule to chelate also Zn2+.
UV–vis titrations of these compounds were carried out with the metal ions with which they form complexes, recording first the UV–vis spectrum of the ligand and then the spectra obtained by mixing solutions of the ligand and metal according to increasing metal/ligand molar ratios (Figure 5a).
Figure 5.

(a) UV–vis titration of ligand 25 with Fe3+. The greatest variations in spectra with increasing amount of metal are observed at 251 and 374 nm, with a reduction of absorbance, and at 318 nm, with an increase of absorbance. There are two isosbestic points at 270 and 350 nm. (b) Job’s plot of compound 25 in the presence of Fe3+: variation of the absorbance (ΔA) at the wavelength of 318 nm, in the ordinate, vs the mole fraction of 25, in the abscissa. X1 (mole fraction that causes the maximum variation of absorbance) = 0.57; X2 = 0.77; n1 (number of ligand molecules per cation) = 1; and n2 = 3.
The stoichiometries of metal–ligand complexes were determined through Job’s method.32,33 In Job’s plot, the values of ΔA, measured at the wavelengths where evident absorbance variations were observed in the titration spectra, are in the ordinate and the mole fractions of the ligand are in the abscissa. The mole fraction X, which causes the maximum variation of absorbance, is extracted from the graph and then the value of the coefficient n, which corresponds to the number of ligand molecules per cation, is obtained (Figure 5b). The UV–vis titration spectra and Job’s plots of all the tested compounds are reported in the Supporting Information (Figures S18–S46), while the data extrapolated from the Job’s plots are summarized in Table 5.
Table 5. Job’s Plot Data for the Tested Compounds in the Presence of Fe3+ and Cu2+a.
| Fe3+ |
Cu2+ |
|||||
|---|---|---|---|---|---|---|
| cmpd | λ (nm) | X | n | λ (nm) | X | n |
| 18 | 375 | 0.69 | 2 | 330 | 0.70 | 2 |
| 19 | 375 | 0.68 | 2 | 330 | 0.51 | 1 |
| 20 | 370 | X1 = 0.53 | n1 = 1 | 330 | 0.53 | 1 |
| X2 = 0.80 | n2 = 4 | |||||
| 25 | 318 | X1 = 0.57 | n1 = 1 | 330 | 0.44 | 1 |
| X2 = 0.77 | n2 = 3 | |||||
| 28 | 316 | X1 = 0.53 | n1 = 1 | 330 | 0.53 | 1 |
| X2 = 0.74 | n2 = 3 | |||||
| 30 | 310 | X1 = 0.53 | n1 = 1 | 312 | 0.50 | 1 |
| X2 = 0.74 | n2 = 3 | |||||
The table shows the wavelength values (λ) in which the absorbance variation was measured, the molar ratios (X) that cause the maximum variation of absorbance, and the coordination values (n) that correspond to the number of ligand molecules per cation.
Antioxidant Activity
To characterize multitarget compounds, antioxidant activity assays were carried out on 18, 19, 20, and 30 since they are the most potent ChEIs among phenolic derivatives, they exert chelating activity on metal ions involved in AD, and they have a structure that could justify an antioxidant activity.
The antioxidant activity assays were carried out according to the 2,2-diphenyl-1-picrylhydrazyl (DPPH) spectrophotometric method.34 Initially, to assess whether the compounds had antioxidant activity and to define the time required by each antioxidant to reach the steady state, the reduction in absorbance over time at 515 nm of a solution of DPPH mixed with the tested compound was recorded until reaching the plateau. From these measurements, compound 19 did not show a significant ability to interact with DPPH since a reduction of the absorbance over time was not observed; otherwise, compounds 18, 20, and 30 possess antioxidant activity as a reduction in absorbance over time, until reaching the plateau (in 1 min for 2 and 90 min for 18 and 30), was observed. Subsequently, the EC50, defined as the ratio of moles of antioxidant that reduce by 50% the initial concentration of DPPH to initial moles of DPPH,35 was determined for compounds 18, 20, and 30. The value of EC50 can be extrapolated by plotting the percentage of residual DPPH at the steady state as a function of the molar ratio of the antioxidant to initial DPPH.
Based on the obtained results reported in Table 6, the more efficient antioxidant, even better than ascorbic acid, which was used as the positive control, is the catechol derivative 20 both for the high reaction rate with DPPH and for the EC50 value equal to 0.1366 ± 0.0060. Compounds 18 and 30, having the 3-methoxy-4-hydroxyphenyl ring, show antioxidant capacity lower than 20 and comparable to each other, with EC50 respectively equal to 0.5997 ± 0.0940 and 0.6419 ± 0.0730.
Table 6. EC50 Values and Times for Reaching the Plateau for the Tested Compounds.
| compound | EC50 ± SDa | reaction time (min) |
|---|---|---|
| 18 | 0.5997 ± 0.0940 | 90 |
| 20 | 0.1366 ± 0.0060 | 1 |
| 30 | 0.6419 ± 0.0730 | 90 |
| ascorbic acid | 0.2650 ± 0.0070 | 2 |
EC50, defined as the ratio of antioxidant moles necessary to reduce the initial concentration of DPPH by 50% to the initial moles of DPPH. Data are the average of three independent assays.
Inhibition of Amyloid and Tau Aggregation
For the most potent ChEIs, the direct anti-amyloid aggregation activity against Aβ42 and tau aggregation was evaluated. The anti-aggregating effect of the tested compounds was monitored by a cell-based assay in intact Escherichia coli cells that overexpress either the Aβ42 peptide or tau protein, which upon overexpression form insoluble inclusion bodies that were stained with thioflavin-S, an amyloid specific dye.36 The percentages of inhibition toward Aβ42 and tau aggregation were determined at an inhibitor concentration of 100 μM, and the obtained results are reported in Table 7. Low percentages of inhibition were found both on Aβ42 (up to 22%) and on tau (up to 17%), so these compounds have a weak anti-aggregating activity when compared with the reference compound DP-128.37 The pyridine derivative 28 is the best inhibitor of aggregation of both Aβ42 and tau, while the pyrimidine 13 showed a similar percentage of inhibition on Aβ42 aggregation but lower toward tau.
Table 7. Inhibition of Aβ42 Aggregation and Tau Aggregation by the Tested Compounds.
| Aβ42 aggregation |
tau
aggregation |
|||
|---|---|---|---|---|
| cmpd | % inhibition [I] = 100 μM | SEMa | % inhibition [I] = 100 μM | SEMa |
| 9 | 5.8 | 1.6 | 2.6 | 6.7 |
| 13 | 21.0 | 4.2 | 12.6 | 6.6 |
| 18 | 1.7 | 2.1 | 1.0 | 4.1 |
| 19 | 16.7 | 2.7 | 9.9 | 3.9 |
| 20 | 7.2 | 2.4 | 4.6 | 1.0 |
| 22 | 9.7 | 1.8 | 6.8 | 6.1 |
| 23 | 10.4 | 3.9 | 7.0 | 4.2 |
| 25 | 16.8 | 2.9 | 6.7 | 2.9 |
| 26 | 7.3 | 3.7 | 7.3 | 4.8 |
| 28 | 22.3 | 3.3 | 17.0 | 6.4 |
| 30 | 13.6 | 5.3 | 12.8 | 2.7 |
| ref | 98.8 | 1.0 | 94.7 | 3.1 |
A minimum of five independent assays (with three replicates for assay) was performed for each tested compound. More assays were performed to obtain a SEM < 5% with a maximum of 10 independent assays. As a reference compound, we have used the known anti-amyloid drug DP-128 at 100 μM.
Computation of Physicochemical Descriptors and ADME Parameters
Physicochemical descriptors and ADME parameters of the most interesting compounds, by virtue of their inhibitory activity toward ChEs, as well as chelating and antioxidant ability, were predicted by means of the SwissADME public server,38 and the obtained data are reported in Table 8. All the studied compounds fit Lipinski’s rule of five (MW ≤ 500; MLogP ≤ 4.15; H bond acceptor ≤ 10; and H bond donor ≤ 5).39 They should be soluble or moderately soluble in water. They should have high gastrointestinal absorption after oral administration. Worthily, these compounds should be BBB accessible, with the exception of 18 and 20.
Table 8. Predicted Physicochemical Properties and ADME Parameters: Molecular Weight (MW); Number of H-Bond Acceptors (H-b Acc); Number of H-Bond Donors (H-b Don); Number of Heavy Atoms (Heavy Atoms); Number of Rotatable Bonds (Rot Bonds); Topological Polar Surface Area in Å2 (TPSA); Octanol/Water Partition Coefficient (MLogP); Water Solubility (LogS ESOL); Water Solubility Class (Sol Class); Gastrointestinal Absorption (GI); Blood–Brain Barrier Permeation (BBB); and Number of Lipinski’s Rule of Five Violations (Lipinski Viol)a.
| cmpd | MW | H-b acc | H-b don | heavy atoms | rot bonds | TPSA | MLogP | logS ESOL | sol class | GI | BBB | Lipinski viol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 9 | 270.37 | 3 | 2 | 20 | 9 | 49.84 | 1.88 | –3.04 | soluble | high | yes | 0 |
| 13 | 284.40 | 3 | 2 | 21 | 10 | 49.84 | 2.12 | –3.26 | soluble | high | yes | 0 |
| 18 | 330.42 | 5 | 3 | 24 | 11 | 79.30 | 1.25 | –3.19 | soluble | high | no | 0 |
| 19 | 300.40 | 4 | 3 | 22 | 10 | 70.07 | 1.56 | –3.12 | soluble | high | yes | 0 |
| 20 | 316.40 | 5 | 4 | 23 | 10 | 90.30 | 1.01 | –3.32 | soluble | high | no | 0 |
| 22 | 323.44 | 3 | 3 | 24 | 10 | 65.63 | 1.87 | –3.63 | soluble | high | yes | 0 |
| 23 | 269.38 | 2 | 2 | 20 | 9 | 36.95 | 2.56 | –3.45 | soluble | high | yes | 0 |
| 25 | 283.41 | 2 | 2 | 21 | 10 | 36.95 | 2.80 | –3.67 | soluble | high | yes | 0 |
| 26 | 362.31 | 2 | 2 | 22 | 10 | 36.95 | 3.42 | –4.57 | moderately soluble | high | yes | 0 |
| 28 | 313.44 | 3 | 2 | 23 | 11 | 46.18 | 2.45 | –3.73 | soluble | high | yes | 0 |
| 30 | 329.44 | 4 | 3 | 24 | 11 | 66.41 | 1.89 | –3.59 | soluble | high | yes | 0 |
Data were obtained from the SwissADME public server.38
Furthermore, all synthesized final compounds were screened using in silico public tools, specified in the Experimental Section, and were not found to be pan assay interference compounds (PAINS),40 except for 20 and 32. Nevertheless, the activities of these compounds are in line with those of the other pyrimidine and pyridine molecules studied and they can be framed in logical SAR, as previously discussed. Moreover, compound 20 showed a strongly different activity toward the related targets EeAChE and eqBChE (percentages of inhibitions at 9 μM equal to 90.5 ± 0.5 and 17.1 ± 1.6, respectively; Table 1) and it was not cytotoxic at concentrations up to 10 μM, as reported in the subsequent section. Overall, these evidences suggested that these molecules do not act via a PAINS mechanism.
Cytotoxicity Assays
A selection of the most active inhibitors of EeAChE and eqBChE (9, 13, 19, 20, 22, 23, 25, 26, and 28) was tested to evaluate the cytotoxic effects on the U-87 MG cell line from the human brain (glioblastoma astrocytoma) at concentrations ranging from 1 to 50 μM. The obtained data, represented in the histogram of Figure 6, suggest that the tested compounds are characterized by low toxicity toward the studied cells, in particular at concentrations up to 10 μM. In general, the pyrimidine derivatives appear to be less cytotoxic than pyridine derivatives, as evidenced by the comparison of pyrimidines 9 and 13 with the corresponding pyridines 23 and 25. The pyridines tested showed a very similar toxicity profile, with the exception of compound 23 (having the spacer chain with five methylene units), which at concentrations of 1 and 5 μM showed a lower cytotoxic effect. Also, among pyrimidine derivatives, compound 9, with five methylene units, exerted a minor effect on cell viability than the corresponding compound 13, with six methylene units. Moreover, among pyrimidine derivatives, compound 22, with an indole group on one side of the aliphatic chain, showed a more pronounced cytotoxic effect, with a toxicity profile similar to the tested pyridines.
Figure 6.

Cell viability assay of U-87 MG cells in the presence of increasing concentrations of studied compounds, evaluated using the MTT method. Data represent the absorbance % (± SD) relative to untreated cells in the same experiment and standardized to 100%. All data points were performed in triplicate and at least three independent experiments.
Conclusions
Series of pyrimidine and pyridine derivatives were designed as potential multifunctional compounds for AD, whose purpose could be to restore the cholinergic tone by inhibition of ChEs, attenuate the dyshomeostasis of the metals mainly involved in the pathology, reduce the oxidative stress, and contrast the toxicity and deposition of the Aβ peptide. These compounds contain two π systems, one of which is represented by a 2-aminopyrimidine or 2-aminopyridine moiety, connected to a small aromatic group by a flexible amino-alkyl linker.
