

Abstract
Identifying previously unknown proteins or detecting the presence of known proteins in research samples is critical to many experiments conducted in life sciences, including dermatology. Sensitive protein detection can help to elucidate new intervention targets and mechanisms of disease, such as in autoimmune blistering skin diseases, atopic eczema, or other conditions. Historically, peptides from highly purified, single proteins were sequenced, with many limitations, by stepwise degradation from the N-terminus to the C-terminus with subsequent identification by UV absorbance spectroscopy of the released amino acids (i.e. Edman degradation). Recently, however, the availability of comprehensive protein databases from different species (derived from high-throughput next-generation sequencing of those organisms’ genomes) and sophisticated bioinformatics analysis tools have facilitated the development and use of mass spectrometry (MS) for identification and global analysis of proteins, summarized as MS-based proteomics. MS is an analytical technique measuring the mass (m) to charge (z) ratio of ionized biological molecules such as peptides. Proteins can be identified by correlating peptide-derived, experimental MS spectra with theoretical spectra predicted from protein databases. Here we briefly describe how this technique works, how it can be used for identification of proteins, and how this knowledge can be applied in elucidating human biology and disease.
Keywords: LC-MS/MS, mass spectrometry, protein sequencing, proteomics, pemphigus
Introduction
Basic dermatological research employing genetic and cellular techniques has resulted in significant advances allowing for precise diagnosis and optimized therapy of skin disease, as illustrated for autoimmune blistering diseases (Kasperkiewicz et al., 2017). Only recently has a more global proteomic picture in dermatologic (and other) conditions emerged, allowing new insights of clinical relevance. For example for pemphigus vulgaris, it was shown how various monoclonal anti-desmoglein 3 autoantibodies (ab) contribute to the polyclonal serum response, and how the amount of each monoclonal ab changes over the course of disease (Chen et al., 2017). In another study, proteomics was used to identify differentially expressed proteins relevant to filaggrin-deficient atopic eczema (Elias et al., 2017), potentially yielding new therapeutic targets. Additionally, previously unknown interaction partners of autoantibodies in dermatologic and other autoimmune conditions were successfully identified by proteomics (Miske et al., 2016, Schepens et al., 2010).
In this review we focus on use of liquid chromatography tandem mass spectrometry (LC-MS/MS) for protein identification because it is currently the most practical means of direct and global protein identification (Domon and Aebersold, 2006).
MS-based proteomics consist of the following stages, which will be briefly described: i) Isolation of the protein sample; ii) Mass-spectrometric analysis; and iii) Analysis and interpretation of MS data using bioinformatic tools. The general steps of a typical LC-MS/MS experiment are summarized in Figure 1.
Figure 1. General steps of a typical LC-MS/MS experiment.
