

Abstract
Studies in tau and Aβ plaque transgenic mouse models demonstrated that brain-penetrant microtubule (MT)-stabilizing compounds, including the 1,2,4-triazolo[1,5-a]pyrimidines, hold promise as candidate treatments for Alzheimer’s disease (AD) and related neurodegenerative tauopathies. Triazolopyrimidines have already been investigated as anti-cancer agents; however, the anti-mitotic activity of these compounds does not always correlate with stabilization of MTs in cells. Indeed, previous studies from our laboratories identified a critical role for the fragment linked at C6 in determining whether triazolopyrimidines promote MT stabilization or, conversely, disrupt MT integrity in cells. To further elucidate the structure-activity relationship (SAR) and to identify potentially improved MT-stabilizing candidates for neurodegenerative disease, a comprehensive set of 68 triazolopyrimidine congeners bearing structural modifications at C6 and/or C7 was designed, synthesized and evaluated. These studies expand upon prior understanding of triazolopyrimidine SAR and enabled the identification of novel analogues that, relative to the existing lead, exhibit improved physicochemical properties, MT-stabilizing activity and pharmacokinetics.
Keywords: Triazolopyrimidine, tau, microtubule, microtubule-stabilizing agents, Alzheimer’s disease, neurodegenerative tauopathies
Graphical Abstract
Introduction.
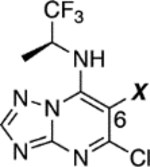
In Alzheimer’s disease (AD) and related neurodegenerative tauopathies,1 the hyperphosphorylation of the microtubule (MT)-associated protein tau promotes its dissociation from MTs and eventual deposition into insoluble aggregates, which are commonly referred to as neurofibrillary tangles (NFTs) and neuropil threads (NTs). Reduced tau binding to MTs is believed to trigger alterations in the normal structure and dynamics of MTs in the axons of neurons that ultimately cause or contribute to impaired axonal transport and resulting axonal dystrophy.2 Given the evidence of neuronal MT abnormalities in AD patients and in tau and Aβ plaque transgenic (Tg) mouse models,3 CNS-active MT-stabilizing agents have been proposed as molecules that may provide therapeutic benefits in AD and related tauopathies.4 Multiple preclinical studies5–8 in tau Tg animals have shown that relatively low and infrequent doses of CNS-active MT-stabilizing natural products, such as epothilone D (1, Figure 1) and dictyostatin (2, Figure 1), can lead to significant improvements in several endpoints including an increase in MT density, a reduction in axonal dystrophy, and a significant lowering of both tau pathology and neuron loss.5–7 More recently, brain-penetrant MT-stabilizing 1,2,4-triazolo[1,5-a]pyrimidines that are structurally simpler and are endowed with generally more favorable drug-like ADME-PK properties than MT-stabilizing natural products9 have also shown promising results both in vitro and in vivo.10–12 Although the MT binding site of the triazolopyrimidines is known to be different than that of taxol, epothilone D and other taxane site binders,13 evaluation of a selected prototype (3, Figure 1) in a mouse model of tauopathy revealed that this compound could exert similar beneficial effects to those previously observed with 1, indicating that triazolopyrimidines hold promise as candidate treatments for neurodegenerative tauopathies.12 More recently, 3 has also been shown to decrease axonal dystrophy, with a resulting reduction of Aβ peptide and Aβ plaque deposition, in a Tg mouse model of senile plaque formation.14
Figure 1.
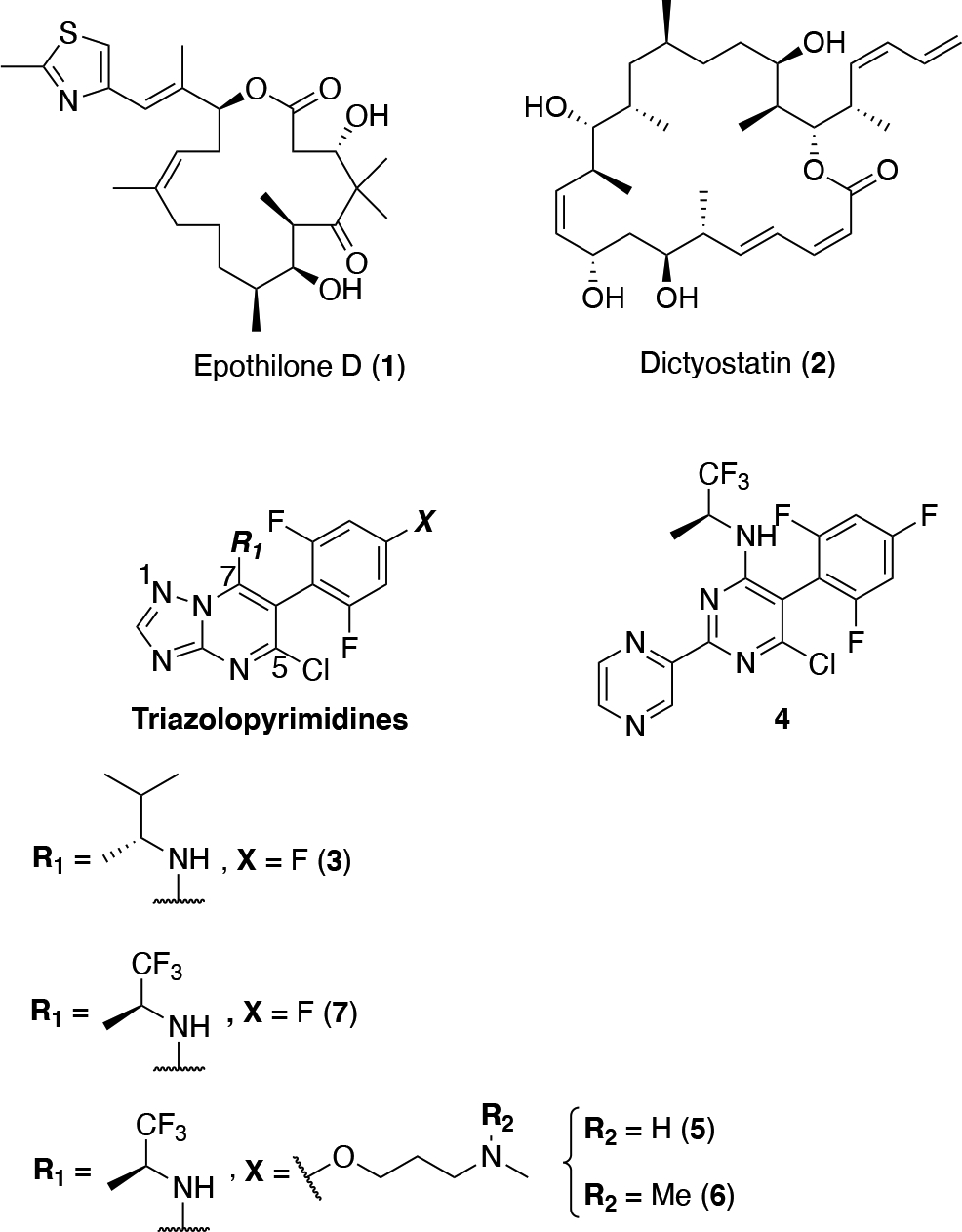
The structure of MT-stabilizing natural products, epothilone D and dictyostatin, and selected examples of MT-binding triazolopyrimidines and phenylpyrimidines.
MT-active triazolopyrimidines and structurally related phenylpyrimidines (e.g., 4) have already been evaluated as candidate treatments for cancer chemotherapy.15, 16 This effort, which culminated in the identification of cevipabulin (5) as a clinical candidate,17, 18 included a comprehensive assessment of the SAR in cytotoxicity assays that used rapidly dividing cancer cell-lines.15 More recent studies from our laboratories suggested that the anti-mitotic activity of these compounds may not always correlate with stabilization of MTs in cells. Indeed, evaluation of representative examples of triazolopyrimidines and related phenylpyrimidines in cell-based assays of MT stabilization revealed that varying the scaffold (i.e., triazolopyrimidine or phenylpyrimidine) or substitution pattern results in molecules that can either promote MT stabilization or, conversely, disrupt MT integrity.11 For example, an evaluation of a selected set of triazolopyrimidines and phenylpyrimidines for their ability to produce elevations in known cellular markers of stable MTs,19 such as acetylated and de-tyrosinated α-tubulin (AcTub and GluTub, respectively), revealed that typically the phenylpyrimidines, such as 4, produce an unusual bell-shaped concentration-response curve in these assays. In addition, these compounds cause a concentration-dependent decrease in total cellular α- and β-tubulin resulting from proteasome-mediated degradation, with a visible reduction of MT networks. Interestingly, although similar observations were made for some triazolopyrimidines, including 5 and its brain-penetrant10 congener 6 (Figure 1), replacement of the alkoxide side-chain of these molecules with a fluorine atom, as in 7 (Figure 1) or 3, had a dramatic impact on the MT-stabilizing properties of the compounds as demonstrated by the fact that the latter molecules caused the desired linear concentration-dependent increase in MT stabilization and MT mass in cells, without decreased cellular tubulin levels or changing MT morphology.11 These findings, combined with the observation that the amine fragment linked at C7 of lead compound, 3, is likely to be the primary site of metabolism,12 led to a more systematic exploration of the SAR of triazolopyrimidines modified at C6 and C7. The results from these studies led to the identification of selected congeners that, relative to the existing lead, 3, exhibit improved physicochemical properties, MT-stabilizing activity and pharmacokinetics.
Chemistry.
As part of these studies a total of 68 compounds (7–75, Scheme 1–5) have been synthesized and tested, including 8 previously described triazolopyrimidines (i.e., 7,15 11,15 25,15 31–33,20 34,12 and 5413); 23 compounds exemplified in the patent literature (i.e., 10, 12, 14, 16, 18, 24, 28–30, 35, 41, 45–48, 50, 51, 52, 57, 60, 61, 64, 65); and 37 structurally novel congeners. In all cases, the triazolopyrimidine ring was accessed via cyclocondensation reaction between the appropriate diethylmalonate (76–92, Scheme 1) and 1H-1,2,4-triazol-5-amine. Next, treatment with phosphorous oxychloride provided the corresponding 5,7-dichloro triazolopyrimidines (93–109, Scheme 1). Chemoselective amination at C7 with (S)-1,1,1-trifluoropropan-2-amine furnished triazolopyrimidines 7–22 (Scheme 1). Reduction of the para nitro derivative 22 provided the corresponding aniline derivative 23 (Scheme 1).
Scheme 1. Reagents and Reaction conditions:
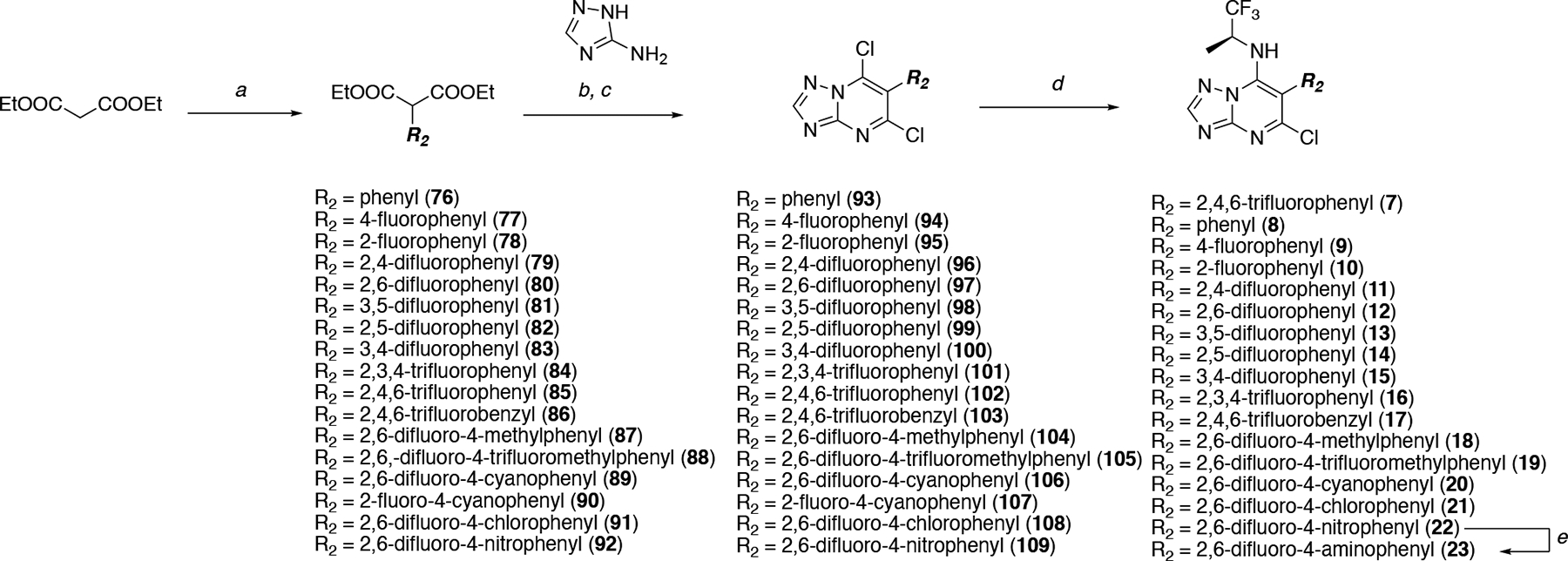
a) For 76–85, 87–89, 91: Ar-X, CuBr, NaH, 1,4-dioxane, 60–100 °C, 12 h, 19–74%; For 86: Ar-CH2Br, NaH, N,N-dimethylformamide, 0 °C, 1 h, 90%; For 90 and 92: Ar-F, K2CO3, N,N-dimethylformamide, 60 °C, 4 h, 84–86%; b) N-tributylamine, 170–180 °C, 2–6 h; c) phosphorus oxychloride, 110–130 °C, 6–16 h, 46–58% over two steps; d) appropriate amine, N,N-dimethylformamide, rt–90 °C, 1–18 h, 12–50%; e) Fe, NH4Cl, H2O/MeOH (40/50), 80 °C, 2 h, 69%.
Scheme 5. Reagents and Reaction conditions:
a) appropriate amine, N,N-dimethylformamide, r.t., 1 h, 56–86%.
Triazolopyrimidine 7 was then used to access additional derivatives bearing different substitutions at the para position of the fluorinated phenyl ring (24–33, Scheme 2). In these cases, the installation of the appropriate alcohol or thiol side-chain was conducted via SNAr followed by HPLC purification of the final product. Sulfoxide (26, 29) and sulfone (27, 30) derivatives were obtained from the corresponding thioethers (i.e., 25, and 28, respectively) upon treatment with m-CPBA (Scheme 2).
Scheme 2. Reagents and Reaction conditions:
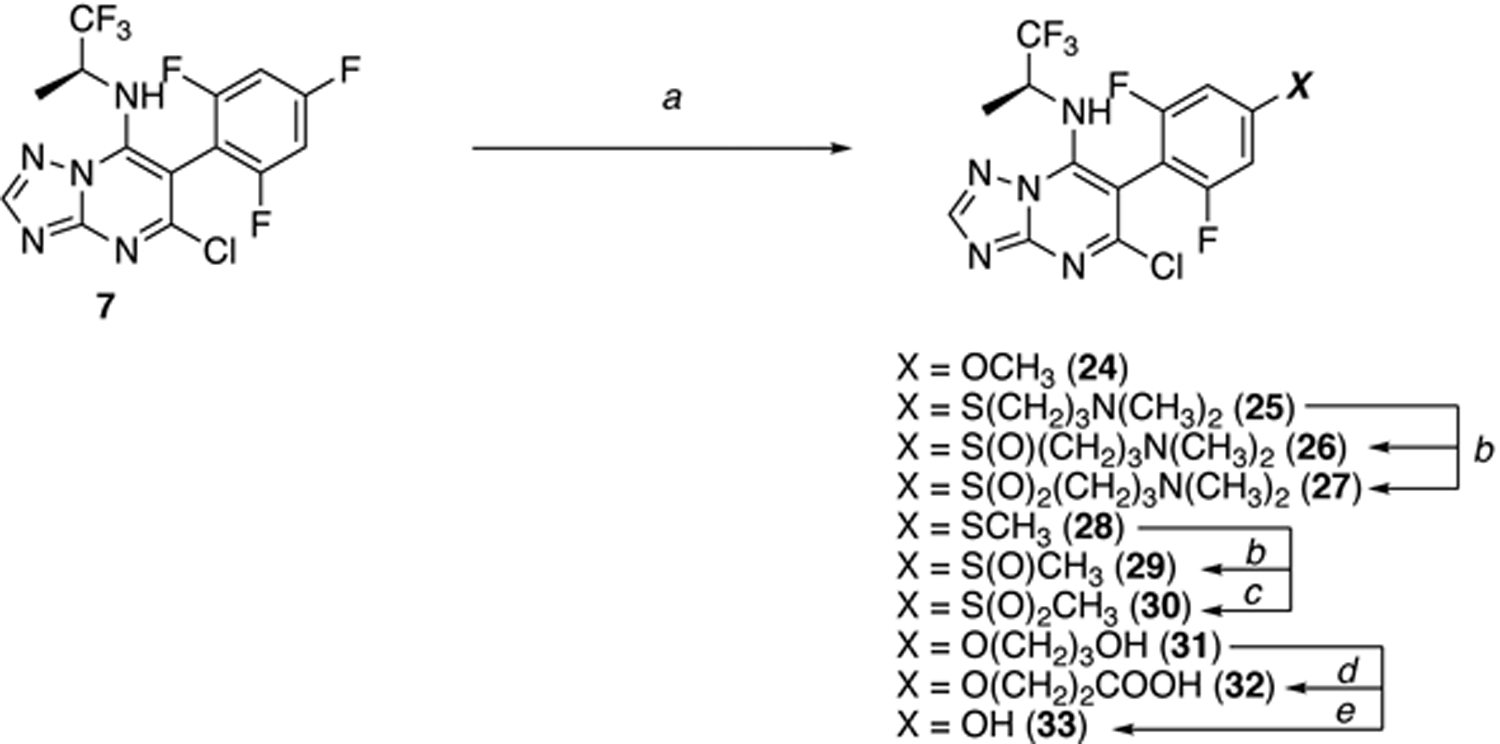
a) For 24: NaOMe, THF, 0 °C to 60 °C, 14 h, 71%; For 25, 31: ROH or RSH, NaH, DMSO, 60 °C, 3 h, 81–95%; For 28: CH3SNa, DMSO, 60 °C, 3 h, quant.; b) m-CPBA (1 equiv), CH2Cl2, 20 °C, 2 h, 32% of 26, 52% of 27, 70% of 29; c) m-CPBA (3 equiv), CH2Cl2, 20 °C, 3 h, 71%; d) TEMPO, BAIB, CH2Cl2/H2O (90/70), 20 °C, 2 h, 59%; e) LiOH, MeOH/H2O (1/1), 20 °C, 16 h, 39%.
Triazolopyrimidine dichloride 102 was used to access a series of congeners modified at position C7 (i.e., 34–61, Scheme 3). In these cases, chemoselective displacement of the chloro substituent at C7 was accomplished in moderate to good yields by treating 102 with the appropriate amine. The synthesis of C7 alkyl derivative 62 (Scheme 4) was initially attempted adopting a known procedure21 by reacting dichloride 102 with the methyl ester of 4,4,4-trifluoro-3-methylbutanoic acid in the presence of LiHMDS to obtain 110, followed by a Krapcho decarboxylation.22 The main product isolated from this synthesis was the tetracyclic compound 111 (Scheme 4) whose structure was confirmed by X-ray crystallography. Desired compound 62 was ultimately obtained employing a similar approach in which alkylation reaction at C7 was conducted using the tert-butyl ester of 4,4,4-trifluoro-3-methylbutanoic acid to obtain intermediate 112, which was then subjected to TFA-mediated cleavage of the ester moiety and in situ decarboxylation (Scheme 4).
Scheme 3. Reagents and Reaction conditions:
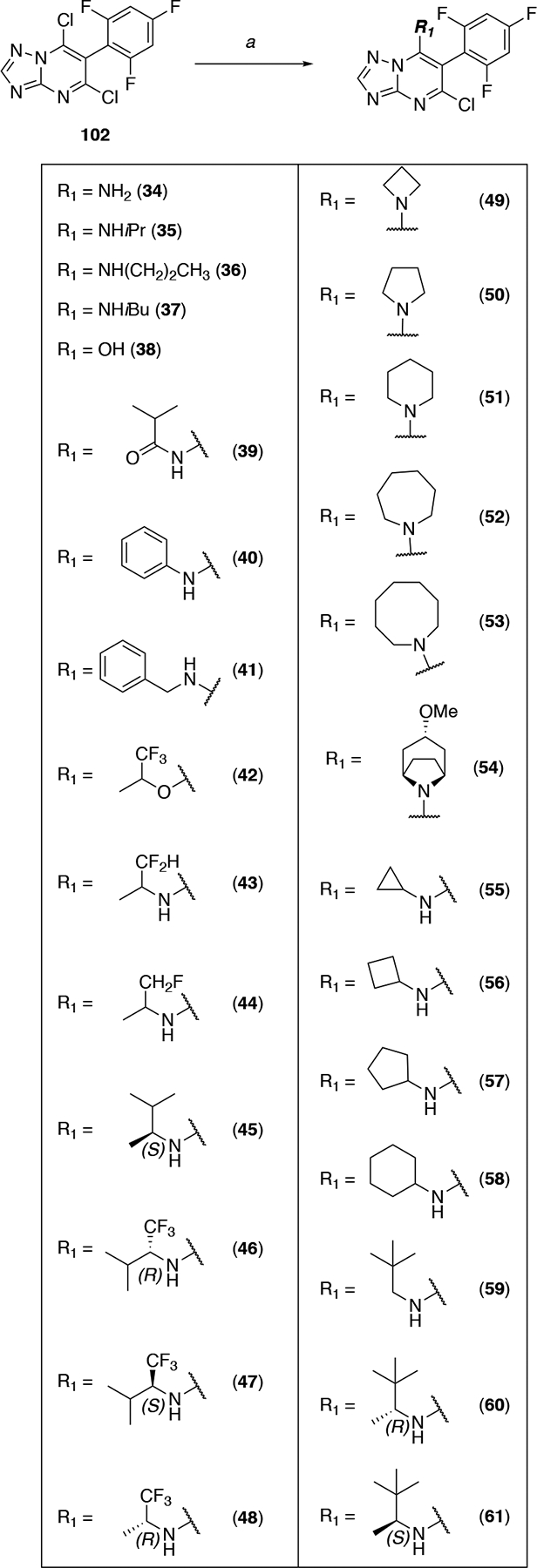
a) appropriate amine, base, N,N-dimethylformamide, 20–90 °C, 1–18 h, 12–50%.
Scheme 4. Reagents and Reaction conditions:
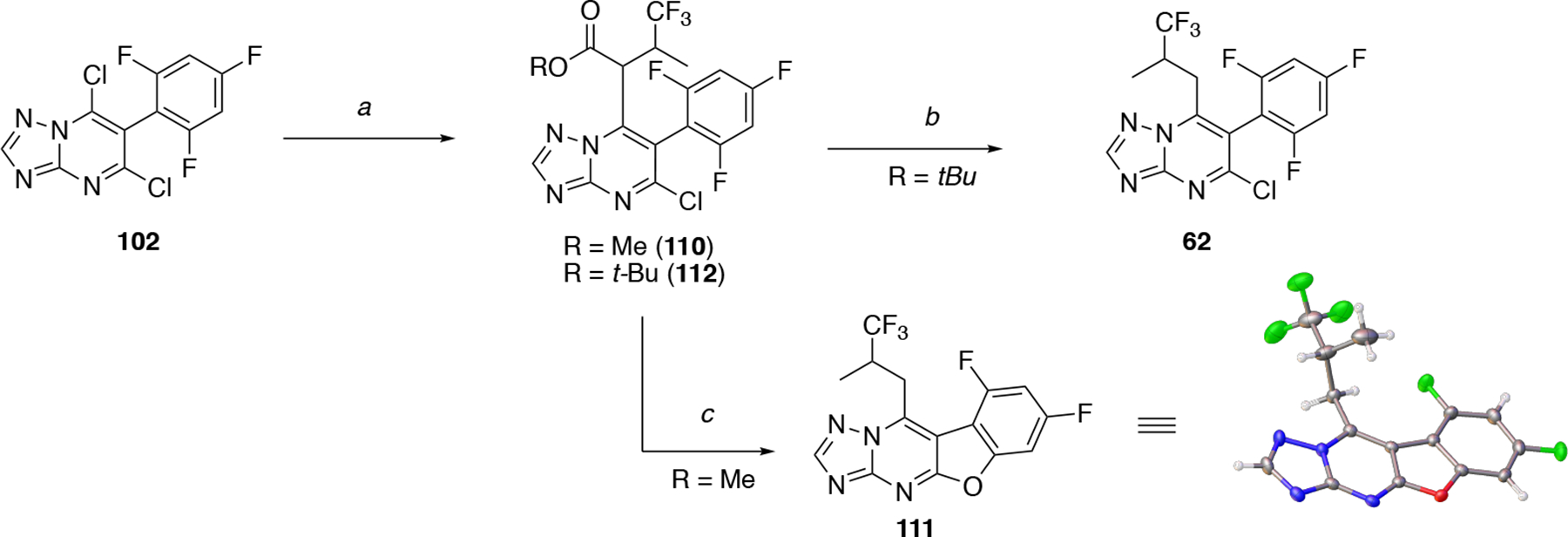
a) appropriate ester of 4,4,4-trifluoro-3-methylbutanoic acid, HMDS, n-BuLi, THF, −78 °C, 2.5 h, 70% for 110, 25% for 112; b) TFA, CH2Cl2, 20 °C, 16 h, 68%; c) LiCl, DMSO, 130 °C (microwave irradiation), 3 h, 76%.
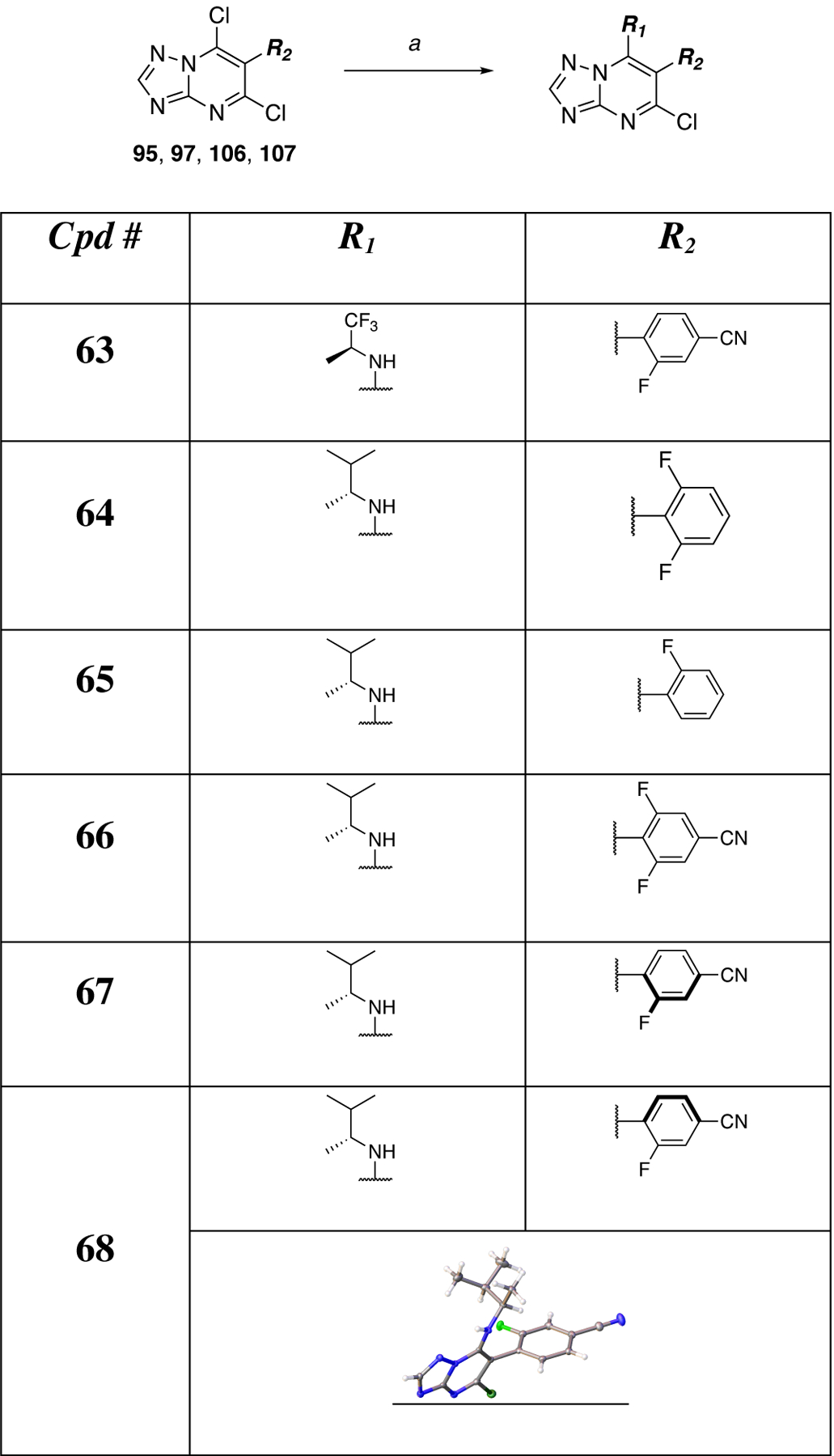
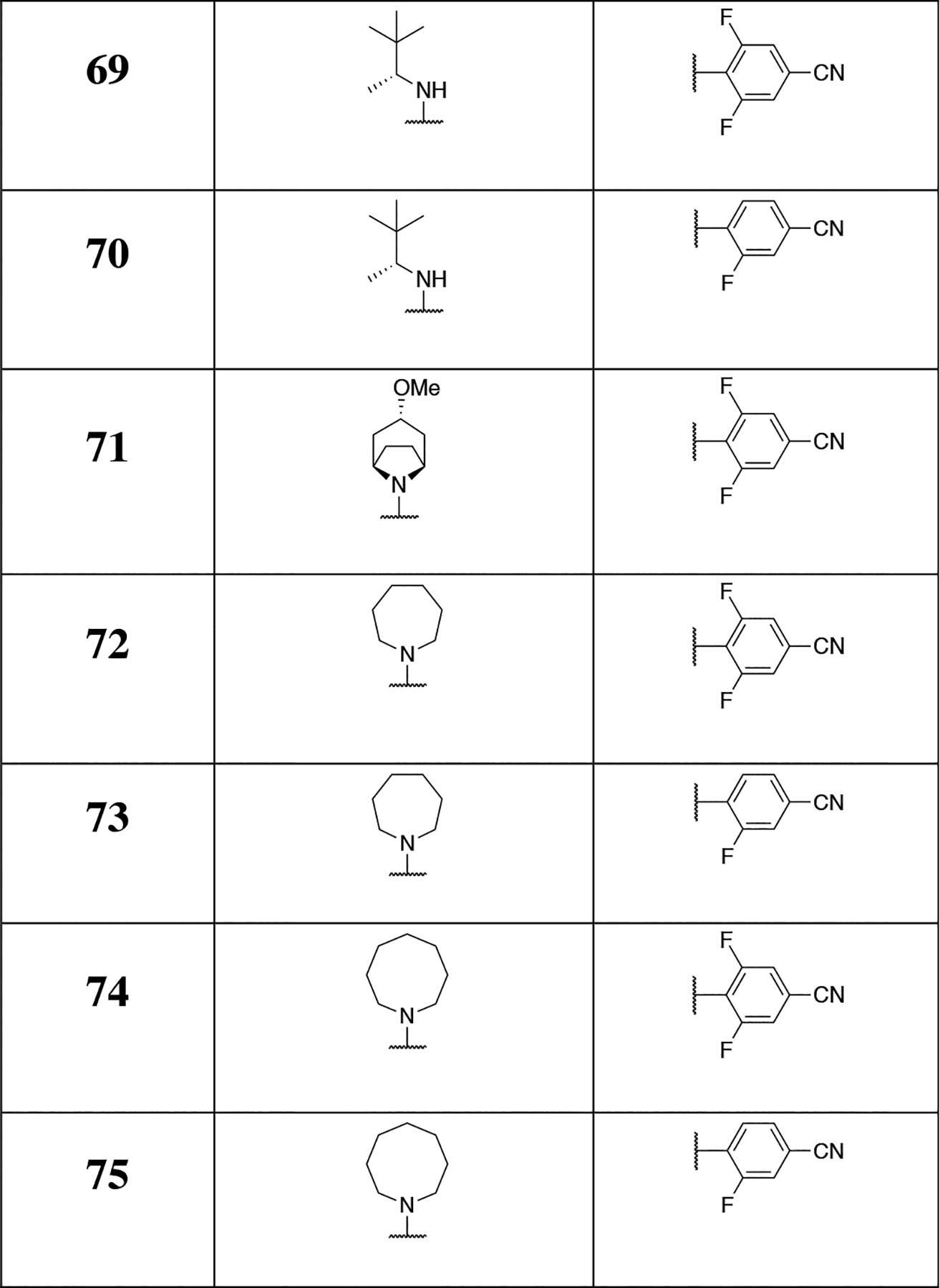
Finally, the synthesis of compounds 63–75 (Scheme 5) was conducted by reacting the appropriate triazolopyrimidine dichlorides (i.e., 95, 97, 106, or 107) with amine fragments (Scheme 5). In selected cases (i.e., 67 and 68) purification via silica gel column chromatography afforded individual atropoisomers, the structures of which could be determined via single crystal X-ray structure determination of 68 (Scheme 5).