On the synthesized compounds, enzymatic inhibition studies were carried out toward EeAChE and eqBChE. The initial determination of the percentages of inhibition at concentrations equal to 9–0.09 μM led to the identification of the most potent inhibitors. For a selection of these compounds, the Ki were determined, obtaining values in the range of 0.312–1.323 μM vs EeAChE and 0.099–3.465 μM vs eqBChE. All the tested compounds result in mixed inhibitors of ChEs, with the only exception of 19, which acts with an uncompetitive mechanism toward EeAChE. The uncompetitive inhibitors might interact with an allosteric modulation site such as PAS, while mixed inhibitors interact simultaneously with both CAS and PAS of the enzyme; these evidences may suggest that these compounds could interfere with the aggregation of Aβ plaques induced by AChE through interactions with PAS.
In general, the pyrimidine amine compounds are more potent on EeAChE than the pyridine analogs. On the contrary, pyridine amine derivatives are more potent on eqBChE than the corresponding pyrimidine derivatives. The greater inhibitory potency toward EeAChE is shown by compounds 9 (Ki = 0.312 ± 0.108 μM) and 13 (Ki = 0.426 ± 0.132 μM), both having the unsubstituted phenyl ring on one side of the aliphatic chain; indeed, modifications of the phenyl group, both adding substituents and changing the type of aromatic ring, reduce the inhibition activity toward EeAChE. Differently, the most potent inhibitor of eqBChE is 22 (Ki = 0.099 ± 0.071 μM), the pyrimidine derivative with the indole ring on one side of the aliphatic chain.
In silico analyses highlighted that the protonated amine group inserted into the linear alkyl chain is the pivotal pharmacophore feature for ChE inhibition due to its strong interaction with the catalytic site and PAS region. Moreover, the pyrimidine diamine scaffold, bearing the indole moiety and 3-methoxy-4-hydroxyphenyl ring, resulted to drive the selectivity of the molecule toward BChE inhibition. On the other hand, phenolic and catecholic rings were responsible for the AChE inhibition selectivity, strongly supported by the interaction with Tyr133 and Glu202 of this enzymatic binding pocket. Additionally, the phenyl ring of compounds 9 and 13, which differ only in the length of the alkyl chain, is well stabilized into the bottom of the gorge. Based on the computational results, the involvement of the pyrimidine ring, protonated amine group, and substituted aromatic moiety of these derivatives in occupying CAS, PAS, and mid-gorge cavities suggested their potential role as dual binding site inhibitors.
Metal chelation studies revealed that all the tested compounds have the ability to chelate Fe3+ and Cu2+ ions; this chelating activity can be attributed to the presence of two adjacent nitrogen atoms in the 2-aminopyrimidine or 2-aminopyridine moieties. The only 2-aminopyrimidine derivative that shows a chelating ability also toward Zn2+ ion is 20, which contains a catechol group.
Moreover, antioxidant activity assays were carried out on compounds 18, 19, 20, and 30 since they are the most potent ChEIs among phenolic derivatives. Catechol 20 is the most efficient antioxidant, even better than ascorbic acid, both for the high reaction rate with DPPH and for its EC50 value. Compounds 18 and 30, having the 3-methoxy-4-hydroxyphenyl ring, show an antioxidant capacity lower than 20 and comparable to each other, while compound 19, having the 3-hydroxyphenyl ring, has no significant antioxidant capacity.
Unfortunately, the tested compounds show low percentages of inhibition toward both Aβ42 and tau aggregation, proving to have a weak anti-aggregating activity in E. coli. However, the low anti-amyloid activity reported could also be due to an unusually low ability of the compounds to cross the bacterial wall. Although it is unusual, this effect has been observed recently and should not be ruled out.41
In conclusion, this research has allowed one to identify interesting lead compounds for the development of new multifunctional molecules useful for AD by virtue of their inhibitory activity toward ChEs, as well as for chelating and antioxidant abilities, low toxicity, and good predicted ADME parameters.
Experimental Section
Chemistry: General
All reagents and solvents were of analytical grade and were purchased from Sigma-Aldrich (Milano, Italy) or from Fluorochem (Hadfield, UK). Dichloromethane was dried by storing it over activated 4 Å molecular sieves (10% m/v). Triethylamine was freshly purified by distillation over potassium hydroxide. Column chromatographies were performed on a silica gel (Merck; 63–200 μm particle size).1H NMR and 13C NMR spectra were acquired at 25 °C, unless otherwise specified, on a Bruker AVANCE-400 spectrometer at 9.4 T operating at 400 MHz (1H NMR) and 100 MHz (13C- MR); chemical shift values (δ) are given in ppm relative to TMS, using the solvent as the internal reference; coupling constants are given in Hz. The following abbreviation were used: s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, ddd = double double doublet, bs = broad singlet, and m = multiplet. Mass spectra were recorded on a ThermoFinnigan LCQ Classic LC/MS/MS ion trap equipped with an ESI source and a syringe pump; samples (10–4–10–5 M in MeOH/H2O 80:20) were infused in the electrospray system at a flow rate of 5–10 μL min–1; when necessary, 50 μL of 10–2 M HCOOH or 10–2 M NH3 was added to the sample solutions to promote the analyte ionization; the ESI-MS data are given as m/z, with mass expressed in amu. Melting points were determined on a FALC Mod. 360 D apparatus and are uncorrected. Infrared spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer equipped with an ATR system. The purity of the compounds was determined by elemental analyses obtained by a PE 2400 (Perkin-Elmer) analyzer, and the analytical results were within ±0.4% of the theoretical values for all compounds.
General Experimental Procedures
General Procedure A (GP-A) for the Synthesis of Pyrimidine Intermediates 1 and 2
To a solution of N-Boc-1,5-diaminopentane (1 equiv) or N-Boc-1,6-diaminohexane (1 equiv) in 20 mL of MeOH, TEA (1 equiv) and 2-chloropyrimidine (1 equiv) were added. The reaction mixture was stirred at reflux for 20 h. Then it was cooled to room temperature and the solvent was removed under reduced pressure. The residue was diluted with a saturated aqueous solution of Na2CO3 (25 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic layer was dried over Na2SO4 and filtered, and the solvent was evaporated under reduced pressure. The crude material was purified by column chromatography on a silica gel (CH2Cl2/MeOH 9.5:0.5). For each compound, Rf, yield (%), 1H NMR, ESI-MS, and elemental analysis are reported.
General Procedure B (GP-B) for the Synthesis of N1-(Pyrimidin-2-yl)pentane-1,5-diamine (3), N1-(Pyrimidin-2-yl)hexane-1,6-diamine (4), N1-(Pyridin-2-yl)pentane-1,5-diamine (7), and N1-(Pyridin-2-yl)hexane-1,6-diamine (8)
Procedure 1
Intermediates 1, 2, 5, or 6 (1 equiv), used for the synthesis of compounds 3, 4, 7, and 8, respectively, were dissolved in 20 mL of dry CH2Cl2, and to the solution, TFA (20 equiv) was added. The mixture was stirred at room temperature for 3.5 h, and then the solvent and the excess of TFA were removed under reduced pressure to give trifluoroacetic salts of compounds 3, 4, 7, or 8 as yellow oils, which were used for the subsequent reactions without further purification.
Procedure 2
To a solution of intermediate 2 or 6 (1 equiv) in 20 mL of dry CH2Cl2, TFA (20 equiv) was added. The mixture was stirred at room temperature for 3.5 h, and then it was extracted twice with 5 N NaOH solution (2 × 20 mL) and once with H2O (20 mL). The organic layer was dried over Na2SO4 and filtered, and the solvent was removed under vacuum to give compounds 4 (77% yield) or 8 (88% yield) as yellow oils, which were used for the subsequent reactions without further purification.
Procedure 3
To a solution of 1,6-diaminohexane (1.516 g, 13.05 mmol) in 10 mL of MeOH, TEA (605 μL, d = 0.726 g/mL, 4.35 mmol) and 2-chloropyrimidine (0.498 g, 4.35 mmol) were added. The reaction mixture was stirred at reflux for 20 h. Then it was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was diluted with a saturated aqueous solution of Na2CO3 (15 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by column chromatography on a silica gel (MeOH/CH2Cl2/TEA 8:2:0.2, Rf = 0.17), and subsequently, the product was filtered with warm acetonitrile to afford compound 4.
General Procedure C (GP-C) for the Synthesis of Pyridine Intermediates 5 and 6
An oven-dried Schlenk flask equipped with a Teflon valve was charged with N-Boc-1,5-diaminopentane or N-Boc-1,6-diaminohexane (1.5 equiv), CuI (0.05 equiv), Cs2CO3 (2 equiv), and a magnetic stir bar. The flask was closed, evacuated, and backfilled with N2 for at least 10 min. Under a counter flow of N2, DMF (0.5 mL), 2-iodopyridine (1 equiv), and finally 2-isobutyrylcyclohexanone (0.2 equiv) were added by a syringe. The mixture was allowed to stir under N2 at 40 °C for 21 h. After this period, the mixture was diluted with ethyl acetate, transferred to a centrifuge tube, and centrifuged to separate the inorganic salts. The solvent was removed under reduced pressure. The crude residue was purified by column chromatography on a silica gel (CH2Cl2/MeOH 9.5:0.5). For each compound, Rf, yield (%), 1H NMR, ESI-MS, and elemental analysis are reported.
General Procedures D (GP-D) for the Synthesis of Pyrimidine and Pyridine Diamine Derivatives 9–33
Procedure 1
Intermediate 3, 4, 7, or 8 (1 equiv), obtained following procedure 1 of GP-B described above, was dissolved in 20 mL of dry CH2Cl2, and to the solution, the opportune aldehyde (1 equiv) and dry K2CO3 (20 equiv) were added. The mixture was stirred at room temperature for 12 h and then filtered. The solvent was removed under reduced pressure, and the residue was dissolved in 30 mL of MeOH and treated with NaBH4 (3 equiv) at room temperature for 2 h. Then 5 mL of H2O was added; after 5 min under stirring, MeOH was evaporated under reduced pressure, H2O (20 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 20 mL). The organic layer was dried over Na2SO4, and the solvent was removed under vacuum. The obtained residue was purified by column chromatography on a silica gel. For each compound chromatography system, Rf, yield (%), 1H NMR, 13C NMR, ESI-MS, IR, melting point (°C), and elemental analysis are reported.
Procedure 2
Intermediate 4 (1 equiv), obtained following procedure 2 of GP-B described above, was dissolved in 20 mL of dry CH2Cl2, and to the solution, the opportune aldehyde (1 equiv) and molecular sieves (4 Å) were added. The mixture was stirred at room temperature for 12 h and then filtered. The solvent was removed under reduced pressure, and the residue was dissolved in 30 mL of MeOH and treated with NaBH4 (3 equiv) at room temperature for 2 h. Then 5 mL of H2O was added; after 5 min under stirring, MeOH was evaporated under reduced pressure, H2O (20 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 20 mL). The organic layer was dried over Na2SO4, and the solvent was removed under vacuum. The obtained residue was purified by column chromatography on a silica gel. For each compound chromatography system, Rf, yield (%), 1H NMR, 13C NMR, ESI-MS, IR, melting point (°C), and elemental analysis are reported.
Procedure 3
Intermediate 4 or 8 (1 equiv), obtained following, respectively, procedure 3 or 2 of GP-B described above, was dissolved in 20 mL of dry CH2Cl2, and to the solution, the opportune aldehyde (1 equiv) and molecular sieves (4 Å) were added. The mixture was stirred at room temperature for 12 h and then filtered. The solvent was removed under reduced pressure, and the residue was dissolved in 30 mL of MeOH and treated with NaBH4 (3 equiv) at room temperature for 2 h. Then 5 mL of 1 M HCl was added; after 5 min under stirring, the solvent was evaporated under reduced pressure, and the residue was diluted with 20 mL of saturated aqueous solution of NaHCO3 and extracted with CH2Cl2 (3 × 20 mL). The organic layer was dried over Na2SO4, and the solvent was removed under vacuum. The obtained residue was purified by column chromatography on a silica gel and/or by crystallization. For each compound chromatography system (and/or crystallization solvent), Rf, yield (%), 1H NMR, 13C NMR, ESI-MS, IR, melting point (°C), and elemental analysis are reported.
tert-Butyl {5-[(Pyrimidin-2-yl)amino]pentyl}carbamate (1)
Compound 1 was prepared using N-Boc-1,5-diaminopentane (0.773 g, 3.82 mmol), TEA (531 μL, d = 0.726 g/mL, 3.82 mmol), and 2-chloropyrimidine (0.438 g, 3.82 mmol) following GP-A. Rf (CH2Cl2/MeOH 9.5:0.5) = 0.34. White solid, 0.539 g, 50% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 6.51 (t, 1H, J = 4.8 Hz, pyrimidine); 5.68 (bs, 1H, pyrimidine-NH-); 5.26 (bs, 1H, Boc-NH-); 3.32 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 3.00 (q, 2H, J = 6.6 Hz, Boc-NH-CH2-); 1.60–1.53 (m, 2H, pyrimidine-NH-CH2-CH2-); 1.49–1.30 (m, 13H, pyrimidine-NH-CH2-CH2-CH2-CH2-CH2-NH-COO-C(CH3)3). ESI-MS (m/z): [M + H]+ = 280.79. Anal. (C14H24N4) C, H, N calcd: C 59.98%, H 8.63%, N 19.98%; found: C 59.89%, H 8.59%, N 19.90%.
tert-Butyl {6-[(Pyrimidin-2-yl)amino]hexyl}carbamate (2)
Compound 2 was prepared using N-Boc-1,6-diaminohexane (0.437 g, 2.02 mmol), TEA (281 μL, d = 0.726 g/mL, 2.02 mmol), and 2-chloropyrimidine (0.231 g, 2.02 mmol) following GP-A. Rf (CH2Cl2/MeOH 9.5:0.5) = 0.5. White solid, 0.358 g, 60% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.4 Hz, pyrimidine); 6.51 (t, 1H, J = 4.2 Hz, pyrimidine); 5.69 (bs, 1H, pyrimidine-NH-); 5.24 (bs, 1H, Boc-NH-); 3.32 (q, 2H, J = 6.5 Hz, pyrimidine-NH-CH2-); 3.00 (q, 2H, J = 6.2 Hz, Boc-NH-CH2-); 1.59–1.52 (m, 2H, pyrimidine-NH-CH2-CH2-); 1.46–1.23 (m, 15H, pyrimidine-NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-COO-C(CH3)3). ESI-MS (m/z): [M + H]+ = 294.73. Anal. (C15H26N4) C, H, N calcd: C 61.20%, H 8.90%, N 19.03%; found: C 60.97%, H 8.86%, N 18.96%.