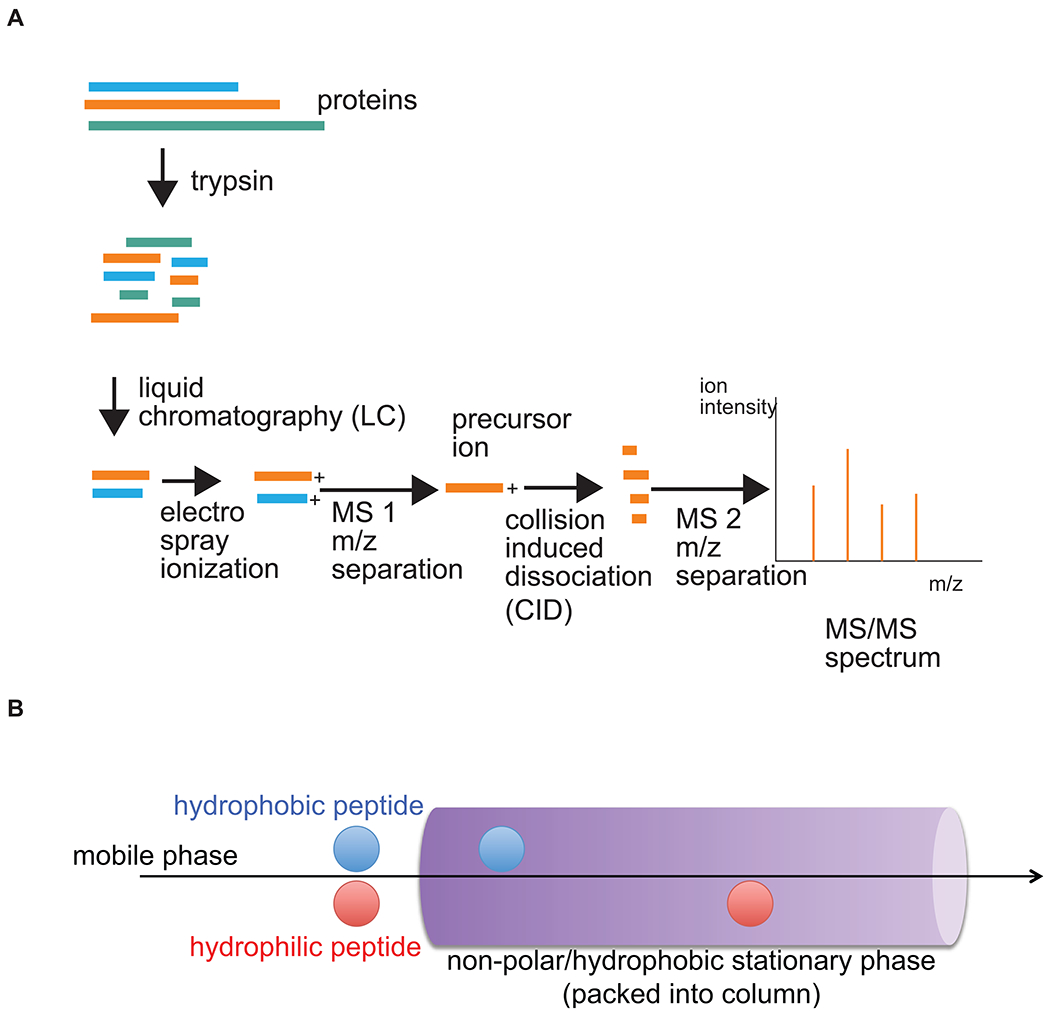
A) After isolation during the experiment of interest, proteins are treated with proteolytic enzymes (e.g. trypsin), then subjected to liquid chromatography (LC; explained in B)). Separated peptides are then ionized (i.e. by exposing drops of peptide-containing eluate from LC to a strong electric field, an atomic gas is formed) and separated by their mass (m)-to-charge (z) ratios in the first mass spectrometer (MS1). Precursor ions of a given m/z are then further fragmented by collision-induced dissociation (CID) and the ion fragments separated again (MS2). Resulting fragment ion spectra are recorded and analyzed as detailed in the text. The basic principle of reverse-phase LC is illustrated in B); the most hydrophobic peptides interact best with the non-polar stationary phase, whereas the least hydrophobic components elute first. Complete elution off the column, including the most non-polar peptides, is ensured by gradually increasing the concentration of non-polar solvents in the mobile phase.
Isolation and Fractionation of the Protein Sample of Interest
A major advantage of LC-MS/MS is that it can identify unknown proteins. Potential sources of such proteins are theoretically unlimited and depend on the research question under investigation. For example, unknown reaction partners of antibodies (e.g. autoantigens in autoimmune diseases) can be immunoprecipitated from cell lysates and subjected to LC-MS/MS for identification and validation (Miske et al., 2016, Schepens et al., 2010). A single protein band can be stained after separation by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), cut out of the gel, digested, and subjected to analysis by LC-MS/MS. However, samples of higher complexity can be studied (and compared) as well to gain a more global view of proteins expressed, under stable or different experimental conditions, or at different time points over the course of disease. Examples of use of this type of analysis include plasma membrane isolates from whole keratinocyte lysates (Blonder et al., 2004), homogenized human epidermal living skin-equivalents in atopic eczema (Elias et al., 2017), or affinity-purified autoantibodies from serum (Chen et al., 2017). These examples are discussed in more detail below.
Since accurate sequence assignment of MS/MS spectra can only be achieved for short linear peptides (approx. 7-50 aa residues), the purified (often SDS-PAGE-separated) proteins are usually treated with proteolytic enzymes (e.g. trypsin) before loading them into the LC-MS/MS instrument (Fig. 1A). To allow for higher resolution in LC-MS/MS, the digested peptide pools are first separated on a LC column, usually by reversed-phase chromatography: This separation technique is based on a column with a hydrophobic stationary phase, with high affinity for hydrophobic peptides (Fig. 1B). By applying a mobile phase that consists of an increasing gradient of non-polar solvents over polar solvents (e.g. acetonitrile over water) with time, hydrophilic peptides are eluted first and hydrophobic peptides last. This elution can take place over time (such as 1-2 hours), with the resultant eluate continuously loaded into the MS/MS analyzer.