Results.
All test compounds were evaluated in a previously described11 cell-based assay of MT-stabilization, in which compound-dependent changes in tubulin polymerization and total tubulin levels were determined after 4 h of incubation of QBI293 cells with 1 and 10 μM compound via quantification of acetylated α-tubulin (AcTub) and total α-tubulin (α-Tub) in cell lysates by ELISA. Although the changes in AcTub levels of compound-treated cells relative to DMSO-treated cells provide a valid assessment of the MT-stabilizing activity of test compounds, day-to-day differences in the responsiveness of cells to MT-stabilizing treatment can result in occasional variability in the fold-change in AcTub levels. Thus, to account for this variability and ultimately enable a ranking of different triazolopyrimidines based on MT-stabilizing activity, compound-dependent changes in AcTub levels relative to DMSO-treated cells (i.e., negative control) were also normalized to the corresponding changes caused by 100 nM of cevipabulin (5), which was routinely used as positive control.10
Previous evaluations of the MT-stabilizing activity of selected examples of triazolopyrimidines in this assay suggested that the nature of the substituent in the para position of the fluorinated phenyl can elicit one of two distinct cellular responses: one characterized by a linear concentration-dependent increase of markers of stable MTs (i.e., acetylated- and detyrosinated-α-tubulin) with no effect on total tubulin levels, which is typically observed with triazolopyrimidines such as 3 and 7; and a second response caused by compounds bearing the alkoxy side-chain (e.g., 5 and 6) that is characterized by a proteasome-dependent degradation of tubulin that is often associated with the appearance of a bell-shaped concentration-response relationship in the AcTub assay.11 To facilitate the presentation and discussion of the SAR results, we will refer here to these two distinct cellular responses as “Class I” and “Class II”, respectively, and triazolopyrimidines 7 and 6 are used as defining examples of these classes.
Since the replacement of the alkoxy side-chain of 5 or 6 (Class II) with a fluorine atom (as in 7, Class I) was found to produce a profound impact in the MT-stabilizing activity of the triazolopyrimidine compounds,11 our investigations of SARs began by synthesizing and testing selected analogues (18–33, Table 1) that could help identify specific stereoelectronic characteristics of the substituent in the para position of the fluorinated phenyl ring that may be necessary/sufficient to impart Class I activity. Evaluation of these compounds in the QBI293 assay of MT-stabilization revealed a possible general trend indicating that active congeners bearing electron-donating groups at the para position generally elicit a significant (i.e., >15%) loss of α-tubulin levels when tested at either 1 and/or 10 μM concentration, which is characteristic of Class II compounds. For example, both para hydroxyl- (33) and methoxy- (24) derivatives, as well as thioalkyl compounds, 25 and 28, were all found to produce >20% reduction in cellular α-tubulin. In contrast, active analogues bearing electron-withdrawing groups, such as cyano (20) and nitro (22) were found to increase AcTub levels without producing a significant reduction in α-tubulin levels (i.e., Class I phenotype). However, in cases such as 21, where the chloro substituent produces opposite mesomeric (+M) and inductive (−I) effects, a comparatively more moderate but still significant loss of α-tubulin (19%) was observed when the compound was tested at 10 μM concentration. These results indicate that the electronic properties of the substituent in para may play an important role in determining the phenotypic response. However, differences in the shape/volume and/or the geometry of hydrogen bonding of the substituent can modulate significantly the MT-stabilizing activity, independent of the phenotype. This is best represented by comparing selected pairs of compounds, such as nitrile (20) and sulfone (30), or alkyl-sulfone (27) and the corresponding alkoxy compound (6). Docking studies (vide infra) indicate that the larger volume of the sulfone moiety is likely to affect negatively the interaction of the triazolopyrimidine with the binding site.
Table 1.
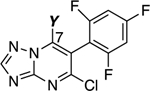
MT-stabilizing activity of triazolopyrimidines modified at C6. Fold-changes in acetylated α-tubulin (AcTub) and total α-tubulin (α-Tub) levels in QBI293 cells after 4 h incubation with test compounds at either 1 or 10 μM. Reported values for AcTub and α-Tub represent the fold-change over control (DMSO)–treated cells (*p<0.05 and**p<0.01 by one-way ANOVA); numbers in parentheses represent the fold-change of AcTub over positive control-treated cells (i.e., 100 nM of 5).
| Cpd # |
|
AcTub | α-Tub | Classa | ||
|---|---|---|---|---|---|---|
| 1 μM | 10 μM | 1 μM | 10 μM | |||
| 6 |
|
13.2 ± 1.8** (1.28) | 1.92 ± 0.14 (0.19) | 0.74 ± 0.05** | 0.40 ± 0.01** | II |
| 7 |
|
2.78 ± 0.17** (0.45) | 5.04 ± 0.45** (0.82) | 1.18 ± 0.09 | 1.22 ± 0.18 | I |
| 8 |
|
1.11 ± 0.04 (0.12) | 2.07 ± 0.10** (0.30) | 1.09 ± 0.03 | 1.03 ± 0.02 | I |
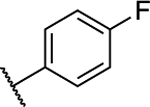
| 9 |
|
1.05 ± 0.05 (0.07) | 2.41 ± 0.13** (0.16) | 1.04 ± 0.06 | 1.13 ± 0.06 | I |
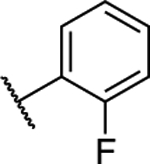
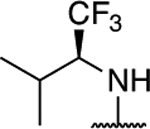
| 10 |
|
1.73 ± 0.02* (0.13) | 3.56 ± 0.08** (0.26) | 1.09 ± 0.01 | 1.05 ± 0.06 | I |
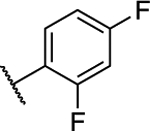
| 11 |
|
1.54 ± 0.05 (0.13) | 3.19 ± 0.18** (0.27) | 0.99 ± 0.02 | 0.86 ± 0.01 | I |
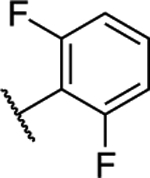
| 12 |
|
2.20 ± 0.13 (0.16) | 5.28 ± 0.54** (0.39) | 1.06 ± 0.14 | 1.02 ± 0.04 | I |
| 13 |
|
1.52 ± 0.074 (0.12) | 3.97 ± 0.82* (0.30) | 1.12 ± 0.13 | 1.12 ± 0.02 | I |
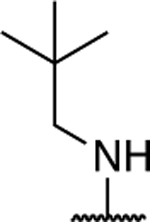
| 14 |
|
1.40 ± 0.38 (0.07) | 4.10 ± 0.44* (0.22) | 1.05 ± 0.11 | 0.90 ± 0.06 | I |
| 15 |
|
1.85 ± 0.09 (0.05) | 4.76 ± 0.62** (0.12) | 1.01 ± 0.08 | 1.13 ± 0.06 | I |
| 16 |
|
1.50 ± 0.03 (0.12) | 4.28 ± 0.33** (0.34) | 1.11 ± 0.05 | 0.88 ± 0.04 | I |
| 17 |
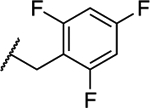
|
1.26 ± 0.06 (0.09) | 1.43 ± 0.02 (0.10) | 1.01 ± 0.07 | 1.05 ± 0.04 | NA |
| 18 |
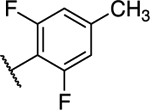
|
1.62 ± 0.18** (0.28) | 2.73 ± 0.18** (0.48) | 0.99 ± 0.09 | 0.78 ± 0.07* | II |
| 19 |
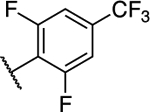
|
1.09 ± 0.04 (0.13) | 1.39 ± 0.11* (0.16) | 1.06 ± 0.02 | 1.02 ± 0.03 | I |
| 20 |
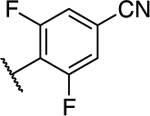
|
1.90 ± 0.06* (0.27) | 3.56 ± 0.31** (0.51) | 0.94 ± 0.02 | 0.95 ± 0.05 | I |
| 21 |
|
1.88 ± 0.03** (0.31) | 4.28 ± 0.17** (0.70) | 0.93 ± 0.02 | 0.81 ± 0.06** | II |
| 22 |
|
3.24 ± 0.36** (0.60) | 6.65 ± 0.45** (1.23) | 0.96 ± 0.01 | 0.94 ± 0.05 | I |
| 23 |
|
0.94 ± 0.27 (0.17) | 0.69 ± 0.37 (0.13) | 0.97 ± 0.03 | 0.96 ± 0.02 | NA |
| 24 |
|
3.16 ± 0.29** (0.64) | 2.07 ± 0.07** (0.42) | 0.72 ± 0.09** | 0.51 ± 0.05** | II |
| 25 |
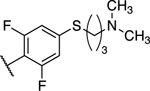
|
3.90 ± 0.66** (0.90) | 1.49 ± 0.48 (0.34) | 0.83 ± 0.05* | 1.07 ± 0.02 | II |
| 26 |
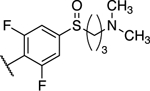
|
1.49 ± 0.48 (0.34) | 1.39 ± 0.11 (0.32) | 1.22 ± 0.06 | 0.98 ± 0.24 | NA |
| 27 |
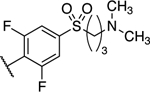
|
1.13 ± 0.10 (0.24) | 1.08 ± 0.11 (0.23) | 0.70 ± 0.58 | 1.07 ± 0.07 | NA |
| 28 |
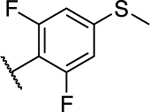
|
1.75 ± 0.37* (0.27) | 2.23 ± 0.15** (0.35) | 0.88 ± 0.05* | 0.56 ± 0.06** | II |
| 29 |
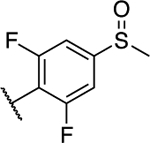
|
0.84 ± 0.21 (0.21) | 0.87 ± 0.21 (0.21) | 0.96 ± 0.02 | 0.93 ± 0.03 | NA |
| 30 |
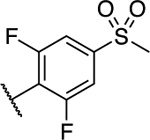
|
1.21 ± 0.38 (0.19) | 1.23 ± 0.37 (0.19) | 1.03 ± 0.02 | 1.02 ± 0.04 | NA |
| 31 |
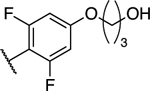
|
7.25 ± 0.23** (1.34) | 1.33 ± 0.15 (0.25) | 0.73 ± 0.06** | 0.37 ± 0.02** | II |
| 33 |
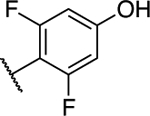
|
1.22 ± 0.73 (0.23) | 4.11 ± 0.76** (0.77) | 0.84 ± 0.07 | 0.57 ± 0.02** | II |
Class I compounds are those producing a concentration-dependent increase in AcTub levels and that do not cause >15% reduction in α-tubulin at either concentration; Class II compounds are those that cause >15% decrease in α-tubulin at either 1 or 10 μM compound concentration. NA = not applicable as the test compound does not cause significant changes in AcTub or total α-tubulin when tested at 1 or 10 μM concentration.
To investigate the effect of the degree and pattern of fluorination of the phenyl ring at C6, compounds 8–16 were evaluated in comparison with Class I control compound, 7. The results of these studies, summarized in Table 1 and Figure 2, show that all compounds within this series maintain MT-stabilizing activity that is qualitatively similar to 7 (i.e., all compounds elicit Class I phenotype); however, depending on the number and position of the fluorine atoms, these triazolopyrimidine congeners can differ significantly in terms of potency. In general, the Class I MT-stabilizing activity appears to increase with the number of fluorine atoms in the ring, with a preferred arrangement within the series being the 2,4,6-trifluorophenyl (Figure 2). Among derivatives bearing a mono-fluorinated phenyl fragment at C6, the ortho fluoro derivative (10) appeared to be more active than the corresponding para substituted compound (9) at both 1 and 10 μM concentrations. Likewise, among triazolopyrimidines bearing a di-fluorinated phenyl at C6 (11–15), the 2,6-difluoro compound (12) was found to be the most active. Finally, homologation of the 2,4,6-trifluorophenyl of 7 to the corresponding 2,4,6-trifluorobenzyl congener (17) led to a drastic reduction of MT-stabilizing activity.
Figure 2.
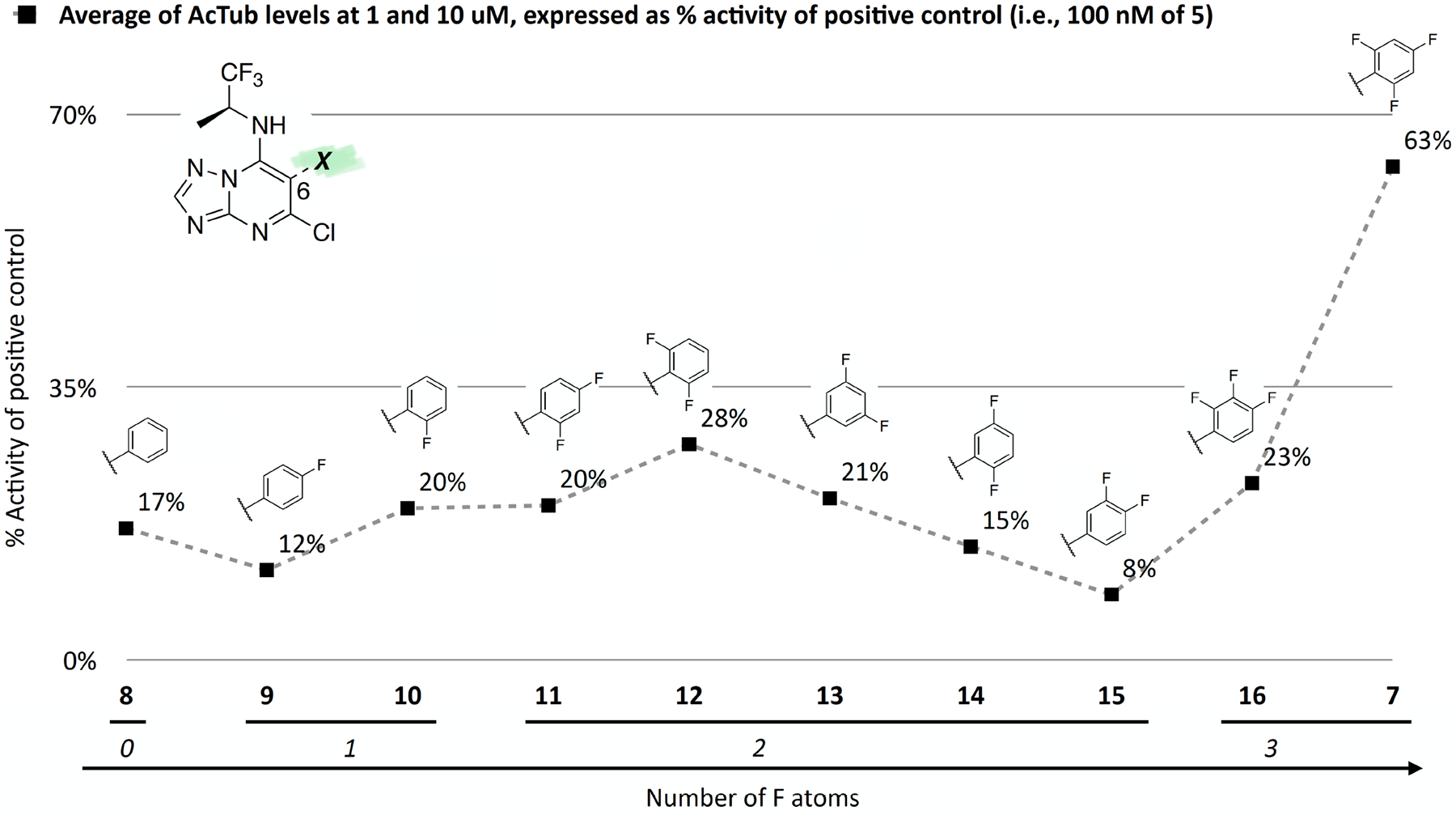
Effect of fluorination of the aryl group at C6 on MT-stabilizing activity of Class I triazolopyrimidines. Black squares indicate the average activity of test compounds at 1 and 10 μM, normalized to the activity of 100 nM of 5 (positive control).
Comparative evaluation of Table 2 compounds (3, 34–62) and 7 in the cell-based MT-stabilization assay helped define several SAR elements of the fragment linked at the C7 position of the triazolopyrimidine ring. Truncation of this fragment as in 34 and 38 led to a dramatic loss of MT-stabilizing activity. Replacement of the nitrogen atom at C7 of control compound, 7, with an oxygen atom (42) revealed that the ether linkage is not permitted as it resulted in a drastic reduction of activity in the cellular assay. Conversely, replacement of the nitrogen atom at C7 with a methylene, as in 62, showed that the alkyl linkage allows for retention of significant activity in the AcTub assay at both 1 and 10 μM. However, this compound was found to cause a significant reduction of α-tubulin (i.e., ~22%) when tested at 10 μM. These observations demonstrate that the nature of the fragment linked at C7 can play an important role in determining both potency as well as cellular phenotype elicited by MT-active triazolopyrimidines, and that the presence of a substituted nitrogen at this position may be a necessary condition for Class I activity.
Table 2.
MT-stabilizing activity of triazolopyrimidines modified at C7. Fold-changes in acetylated α-tubulin (AcTub) and α-tubulin (α-Tub) levels in QBI293 cells after 4 h incubation with test compounds at either 1 or 10 μM. Reported values for AcTub and α-Tub represent the fold-change over control (DMSO)–treated cells (*p<0.05 and**p<0.01 by one-way ANOVA); numbers in parentheses represent the fold-change of AcTub over positive control-treated cells (i.e., 100 nM of 5).
| cpd # |
|
AcTub | α-Tub | Classa | ||
|---|---|---|---|---|---|---|
| 1 μM | 10 μM | 1 μM | 10 μM | |||
| 3 |
|
2.14 ± 0.15* (0.59) | 2.57 ± 0.15* (0.71) | 1.10 ± 0.17 | 0.90 ± 0.16 | I |
| 34 |
|
1.18 ± 0.01 (0.09) | 1.12 ± 0.08 (0.08) | 1.08 ± 0.07 | 1.05 ± 0.02 | NA |
| 35 |
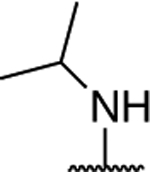
|
1.15 ± 0.15 (0.15) | 2.01 ± 0.17** (0.27) | 0.92 ± 0.10 | 0.98 ± 0.03 | I |
| 36 |
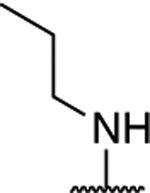
|
1.15 ± 0.07 (0.27) | 1.14 ± 0.14 (0.26) | 1.14 ± 0.23 | 0.86 ± 0.24 | NA |
| 37 |
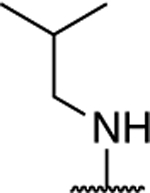
|
1.63 ± 0.14** (0.38) | 2.28 ± 0.17** (0.53) | 1.30 ± 0.04 | 0.99 ± 0.25 | I |
| 38 |
|
1.04 ± 0.04 (0.17) | 1.03 ± 0.08 (0.17) | 1.06 ± 0.03 | 1.02 ± 0.05 | NA |
| 39 |
|
1.09 ± 0.01 (0.07) | 1.31 ± 0.47 (0.09) | 1.00 ± 0.03 | 0.97 ± 0.07 | NA |
| 40 |
|
1.14 ± 0.26 (0.09) | 1.05 ± 0.06 (0.08) | 1.10 ± 0.17 | 0.96 ± 0.01 | NA |
| 41 |
|
1.05 ± 0.16 (0.16) | 1.49 ± 0.04* (0.22) | 1.00 ± 0.12 | 1.06 ± 0.06 | I |
| 42 |
|
0.91 ± 0.04 (0.19) | 1.01 ± 0.07 (0.21) | 1.03 ± 0.04 | 1.04 ± 0.05 | NA |
| 43 |
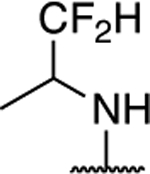
|
1.28 ± 0.09 (0.30) | 1.82 ± 0.10** (0.35) | 1.19 ± 0.13 | 1.17 ± 0.01 | I |
| 44 |
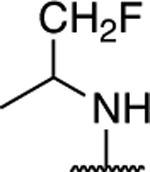
|
1.32 ± 0.26 (0.30) | 1.50 ± 0.09* (0.35) | 0.92 ± 0.12 | 1.09 ± 0.03 | I |
| 45 |
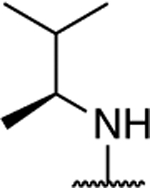
|
1.32 ± 0.14 (0.06) | 3.49 ± 0.07** (0.15) | 0.95 ± 0.01 | 0.89 ± 0.01* | I |
| 46 |
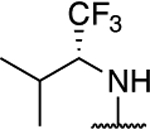
|
3.12 ± 0.25** (0.17) | 6.78 ± 0.18** (0.38) | 1.04 ± 0.01 | 1.06 ± 0.01 | I |
| 47 |
|
2.79 ± 0.04** (0.16) | 13.0 ± 0.54** (0.73) | 1.10 ± 0.09 | 0.61 ± 0.02** | II |
| 48 |
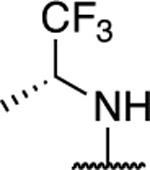
|
1.39 ± 0.11 (0.24) | 2.47 ± 0.10* (0.43) | 1.23 ± 0.21 | 1.47 ± 0.06* | I |
| 49 |
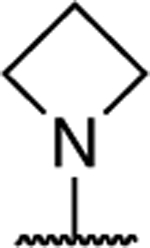
|
0.98 ± 0.07 (0.26) | 1.22 ± 0.02* (0.32) | 0.96 ± 0.07 | 1.17 ± 0.05 | I |
| 50 |
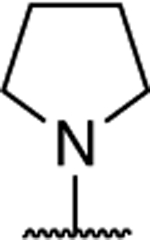
|
1.14 ± 0.37 (0.07) | 2.74 ± 0.23* (0.16) | 1.12 ± 0.31 | 1.18 ± 0.10 | I |
| 51 |
|
2.21 ± 0.36 (0.13) | 5.84 ± 1.38** (0.33) | 1.23 ± 0.05 | 1.20 ± 0.22 | I |
| 52 |
|
4.16 ± 0.97** (0.45) | 6.87 ± 0.04** (0.74) | 0.99 ± 0.03 | 0.99 ± 0.04 | I |
| 53 |
|
3.70 ± 0.32** (0.36) | 7.16 ± 0.11** (0.70) | 1.01 ± 0.14 | 1.28 ± 0.03** | I |
| 54 |
|
3.65 ± 0.15** (0.54) | 7.43 ± 0.93** (1.11) | 1.12 ± 0.08 | 1.38 ± 0.19** | I |
| 55 |
|
1.17 ± 0.15 (0.07) | 1.27 ± 0.06 (0.07) | 1.20 ± 0.03 | 1.26 ± 0.03 | NA |
| 56 |
|
0.97 ± 0.15 (0.13) | 1.75 ± 0.03** (0.23) | 0.94 ± 0.01 | 0.95 ± 0.02 | I |
| 57 |
|
2.03 ± 0.04* (0.27) | 4.83 ± 0.04** (0.45) | 1.03 ± 0.02 | 0.98 ± 0.02 | I |
| 58 |
|
1.13 ± 0.11 (0.07) | 3.51 ± 0.19** (0.23) | 1.07 ± 0.05 | 0.99 ± 0.05 | I |
| 59 |
|
2.04 ± 0.11* (0.30) | 4.79 ± 0.74** (0.71) | 1.03 ± 0.07 | 1.06 ± 0.08 | I |
| 60 |
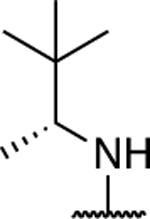
|
5.70 ± 0.27** (0.56) | 8.42 ± 0.20** (0.83) | 0.97 ± 0.02 | 1.08 ± 0.03 | I |
| 61 § |
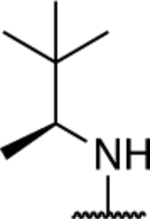
|
3.36 ± 0.13** (10 μM) (0.43) | 4.29 ± 0.04** (30 μM) (0.55) | 1.08 ± 0.05 (10 μM) | 1.22 ± 0.03 (30 μM) | I |
| 62 |
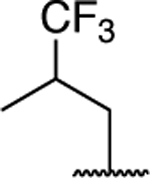
|
2.30 ± 0.59* (0.53) | 2.29 ± 0.38* (0.53) | 1.03 ± 0.07 | 0.78 ± 0.05 | II |
Class I compounds are those producing a concentration-dependent increase in AcTub levels and that do not cause >15% reduction in α-tubulin at either concentration; Class II compounds are those that cause >15% decrease in α-tubulin at either 1 or 10 μM compound concentration. NA = not applicable as the test compound does not cause significant changes in AcTub or total α-Tub when tested at 1 or 10 μM concentration.
Test compound was tested at 10 and 30 μM.
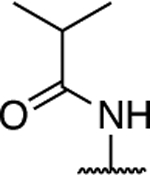
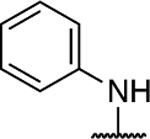
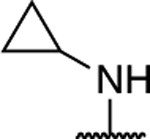
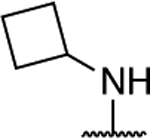
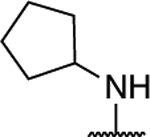
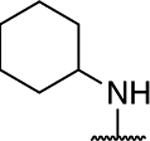
Further investigation of the SAR of triazolopyrimidines featuring a substituted nitrogen at C7 included a comparison between selected aliphatic and aromatic amines, as well as amides. Comparison of lead compound 3 with amide derivative, 39, indicates that aliphatic amines may be preferred over amides. Likewise, comparison of aniline derivative, 40, with the corresponding cyclohexyl amine congener, 58, shows that whereas the former is essentially devoid of activity at both 1 and 10 μM, the aliphatic derivative, 58, is significantly active at 10 μM without decreasing α-tubulin levels. These results suggest that aliphatic amines at C7 may be generally preferred and, as a result, further exploration of the SAR focused on identifying the key requisites of the aliphatic amine fragment, which included an evaluation of linear, branched, and cyclic amines of different sizes. Comparison of the in vitro MT-stabilizing activity of lead compound, 3, with congeners featuring amine substituents at C7 that are either substructures (i.e., 35–37) or that are incrementally larger fragments than the C7 substituent of 3 (e.g., 60) clearly suggest that the MT-stabilizing activity of triazolopyrimidines may increase with the size and branching of the aliphatic fragment. As highlighted in Figure 3, the extreme examples within this series are the linear amine derivative, 36, which is devoid of MT-stabilizing activity in the QBI293 assay, and the t-butyl derivative, 60, which exhibits relatively potent activity as revealed by the marked increases in AcTub levels at both 1 and 10 μM concentrations. A similar trend also emerges from the comparison of series of homologs featuring different endo- (i.e., 49–54) or exo-cyclic (i.e., 55–58) amines. Within each of these series, relatively bulky fragments led to marked elevations of AcTub levels. However, comparison of the average MT-stabilizing activity at 1 and 10 μM, relative to positive control, suggested that the optimal ring size in the endo- and exo-cyclic amine series may be different. Whereas in the exocyclic amine series, the 5-membered ring (57) seemed optimal, in the case of endocyclic amines, the presence of a 7-membered ring system (52) or a fused bicyclic system (54) produced the highest activity (Figure 3). Finally, an evaluation of the effect of fluorination and chiral configuration of the branched amine fragment at C7 was also conducted. Comparison of 7, which features a fluorinated isopropyl amine at C7, with corresponding analogues 43, 44, and 35 that exhibit either 2, 1 or no fluorine substituents in the amine fragment indicates the general trend linking the degree of fluorination of the amine fragment with potency. Similarly, fluorination of 45 resulted in a comparatively more potent congener (46). With respect to the chiral configuration, in the majority of cases examined the chirality of the amine was found to influence the potency of the compound with one enantiomer being typically more active than the other (cf., 3 and 45; 7 and 48; 60 and 61). In one case, however, the chiral configuration of the amine substituent of 46 (R) and 47 (S) was also found to impact significantly the cellular phenotype. Whereas the R-enantiomer was found to be relatively potent in the AcTub QBI293 assay without evidence of a reduction in α-tubulin levels, the opposite enantiomer (S) produced a marked reduction in cellular α-tubulin of approximately ~39% when tested at 10 μM, suggesting that 47 is likely to negatively impact MT-integrity. Notwithstanding this notable exception, all other MT-active triazolopyrimidines shown in Table 2 that feature an amine fragment at C7 were found to be MT-stabilizing without producing losses in α-tubulin.
Figure 3.
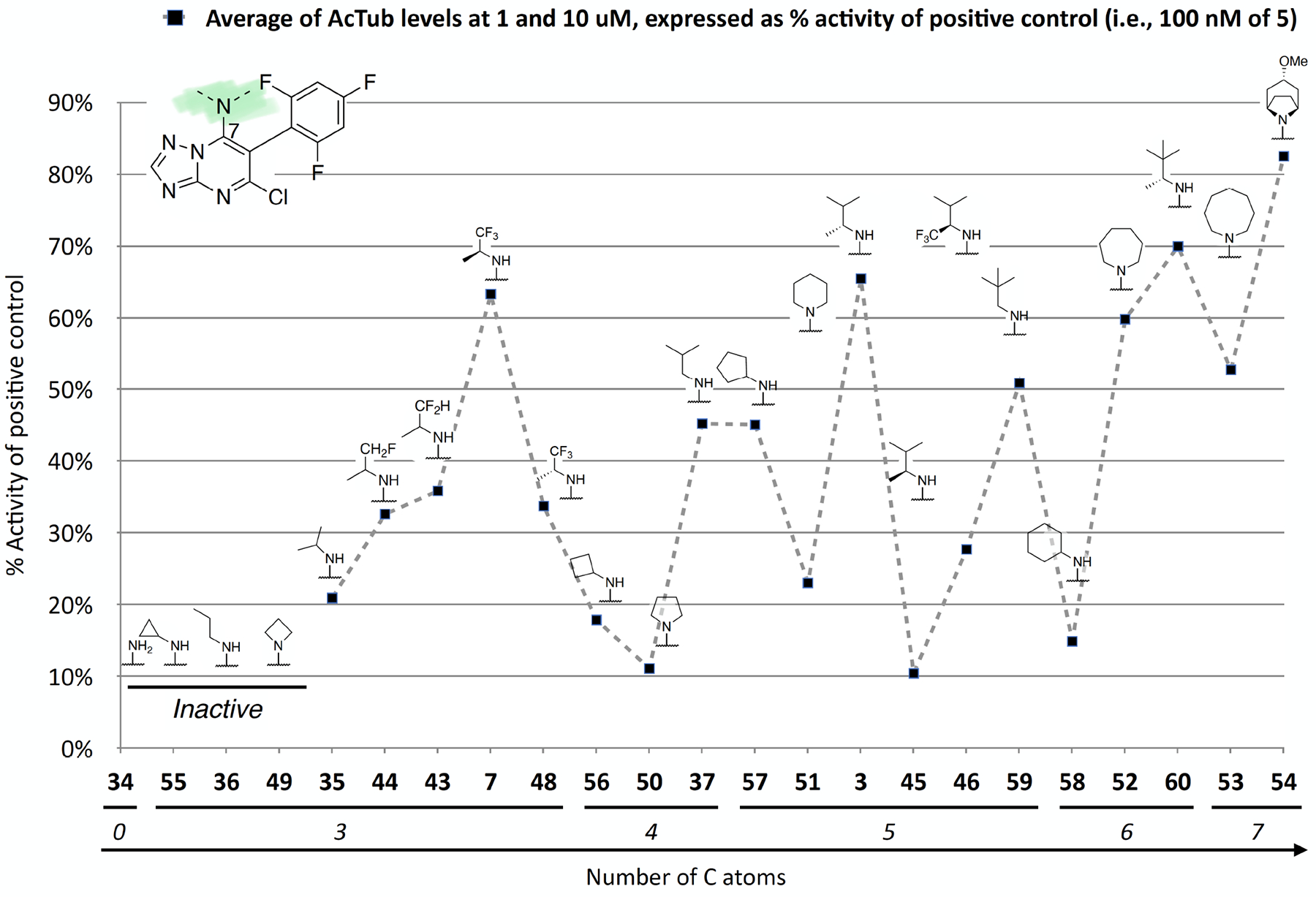
The effect of both branching of aliphatic acyclic amines and homologation of endo- and exo-cyclic amines on MT-stabilizing activity of Class I triazolopyrimidines. Test compounds are ordered along the X-axis based on the number of carbon atoms in the amine fragment. Black squares indicate the average activity of test compounds at 1 and 10 μM, normalized to the activity of 100 nM of 5 (positive control). Compounds that did not produced a statistically significant elevation in AcTub at either 1 or 10 μM are not plotted and marked as inactive.