N1-(Pyrimidin-2-yl)pentane-1,5-diamine (3)
Compound 3 was prepared following procedure 1 of GP-B as trifluoroacetic salt and used for the subsequent reactions without purification.
N1-(Pyrimidin-2-yl)hexane-1,6-diamine (4)
Compound 4 was prepared following procedure 1 of GP-B as trifluoroacetic salt (used for the subsequent reactions without purification) or following procedure 2 or 3 of GP-B as free amine. Yellow wax, 0.590 g, 70% yield. 1H NMR (400 MHz) (MeOD) δ (ppm): 8.24 (d, 2H, J = 4.8 Hz, pyrimidine); 6.57 (t, 1H, J = 4.9 Hz, pyrimidine); 3.35 (t, 2H, J = 7.1 Hz, pyrimidine-NH-CH2-); 2.71 (t, 2H, J = 7.2 Hz; -CH2-NH2); 1.66–1.40 (m, 8H, pyrimidine-NH-CH2-CH2-CH2-CH2-CH2-CH2-NH2). ESI-MS (m/z): [M + H]+ = 194.70. Anal. (C10H18N4) C, H, N calcd: C 61.82%, H 9.34%, N 28.84%; found: C 61.58%, H 9.30%, N 28.73%.
tert-Butyl {5-[(Pyridin-2-yl)amino]pentyl}carbamate (5)
Compound 5 was prepared using N-Boc-1,5-diaminopentane (0.303 g, 1.5 mmol), CuI (0.095 g, 0.05 mmol), Cs2CO3 (0.652 g, 2 mmol), DMF (0.5 mL), 2-iodopyridine (107 μL, d = 1.928 g/mL, 1 mmol), and 2-isobutyrylcyclohexanone (33 μL, d = 1.008 g/mL, 0.2 mmol) following GP-C. Rf (CH2Cl2/MeOH 9.5:0.5) = 0.38. Yellowish solid, 0.279 g, 100% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.97 (d, 1H, J = 4.1 Hz, pyridine); 7.39–7.35 (m, 1H, pyridine); 6.50–6.47 (m, 1H, pyridine); 6.41 (d, 1H, J = 8.4 Hz, pyridine); 5.26 (bs, 1H, Boc-NH-); 5.09 (bs, 1H, pyridine-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 3.01 (q, 2H, J = 6.6 Hz, Boc-NH-CH2-); 1.60–1.53 (m, 2H, pyridine-NH-CH2-CH2-); 1.50–1.31 (m, 13H, pyridine-NH-CH2-CH2-CH2-CH2-CH2-NH-COO-C(CH3)3). ESI-MS (m/z): [M + H]+ = 280.00. Anal. (C15H25N3) C, H, N calcd: C 64.49%, H 9.02%, N 15.04%; found: C 64.25%, H 8.98%, N 14.98%.
tert-Butyl {6-[(Pyridin-2-yl)amino]hexyl}carbamate (6)
Compound 6 was prepared using N-Boc-1,6-diaminohexane (0.811 g, 3.75 mmol), CuI (0.024 g, 0.125 mmol), Cs2CO3 (1.63 g, 5 mmol), DMF (1.25 mL), 2-iodopyridine (267 μL, d = 1.928 g/mL, 2.5 mmol), and 2-isobutyrylcyclohexanone (83 μL, d = 1.008 g/mL, 0.5 mmol) following GP-C. Rf (CH2Cl2/MeOH 9.5:0.5) = 0.38. Yellowish solid, 0.734 g, 100% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.97 (dd, 1H, J1 = 5.0 Hz, J2 = 1.1 Hz, pyridine); 7.39–7.35 (m, 1H, pyridine); 6.50–6.47 (m, 1H, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.25 (bs, 1H, Boc-NH-); 5.09 (bs, 1H, pyridine-NH-); 3.23 (q, 2H, J = 7.0 Hz, pyridine-NH-CH2-); 3.00 (q, 2H, J = 6.6 Hz, Boc-NH-CH2-); 1.59–1.52 (m, 2H, pyridine-NH-CH2-CH2-); 1.46–1.27 (m, 15H, pyridine-NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-COO-C(CH3)3). ESI-MS (m/z): [M + H]+ = 293.87. Anal. (C16H27N3) C, H, N calcd: C 65.50%, H 9.28%, N 14.32%; found: C 65.24%, H 9.24%, N 14.26%.
N1-(Pyridin-2-yl)pentane-1,5-diamine (7)
Compound 7 was prepared following procedure 1 of GP-B as trifluoroacetic salt and used for the subsequent reactions without purification.
N1-(Pyridin-2-yl)hexane-1,6-diamine (8)
Compound 8 was prepared following procedure 1 of GP-B as trifluoroacetic salt or following procedure 2 of GP-B as free amine and used for the subsequent reactions without purification.
N1-Benzyl-N5-(pyrimidin-2-yl)pentane-1,5-diamine (9)
Compound 9 was prepared using N1-(pyrimidin-2-yl)pentane-1,5-diamine (3) (0.108 g, 0.60 mmol), benzaldehyde (61 μL, d = 1.045 g/mL, 0.60 mmol), K2CO3 (1.659 g, 12.0 mmol), and NaBH4 (0.068 g, 1.8 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.4. White solid, 0.093 g, 57% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.7 Hz, pyrimidine); 7.31–7.20 (m, 5H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.69 (bs, 1H, pyrimidine-NH-); 3.71 (s, 2H, Ar-CH2-NH-); 3.32 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.54 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.45 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-NH-); 1.41–1.34 (m, 2H, -NH-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 163.7; 158.9; 142.4; 129.1; 128.9; 127.5; 111.0; 54.3; 49.9; 41.8; 30.5; 30.1; 25.4. ESI-MS (m/z): [M + H]+ = 270.87. I.R. (cm–1): 3255; 1124; 1100; 1074; 1027. m.p. = 58.0–59.8 °C. Anal. (C16H22N4) C, H, N calcd: C 71.08%, H 8.20%, N 20.72%; found: C 70.98%, H 8.23%, N 20.79%.
N1-(4-Bromobenzyl)-N5-(pyrimidin-2-yl)pentane-1,5-diamine (10)
Compound 10 was prepared using N1-(pyrimidin-2-yl)pentane-1,5-diamine (3) (0.097 g, 0.54 mmol), 4-bromobenzaldehyde (0.100 g, 0.54 mmol), K2CO3 (1.493 g, 10.8 mmol), and NaBH4 (0.061 g, 1.62 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.5. White solid, 0.093 g, 50% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 7.45 (m, 2H, aromatic); 7.25 (m, 2H, aromatic); 6.51 (t, 1H, J = 4.8 Hz, pyrimidine); 5.68 (bs, 1H, pyrimidine-NH-); 3.68 (s, 2H, Ar-CH2-NH-); 3.32 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.52 (t, 2H, J = 6.8 Hz, Ar-CH2-NH-CH2-); 1.59–1.44 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-NH-); 1.41–1.33 (m, 2H, -NH-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 140.0; 132.5; 131.5; 121.8; 111.0; 53.7; 50.0; 42.0; 30.3; 30.1; 25.7. ESI-MS (m/z): [M + H]+ = 348.80 (95); 350.80 (100). I.R. (cm–1): 3235; 1265; 1129; 1066; 1007. m.p. = 75.2–77.0 °C. Anal. (C16H21BrN4) C, H, N calcd: C 55.02%, H 6.06%, N 16.04%; found: C 54.95%, H 6.08%, N 16.09%.
N1-(2-Methoxybenzyl)-N5-(pyrimidin-2-yl)pentane-1,5-diamine (11)
Compound 11 was prepared using N1-(pyrimidin-2-yl)pentane-1,5-diamine (3) (0.108 g, 0.60 mmol), 2-methoxybenzaldehyde (0.082 g, 0.60 mmol), K2CO3 (1.659 g, 12.0 mmol), and NaBH4 (0.068 g, 1.8 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.3. Pale yellow solid, 0.091 g, 50% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 7.26–7.20 (m, 2H, aromatic); 6.94–6.87 (m, 2H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.68 (bs, 1H, pyrimidine-NH-); 3.80 (s, 3H, -O-CH3); 3.68 (s, 2H, Ar-CH2-NH-); 3.32 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.53 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.45 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-NH-); 1.40–1.33 (m, 2H, -NH-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.23; 159.19; 131.2; 129.8; 128.2; 121.5; 111.5; 111.0; 55.8; 49.8; 49.6; 42.0; 30.3; 30.0; 25.7. ESI-MS (m/z): [M + H]+ = 301.02. I.R. (cm–1): 3247; 1293; 1266; 1235; 1100; 1050; 1023; 932. m.p. = 44.8–46.8 °C. Anal. (C17H24N4O) C, H, N calcd: C 67.97%, H 8.05%, N 18.65%; found: C 67.98%, H 8.07%, N 18.60%.
N1-((2,6-Dichloropyridin-4-yl)methyl)-N5-(pyrimidin-2-yl)pentane-1,5-diamine (12)
Compound 12 was prepared using N1-(pyrimidin-2-yl)pentane-1,5-diamine (3) (0.090 g, 0.50 mmol), 2,6-dichloropyridine-4-carbaldehyde (0.088 g, 0.50 mmol), K2CO3 (1.382 g, 10.0 mmol), and NaBH4 (0.057 g, 1.5 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH 1:1, Rf = 0.5. Pale yellow solid, 0.094 g, 55% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.7 Hz, pyrimidine); 7.36 (s, 2H, pyridine); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.67 (bs, 1H, pyrimidine-NH-); 3.75 (s, 2H, pyridine-CH2-NH-); 3.32 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.52 (t, 2H, J = 6.8 Hz, pyridine-CH2-NH-CH2-); 1.60–1.45 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-NH-); 1.42–1.34 (m, 2H -NH-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 157.7; 151.4; 123.6; 111.0; 52.4; 50.1; 42.1; 30.5; 30.6; 25.9. ESI-MS (m/z): [M + H]+ = 340.09 (100); 342.08 (65); 344.06 (10). Anal. (C15H19Cl2N5) C, H, N calcd: C 52.95%, H 5.63%, N 20.58%; found: C 52.91%, H 5.62%, N 20.61%.
N1-Benzyl-N6-(pyrimidin-2-yl)hexane-1,6-diamine (13)
Compound 13 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.103 g, 0.53 mmol), benzaldehyde (54 μL, d = 1.045 g/mL, 0.53 mmol), K2CO3 (1.465 g, 10.6 mmol), and NaBH4 (0.060 g, 1.59 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 7:3, Rf = 0.42. White solid, 0.066 g, 43% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 7.33–7.20 (m, 5H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.74 (bs, 1H, pyrimidine-NH-); 3.71 (s, 2H, Ar-CH2-NH-); 3.31 (q, 2H, J = 6.8 Hz, pyrimidine-NH-CH2-); 2.53 (t, 2H, J = 7.0 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.49–1.41 (m, 2H, -NH-CH2-CH2-); 1.38–1.27 (m, 4H -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 163.7; 158.9; 142.2; 129.1; 129.0; 127.5; 111.0; 54.3; 49.9; 41.8; 30.6; 30.2; 27.8; 27.5. ESI-MS (m/z): [M + H]+ = 285.13. I.R. (cm–1): 3301; 3267; 1323; 1258; 1129; 1027; 984; 975. m.p. = 51.8–54.4 °C. Anal. (C17H24N4) C, H, N calcd: C 71.79%, H 8.51%, N 19.70%; found: C 71.89%, H 8.47%, N 19.64%.
N1-(4-Bromobenzyl)-N6-(pyrimidin-2-yl)hexane-1,6-diamine (14)
Compound 14 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.136 g, 0.70 mmol), 4-bromobenzaldehyde (0.130 g, 0.70 mmol), K2CO3 (1.935 g, 14.0 mmol), and NaBH4 (0.079 g, 2.1 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.4. White solid, 0.152 g, 60% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.21 (d, 2H, J = 4.7 Hz, pyrimidine); 7.45 (m, 2H, aromatic); 7.24 (m, 2H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.77 (bs, 1H, pyrimidine-NH-); 3.67 (s, 2H, Ar-CH2-NH-); 3.31 (q, 2H, J = 6.7 Hz, pyrimidine-NH-CH2-); 2.50 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.58–1.51 (m, 2H, -NH-CH2-CH2-); 1.47–1.41 (m, 2H, -NH-CH2-CH2-); 1.37–1.26 (m, 4H -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 163.7; 158.9; 141.8; 132.0; 130.9; 120.6; 111.0; 53.5; 49.8; 41.8; 30.7; 30.2; 27.7; 27.5. ESI-MS (m/z): [M + H]+ = 363.07 (95); 365.00 (100). I.R. (cm–1): 3267; 1255; 1132; 1068; 1010. m.p. = 43–46 °C. Anal. (C17H23BrN4) C, H, N calcd: C 56.20%, H 6.38%, N 15.42%; found: C 56.21%, H 6.39%, N 15.45%.