Mass-spectrometric Analysis
The instrument utilized for MS analysis consists of an ionizer, a mass analyzer, and a detector. MS analysis of peptides (usually derived from trypsinization of a protein) after ionization is based on their migration in an electromagnetic field, which is a function of their mass (m) and charge (z). To reliably differentiate distinct peptides with equal mass and charge (i.e. with same m/z), reversed-phase LC is used first because such peptides will most likely elute at different retention times through the LC column, based on their hydrophobicity (Fig. 1B). As each peptide comes off the column it is ionized and analyzed in the first mass analyzer of a tandem-in-space mass spectrometer. Then each precursor peptide with a defined m/z is fragmented by collision-induced dissociation (CID) in a collision cell (Fig. 1A). The resulting fragment ions of that precursor ion are then analyzed in a second mass analyzer, and a fragment ion spectrum is recorded. Alternatively, a tandem-in-time mass spectrometer can perform both MS scans in one trapping mass analyzer. This process is repeated throughout the LC separation process to allow aa sequence determination of most of the peptides in the digest (Boström, 2014). During CID fragmentation the most common bonds cut are the peptide bonds (Fig. 2). From each cleavage two ions result, the C-terminal fragment, called the y-ion and the N-terminal fragment, called the b-ion. To be detected by MS, the fragments must be charged. The usual site of charge is at the cleaved peptide bond of the fragment which results in one charge (i.e. z=1), but additional charges may be introduced on other parts of the peptide (z>1), such as the amino group side chain of lysine and arginine, and the imidazole ring of histidine. For ions with the same charge, the differences between the peaks in the ion spectrum measures the difference in mass of the ions. Since the mass of each aa is known, the aa cleaved off the peptide by CID can be deduced from the loss of that given mass, allowing sequence determination (Figures 3A and 3B).
Figure 2. Peptide fragmentation by CID.
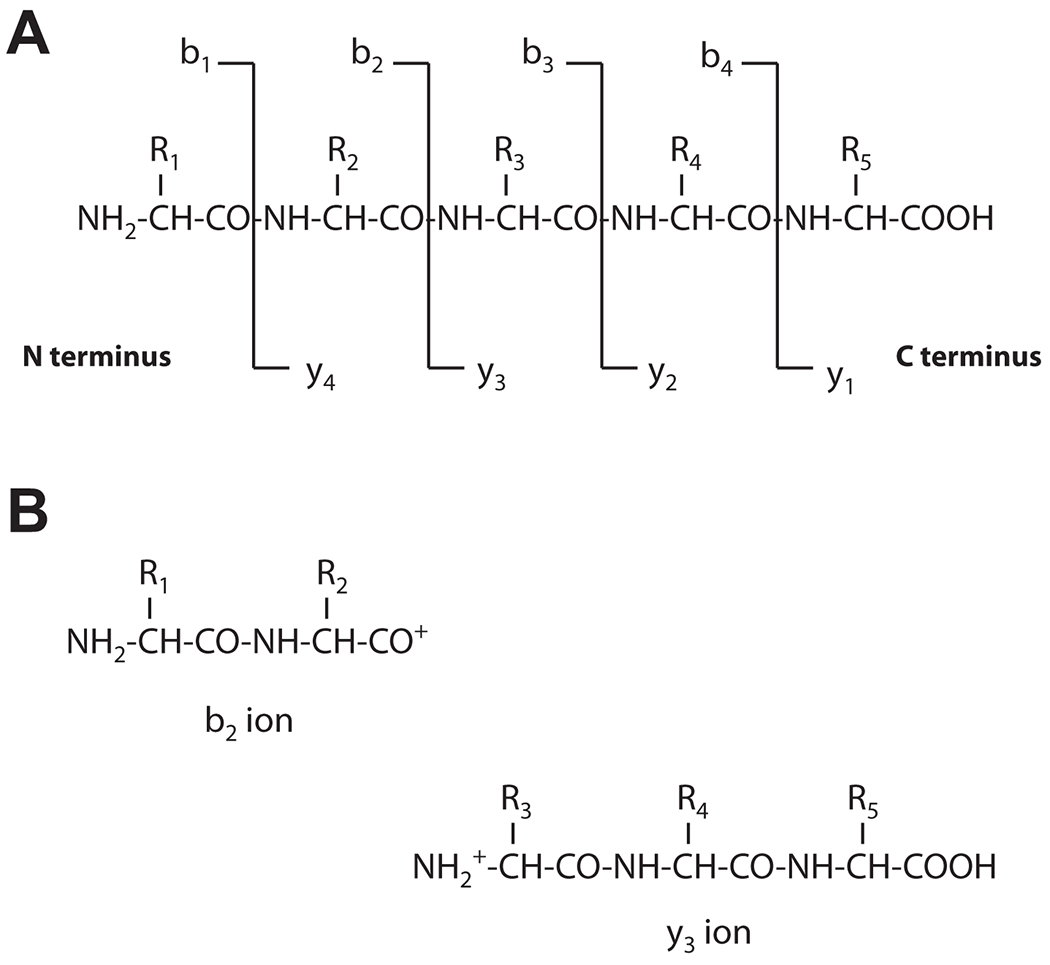
An example of fragmentation for a 5 aa long peptide with amino acid residues R1 to R5 is shown in (A). During CID, peptides usually break at the peptide bond (CO-NH). B) Resulting peptides are termed b ion (charged N-terminal fragment, shown on the left) or y ion (charged C-terminal fragment, shown on the right). The symbol “+” represents a proton. Peptides can also break at positions other than the peptide bonds, resulting in the a/x and the c/z series ions (not shown).
Figure 3. Basic concept of interpretation of LC-MS/MS spectra.