Having identified the main characteristics of both the C6 and C7 fragments that may be required to obtain Class I MT-stabilizing triazolopyrimidines, a focused series of derivatives was designed by combining the most promising C6 and C7 fragments. In particular, since the C6 fragment SAR studies highlighted the importance of a fluorine atom in the ortho position of the phenyl ring, whereas the para position may be left unsubstituted or occupied by an electron-withdrawing group, such as a fluorine, a nitro or a nitrile, we chose, in addition to the 2,4,6-trifluorophenyl fragment, the corresponding 2-fluoro-, 2,6-difluoro-, 2-fluoro-4-cyano-, and 2,6-difluoro-4-cyano-phenyl C6 fragments. Likewise, among the amine fragments at C7, all instances that by matched molecular pair analysis were found to produce comparable or better activity than 3 (i.e., 7, 52–54 and 60) were selected. By systematically combining the preferred set of C6 and C7 fragments, a panel of 25 compounds could be designed that based on SAR findings would be expected to exhibit relatively potent Class I MT-stabilizing activity. As summarized in Table 3, several examples (13) from this set were synthesized and tested. In all cases, these compounds were confirmed to be active Class I triazolopyrimidines. Nine analogues exhibited comparable (i.e., 66, 67, 73, and 74) or improved (i.e., 64, 69 and 70–72) in vitro potency compared to lead compound 3, with 72 being particularly effective in the AcTub assay when tested at either 10 or 1 μM. Evaluation of individual atropoisomers, 67 and 68, indicated that the former may be more potent than the latter. Among derivatives bearing fluorinated cyanobenzene fragments at C6, evaluation of a representative example (69) in primary neurons confirmed that compounds of this type increase AcTub levels and prevent the characteristic loss of neuronal MTs that is observed after incubation with the phosphatase inhibitor, okadaic acid (Figure 4). Although the cellular data do not allow for resolution of detailed MT structure, the AcTub staining clearly shows that incubation with 69 protects the MT network from okadaic acid-induced collapse (Figure 4).
Table 3.
MT-stabilizing activity of triazolopyrimidines modified at C7 and/or C6. Fold-changes in acetylated α-tubulin (AcTub) and α-tubulin (α-Tub) levels in QBI293 cells after 4 h incubation with test compounds at either 1 or 10 μM. Reported values for AcTub and α-Tub represent the fold-change over control (DMSO)–treated cells (*p<0.05 and**p<0.01 by one-way ANOVA); numbers in parentheses represent the fold-change of AcTub over positive control-treated cells (i.e., 100 nM of 5).
| Cpd # | Structure | AcTub | α-Tub | ||
|---|---|---|---|---|---|
| 1 μM | 10 μM | 1 μM | 10 μM | ||
| 63 |
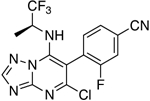
|
2.01 ± 0.38* (0.37) | 3.64 ± 0.26** (0.67) | 1.00 ± 0.01 | 1.00 ± 0.01 |
| 64 |
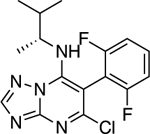
|
4.36 ± 0.72** (0.96) | 4.88 ± 0.72** (1.07) | 1.00 ± 0.02 | 0.96 ± 0.01 |
| 65 |
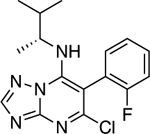
|
0.95 ± 0.26 (0.20) | 2.10 ± 0.31** (0.43) | 1.00 ± 0.04 | 1.04 ± 0.07 |
| 66 |
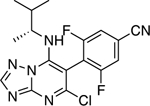
|
3.80 ± 0.15* (0.62) | 4.28 ± 0.14** (0.69) | 0.93 ± 0.02 | 0.94 ± 0.02 |
| 67 |
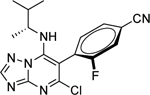
|
2.79 ± 1.11 (0.61) | 3.42 ± 0.55* (0.75) | 0.99 ± 0.02 | 0.93 ± 0.02 |
| 68 |
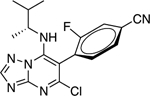
|
2.15 ± 0.36* (0.47) | 2.29 ± 0.30* (0.50) | 0.95 ± 0.02 | 0.92 ± 0.01 |
| 69 |
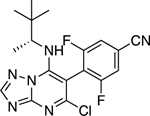
|
2.83 ± 0.33** (0.68) | 4.60 ± 0.35** (1.10) | 0.98 ± 0.03 | 1.03 ± 0.03 |
| 70 |
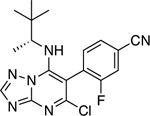
|
4.41 ± 0.27** (0.71) | 7.67 ± 0.70** (1.24) | 0.86 ± 0.03 | 0.93 ± 0.08 |
| 71 |
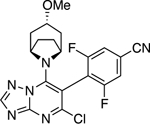
|
2.61 ± 0.26** (0.64) | 4.32 ± 0.29** (1.06) | 0.99 ± 0.01 | 1.00 ± 0.03 |
| 72 |
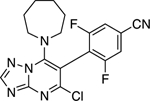
|
6.40 ± 0.61** (1.04) | 11.2 ± 0.77** (1.82) | 0.98 ± 0.03 | 0.99 ± 0.02 |
| 73 |
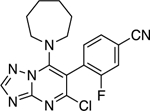
|
7.17 ± 1.09** (0.55) | 12.6 ± 2.8** (0.97) | 1.04 ± 0.05 | 0.99 ± 0.03 |
| 74 |
|
3.24 ± 0.80** (0.58) | 4.40 ± 0.34** (0.79) | 1.01 ± 0.02 | 0.99 ± 0.02 |
| 75 |
|
1.09 ± 0.22 (0.22) | 2.89 ± 0.56** (0.59) | 0.97 ± 0.02 | 0.95 ± 0.07 |
Figure 4.
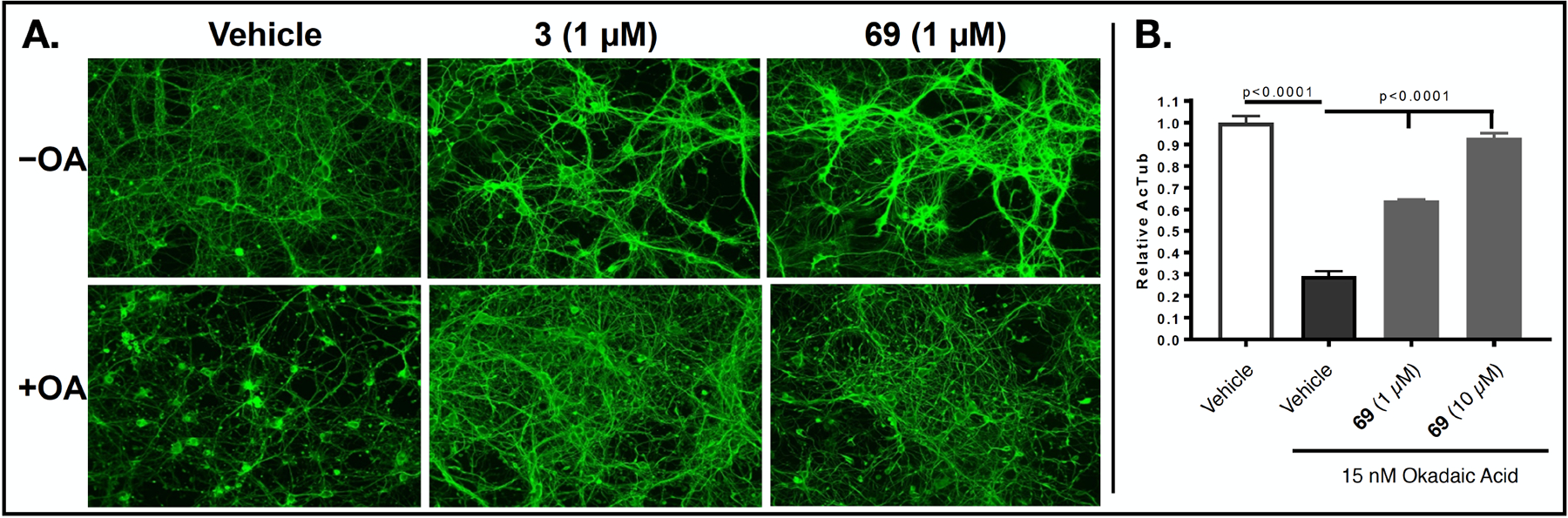
To confirm that the nitrile containing triazolopyrimidine derivatives identified in these studies can stabilize neuronal MTs under conditions of tau loss-of-function, we examined the ability of representative compound, 69, to prevent the MT collapse that occurs from reduced binding of hyperphosphorylated tau to axonal MTs after treatment of neuron cultures with the phosphatase inhibitor, okadaic acid (OA). A. Primary rat cortical neurons treated with 1 μM reference compound 3, or 69 in the absence of OA (−OA) show increased axonal acetyl-tubulin staining relative to those receiving vehicle only. Upon treatment with OA (+OA) in the absence of compound (Vehicle), there is a dramatic reduction in axonal AcTub staining with fragmentation of MTs and neuronal processes (also see11). Co-addition of 3 or 69 (1 μM) with OA results in normalization of AcTub staining and axonal processes. B. ELISA determination of AcTub levels in homogenates from primary mouse cortical neurons treated with 1 or 10 μM of 69, or vehicle, in the presence of OA. The higher concentration of 69 resulted in AcTub levels comparable to those in neurons without OA treatment.
Interestingly, an assessment of lipophilicity of several compounds (Figure 5), as determined by experimental determinations of the distribution coefficient between n-octanol and water at pH 7.4 (i.e., logD7.4 values), revealed that the presence of a nitrile moiety in the para position of the phenyl ring at C6 produces a significant lowering of logD7.4 values that becomes more pronounced in 2,6-difluorophenyl congeners, such as 66, 69 and 72.
Figure 5.
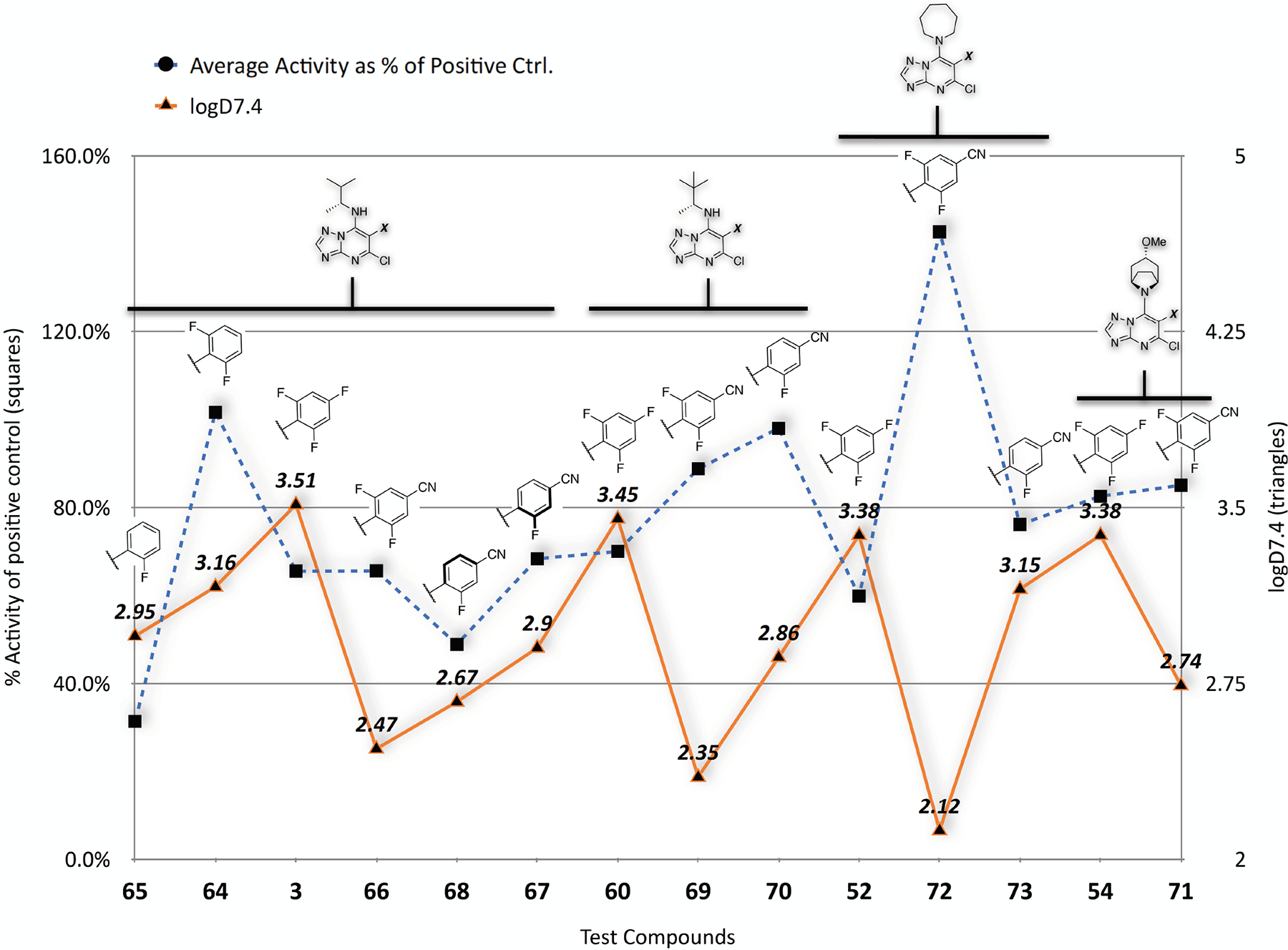
Comparison of selected compounds based on experimental logD7.4 values (triangles) and MT-stabilizing activity (squares) expressed as the average activity in the AcTub assay at 1 and 10 μM normalized to positive control (i.e., 100 nM 5). LogD7.4 values were determined via shake flask method (experiments run by Analiza, Inc.)
Importantly, an evaluation of brain-to-plasma (B/P) ratios of selected examples 1 h after administration to mice confirmed that the nitrile derivatives 66, 69 and 72 are readily brain penetrant with B/P values > 0.6 (Table 4). This result was confirmed by a 16 h brain and plasma PK study with 69 (Figure 6A). Moreover, a comparison of plasma PK of lead compound 3 and 69 indicates a greater metabolic stability of the nitrile-containing congener (Figure 6B).
Table 4.
The ratio between brain and plasma compound concentration 1 h after administration (i.p. injection) of test compound.
| Cpd # | i.p. dose | B/P |
|---|---|---|
| 3 | 5 mg/kg | 2.7 |
| 66 * | 2.5 mg/kg | 0.7 |
| 60 | 5 mg/kg | 2.1 |
| 69 * | 2.5 mg/kg | 0.7 |
| 52 | 5 mg/kg | 1.4 |
| 72 * | 2.5 mg/kg | 0.7 |
Cassette dosing of three compounds (66, 69, and 72)
Figure 6.
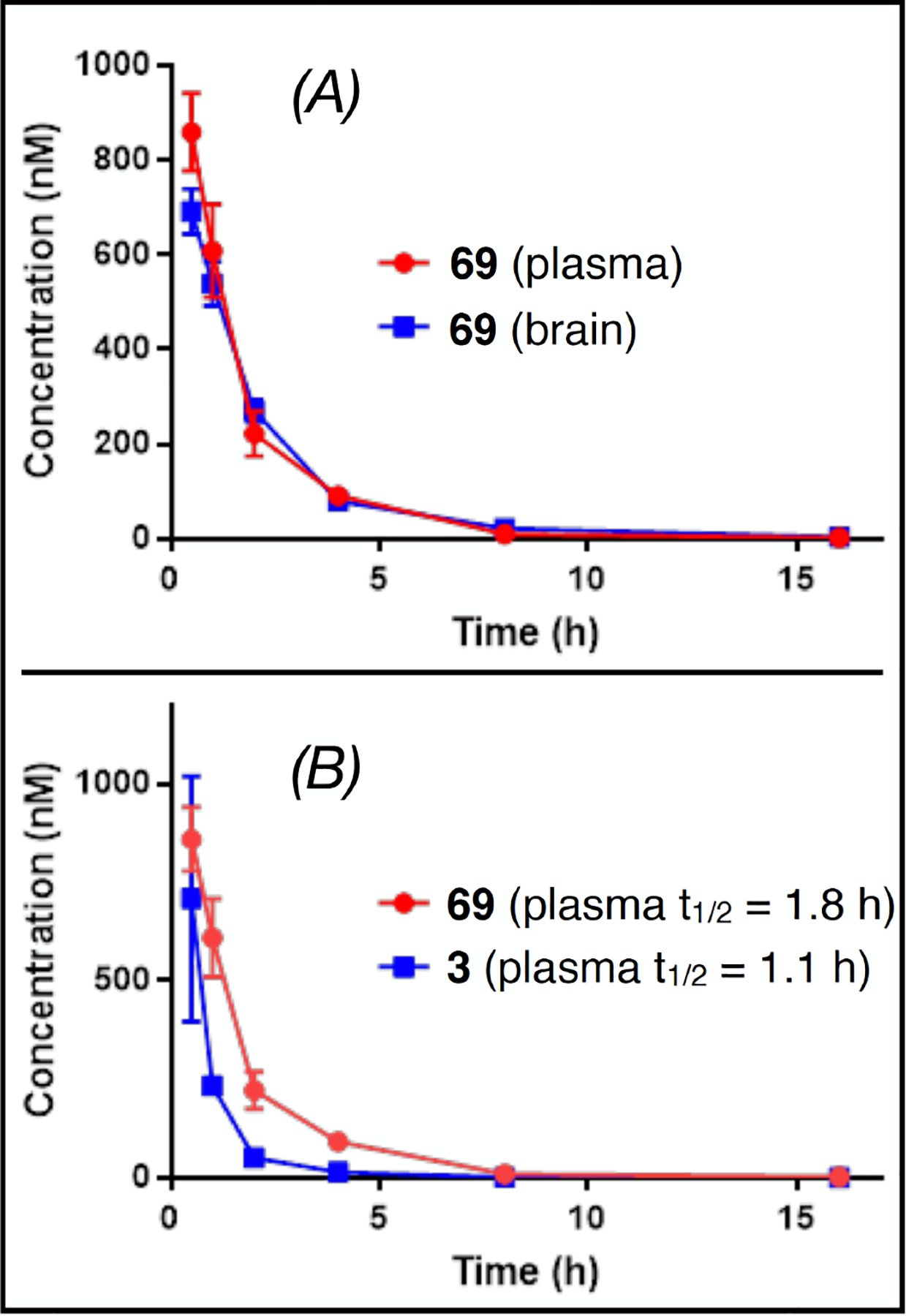
Brain and plasma pharmacokinetics of 69 after 5 mg/kg i.p. dosing to CD1 mice (A), and comparison of plasma pharmacokinetics of 69 and 3.12
Computational Studies.
To investigate the nature of the interaction of Class I triazolopyrimidines with tubulin and to develop an in silico model that may ultimately be predictive of MT-stabilizing activity, we conducted docking studies with both active and inactive triazolopyrimidine congeners using the X-ray co-crystal structure of a tubulin preparation in complex with Class I triazolopyrimidine 5413, 23 (PDB: 5NJH; Figure 7A). The X-ray studies highlighted two key π-π interactions between 54 and tubulin: one involving the pyrimidine core and Tyr224 of α-tubulin, and a second between the trifluorophenyl group and Tyr210 (α-tubulin).13 Both of these interactions are likely essential for a correct orientation of the triazolopyrimidine derivative within the binding pocket and substituents that interfere with these critical interactions are likely to result in reduced binding and biological activity. Indeed, docking of inactive derivative, 30, within the triazolopyrimidine binding site suggests that the relatively large volume of the sulfone moiety disrupts the critical π-π stacking interaction with the two tyrosine residues, leading to a different binding pose (Figure 7B) and, ultimately, a dramatic loss in MT-stabilizing activity. In the case of active Class I compounds, including lead compound 3 (Figure 7C), docking studies generally identify very similar binding poses and sets of interactions as seen in the co-crystal structure of 54. However, for derivatives bearing a nitro or a nitrile at the para position of the fluorinated ring at C6, an additional H-bond interaction with the backbone of Glu207 (α-tubulin) is also observed, which in some cases (e.g., 72) may contribute to better ΔG of binding (Figure 7D) and MT-stabilizing activity (see Figure 5).
Figure 7.
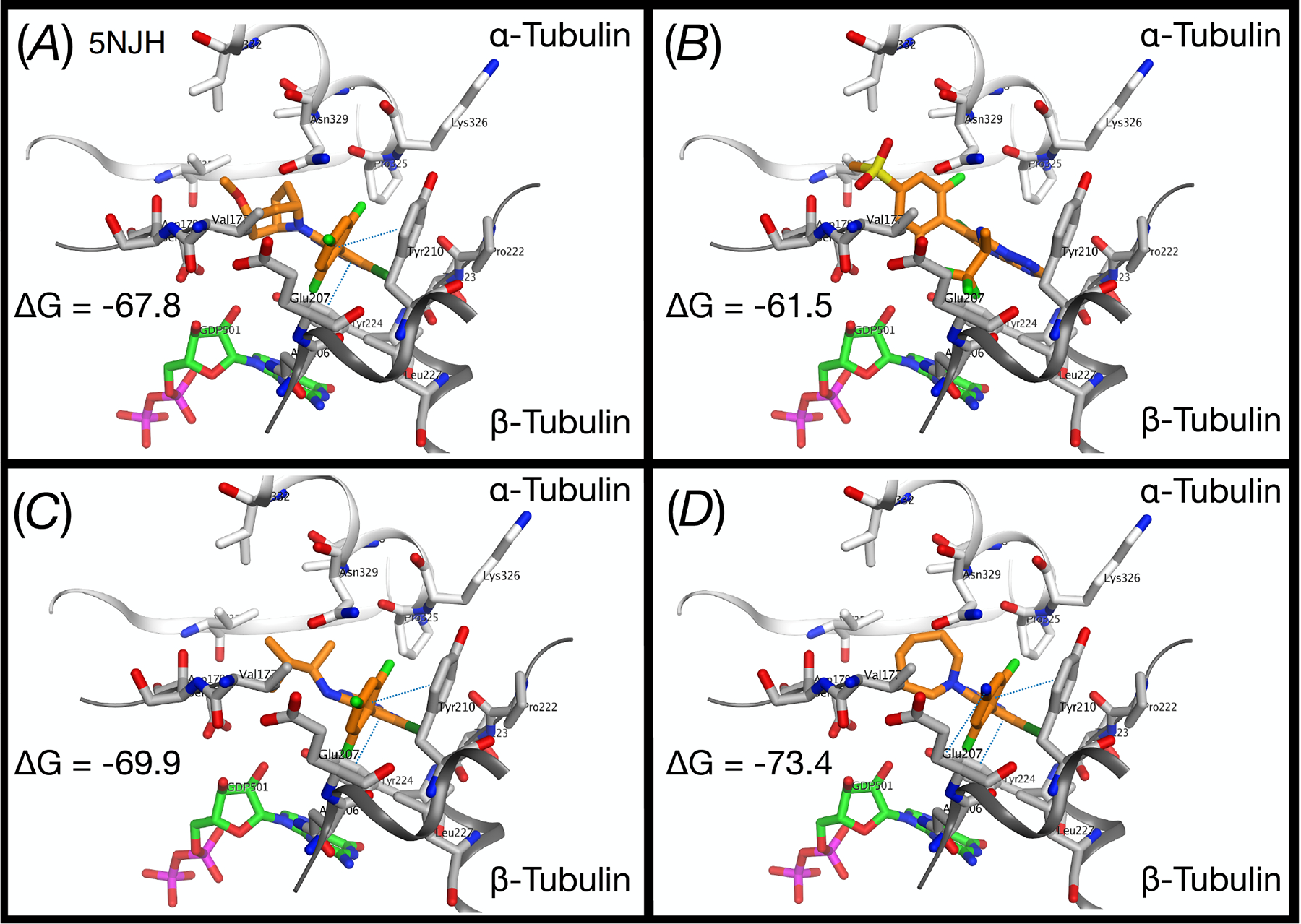
Different views of the co-crystal structure (PBD: 5NJH) of 54 bound within a tubulin preparation (A); and the docked structures of 30 (B), 3 (C) and 72 (D) within the triazolopyrimidine binding site.
The binding modes of the different compounds were further evaluated by calculating the free energy of binding via a “molecular mechanics generalized Born surface area” (MMGBSA) approach.24 Interestingly, by plotting the computed MMGBSA free energetics of the Class I ligands in complex with tubulin against the MT-stabilizing activity (expressed as the average of the activity at 1 and 10 μM compound concentration in the QBI293 AcTub assay, relative to positive control), a moderately high Pearson correlation was found [r = −0.70, P-value (two-tailed) < 0.0001, Figure 8].
Figure 8.
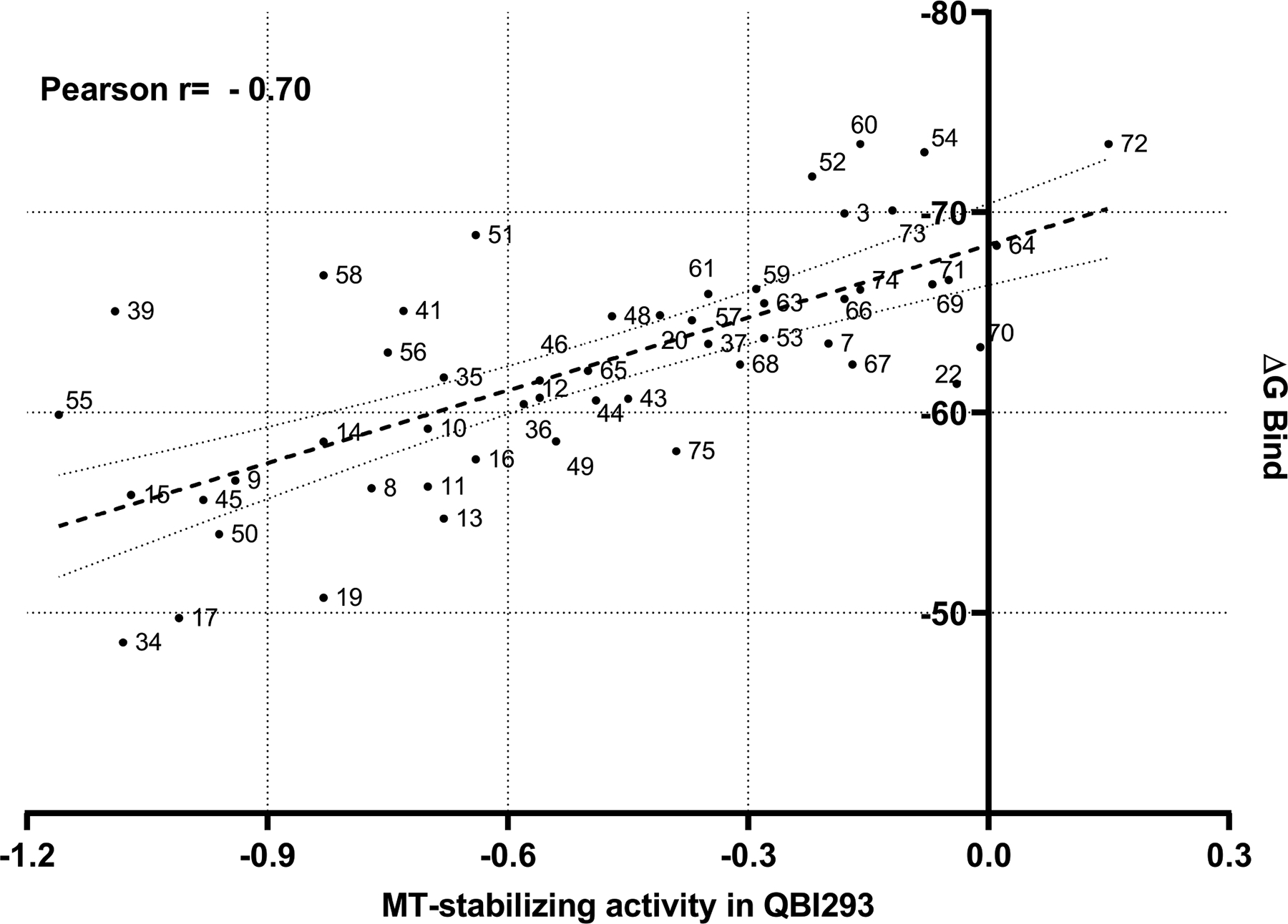
MMGBSA scores for the Class I compounds vs MT-stabilizing activity in QBI293 cells expressed as the log of the average of the activity at 1 and 10 μM in the AcTub assay, relative to positive control, .
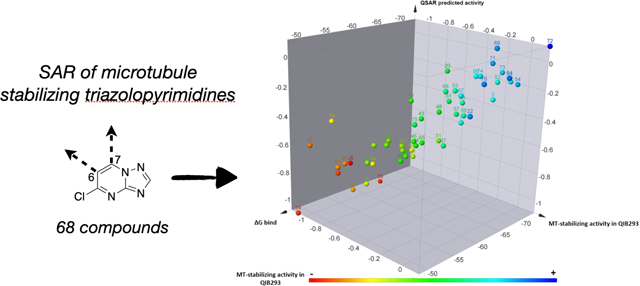
A 3D-QSAR study was also performed through a “field points” template, obtained from the co-crystallized compound. These field points define the shape, electrostatic and hydrophobic property of the molecule and their spatial distribution.25 For this study, 52 active and inactive Class I compounds were randomly partitioned to a training set (47) and a test set (5), while the experimental in vitro activity was computed using the normalized activity, expressed as the log of the average of the activity at 1 and 10 μM in the AcTub assay relative to positive control (5), and defined as a dependent variable. In this case, the model shows a comparatively better correlation with a Pearson r = 0.90 (Figure 9A), which was cross-validated by leave-one-out (LOO) technique, q2= 0.62. Interestingly, the 3D-QSAR model highlighted the following steric and electrostatic contributions to the predicted activity (Figure 9B): a favorable electrostatic contribution (green) near the para position of the phenyl ring at C6, and strong steric contributions of the amine fragment at C7, as indicated by the large size of the dark teal field point (Figure 9B). Importantly, the combination of the binding free energies and the QSAR models (Figure 10) appeared to accurately place all of the most active MT-stabilizing compounds identified in this study within a well-defined area of lower ΔG of binding and higher QSAR predicted activity. Thus, the combinations of these models may provide a valuable tool to guide future design of novel potent MT-stabilizing triazolopyrimidines.
Figure 9.
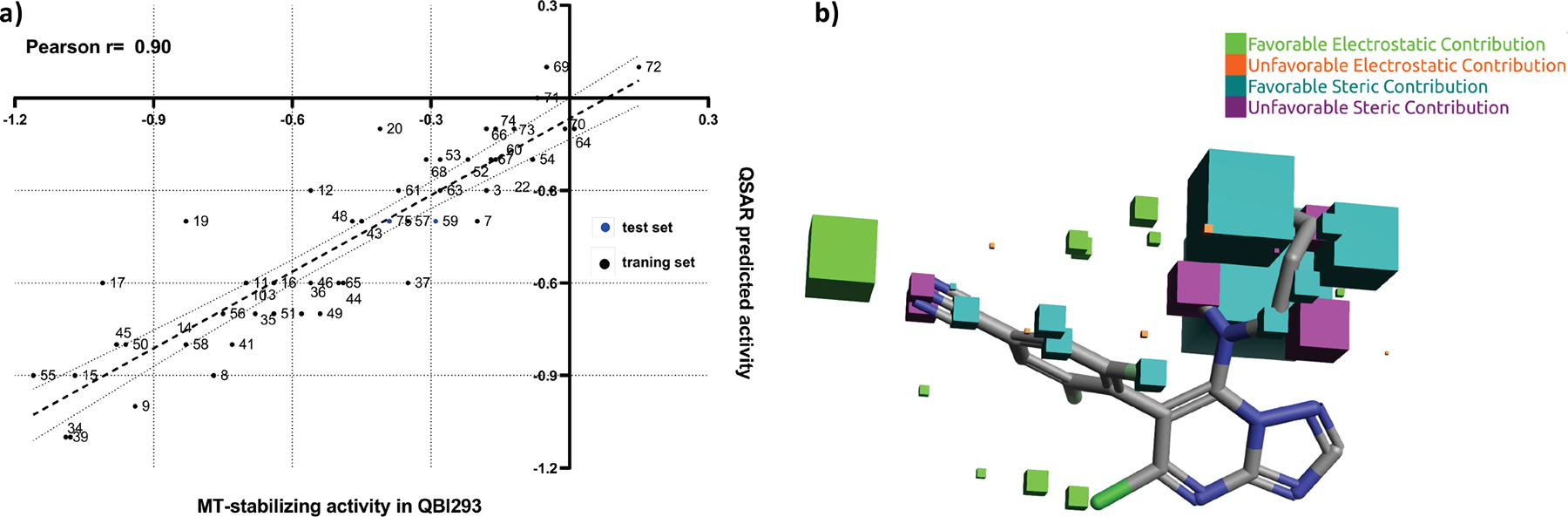
(A) QSAR predicted vs experimental MT-stabilizing activity in QBI293 cells for training and test sets. In both cases, MT-stabilizing activity is expressed as the log of the average of the activity at 1 and 10 μM in the AcTub assay, relative to positive control, . (B) The visual representation of the field and steric contributions to predicted activity for compound 72.
Figure 10.