N1-(4-Chlorobenzyl)-N6-(pyrimidin-2-yl)hexane-1,6-diamine (15)
Compound 15 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.107 g, 0.55 mmol), 4-chlorobenzaldehyde (0.077 g, 0.55 mmol), K2CO3 (1.520 g, 11.0 mmol), and NaBH4 (0.062 g, 1.65 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 7:3, Rf = 0.53. White solid, 0.085 g, 50% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 7.31 (s, 4H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.71 (bs, 1H, pyrimidine-NH-); 3.69 (s, 2H, Ar-CH2-NH-); 3.31 (q, 2H, J = 6.8 Hz, pyrimidine-NH-CH2-); 2.51 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.58–1.51 (m, 2H, -NH-CH2-CH2-); 1.48–1.41 (m, 2H, -NH-CH2-CH2-); 1.38–1.27 (m, 4H -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 163.7; 158.9; 141.3; 132.5; 130.6; 129.0; 111.0; 53.5; 49.8; 41.8; 30.6; 30.2; 27.7; 27.5. ESI-MS (m/z): [M + H]+ = 318.87 (100); 320.87 (30). I.R. (cm–1): 3264; 1256; 1132; 1123; 1088; 1012; 987. m.p. = 53–56 °C. Anal. (C17H23ClN4) C, H, N calcd: C 64.04%, H 7.27%, N 17.57%; found: C 64.08%, H 7.26%, N 17.52%.
N1-(2-Methoxybenzyl)-N6-(pyrimidin-2-yl)hexane-1,6-diamine (16)
Compound 16 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.185 g, 0.95 mmol), 2-methoxybenzaldehyde (0.129 g, 0.95 mmol), K2CO3 (2.626 g, 19.0 mmol), and NaBH4 (0.108 g, 2.85 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.23. Pale yellow oil, 0.184 g, 62% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.22 (d, 2H, J = 4.8 Hz, pyrimidine); 7.28–7.21 (m, 2H, aromatic); 6.94–6.88 (m, 2H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.70 (bs, 1H, pyrimidine-NH-); 3.81 (s, 3H, -O-CH3); 3.71 (s, 2H, Ar-CH2-NH-); 3.32 (q, 2H, J = 6.8 Hz, pyrimidine-NH-CH2-); 2.54 (t, 2H, J = 7.0 Hz, Ar-CH2-NH-CH2-); 1.58–1.44 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-); 1.40–1.28 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 163.9; 159.0; 158.8; 130.5; 129.6; 129.1; 121.3; 111.6; 111.1; 56.1; 49.9; 49.3; 42.0; 30.6; 30.3; 27.9; 27.6. ESI-MS (m/z): [M + H]+ = 315.13. I.R. (cm–1): 3265; 1240; 1104; 1031. Anal. (C18H26N4O) C, H, N calcd: C 68.76%, H 8.33%, N 17.82%; found: C 68.70%, H 8.30%, N 17.78%.
N1-(Pyrimidin-2-yl)-N6-(3,4,5-trimethoxybenzyl)hexane-1,6-diamine (17)
Compound 17 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.101 g, 0.52 mmol), 3,4,5-trimethoxybenzaldehyde (0.102 g, 0.52 mmol), and NaBH4 (0.059 g, 1.56 mmol) following procedure 3 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH 1:1, Rf = 0.23. Yellow oil, 0.138 g, 71% yield. 1H NMR (400 MHz) (acetone-d6) δ (ppm): 8.22 (d, 2H, J = 4.7 Hz, pyrimidine); 6.67 (s, 2H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 6.26 (bs, 1H, pyrimidine-NH-); 3.79 (s, 6H, Ar-(OCH3)2); 3.69 (s, 3H, Ar-OCH3); 3.68 (s, 2H, Ar-CH2-NH-); 3.40–3.35 (m, 2H, pyrimidine-NH-CH2-); 2.57 (t, 2H, J = 6.8 Hz, Ar-CH2-NH-CH2-); 1.64–1.57 (m, 2H, -NH-CH2-CH2-); 1.54–1.47 (m, 2H, -NH-CH2-CH2-); 1.43–1.37 (m, 4H, -NH-CH2-CH2-CH2–CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 154.5; 138.2; 136.3; 111.0; 106.9; 61.1; 56.6; 54.5; 49.8; 42.1; 30.4; 30.1; 28.2; 27.9. ESI-MS (m/z): [M + H]+ = 374.67. I.R. (cm–1): 3276; 1233; 1121; 1007. Anal. (C20H30N4O3) C, H, N calcd: C 64.15%, H 8.07%, N 14.96%; found: C 64.09%, H 8.09%, N 15.01%.
2-Methoxy-4-(((6-(pyrimidin-2-ylamino)hexyl)amino)methyl)phenol (18)
Compound 18 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.117 g, 0.60 mmol), 4-hydroxy-3-methoxybenzaldehyde (0.091 g, 0.60 mmol), and NaBH4 (0.068 g, 1.80 mmol) following procedure 3 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH/TEA 5:5:0.1, Rf = 0.37. White solid, 0.135 g, 68% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.21 (d, 2H, J = 4.8 Hz, pyrimidine); 6.91 (s, 1H, aromatic); 6.73 (s, 2H, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.72 (bs, 1H, pyrimidine-NH-); 3.80 (s, 3H, Ar-OCH3); 3.63 (s, 2H, Ar-CH2-NH-); 3.31 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.53 (t, 2H, J = 7.0 Hz, Ar-CH2-NH-CH2-); 1.58–1.51 (m, 2H, -NH-CH2-CH2-); 1.49–1.42 (m, 2H, -NH-CH2-CH2-); 1.39–1.32 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 149.2; 147.9; 128.2; 123.1; 116.3; 113.7; 111.0; 56.4; 53.4; 49.0; 42.0; 30.3; 28.8; 27.8; 27.7. ESI-MS (m/z): [M + H]+ = 330.60. I.R. (cm–1): 3261; 1284; 1258; 1158; 1130; 1034. m.p. = 87–89 °C. Anal. (C18H26N4O2) C, H, N calcd: C 65.43%, H 7.93%, N 16.96%; found: C 65.41%, H 7.92%, N 17.00%.
3-(((6-(Pyrimidin-2-ylamino)hexyl)amino)methyl)phenol (19)
Compound 19 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.117 g, 0.60 mmol), 3-hydroxybenzaldehyde (0.073 g, 0.60 mmol), and NaBH4 (0.068 g, 1.80 mmol) following procedure 3 of GP-D described above. In this case, the extraction was carried out with AcOEt instead of CH2Cl2. Column chromatography: silica gel, initially CH2Cl2/MeOH 1:1 (until the separation of the spot with Rf = 0.9) and subsequently CH2Cl2/MeOH/TEA 5:5:0.1, Rf = 0.67. After the chromatography, the compound was purified by crystallization in CH3CN. Pinkish solid, 0.104 g, 57% yield. 1H NMR (400 MHz) (acetone-d6) δ (ppm): 8.22 (d, 2H, J = 4.6 Hz, pyrimidine); 7.09 (t, 1H, J = 7.8 Hz, aromatic); 6.85 (s, 1H, aromatic); 6.79 (d, 1H, J = 7.5 Hz, aromatic); 6.67 (dd, 1H, J1 = 8.0 Hz, J2 = 1.8 Hz, aromatic); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 6.22 (bs, 1H, pyrimidine-NH-); 3.67 (s, 2H, Ar-CH2-NH-); 3.40–3.36 (m, 2H, pyrimidine-NH-CH2-); 2.56 (t, 2H, 6.88 Hz, Ar-CH2-NH-CH2-); 1.64–1.57 (m, 2H, -NH-CH2-CH2-); 1.53–1.45 (m, 2H, -NH-CH2-CH2-); 1.43–1.37 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 158.8; 141.8; 130.5; 120.5; 116.4; 115.2; 111.0; 54.3; 49.7; 42.1; 30.4; 30.2; 28.1; 27.8. ESI-MS (m/z): [M + H]+ = 301.02. I.R. (cm–1): 3289; 1287; 1239; 1228; 1160; 927. m.p. = 75–77 °C. Anal. (C17H24N4O) C, H, N calcd: C 67.97%, H 8.05%, N 18.65%; found: C 68.03%, H 8.04%, N 18.61%.
3-(((6-(Pyrimidin-2-ylamino)hexyl)amino)methyl)benzene-1,2-diol (20)
Compound 20 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.152 g, 0.78 mmol), 2,3-dihydroxybenzaldehyde (0.108 g, 0.78 mmol), and NaBH4 (0.089 g, 2.34 mmol) following procedure 3 of GP-D described above. The compound was purified by crystallization in CH3CN. Pinkish solid, 0.145 g, 59% yield. 1H NMR (400 MHz) (d6-DMSO) δ (ppm): 8.22 (d, 2H, J = 4.7 Hz, pyrimidine); 7.08 (t, 1H, J = 5.6 Hz, pyrimidine-NH-); 6.61 (dd, 1H, J1 = 7.5 Hz, J2 = 1.9 Hz, aromatic); 6.52–6.46 (m, 3H, 2H aromatic and 1H pyrimidine); 3.80 (s, 2H, Ar-CH2-NH-); 3.24 (q, 2H, J = 6.5 Hz, pyrimidine-NH-CH2-); 1.53–1.41 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-); 1.31–1.29 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). *Ar-CH2-NH-CH2- covered by d6-DMSO. 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 147.7; 146.9; 123.6; 121.1; 119.4; 115.6; 111.0; 51.4; 48.9; 42.0; 30.3; 29.5; 27.8; 27.6. ESI-MS (m/z): [M + H]+ = 316.87. I.R. (cm–1): 1289; 1194; 1102; 1068; 936. m.p. = 99–101 °C. Anal. (C17H24N4O2) C, H, N calcd: C 64.53%, H 7.65%, N 17.71%; found: C 64.56%, H 7.68%, N 17.67%.
N1-((2,6-Dichloropyridin-4-yl)methyl)-N6-(pyrimidin-2-yl)hexane-1,6-diamine (21)
Compound 21 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.117 g, 0.60 mmol), 2,6-dichloropyridine-4-carbaldehyde (0.106 g, 0.60 mmol), K2CO3 (1.659 g, 12.0 mmol), and NaBH4 (0.068 g, 1.8 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH 9:1, Rf = 0.4. White solid, 0.119 g, 57% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 8.21 (d, 2H, J = 4.8 Hz, pyrimidine); 7.36 (s, 2H, pyridine); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.69 (bs, 1H, pyrimidine-NH-); 3.74 (s, 2H, pyridine-CH2-NH-); 3.32 (q, 2H, J = 6.8 Hz, pyrimidine-NH-CH2-); 2.51 (t, 2H, J = 6.9 Hz, pyridine-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.49–1.42 (m, 2H, -NH-CH2-CH2-); 1.39–1.33 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 163.5; 159.2; 157.8; 151.5; 123.6; 111.0; 52.4; 50.1; 42.1; 30.5; 30.4; 28.1; 27.9. ESI-MS (m/z): [M + H]+ = 353.98 (100); 355.91 (65); 356.97 (10). I.R. (cm–1): 3235; 1216; 1155; 1129; 987. m.p. = 68–70 °C. Anal. (C16H21Cl2N5) C, H, N calcd: C 54.24%, H 5.97%, N 19.77%; found: C 54.17%, H 5.95%, N 19.82%.
N1-((1H-Indol-3-yl)methyl)-N6-(pyrimidin-2-yl)hexane-1,6-diamine (22)
Compound 22 was prepared using N1-(pyrimidin-2-yl)hexane-1,6-diamine (4) (0.076 g, 0.39 mmol), 1H-indole-3-carbaldehyde (0.057 g, 0.39 mmol), and NaBH4 (0.044 g, 1.17 mmol) following procedure 2 of GP-D described above. Column chromatography: silica gel, initially MeOH (until the separation of the spot with Rf = 0.77) and subsequently MeOH/TEA 10:0.2, Rf = 0.20. After the chromatography, the compound was filtered with CH2Cl2 to remove silica. Orange oil, 0.044 g, 36% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 9.18 (bs, 1H, indole-NH-); 8.21 (d, 2H, J = 4.8 Hz, pyrimidine); 7.62 (d, 1H, J = 7.9 Hz, indole); 7.38 (d, 1H, J = 8.1 Hz, indole); 7.15–7.10 (m, 2H, indole); 7.05–7.00 (m, 1H, indole); 6.50 (t, 1H, J = 4.8 Hz, pyrimidine); 5.70 (bs, 1H, pyrimidine-NH-); 3.89 (s, 2H, indole-CH2-NH-); 3.30 (q, 2H, J = 6.9 Hz, pyrimidine-NH-CH2-); 2.60 (t, 2H, J = 7.0 Hz, indole-CH2-NH-CH2-); 1.58–1.44 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-); 1.39–1.32 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CDCl3) δ (ppm): 162.5; 158.1; 136.4; 127.2; 123.4; 122.2; 119.7; 118.7; 113.5; 111.4; 110.4; 49.0; 44.3; 41.5; 29.5; 29.4; 27.1; 26.8. ESI-MS (m/z): [M + H]+ = 323.67. I.R. (cm–1): 3259; 1236; 1100; 1074; 1010; 983. Anal. (C19H25N5) C, H, N calcd: C 70.56%, H 7.79%, N 21.65%; found: C 70.47%, H 7.81%, N 21.72%.