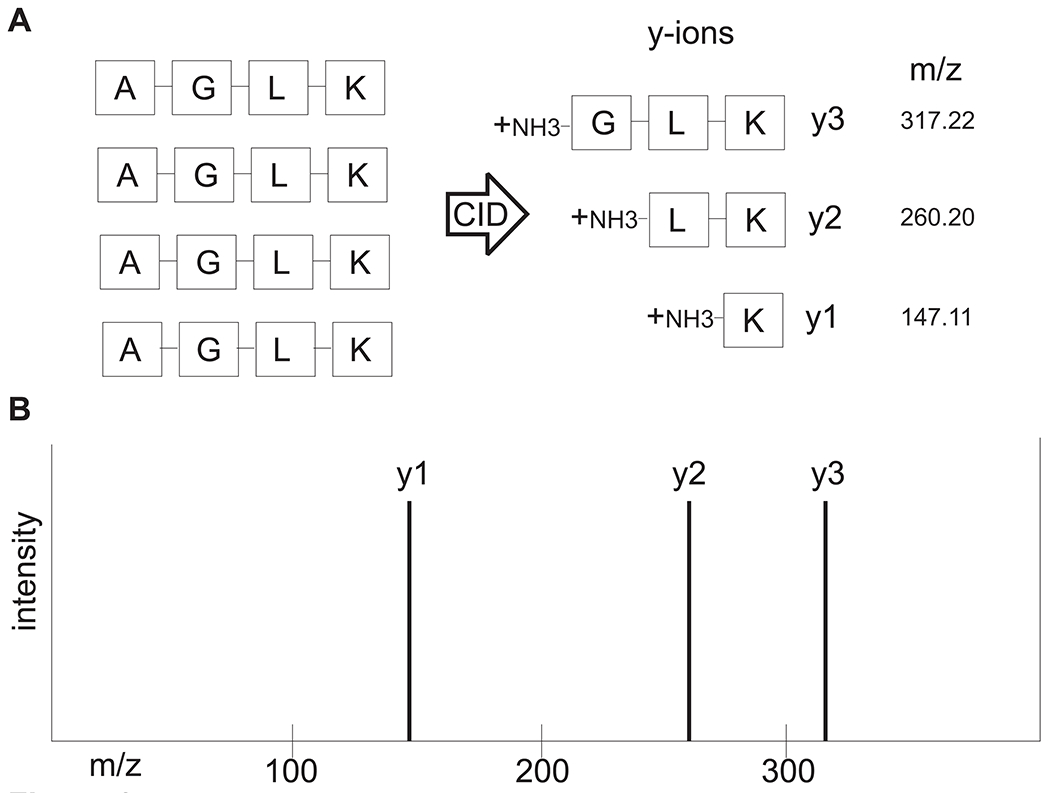
(A) A precursor peptide consisting of amino acids Alanine-Glycine-Leucine-Lysine is fragmented by CID into b and y ions with mass-to-charge ratios (m/z). For the sake of simplicity, only y ions resulting from CID are shown. The aa sequence can now be deduced from the idealized ladder of y ions, as shown in (B). The mass difference between y3 and y2 is 57.02 (which is the residue mass of glycine), and the mass difference between y2 and y1 is 113.09 (which is the residue mass of leucine).
Bioinformatic Analysis and Interpretation of MS-data
As resulting fragment ion spectra rarely contain all possible fragment ions and de novo interpretation of such spectra is time consuming and error prone, experimental spectra are searched, by bioinformatic means and with the help of search engines such as Mascot, Sequest or Andromeda, against theoretical spectra generated from in silico digestion of theoretical input proteins to identify matches of experimental to theoretical spectra. If those input proteins are unknown, public databases that include all known relevant proteins (e.g. all human proteins, see Fig. 4 as an application example) can be used by the software to generate theoretical spectra (see www.uniprot.org for an example of such databases, and (Magrane and UniProt, 2011)). In some studies, such as studies of antibodies which differ in each individual, custom-made databases must be produced. For example, next-generation sequencing of B cell-derived transcripts coding for antibodies can be used to deduce a database of possible antibody aa sequences (Figure 5). Peptides identified by matching the LC-MS/MS spectra to such databases are reported with a probability score as a measure of the reliability of their identification (Boström, 2014). By spiking in internal standards to the sample being analyzed, confirmation of the spectra identifying that particular peptide, and even its absolute quantitation, becomes possible. This is accomplished by using synthetic heavy isotope-labeled peptides with the same sequence as the deduced light peptide from the sample (Domon and Aebersold, 2006).
Figure 4. Cell adhesion proteins identified from the keratinocyte plasma membrane by in-solution LC-MS/MS.