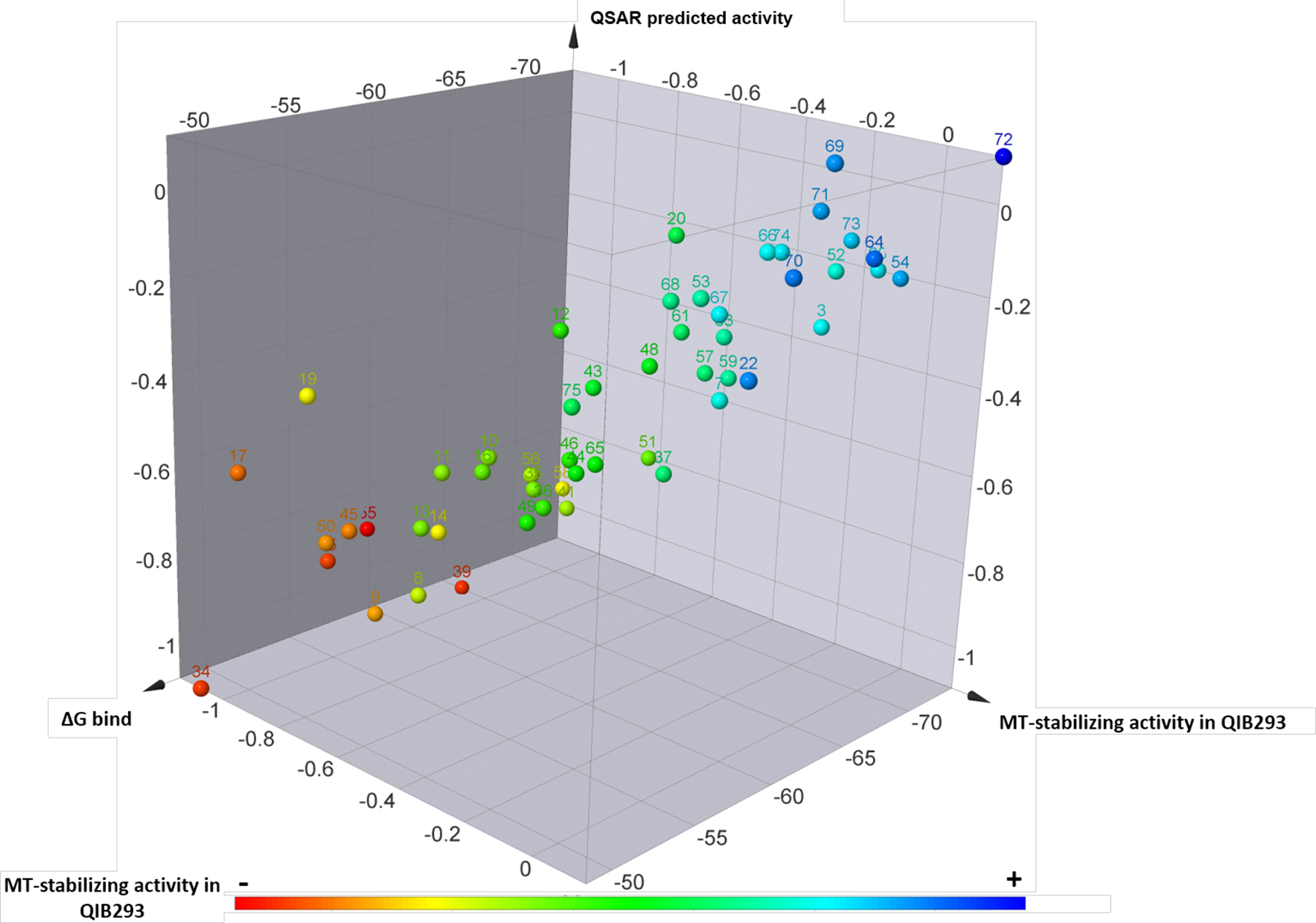
3D summary plot of Class I triazolopyrimidines showing the experimental MT-stabilizing activity in the QBI293 assay, the MMGBSA score value (ΔG bind), and the QSAR predicted activity. The experimental MT-stabilizing activity of test compounds is plotted both via color coding and as the log of the average of the activity at 1 and 10 μM in the AcTub assay, relative to positive control, , see Experimental for further details.
Discussion.
CNS-active MT-stabilizing compounds have been proposed as promising therapeutic candidates to treat AD and related neurodegenerative tauopathies.26 In this context, the essential prerequisites for compound selection and advancement are both the ability to cross the blood-brain barrier (BBB) and normalize the dynamics of MTs in the axons of neurons, such that an efficient axonal transport could be maintained or restored. MT-stabilizing triazolopyrimidines have a number of potentially attractive features as MT-stabilizing compounds for CNS indications,9 including synthetic tractability, a generally good brain penetration and oral bioavailability. Selected members from this class of compounds have already been the focus of mechanism-of-action13 and SAR studies that were based largely on cell-free experiments and cytotoxicity studies in cancer cell-lines.15, 16, 27 Although clearly informative, such studies did not provide particular insight into the effects that triazolopyrimidines can have on cellular MTs. Studies from our laboratories in which the activity of MT-active triazolopyrimidines was evaluated by monitoring compound-dependent changes in cellular levels of tubulin, as well as changes in levels of known markers of MT-stabilization, such as acetylated- and detyrosinated-α-tubulin, showed that different congeners can generally elicit one of two cellular responses discriminated by whether (Class II) or not (Class I) these compounds reduce tubulin levels.11 Furthermore, although one selected prototype from Class I triazolopyrimidines, 3, has proven useful in obtaining validation of this class in mouse models of AD, further characterization of the structural determinants that may be most important to impart the desired MT-stabilizing effect in cells is a necessary step that could facilitate the identification of candidates for clinical development.
Given that our previous studies showed that the substituent in the para position of the phenyl ring (C6 fragment) of triazolopyrimidines can play an important role in determining the particular cellular response (i.e., Class I or II) elicited by these compounds, whereas the C7 fragment may be especially important for potency as well as ADME-PK properties,11, 12 our exploration of SAR of triazolopyrimidines focused primarily on identifying specific features (e.g., electrostatic, hydrophobic, shape) of the C6 and C7 fragments that may be preferred for Class I MT-stabilizing triazolopyrimidines. Data from the cell-based MT-stabilization assays revealed different structural and property features that appear to have a relatively high impact on the biological activity. In particular, a comparison of active congeners of either class suggests that the electrostatic characteristics of the substituent in the para position of the phenyl ring at C6 play an important role in determining the cellular phenotype, while the degree and pattern of fluorination can modulate potency (Figure 2). Indeed, an analysis of in vitro biological activity vs the Hammett σ values of the different substituents in para suggests a general trend, whereby with the sole exception of the para-chloro derivative, 21, positive σ values (e.g., fluorine, nitro or nitrile) are typically conducive to Class I activity, whereas σ values < 0 (e.g., hydroxyl or methoxy groups) appear to be associated with Class II phenotype (Figure 11). Thus, the SAR data of the C6 fragment of triazolopyrimidines indicate that the desired Class I MT-stabilizing activity generally requires the presence of an electron-poor phenyl ring. Different fluorinated phenyl fragments can be tolerated, especially those bearing either one or two fluorine atoms in the ortho positions, and a para position that is either unsubstituted or substituted with an additional fluorine or other electron-withdrawing groups, such as nitro and nitrile.
Figure 11.
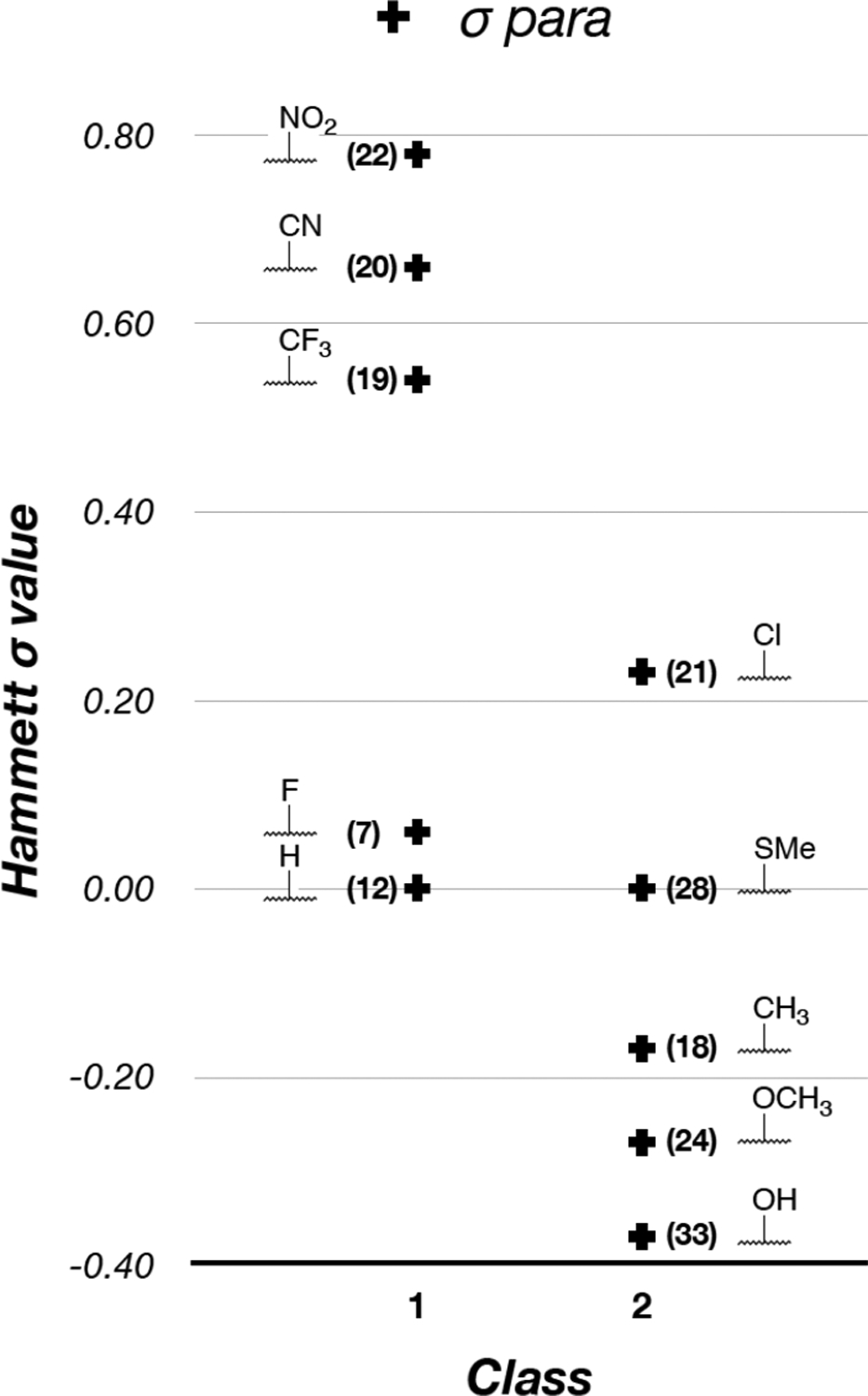
The effect of the stereo-electronic properties of the substituent in para. Active Class I and II compounds are ranked based on the Hammett σp values.28
With respect to the SAR of the fragment linked at C7, the data presented in Table 2 revealed a number of structural and property requirements for optimal MT-stabilizing activity. In particular, in addition to aliphatic amines being typically preferred (e.g., over ethers or aromatic amines), a general trend emerged linking MT-stabilizing activity in cells with the lipophilic character and size of the C7 fragment (Figure 3). An interesting observation made during these studies was the fact that in specific instances the chirality of the aliphatic amine fragment can play a role in determining the cellular phenotype elicited by the triazolopyrimidine. Although the configuration of the chiral center of the amine fragment of 3 or 7 appears to impact the potency of these compounds in the AcTub assay without affecting α-tubulin levels (cf., 7 with 48, and 3 with 45), in selected cases, such as compounds 46 and 47, the inversion of chiral configuration was found to cause a shift in cellular phenotype, with 46 being a Class I compound while its enantiomer, 47, clearly produced a marked reduction (~40%) in α-Tub (Class II). Interestingly, docking studies with 46 and 47 did not reveal significant differences in binding modes that could help explain the different cellular phenotype elicited by the two enantiomers. In addition, side-by-side comparison of the cytotoxicity of enantiomers 46 (Class I) and 47 (Class II) in rapidly dividing QBI293 or HeLa cells did not reveal significant differences in IC50 values (Supporting Information), suggesting that both Class I and II triazolopyrimidines exhibit comparable anti-mitotic effects. These observations indicate that neither docking nor IC50 values in cell cytotoxicity assays should be used to predict whether a triazolopyrimidine would stabilize or disrupt the MT network. Given that such a distinction is an essential aspect driving the prioritization of candidates for neurodegenerative tauopathies, an evaluation of compounds in a cellular assay that assesses compound effect on α/β-tubulin levels as well as increases in markers of MT stabilization is ultimately a necessary step. Although such assays have some limitations, such as a relatively low-throughput and possible day-to-day variability that can be minimized by normalizing the activity of test compounds to a positive control, the SAR data generated in this study led to the identification of several characteristics that are important for Class I MT-stabilizing triazolopyrimidines. Indeed, a qualitative assessment of the combined C6 and C7 SAR data of all Class I triazolopyrimidines tested in this study, which was conducted using Activity atlas analysis,29 identified distinct regions/characteristics that are shared by the vast majority of Class I congeners (Figure 12 A&B); these include a positive electrostatic region in the proximity of the amine fragment at C7 (indicated in red, Figure 12A), a negative electrostatic region in the proximity of the para position of the aryl group at C6 (cyan, Figure 12A), and two hydrophobic interaction regions near the fluorinated phenyl at C6 and the aliphatic amine fragment at C7 (yellow, Figure 12B). Furthermore, an activity cliff analysis (Figure 12C), which highlights structural differences of similar compounds that produce a disproportionally high impact on the biological activity (i.e., activity cliff regions),30 revealed that an increase of negative electrostatic field at the para position of the phenyl ring (cyan), as associated with substituents like nitro and nitrile, as well as the presence of a favorable hydrophobic region in C7 (green), produce a drastic increase in MT-stabilizing activity. Collectively, these findings, combined with the development of relatively predictive models that are based on calculated binding free energies and QSAR, provide valuable insights that could facilitate the design of Class I triazolopyrimidines.
Figure 12.
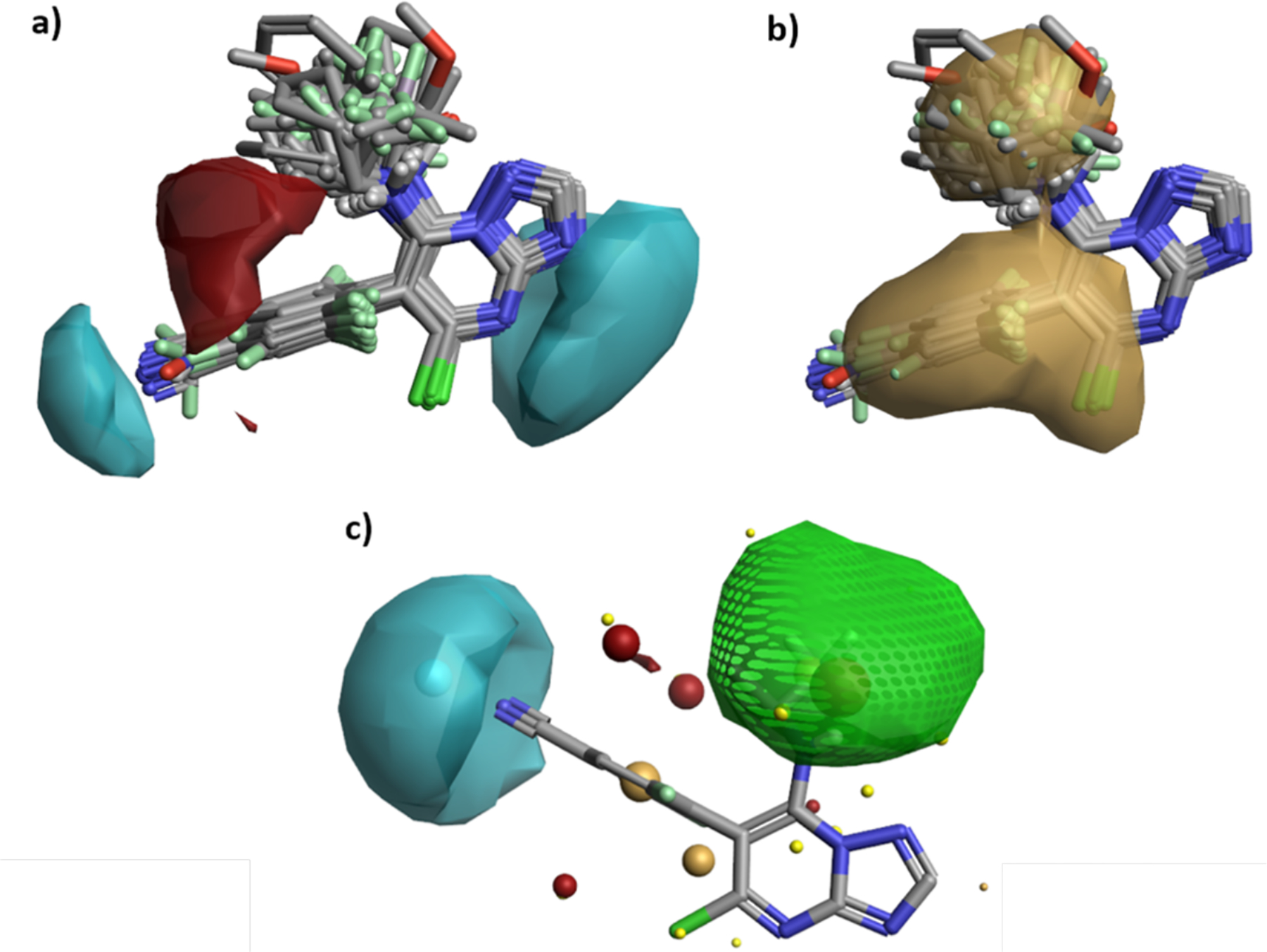
Activity atlas analysis revealing key features of Class I triazolopyrimidines that are necessary for MT-stabilizing activity: (a) red and cyan colors show positive and negative field regions respectively; (b) brown beige regions show hydrophobic interaction sites required for activity; (c) Activity cliffs analysis revealing a favorable hydrophobic region (green) and a favorable negative electrostatic region (cyan).
Importantly, by combining the preferred C6 and C7 fragments found in the course of these SAR studies, several new Class I triazolopyrimidine analogues have been identified (Table 3), including multiple examples (64, 69 and 70–72) that exhibit both improved in vitro activity as well as reduced lipophilic character compared to lead compound 3. In primary cortical neurons, 69 was found to protect the MT-network against okadaic acid-induced collapse in a manner comparable to 3 (Figure 4) suggesting that these new congeners, like 3, may be beneficial in normalizing MT-deficits in tauopathy neurons. Interestingly, although the effect of the nitrile moiety in lowering lipophilicity is well documented,31 direct comparison of experimentally determined logD7.4 values of matched paired compounds bearing a nitrile moiety in the para position of the ring at C6 (cf., 66 with 67/68; 69 with 70; and 72 with 73) demonstrates that the fluorination state of the aromatic ring plays an equally important role as evidenced by the significantly lower logD7.4 values observed when both ortho positions are fluorinated (Figure 5). Of notice, in spite of the reduced lipophilic character of 66, 69 and 72 compared to the corresponding 2,4,6-trifluorophenyl congeners (3, 60, and 52), these compounds retained excellent brain penetration (Table 4). Furthermore, evaluation of brain/plasma PK of 69 after i.p. injection revealed that this compound exhibits greater metabolic stability (i.e., reduced clearance and increased plasma half-life) compared to existing lead (3). Thus, taken together, our results show that an appropriate combination of relatively bulky/lipophilic aliphatic amines at C7 with polar fluorinated cyanobenzene fragments at C6 results in Class I congeners that exhibit both improved MT-stabilizing activity in cells, as well as improved lipophilic efficiency and metabolic stability.
Conclusions.
Brain penetrant, MT-stabilizing triazolopyrimidines have produced promising results in different mouse models of AD, suggesting that compounds from this class may be considered as candidate therapeutics for neurodegenerative tauopathies. However, depending on the substitution pattern, this type of compounds can elicit very different cellular responses by either promoting MT stabilization or, conversely, disrupting MT integrity in cells. The studies presented here define the most important stereoelectronic characteristics of the C6 and C7 fragments of MT-active triazolopyrimidines that can lead to relatively potent MT-stabilizing activity in cells without reduction in cellular tubulin levels. These results, which provide additional insight into the SAR of MT-stabilizing triazolopyrimidines, enabled both the development of predictive models as well as the design of congeners exhibiting improved properties and in vitro biological activity. Compounds of this type hold promise as candidate treatments for neurodegenerative tauopathies.
Experimental Section.
Materials and Methods.
All solvents were reagent grade. All reagents were purchased from Aldrich or Acros and used as received. Thin layer chromatography (TLC) was performed with 0.25 mm E. Merck precoated silica gel plates. Silica gel column chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm) supplied by Silicycle and Sorbent Technologies. TLC spots were detected by viewing under a UV light. Melting points (mp) were acquired on a Mel-Temp II (model : 1001) and are uncorrected. Infrared (IR) spectra were recorded on a Bruker, model Alpha spectrometer (part number 1003271/03). Proton (1H) and carbon (13C) NMR spectra were recorded on a 500 MHz Bruker AMX-500 spectrometer or 600 MHz Bruker Avance III spectrometer. Fluorine (19F) NMR spectra were recorded on a 500 MHz Bruker Avance II spectrometer. Chemical shifts were reported relative to solvents, Data for 1H-NMR spectra are reported as follows: chemical shift (ppm, referenced to protium; s = singlet, d = doublet, t = triplet, q= quartet, quint = quintet, dd = doublet of doublets, td = triplet of doublets, ddd = doublet of doublet of doublets, bs = broad singlet, m = multiplet, coupling constant (Hz), and integration). High-resolution mass spectra were measured using an Agilent 6230 time-of-flight mass spectrometer (TOFMS) with Jet stream electrospray ionisation source (ESI). Single-crystal X-ray structure determinations were performed Bruker MicroStar with an APEX II detector, double-bounce micro-focus optics and a Cu rotating anode source. Analytical reverse-phase (Sunfire C18; 4.6 mm × 50 mm, 5 mL) high-performance liquid chromatography (HPLC) was performed with a Gilson HPLC equipped with UV and mass detector. All samples were analyzed employing a linear gradient from 10% to 90% of ACN in H2O over 8 min and flow rate of 1 mL/min. Preparative reverse-phase HPLC purifications were performed on a Gilson instrument employing Waters SunFire preparative C18 OBD columns (5 μm 19 mm × 50 mm or 19 mm × 100 mm). Purifications were carried out employing a linear gradient from 10% to 90% of ACN in H2O for 15 min with a flow rate of 20 mL/min. All final compounds were found to be >95% pure by HPLC.
General Procedure A (synthesis of diethylmalonate derivatives):
To a suspension of NaH (60 wt % in mineral oil) (1.00 equiv) in anhydrous 1,4-dioxane (previously degassed with N2) at 60 °C under N2 was slowly added diethyl malonate (3.00 equiv) and the resulting mixture was stirred for 10 min. CuBr (1.20 equiv) and aryl bromide derivate were then added and the reaction mixture (1.75 mol/L of aryl bromide derivate) was heated to reflux overnight. The reaction mixture was then cooled to r.t., quenched with HCl 12N (1.40 equiv), filtered and washed with H2O. The filtrate was extracted with EtOAc (×3). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification via silica gel column chromatography or preparative reverse-phase HPLC provided the desired diethylmalonate derivatives.
General procedure B (synthesis of dichloro-triazolopyrimidine derivatives):
A mixture of the appropriate diethylmalonate derivate (1.00 equiv), 3-amino-1,2,4-triazole (1.05 equiv) and tributylamine (1.05 equiv) in a sealed tube was stirred at 170 °C for 2 h. After cooling to 130 °C, the reaction mixture was diluted with toluene to reach approximately a concentration of 1.00 M of diethylmalonate derivative. The reaction was then cooled to 50 °C and an aqueous solution of NaOH (50% wt) (0.160 mL/mmol of diethylmalonate derivate) was added. The resulting mixture was stirred at 0 °C for 10 min and then filtered to obtain the triazolopyrimidine disodic salt intermediate as a solid, which was washed with cold toluene, dried and used directly without further purification. This disodic derivate (1.00 equiv) and POCl3 (17.80 equiv) were mixed in a sealed tube and heated to 130 °C for 6 h. The reaction was then quenched with H2O and extracted with EtOAc (×2). The combined organic extracts were washed with H2O (×5), then brine, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting dichloro-triazolopyrimidine was used directly without further purification.
General procedure C (addition of the amine):
According to a reported procedure,15 to a solution of 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (1.0 equiv) in DMF (0.1 M) at r.t. was added the appropriate amine (1.5 – 3.0 equiv either as HCl salt or free base) followed by i-Pr2NEt (3.0 equiv) or Et3N (3.0 equiv), if necessary. The reaction mixture was stirred for 0.5–16 h and then diluted with H2O. The organic layer was washed with a 1N HCl (×2), the aqueous phase was extracted with EtOAc (×3) and the combined organic layers were washed with brine (×2), dried over MgSO4, filtered, and concentrated. Finally, purification via silica gel flash chromatography or by reverse-phase HPLC yielded the desired product.
General procedure D (addition of the side chain):
According to reported procedures,15, 16 to a suspension of NaH (4.0 equiv) in a 2:1 mixture of DMSO and THF (0.35 M) was added the appropriate aminoalcohol (4.0 equiv), and the mixture was heated to 60 °C for 1 h. The resulting solution was treated with a solution of trifluoroarene (1.0 equiv) in a 1:1 mixture DMSO and THF (0.5 M). The reaction mixture was stirred at 60 °C for 3 h and monitored by LCMS. If the starting material remained after 3 h, additional aminoalcohol (4.0 equiv) and NaH (4.0 equiv) were added, sequentially, and the reaction mixture was heated for 16 h. Following complete consumption of the starting material, the reaction mixture was cooled to r.t. and diluted with H2O and EtOAc. The organic layer was washed with H2O and brine, and the combined aqueous layers were extracted with EtOAc (×3). The combined organic layers were dried over MgSO4, filtered, and concentrated. Purification by reverse-phase HPLC provided the desired product.
(S)-5-Chloro-6-phenyl-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (8).
Following General Procedure C using 5,7-dichloro-6-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine (0.500 g, 1.88 mmol) (93) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.840 g, 5.64 mmol). Purification via preparative reverse-phase chromatography provided the title compound as a white powder (0.066 g, 0.19 mmol, 10%).1H NMR (500 MHz, CDCl3) δ 8.40 (s, 1H), 7.62 – 7.52 (m, 3H), 7.42 – 7.36 (m, 1H), 7.35 – 7.28 (m, 1H), 5.68 (s, 1H), 4.63 (s, 1H), 1.29 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.17, 155.44, 145.72, 131.67, 131.51, 130.54, 130.09, 129.96, 129.82, 124.90 (q, J = 282.4 Hz), 105.26, 50.67 (q, J = 31.5 Hz), 15.02 (q, J = 1.7 Hz) ppm; IR (film) ν 3435, 1619, 1577, 1491, 1460, 1384, 1366, 1338, 1264, 1248, 1179, 1145, 1085, 1024, 955, 906, 767, 742, 702, 652, 615 cm−1; HRMS (ES+) calculated for C14H12ClF3N5 [M + H]+: 342.0728, found 342.0726.
(S)-5-Chloro-6-(4-fluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (9).
Following General Procedure C using 5,7-dichloro-4-fluorophenyl-[1,2,4]triazolo[1,5-a]pyrimidine (0.300 g, 1.04 mmol) (94) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.465 g, 3.11 mmol). Purification via preparative reverse-phase chromatography provided the title compound as a white powder (0.126 g, 0.35 mmol, 34%). 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 1H), 7.42 – 7.35 (m, 1H), 7.36 – 7.24 (m, 3H), 5.65 (s, 1H), 4.77 (s, 1H), 1.34 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.57 (d, J = 251.3 Hz), 157.29, 155.56, 153.93, 145.86, 133.63 (d, J = 8.4 Hz), 132.61 (d, J = 8.4 Hz), 127.57 (d, J = 3.7 Hz), 124.94 (q, J = 282.4 Hz), 117.34 (d, J = 22.0 Hz), 117.16 (d, J = 22.0 Hz), 104.31, 50.83 (q, J = 31.6 Hz), 15.04 (d, J = 2.1 Hz) ppm; IR (film) ν 3421, 1619, 1574, 1504, 1461, 1366, 1337, 1247, 1180, 1146, 1096, 1024, 957, 841, 766, 738, 610 cm−1; HRMS (ES+) calculated for C14H10ClF4N5Na [M + Na]+: 382.0459, found 382.0464.
5-Chloro-6-(2-fluorophenyl)-N-(−1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (10).
Following General Procedure C using 5,7-dichloro-6-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.300 g, 1.04 mmol) (95) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.465 g, 3.11 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.060 g, 0.17 mmol, 16%). 1H NMR (500 MHz, CDCl3) mixture of diastereomers δ 8.39 (s, 1H), 7.61 – 7.52 (m, 1H), 7.40 – 7.27 (m, 3H), 6.08 – 5.49 (m, 1H), 4.95 – 4.22 (m, 1H), 1.34 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 161.79, 161.59, 159.80, 159.61, 157.71, 157.50, 155.50, 155.43, 154.20, 153.83, 145.83, 145.41, 133.41, 133.39, 132.79, 132.72, 132.66, 132.35, 132.34, 128.18, 128.00, 125.93, 125.76, 125.50, 125.47, 125.44, 125.41, 123.69, 123.51, 121.45, 121.27, 119.42, 119.30, 119.18, 117.21, 117.04, 116.70, 116.53, 99.20, 98.22, 51.26, 51.00, 50.75, 50.49, 50.24, 15.18, 15.16, 15.07, 15.05 ppm; IR (film) ν 3435, 1618, 1554, 1491, 1460, 1368, 1248, 1179, 1144, 1094, 1025, 960, 759, 651, 616 cm−1; HRMS (ES+) calculated for C14H10ClF4N5Na [M + Na]+: 382.0459, found 382.0441.
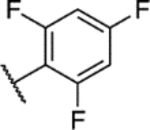
(S)-5-Chloro-6-(2,6-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (12).
Following General Procedure C using 5,7-dichloro-6-(2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.280 g, 0.93 mmol) (97) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.417 g, 2.79 mmol). Purification via preparative reverse-phase chromatography provided the title compound as a white powder (0.024 g, 0.06 mmol, 7%). 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 1H), 7.54 – 7.45 (m, 1H), 7.10 – 7.00 (m, 2H), 6.19 – 6.10 (m, 1H), 4.76 (s, 1H), 1.39 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 161.12 (dd, J = 252.3, 5.5 Hz), 160.82 (dd, J = 250.9, 5.3 Hz), 157.72, 155.26, 154.16, 145.89, 133.01 (t, J = 10.0 Hz), 124.59 (dd, J = 564.1, 282.1 Hz), 112.43 (dd, J = 21.5, 3.4 Hz), 112.07 (dd, J = 21.5, 3.6 Hz), 108.90 (t, J = 20.2 Hz), 92.39, 50.86 (q, J = 32.3 Hz), 14.98 ppm. IR (film) ν 3227, 1630, 1602, 1460, 1383, 1266, 1239, 1195, 1126, 1003, 900, 809, 765, 652, 622 cm−1; HRMS (ES+) calculated for C14H10ClF5N5 [M + H]+: 378.0539, found 378.0556.
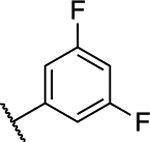
(S)-5-Chloro-6-(3,5-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (13).
Following General Procedure C using 5,7-dichloro-6-(3,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.350 g, 1.16 mmol) (98) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.521 g, 3.49 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.148 g, 0.39 mmol, 34%). 1H NMR (500 MHz, CDCl3) δ 8.40 (s, 1H), 7.03 (dt, J = 8.6, 2.3 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 5.64 (s, 1H), 4.82 (s, 1H), 1.38 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.72 (dd, J = 253.2, 13.0 Hz), 163.66 (dd, J = 251.7, 11.7 Hz), 156.45, 155.71, 153.96, 145.57, 134.65 (t, J = 10.0 Hz), 124.85 (q, J = 282.4 Hz), 115.03 (dd, J = 21.9, 3.7 Hz), 114.04 (dd, J = 21.9, 3.7 Hz), 105.99 (t, J = 24.8 Hz), 103.18, 51.07 (q, J = 31.6 Hz), 15.02 (d, J = 1.9 Hz) ppm; IR (film) ν 3435, 1620, 1590, 1521, 1493, 1460, 1434, 1384, 1362, 1333, 1264, 1192, 1159, 1125, 1081, 1028, 987, 956, 906, 855, 763, 670, 653, 628 cm−1; HRMS (ES−) calculated for C14H8ClF5N5 [M − H]− 376.0394, found 376.0376.
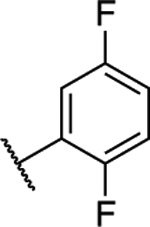
(S)-5-Chloro-6-(2,5-difluorophenyl)-N-(−1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (14).
Following General Procedure C using 5,7-dichloro-6-(2,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.400 g, 1.33 mmol) (99) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.596 g, 3.99 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.020 g, 0.05 mmol, 4%). 1H NMR (500 MHz, CDCl3) mixture of diastereomers δ 8.40 (s, 1H), 7.32 – 7.24 (m, 2H), 7.15 – 7.02 (m, 1H), 6.07 – 5.46 (m, 1H), 4.71 (d, J = 156.9 Hz, 1H), 1.39 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) mixture of diastereomers δ 159.86, 159.84, 157.90, 157.88, 157.71, 157.68, 157.28, 157.15, 155.95, 155.93, 155.76, 155.73, 155.64, 155.58, 154.21, 153.94, 145.78, 145.47, 128.12, 127.98, 125.88, 125.74, 123.64, 123.49, 121.39, 121.25, 120.68, 120.62, 120.56, 120.54, 120.50, 120.47, 120.41, 120.35, 119.99, 119.97, 119.80, 119.78, 119.54, 119.51, 119.47, 119.44, 119.35, 119.32, 119.28, 119.25, 118.91, 118.89, 118.72, 118.70, 118.52, 118.45, 118.33, 118.26, 118.00, 117.93, 117.81, 117.74, 98.19, 97.30, 51.46, 51.25, 51.21, 50.99, 50.95, 50.74, 50.70, 50.49, 15.19, 15.18, 15.09, 15.08 ppm; IR (film) ν 3441, 1622, 1494, 1461, 1424, 1383, 1251, 1182, 1145, 1092, 1025, 979, 877, 821, 775 cm−1; HRMS (ES+) calculated for C14H10ClF5N5 [M + H]+: 378.0539, found 378.0509.