N1-Benzyl-N5-(pyridin-2-yl)pentane-1,5-diamine (23)
Compound 23 was prepared using N1-(pyridin-2-yl)pentane-1,5-diamine (7) (0.086 g, 0.48 mmol), benzaldehyde (49 μL, d = 1.045 g/mL, 0.48 mmol), K2CO3 (1.327 g, 9.6 mmol), and NaBH4 (0.054 g, 1.44 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.36. Yellow oil, 0.071 g, 55% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.0 Hz, pyridine); 7.39–7.34 (m, 1H, pyridine); 7.33–7.20 (m, 5H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.09 (s, 1H, pyridine-NH-); 3.72 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.55 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.46 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-NH-); 1.43–1.35 (m, 2H, -NH-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.4; 147.8; 140.4; 138.7; 129.6; 129.5; 128.2; 112.9; 109.7; 54.4; 49.8; 42.5; 30.3; 30.0; 25.8. ESI-MS (m/z): [M + H]+ = 270.00. I.R. (cm–1): 3286; 1289; 1152; 982. Anal. (C17H23N3) C, H, N calcd: C 75.80%, H 8.61%, N 15.60%; found: C 75.86%, H 8.59%, N 15.55%.
N1-(4-Bromobenzyl)-N5-(pyridin-2-yl)pentane-1,5-diamine (24)
Compound 24 was prepared using N1-(pyridin-2-yl)pentane-1,5-diamine (7) (0.075 g, 0.42 mmol), 4-bromobenzaldehyde (0.078 g, 0.42 mmol), K2CO3 (1.161 g, 8.4 mmol), and NaBH4 (0.048 g, 1.26 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH 1:1, Rf = 0.33. White solid, 0.070 g, 48% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 4.9 Hz, J2 = 1.0 Hz, pyridine); 7.46 (m, 2H, aromatic); 7.39–7.34 (m, 1H, pyridine); 7.25 (m, 2H, aromatic); 6.50–6.47 (m, 1H, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.07 (bs, 1H, pyridine-NH-); 3.68 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.53 (t, 2H, J = 6.8 Hz, Ar-CH2-NH-CH2-); 1.59–1.35 (m, 6H, -NH- CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.4; 147.8; 140.0; 138.7; 132.5; 131.5; 121.8; 112.9; 109.7; 53.7; 49.8; 42.5; 30.3; 30.2; 25.8. ESI-MS (m/z): [M + H]+ = 347.54 (100); 349.60 (90). I.R. (cm–1): 3284; 1287; 1156; 1135; 1069; 1009; 980. m.p. = 65–68 °C. Anal. (C17H22BrN3) C, H, N calcd: C 58.63%, H 6.37%, N 12.07%; found: C 58.59%, H 6.36%, N 12.04%.
N1-Benzyl-N6-(pyridin-2-yl)hexane-1,6-diamine (25)
Compound 25 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.106 g, 0.55 mmol), benzaldehyde (56 μL, d = 1.045 g/mL, 0.55 mmol), K2CO3 (1.520 g, 11.0 mmol), and NaBH4 (0.062 g, 1.65 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 7:3, Rf = 0.3. Yellow oil, 0.083 g, 53% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.1 Hz, pyridine); 7.39–7.34 (m, 1H, pyridine); 7.33–7.20 (m, 5H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.1 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.10 (bs, 1H, pyridine-NH-); 3.72 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.54 (t, 2H, J = 7.0 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.50–1.43 (m, 2H, -NH-CH2-CH2-); 1.40–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 160.2; 148.8; 142.2; 137.8; 129.1; 129.0; 127.5; 112.8; 108.1; 54.3; 49.9; 42.2; 30.7; 30.2; 27.8; 27.6. ESI-MS (m/z): [M + H]+ = 283.98. I.R. (cm–1): 3279; 1288; 1152; 982. Anal. (C18H25N3) C, H, N calcd: C 76.28%, H 8.89%, N 14.83%; found: C 76.33%, H 8.87%, N 14.80%.
N1-(4-Bromobenzyl)-N6-(pyridin-2-yl)hexane-1,6-diamine (26)
Compound 26 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.110 g, 0.57 mmol), 4-bromobenzaldehyde (0.106 g, 0.57 mmol), K2CO3 (1.576 g, 11.4 mmol), and NaBH4 (0.065 g, 1.71 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.38. Yellow solid, 0.084 g, 40% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.1 Hz, pyridine); 7.46 (m, 2H, aromatic); 7.39–7.34 (m, 1H, pyridine); 7.25 (m, 2H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.08 (bs, 1H, pyridine-NH-); 3.68 (s, 2H, Ar-CH2-NH-); 3.22 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.51 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.51 (m, 2H, -NH-CH2-CH2-); 1.49–1.42 (m, 2H, -NH-CH2-CH2-); 1.40–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 160.2; 148.8; 141.8; 137.8; 132.0; 131.0; 120.6; 112.8; 108.1; 53.5; 49.8; 42.2; 30.7; 30.2; 27.8; 27.6. ESI-MS (m/z): [M + H]+ = 361.76 (90), 363.95 (100). I.R. (cm–1): 3269; 1156; 1109; 1067; 1011; 979. m.p. = 42–44 °C. Anal. (C18H24BrN3) C, H, N calcd: C 59.67%, H 6.68%, N 11.60%; found: C 59.69%, H 6.69%, N 11.63%.
N1-(4-Chlorobenzyl)-N6-(pyridin-2-yl)hexane-1,6-diamine (27)
Compound 27 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.108 g, 0.56 mmol), 4-chlorobenzaldehyde (0.079 g, 0.56 mmol), K2CO3 (1.548 g, 11.2 mmol), and NaBH4 (0.064 g, 1.68 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 1:1, Rf = 0.31. Yellow solid, 0.090 g, 51% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (d, 1H, J = 5.0 Hz, pyridine); 7.36 (t, 1H, J = 8.0 Hz, pyridine); 7.31 (s, 4H, aromatic); 6.48 (t, 1H, J = 6.0 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.07 (bs, 1H, pyridine-NH-); 3.70 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.6 Hz, pyridine-NH-CH2-); 2.52 (t, 2H, J = 6.8 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.49–1.42 (m, 2H, -NH-CH2-CH2-); 1.40–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 160.2; 148.8; 141.3; 137.8; 132.6; 130.6; 129.0; 112.8; 108.1; 53.5; 49.8; 42.2; 30.7; 30.1; 27.8; 27.6. ESI-MS (m/z): [M + H]+ = 318.13 (100); 320.13 (35). I.R. (cm–1): 3261; 1296; 1156; 1121; 1091; 1084; 1015; 982. m.p. = 41–43 °C. Anal. (C18H24ClN3) C, H, N calcd: C 68.02%, H 7.61%, N 13.22%; found: C 67.97%, H 7.59%, N 13.26%.
N1-(2-Methoxybenzyl)-N6-(pyridin-2-yl)hexane-1,6-diamine (28)
Compound 28 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.106 g, 0.55 mmol), 2-methoxybenzaldehyde (0.075 g, 0.55 mmol), K2CO3 (1.520 g, 11.0 mmol), and NaBH4 (0.062 g, 1.65 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, CH2Cl2/MeOH 7:3, Rf = 0.2. Yellow oil, 0.112 g, 65% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.1 Hz, pyridine); 7.38–7.34 (m, 1H, pyridine); 7.26–7.20 (m, 2H, aromatic); 6.94–6.88 (m, 2H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.07 (bs, 1H, pyridine-NH-); 3.80 (s, 3H, Ar-OCH3); 3.68 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.52 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.49–1.42 (m, 2H, -NH-CH2-CH2-); 1.40–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (CD3CN) δ (ppm): 160.2; 158.5; 148.8; 137.8; 130.2; 129.9; 128.8; 121.1; 112.8; 111.3; 108.1; 55.9; 49.9; 49.3; 42.2; 30.7; 30.2; 27.8; 27.7. ESI-MS (m/z): [M + H]+ = 313.88. I.R. (cm–1): 3281; 1289; 1239; 1151; 1100; 1050; 1029; 982. Anal. (C19H27N3O) C, H, N calcd: C 72.81%, H 8.68%, N 13.41%; found: C 72.82%, H 8.71%, N 13.38%.
N1-(Pyridin-2-yl)-N6-(3,4,5-trimethoxybenzyl)hexane-1,6-diamine (29)
Compound 29 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.102 g, 0.53 mmol), 3,4,5-trimethoxybenzaldehyde (0.104 g, 0.53 mmol), and NaBH4 (0.060 g, 1.59 mmol) following procedure 3 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH/TEA 5:5:0.2, Rf = 0.53. Orange oil, 0.133 g, 67% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.97–7.96 (m, 1H, pyridine); 7.38–7.34 (m, 1H, pyridine); 6.62 (s, 2H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.07 (bs, 1H, pyridine-NH-); 3.79 (s, 6H, Ar-(OCH3)2); 3.68 (s, 3H, -OCH3); 3.66 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.55 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.51–1.45 (m, 2H, -NH-CH2-CH2-); 1.41–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.5; 154.6; 147.8; 138.7; 138.5; 134.9; 112.9; 109.7; 107.1; 61.1; 56.6; 54.2; 49.5; 42.5; 30.3; 29.6; 28.0; 27.9. ESI-MS (m/z): [M + H]+ = 373.90. I.R. (cm–1): 3295; 1234; 1121; 1006. Anal. (C21H31N3O3) C, H, N calcd: C 67.53%, H 8.37%, N 11.25%; found: C 67.57%, H 8.34%, N 11.23%.
2-Methoxy-4-(((6-(pyridin-2-ylamino)hexyl)amino)methyl)phenol (30)
Compound 30 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.102 g, 0.53 mmol), 4-hydroxy-3-methoxybenzaldehyde (0.081 g, 0.53 mmol), and NaBH4 (0.060 g, 1.59 mmol) following procedure 3 of GP-D with some modifications: after the reaction with NaBH4, the addition of HCl, and the removal of the solvent, the residue was washed with warm AcOEt and then the solid was diluted with 20 mL of a saturated aqueous solution of NaHCO3 and extracted with AcOEt (3 × 20 mL). The organic layer was dried over Na2SO4, and the solvent was removed under vacuum. Yellow oil, 0.140 g, 80% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.1 Hz, pyridine); 7.39–7.34 (m, 1H, pyridine); 6.92 (s, 1H, aromatic); 6.74 (s, 2H, aromatic); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.08 (bs, 1H, pyridine-NH-); 3.82 (s, 3H, -OCH3); 3.63 (s, 2H, Ar-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.53 (t, 2H, J = 7.0 Hz, Ar-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.50–1.43 (m, 2H, -NH-CH2-CH2-); 1.40–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.5; 149.1; 147.8; 147.3; 138.7; 130.4; 122.7; 116.2; 113.4; 112.9; 109.7; 56.4; 54.0; 49.4; 42.5; 30.3; 29.7; 28.1; 27.9. ESI-MS (m/z): [M + H]+ = 330.20. I.R. (cm–1): 1274; 1152; 1125; 1033; 982. Anal. (C19H27N3O2) C, H, N calcd: C 69.27%, H 8.26%, N 12.76%; found: C 69.19%, H 8.28%, N 12.80%.
3-(((6-(Pyridin-2-ylamino)hexyl)amino)methyl)phenol (31)
Compound 31 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.075 g, 0.39 mmol), 3-hydroxybenzaldehyde (0.048 g, 0.39 mmol), and NaBH4 (0.044 g, 1.17 mmol) following procedure 3 of GP-D described above. In this case, the extraction was carried out with AcOEt instead of CH2Cl2. Column chromatography: silica gel, AcOEt/MeOH/TEA 5:5:0.1, Rf = 0.22. White solid, 0.062 g, 53% yield. 1H NMR (400 MHz) (acetone-d6) δ (ppm): 7.98–7.96 (m, 1H, pyridine); 7.36–7.32 (m, 1H, pyridine); 7.09 (t, 1H, J = 7.8 Hz, aromatic); 6.86 (s, 1H, aromatic); 6.79 (d, 1H, J = 7.5, aromatic); 6.67 (dd, 1H, J1 = 7.8 Hz, J2 = 1.8 Hz, aromatic); 6.46–6.43 (m, 2H, pyridine); 5.64 (bs, 1H, pyridine-NH-); 3.67 (s, 2H, Ar-CH2-NH-); 3.33–3.28 (m, 2H, pyridine-NH-CH2-); 2.56 (t, 2H, J = 6.9 Hz, Ar-CH2-NH-CH2-); 1.63–1.56 (m, 2H, -NH-CH2-CH2-); 1.53–1.46 (m, 2H, -NH-CH2-CH2-); 1.43–1.34 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.4; 158.9; 147.8; 140.4; 138.7; 130.6; 120.7; 116.6; 115.6; 112.9; 109.7; 53.9; 49.5; 42.5; 30.3; 29.6; 28.0; 27.9. ESI-MS (m/z): [M + H]+ = 300.40. I.R. (cm–1): 3270; 1281; 1153; 1112; 926. m.p. = 86–88 °C. Anal. (C18H25N3O) C, H, N calcd: C 72.21%, H 8.42%, N 14.03%; found: C 72.17%, H 8.44%, N 14.06%.