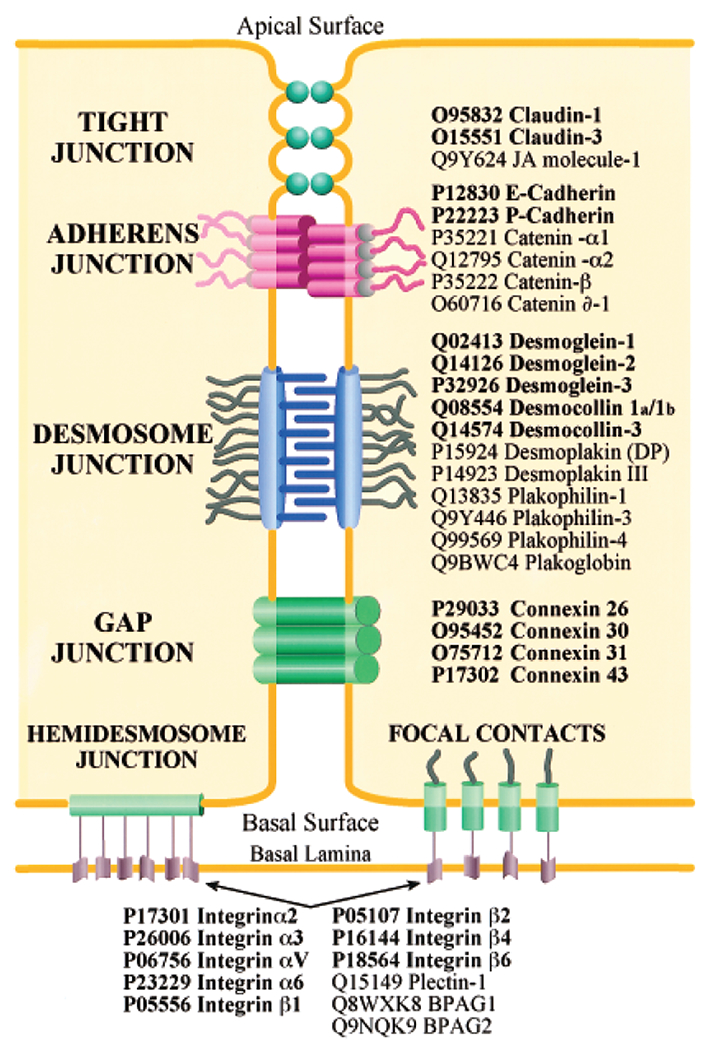
Transmembrane linkers are printed in bold font and attachment proteins in normal font. Shown are proteins of tight junctions, adherens junctions, desmosomes, gap junctions, hemidesmosomes, and focal contacts. Reprinted with permission from Blonder et al. (2004).
Figure 5. Use of LC-MS/MS to identify circulating pemphigus anti-desmoglein (dsg) antibodies (abs).
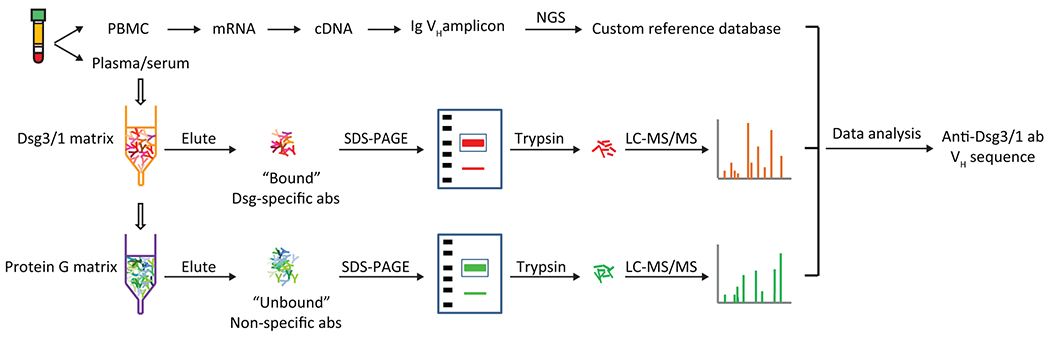
IgG heavy chains from dsg-binding abs and from abs which do not bind to dsg are analyzed by LC-MS/MS. Resultant spectra are compared against a custom database of all variable heavy chain (VH) amino acid (aa) sequences from the same patient to identify ab peptides (for database construction, VH-mRNA transcripts were PCR amplified, sequenced by high-throughput sequencing, and translated into aa to create a VH-specific database of that patient). Peptides that match heavy chain-complementarity region 3 amino acid sequences in the database and that are found only in the bound, but not the unbound pool are informative, allowing definition of the specific clonotype profile in the antigen-specific (dsg-binding) population. NGS, next-generation sequencing; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis. Taken under the CC BY-NC-ND license and under retained rights of the authors from reference Chen et al. (2017).
Examples of Applications of Mass Spectrometry in Immunology and Investigative Dermatology
In an attempt to globally characterize plasma membrane proteins of human epidermal keratinocytes, LC-MS/MS has been successfully used to identify 496 proteins, including many of those already previously identified (by genetic methods, immunoprecipitation, and other approaches), thus independently confirming their expression and membrane localization (Blonder et al., 2004) (Figure 4). LC-MS/MS of whole epidermis extracts has led to identification of new proteins potentially relevant to the pathogenesis (and, importantly, novel therapeutic options) of atopic eczema by comparing normal to filaggrin-deficient skin (of note, expression changes of some of those proteins were not correlated to changes in mRNA expression profiles, highlighting the importance of complementing genetic analyses using proteomic approaches) (Elias et al., 2017).