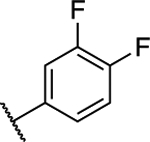
(S)-5-Chloro-6-(3,4-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (15).
Following General Procedure C using 5,7-dichloro-6-(3,4-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.150 g, 0.50 mmol) (100) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.223 g, 1.49 mmol). Purification via preparative reverse-phase chromatography provided the title compound as a white powder (0.007 g, 0.02 mmol, 4%).1H NMR (500 MHz, CDCl3) mixture of diastereomers δ 8.42 (s, 1H), 7.45 – 7.34 (m, 1H), 7.22 – 7.05 (m, 2H), 5.71 – 5.47 (m, 1H), 5.02 – 4.63 (m, 1H), 1.37 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) mixture of diastereomers δ 157.04, 157.01, 156.89, 156.88, 155.78, 155.73, 155.72, 155.66, 152.56, 152.50, 152.43, 152.38, 152.33, 152.20, 152.09, 151.96, 150.51, 150.46, 150.38, 150.35, 150.17, 150.06, 149.94, 145.95, 145.91, 145.82, 145.78, 145.74, 128.38, 128.34, 128.30, 127.36, 127.31, 127.28, 126.04, 125.99, 123.80, 123.75, 121.58, 121.55, 121.18, 121.05, 120.19, 120.17, 120.06, 120.05, 119.34, 119.29, 119.24, 119.19, 119.16, 119.02, 103.47, 103.35, 51.42, 51.35, 51.16, 51.10, 50.91, 50.84, 50.65, 50.59, 15.08 ppm; IR (film) ν 3436, 2923, 2853, 1632, 1463, 1383, 1270, 1147 cm−1; HRMS (ES+) calculated for C14H10ClF5N5 [M + H]+: 378.0539, found 378.0550.
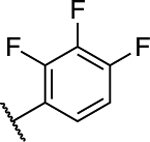
(S)-5-Chloro-6-(2,3,4-trifluorophenyl)-N-(−1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (16).
Following General Procedure C using 5,7-dichloro-6-(2,3,4-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.300 g, 0.94 mmol) (101) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.422 g, 2.82 mmol). Purification via preparative reverse-phase chromatography provided the title compound as a white powder (0.050 g, 0.13 mmol, 13%). 1H NMR (500 MHz, CDCl3) mixture of diastereomers δ 8.37 (s, 1H), 7.24 – 7.05 (m, 2H), 6.05 – 5.61 (m, 1H), 5.20 – 4.47 (m, 1H), 1.42 (d, J = 6.3 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) mixture of diastereomers δ 157.56, 157.24, 155.75, 155.67, 154.40, 153.95, 153.77, 153.74, 153.69, 153.67, 153.62, 153.59, 151.74, 151.71, 151.66, 151.63, 151.58, 151.56, 151.39, 151.36, 151.31, 151.28, 151.06, 151.03, 150.98, 150.95, 149.37, 149.34, 149.29, 149.26, 149.05, 149.02, 148.97, 148.94, 146.17, 145.67, 142.19, 142.07, 141.95, 141.86, 141.74, 141.62, 140.16, 140.04, 139.91, 139.83, 139.70, 139.58, 128.20, 127.95, 127.23, 127.20, 127.18, 127.17, 127.14, 126.15, 126.14, 126.11, 126.10, 126.09, 126.08, 126.05, 125.95, 125.70, 123.71, 123.46, 121.47, 121.22, 117.07, 117.04, 116.96, 116.93, 116.91, 116.84, 116.80, 113.96, 113.92, 113.91, 113.88, 113.81, 113.78, 113.77, 113.74, 97.55, 96.47, 51.59, 51.34, 51.31, 51.08, 51.05, 50.83, 50.80, 50.55, 15.21, 15.19, 15.07, 15.06 ppm. IR (film) ν 3388, 3338, 1619, 1553, 1509, 1486, 1463, 1367, 1352, 1251, 1181, 1143, 1090, 1041, 1025, 987, 957, 906, 878, 808, 766, 738, 698, 680, 653, 619 cm−1; HRMS (ES+) calculated for C14H9ClF6N5 [M + H]+: 396.0445, found 396.0464.
(S)-5-Chloro-6-(2,4,6-trifluorobenzyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (17).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorobenzyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.300 g, 0.90 mmol) (103) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.404 g, 2.70 mmol). Purification via silica gel column chromatography (0–25% EtOAc in hexanes) provided the title compound as a white powder (0.085 g, 0.21 mmol, 23%). 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 6.73 (t, J = 8.3 Hz, 2H), 6.32 – 6.12 (m, 1H), 5.23 (d, J = 10.2 Hz, 1H), 4.27 – 4.08 (m, 2H), 1.50 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 162.21 (dt, J = 251.1, 16.0 Hz), 161.52 (dd, J = 247.9, 10.9 Hz), 161.41 (dd, J = 248.0, 10.8 Hz), 157.36, 155.42, 154.39, 146.49, 125.14 (q, J = 282.1 Hz), 108.38 (td, J = 19.0, 4.8 Hz), 101.31 – 100.77 (m), 100.73, 51.82 (q, J = 31.5 Hz), 21.06, 15.30 (d, J = 2.2 Hz) ppm; IR (film) ν 3427, 3276, 3143, 3001, 2926, 2855, 1621, 1552, 1496, 1464, 1443, 1378, 1337, 1274, 1244, 1182, 1145, 1118, 1091, 1035, 999, 959, 910, 842, 804, 762, 734, 680, 648, 613 cm−1; HRMS (ES+) calculated for C15H11ClF6N5 [M + H]+: 410.0602, found 410.0608.
(S)-5-Chloro-6-(2,6-difluoro-4-methylphenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (18).
Following general procedure C using 5,7-dichloro-6-(2,6-difluoro-4-methylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.050 g, 0.16 mmol) (104) and (S)-1,1,1-trifluoropropan-2-amine (0.038 g, 0.33 mmol). Purification via silica gel column chromatography (0–25% EtOAc in hexanes) provided the title compound as a white powder (0.028 g, 0.071 mmol, 65%). 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 6.91 (d, J = 9.0 Hz, 2H), 6.00 – 5.94 (m, 1H), 4.73 (s, 1H), 2.45 (s, 3H), 1.39 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.75 (dd, J = 251.4, 7.0 Hz), 160.50 (dd, J = 249.9, 7.3 Hz), 158.07, 155.34, 154.11, 145.83, 144.86 (t, J = 10.2 Hz), 124.59 (q, J = 282.1 Hz), 112.93 (ddd, J = 65.8, 21.3, 4.0 Hz), 105.67 (t, J = 20.9 Hz), 92.50, 22.72 – 20.28 (m), 15.19 ppm; IR (film) ν 1618, 1559, 1143, 840 cm−1; HRMS (ES+) calculated for C15H12N5ClF5 [M + H]+: 392.0696, found 392.0693.
(S)-5-Chloro-6-(2,6-difluoro-4-(trifluoromethyl)phenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (19).
Following General Procedure C using 5,7-dichloro-6-(2,6-difluoro-4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.350 g, 1.16 mmol) (105) and (S)-2-amino-1,1,1-trifluoropropane hydrochloride (0.389 g, 2.60 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.240 g, 0.54 mmol, 62%). 1H NMR (500 MHz, CDCl3) δ 8.42 (s, 1H), 7.42 (d, J = 7.3 Hz, 2H), 5.87 (d, J = 10.5 Hz, 1H), 4.74 (s, 1H), 1.44 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 161.32 (dd, J = 255.5, 5.5 Hz), 160.96 (dd, J = 253.8, 5.6 Hz), 157.25, 155.74, 154.36, 145.83, 135.62 (qt, J = 35.0, 9.8 Hz), 124.53 (q, J = 280.6 Hz), 122.33 (qt, J = 273.4, 2.9 Hz), 112.99 (t, J = 20.1 Hz), 110.42 (dp, J = 24.9, 3.7 Hz), 109.93 (dp, J = 25.0, 3.8 Hz), 91.17, 51.21 (q, J = 32.2 Hz), 15.31 (d, J = 1.9 Hz) ppm; IR (film) ν 3208, 2925, 2855, 1620, 1555, 1461, 1436, 1369, 1249, 1179, 1143, 1111, 1090, 1037, 963, 909, 896, 868, 819, 767, 734, 680, 652, 612 cm−1; HRMS (ES+) calculated for C15H9ClF8N5 [M + H]+: 446.0413, found 446.0438.
(S)-4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (20).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.030 g, 0.09 mmol) (106) and (S)-1,1,1-trifluoropropan-2-amine (0.022 g, 0.19 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.020 g, 0.05 mmol, 54%). 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 7.44 (t, J = 7.1 Hz, 2H), 6.00 (d, J = 10.7 Hz, 1H), 4.77 (s, 1H), 1.45 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.28 (dd, J = 256.4, 5.9 Hz), 160.87 (dd, J = 254.7, 6.0 Hz), 156.84, 155.68, 154.32, 124.46 (q, J = 282.1 Hz), 116.58 (t, J = 11.8 Hz), 116.47 (ddd, J = 67.6, 25.4, 4.1 Hz), 115.79 (d, J = 3.5 Hz), 114.93 (t, J = 20.0 Hz), 90.61, 51.27 (q, J = 32.2 Hz), 29.83, 15.14 ppm; 19F NMR (469 MHz) δ −78.31, −102.98, −105.00 ppm; IR (KBr): 2923, 2239, 1617, 1556, 1141 cm−1; HRMS (ES+) calculated for C15H9N6ClF5 [M + H]+: 403.0492, found 403.0486.
(S)-5-Chloro-6-(4-chloro-2,6-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (21).
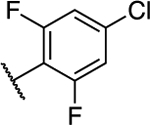
Following general procedure C using 5,7-dichloro-6-(4-chloro-2,6difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.050 g, 0.15 mmol) (108) and (S)-1,1,1-trifluoropropan-2-amine (0.035 g, 0.31 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.035 g, 0.085 mmol, 57%). 1H NMR (600 MHz, CDCl3) δ 8.40 (s, 1H), 7.18 – 7.16 (m, 2H), 5.93 – 5.88 (m, 1H), 4.75 (s, 1H), 1.43 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.84 (ddd, J = 253.4, 48.9, 9.1 Hz), 157.67, 155.57, 154.21, 145.82, 138.62 (t, J = 14.0 Hz), 124.53 (q, J = 280.5 Hz), 114.47 – 113.17 (m), 51.00 (q, J = 32.9 Hz), 15.27 ppm. IR (film) ν 3000, 1571, 1517, 1142 cm−1; HRMS (ES+) calculated for C14H9Cl2F5N5 [M + H]+: 412.0150, found 412.0146.
(S)-5-Chloro-6-(2,6-difluoro-4-nitrophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (22).
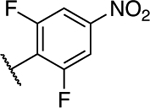
Following general procedure C 5,7-dichloro-6-(2,6-difluoro-4-nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.070 g, 0.21 mmol) (109) and (S)-1,1,1-trifluoropropan-2-amine (0.048 g, 0.42 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.075 g, 0.18 mmol, 88%). 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 8.00 – 7.98 (m, 2H), 5.99 (d, J = 10.6 Hz, 1H), 4.78 (s, 1H), 1.46 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.15 (dd, J = 257.2, 5.8 Hz), 160.73 (dd, J = 255.3, 6.1 Hz), 156.78, 155.75, 154.37, 150.43 (t, J = 10.5 Hz), 145.80, 124.47 (q, J = 282.2 Hz), 116.00 (t, J = 20.5 Hz), 108.53 (ddd, J = 61.2, 27.3, 4.0 Hz), 90.62, 51.38 (q, J = 32.1 Hz), 15.20 (d, J = 1.5 Hz) ppm; IR (film) ν 1618, 1530, 1489, 1143, 1043 cm−1; HRMS (ES+) calculated for C14H9ClF5N6O2 [M + H]+: 423.0390, found 423.0384.
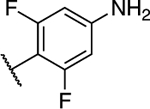
(S)-6-(4-Amino-2,6-difluorophenyl)-5-chloro-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (23).
To a solution of (S)-5-chloro-6-(2,6-difluoro-4-nitrophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.025 g, 0.059 mmol) (22) in 0.100 ml of H2O/MeOH (1:1) was added iron powder (0.017 g, 0.30 mmol) and ammonium chloride (0.013 g, 0.24 mmol). The mixture was stirred at 80 °C for 2 h and then filtered and rinsed with hot methanol. The filtrate was evaporated under reduced pressure, and the resulting mixture was purified by HPLC to afford the product as a white solid (0.016 g, 0.04 mmol, 69%). 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 6.37 (t, J = 9.8 Hz, 2H), 5.93 (s, 1H), 4.86 (s, 1H), 4.28 (s, 2H), 2.17 (s, 1H), 1.41 – 1.39 (m, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.86 (ddd, J = 246.8, 32.1, 8.7 Hz), 158.99, 155.32, 154.09, 151.02 (t, J = 13.9 Hz), 146.15, 130.31, 128.98, 124.72 (q, J = 281.8 Hz), 98.19 (ddd, J = 63.6, 25.1, 3.2 Hz), 97.13 (t, J = 21.1 Hz), 92.99, 50.71 (q, J = 32.4 Hz), 31.08, 15.35 ppm; IR (film) ν 3341, 1651, 1614, 1587, 1346 cm−1; HRMS (ES+) calculated for C14H11ClF5N6 [M + H]+: 393.0648, found 393.0646.
(S)-5-chloro-6-(2,6-difluoro-4-methoxyphenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (24).
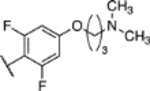
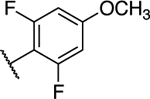
To a solution of (S)-5-chloro-6-(2,4,6-trifluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.020 g, 0.05 mmol) (7) in 0.3 mL of THF, was added 0.04 ml of a 30% sodium methoxide solution in methanol at 0 °C. The reaction mixture was heated for 2 h at 60 °C in a microwave reactor, then cooled to r.t. and diluted with aqueous ammonium chloride solution. The aqueous phase was extracted with EtOAc (×3). The combined organic layers were washed with brine (×2), dried (MgSO4), filtered, and concentrated. Purification by reverse-phase HPLC provided the title compound as a white solid (0.015 g, 0.036 mmol, 71% yield). 1H-NMR (500 MHz; CDCl3): δ 8.45 (s, 1H), 6.67 – 6.65 (m, 2H), 5.94 – 5.93 (m, 1H), 4.77 – 4.74 (m, 1H), 3.90 (s, 3H), 1.41 (d, J = 6.8 Hz, 3H) ppm; 13C-NMR (126 MHz; CDCl3): δ 163.48 (t, J = 13.9 Hz), 161.80 (dd, J = 249.9, 9.0 Hz), 161.54 (dd, J = 248.4, 8.9 Hz), 159.55 (q, J = 40.7 Hz), 158.79, 155.09, 146.15, 124.63 (q, J = 282.0 Hz), 100.62 (t, J = 21.1 Hz), 98.93 (td, J = 26.8, 3.0 Hz), 92.71, 56.30, 50.87 (q, J = 32.1 Hz), 15.32 ppm; IR (film) ν 3340, 2953, 2924, 2848, 1642, 1618, 1579, 1553, 1147 cm−1; HRMS (ES+) calculated for C15H12ClF5N5O [M + H]+: 408.0651, found 408.0652.
(S)-5-Chloro-6-(4-((3-(dimethylamino)propyl)sulfinyl)-2,6-difluorophenyl)-N-((S)-1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (26) and (S)-5-Chloro-6-(4-((3-(dimethylamino)propyl)sulfonyl)-2,6-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (27).
To a solution of (S)-5-chloro-6-(4-((3-(dimethylamino)propyl)thio)-2,6-difluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.050 g, 0.100 mmol) (25) in CH2Cl2 (1.00 ml) was added m-CPBA (70%, 0.025 g, 0.100 mmol). After 2 h, the reaction mixture was concentrated under reduced pressure and purified by reverse-phase HPLC to obtain 26 (0.017 g, 0.03 mmol, 32%) and 27 (0.027 g, 0.05 mmol, 52%). (26): 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.19 (s, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.69 (s, 1H), 5.41 (s, 1H), 3.76 – 3.67 (m, 2H), 3.40 (d, J = 10.2 Hz, 7H), 3.22 – 3.15 (m, 2H), 2.44 – 2.37 (m, 2H), 1.43 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 166.95, 161.56 (dd, J = 253.3, 6.8 Hz), 160.98 (dd, J = 252.6, 6.6 Hz), 157.65, 155.36, 154.85, 146.84, 125.91 (q), 112.00 – 110.04 (m), 107.56 – 105.57 (m), 92.69, 68.14, 57.73 (d, J = 11.8 Hz), 51.16 (q, J = 31.8 Hz), 29.62 (d, J = 2.0 Hz), 22.63 (d, J = 1.7 Hz), 14.82 ppm; IR (film) ν 3053, 1589, 1535, 1178 cm−1; HRMS (ES+) calculated for C19H21ClF5N6OS [M + H]+: 511.1101, found 511.1100. (27): 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.38 (s, 1H), 8.11 (s, 1H), 7.50 (dd, J = 16.6, 7.4 Hz, 1H), 7.17 (d, J = 7.0 Hz, 0.5H), 5.88 (s, 0.5H), 5.59 (s, 0.5H), 3.86 – 3.78 (m, 1H), 3.77 – 3.70 (m, 0.5H), 3.69 – 3.62 (m, 1H), 3.36 (dd, J = 25.9, 3.6 Hz, 6H), 3.40 – 3.27 (m, 0.5H), 2.99 – 2.90 (m, 1H), 2.51 – 2.40 (m, 1H), 2.36 – 2.26 (m, 1H), 1.42 (dd, J = 6.9, 2.9 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 167.52, 163.09 – 161.10 (m), 161.44 (ddd, J = 256.9, 21.4, 4.9 Hz), 157.00, 156.86, 155.45 – 155.33 (m), 155.12, 129.28 – 119.96 (m), 112.17 (td, J = 20.5, 4.6 Hz), 109.28 – 107.40 (m), 92.88, 92.30, 67.84, 67.54, 58.59, 58.51, 57.34, 56.93, 52.19, 51.77, 51.29 (td, J = 31.8, 17.1 Hz), 15.61, 15.10, 14.68, 14.46 (d, J = 1.7 Hz) ppm; IR (film) ν 3285, 1589, 1535, 1142 cm−1; HRMS (ES+) calculated for C19H21ClF5N6O2S [M + H]+: 527.1050, found 527.1045.
(S)-5-Chloro-6-(2,6-difluoro-4-(methylthio)phenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (28).
To a suspension of sodium methanethiolate (0.331 mL, 0.71 mmol, 4 equiv) in DMSO (2 mL) at r.t. was added (S)-5-chloro-6-(2,4,6-trifluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.070 g, 0.18 mmol, 1 equiv) (7) and the mixture was heated for 3 h at 60 °C before it was quenched at r.t. with a saturated NH4Cl solution. The mixture was extracted with EtOAc and the combined organic layers were washed 5 times with H2O, dried and concentrated under reduced pressure to furnish the title compound as a white solid (0.070 g, 0.17 mmol, 93 %). 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 6.94 – 6.92 (m, 2H), 5.92 (bs, 1H), 4.79 (bs, 1H), 2.56 (s, 3H), 1.41 (d, J = 6.0 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.79 (dd, J = 40.77, 6.0 Hz), 160.11 (dd, J = 39.3, 7.6 Hz), 158.15, 155.51, 154.22, 146.81 (t, J = 10.6 Hz), 145.95, 124.63 (q, J = 282.4 Hz), 109.06 (dd, J = 24.2, 3.0 Hz), 108.69 (dd, J = 24.9, 3.0 Hz), 104.03 (t, J = 9.0 Hz), 92.28, 50.81 (q, J = 18.1 Hz), 15.31, 15.10 ppm; HRMS (ES+) calculated for C14H8N4ClF6O [M + H]+: 397.0285, found 397.0279.
5-Chloro-6-(2,6-difluoro-4-(methylsulfinyl)phenyl)-N-((S)-1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (29).
To a solution of (S)-5-chloro-6-(2,6-difluoro-4-(methylthio)phenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.029 g, 0.07 mmol, 1 equiv) (28) in CH2Cl2 (0.7 mL) at r.t. was added m-CPBA (0.012 g, 0.07 mmol, 1 equiv). After 1h the reaction was quenched with H2O and extracted twice with EtOAc. The combined organic fractions were washed with a sat. aq. Na2S2O3 solution, dried and concentrated under vacuum. The crude yellow material was purified by reverse phase HPLC (C18 column, H2O/ACN 90/10 to 10/90, 15 min ramp) and lyophilized to furnish the title compound (0.021 g, 0.05 mmol, 70 %) as a white solid. 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.43 (s, 1H), 8.08 (s, 1H), 7.98 (d, J = 6.0 Hz, 1H), 7.58 (d, J = 6.0 Hz, 1.6H), 7.51 (d, J = 6.0 Hz, 0.4H), 7.42 (d, J = 9.0 Hz, 1H), 7.38 (d, J = 6.0 Hz, 0.4H), 7.33 (d, J = 6.0 Hz, 0.6H), 5.93 (bs, 0.5H), 5.87 (bs, 0.3H), 4.75 (bs, 0.4H), 4.62 (bs, 0.5H), 2.88 (s, 3H), 1.44 – 1.42 (m, 3H) ppm; HRMS (ES+) calculated for C14H8N4ClF6O [M + H]+: 440.0366, found 440.0365.
(S)-5-Chloro-6-(2,6-difluoro-4-(methylsulfonyl)phenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (30).
To a solution of (S)-5-chloro-6-(2,6-difluoro-4-(methylthio)phenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.017 g, 0.04 mmol, 1 equiv) (28) in CH2Cl2 (0.7 mL) at r.t. was added 3-chlorobenzoperoxoic acid (0.021 g, 0.12 mmol, 3 equiv). After 1h the reaction was quenched with H2O and extracted twice with EtOAc. The combined organic fractions were washed with a sat. aq. Na2S2O3 solution, dried and concentrated under vacuum. Purification by reverse-phase HPLC (C18 column, H2O/ACN 90/10 to 10/90, 15 min ramp) provided the title compound (0.013 g, 0.03 mmol, 71 %) as a white solid. 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 7.73 (d, J = 6.0 Hz, 2H), 5.82 (d, J = 6.0 Hz, 1H), 4.78 (bs, 1H), 3.21 (s, 3H), 1.45 (d, J = 6.0Hz, 3H) ppm; HRMS (ES+) calculated for C145H12N45ClF5O2S [M + H]+: 456.0315, found 456.0311.
5-Chloro-N-isopropyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (35).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102) and propan-2-amine (0.036 g, 0.60 mmol), i-Pr2NEt (0.078 g, 0.60 mmol) and, reverse-phase HPLC purification afforded the title compound as a white solid (0.030 g, 0.09 mmol, 44% yield). 1H-NMR (500 MHz; CDCl3): δ 8.46 (s, 1H), 6.90 – 6.85 (m, 2H), 6.32 (d, J = 8.7 Hz, 1H), 3.62 – 3.60 (m, 1H), 1.19 (d, J = 6.4 Hz, 6H) ppm; 13C-NMR (126 MHz; CDCl3): δ 164.27 (dt, J = 254.4, 15.2 Hz), 161.64 (dd, J = 251.0, 8.4 Hz), 161.53 (dd, J = 251.0, 8.4 Hz), 159.04, 153.77, 145.89, 106.92 (td, J = 20.7, 4.9 Hz), 101.46 – 101.02 (m), 90.00, 46.34, 23.82 ppm; IR (film) ν 3344, 2982, 2936, 2873, 1615, 1579, 1264, 1208, 1171, 1124 cm−1; HRMS (ES+) calculated for C14H12ClF3N5 [M + H]+: 342.0733, found 342.0733.
5-Chloro-N-propyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (36).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.030 g, 0.09 mmol) (102) and propan-1-amine (0.012 g, 0.20 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.020 g, 0.06 mmol, 62%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 6.84 – 6.82 (m, 2H), 6.48 (t, J = 5.9 Hz, 1H), 2.98 (q, J = 6.7 Hz, 2H), 1.57 (h, J = 7.3 Hz, 2H), 0.85 (t, J = 7.4 Hz, 4H) ppm; 13C NMR (150 MHz, CDCl3) δ 163.93 (dt, J = 253.7, 15.4 Hz), 161.77 (ddd, J = 250.6, 14.9, 8.5 Hz), 158.16, 155.07, 153.65, 146.36, 107.18 (td, J = 20.8, 4.9 Hz), 101.12 – 100.62 (m), 88.91, 45.31, 29.83, 23.33, 11.04 ppm; IR (film) ν 2923, 1573, 1439, 1122 cm−1; HRMS (ES+) calculated for C14H12N5ClF3 [M + H]+: 342.0728, found 342.0725.
5-Chloro-N-isobutyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (37).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.030 g, 0.09 mmol) (102) and 2-methylpropan-1-amine (0.014 g, 0.20 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.027 g, 0.08 mmol, 81%). 1H NMR (600 MHz, CDCl3) δ 8.33 (s, 1H), 6.88 – 6.81 (m, 2H), 6.56 (t, J = 6.1 Hz, 1H), 2.84 (t, J = 6.5 Hz, 2H), 1.77 – 1.70 (m, 1H), 0.84 (d, J = 6.7 Hz, 8H) ppm; 13C NMR (150 MHz, CDCl3) δ 164.05 (dt, J = 253.8, 15.1 Hz), 161.72 (ddd, J = 250.4, 14.9, 8.4 Hz), 158.13, 155.05, 153.63, 146.42, 107.31 (td, J = 20.8, 4.8 Hz), 102.26 – 98.14 (m), 88.95, 50.83, 29.05, 19.79 ppm; IR (film) ν 2961, 2924, 1637, 1577, 1434 cm−1; HRMS (ES+) calculated for C15H14N5ClF3 [M + H]+: 356.0884, found 356.0887.
5-Chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (38).
This compound has been obtained as a side product form the attempt of synthetizing 5-chloro-N-isobutyl-N-(2,2,2-trifluoroethyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine. Following General Procedure C using5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102), 2-methyl-N-(2,2,2-trifluoroethyl)propan-1-amine hydrochloride (0.115 g, 0.60 mmol) and i-Pr2NEt (0.139 mL, 0.80 mmol). Purification via preparative reverse-phase HPLC provided the title compound (0.023 g, 0.08 mmol, 38%). 1H-NMR (500 MHz; MeOD): δ 8.74 (s, 1H), 6.97 (dd, J = 8.7, 7.8 Hz, 2H) ppm; 13C NMR (126 MHz; MeOD): δ 164.78 (dt, J = 250.7, 16.4 Hz), 162.77 (ddd, J = 248.8, 15.2, 9.5 Hz), 159.40, 156.62, 108.75 (td, J = 21.2, 4.8 Hz), 102.66, 101.52 – 101.06 (m) ppm; IR (film) ν 3446, 2789, 2684, 2549, 1683, 1667, 1655, 1635, 1616, 1600, 1558, 1532 cm−1.
N-(5-Chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)isobutyramide (39).
To a solution of 5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (0.035 g, 0.12 mmol, 1.00 equiv) (102) in anhydrous CH2Cl2 (0.450 mL) was added Et3N (0.022 mL, 0.15 mmol, 1.30 equiv) and DMAP (0.001 g, 0.01 mmol, 0.10 equiv) under N2 and the resulting solution was stirred at 0 °C. Isobutyryl chloride (0.015 mL, 0.14 mmol, 1.20 equiv) was slowly added and the reaction was stirred at 0 °C for 30 min and r.t. for 18 h. Then, Et3N (0.022 mL, 0.15 mmol, 1.30 equiv) and isobutyryl chloride (0.015 mL, 0.14 mmol, 1.20 equiv) were added and the reaction was stirred at r.t. for 2 h. The reaction mixture was then concentrated under reduced pressure. Purification via preparative reverse-phase HPLC provided the title compound (0.023 g, 0.06 mmol, 53%). 1H NMR (500 MHz, CDCl3) δ 8.72 (s, 1H), 8.47 (s, 1H), 6.81 (t, J = 8.1 Hz, 2H), 2.66 (hept, J = 6.9 Hz, 1H), 1.12 (d, J = 6.9 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 173.47, 163.85 (dt, J = 252.8, 15.4 Hz), 160.51 (dd, J = 251.6, 9.0 Hz), 160.39 (dd, J = 251.7, 8.9 Hz), 158.48, 156.09, 153.73, 141.44, 107.40 (td, J = 19.8, 4.9 Hz), 104.48, 100.85 (td, J = 26.1, 3.4 Hz), 36.73, 19.10 ppm; IR (film) ν 3415, 2974, 1732, 1618, 1515, 1487, 1440, 1384, 1263, 1230, 1153, 1126, 1040, 997, 842, 738, 669 cm−1; HRMS (ES+) calculated for C15H11ClF3N5NaO [M + Na]+: 392.0502, found 392.0488.
5-Chloro-N-phenyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (40).
To a solution of 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.053 g, 0.17 mmol) (102) and aniline (0.015 mL, 0.17 mmol) in DMF (1.67 mL) was added K2CO3 (0.046 g, 0.33 mmol) and the resulting mixture was stirred for 24 h at 30 °C. The reaction mixture was then diluted with H2O. The organic layer was washed with a 1N HCl (×2), the aqueous phase was extracted with EtOAc (×3) and the combined organic layers were washed with brine (×2), dried over MgSO4, filtered, and concentrated. Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.049 g, 0.13 mmol, 78%). 1H NMR (500 MHz, CDCl3) δ 8.43 (s, 1H), 8.24 (s, 1H), 7.16 – 7.11 (m, 3H), 7.10 – 7.00 (m, 2H), 6.36 (t, J = 8.2 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.69 (dt, J = 255.2, 15.1 Hz), 161.57 (ddd, J = 253.8, 15.0, 8.2 Hz), 161.32 (ddd, J = 251.3, 14.9, 8.2 Hz), 158.29, 155.56, 153.98, 146.56, 124.53 (q, J = 283.5 Hz), 101.86 (td, J = 25.8, 4.1 Hz), 101.36 (td, J = 25.2, 2.4 Hz), 90.99, 58.99 (q, J = 29.4 Hz), 28.34, 19.81, 16.74 ppm; IR (film) ν 3423, 1638, 1613, 1563, 1491, 1440, 1335, 1261, 1197, 1122, 1036, 998, 940, 840, 766, 694, 668 cm−1; HRMS (ES+) calculated for C17H10ClF3N5 [M + H]+: 376.0571, found 376.0571.
N-Benzyl-5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (41).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102) and benzylamine (0.064 g, 0.60 mmol), reverse-phase HPLC purification afforded the title compound as a yellow solid (0.024 g, 0.06 mmol, 31% yield). 1H-NMR (500 MHz; CDCl3): δ 8.45 (s, 1H), 7.33 – 7.31 (m, 3H), 7.08 – 7.06 (m, J = 6.5, 2.7 Hz, 2H), 6.89 – 6.88 (m, 1H), 6.89 – 6.86 (m, 1H), 6.72 (dd, J = 8.5, 6.8 Hz, 2H), 4.33 (d, J = 5.9 Hz, 2H) ppm; 13C-NMR (126 MHz; CDCl3): δ 164.32 (dt, J = 254.1, 15.1 Hz), 164.01, 161.77 (ddd, J = 250.8, 14.9, 8.2 Hz), 159.63 (q, J = 40.8 Hz), 159.26, 154.03, 146.40, 135.30, 129.28, 128.75, 126.77, 106.56 (td, J = 20.7, 4.8 Hz), 101.15 – 100.70 (m), 90.43, 47.76 ppm; IR (film) ν 3352, 3268, 3113, 3067, 3033, 2928, 1642, 1617, 1595, 1575, 1495, 1441, 1124, 1036 cm−1; HRMS (ES+) calculated for C18H12ClF3N5 [M + H]+: 390.0733, found 390.0730.
5-Chloro-6-(2,4,6-trifluorophenyl)-7-((1,1,1-trifluoropropan-2-yl)oxy)-[1,2,4]triazolo[1,5-a]pyrimidine (42).
To sodium hydride (60%, 0.015 g, 0.38 mmol) suspended in THF (3 ml) was added 1,1,1-trifluoropropan-2-ol (0.034 mL, 0.38 mmol) and the resulting mixture was stirred for 2 h while heated to reflux. 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.100 g, 0.31 mmol) (102) was then added at r.t.. The reaction mixture was stirred for 20 min before the reaction was quenched with H2O (5 ml) and extracted with EtOAc (×3). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound (0.046 g, 0.11 mmol, 37%). 1H NMR (600 MHz, CDCl3) δ 8.51 (s, 1H), 6.89 – 6.77 (m, 2H), 6.21 – 6.12 (m, 1H), 1.63 (d, J = 6.6 Hz, 4H) ppm; 13C NMR (150 MHz, CDCl3) δ 164.14 (dt, J = 253.1, 15.2 Hz), 161.12 (ddd, J = 252.7, 15.1, 8.7 Hz), 160.86 (ddd, J = 251.3, 15.0, 8.6 Hz), 158.43, 156.71, 155.59 (d, J = 43.8 Hz), 151.92, 123.23 (q, J = 281.1 Hz), 104.10 (td, J = 20.5, 4.9 Hz), 102.40 – 98.42 (m), 76.02 (q, J = 32.7, 31.8 Hz), 14.07, 12.80 ppm; IR (film) ν 2925, 2854, 1606, 1517, 1493 cm−1; HRMS (ES+) calculated for C14H8N4ClF6O [M + H]+: 397.0285, found 397.0279.
5-Chloro-N-(1,1-difluoropropan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (43).