3-(((6-(Pyridin-2-ylamino)hexyl)amino)methyl)benzene-1,2-diol (32)
Compound 32 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.102 g, 0.53 mmol), 2,3-dihydroxybenzaldehyde (0.073 g, 0.53 mmol), and NaBH4 (0.060 g, 1.59 mmol) following procedure 1 of GP-D described above. The compound was purified by washing with CH3CN. White solid, 0.084 g, 50% yield. 1H NMR (400 MHz) (MeOD) δ (ppm): 7.89–7.88 (m, 1H, pyridine); 7.42–7.38 (m, 1H, pyridine); 6.69 (t, 1H, J = 4.6 Hz, aromatic); 6.56 (d, 2H, J = 4.7 Hz, aromatic); 6.52–6.48 (m, 2H, pyridine); 3.90 (s, 2H, Ar-CH2-NH-); 3.24 (t, 2H, J = 7.0 Hz, pyridine-NH-CH2-); 2.67 (t, 2H, J = 7.2 Hz, Ar-CH2-NH-CH2-); 1.64–1.55 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-); 1.42–1.40 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (d6-DMSO) δ (ppm): 159.0; 147.6; 146.0; 145.2; 136.5; 123.7; 118.8; 118.0; 114.3; 111.2; 107.9; 51.1; 48.0; 40.7; 29.0; 28.9; 26.59; 26.55. ESI-MS (m/z): [M + H]+ = 315.92. I.R. (cm–1): 1292; 1280; 1199; 1067. m.p. = 112–114 °C. Anal. (C18H25N3O2) C, H, N calcd: C 68.54%, H 7.99%, N 13.32%; found: C 68.61%, H 7.98%, N 13.28%.
N1-((2,6-Dichloropyridin-4-yl)methyl)-N6-(pyridin-2-yl)hexane-1,6-diamine (33)
Compound 33 was prepared using N1-(pyridin-2-yl)hexane-1,6-diamine (8) (0.108 g, 0.56 mmol), 2,6-dichloropyridine-4-carbaldehyde (0.099 g, 0.56 mmol), K2CO3 (1.548 g, 11.2 mmol), and NaBH4 (0.064 g, 1.68 mmol) following procedure 1 of GP-D described above. Column chromatography: silica gel, AcOEt/MeOH 9.5:0.5, Rf = 0.32. White solid, 0.131 g, 66% yield. 1H NMR (400 MHz) (CD3CN) δ (ppm): 7.96 (dd, 1H, J1 = 5.0 Hz, J2 = 1.0 Hz, pyridine); 7.39–7.24 (m, 3H, 1H pyridine and 2H 2,6-dichloropyridine); 6.48 (ddd, 1H, J1 = 7.0 Hz, J2 = 5.0 Hz, J3 = 0.8 Hz, pyridine); 6.40 (d, 1H, J = 8.4 Hz, pyridine); 5.07 (bs, 1H, pyridine-NH-); 3.75 (s, 2H, 2,6-dichloropyridine-CH2-NH-); 3.23 (q, 2H, J = 6.9 Hz, pyridine-NH-CH2-); 2.51 (t, 2H, J = 6.8 Hz, 2,6-dichloropyridine-CH2-NH-CH2-); 1.59–1.52 (m, 2H, -NH-CH2-CH2-); 1.49–1.43 (m, 2H, -NH-CH2-CH2-); 1.41–1.27 (m, 4H, -NH-CH2-CH2-CH2-CH2-CH2-CH2-NH-). 13C NMR (100 MHz) (MeOD) δ (ppm): 160.5; 157.8; 151.5; 147.8; 138.7; 123.6; 112.9; 109.7; 52.4; 50.1; 42.6; 30.5; 30.4; 28.1; 28.0. ESI-MS (m/z): [M + H]+ = 353.05 (90), 354.98 (100), 356.91 (15). I.R. (cm–1): 3237; 1246; 1212; 1154; 1128; 1085; 986. m.p. = 61–64 °C. Anal. (C17H22Cl2N4) C, H, N calcd: C 57.79%, H 6.28%, N 15.86%; found: C 57.83%, H 6.30%, N 15.81%.
Enzymatic Assays
Materials and Methods
Electric eel AChE (EeAChE, EC 3.1.1.7), equine BChE (EC 3.1.1.8), acetylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), tacrine, and donepezil, used as the reference standard, were purchased from Sigma-Aldrich (Milan, Italy). All other chemical and biological reagents and solvents used were of the highest analytical, commercially available grade. The water, utilized for the preparation of the phosphate buffer and of the compound solutions, was distilled and filtered on nylon membrane filters with a 0.2 μm pore size with the Millipore Filtration apparatus before each use. Micropipettes Labmate (BRAND Dig. 10–100 μL; Dig. 100–1000 μL; Dig. 0.1–2 μL) and Transferpette (HIGH TECH LAB LM200: 20–200 μL; LM5000: 1000–5000 μL) were used to collect the samples. The assays were carried out by a double beam UV–vis Lambda 40 Perkin Elmer spectrophotometer using optical polystyrene cuvettes (10 × 10 × 45 mm, 340–800 nm optical transparency); each measure was repeated at least in triplicate. For data processing, UV-WIN Lab version 2.0 (Perkin Elmer Corporation) and SigmaPlot version 8.02 were used. The spectrophotometric method of Ellman25 with minor modifications was used to evaluate the inhibition of ChEs. This method is based on the reaction of released thiocholine to give a colored product, at a wavelength of 412 nm, with the chromogenic reagent DTNB. The absorbance was recorded at 412 nm between 0 and 1.6 min, and the absorbance variation, utilized for the kinetic assay, was measured between 0.5 and 1.5 min to allow the stabilization of the UV–vis lamp and of the solution. The method is extremely sensitive to variations in the order of microliters: the standard deviations (less than 5%) of the values obtained are compatible with the experimental errors associated with the use of micropipettes. Each tested compound was dissolved in the opportune quantity of DMSO to obtain a final cuvette DMSO content <0.033%, which does not affect the enzyme activity. EeAChE and eqBChE were periodically tested to evaluate the effective enzymatic activity.
Determination of Kinetic Parameters of EeAChE and eqBChE
The Michaelis constant (Km) and the maximum reaction velocity (Vmax) of EeAChE and eqBChE were determined using acetylthiocholine as substrate. For the assay, five different concentrations of acetylthiocholine were used (range 50–600 μM for EeAChE and 100–800 μM for eqBChE).
Three milliliters of 0.1 M phosphate buffer (pH = 7.4) containing DTNB (0.25 mM) and EeAChE (0.083 UmL–1) or eqBChE (0.083 UmL–1) was placed in a polystyrene cuvette of 1.0 cm path length. With this solution, the blank was made. To start the reaction, the opportune volume of a solution in 0.1 M phosphate buffer (pH = 7.4) of acetylthiocholine (10 mM or 20 mM, to introduce in the cuvette volumes ranging from 15 to 120 μL) was added to obtain the desired final concentration of substrate. Each determination was repeated for five times. The absorbance variations were measured at 412 nm at 25 °C between 0.5 and 1.5 min. From these data, the values of the enzymatic hydrolysis rate, expressed as μmol of substrate hydrolyzed in 1 min by an enzymatic unit, were obtained. The recorded data were analyzed by the enzyme kinetic module of SigmaPlot (version 8.02) based on the Michaelis–Menten equation.
Km = 145.8 ± 11.5 μM and Vmax = 348.6 ± 9.9 μmol/min, with R2 = 0.977, were determined for EeAChE. Km = 443.9 ± 16.5 μM and Vmax = 178.5 ± 3.1 μmol/min, with R2 = 0.997, were determined for eqBChE.
Percent Inhibition of EeAChE and eqBChE
For all the synthesized compounds, the percentages of inhibition toward AChE of Electrophorus electricus (EeAChE) and equine BChE (eqBChE) were evaluated.
Three milliliters of a solution in 0.1 M phosphate buffer (pH = 7.4) containing DTNB (0.25 mM) and EeAChE (0.083 UmL–1) or eqBChE (0.083 UmL–1) was placed in a polystyrene cuvette of 1.0 cm path length; 1 μL of a solution in DMSO of the tested compound was added to obtain a cuvette concentration range of 9–0.09 μM. With this solution, the blank was made. To start the reaction, 30 μL of a solution in 0.1 M phosphate buffer (pH = 7.4) of acetylthiocholine (10 mM) was added to obtain a final concentration of acetylthiocholine equal to 100 μM. The increase in the absorbance, due to the production of the yellow 2-nitro-5-thiobenzoic anion, was recorded at 412 nm at 25 °C, and the absorbance variation was measured between 0.5 and 1.5 min. As a control, an identical solution of the enzyme without the inhibitor was processed following the same protocol to determine 100% of the enzyme activity. Each experiment was repeated at least in triplicate. The potency of each compound to inhibit EeAChE or eqBChE activity was expressed as percent inhibition calculated using the following equation:
where Ai and Ac represent the average of the absorbance variation in the presence of inhibitor and without inhibitor, respectively.
Determination of Constant and Mechanism of Inhibition vs EeAChE and eqBChE
The constants and the mechanisms of inhibition were determined for a selection of compounds that are among the most potent as inhibitors of EeAChE and eqBChE (9, 13, 19, 23, 25, and 28 toward EeAChE; 18, 22, 25, 26, 28, and 30 toward eqBChE). For each compound, a 500 μM stock solution was prepared in H2O/DMSO or in 10–2 M HCl and diluted in water to prepare solutions of opportune concentrations to introduce in the cuvette volumes ranging from 30 to 100 μL, to obtain the desired final concentrations. The maximum amount of DMSO used for the preparation of the stock solution was calculated to have a DMSO content in cuvette <0.033%.
Three milliliters of 0.1 M phosphate buffer (pH = 7.4) containing DTNB (0.25 mM) and EeAChE (0.083 UmL–1) or eqBChE (0.083 UmL–1) was mixed with the opportune volume of the inhibitor solution to obtain a final inhibitor concentration in cuvette between 90 and 1800 nM. With this solution, the blank was made. The reaction was started by adding to the enzyme–inhibitor mixture the proper volume of a solution in 0.1 M phosphate buffer (pH = 7.4) of acetylthiocholine (10 mM) to obtain a final concentration of substrate equal to 100–400 μM. Each determination was repeated five times. The absorbance variations were measured at 412 nm at 25 °C between 0.5 and 1.5 min. From these data, the values of the enzymatic hydrolysis rate, expressed as μmol of substrate hydrolyzed in 1 min by an enzymatic unit, were obtained.
To determine the inhibition constants (Ki) and the inhibition mechanisms, the rates of hydrolysis at three different concentrations of substrate in the presence of five different concentrations of inhibitor were measured. The recorded data were analyzed by the enzyme kinetic module of SigmaPlot (version 8.02) plotting the reciprocal of the rate of hydrolysis (1/v, min/μM) vs the concentration of the inhibitor (nM) according to Dixon’s method26 to find the best fitting model of inhibition, as indicated by the calculated linear regression coefficient R2.
Determination of IC50vs EeAChE for Compound 20, Tacrine, and Donepezil
The IC50 value of compound 20vs EeAChE and the IC50 values of tacrine and donepezil vs both ChEs were calculated. For this purpose, eight different inhibitor solutions were prepared. For compound 20, solutions in 10–2 M HCl were prepared, having suitable concentrations, so as to add 20 μL of these solutions to the assay mixture to obtain a final concentration between 0.05 and 10 μM; for tacrine and donepezil, solutions in DMSO were prepared, having suitable concentrations, so as to add 1 μL of these solutions to the assay mixture to obtain a final concentration between 0.5 and 27,000 nM. For each inhibitor concentration, the percentage of inhibition in the presence of EeAChE or eqBChE (0.0833 U mL–1) and acetylthiocholine (100 μM) was measured as described above. The recorded data were analyzed by the enzyme kinetic module of SigmaPlot plotting the percentages of inhibition as a function of the different concentrations of the inhibitor, expressed in logarithmic scale, to obtain the sigmoid graph, from which it was possible to determine the IC50 value of the tested compound. The IC50 was confirmed by repeating the experiment twice.
Computational Studies and ADME Prediction
All computational studies were carried out by means of Schrödinger Suite 2018-1.42 The X-ray crystallographic structures of the hAChE in complex to donepezil (PDB code:4EY7)13 and the hBChE in complex to the naphthamide derivative (PDB code:5NN0)27 were used. hChEs structures were prepared by using the Maestro Protein Preparation Wizard43 tool and were refined to optimize hydrogen bonds and energy minimized by using OPLS_2005 as force field at pH 7.4.44−46 The homology between the human and not human ChE isoforms used for the enzymatic assays was assessed by the sequence alignment performed by Prime.47
All derivatives were submitted to 5000 steps of Monte Carlo conformational search applied to all rotatable bonds. The water solvent effect was considered using the implicit model GB/SA.48 The global minimum energy structures were used to carry out molecular docking simulations.