The identification of the p170 paraneoplastic antigen as alpha-2-macroglobulin-like-1 was possible by analyzing an unknown band in a SDS-PAGE gel following immunoprecipitation (Schepens et al., 2010). Similarly, in a neurologic patient with a suspected autoimmune condition, serum was incubated with cryosections of nerve tissue, followed by extraction and precipitation of immunocomplexes and LC-MS/MS, resulting in identification of the autoantigen (Miske et al., 2016). By using skin cryosections, this histoimmunoprecipitation approach could be translated to a subgroup of dermatological patients that show skin blisters and bound, keratinocyte-specific autoantibodies in direct immunofluorescence studies on skin biopsies, but that do not show reactivity of serum autoantibodies by routine ELISA or blotting techniques with any of the major keratinocyte autoantigens described and extensively validated so far (e.g. desmogleins 3/1, BP180/230, laminin 332, collagen VII), resulting in diagnostic difficulties and uncertain final diagnoses (Giurdanella et al., 2016, and personal observation).
LC-MS/MS also allows characterization of serum antibodies (abs). Antibody responses have historically been analyzed mostly by genetic studies of the B cells that encode the antibodies, while few studies have characterized circulating antibodies (Wine et al., 2015). This is an important distinction because, while B cells may or may not differentiate to secrete abs, the actual circulating abs are what protect from infection or result in autoantibody-mediated disease. It is possible to affinity purify most, or all, of the serum abs, but it is much more difficult to comprehensively analyze antigen-specific B cells. Using LC-MS/MS to characterize peptides encompassing the heavy chain complementarity determining region 3 (H-CDR3) of serum antibodies (which defines the antibody’s B-cell clonal origin), researchers were recently able to characterize the serum ab response after immunization (Lavinder et al., 2014, Lee et al., 2016, Wine et al., 2013). Using similar techniques we have characterized the autoantibody response in human patients with pemphigus, a prototypic organ-specific autoimmune disease with serum abs against desmogleins (Dsgs) (Chen et al., 2017). Contributing to new insights of pathophysiology, we showed that the serum autoantibody repertoire in pemphigus was much more diverse and dynamic than had previously been indicated by genetic studies of B cells (Figure 6): We found, studying the same patients’ abs genetically and by proteomics, that most serum abs are not identified by genetic cloning methods, and conversely, that many genetically identified clones are not identified as serum abs. We also demonstrated, by proteomics, that, although the anti-Dsg response is polyclonal, that a dominant few clones produce most of the circulating serum abs, and that individual serum ab clones can persist in patients over years, with variations in their expression levels. The latter finding may explain why anti-Dsg ELISA titers do not always correlate with observed clinical disease activity, because under the same total titer the serum distribution of pathogenic to non-pathogenic ab clones may change.
Figure 6. Use of LC-MS/MS to trace anti-desmoglein clonotypes over time.
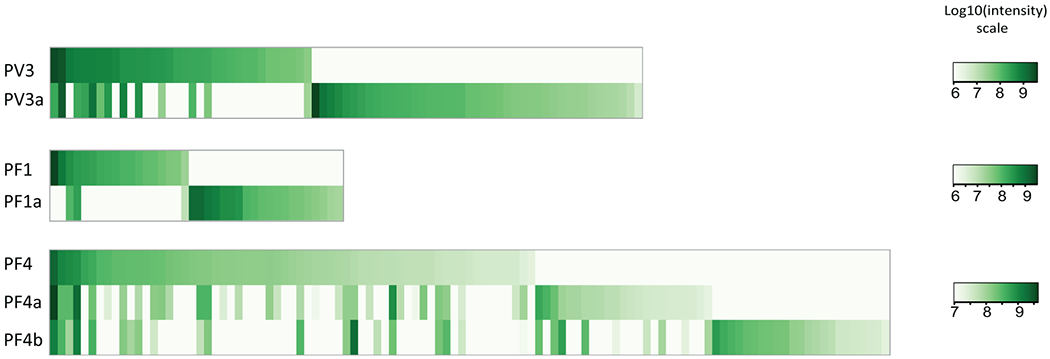
Each green column represents one antibody clone, whereas the color intensity represents the expression level of any given clone at one point in time. These data indicate that in both a pemphigus vulgaris patient (PV3 at first time point, PV3a at second time point 6 years later) and in two pemphigus foliaceus patients (PF1/1a, PF4/4a/4b) some antibody clonotypes persist, with varying antibody production (vertical comparison), and that the overall landscape of clones changes over time (columns only found at one, but not at another time point). This finding can explain the clinical observation that ELISA anti-desmoglein titers do not always correlate with disease activity, presumably because of differential expression of pathogenic and non-pathogenic antibodies. Taken under the CC BY-NC-ND license and under retained rights of the authors from reference Chen et al. (2017).