Following General Procedure C using 5,7-dichloro-6-(2,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.053 g, 0.17 mmol) (102), 1,1-difluoropropan-2-amine hydrochloride (0.045 g, 0.34 mmol) and Et3N (0.093 mL, 0.67 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.035 g, 0.09 mmol, 56%). 1H NMR (500 MHz, CDCl3) δ 8.37 (s, 1H), 6.89 (t, J = 7.9 Hz, 2H), 6.00 (d, J = 8.2 Hz, 1H), 5.75 (td, J = 55.4, 1.6 Hz, 1H), 4.32 (bs, 1H), 1.31 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (151 MHz, CDCl3) δ 164.46 (dt, J = 254.9, 15.1 Hz), 161.5 (ddd, J = 250.66, 8.4, 5.4 Hz), 161.41 (ddd, J = 252.17, 8.3, 5.3 Hz), 157.94, 155.46, 154.22, 114.59 (t, J = 246.2 Hz), 105.90 (td, J = 20.5, 4.8 Hz), 101.54 (dtd, J = 29.9, 25.9, 4.1 Hz), 90.88, 51.09 (t, J = 24.16 Hz), 42.38 (t, J = 552.1 Hz), 29.82, 14.61 (t, J = 3.8 Hz) ppm; IR (film) ν 1613, 1578, 1552, 1527, 1492, 1461, 1440, 1360, 1263, 1124, 1208, 1171, 1134, 1122, 1100, 1083 cm−1; HRMS (ES+) calculated for C15H10ClF5N4 [M + H]+: 378.0539, found 378.0537.
5-Chloro-N-(1-fluoropropan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (44).
Following General Procedure C using 5,7-dichloro-6-(2,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.073 g, 0.23 mmol) (102), 1-fluoropropan-2-amine hydrochloride (0.053 g, 0.47 mmol) and Et3N (0.130 mL, 0.91 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.059 g, 0.16 mmol, 72%). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 6.93 – 6.83 (m, 2H), 6.26 (d, J = 8.1 Hz, 1H), 4.37 (ddd, J = 66, 9.6, 3.5 Hz, 1H), 4.29 (ddd, J = 66, 9.6, 4.3 Hz, 1H), 4.01 (bs, 1H), 1.26 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (151 MHz, CDCl3) δ 164.29 (dt, J = 254.6, 15.2 Hz), 161.53 (ddd, J = 250.7, 14.9, 8.3, Hz), 161.52 (ddd, J = 249.9, 14.9, 8.3, Hz), 157.98, 155.28, 154.08, 146.19, 106.53 (td, J = 20.7, 4.7 Hz), 101.39 (tdd, J = 25.9, 9.1, 4.1 Hz), 90.03, 85.42 (d, J = 175.2 Hz), 49.79 (d, J = 19.6 Hz), 17.59 (d, J = 5.3 Hz), 14.32 ppm; IR (film) ν 1639, 1611, 1594, 1580, 1492, 1468, 1440, 1358, 1239, 1207, 1171 cm−1; HRMS (ES+) calculated for C15H11ClF4N4 [M + H]+: 360.0634, found 360.0636.
(S)-5-chloro-N-(3-methylbutan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (45).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.119 g, 0.37 mmol) (102) and (S)-3-methylbutan-2-amine (0.090 mL, 0.78 mmol). Purification via silica gel column chromatography (0–15% EtOAc in hexanes) provided the title compound as a white powder (0.099 g, 0.27 mmol, 72%). The spectroscopic properties of the compound were identical to those reported for its enantiomer, 3.11
(R)-5-Chloro-N-(1,1,1-trifluoro-3-methylbutan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (46).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.123 g, 0.38 mmol) (102) and (R)-1,1,1-trifluoro-3-methyl-2-butylamine (0.152 mL, 1.15 mmol). Purification via silica gel column chromatography (0–15% EtOAc in hexanes) provided the title compound as a white powder (0.056 g, 0.13 mmol, 35%). 1H NMR (500 MHz, CDCl3) δ 8.41 (s, 1H), 7.01 – 6.77 (m, 2H), 6.28 (s, 1H), 3.96 (s, 1H), 2.26 – 2.09 (m, 1H), 1.03 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.69 (dt, J = 255.2, 15.1 Hz), 161.57 (ddd, J = 253.8, 15.0, 8.2 Hz), 161.32 (ddd, J = 251.3, 14.9, 8.2 Hz), 158.29, 155.56, 153.98, 146.56, 124.53 (q, J = 283.5 Hz), 101.86 (td, J = 25.8, 4.1 Hz), 101.36 (td, J = 25.2, 2.4 Hz), 90.99, 58.99 (q, J = 29.4 Hz), 28.34, 19.81, 16.74 ppm; IR (film) ν 3339, 2972, 2928, 1618, 1562, 1493, 1442, 1364, 1264, 1170, 1126, 1090, 1039, 999, 937, 911, 846, 768, 734, 705, 653 cm−1; HRMS (ES+) calculated for C16H12ClF6N5Na [M + Na]+: 446.0583, found 446.0598.
(S)-5-Chloro-N-(1,1,1-trifluoro-3-methylbutan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (47).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.123 g, 0.38 mmol) (102) and (S)-1,1,1-trifluoro-3-methyl-2-butylamine (0.152 mL, 1.15 mmol). Purification via silica gel column chromatography (0–15% EtOAc in hexanes) provided the title compound as a white powder (0.039 g, 0.09 mmol, 24%). 1H NMR (500 MHz, CDCl3) δ 8.41 (s, 1H), 6.99 – 6.84 (m, 2H), 6.29 (s, 1H), 3.98 (s, 1H), 2.24 – 2.10 (m, J = 6.7 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.63 (dt, J = 255.2, 15.0 Hz), 161.50 (ddd, J = 253.8, 14.9, 8.2 Hz), 161.25 (ddd, J = 251.3, 14.9, 8.2 Hz), 158.23, 155.49, 153.90, 146.50, 124.47 (q, J = 283.5 Hz), 101.80 (td, J = 25.8, 4.2 Hz), 101.30 (td, J = 27.1, 26.1, 3.7 Hz), 90.92, 58.93 (q, J = 29.4 Hz), 28.28, 19.75, 16.68 ppm; IR (film) ν 3339, 2972, 2930, 1618, 1562, 1493, 1459, 1442, 1364, 1264, 1170, 1126, 1090, 1066, 1038, 999, 938, 846, 768, 738, 704, 628 cm−1; HRMS (ES+) calculated for C16H13ClF6N5 [M + H]+: 424.0758, found 424.0762.
(R)-5-Chloro-6-(2,4,6-trifluorophenyl)-N-(1,1,1-trifluoropropan-2-yl)-[1,2,4]triazolo[1,5- a]pyrimidin-7-amine (48).
Following General Procedure A using (R)-1,1,1-trifluoropropan-2-amine hydrochloride (0.090 g, 0.60 mmol), i-Pr2NEt (0.078 g, 0.60 mmol) and 5,7-dichloro-6-(2,4,6trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102). Purification via reverse-phase HPLC afforded the title compound as a white solid (0.017 g, 0.04 mmol, 21% yield). 1H-NMR (500 MHz; CDCl3): δ 8.39 (s, 1H), 6.91 (dd, J = 8.4, 7.1 Hz, 2H), 5.92 (d, J = 10.3 Hz, 1H), 4.73 (s, 1H), 1.43 (d, J = 6.9 Hz, 3H) ppm; 13C-NMR (126 MHz; CDCl3): δ 164.64 (dt, J = 255.3, 15.1 Hz), 161.65 (ddd, J = 253.4, 15.1, 8.2 Hz), 161.32 (ddd, J = 251.7, 14.9, 8.2 Hz), 157.97, 155.60, 154.26, 145.97, 124.57 (q, J = 282.0 Hz), 105.51 (td, J = 20.7, 4.8 Hz), 101.61 (dtd, J = 52.6, 26.1, 4.0 Hz), 91.50, 51.02 (q, J = 32.2 Hz), 15.30 ppm; IR (film) ν 3335, 3205, 2924, 2852, 1621, 1558, 1272, 1251, 1175, 1145, 1120 cm−1; HRMS (ES+) calculated for C14H9ClF6N5 [M + H]+: 396.0451, found 396.0451.
7-(Azetidin-1-yl)-5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (49).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102) and azetidine (0.034 g, 0.60 mmol). Purification via reverse-phase HPLC afforded the title compound as a white solid (0.005 g, 0.02 mmol, 8% yield). 1H-NMR (500 MHz; CDCl3): δ 8.34 (s, 1H), 6.84 – 6.79 (m, 2H), 4.49 – 4.45 (m, 4H), 2.37 (quintet, J = 7.9 Hz, 2H) ppm; 13C-NMR (126 MHz; CDCl3): δ 164.10 (dt, J = 253.9, 15.2 Hz), 162.35 (dd, J = 249.8, 8.2 Hz), 162.23 (dd, J = 249.7, 8.4 Hz), 158.12, 155.06, 155.04, 154.45, 146.35, 106.96 (td, J = 21.0, 4.6 Hz), 100.80 – 100.36 (m), 89.00, 56.73, 29.86, 17.36 ppm; IR (film) ν 2957, 2924, 2852, 1594, 1558, 1455, 1255, 1167, 1120, 1041 cm−1; HRMS (ES+) calculated for C14H9ClF3N5 [M + H]+: 340.0577, found 340.0573.
5-Chloro-7-(pyrrolidin-1-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (50).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102) and pyrrolidine (0.043 g, 0.60 mmol). Purification by reverse-phase HPLC afforded the title compound as a yellow solid (0.009 g, 0.03 mmol, 14% yield). 1H-NMR (500 MHz; CDCl3): δ 8.37 (s, 1H), 6.81 (dd, J = 8.4, 6.9 Hz, 2H), 3.71 (t, J = 6.5 Hz, 4H), 1.91 (dt, J = 6.2, 3.3 Hz, 4H) ppm; 13C-NMR (126 MHz; CDCl3): δ 163.80 (dt, J = 252.3, 14.5 Hz), 161.66 (dd, J = 248.1, 7.1 Hz), 161.55 (dd, J = 249.6, 8.3 Hz), 158.54, 155.74, 154.26, 148.57, 109.66 (td, J = 20.7, 4.9 Hz), 100.94 – 100.50 (m), 92.85, 53.27, 25.94 ppm; IR (film) ν 3444, 1636, 1594, 1537, 1455, 1333, 1253, 1123 cm−1. HRMS (ES+) calculated for C15H12ClF3N5 [M + H] +: 354.0733, found 354.0723.
5-Chloro-7-(piperidin-1-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (51).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102) and piperidine (0.051 g, 0.60 mmol). Purification via reverse-phase HPLC afforded the title compound as a yellow solid (0.012 g, 0.03 mmol, 17% yield). 1H-NMR (500 MHz; CDCl3): δ 8.40 (s, 1H), 6.86 (dd, J = 8.4, 7.3 Hz, 2H), 3.27 (bs, 4H), 1.64 (bs, 6H) ppm; 13C-NMR (126 MHz; CDCl3): δ 163.71 (dt, J = 252.8, 15.4 Hz), 161.19 (dd, J = 250.3, 8.7 Hz), 161.08 (dd, J = 250.1, 8.8 Hz), 158.24, 155.29, 151.15, 108.64 (td, J = 20.2, 4.8 Hz), 101.30 – 100.86 (m), 98.10, 51.13, 25.98, 23.71 ppm; IR (film) ν 2945, 2929, 2857, 1595, 1558, 1538, 1506, 1446, 1361, 1121, 1035 cm−1; HRMS (ES+) calculated for C16H14ClF3N5 [M + H]+: 368.0890, found 368.0877.
7-(Azepan-1-yl)-5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (52).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.197 g, 0.62 mmol) (102) and azepane (0.184 g, 1.85 mmol). Purification via silica gel column chromatography (0–15% EtOAc in hexanes) provided the title compound as a white powder (0.057 g, 0.15 mmol, 24%). 1H NMR (500 MHz, CDCl3) δ 8.36 (s, 1H), 6.85 (t, J = 8.1 Hz, 2H), 3.45 – 3.39 (m, 4H), 1.76 – 1.67 (m, 4H), 1.65 – 1.58 (m, 4H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.64 (dt, J = 253.0, 15.4 Hz), 161.10 (dd, J = 250.2, 8.8 Hz), 160.98 (dd, J = 250.3, 8.8 Hz), 158.21, 155.45, 155.08, 152.31, 109.08 (td, J = 20.4, 5.0 Hz), 102.17 – 100.48 (m), 98.50, 53.70, 28.28, 28.23 ppm; IR (film) ν 3436, 2932, 2858, 1637, 1593, 1525, 1492, 1443, 1357, 1284, 1259, 1231, 1200, 1171, 1152, 1125, 1100, 1035, 998, 968, 931, 842, 776, 760, 732, 656, 636, 614 cm−1; HRMS (ES+) calculated for C17H16ClF3N5 [M + H]+: 382.1041, found 382.1031.
7-(Azocan-1-yl)-5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (53).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.191 g, 0.60 mmol) (102) and azocane (0.142 g, 1.26 mmol). Purification via silica gel column chromatography (0–20% EtOAc in hexanes) provided the title compound as a white powder (0.177 g, 0.45 mmol, 75%). 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 6.81 (t, J = 7.9 Hz, 2H), 3.49 – 3.38 (m, 4H), 1.78 – 1.15 (m, 10H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.63 (dt, J = 253.5, 15.2 Hz), 160.93 (dd, J = 250.6, 8.9 Hz), 160.81 (dd, J = 250.5, 8.7 Hz), 158.19, 155.50, 154.73, 151.18, 108.91 (td, J = 20.2, 4.8 Hz), 102.27 – 100.10 (m), 98.25, 51.91, 27.67, 26.77, 24.36 ppm; IR (film) ν 3450, 2927, 2860, 1637, 1593, 1529, 1494, 1440, 1375, 1356, 1266, 1230, 1212, 1171, 1151, 1124, 1091, 1035, 998, 967, 914, 846, 761, 735, 656, 636, 612 cm−1; HRMS (ES+) calculated for C18H18ClF3N5 [M + H]+: 396.1197, found 396.1201.
5-Chloro-N-cyclopropyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (55).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.064 g, 0.20 mmol) (102), cyclopropanamine (0.057 g, 0.60 mmol) and i-Pr2NEt (0.078 g, 0.60 mmol). Purification via reverse-phase HPLC afforded the title compound as a beige solid (0.021 g, 0.06 mmol, 30% yield). 1H-NMR (500 MHz; CDCl3): δ 8.34 (s, 1H), 6.84 – 6.80 (m, 3H), 2.36 – 2.33 (m, 1H), 0.68 – 0.65 (m, 2H), 0.48 – 0.44 (m, 2H) ppm; 13C-NMR (126 MHz; CDCl3): δ 163.93 (dt, J = 252.6, 13.5 Hz), 162.00 (dd, J = 249.8, 8.5 Hz), 161.88 (dd, J = 250.1, 9.0 Hz), 158.41, 155.18, 153.53, 147.23, 147.17, 108.06 (td, J = 20.8, 5.0 Hz), 100.71 – 100.27 (m), 89.74, 89.70, 25.70, 25.56, 9.00, 8.96 ppm; IR (film) ν 3361, 3260, 3104, 2919, 1597, 1576, 1494, 1441, 1264, 1123, 1032 cm−1; HRMS (ES+) calculated for C14H19ClF3N5 [M + H]+: 340.0577, found 340.0574.
5-Chloro-N-cyclobutyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (56).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.030 g, 0.09 mmol) (102) and cyclobutanamine (0.014 g, 0.20 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.025 g, 0.07 mmol, 75%). 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 6.85 – 6.81 (m, 2H), 6.61 (d, J = 7.5 Hz, 1H), 3.71 (q, J = 7.7 Hz, 1H), 2.08 – 1.94 (m, 4H), 1.80 – 1.73 (m, 1H), 1.60 – 1.51 (m, 1H), 1.24 (s, 1H) ppm; 13C NMR (150 MHz, CDCl3) δ 164.03 (dt, J = 253.8, 15.4 Hz), 161.71 (ddd, J = 250.5, 15.1, 8.4 Hz), 158.22, 155.04, 145.33, 120.10, 102.32 – 99.71 (m), 89.07, 48.80, 31.96, 14.62 ppm; IR (film) 3285, 2838, 2178, 1571 cm−1; HRMS (ES+) calculated for C15H12ClF3N5 [M + H]+: 354.0728, found 354.0726.
5-Chloro-N-cyclopentyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (57).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.030 g, 0.09 mmol) (102) and cyclopentanamine (0.017 g, 0.20 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.027 g, 0.07 mmol, 78%). 1H NMR (600 MHz, CDCl3) δ 8.31 (s, 1H), 6.84 – 6.81 (m, 2H), 6.42 (d, J = 8.3 Hz, 1H), 3.63 (s, 1H), 1.82 – 1.59 (m, 4H), 1.62 – 1.38 (m, 4H) ppm. 13C NMR (150 MHz, CDCl3) δ 163.99 (dt, J = 253.7, 15.2 Hz), 161.66 (ddd, J = 250.5, 15.0, 8.4 Hz), 158.19, 154.93, 145.81, 101.30 – 100.53 (m), 88.98, 55.11, 34.64, 23.92 ppm; IR (film) ν 3195, 2838, 1678, 1553 cm−1; HRMS (ES+) calculated for C16H14N5ClF3 [M + H]+: 368.0884, found 368.0881.
5-Chloro-N-cyclohexyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (58).
Following general procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.100 g, 0.31 mmol) (102) and cyclohexanamine (0.094 g, 0.94 mmol). Purification via silica gel column chromatography (0–40% EtOAc in hexanes) provided the title compound as a white powder (0.119 g, 0.31 mmol, 99%). 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 1H), 6.92 – 6.81 (m, 2H), 6.37 (d, J = 8.7 Hz, 1H), 3.14 – 2.98 (m, 1H), 1.86 – 1.77 (m, 2H), 1.74 – 1.64 (m, 2H), 1.53 (dt, J = 13.2, 3.8 Hz, 1H), 1.30 – 1.18 (m, 2H), 1.12 (tdd, J = 12.9, 9.8, 6.1 Hz, 1H), 1.01 – 0.85 (m, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.08 (dt, J = 253.8, 15.1 Hz), 161.76 (dd, J = 250.7, 8.5 Hz), 161.65 (dd, J = 250.7, 8.4 Hz), 158.12, 155.00, 153.74, 145.59, 107.50 (td, J = 20.7, 4.8 Hz), 102.70 – 97.55 (m), 88.82, 52.94, 34.06, 24.92, 24.75 ppm; IR (film) ν 3341, 3103, 2934, 2856, 2239, 1636, 1610, 1578, 1492, 1439, 1367, 1341, 1313, 1263, 1207, 1159, 1125, 1036, 998, 976, 957, 930, 844, 766, 733, 655, 627 cm−1; HRMS (ES+) calculated for C17H16ClF3N5 [M + H]+: 382.1041, found 382.1054.
5-Chloro-N-neopentyl-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (59).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.050 g, 0.16 mmol) (102) and 2,2-dimethylpropan-1-amine (0.039 mL, 0.33 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.033 g, 0.09 mmol, 57%). 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 1H), 6.85 (dd, J = 8.0, 7.2 Hz, 2H), 6.45 (s, 1H), 2.80 (d, J = 5.7 Hz, 2H), 0.88 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.63 (dt, J = 255.2, 15.0 Hz), 161.50 (ddd, J = 253.8, 14.9, 8.2 Hz), 161.25 (ddd, J = 251.3, 14.9, 8.2 Hz), 158.23, 155.49, 153.90, 146.50, 124.47 (q, J = 283.5 Hz), 101.80 (td, J = 25.8, 4.2 Hz), 101.30 (td, J = 27.1, 26.1, 3.7 Hz), 90.92, 58.93 (q, J = 29.4 Hz), 28.28, 19.75, 16.68 ppm; IR (film) ν 2959, 1636, 1611, 1573, 1495, 1438, 1358, 1263, 1250, 1124, 1036, 998, 843 766, 533 cm−1; HRMS (ES+) calculated for C16H15ClF3N5 [M + H]+: 370.1041, found 370.1039.
(S)-5-Chloro-N-(3,3-dimethylbutan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]-pyrimidin7-amine (60).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.100 g, 0.31 mmol) (102) and (S)-3,3-dimethylbutan-2-amine (0.088 mL, 0.66 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.094 g, 0.24 mmol, 78%). 1H NMR (500 MHz, CDCl3) δ 8.31 (s, 1H), 6.92 – 6.74 (m, 2H), 6.44 (s, 1H), 3.08 (s, 1H), 1.00 (d, J = 6.7 Hz, 4H), 0.82 (s, 11H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.07 (dt, J = 254.0, 15.2 Hz), 161.52 (ddd, J = 250.8, 14.8, 8.4 Hz), 161.27 (ddd, J = 250.6, 14.9, 8.4 Hz), 158.13, 154.94, 153.61, 146.25, 107.54 (td, J = 20.6, 5.1 Hz), 101.51 – 100.58 (m), 88.60, 58.07, 34.72, 25.78, 16.58 ppm; IR (film) ν 1608, 1578, 1490, 1438, 1354, 1264, 1252, 1124, 999, 767, 623, 540 cm−1; HRMS (ES+) calculated for C17H17ClF3N5 [M + H]+: 384.1197, found 384.1198.
(R)-5-Chloro-N-(3,3-dimethylbutan-2-yl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin7-amine (61).
Following General Procedure C using 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.150 g, 0.47 mmol) (102) and (R)-3,3-dimethylbutan-2-amine (0.132 mL, 0.99 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.157 g, 0.86 mmol, 87%). The spectroscopic properties of the compound were identical to those reported for 60.
5-Chloro-7-(3,3,3-trifluoro-2-methylpropyl)-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (62).
To a solution of tert-butyl 2-(5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-4,4,4-trifluoro-3-methylbutanoate (0.026 g, 0.05 mmol, 1 equiv) (112) in CH2Cl2 (2 mL) at r.t. was added 2,2,2-trifluoroacetic acid (0.125 mL, 1.60 mmol, 30 equiv). The mixture was stirred 16 h at r.t. and then diluted with CH2Cl2 and quenched with a saturated NaHCO3 solution. The aqueous layer was then extracted with CH2Cl2 and the combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification via preparative reverse-phase HPLC (C18 column, 20 ml/min, 10 min ramp, 10 to 90% ACN/H2O + 0.1% formic acid, tr = 8.5 min) provided the title compound (0.014 g, 0.04 mmol, 68 %) as a tan solid. 1H NMR (600 MHz, CDCl3) δ 8.56 (s, 1H), 6.92 – 6.88 (m, 2H), 3.34 (dd, J = 13.9, 6.2 Hz, 1H), 3.22 (m, 1H), 3.05 (dd, J = 13.9, 8.4 Hz, 1H), 1.03 (d, J = 7.0 Hz, 4H) ppm; 13C NMR (151 MHz, CDCl3) δ 164.45 (dt, 256.7, 18.1 Hz), 160.86 (ddd, 252.2, 15.1, 9.1 Hz), 160.61 (ddd, 250.7, 15.1, 9.06 Hz), 157.19, 157.02, 154.03, 148.77, 127.25 (q, 280.11 Hz) 112.29, 106.39 (td, 21.14, 4.9 Hz), 101.48 (dtd, J = 30.1, 25.9, 4.1 Hz), 35.37 (q, J = 27.6 Hz), 30.65, 12.88 ppm; IR (film) ν 1639, 1609, 1599, 1516, 1495, 1442, 1278, 1265, 1207, 1186, 1172, 1146, 1122, 1039, 999 cm−1; HRMS (ES+) calculated for C15H9ClF6N4 [M + H]+: 395.0493, found 395.0486.
(S)-4-(5-Chloro-7-((1,1,1-trifluoropropan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (63).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (0.040 g, 0.13 mmol) (107) and (S)-1,1,1-trifluoropropan-2-amine (0.031 g, 0.27 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.031 g, 0.07 mmol, 54%). 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.41 (s, 1H), 7.68 (ddd, J = 7.8, 3.8, 1.5 Hz, 1H), 7.61 (ddd, J = 8.4, 3.8, 1.5 Hz, 1H), 7.57 (t, J = 7.4 Hz, 1H), 7.50 (t, J = 7.3 Hz, 1H), 5.94 (d, J = 11.0 Hz, 1H), 5.57 (d, J = 10.8 Hz, 1H), 4.97 (s, 1H), 4.50 (s, 1H), 1.40 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.29 (d, J = 52.0 Hz), 159.61 (d, J = 50.4 Hz), 156.65 (d, J = 24.9 Hz), 155.72 (d, J = 11.0 Hz), 154.20 (d, J = 57.5 Hz), 145.65 (d, J = 62.8 Hz), 135.00 (d, J = 2.0 Hz), 133.84 (d, J = 2.2 Hz), 129.21 (d, J = 4.2 Hz), 124.97 (dd, J = 16.2, 13.1 Hz), 124.67 (qd, J = 282.3, 34.7 Hz), 120.62 (dd, J = 74.3, 25.2 Hz), 116.67 (d, J = 2.8 Hz), 116.33 (dd, J = 9.3, 5.2 Hz), 97.66, 96.61, 51.26 (dq, J = 37.9, 31.8 Hz), 15.13, 14.99 ppm; HRMS (ES+) calculated for C15H10ClF4N6 [M + H]+: 385.0586, found 385.0589.
(R)-5-Chloro-6-(2,6-difluorophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (64).
Following General Procedure C using 5,7-dichloro-6-(3,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.048 g, 0.16 mmol) (97) and (R)-3-methylbutan-2-amine (0.028 g, 0.32 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.049 g, 0.14 mmol, 87%). 1H NMR (600 MHz, CDCl3) δ 8.32 (s, 1H), 7.54 – 7.49 (m, 1H), 7.09 – 7.06 (m, 2H), 6.34 (bs, 1H), 3.12 (bs, 1H), 1.61 (oct, J = 6 Hz, 1H), 1.03 (d, J = 6 Hz, 3H), 0.77 (d, J = 6 Hz, 3H), 0.74 (d, J = 6 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.77 (dt, J = 250.66, 5.3 Hz), 158.04, 154.89 (d, J = 4.5 Hz), 153.65, 145.94, 132.38 – 132.22 (m), 110.93 (t, J = 21.1 Hz), 112.08 – 111.81 (m), 89.78, 54.76, 33.65, 33.63, 18.21 (q, 3Hz), 18.00 (q, J = 7.6 Hz), 17.80 (q, J = 3 Hz) ppm; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 352.1135, found 352.1135.
(R)-5-chloro-6-(2-fluorophenyl)-N-(3-methylbutan-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (65).
Following General Procedure C using 5,7-dichloro-6-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.046 g, 0.16 mmol) (95) and (R)-3-methylbutan-2-amine (0.028 g, 0.32 mmol). Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white powder (0.017 g, 0.051 mmol, 31%). 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.33 (s, 1H), 7.52 (q, J = 6 Hz, 1H), 7.36 – 7.29 (m, 2H), 7.24 (t, J = 6 Hz, 1H), 6.19 (bs, 1H), 3.15 (bs, 1H), 1.02 (d, J = 6 Hz, 1.5H), 0.96 (d, J = 6 Hz, 1.5H), 0.77 (t, J = 6 Hz, 3H), 0.74 (d, J = 6 Hz, 1.5H), 0.72 (d, J = 6 Hz, 1.5H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.83 (dd, J = 250.66, 9 Hz), 158.26 (d, J = 12.1 Hz), 153.91, 152.70, 146.02, 133.61, 133.61, 132.98, 132.06 (t, J = 8.3 Hz), 124.94 (d, J = 4.5 Hz), 124.89 (d, J = 3.0 Hz), 120.96 (d, J = 25.7 Hz), 120.85 (dd, J = 21.9, 1.6 Hz), 97.14, 96.99, 54.75, 54.59, 33.77, 33.47, 18.28, 17.98, 17.81, 17.73, 17.62 ppm; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 334.1229, found 334.1227.
(R)-4-(5-Chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (66).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.030 g, 0.09 mmol) (106) and (R)-3-methylbutan-2-amine (0.017 g, 0.19 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.028 g, 0.07 mmol, 81%). 1H NMR (600 MHz, DMSO-d6) δ 8.62 (s, 1H), 8.11 (t, J = 7.8 Hz, 2H), 7.88 (s, 1H), 1.78 (dq, J = 13.9, 6.8 Hz, 1H), 1.10 (d, J = 6.6 Hz, 3H), 0.74 (dd, J = 6.8, 2.9 Hz, 6H) ppm; 13C NMR (151 MHz, DMSO-d6) δ 160.71 (dd, J = 249.2, 6.8 Hz), 160.38 (dd, J = 249.5, 6.3 Hz), 155.02, 146.81, 117.00 (ddd, J = 26.3, 12.6, 3.9 Hz), 116.48 (d, J = 3.5 Hz), 114.83 (t, J = 12.8 Hz) ppm; 19F NMR (469 MHz) δ −103.93 (d, J = 9.9 Hz), −104.42 (d, J = 9.9 Hz) ppm; IR (film) ν 2966, 2220, 1690, 1562, 1422, 1204 cm−1; HRMS (ES+) calculated for C17H16ClF2N6 [M + H]+: 377.1088, found 377.1089.
(R)-4-(5-chloro-7-((3-methylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3fluorobenzonitrile (67) and (68).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (0.048 g, 0.15 mmol) (107) and (R)-3-methylbutan-2-amine (0.037 g, 0.42 mmol). Purification via silica gel column chromatography provided atropoisomer 67 as a white solid (0.029 g, 0.08 mmol, 39%) and atroposiomer 68 (0.023 g, 0.06 mmol, 30%). Recrystallization of 68 from CH2Cl2, followed by single crystal diffraction (m.p. 143.7 – 145.2) permitted the structural assignment of the two atropoisomers (see Supporting Material). 68 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.63 (dd, J = 12.0, 3.0 Hz, 1H), 7.56 (dd, J = 12.0, 3.0 Hz, 1H), 7.53 (t, J = 9.0 Hz, 1H), 6.29 (d, J = 12.0 Hz, 1H), 3.00 (bs, 1H), 1.64 (oct, J = 6.0 Hz, 1H), 0.99 (d, J = 6 Hz, 3H), 0.80 (d, J = 6 Hz, 3H), 0.77 (d, J = 6 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.37, 159.70, 156.91, 155.15, 153.15, 145.67, 134.39, 128.71, 128.69, 127.13 (d, J = 16.6 Hz), 120.02 (d, J = 25.7 Hz), 116.98, 115.50 (d, J = 9.1 Hz), 94.53, 55.04, 33.48, 18.16, 17.80, 17.14 ppm. 67 1H NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.63 (dd, J = 12.0, 3.0 Hz, 1H), 7.56 (dd, J = 12.0, 3.0 Hz, 1H), 7.52 (t, J = 9.0 Hz, 1H), 6.30 (bs, 1H), 3.05 (bs, 1H), 1.61 (oct, J = 6.0 Hz, 1H), 1.05 (d, J = 6 Hz, 3H), 0.79 (d, J = 6 Hz, 3H), 0.75 (d, J = 6 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.40, 159.73, 156.90, 155.19, 153,57, 145.67, 135.08, 134.40 (d, J = 3.0 Hz), 128.55 (d, J = 4.5 Hz), 127.03 (d, J = 15.1 Hz), 120.22 (d, J = 25.7 Hz), 117.01, 115.45 (d, J = 9.1 Hz), 94.37, 54.85, 33.80, 18.20, 17.90, 17.82 ppm; IR (film) ν 2963, 2220, 1608, 1570, 1260, 1156 cm−1; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 359.1182, found 359.1184.
(R)-4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (69).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.020 g, 0.06 mmol) (106) and (R)-3,3-dimethylbutan-2-amine (0.013 g, 0.13 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.018 g, 0.05 mmol, 75%). 1H NMR (600 MHz, CDCl3) δ 8.36 (s, 1H), 7.45 – 7.41 (m, 2H), 6.52 (d, J = 10.8 Hz, 1H), 2.94 (s, 1H), 1.02 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.33 (dd, J = 253.8, 6.2 Hz), 161.05 (dd, J = 253.8, 6.2 Hz), 157.12, 155.22, 146.03, 116.22 (d, J = 4.0 Hz), 116.13 – 115.95 (m), 115.95 – 115.82 (m), 58.57, 34.93, 25.86, 16.56 ppm; 19F NMR (469 MHz) δ −103.70 (d, J = 4.7 Hz), −104.31 (d, J = 4.7 Hz) ppm; HRMS (ES+) calculated for C18H18ClF2N6 [M + H]+: 391.1244, found 391.1241.
(R)-4-(5-Chloro-7-((3,3-dimethylbutan-2-yl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3fluorobenzonitrile (70).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (0.040 g, 0.13 mmol) (107) and (R)-3,3-dimethylbutan-2-amine (0.028 g, 0.27 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.027 g, 0.07 mmol, 56%). 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.41 – 8.34 (m, 1H), 7.67 – 7.61 (m, 1H), 7.60 – 7.55 (m, 1H), 7.55 – 7.49 (m, 1H), 6.37 (s, 1H), 2.94 – 2.89 (m, 1H), 1.01 (d, J = 6.7 Hz, 1H), 0.94 (d, J = 6.7 Hz, 2H), 0.82 (s, 5H), 0.81 (s, 4H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.71 – 159.28 (m), 156.76, 156.67, 145.96, 135.28 (d, J = 2.4 Hz), 133.96 (d, J = 2.3 Hz), 128.65 (d, J = 4.1 Hz), 128.43 (d, J = 4.1 Hz), 119.96 (dd, J = 37.6, 25.4 Hz), 116.86 (d, J = 2.8 Hz), 115.42 (dd, J = 16.7, 9.3 Hz), 94.27, 94.14, 58.27, 58.07, 35.01, 34.82, 25.77 (d, J = 1.9 Hz), 16.42 (d, J = 2.2 Hz) ppm; HRMS (ES+) calculated for C18H19ClFN6 [M + H]+: 373.1338, found 373.1338.