Then, target binding sites were defined by means of a regular grid box of about 27,000 Å3 centered on the catalytic serine residues. All docking simulations were computed using the Glide49 ligand flexible algorithm at the standard-precision (SP) level. The best docked poses of the most potent inhibitors resulted by in vitro assays of pyrimidine and pyridine derivatives were submitted to 250 ns of molecular dynamic (MD) simulations by using Desmond ver. 4.2.50 The system was solvated in the SPC explicit solvent model, and counter ions were added to neutralize the system net charge. After optimization of the solvated model, we relaxed the system with the Martyna–Tobias–Klein isobaric–isothermal ensemble (MTK_NPT). This preliminary stage included two energy minimizations of 2000 steps: in the first run, the system was restrained with a force constant of 50 kcal mol–1 A–1, while in the second one, all the system was released without any restrains. The following conditions for MDs were used: the NPT ensemble, a temperature of 300 K, a pressure of 1 bar, a Berendsen thermostat–barostat, and a recording interval equal to 250 ps both for energy and for trajectory collecting 1000 frames for each simulation.
To identify possible PAINS, all synthesized final molecules were screened using in silico public tools available at https://zinc15.docking.org/patterns/home/ and http://www.swissadme.ch/.
ADME descriptors of the most potent compounds were predicted using the SwissADME public server.38 The following ADME parameters have been selected: molecular weight (MW); number of H-bond acceptors (H-b acc); number of H-bond donors (H-b don); number of heavy atoms (heavy atoms); number of rotatable bonds (rot bonds); topological polar surface area in Å2 (TPSA); octanol/water partition coefficient (MLogP); water solubility (LogS ESOL); water solubility class (Sol class); gastrointestinal absorption (GI); blood–brain barrier permeation (BBB); and number of Lipinski’s rule of five violations (Lipinski viol).39
Chelation Studies
Materials and Methods
FeCl3·6H2O, CuSO4·5H2O, Zn(NO3)2·6H2O, and MeOH used for chelation studies were purchased from Sigma-Aldrich. Micropipettes Labmate (BRAND Dig. 10–100 μL; Dig. 100–1000 μL) and Transferpette (HIGH TECH LAB LM200: 20–200 μL; LM5000: 1000–5000 μL) were used to collect the samples. The assays were carried out by a double beam UV–vis Lambda 40 Perkin Elmer spectrophotometer using quartz cuvettes (Optech, type S/Q/10). Data were elaborated using UV-WIN Lab Version 2.0 (Perkin Elmer Corporation), Microsoft Excel 2010, and Spekwin32. For each measurement, the absorbance values were recorded immediately after mixing the solutions.
UV–Vis Titration
For the UV–vis titration, ligand solutions in MeOH were prepared: 10–3 M for compounds 9, 13, 20, 25, 28, and 30; 5 × 10–3 M for compounds 18 and 19. Stock solutions (1 M) of CuSO4·5H2O and Zn(NO3)2·6H2O in water and 0.1 M of FeCl3·6H2O in MeOH were prepared and diluted with MeOH to obtain different solutions, having suitable concentrations, so as to add 2.85 mL of these solutions to 150 μL of the ligand solution to obtain in 3 mL of the assay mixture a final molar ratio of metal ion to ligand between 0.1 and 20. UV–vis spectra were recorded from 210 to 450 nm.
For the first measure, 150 μL of the ligand solution was diluted in 2.85 mL of MeOH into a sample cuvette, while only MeOH was placed in the reference cuvette and the UV–vis spectrum was recorded. For the subsequent measurements, 2.85 mL of the opportune metal solution was placed in both the sample cuvette and reference cuvette; then 150 μL of the ligand solution was added to the sample cuvette, while 150 μL of MeOH was added to the reference cuvette, and the UV–vis spectra were recorded. The spectra thus obtained were superimposed to observe the variations present between the ligand alone and in the presence of an increasing amount of metal.
Job’s Plot
To determine the stoichiometric coefficients of the complexes, Job’s method33 was used, which requires to mix, in appropriate proportions, an equimolar solution of metal ion and ligand so that the final volume and the total moles present in the cuvette are equal for each measurement. The absorbance values were recorded at the wavelengths, extrapolated from the UV–vis titration spectra, where the maximum variation of absorbance was observed. At the same time, two solutions in MeOH of equal concentration were prepared, one of ligand and one of metal ion, diluting the solutions 1 M of CuSO4·5H2O and Zn(NO3)2·6H2O and 0.1 M of FeCl3·6H2O previously prepared. The concentrations of the solutions used and the wavelengths in which the absorbance was recorded were as follows: 4.96 × 10–4 M for 18 and Fe3+ −258, 375 nm; 5.02 × 10–4 M for 18 and Cu2+ −330 nm; 5.66 × 10–4 M for 19 and Fe3+ −292, 375 nm; 5.66 × 10–4 M for 19 and Cu2+ −312, 330 nm; 3.10 × 10–4 M for 20 and Fe3+ −370 nm; 3.35 × 10–4 M for 20 and Cu2+ −330 nm; 3.00 × 10–4 M for 25 and Fe3+ −251, 318 nm; 3.99 × 10–4 M for 25 and Cu2+ −330 nm; 3.00 × 10–4 M for 28 and Fe3+ −316, 374 nm; 3.99 × 10–4 M for 28 and Cu2+ −330 nm; 3.28 × 10–4 M for 30 and Fe3+ −310, 372 nm; 3.58 × 10–4 M for 30 and Cu2+ −312, 330 nm.
For each determination, 9–24 measurements were made, introducing appropriate volumes of metal and ligand solutions in the sample cuvette to obtain mole fractions of the ligand in the range of 0.1–0.95, while appropriate volumes of ligand solution and MeOH were placed in the reference cuvette, to obtain the same concentration of the ligand in the sample cuvette. The absorbance value of the metal ion was calculated by the Lambert–Beer relation, being known its extinction coefficients ε at used wavelengths; for each measure, the nominal absorbance due to the metal ion was algebraically subtracted from the recorded absorbance values to obtain the exclusive absorbance variations due to the complex formation. The resulting ΔA values were reported in a graph as a function of the mole fraction of the ligand, and through the intersections of the linear regression lines, the mole fraction X, which caused the maximum variation in absorbance, was determined and used to calculated the value of the coefficient n, which corresponds to the number of ligand molecules per cation, applying the following equation:
Antioxidant Activity
Materials and Methods
2,2-Diphenyl-1-picrylhydrazyl (DPPH), MeOH, and ascorbic acid, used as the reference standard, were purchased from Sigma-Aldrich. Micropipettes Labmate (BRAND Dig. 10–100 μL; Dig. 100–1000 μL) and Transferpette (HIGH TECH LAB LM200: 20–200 μL; LM5000: 1000–5000 μL) were used to collect the samples. The assays were carried out by a double beam UV–vis Lambda 40 Perkin Elmer spectrophotometer using optical polystyrene cuvettes (10 × 10 × 45 mm, 340–800 nm optical transparency); each measure was repeated at least in triplicate. For data processing, UV-WIN Lab version 2.0 (Perkin Elmer Corporation) was used. The antioxidant activity of the tested compounds was evaluated by the DPPH method34 based on the ability of antioxidant compounds to react with the stable radical DPPH, which has a maximum absorption at about 515 nm. Thus, by measuring the decrease in absorbance over time of DPPH solutions to which increasing amounts of the tested compound are added, the antioxidant activity of the compound can be determined.
Antioxidant Activity Tests
To assess the antioxidant activity of 18, 19, 20, and 30, at first, the decrease in absorbance over time at 515 nm of a solution of DPPH and the tested compound was recorded until the completion of the reaction. To 2.90 mL of MeOH, 60 μL of a DPPH solution in MeOH (∼1.2 mM) and 40 μL of an antioxidant solution in MeOH (4.5 mM) were added to have in the cuvette concentrations of DPPH equal to about 25 μM and of antioxidant equal to 60 μM. The exact DPPH concentration was calculated through the Lambert–Beer relation, known the extinction coefficient of DPPH at 515 nm, equal to 1.25 × 104.34 The absorbance was recorded until the plateau was reached.
Subsequently, for compounds that showed antioxidant activity (18, 20, and 30), the EC50 values were determined. To obtain the initial DPPH concentration, 2.94 mL of MeOH and 60 μL of the DPPH solution in MeOH (∼1.2 mM) were placed in a polystyrene cuvette and the absorbance was recorded at 515 nm. The exact initial DPPH concentration was calculated through the Lambert–Beer relation. For subsequent measurements, the cuvettes were set up by introducing the opportune volume of MeOH to reach a total volume of 3 mL, 60 μL of the DPPH solution in MeOH (∼1.2 mM, to have a final concentration of ∼25 μM), and the appropriate volumes of the antioxidant stock solution in MeOH to have final concentrations between 1.5 and 12 μM for 20 and between 5 and 60 μM for 18 and 30; the absorbance was recorded at 515 nm at the plateau and then after 1 min for compound 20 and after 90 min for compounds 18 and 30. Each measure was repeated four times. With absorbance data, the percentages of residual DPPH at steady state were obtained using the following equation:
where Ac is the average of absorbance in the presence of antioxidant at plateau and A0 is the average of the initial absorbance of DPPH.
By plotting the percentage of residual DPPH at the steady state as a function of the molar ratio of the antioxidant to initial DPPH, it is possible to obtain the value of EC50, defined by the ratio
were nao is the moles of antioxidant necessary to reduce the initial concentration of DPPH by 50% and nDPPH is the initial moles of DPPH.35 The same procedure was performed using ascorbic acid as the antioxidant at cuvette concentrations between 1.5 and 18 μM, recording the absorbance at 515 nm after 2 min from the preparation of the cuvettes. In this way, an EC50 equal to 0.265 ± 0.007 was measured, in accordance with literature.35
Inhibition of Amyloid and Tau Aggregation
Cloning and Overexpression of the Aβ42 Peptide
E. coli competent cells BL21 (DE3) were transformed with the pET28a vector (Novagen, Inc., Madison, WI, USA) carrying the DNA sequence of Aβ42. Because of the addition of the initiation codon ATG in front of both genes, the overexpressed peptide contains an additional methionine residue at its N terminus. For overnight culture preparation, an amount of 10 mL of the M9 minimal medium containing 50 μg·mL–1 of kanamycin was inoculated with a colony of BL21 (DE3) bearing the plasmid to be expressed at 37 °C. For expression of the Aβ42 peptide, the required volume of overnight culture to obtain 1:500 dilution was added into the fresh M9 minimal medium containing 50 μg·mL–1 of kanamycin and 250 μM Th-S. The bacterial culture was grown at 37 °C and 250 rpm. When the cell density reached OD600 = 0.6, an amount of 980 μL of culture was transferred into Eppendorf tubes of 1.5 mL with 10 μL of each compound to be tested in DMSO and 10 μL of isopropyl 1-thio-β-d-galactopyranoside (IPTG) at 100 mM. The final concentration of the drug was fixed at 100 μM. The samples were grown overnight at 37 °C and 1400 rpm using a ThermoMixer (Eppendorf, Hamburg, Germany). As negative control (maximal amyloid presence), the same amount of DMSO without the drug was added in the sample. In parallel, non-induced samples (in the absence of IPTG) were also prepared and used as positive controls (non-amyloid presence). In addition, these samples were used to assess the potential intrinsic toxicity of the compounds and to confirm the correct bacterial growth.
Cloning and Overexpression of the Tau Protein
E. coli BL21 (DE3) competent cells were transformed with pTARA containing the RNA-polymerase gene of T7 phage (T7RP) under the control of the promoter PBAD. E. coli BL21 (DE3) with pTARA competent cells were transformed with the pRKT42 vector encoding four repeats of the tau protein in two inserts. For overnight culture preparation, 10 mL of the M9 medium containing 0.5% of glucose, 50 μg·mL–1 of ampicillin, and 12.5 μg·mL–1 of chloramphenicol was inoculated with a colony of BL21 (DE3) bearing the plasmids to be expressed at 37 °C. For expression of the tau protein, the required volume of overnight culture to obtain 1:500 dilution was added to the fresh M9 minimal medium containing 0.5% of glucose, 50 μg·mL–1 of ampicillin, 12.5 μg·mL–1 of chloramphenicol, and 250 μM Th-S. The bacterial culture was grown at 37 °C and 250 rpm. When the cell density reached OD600 = 0.6, an amount of 980 μL of culture was transferred into Eppendorf tubes of 1.5 mL with 10 μL of each compound to be tested in DMSO and 10 μL of arabinose at 25%. The final concentration of the drug was fixed at 100 μM. The samples were grown overnight at 37 °C and 1400 rpm using a ThermoMixer (Eppendorf, Hamburg, Germany). As negative control (maximal presence of tau), the same amount of DMSO without the drug was added in the sample. In parallel, non-induced samples (in the absence of arabinose) were also prepared and used as positive controls (absence of tau). In addition, these samples were used to assess the potential intrinsic toxicity of the compounds and to confirm the correct bacterial growth.
Thioflavin S (Th-S) Steady-State Fluorescence
Th-S (T1892) and other chemical reagents were purchased from Sigma (St. Louis, MO). The Th-S stock solution (2500 mM) was prepared in double-distilled water purified through a Milli-Q system (Millipore, USA). Th-S fluorescence and absorbance were tracked using a DTX 800 plate reader Multimode Detector equipped with a Multimode Analysis Software (Beckman-Coulter, USA). Filters of 430/35 and 485/20 nm were used for the excitation and emission wavelengths, respectively. Filters of 535/25 nm were also used for the absorbance determination. To normalize the Th-S fluorescence as a function of the bacterial concentration, OD600 was obtained using a Shimadzu UV-2401 PC UV–vis spectrophotometer (Shimadzu, Japan). Note that the fluorescence normalization was carried out considering as 100% the Th-S fluorescence of the bacterial cells expressing the peptide or protein in the absence of drug and 0% the Th-S fluorescence of the bacterial cells not expressing the peptide or protein. A minimum of five independent assays (with three replicates for assay) was performed for each tested compound. More assays were performed to obtain a SEM < 5% with a maximum of 10 independent assays.