Limitations
As outlined, LC-MS/MS is a powerful and versatile technique that directly identifies proteins/peptides produced by cells. By contrast, immunohistochemistry approaches need well-characterized antibodies against known proteins, and RNA sequencing or microarray techniques identify genetic sequences that may or may not be produced as proteins in cells.
This field is advancing and improving rapidly, but there remain substantial limitations that should be taken into account, as highlighted in the following (not comprehensive) examples. In most cases, MS protein identification involves enzymatic digestion of protein samples into peptides, and subsequent analysis of the resulting peptides by tandem MS. This peptide-centric approach results in the fundamental issue of protein inference especially for complex proteomes. The presence of degenerate peptides, i.e. identical peptide sequences that are found in multiple homologous proteins or protein isoforms, makes it difficult to accurately reassemble peptides to proteins for identification. The protein inference issue is exacerbated by significant protein sequence redundancy in databases that is caused by polymorphisms and DNA sequencing errors that produced partial or nearly identical sequences. Therefore, it can be difficult to determine whether all related protein isoforms are present in a sample or only some are truly present, and it is important to differentiate those because these related isoforms often have distinct structural or functional roles in vivo.
Current quantitative LC-MS/MS proteomic approaches also rely on the assumption that proteins are completely digested into peptides that are all reproducibly detected by MS analysis. In practice, this is not always true as incomplete digestion or recovery can occur, and unpredictable interferences from sample matrix can result in ion suppression or variable peptide signal intensity. In addition, peptide bonds have different structural labilities, and fragmentation may thus be skewed towards more labile bonds such as the N-terminal side of proline, resulting in poor MS/MS spectra that are difficult to interpret. Finally, some aa have the same mass (Leu, Ile) or nearly exactly half of the mass of others (Gly with 57.02146 Da vs. Asn with 114.04293 Da), and peptides containing combinations of these residues will be impossible to distinguish, resulting in ambiguous sequence assignment. These examples point to the need for skilled scientists trained specifically in proteomic analysis. Successful proteomics analysis requires collaborative efforts between cell biologists, biochemists, and bioinformaticians.
Summary points.
What mass spectrometry (MS) for analysis of proteins does:
Enables direct analysis of protein amino acid sequences, allowing for identification of unknown proteins (e.g. new autoantigens in disease)
Enables analysis of changes in global protein expression, for example in epidermis or other organs, under different experimental conditions
Limitations:
Limits in detection of proteins in very complex samples, requiring reduction in complexity of samples of interest (e.g. by affinity purification)
Non-detection of a protein of interest in complex samples does not exclude presence of the protein, and detection of a peptide characteristic for one protein may not be specific for this protein as peptides can be shared between proteins (i.e. protein interference)
Experienced bioinformaticians are needed to interpret the results
5 Multiple choice questions (and answers).
-
In analysis and interpretation of MS/MS data, which of the following statements is not correct? (C)
- Theoretical MS/MS spectra are generated from in silico analysis of predicted digestion products of known proteins
- To interpret MS spectra of human antibodies (and their clonalities), a custom database or de novo interpretation is required
- Precursor ion spectra are correlated with theoretical MS/MS spectra generated from protein databases
- Public protein databases serve as input to guide in silico analysis of proteins into predicted digestion products
Explanation: Precursor ion spectra are not correlated with theoretical MS/MS spectra generated from protein databases, but fragment ion spectra, which stem from collision-induced fragmentation of precursor ions. All other answers are correct.
-
In the fields of immunology and investigative dermatology, LC-MS/MS has been successfully used to… (D)
- …identify an unknown protein from immunoprecipitation
- …characterize plasma membrane proteins within the human epidermis
- …characterize the circulating antibody response in an autoimmune disease and/or after immunization
- All of the above
Explanation: All of the above answers are correct. References to the above examples can be found within the manuscript.
-
Which of the following statements about potential limitations of LC-MS/MS is not correct? (B)
- Ambiguity in protein inference can be introduced by the use of proteolytic enzymes and by redundancy phenomena in the databases used for comparison
- Unknown proteins can be easily identified without the use of protein databases
- Different structural labilities of peptide bonds can make interpretation of MS/MS spectra difficult
- De novo interpretation of fragment ion spectra is time consuming and error prone
Explanation: B) is not correct, because for a seamless identification of unknown proteins a protein database is of great help, providing the input for in silico generation of theoretical peptide spectra that can be matched computationally to the experimental spectra obtained. Some advances have been made towards de novo interpretation of fragment ion spectra, but this approach is error prone and not used in routine proteomic analyses.