4-(5-chloro-7-((1R,3s,5S)-3-methoxy-8-azabicyclo[3.2.1]octan-8-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (71).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.100 g, 0.31 mmol) (106), (1R,3R,5S)-3-methoxy-8-azabicyclo[3.2.1]octane hydrochloride (0.082 g, 0.46 mmol) and Et3N (0.094 g, 0.92 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.112 g, 0.26 mmol, 85%). 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 7.39 (d, J = 5.9 Hz, 2H), 4.57 (s, 2H), 3.45 (s, 1H), 3.23 (s, 3H), 2.13 (d, J = 7.4 Hz, 2H), 2.00 (d, J = 14.9 Hz, 2H), 1.92 (d, J = 14.7 Hz, 2H), 1.80 – 1.74 (m, 2H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.66 (d, J = 6.6 Hz), 159.98 (d, J = 6.6 Hz), 156.99, 155.05, 146.39, 118.79, 116.34, 116.30, 116.18, 116.15, 116.11, 115.16 (t, J = 11.8 Hz), 92.08, 73.33, 62.34, 59.30, 56.53, 36.34, 27.98 ppm; HRMS (ES+) calculated for C20H18ClF2N6O [M + H]+: 431.1188, found 431.1193.
4-(7-(Azepan-1-yl)-5-chloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (72).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.020 g, 0.06 mmol) (106) and azepane (0.013 g, 0.13 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.015 g, 0.04 mmol, 63%). 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 7.40 (d, J = 6.0 Hz, 2H), 3.45 – 3.39 (m, 4H), 1.78 – 1.70 (m, 4H), 1.64 – 1.60 (m, 4H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.74 (dd, J = 253.4, 6.6 Hz), 157.14, 155.26, 152.10, 118.61 (t, J = 19.7 Hz), 116.30 – 115.97 (m), 115.12 (t, J = 11.9 Hz), 97.32, 53.99, 51.22, 28.27, 28.17, 27.28, 27.07 ppm; 19F NMR (469 MHz) δ −105.22 ppm; IR (film) ν 2928, 2857, 2238, 1589, 1515, 1422 cm−1; HRMS (ES+) calculated for C18H16ClF2N6 [M + H]+: 389.1088, found 389.1085.
4-(7-(Azepan-1-yl)-5-chloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (73).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (0.040 g, 0.13 mmol) (107) and azepane (0.027 g, 0.27 mmol). Purification by reverse-phase HPLC provided the title compound as a white powder (0.027 g, 0.07 mmol, 56%). 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 1H), 7.61 (dd, J = 7.9, 1.6 Hz, 1H), 7.53 (dd, J = 8.7, 1.6 Hz, 1H), 7.48 (t, J = 7.5 Hz, 1H), 3.38 – 3.33 (m, 4H), 1.71 (dq, J = 8.0, 4.2 Hz, 4H), 1.60 (p, J = 3.0 Hz, 4H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.85, 159.18, 156.93, 155.27, 155.18, 151.61, 134.44 (d, J = 2.8 Hz), 128.60 (d, J = 3.9 Hz), 128.48, 120.11, 119.94, 117.09 (d, J = 2.8 Hz), 114.68 (d, J = 9.2 Hz), 103.59, 54.05, 28.27, 28.03 ppm; IR (film) ν 2926, 2235, 1589, 1519, 1446 cm−1; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 371.1182, found 371.1180.
4-(7-(Azocan-1-yl)-5-chloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (74).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (0.054 g, 0.17 mmol) (106) and azocane (0.019 g, 0.17 mmol). Purification via silica gel column chromatography provided the title compound as a white solid (0.031 g, 0.08 mmol, 46%). 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1H), 7.30 (d, J = 6.0 Hz, 2H), 3.36 (t, J = 5.7 Hz, 5H), 1.62 (t, J = 5.8 Hz, 5H), 1.52 (t, J = 5.5 Hz, 6H), 1.44 (d, J = 6.0 Hz, 2H) ppm; 13C NMR (151 MHz, CDCl3) δ 160.68 (dd, J = 253.6, 6.5 Hz), 157.23, 155.73, 155.12, 151.06, 118.55 (t, J = 19.8 Hz), 116.38, 116.34, 116.23, 116.19, 115.23 (t, J = 11.8 Hz), 97.29, 52.24, 27.72, 26.80, 24.44 ppm; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 403.1244, found 403.1240.
4-(7-(Azocan-1-yl)-5-chloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (75).
Following general procedure C using 4-(5,7-dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (0.048 g, 0.15 mmol) (107) and azocane (0.035 g, 0.30 mmol). Purification via silica gel column chromatography provided the title compound as a white solid (0.032 g, 0.08 mmol, 53%). 1H NMR (600 MHz, CDCl3,) mixture of diastereomers δ 8.59 (s, 0.4H), 8.42 (s, 0.3H), 7.80 – 7.77 (m, 0.6H), 7.30 (d, J = 6 Hz, 0.3H), 7.16 (d, J = 12 Hz, 0.4H), 3.41 (t, J = 6 Hz, 1.2H), 3.32 (bs, 1.8H), 3.04 (bs, 1.8H), 1.70 – 1.41 (m, 10H) ppm; HRMS (ES+) calculated for C18H17ClFN6 [M + H]+: 385.1338, found 385.1332.
Diethyl 2-phenylmalonate (76).
Following General Procedure A using diethyl malonate (6.122 g, 38.20 mmol) and bromobenzene (2.002 g, 12.70mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a colorless oil (0.833 g, 3.53 mmol, 28%). 1H NMR (500 MHz, CDCl3) δ 7.50 – 7.31 (m, 5H), 4.61 (s, 1H), 4.45 – 4.01 (m, 4H), 1.26 (t, J = 7.1 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 168.30, 132.94, 129.40, 128.73, 128.33, 61.94, 58.12, 14.16 ppm; IR (film) ν 3450, 1731, 1638, 1455, 1368, 1305, 1217, 1147, 1029, 700 cm−1; HRMS (ES+) calculated for C13H16NaO4 [M + Na]+: 259.0946, found 259.0952.
Diethyl 2-(4-fluorophenyl)malonate (77).
Following General Procedure A using diethyl malonate (5.491 g, 34.30 mmol) and 1-bromo-4-fluorobenzene (2.000 g, 11.40 mmol). Purification via silica gel column chromatography (8/8/84 EtOAc/CH2Cl2/hexanes) provided the title compound as a colorless oil (0.818 g, 3.22 mmol, 28%). 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.36 (m, 2H), 7.07 – 7.01 (m, 2H), 4.59 (s, 1H), 4.27 – 4.14 (m, 4H), 1.25 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 168.10, 162.72 (d, J = 246.9 Hz), 131.11 (d, J = 8.3 Hz), 128.70 (d, J = 3.4 Hz), 115.59 (d, J = 21.6 Hz), 61.99, 57.18, 14.07 ppm; IR (film) ν 3451, 2985, 2940, 1733, 1607, 1467, 1447, 1369, 1302, 1223, 1157, 1096, 1031, 841, 806, 753 cm−1; HRMS (ES+) calculated for C13H15FNaO4 [M + Na]+: 277.0852, found 277.0861.
Diethyl 2-(2-fluorophenyl)malonate (78).
Following General Procedure A using diethyl malonate (5.491 g, 34.30 mmol) and 1-bromo-2-fluorobenzene (2.000 g, 11.40 mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a colorless oil (1.411 g, 5.55 mmol, 49%). 1H NMR (500 MHz, CDCl3) δ 7.47 (td, J = 7.6, 1.8 Hz, 1H), 7.34 – 7.28 (m, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.07 (t, J = 9.1 Hz, 1H), 4.98 (s, 1H), 4.23 (p, J = 7.0 Hz, 4H), 1.27 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.66, 160.59 (d, J = 247.6 Hz), 130.54 (d, J = 3.0 Hz), 130.04 (d, J = 8.3 Hz), 124.34 (d, J = 3.7 Hz), 120.51 (d, J = 14.5 Hz), 115.51 (d, J = 22.2 Hz), 62.09, 50.61 (d, J = 3.3 Hz), 14.07 ppm; IR (film) ν 3458, 2984, 2940, 2907, 1737, 1617, 1589, 1494, 1458, 1369, 1303, 1232, 1149, 1094, 1030, 952, 860, 815, 756, 697 cm−1; HRMS (ES+) calculated for C13H15FNaO4 [M + Na]+: 277.0852, found 277.0858.
Diethyl 2-(2,4-difluorophenyl)malonate (79).
Following General Procedure A using diethyl malonate (5.000 g, 31.10 mmol) and 1-bromo-2,4-difluorobenzene (2.000 g, 10.30 mmol). Purification via silica gel column chromatography (0–5% EtOAc in hexanes) provided the title compound as a colorless oil (1.907 g, 6.98 mmol, 68%). 1H NMR (500 MHz, CDCl3) δ 7.47 (td, J = 8.5, 6.3 Hz, 1H), 6.89 (td, J = 7.4, 3.8 Hz, 1H), 6.82 (td, J = 8.5, 2.5 Hz, 1H), 4.91 (s, 1H), 4.29 – 4.15 (m, 4H), 1.25 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.46, 162.83 (dd, J = 249.9, 12.2 Hz), 160.68 (dd, J = 250.0, 12.1 Hz), 131.58 (dd, J = 9.8, 4.5 Hz), 116.52 (dd, J = 14.6, 4.0 Hz), 111.64 (dd, J = 21.4, 3.7 Hz), 103.88 (t, J = 25.8 Hz), 62.17, 49.97 (d, J = 2.7 Hz), 14.01 ppm; IR (film) ν 3449, 1737, 1624, 1508, 1291, 1220, 1144, 1031, 970, 851 cm−1; HRMS (ES+) calculated for C13H15F2O4 [M + H]+: 273.0938, found 273.0946.
Diethyl 2-(2,6-difluorophenyl)malonate (80).
Following General Procedure A using diethyl malonate (4.981 g, 31.10 mmol) and 2-bromo-1,3-difluorobenzene (2.000 g, 10.30 mmol). Purification via preparative reverse-phase HPLC provided the title compound as a colorless oil (2.082 g, 7.64 mmol, 74%). 1H NMR (500 MHz, CDCl3) δ 7.34 – 7.27 (m, 1H), 6.93 (t, J = 8.1 Hz, 2H), 4.97 (s, 1H), 4.26 (q, J = 7.1 Hz, 4H), 1.27 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.05, 161.29 (dd, J = 250.6, 7.1 Hz), 130.27 (t, J = 11.9 Hz), 111.75 (t, J = 25.6 Hz), 111.65 (dd, J = 20.6, 4.6 Hz), 62.38, 47.32, 14.05 ppm; IR (film) ν 3460, 2985, 1746, 1628, 1595, 1471, 1370, 1303, 1274, 1236, 1157, 1096, 1035, 1003, 788, 698 cm−1; HRMS (ES+) calculated for C13H14F2NaO4 [M + Na]+: 295.0758, found 295.0757.
Diethyl 2-(3,5-difluorophenyl)malonate (81).
Following General Procedure A using diethyl malonate (5.000 g, 31.10 mmol) and 1-bromo-3,5-difluorobenzene (2.000 g, 10.30 mmol). Purification via silica gel column chromatography (0–5% EtOAc in hexanes) provided the title compound as a colorless oil (1.462 g, 5.36 mmol, 52%). 1H NMR (500 MHz, CDCl3) δ 6.99 – 6.93 (m, 2H), 6.76 (tt, J = 8.9, 2.4 Hz, 1H), 4.57 (s, 1H), 4.28 – 4.13 (m, 4H), 1.25 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.20, 162.86 (dd, J = 248.7, 12.9 Hz), 136.08 (t, J = 9.9 Hz), 112.63 (dd, J = 20.7, 6.0 Hz), 103.87 (t, J = 25.2 Hz), 62.26, 57.35 (t, J = 1.9 Hz), 14.00 ppm; IR (film) ν 3450, 2986, 1736, 1626, 1601, 1465, 1369, 1305, 1234, 1154, 1121, 1037, 995, 867, 738, 675 cm−1; HRMS (ES+) calculated for C13H14F2NaO4 [M + Na]+: 295.0758, found 295.0745.
Diethyl 2-(2,5-difluorophenyl)malonate (82).
Following General Procedure A using diethyl malonate (5.000 g, 31.10 mmol) and 2-bromo-1,4-difluorobenzene (2.000 g, 10.30 mmol). Purification via silica gel column chromatography (0–10% EtOAc in hexanes) provided the title compound as a colorless oil (1.767 g, 6.46 mmol, 62%). 1H NMR (500 MHz, CDCl3) δ 7.25 – 7.19 (m, 1H), 7.06 – 6.95 (m, 2H), 4.94 (s, 1H), 4.29 – 4.17 (m, 4H), 1.26 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.10, 158.54 (dd, J = 242.3, 2.5 Hz), 156.62 (dd, J = 243.9, 2.5 Hz), 121.84 (dd, J = 17.0, 8.4 Hz), 117.20 (dd, J = 25.5, 3.2 Hz), 116.59 (dd, J = 15.0, 8.8 Hz), 116.39 (dd, J = 16.3, 8.6 Hz), 62.29, 50.35 (d, J = 2.7 Hz), 14.02 ppm; IR (film) ν 3452, 2986, 1737, 1631, 1499, 1369, 1303, 1226, 1154, 1031, 877, 819, 739 cm−1; HRMS (ES+) calculated for C13H14F2NaO4 [M + Na]+: 295.0758, found 295.0755.
Diethyl 2-(3,4-difluorophenyl)malonate (83).
Following General Procedure A using diethyl malonate (5.002 g, 31.10 mmol) and 4-bromo-1,2-difluorobenzene (2.001 g, 10.30 mmol). Purification via preparative reverse-phase HPLC provided the title compound as an orange oil (0.530 g, 1.95 mmol, 19%). 1H NMR (500 MHz, CDCl3) δ 7.33 – 7.28 (m, 1H), 7.18 – 7.07 (m, 2H), 4.56 (d, J = 2.9 Hz, 1H), 4.28 – 4.17 (m, 4H), 1.27 (td, J = 7.3, 2.2 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 167.67, 150.35 (dd, J = 272.8, 12.7 Hz), 150.35 (dd, J = 224.7, 12.6 Hz), 129.67 (dd, J = 6.2, 4.0 Hz), 125.77 (dd, J = 6.5, 3.7 Hz), 118.63 (d, J = 18.4 Hz), 117.34 (d, J = 17.4 Hz), 62.24, 57.00, 14.05 ppm; IR (film) ν 3450, 2986, 1734, 1613, 1520, 1441, 1369, 1287, 1154, 1032, 773 cm−1; HRMS (ES+) calculated for C13H15F2O4 [M + H]+: 273.0938, found 273.0941.
Diethyl 2-(2,3,4-trifluorophenyl)malonate (84).
Following General Procedure A using diethyl malonate (4.560 g, 28.44 mmol) and 1-bromo-2,3,4-trifluorobenzene (2.000 g, 9.48 mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a colorless oil (1.621 g, 5.58 mmol, 59%). 1H NMR (500 MHz, CDCl3) δ 7.28 – 7.19 (m, 1H), 7.00 (tdd, J = 9.2, 7.0, 2.1 Hz, 1H), 4.90 (s, 1H), 4.29 – 4.19 (m, 4H), 1.28 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.98, 151.11 (ddd, J = 251.2, 9.8, 3.2 Hz), 149.78 (ddd, J = 251.2, 10.7, 3.4 Hz), 139.90 (dt, J = 251.5, 15.5 Hz), 124.30 (dt, J = 7.9, 3.9 Hz), 118.08 (dd, J = 11.8, 4.0 Hz), 112.31 (dd, J = 17.6, 3.9 Hz), 62.40, 50.06, 14.01 ppm; IR (film) ν 3405, 2986, 1738, 1617, 1513, 1490, 1370, 1305, 1243, 1151, 1033, 992, 865, 810, 672 cm−1; HRMS (ES+) calculated for C13H13F3NaO4 [M + Na]+: 313.0664, found 313.0663.
Diethyl 2-(2,4,6-trifluorophenyl)malonate (85).
Following General Procedure A using diethyl malonate (8.408 g, 52.5 mmol) and 2-bromo-1,3,5-trifluorobenzene (3.692 g, 17.5 mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a white powder (2.542 g, 8.75 mmol, 50% yield). 1H-NMR (500 MHz; CDCl3): δ 6.74 – 6.68 (m, 2H), 4.89 (s, 1H), 4.29 – 4.23 (m, 4H), 1.28 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.68, 162.78 (dt, J = 250.7, 15.1 Hz), 161.5 (dd, J = 250.7, 15.1 Hz), 161.44 (dd, J = 252.2, 15.1 Hz), 107.25 (dt, J = 18.1, 4.5 Hz), 100.81 – 100.43 (m), 62.41, 46.95, 14.04 ppm; LCMS: [M + H]+: 291.
Diethyl 2-(2,4,6-trifluorobenzyl)malonate (86).
To a suspension of NaH (60 wt % in mineral oil) (0.462 g, 11.56 mmol, 1.30 equiv) in anhydrous DMF (8.5 mL) at 0 °C under N2 was slowly added diethyl malonate (1.712 g, 10.67 mmol, 1.20 equiv). After addition, the reaction was stirred at 0 °C for 10 min. Then, 2-(bromomethyl)-1,3,5-trifluorobenzene (2.001 g, 8.89 mmol, 1.00 equiv) in solution in anhydrous DMF (8.5 mL) was slowly added and the reaction was stirred at 0 °C for 1 h. Then, the reaction was quenched with saturated solution of NH4Cl and H2O (85 mL) was added. The mixture was extracted with EtOAc (×3). The combined organic extracts were washed with brine, then dried over MgSO4, filtered and concentrated under reduced pressure. Purification via silica gel column chromatography (0–5% EtOAc in hexanes) provided the title compound as a colorless oil (2.43 g, 7.99 mmol, 90%). 1H NMR (500 MHz, CDCl3) δ 7.28 – 7.19 (m, 1H), 7.04 – 6.95 (m, 1H), 4.90 (s, 1H), 4.29 – 4.19 (m, 4H), 1.28 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 168.33, 161.72 (dd, J = 248.9, 11.3 Hz), 161.70 (dt, J = 248.9, 16.0, 15.5 Hz), 161.60 (dd, J = 249.0, 11.2 Hz), 109.78 (td, J = 20.1, 4.7 Hz), 100.35 – 99.84 (m), 61.63, 51.10, 21.45 (t, J = 2.3 Hz), 13.90 ppm; IR (film) ν 3437, 1733, 1639, 1499, 1444, 1371, 1304, 1239, 1153, 1048, 838, 617 cm−1; HRMS (ES+) calculated for C14H15F3NaO4 [M + Na]+: 327.0820, found 327.0818.
Diethyl 2-(2,6-difluoro-4-methylphenyl)malonate (87).
Following General Procedure A using diethyl malonate (4.180 g, 26.10 mmol) and 2-bromo-1,3-difluoro-5-methylbenzene (1.800 g, 8.69 mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a colorless oil (2.005 g, 6.99 mmol, 80%). 1H NMR (600 MHz, CDCl3) δ 6.67 – 6.65 (m, 2H), 4.83 (d, J = 3.0 Hz, 1H), 4.20 – 4.14 (m, 4H), 2.27 (d, J = 3.2 Hz, 3H), 1.22 – 1.17 (m, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 166.89, 160.75 (dd, J = 249.1, 8.1 Hz), 141.32, 115.89 – 110.83 (m), 107.42 (t, J = 18.8 Hz), 61.99, 46.98, 21.23 (d, J = 1.9 Hz), 13.87 ppm; IR (film) ν 3000, 1678, 1106, 1017 cm−1. LCMS: [M + H]+: 287.
Diethyl 2-(2,6-difluoro-4-(trifluoromethyl)phenyl)malonate (88).
Following General Procedure A using diethyl malonate (2.160 g, 13.50 mmol) and 1,2,3-trifluoro-5-(trifluoromethyl)benzene (0.900 g, 4.50 mmol). Purification via silica gel column chromatography (0–5% EtOAc in hexanes) provided the title compound as a colorless oil (0.620 g, 1.82 mmol, 40%). 1H NMR (500 MHz, CDCl3) δ 7.23 (d, J = 7.3 Hz, 2H), 4.99 (s, 1H), 4.27 (dt, J = 7.4, 6.5 Hz, 4H), 1.29 (t, J = 7.1 Hz, 6H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.12, 161.21 (dd, J = 252.9, 7.4 Hz), 132.89 (qt, J = 34.8, 10.3 Hz), 122.63 (qt, J = 272.6, 3.3 Hz), 114.85 (t, J = 18.5 Hz), 109.41 (ddt, J = 26.3, 7.4, 3.8 Hz), 62.69, 47.34, 14.07 ppm; IR (film) ν 3083, 2987, 1755, 1061, 1442, 1365, 1306, 1240, 1177, 1138, 1080, 1031, 910, 867, 719, 702 cm−1; HRMS (ES+) calculated for C14H13F5NaO4 [M + Na]+: 363.0632, found 363.0631.
Diethyl 2-(4-cyano-2,6-difluorophenyl)malonate (89).
A mixture of 3,4,5-trifluorobenzonitrile (1.000 g, 6.37 mmol, 1 equiv), potassium carbonate (1.760 g, 12.70 mmol, 2.00 equiv), and diethyl malonate (1.031 g, 6.43 mmol, 1.01 equiv) in DMF (6.37 ml) under N2 was stirred at 65 °C until starting material was consumed as determined by TLC. The reaction mixture was cooled to r.t., washed with 1M HCl (50 mL), and extracted with EtOAc (×3). The organic layers were combined, washed with satd. aq. NaCl, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white solid (1.408 g, 4.69 mmol, 73%). 1H NMR (600 MHz, CDCl3) δ 7.30 (d, J = 7.0 Hz, 2H), 5.01 (s, 1H), 4.31 – 4.24 (m, 4H), 1.29 (t, J = 7.4 Hz, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 165.54, 161.02 (dd, J = 254.0, 7.8 Hz), 116.78 (t, J = 18.5 Hz), 116.20 (t, J = 3.4 Hz), 115.94 – 115.48 (m), 113.88 (t, J = 12.4 Hz), 62.60, 47.24, 13.87 ppm; IR (film) ν 2856, 2178, 1695 cm−1; LCMS: [M + H]+: 298.
Diethyl 2-(4-cyano-2-fluorophenyl)malonate (90).
To a dry flask, under nitrogen, was added 3,4-difluorobenzonitrile (4.000 g, 28.81 mmol, 1 equiv), K2CO3 (7.954 g, 57.51 mmol, 2.00 equiv), diethyl malonate (4.660 g, 29.10 mmol, 1.01 equiv), and anhydrous DMF (24 ml) and was heated at 65 °C until starting material was consumed based on TLC. The reaction mixture was cooled to r.t. and added to a separatory funnel containing 50 ml of 1 N HCl. The mixture was extracted with EtOAc (×3), and the combined organic layers were washed with H2O and satd. aq. NaCl. The organic layer was dried over anh. Na2SO4, filtered, and concentrated under reduced pressure. Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a colorless oil (6.734 g, 24.10 mmol, 84%). 1H NMR (600 MHz, CDCl3) δ 7.64 (t, J = 7.6 Hz, 1H), 7.47 (dd, J = 8.0, 1.6 Hz, 1H), 7.38 (dd, J = 9.2, 1.6 Hz, 1H), 4.98 (s, 1H), 4.28 – 4.18 (m, 4H), 1.26 (t, J = 7.2 Hz, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 166.55, 160.04 (d, J = 252.0 Hz), 131.98 (d, J = 3.5 Hz), 128.26 (d, J = 4.0 Hz), 126.21 (d, J = 14.4 Hz), 119.23 (d, J = 26.0 Hz), 117.28 (d, J = 2.9 Hz), 113.71 (d, J = 9.8 Hz), 62.55, 50.44 (d, J = 2.9 Hz), 14.00 ppm; IR (film) ν 2984, 2236, 1733, 1219 cm−1; LCMS: [M + H]+: 280.
Diethyl 2-(4-chloro-2,6-difluorophenyl)malonate (91).
Following General Procedure A using diethyl malonate (6.390 g, 39.57 mmol) and 2-bromo-5-chloro-1,3-difluorobenzene (3.000 g, 13.19 mmol). Purification via silica gel column chromatography (0–8% EtOAc in hexanes) provided the title compound as a colorless oil (1.424 g, 4.63 mmol, 35%). 1H NMR (600 MHz, CDCl3) δ 6.96 – 6.93 (m, 2H), 4.89 (s, 1H), 4.24 (q, J = 7.2 Hz, 4H), 1.28 – 1.24 (m, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 166.40, 161.05 (dd, J = 252.7, 8.8 Hz), 135.40 (t, J = 13.4 Hz), 113.18 – 112.42 (m), 109.85 (t, J = 18.8 Hz), 62.4, 47.07, 14.01 ppm; IR (film) ν 2838, 1678, 1642, 1249 cm−1; LCMS: [M + H]+: 308.
Diethyl 2-(2,6-difluoro-4-nitrophenyl)malonate (92).
A mixture of 1,2,3-trifluoro-5-nitrobenzene (1.000 g, 5.65 mmol, 1 equiv), potassium carbonate (1.562 g, 11.29 mmol, 2.00 equiv), and diethyl malonate (0.914 g, 5.71 mmol, 1.01 equiv), in DMF (6.37 ml) under N2 was stirred at 65 °C until starting material was consumed as indicated by TLC. The reaction mixture was cooled to r.t. and washed with 1N HCl (50 mL) and extracted with EtOAc (×3). The organic layers were combined, washed with satd. aq. NaCl, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification via silica gel column chromatography (0–30% EtOAc in hexanes) provided the title compound as a white solid (1.536 g, 4.85 mmol, 86%). 1H NMR (600 MHz, CDCl3) δ 7.84 (d, J = 7.2 Hz, 2H), 5.00 (s, 1H), 4.27 (q, J = 7.1 Hz, 4H), 1.28 (t, J = 7.2 Hz, 6H) ppm; 13C NMR (150 MHz, CDCl3) δ 165.55, 160.93 (dd, J = 254.8, 7.7 Hz), 148.60 (t, J = 11.2 Hz), 118.05 (t, J = 18.7 Hz), 108.15 – 107.61 (m), 62.84, 47.49, 14.04 ppm.
5,7-Dichloro-6-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine (93).
Following General Procedure B using diethyl 2-phenylmalonate (0.780 g, 3.30 mmol) (76) and 3-amino-1,2,4-triazole (0.292 g, 3.47 mmol). Then using the intermediate sodium 6-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.740 g, 2.71 mmol) and POCl3 (7.410 g, 48.46 mmol) to obtain the title compound as a brown solid (0.527 g, 1.99 mmol, 73%). 1H NMR (500 MHz, CDCl3) δ 8.56 (s, 1H), 7.58 – 7.51 (m, 3H), 7.38 – 7.34 (m, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 156.93, 156.65, 153.35, 139.90, 131.72, 130.07, 129.95, 129.16, 124.01 ppm; IR (film) ν 3437, 1637, 1458, 1383, 1267, 1205, 1180, 867, 802, 764, 740, 698, 652 cm−1; HRMS (ES+) calculated for C11H7Cl2N4 [M + H]+: 265.0042, found 265.0043.
5,7-Dichloro-6-(4-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (94).
Following General Procedure B using diethyl 2-(4-fluorophenyl)malonate (0.700 g, 2.75 mmol) (77) and 3-amino-1,2,4-triazole (0.242 g, 2.89 mmol). Then using the intermediate sodium 6-(4-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.760 g, 2.61 mmol) and POCl3 (7.150 g, 46.45 mmol) to obtain the title compound as a brown solid (0.481 g, 1.70 mmol, 67%). 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H), 7.36 (dd, J = 8.4, 5.2 Hz, 2H), 7.29 – 7.20 (m, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.47 (d, J = 250.9 Hz), 156.95, 156.53, 153.29, 140.05, 132.18 (d, J = 8.6 Hz), 127.59 (d, J = 3.7 Hz), 123.03, 116.43 (d, J = 21.9 Hz) ppm; IR (film) ν 3421, 1600, 1529, 1504, 1460, 1330, 1267, 1229, 1203, 1183, 1160, 1089, 1024, 899, 837, 790 cm−1; HRMS (ES+) calculated for C11H6Cl2FN4 [M + H]+: 282.9948, found 282.9945.
5,7-Dichloro-6-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (95).
Following General Procedure B using diethyl 2-(2-fluorophenyl)malonate (0.700 g, 2.75 mmol) (78) and 3-amino-1,2,4-triazole (0.242 g, 2.89 mmol). Then using the intermediate sodium 6-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.760 g, 2.61 mmol) and POCl3 (7.150 g, 46.45 mmol) to obtain the title compound as a brown solid (0.572 g, 2.02 mmol, 77%). 1H NMR (500 MHz, CDCl3) δ 8.54 (t, J = 3.3 Hz, 1H), 7.59 – 7.51 (m, 1H), 7.38 – 7.29 (m, 2H), 7.28 – 7.20 (m, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ 159.79 (d, J = 249.6 Hz), 156.94, 156.57, 153.53, 140.57, 132.48 (d, J = 8.2 Hz), 131.78 (d, J = 1.8 Hz), 124.85 (d, J = 3.7 Hz), 119.39 (d, J = 15.9 Hz), 118.45, 116.38 (d, J = 21.1 Hz) ppm; IR (film) ν 3436, 1598, 1492, 1463, 1425, 1366, 1330, 1263, 1221, 1196, 1131, 1099, 1033, 900, 822, 792, 755, 652 cm−1; HRMS (ES+) calculated for C11H5Cl2FN4Na [M + Na]+: 304.9768, found 304.9772.
5,7-Dichloro-6-(2,4-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (96).
Following General Procedure B using diethyl 2-(2,4-difluorophenyl)malonate (0.800 g, 2.94 mmol) (79) and 3-amino-1,2,4-triazole (0.260 g, 3.09 mmol). Then using the intermediate sodium 6-(2,4-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.800 g, 2.60 mmol) and POCl3 (7.080 g, 46.23 mmol) to obtain the title compound as a brown solid (0.455 g, 1.51 mmol, 58%). 1H NMR (500 MHz, CDCl3) δ 8.50 (s, 1H), 7.39 – 7.32 (m, 1H), 7.06 (td, J = 8.3, 2.5 Hz, 1H), 6.99 (td, J = 9.1, 2.5 Hz, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.23 (dd, J = 253.6, 11.8 Hz), 160.18 (dd, J = 252.2, 12.3 Hz), 156.94, 156.47, 153.47, 140.79, 132.91 (dd, J = 10.1, 3.4 Hz), 117.55, 115.54 (dd, J = 16.1, 4.1 Hz), 112.47 (dd, J = 21.9, 3.7 Hz), 104.96 (t, J = 25.4 Hz) ppm; IR (film) ν 3451, 1600, 1503, 1464, 1424, 1368, 1335, 1267, 1196, 1143, 1128, 1092, 1022, 969, 890, 851, 796, 765, 735, 652, 631 cm−1; HRMS (ES+) calculated for C11H5Cl2F2N4 [M + H]+: 300.9854, found 300.9850.
5,7-Dichloro-6-(2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (97).
Following General Procedure B using diethyl 2-(2,6-difluorophenyl)malonate (0.800 g, 2.94 mmol) (80) and 3-amino-1,2,4-triazole (0.260 g, 3.09 mmol). Then using the intermediate sodium 6-(2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.600 g, 1.90 mmol) and POCl3 (5.310 g, 34.70 mmol) to obtain the title compound as a brown solid (0.290 g, 0.96 mmol, 49%). 1H NMR (500 MHz, CDCl3) δ 8.59 (d, J = 1.4 Hz, 1H), 7.61 – 7.52 (m, 1H), 7.11 (t, J = 7.9 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 160.23 (dd, J = 252.0, 5.7 Hz), 157.17, 156.65, 153.87, 141.38, 133.06 (t, J = 10.2 Hz), 113.14, 112.16 (dd, J = 20.4, 4.2 Hz), 109.07 (t, J = 19.9 Hz) ppm; IR (film) ν 3427, 1629, 1602, 1460, 1384, 1266, 1194, 1003899, 806, 765, 652 cm−1; HRMS (ES+) calculated for C11H5Cl2F2N4 [M + H]+: 300.9854, found 300.9866.
5,7-Dichloro-6-(3,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (98).
Following General Procedure B using diethyl 2-(3,5-difluorophenyl)malonate (0.700 g, 2.57 mmol) (81) and 3-amino-1,2,4-triazole (0.227 g, 2.70 mmol). Then using the intermediate sodium 6-(3,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.700 g, 2.27 mmol) and POCl3 (6.200 g, 40.51 mmol) to obtain the title compound as a brown solid (0.422 g, 1.40 mmol, 62%). 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 7.02 (tt, J = 8.8, 2.4 Hz, 1H), 6.97 – 6.89 (m, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.29 (dd, J = 251.5, 12.6 Hz), 157.31, 155.74, 153.47, 140.16, 134.33 (t, J = 10.3 Hz), 121.84, 113.71 (dd, J = 19.6, 6.9 Hz), 105.89 (t, J = 25.0 Hz) ppm; IR (film) ν 3434, 1628, 1595, 1460, 1435, 1121, 987, 833 cm−1; HRMS (ES+) calculated for C11H5Cl2F2N4 [M + H]+: 300.9854, found 300.9871.
5,7-Dichloro-6-(2,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (99).