Cytotoxicity Assays
The compounds 9, 13, 19, 20, 22, 23, 25, 26, and 28 were evaluated for cell viability effects using the MTT assay. Briefly, the U-87 MG cell line from the human brain (glioblastoma astrocytoma) (ATCC, Manassas, VA, USA) was seeded into 96-well microtiter plates (Nunclon, Nunc, Germany) at a density of 1.5 × 104 cells/well.51,52 After 24 h, cells were exposed to increased concentrations of tested compounds (1, 5, 10, and 50 μM) in the cell culture medium. After an incubation time of 24 h, the medium was replaced by a new fresh medium containing 0.5 mg/mL MTT (Sigma, Deisenhofen, Germany). After 2 h at 37 °C in 5% CO2, unreacted dye was removed and cells were dissolved by DMSO (Merck, Darmstadt, Germany). Absorbance was read at 570 nm by a microtiter spectrophotometer plate reader. The data were expressed as absorbance relative to untreated cells in the same experiment and standardized to 100%. All data points were performed in triplicate and at least three independent experiments.
Acknowledgments
This work was financially supported by the Italian Ministry for Education, Universities and Research (MIUR), Sapienza University of Rome, and Istituto Superiore di Sanità, ISS (Ministry of Health, ISS funding).
Glossary
Abbreviations Used
- AD
Alzheimer’s disease
- ACh
acetylcholine
- Aβ
β-amyloid peptide
- ChAT
choline acetyl transferase
- ChEIs
cholinesterase inhibitors
- ChEs
cholinesterases
- FDA
Food and Drug Administration
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- CAS
catalytic active site
- PAS
peripheral anionic site
- hAChE
human AChE
- hBChE
human BChE
- TEA
triethylamine
- TFA
trifluoroacetic acid
- DMF
dimethylformamide
- EeAChE
Electrophorus electricus AChE
- eqBChE
equine BChE
- Ki
inhibition constant
- RMSD
root mean square deviation
- MD
molecular dynamics
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- TPSA
topological polar surface area in Å2
- MLogP
octanol/water partition coefficient
- LogS ESOL
water solubility
- GI
gastrointestinal absorption
- BBB
blood–brain barrier permeation
- PAINS
pan assay interference compounds
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00485.
4EY7_9 (PDB)
4EY7_13 (PDB)
4EY7_19 (PDB)
4EY7_20 (PDB)
4EY7_22 (PDB)
4EY7_30 (PDB)
Molecular formula strings and some data (CSV)
The plots of enzyme inhibition studies on selected compounds, additional figures of molecular docking studies, UV–vis titration spectra, and Job’s plots for selected compounds (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Alzheimer’s Disease Statistics. https://alzheimersnewstoday.com/alzheimers-disease-statistics/.
- Wimo A.; Guerchet M.; Ali G. C.; Wu Y. T.; Prina A. M.; Winblad B.; Jönsson L.; Liu Z.; Prince M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dementia 2017, 13, 1–7. 10.1016/j.jalz.2016.07.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M. J.; Wimo A.; Guerchet M. M.; Ali G. C.; Wu Y.-T.; Prina M.. World Alzheimer Report 2015-The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends; Alzheimer’s Disease International: 2015. [Google Scholar]
- Du X.; Wang X.; Geng M. Alzheimer’s disease hypothesis and related therapies. Transl. neurodegener. 2018, 7, 1–7. 10.1186/s40035-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle J. T.; Price D. L.; Delong M. R. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Whitehouse P. J.; Price D. L.; Clark A. W.; Coyle J. T.; DeLong M. R. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Annal. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1981, 10, 122–126. 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- Bowen D. M.; Smith C. B.; White P.; Davison A. N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain: J. neurol. 1976, 99, 459–496. 10.1093/brain/99.3.459. [DOI] [PubMed] [Google Scholar]
- Drachman D. A.; Leavitt J. Human memory and the cholinergic system: a relationship to aging?. Arch. neurol. 1974, 30, 113–121. 10.1001/archneur.1974.00490320001001. [DOI] [PubMed] [Google Scholar]
- Marucci G.; Buccioni M.; Dal Ben D.; Lambertucci C.; Volpini R.; Amenta F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. 10.1016/j.neuropharm.2020.108352. [DOI] [PubMed] [Google Scholar]
- Greig N. H.; Utsuki T.; Yu Q.-s.; Zhu X.; Holloway H. W.; Perry T.; Lee B.; Ingram D. K.; Lahiri D. K. A new therapeutic target in Alzheimer’s disease treatment: attention to butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. 10.1185/03007990152673800. [DOI] [PubMed] [Google Scholar]
- Mesulam M.-M.; Guillozet A.; Shaw P.; Levey A.; Duysen E. G.; Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. 10.1016/S0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- Brus B.; Košak U.; Turk S.; Pišlar A.; Coquelle N.; Kos J.; Stojan J.; Colletier J.-P.; Gobec S. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2014, 57, 8167–8179. 10.1021/jm501195e. [DOI] [PubMed] [Google Scholar]
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Rosenberry T. L.; Brazzolotto X.; Macdonald I. R.; Wandhammer M.; Trovaslet-Leroy M.; Darvesh S.; Nachon F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. 10.3390/molecules22122098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silman I.; Sussman J. L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Cavrini V.; Andrisano V. β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. 10.1016/S0006-2952(02)01514-9. [DOI] [PubMed] [Google Scholar]
- Rees T.; Hammond P.; Soreq H.; Younkin S.; Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol. aging 2003, 24, 777–787. 10.1016/S0197-4580(02)00230-0. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zhang H. Reconsideration of anticholinesterase therapeutic strategies against Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 852–862. 10.1021/acschemneuro.8b00391. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Xu S.; Zhu Z.; Xu J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. 10.1016/j.ejmech.2019.05.020. [DOI] [PubMed] [Google Scholar]
- Bortolami M.; Rocco D.; Messore A.; Di Santo R.; Costi R.; Madia V. N.; Scipione L.; Pandolfi F. Acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease-a patent review (2016-present). Expert Opin. Ther. Pat. 2021, 31, 399. 10.1080/13543776.2021.1874344. [DOI] [PubMed] [Google Scholar]
- Maramai S.; Benchekroun M.; Gabr M. T.; Yahiaoui S. Multitarget therapeutic strategies for Alzheimer’s disease: Review on emerging target combinations. BioMed Research International 2020, 2020, 1. 10.1155/2020/5120230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi F.; De Vita D.; Bortolami M.; Coluccia A.; Di Santo R.; Costi R.; Andrisano V.; Alabiso F.; Bergamini C.; Fato R.; Bartolini M.; Scipione L. New pyridine derivatives as inhibitors of acetylcholinesterase and amyloid aggregation. Eur. J. Med. Chem. 2017, 141, 197–210. 10.1016/j.ejmech.2017.09.022. [DOI] [PubMed] [Google Scholar]
- Bortolami M.; Pandolfi F.; De Vita D.; Carafa C.; Messore A.; Di Santo R.; Feroci M.; Costi R.; Chiarotto I.; Bagetta D.; Alcaro S.; Colone M.; Stringaro A.; Scipione L. New deferiprone derivatives as multi-functional cholinesterase inhibitors: design, synthesis and in vitro evaluation. Eur. J. Med. Chem. 2020, 198, 112350. 10.1016/j.ejmech.2020.112350. [DOI] [PubMed] [Google Scholar]
- Shafir A.; Buchwald S. L. Highly selective room-temperature copper-catalyzed C– N coupling reactions. J. Am. Chem. Soc. 2006, 128, 8742–8743. 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V. Jr.; Featherstone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Dixon M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Košak U.; Brus B.; Knez D.; Zakelj S.; Trontelj J.; Pišlar A.; Šink R.; Jukič M.; Živin M.; Podkowa A.; Nachon F.; Nachon F.; Brazzolotto X.; Stojan J.; Kros J.; Coquelle N.; Sałat K.; Colletier J.-P.; Gobec S. The magic of crystal structure-based inhibitor optimization: development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J. Med. Chem. 2018, 61, 119–139. 10.1021/acs.jmedchem.7b01086. [DOI] [PubMed] [Google Scholar]
- Warren G. L.; Do T. D.; Kelley B. P.; Nicholls A.; Warren S. D. Essential considerations for using protein–ligand structures in drug discovery. Drug Discovery Today 2012, 17, 1270–1281. 10.1016/j.drudis.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Warren G. L.; Andrews C. W.; Capelli A.-M.; Clarke B.; LaLonde J.; Lambert M. H.; Lindvall M.; Nevins N.; Semus S. F.; Senger S.; Tedesco G.; Wall I. D.; Woolven J. M.; Peishoff C. E.; Head M. S. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Saccoliti F.; Madia V. N.; Tudino V.; De Leo A.; Pescatori L.; Messore A.; De Vita D.; Scipione L.; Brun R.; Kaiser M. Design, synthesis, and biological evaluation of new 1-(Aryl-1 H-pyrrolyl)(phenyl) methyl-1 H-imidazole derivatives as antiprotozoal agents. J. Med. Chem. 2019, 62, 1330–1347. 10.1021/acs.jmedchem.8b01464. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K. Quantum chemical insights into Alzheimer’s disease: Curcumin’s chelation with Cu (II), Zn (II), and Pd (II) as a mechanism for its prevention. Int. J. Quantum Chem. 2016, 116, 1107–1119. 10.1002/qua.25145. [DOI] [Google Scholar]
- Catapano M. C.; Tvrdý V.; Karlíčková J.; Migkos T.; Valentová K.; Křen V.; Mladěnka P. The stoichiometry of isoquercitrin complex with iron or copper is highly dependent on experimental conditions. Nutrients 2017, 9, 1193. 10.3390/nu9111193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job P. Formation and Stability of Inorganic Complexes in Solution. Ann. Chim. 1928, 9, 113–203. [Google Scholar]
- Molyneux P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J. sci. technol. 2004, 26, 211–219. [Google Scholar]
- Brand-Williams W.; Cuvelier M.-E.; Berset C. Use of a free radical method to evaluate antioxidant activity. LWT–Food Sci. Technol. 1995, 28, 25–30. 10.1016/S0023-6438(95)80008-5. [DOI] [Google Scholar]
- Espargaró A.; Medina A.; Di Pietro O.; Muñoz-Torrero D.; Sabate R. Ultra rapid in vivo screening for anti-Alzheimer anti-amyloid drugs. Sci. Rep. 2016, 6, 23349–23348. 10.1038/srep23349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espargaro A.; Pont C.; Gamez P.; Muñoz-Torrero D.; Sabate R. Amyloid pan-inhibitors: One family of compounds to cope with all conformational diseases. ACS Chem. Neurosci. 2019, 10, 1311–1317. 10.1021/acschemneuro.8b00398. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42713. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Baell J.; Walters M. A. Chemistry: Chemical con artists foil drug discovery. Nat. News 2014, 513, 481. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Kowalczyk J.; Grapsi E.; Espargaró A.; Caballero A. B.; Juárez-Jiménez J.; Busquets M. A.; Gamez P.; Sabate R.; Estelrich J. Dual Effect of Prussian Blue Nanoparticles on Aβ40 Aggregation: β-Sheet Fibril Reduction and Copper Dyshomeostasis Regulation. Biomacromolecules 2021, 22, 430–440. 10.1021/acs.biomac.0c01290. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. L. C., New York, USA, 2012. LigPrep; 2018.
- Protein Preparation Wizard; Schrödinger LLC: New York (USA), 2018. [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Crucitti G. C.; Pescatori L.; Messore A.; Madia V. N.; Pupo G.; Saccoliti F.; Scipione L.; Tortorella S.; Di Leva F. S.; Cosconati S. Discovery of N-aryl-naphthylamines as in vitro inhibitors of the interaction between HIV integrase and the cofactor LEDGF/p75. Eur. J. Med. Chem. 2015, 101, 288–294. 10.1016/j.ejmech.2015.06.036. [DOI] [PubMed] [Google Scholar]
- Prime; Schrödinger LLC, New York (USA), 2018. [Google Scholar]
- Hasel W.; Hendrickson T. F.; Still W. C. A rapid approximation to the solvent accessible surface areas of atoms. Tetrahedron Computer Methodology 1988, 1, 103–116. 10.1016/0898-5529(88)90015-2. [DOI] [Google Scholar]
- Glide; Schrödinger LLC: New York (USA), 2018. [Google Scholar]
- Desmond Molecular Dynamics System; D. E. Shaw Research: New York (USA), 2018. [Google Scholar]
- Taglieri L.; Saccoliti F.; Nicolai A.; Peruzzi G.; Madia V. N.; Tudino V.; Messore A.; Di Santo R.; Artico M.; Taurone S. Discovery of a pyrimidine compound endowed with antitumor activity. Invest. New Drugs 2020, 38, 39–49. 10.1007/s10637-019-00762-y. [DOI] [PubMed] [Google Scholar]
- Madia V. N.; Nicolai A.; Messore A.; De Leo A.; Ialongo D.; Tudino V.; Saccoliti F.; De Vita D.; Scipione L.; Artico M. Design, Synthesis and Biological Evaluation of New Pyrimidine Derivatives as Anticancer Agents. Molecules 2021, 26, 771. 10.3390/molecules26030771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.