-
What are the main underlying principles that allow for separation of peptides by reverse-phase liquid chromatography (LC) and mass spectrometry (MS), respectively? (D)
- Peptide hydrophobicity, only the charge of the peptide
- Peptide’s charge and mass in both LC and MS
- Only the mass in LC, the mass and the charge in MS
- Peptide hydrophobicity, the mass and the charge of the peptide
Explanation: Reverse-phase liquid chromatography is mainly based on the hydrophobicity of peptides; the non-polar stationary phase interacts well with hydrophobic peptides and leads to long retention times before these get finally eluted. In mass spectrometry, the mass (m) and the charge (z), summarized as the mass-to-charge ratio m/z, are critical variables for separation of peptides in electrical fields.
-
Which of the following statements is correct? (D)
- The proteolytic enzyme trypsin cuts proteins after amino acids arginine and lysine
- The heavy chain complementarity determining region 3 (H-CDR3) is a unique identifier of an antibody and can be detected by LC-MS/MS experimentation
- Collision-induced dissociation (CID) describes fragmentation of precursor ions in a collision cell and does not always result in all potential fragment ions (e.g., b/y ions) of a given peptide
- All of the above
Explanation: D is correct. Trypsin normally cuts after amino acids arginine and lysine. However, sequences of Arg-Pro and Lys-Pro are resistant to trypsin cleavage. The H-CDR3 is a unique “fingerprint” of an antibody and defines a clone (and its clonal progeny and ancestry). In CID, b and y ions are typically observed, the alternative a/x and c/z series mainly occur with fragmenting techniques different from CID.
Acknowledgements
This work was supported by grants from the National Institutes of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health (J.R.S., R01-AR052672); grants from the DFG (C.M.H. and S.E., GRK1727); support from the Section of Medicine at the University of Luebeck (J03-2015) to C.M.H.; National Cancer Institute grant R50CA221838 to H.-Y. T.; and support of the Wistar Proteomics and Metabolomics Core Facility was provided by Cancer Center Support Grant CA010815 to the Wistar Institute.
Abbreviations used:
- aa
amino acid
- CID
collision-induced dissociation
- dsg
desmoglein
- H-CDR3
heavy chain complementarity determining region 3
- LC
liquid chromatography
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- m
mass
- MS
mass spectrometry
- NGS
next-generation sequencing
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- UV
ultraviolet light
- z
charge
Footnotes
Conflict of Interest
The authors do not have any conflicts of interest.
References
- Blonder J, Terunuma A, Conrads TP, Chan KC, Yee C, Lucas DA, et al. A proteomic characterization of the plasma membrane of human epidermis by high-throughput mass spectrometry. The Journal of investigative dermatology 2004;123(4):691–9. [DOI] [PubMed] [Google Scholar]
- Boström T High-throughput protein analysis using mass spectrometry-based methods. School of Biotechnology, Division of Protein Technology: KTH Royal Institute of Technology; 2014. [Google Scholar]
- Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang HY, et al. Proteomic Analysis of Pemphigus Autoantibodies Indicates a Larger, More Diverse, and More Dynamic Repertoire than Determined by B Cell Genetics. Cell reports 2017;18(1):237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Aebersold R. Mass spectrometry and protein analysis. Science 2006;312(5771):212–7. [DOI] [PubMed] [Google Scholar]
- Elias MS, Long HA, Newman CF, Wilson PA, West A, McGill PJ, et al. Proteomic analysis of filaggrin deficiency identifies molecular signatures characteristic of atopic eczema. The Journal of allergy and clinical immunology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurdanella F, Diercks GF, Jonkman MF, Pas HH. Laboratory diagnosis of pemphigus: direct immunofluorescence remains the gold standard. The British journal of dermatology 2016;175(1):185–6. [DOI] [PubMed] [Google Scholar]
- Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nature reviews Disease primers 2017;3:17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proceedings of the National Academy of Sciences of the United States of America 2014;111(6):2259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Boutz DR, Chromikova V, Joyce MG, Vollmers C, Leung K, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nature medicine 2016;22(12):1456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, UniProt C. UniProt Knowledgebase: a hub of integrated protein data. Database : the journal of biological databases and curation 2011;2011:DOI 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miske R, Hahn S, Rosenkranz T, Muller M, Dettmann IM, Mindorf S, et al. Autoantibodies against glutamate receptor delta2 after allogenic stem cell transplantation. Neurology(R) neuroimmunology & neuroinflammation 2016;3(4):e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepens I, Jaunin F, Begre N, Laderach U, Marcus K, Hashimoto T, et al. The protease inhibitor alpha-2-macroglobulin-like-1 is the p170 antigen recognized by paraneoplastic pemphigus autoantibodies in human. PLoS One 2010;5(8):e12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Boutz DR, Lavinder JJ, Miklos AE, Hughes RA, Hoi KH, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proceedings of the National Academy of Sciences of the United States of America 2013;110(8):2993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Horton AP, Ippolito GC, Georgiou G. Serology in the 21st century: the molecular-level analysis of the serum antibody repertoire. Current opinion in immunology 2015;35:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]