Following General Procedure B using diethyl 2-(2,5-difluorophenyl)malonate (0.800 g, 2.94 mmol) (82) and 3-amino-1,2,4-triazole (0.259 g, 3.09 mmol). Then using the intermediate sodium 6-(2,5-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.800 g, 2.60 mmol) and POCl3 (7.080 g, 46.23 mmol) to obtain the title compound as a brown solid (0.443 g, 1.47 mmol, 57%). 1H NMR (500 MHz, CDCl3) δ 8.58 (s, 1H), 7.28 – 7.23 (m, 2H), 7.11 – 7.07 (m, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.60 (dd, J = 245.1, 2.5 Hz), 157.24, 156.21, 156.03 (dd, J = 245.9, 2.7 Hz), 153.68, 140.80, 120.50 (dd, J = 18.7, 8.8 Hz), 119.24 (dd, J = 23.9, 8.4 Hz), 118.42 (dd, J = 25.1, 2.2 Hz), 117.80 (dd, J = 24.2, 8.7 Hz), 117.43 ppm; IR (film) ν 3421, 1642, 1492, 1458, 1432, 1383, 1253, 1203, 1092, 1025, 858, 822, 775, 699, 651 cm−1; HRMS (ES+) calculated for C11H5Cl2F2N4 [M + H]+: 300.9854, found 300.9857.
5,7-Dichloro-6-(3,4-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (100).
Following General Procedure B using diethyl 2-(3,4-difluorophenyl)malonate (0.456 g, 1.67 mmol) (83) and 3-amino-1,2,4-triazole (0.147 g, 1.73 mmol). Then using the intermediate sodium 6-(3,4-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.400 g, 1.30 mmol) and POCl3 (2.982 g, 23.11 mmol) to obtain the title compound as a brown solid (0.183 g, 0.61 mmol, 40%). 1H NMR (500 MHz, CDCl3) δ 8.57 (s, 1H), 7.41 – 7.32 (m, 1H), 7.26 – 7.21 (m, 1H), 7.16 – 7.11 (m, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.20, 156.17, 153.39, 151.41 (dd, J = 252.7, 11.8 Hz), 150.60 (dd, J = 251.2, 12.8 Hz), 140.27, 128.21 (dd, J = 6.6, 4.6 Hz), 126.97 (dd, J = 6.7, 3.9 Hz), 122.03, 119.74 (d, J = 18.4 Hz), 118.49 (d, J = 17.9 Hz) ppm; IR (film) ν 3377, 2918, 1606, 1460, 1383, 1269, 1180, 1120, 861, 793, 652 cm−1; HRMS (ES+) calculated for C11H5Cl2F2N4 [M + H]+: 300.9854, found 300.9859.
5,7-Dichloro-6-(2,3,4-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (101).
Following General Procedure B using diethyl 2-(2,3,4-trifluorophenyl)malonate (0.800 g, 2.75 mmol) (84) and 3-amino-1,2,4-triazole (0.242 g, 2.89 mmol). Then using the intermediate sodium 6-(2,3,4-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.740 g, 2.67 mmol) and POCl3 (6.151 g, 40.33 mmol) to obtain the title compound as a brown solid (0.367 g, 1.15 mmol, 43%). 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 1H), 7.24 – 7.16 (m, 1H), 7.16 – 7.09 (m, 1H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.39, 156.24, 153.75, 152.71 (ddd, J = 254.7, 9.8, 3.0 Hz), 149.54 (ddd, J = 253.6, 10.9, 3.6 Hz), 141.10, 140.64 (dt, J = 254.7, 15.1 Hz), 125.78 (ddd, J = 8.0, 4.4, 1.7 Hz), 117.08 (dd, J = 12.9, 4.0 Hz), 116.70 (d, J = 2.3 Hz), 113.42 (dd, J = 18.0, 3.9 Hz) ppm; IR (film) ν 3387, 1601, 1485, 1458, 1383, 1343, 1274, 12010, 1180, 1053, 1024, 963, 915, 859, 820, 790, 761, 689, 650 cm−1; HRMS (ES+) calculated for C11H4Cl2F3N4 [M + H]+: 318.9760, found 319.9764.
5,7-Dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (102).
Following General Procedure B using diethyl 2-(2,4,6-trifluorophenyl)malonate (3.66 g, 12.58 mmol) (85) and 3-amino-1,2,4-triazole (1.07 g, 12.83 mmol). Then using the intermediate sodium 6-(2,3,4-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (2.000 g, 6.29 mmol) and POCl3 (17.20 g, 112.0 mmol) to obtain the title compound as a brown solid (1.00 g, 3.14 mmol, 50%). 1H NMR (500 MHz, CDCl3) δ 8.62(s, 1H), 6.91 (t, J = 6.0 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 164.60 (dt, J = 253.7, 16.6 Hz), 160.77 (dd, J = 252.9, 9.1 Hz), 160.67 (dd, J = 252.9, 9.1 Hz), 157.36, 156.69, 141.67, 112.36, 105.67 (dt, J = 19.6, 4.5 Hz), 101.63 – 101.26 (m) ppm.
5,7-Dichloro-6-(2,4,6-trifluorobenzyl)-[1,2,4]triazolo[1,5-a]pyrimidine (103).
Following General Procedure B using diethyl 2-(2,4,6-trifluorobenzyl)malonate (0.800 g, 2.63 mmol) (86) and 3-amino-1,2,4-triazole (0.232 g, 2.76 mmol). Then using the intermediate sodium 6-(2,4,6-trifluorobenzyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.700 g, 2.06 mmol) and POCl3 (5.610 g, 36.60 mmol) to obtain the title compound as a brown solid (0.355 g, 1.07 mmol, 52%). 1H NMR (500 MHz, CDCl3) δ 8.40 (s, 1H), 6.59 (t, J = 8.5 Hz, 2H), 4.27 (s, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 161.93 (dt, J = 250.5, 16.0 Hz), 161.44 (dd, J = 249.9, 10.6 Hz), 161.33 (dd, J = 249.8, 10.7 Hz), 156.68, 156.48, 152.67, 140.34, 118.92, 108.12 (td, J = 18.5, 4.8 Hz), 102.62 – 96.83 (m), 23.49 (t, J = 2.4 Hz) ppm; IR (film) ν 3408, 1633, 1471, 1266, 1118, 1038 cm−1; HRMS (ES+) calculated for C12H6Cl2F3N4 [M + H]+: 332.9916, found 332.9906.
5,7-Dichloro-6-(2,6-difluoro-4-methylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (104).
Following General Procedure B using diethyl 2-(2,6-difluoro-4-methylphenyl)malonate (0.800 g, 2.79 mmol) (87) and 3-amino-1,2,4-triazole (0.247 g, 2.93 mmol). Then using the intermediate sodium 6-(2,6-difluoro-4-methylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.500 g, 1.55 mmol) and POCl3 (4.240 g, 27.60 mmol) to obtain the title compound as a brown solid (0.334 g, 1.06 mmol, 68%). 1H NMR (600 MHz, CDCl3) δ 8.59 (s, 1H), 6.92 (d, J = 8.5 Hz, 2H), 2.46 (s, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 159.92 (dd, J = 251.1, 6.6 Hz), 157.07, 157.01, 153.85, 144.81 (d, J = 19.7 Hz), 141.44, 113.44, 112.74 (dd, J = 20.7, 3.5 Hz), 106.01 (d, J = 39.9 Hz), 21.88 (d, J = 1.9 Hz) ppm; IR (film) ν 3071, 1606, 1571, 1356 cm−1; LCMS: [M + H]+: 317.
5,7-Dichloro-6-(2,6-difluoro-4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (105).
Following General Procedure B using diethyl 2-(2,6-difluoro-4-(trifluoromethyl)phenyl)malonate (0.600 g, 1.76 mmol) (88) and 3-amino-1,2,4-triazole (0.156 g, 1.85 mmol). Then using the intermediate sodium 6-(2,6-difluoro-4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.600 g, 1.60 mmol) and POCl3 (4.350 g, 28.41 mmol) to obtain the title compound as a brown solid (0.392 g, 1.07 mmol, 66%). 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H), 7.47 (d, J = 7.2 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 160.16 (dd, J = 254.8, 5.8 Hz), 157.22, 155.50, 153.81, 141.35, 135.19 (qt, J = 35.3, 34.6, 9.5 Hz), 122.14 (qt, J = 273.3, 3.2 Hz), 112.72 (t, J = 19.7 Hz), 111.49, 110.18 – 109.25 (m) ppm; IR (film) ν 3435, 1633, 1603, 1466, 1437, 1364, 1266, 1250, 1179, 1137, 1038, 916, 868, 801, 766, 705, 653 cm−1; HRMS (ES−) calculated for C12H3Cl2F4N4 [M − F]−: 348.9676, found 348.9662.
4-(5,7-Dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3,5-difluorobenzonitrile (106).
Following General Procedure B using diethyl 2-(4-cyano-2,6-difluorophenyl)malonate (0.500 g, 1.68 mmol) (89) and 3-amino-1,2,4-triazole (0.148 g, 1.77 mmol). Then using the intermediate sodium 6-(4-cyano-2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.400 g, 1.20 mmol) and POCl3 (3.280 g, 21.41 mmol) to obtain the title compound as a brown solid (0.210 g, 1.20 mmol, 53%). 1H NMR (600 MHz, CDCl3) δ 8.65 (s, 1H), 7.47 (d, J = 6.2 Hz, 2H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.21 (d, J = 6.4 Hz), 159.51 (d, J = 6.2 Hz), 157.66, 155.57, 116.85, 116.62 – 116.30 (m), 115.86 (t, J = 3.5 Hz), 114.64 (t, J = 19.6 Hz) ppm; IR (film) ν 3087, 2195, 1571, 1338, 1195 cm−1; LCMS: [M + H]+: 327.
4-(5,7-Dichloro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)-3-fluorobenzonitrile (107).
Following General Procedure B using diethyl 2-(4-cyano-2,6-difluorophenyl)malonate (0.500 g, 1.68 mmol) (90) and 3-amino-1,2,4-triazole (0.148 g, 1.77 mmol). Then using the intermediate sodium 6-(4-cyano-2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (3.000 g, 9.52 mmol) and POCl3 (26.000 g, 169.00 mmol) to obtain the title compound as a brown solid (1.970 g, 9.52 mmol, 67%). 1H NMR (600 MHz, CDCl3) δ 8.61 (s, 1H), 7.68 (dd, J = 7.9, 1.5 Hz, 1H), 7.61 (dd, J = 8.6, 1.5 Hz, 1H), 7.54 (t, J = 7.4 Hz, 1H) ppm;13C NMR (150 MHz, CDCl3) δ 160.47, 158.79, 157.48, 155.52, 153.79, 140.73, 133.32, 128.86 (d, J = 4.6 Hz), 124.68 (d, J = 15.7 Hz), 120.42 (d, J = 24.9 Hz), 116.76 (d, J = 2.9 Hz), 116.69, 116.36 (d, J = 9.3 Hz) ppm.
5,7-Dichloro-6-(4-chloro-2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (108).
Following General Procedure B using diethyl 2-(4-chloro-2,6-difluorophenyl)malonate (1.2 g, 3.91 mmol) (91) and 3-amino-1,2,4-triazole (0.350 g, 4.10 mmol). Then using the intermediate sodium 6-(4-chloro-2,6-difluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (1.000 g, 2.92 mmol) and POCl3 (7.970 g, 52.00 mmol) to obtain the title compound as a brown solid (0.710 g, 2.12 mmol, 73%). 1H NMR (600 MHz, CDCl3) δ 8.62 (s, 1H), 7.18 (d, J = 7.1 Hz, 2H) ppm; 13C NMR (150 MHz, CDCl3) δ 161.29 – 159.09 (m), 157.36, 156.45, 153.95, 141.55, 138.63 (t, J = 13.0 Hz), 113.78 – 113.35 (m), 112.29, 108.08 (t, J = 20.0 Hz) ppm; IR (film) ν 3106, 1642, 1427, 1391, 1071 cm−1; LCMS: [M + H]+: 336.
5,7-Dichloro-6-(2,6-difluoro-4-nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (109).
Following General Procedure B using diethyl 2-(2,6-difluoro-4-nitrophenyl)malonate (0.500 g, 1.58 mmol) (92) and 3-amino-1,2,4-triazole (0.139 g, 1.65 mmol). Then using the intermediate sodium 6-(2,6-difluoro-4nitrophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine-5,7-bis(olate) (0.150 g, 0.43 mmol) and POCl3 (1.160 g, 7.56 mmol) to obtain the title compound as a brown solid (0.100 g, 0.29 mmol, 68%). 1H NMR (600 MHz, CDCl3) δ 8.64 (s, 1H), 8.04 (d, J = 6.8 Hz, 2H) ppm; 13C NMR (150 MHz, CDCl3) δ 160.22 (dd), 157.70, 156.90, 155.39, 154.08, 150.51 (d, J = 10.5 Hz), 141.48, 116.12 – 115.19 (m), 111.63, 108.69 – 108.36 (m) ppm; IR (film) ν 2916, 2848, 1698, 1601, 1532 cm−1; LCMS: [M + H]+: 347.
Methyl 2-(5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-4,4,4-trifluoro-3-methylbutanoate (110).
To a solution of HMDS (0.101 g, 0.63 mmol, 1 equiv) in THF (0.632 mL) at −78 °C was added n-butyllithium (0.369 mL, 0.63 mmol, 1 equiv) and the mixture was stirred at 0 °C for 1 h. Then methyl 4,4,4-trifluoro-3-methylbutanoate (0.117 g, 0.69 mmol, 1.1 equiv) was added dropwise at −78 °C and the reaction mixture was stirred for 1 h. Then a solution of 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.200 g, 0.63 mmol, 1 equiv) (102) in THF (0.500 mL) was added at −78 °C and resulting mixture was stirred for 2.5 h prior to quenching of the reaction with 1M HCl. The aqueous layer was then extracted EtOAc (×2) and washed with sat. aq. NaCl, dried and concentrated under reduced pressure. Purification via silica gel chromatography using hexanes /EtOAc (5–20%) provided the product (0.200 g, 0.44 mmol, 70 %) as a white solid. 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.53 (d, J = 6.6 Hz, 1H), 6.94 – 6.86 (m, 2H), 4.04 (d, J = 7.4 Hz, 1H), 3.92 – 3.85 (m, 1H), 3.62 (d, J = 10.4 Hz, 3H), 1.38 (d, J = 6.6 Hz, 1H), 0.80 (d, J = 7.4 Hz, 2H) ppm; HRMS (ES+) calculated for C17H12ClF6N4O2 [M + H]+: 453.0547, found 453.0542.
7,9-Difluoro-10-(3,3,3-trifluoro-2-methylpropyl)benzofuro[2,3-d][1,2,4]triazolo[1,5-a]pyrimidine (111).
Methyl 2-(5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo-[1,5-a]pyrimidin-7-yl)-4,4,4-trifluoro-3-methylbutanoate (0.020 g, 0.04 mmol, 1 equiv) (110), LiCl (0.002 g, 0.04 mmol, 1 equiv), and DMSO (0.100 mL) were stirred at 130 °C for 3 h (microwave irradiation). The reaction mixture was then diluted in water and extracted with EtOAc (×3) and the combined organic layers were washed with brine (×2), dried over MgSO4, filtered, and concentrated. Purification by reverse-phase HPLC provided the title compound as a white solid (0.012 g, 0.03 mmol, 76%). X-ray quality crystals were obtained by slow evaporation from a CH2Cl2/Pentane solution: m.p. (CH2Cl2/Pentane) 103.4–107.4. 1H NMR (600 MHz, CDCl3) δ 8.55 (s, 1H), 8.01 (s, 1H), 7.01 (ddd, J = 11.0, 9.3, 2.2 Hz, 1H), 6.86 – 6.79 (m, 3H), 4.11 – 4.05 (m, 2H), 3.88 – 3.78 (m, 2H), 3.30 (p, J = 7.3 Hz, 1H), 3.20 (dd, J = 13.9, 5.8 Hz, 2H), 3.05 (dt, J = 15.1, 7.6 Hz, 2H), 2.79 (dd, J = 14.0, 8.7 Hz, 2H), 1.20 (d, J = 7.0 Hz, 3H), 1.02 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (150 MHz, CDCl3) δ 165.15 – 164.48 (m), 163.01 (d, J = 15.1 Hz), 162.26 – 160.00 (m), 158.41, 156.38, 152.47, 150.91, 145.88, 143.80, 127.47 (dd, J = 279.5, 28.1 Hz), 108.69, 106.96, 106.21 – 104.64 (m), 102.08 – 100.00 (m), 97.85 (dd, J = 27.4, 4.6 Hz), 36.01 (q, J = 28.3, 27.6 Hz), 30.84, 29.86 ppm; HRMS (ES+) calculated for C15H9F5N4O [M + H]+: 357.0769, found 357.0768.
tert-Butyl-2-(5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-4,4,4-trifluoro-3-methylbutanoate (112).
To a solution of HMDS (0.054 g, 0.33 mmol, 1.6 equiv) in THF (3 mL) at −78 °C was added n-butyllithium (0.180 mL, 0.33 mmol, 1.6 equiv) and the mixture was stirred at 0 °C for 30 min. Then tert-butyl 4,4,4-trifluoro-3-methylbutanoate (0.066 g, 0.31 mmol, 1.5 equiv) was added dropwise at −78 °C and the mixture was stirred at this temperature for 1 h. Finally, a solution of 5,7-dichloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (0.066 g, 0.21 mmol, 1 equiv) (102) in THF (0.500 mL) was added at −78 °C. After stirring for 2.5 h at this temperature, the reaction was quenched with a 1M HCl solution. The aqueous layer was then extracted twice with EtOAc and the combined organic layers were washed with brine, dried and concentrated under reduced pressure. Purification via silica gel chromatography using a hexanes/EtOAc gradient (95/05 to 80/20) provided tert-butyl 2-(5-chloro-6-(2,4,6-trifluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-4,4,4-trifluoro-3-methylbutanoate (0.026 g, 0.05 mmol, 25 %) as a white solid. 1H NMR (600 MHz, CDCl3) mixture of diastereomers δ 8.56 (s, 0.6H), 8.55 (s, 0.4H), 6.91 – 6.87 (m, 2H), 4.14 – 4.04 (m, 0.4H), 3.95 – 3.83 (m, 1.2H), 3.72 (d, 0.4H), 1.38 (d, J = 6.7 Hz, 1.3H), 1.27 (s, 3.5H), 1.25 (s, 6.8H), 0.80 (d, J = 7.0 Hz, 2H) ppm; IR (film) ν 2922, 2852, 1598, 1736, 1493, 1460, 1441, 1371, 1271, 1252, 1201, 1182, 1173, 1142, 1040 cm−1; HRMS (ES+) calculated for C20H18ClF6N4O2 [M + H]+: 495.1017, found 495.1010.
Computational Studies.
The X-ray crystal structure of the tubulin in complex with a triazolopyrimidine was downloaded from the Protein Data Bank and completed with MAESTRO (Schrödinger Release 2019–3),32 waters and other co-crystallised molecules were removed, except for the ligand and GDP. Predicting protonation states of protein residues was calculated considering a temperature of 300K and a pH of 7, while the molecules were prepared considering the ionization states at pH 7±2. A 12 Å docking grid (inner-box 10 Å and outer-box 20 Å) was prepared using as centroid the co-crystallised ligand. The docking studies were performed using Glide SP precision keeping the default parameters and setting, and it was combined with “molecular mechanics generalized Born surface area” (MMGBSA), implemented in the Prime module from Maestro, to re-score the 3 output docking poses of each compound. Only the best MMGBSA score and pose for each compound were plotted in the graph, and correlated with the normalized activity, expressed as the log of the average of the activity at 1 and 10 μM in the AcTub assay, relative to positive control (5).
The 3D-QSAR field-based and activity atlas model were constructed using Forge software (Cresset Inc., Cambridgeshire, UK).33 Firstly, the molecular structures of each compound were subjected to a field-based alignment to the co-crystallized compound, which was used as reference structure. This method is based on 3D-shape and electrostatic potential similarity calculated by the alignment and superposition between the reference compound and the compounds in the database according to their electrostatic distribution and volume occupied. Field point-based descriptors were used for building the 3D-QSAR model after the alignment of 52 Class I compounds with known activity. The activity was computed using the log of the average of the activity at 1 and 10 μM in the AcTub assay and defined as the dependent variable. The derived QSAR model was assessed by the leave-one-out (LOO) technique to optimize the activity-prediction model. Activity atlas was used to acquire qualitative information of the field and steric contributions significant for the activity.
Molecular Operating Environment (MOE) 2019.10 was used to visualize the structures and acquire the images,34 while GraphPad software35 and DataWarrior36 were used for the statistical analysis.
QBI293 Cell and neuronal Acetyl-tubulin and α-Tubulin Determinations.
Compound-induced changes in acetylated-tubulin and α-tubulin in QBI293 cells or primary mouse neurons was as previously described.5, 11 Briefly, QBI293 cells were maintained in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum (FBS), 2 mM L-glutamine (Mediatech), 50 units/ml penicillin, and 50 μg/ml streptomycin. For compound testing, cells were dissociated with trypsin/EDTA and plated at a density of 6 × 105 cells/well in 6-well plates. After overnight incubation, the medium was aspirated and fresh medium containing vehicle or test compound was added. After 4 h incubation, whole-cell extracts were prepared from the QBI293 cells as described.5, 11 The supernatant fraction from each sample was collected and analyzed for protein content by BCA assay.11 The acetyl-tubulin and α-tubulin enzyme-linked immunosorbent assays (ELISA) were performed on the supernatant fraction as previously described.11 The amount of acetyl- and α-tubulin protein in each sample was extrapolated using standard curves generated from serial dilutions with known acetyl- or α-tubulin preparations of known concentrations. Acetyl-tubulin levels were determined in a similar fashion in mouse cortical neuron cultures plated at 6 × 105 cells/well in 6-well plates, essentially as previously described for rat cortical neurons.11 After 10 days of growth, the neurons were treated for 8 hours with 15 nM okadaic acid in the presence or absence of test compound (1 or 10 μM) or vehicle (0.25% DMSO), and homogenates were prepared for determination of acetyl-tubulin levels as previously described.11
Acetyl-tubulin staining of rat cortical neuron cultures treated with test compounds in the absence or presence of okadaic acid.
Rat cortical neurons were grown in 24-well plates on coverslips for 10 days as previously described.11 The cultures were then treated for 8 hours with or without 15 nM okadaic acid in the presence or absence of test compounds (1 μM), with methods as described.11 Finally, the cultures were prepared for acetyl-tubulin staining as previously discussed.11
Determination of Plasma and Brain Drug Concentrations.
All animal protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). Test compounds were administered to groups of three 2–3-month old female CD-1 mice (Charles River). For standard single time-point brain and plasma determinations, mice were injected i.p. with a single dose of 5 mg/kg compound dissolved in DMSO. In some instances, mice were cassette-dosed via i.p. administration with 2–3 compounds concurrently, each at 2.5 mg/kg. One hour following compound administration, mice were euthanized following an IACUC-approved protocol. Whole brain hemispheres were homogenized in 10 mM ammonium acetate, pH 5.7 (50%, w/v), using a hand-held sonic homogenizer. Plasma was obtained from blood collected in 0.5 M EDTA solution and centrifuged for 10 min at 4,500 × g at 4 °C. Compound levels with plasma and brain homogenates were determined essentially as previously described10, 11 either in-house or at a contract laboratory (Inotiv, Inc.).
Supplementary Material
Acknowledgments.
Financial support for this work has been provided by the NIH/NIA (AG061173, AG044332).
Abbreviations Used:
- AcTub
acetylated α-tubulin
- m-CPBA
meta-chloroperbenzoic acid
- MMGBSA
molecular mechanics generalized Born surface-area
- MT
microtubule
- NFTs
neurofibrillary tangles
- NTs
neuropil threads
- Tg
transgenic
Footnotes
Associated Content: NMR spectra of test compounds; X-ray crystal structures of compounds 68 (CCDC 1995770) and 111 (CCDC 2003391), Authors will release the atomic coordinates and experimental data upon article publication; the SMILES string structures along the full data set in tabular form (csv file format). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lee VMY; Goedert M; Trojanowski JQ Neurodegenerative tauopathies. Annu. Rev. Neurosci 2001, 24, 1121–1159. [DOI] [PubMed] [Google Scholar]
- 2.Ballatore C; Lee VMY; Trojanowski JQ Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci 2007, 8, 663–672. [DOI] [PubMed] [Google Scholar]
- 3.Sadleir KR; Kandalepas PC; Buggia-Prevot V; Nicholson DA; Thinakaran G; Vassar R Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta neuropathol. 2016, 132, 235–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee VMY; Daughenbaugh R; Trojanowski JQ Microtubule stabilizing drugs for the treatment of Alzheimers-disease. Neurobiol. Aging 1994, 15, S87–S89. [DOI] [PubMed] [Google Scholar]
- 5.Brunden KR; Zhang B; Carroll J; Yao Y; Potuzak JS; Hogan AM; Iba M; James MJ; Xie SX; Ballatore C; Smith AB III; Lee VM; Trojanowski JQ Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci 2010, 30, 13861–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makani V; Zhang B; Han H; Yao Y; Lassalas P; Lou K; Paterson I; Lee VMY; Trojanowski JQ; Ballatore C; Smith AB III; Brunden KR Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol. Commun 2016, 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B; Carroll J; Trojanowski JQ; Yao Y; Iba M; Potuzak JS; Hogan AM; Xie SX; Ballatore C; Smith AB III; Lee VM; Brunden KR The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci 2012, 32, 3601–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barten DM; Fanara P; Andorfer C; Hoque N; Wong PYA; Husted KH; Cadelina GW; DeCarr LB; Yang L; Liu L; Fessler C; Protassio J; Riff T; Turner H; Janus CG; Sankaranarayanan S; Polson C; Meredith JE; Gray G; Hanna A; Olson RE; Kim SH; Vite GD; Lee FY; Albright CF Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027 J. Neurosci 2012, 32, 7137–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballatore C; Brunden KR; Trojanowski JQ; Lee VM; Smith AB III. Non-naturally occurring small molecule microtubule-stabilizing agents: a potential tactic for CNS-directed therapies. ACS Chem. Neurosci 2017, 8, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou K; Yao Y; Hoye AT; James MJ; Cornec AS; Hyde E; Gay B; Lee VM; Trojanowski JQ; Smith AB III; Brunden KR; Ballatore C Brain-penetrant, orally bioavailable microtubule-stabilizing small molecules are potential candidate therapeutics for Alzheimer’s disease and related tauopathies. J. Med. Chem 2014, 57, 6116–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovalevich J; Cornec AS; Yao Y; James M; Crowe A; Lee VM; Trojanowski JQ; Smith AB; Ballatore C; Brunden KR Characterization of brain-penetrant pyrimidine-containing molecules with differential microtubule-stabilizing activities developed as potential therapeutic agents for Alzheimer’s disease and related tauopathies. J. Pharmacol. Exp. Ther 2016, 357, 432–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B; Yao Y; Cornec A-S; Oukoloff K; James MJ; Koivula P; Trojanowski JQ; Smith AB III; Lee VM-Y; Ballatore C; Brunden KR A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol. Neurodegener 2018, 13, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sáez-Calvo G; Sharma A; Balaguer F. d. A.; Barasoain I; Rodríguez-Salarichs J; Olieric N; Muñoz-Hernández H; Berbís MÁ; Wendeborn S; Peñalva MA; Matesanz R; Canales Á; Prota AE; Jímenez-Barbero J; Andreu JM; Lamberth C; Steinmetz MO; Díaz JF Triazolopyrimidines are microtubule-stabilizing agents that bind the vinca inhibitor site of tubulin. Cell Chem. Biol 2017, 24, 737–750 e6. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y; Nzou G; Alle T; Tsering W; Maimaiti S; Trojanowski JQ; Lee VM; Ballatore C; Brunden K Correction of microtubule defects within Aβ plaque-associated dystrophic axons results in lowered Aβ release and plaque deposition. Alzheimer’s Dement. 2020, 16, 1345–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang N; Ayral-Kaloustian S; Nguyen T; Afragola J; Hernandez R; Lucas J; Gibbons J; Beyer C Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J. Med. Chem 2007, 50, 319–327. [DOI] [PubMed] [Google Scholar]
- 16.Zhang N; Ayral-Kaloustian S; Nguyen T; Hernandez R; Lucas J; Discafani C; Beyer C Synthesis and SAR of 6-chloro-4-fluoroalkylamino-2-heteroaryl-5-(substituted)phenylpyrimidines as anti-cancer agents. Bioorg. Med. Chem 2009, 17, 111–118. [DOI] [PubMed] [Google Scholar]
- 17.Ayral-Kaloustian S; Zhang N; Beyer C Cevipabulin (TTI-237): preclinical and clinical results for a novel antimicrotubule agent. Methods Find. Exp. Clin. Pharmacol 2009, 31, 443–447. [DOI] [PubMed] [Google Scholar]
- 18.Wang-Gillam A; Arnold SM; Bukowski RM; Rothenberg ML; Cooper W; Wang KK; Gauthier E; Lockhart AC A phase I dose escalation study of TTI-237 in patients with advanced malignant solid tumors. Invest. New Drugs 2012, 30, 266–272. [DOI] [PubMed] [Google Scholar]
- 19.Fukushima N; Furuta D; Hidaka Y; Moriyama R; Tsujiuchi T Post-translational modifications of tubulin in the nervous system. J. Neurochem 2009, 109, 683–693. [DOI] [PubMed] [Google Scholar]
- 20.Cornec AS; James MJ; Kovalevich J; Trojanowski JQ; Lee VM; Smith AB III; Ballatore C; Brunden KR Pharmacokinetic, pharmacodynamic and metabolic characterization of a brain retentive microtubule (MT)-stabilizing triazolopyrimidine. Bioorg. Med. Chem. Lett 2015, 25, 4980–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez L; Massari ME; Vickers T; Freestone G; Vernier W; Ly K; Xu R; McCarrick M; Marrone T; Metz M; Yan YG; Yoder ZW; Lemus R; Broadbent NJ; Barido R; Warren N; Schmelzer K; Neul D; Lee D; Andersen CB; Sebring K; Aertgeerts K; Zhou X; Tabatabaei A; Peters M; Breitenbucher JG Design and synthesis of novel and selective phosphodiesterase 2 (PDE2a) inhibitors for the treatment of memory disorders. J. Med. Chem 2017, 60, 2037–2051. [DOI] [PubMed] [Google Scholar]
- 22.Krapcho PA; E., C. The Krapcho dealkoxycarbonylation reaction of esters with α-electron-withdrawing substituents. Org. React 2013, 81, 1–536. [Google Scholar]
- 23.Oukoloff K; Kovalevich J; Cornec AS; Yao Y; Owyang ZA; James M; Trojanowski JQ; Lee VM; Smith AB III; Brunden KR; Ballatore C Design, synthesis and evaluation of photoactivatable derivatives of microtubule (MT)-active [1,2,4]triazolo[1,5-a]pyrimidines. Bioorg. Med. Chem. Lett 2018, 28, 2180–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang E; Sun H; Wang J; Wang Z; Liu H; Zhang JZH; Hou T End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev 2019, 119, 9478–9508. [DOI] [PubMed] [Google Scholar]
- 25.Tosco P; Mackey M Lessons and successes in the use of molecular fields. In Comprehensive medicinal chemistry III; Chackalamannil S, Rotella D, Ward SE, Eds.; Elsevier: 2017; Vol. 3–8, p 253–296. [Google Scholar]
- 26.Ballatore C; Brunden KR; Huryn DM; Trojanowski JQ; Lee VMY; Smith AB III Microtubule stabilizing agents as potential treatment for alzheimer’s disease and related neurodegenerative tauopathies. J. Med. Chem 2012, 55, 8979–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beyer CF; Zhang N; Hernandez R; Vitale D; Lucas J; Nguyen T; Discafani C; Ayral-Kaloustian S; Gibbons JJ TTI-237: a novel microtubule-active compound with in vivo antitumor activity. Cancer Res. 2008, 68, 2292–300. [DOI] [PubMed] [Google Scholar]
- 28.Hansch C; Leo A; Taft RW A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- 29.Activity Atlas, Forge, Version 10.6.0; Cresset: Litlington, Cambridgeshire, https://www.cresset-group.com/software/forge/ (accessed November 17, 2020). [Google Scholar]
- 30.Stumpfe D; Bajorath J Exploring activity cliffs in medicinal chemistry. J. Med. Chem 2012, 55, 2932–2942. [DOI] [PubMed] [Google Scholar]
- 31.Fleming FF; Yao L; Ravikumar PC; Funk L; Shook BC Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J. Med. Chem 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrödinger L Schrödinger, LLC, New York, NY, 2019. https://www.schrodinger.com (accessed Nov 17, 2020). [Google Scholar]
- 33.3D QSAR, Forge, Version 10.6.0; Cresset: Litlington, Cambridgeshire: https://www.cresset-group.com/software/forge/ (accessed Nov 17, 2020). [Google Scholar]
- 34.Molecular Operating Environment (MOE), Montreal, QC, Canada, H3A 2R7, 2019. http://www.chemcomp.com (acessed Nov 17, 2020). [Google Scholar]
- 35.GraphPad Prism Version 8.00 for Windows, GraphPad Software, La Jolla California USA, https://www.graphpad.com (acessed Nov 17, 2020). [Google Scholar]
- 36.Sander T; Freyss J; Von Korff M; Rufener C DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model 2015, 55, 460–473. [DOI] [PubMed] [Google Scholar]
- 37.Brunden KR; Yao Y; Potuzak JS; Ferrer NI; Ballatore C; James MJ; Hogan AM; Trojanowski JQ; Smith AB III; Lee VM The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol. Res 2011, 63, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.