

Abstract
Background:
The cellular mechanism of focal, idiopathic right ventricular outflow tract (RVOT) tachycardia is thought to be due to cAMP-mediated triggered activity. A potential molecular mechanism has not yet been determined. We identified and characterized a novel missense somatic mutation in the gene (GNAS) encoding the stimulatory G protein (Gsα) from a patient with RVOT tachycardia that is proposed to be the etiology of the clinical tachycardia.
Methods:
Percutaneous endomyocardial biopsies were obtained from multiple sites in a patient with non-exertional, repetitive monomorphic RVOT tachycardia. Sequencing of extracted genomic DNA identified a Gsα W234R variant only at the site of tachycardia origin. Functional studies using in vitro transfection with S49 cyc− murine lymphoma cells and measurement of cyclic AMP levels were performed. A trypsin protection assay assessed GTP binding kinetics and structural modeling predicted the impact of the mutation on protein-protein interactions. Whole cell patch clamp experiments of transfected CHO cells assessed the downstream effects of the mutation.
Results:
In vitro studies of the GNAS mutation (W234R) demonstrated basal levels of cAMP ~16-fold higher than wild-type cells, consistent with constitutive stimulation of Gsα. Mutant Gsα was partially protected from proteolysis after incubation with GTP, indicating diminished GTPase activity and reduced GTP hydrolysis as the mechanism for increased basal intracellular cAMP levels. Transfected mutant CHO cells increased unstimulated mean peak L-type calcium channel current density by ~50% and in silico modeling demonstrated spontaneous delayed afterdepolarizations and triggered activity.
Conclusions:
We identified a novel somatic mutation in GNAS associated with RVOT tachycardia. The mutation results in constitutive activation of Gsα, impairs GTP hydrolysis, and elevates basal cAMP levels, leading to enhanced L-type calcium current and triggered activity. These findings confirm that RVOT tachycardia can be caused by somatic mutations in signal transduction proteins that regulate intracellular cAMP and its downstream effectors.
Journal Subject Terms: arrhythmias, electrophysiology, mechanisms, translational studies
Keywords: arrhythmia, GTP - binding proteins, mutation, signal transduction, ventricular tachycardia
Introduction
Approximately 10% of patients with ventricular tachycardia (VT) have no apparent structural abnormalities. The cellular and molecular mechanisms of ventricular tachycardia (VT) in the absence of structural heart disease, commonly referred to as idiopathic VT, are incompletely understood. Genomic mutations have been identified in ion channels and structural proteins that cause some of these ventricular arrhythmias, but the etiology often remains elusive for the most common form of idiopathic VT, a non-familial tachycardia that originates from a discrete focal site within the right ventricular outflow tract (RVOT).1 Affected individuals present with either exercise-induced sustained VT and/or resting repetitive (nonsustained) monomorphic VT. The etiology of this arrhythmia has been thought in some patients to reflect a somatic mutation.2,3 An inability to consistently access the minute focus of myocardial tissue that causes these arrhythmias has impeded efforts to identify other potential somatic mutations.
Idiopathic RVOT tachycardia is sensitive to perturbations that lower intracellular calcium and stimulated levels of cAMP via a G protein-coupled mechanism. Therefore, the tachycardia terminates with adenosine (through inactivation of adenylyl cyclase via the A1 adenosine receptor [A1AR] and Giα), with beta-blockers (via competitive inhibition of the beta-adrenergic receptor), or with edrophonium/vagal maneuvers (through activation of the M2 muscarinic receptor). RVOT tachycardia also terminates via direct blockade of the L-type calcium channel. These composite responses suggest that RVOT tachycardia is caused by cAMP-mediated intracellular calcium overload that gives rise to delayed afterdepolarizations and triggered activity.1,4–6 Based on these data and our previous findings of myocardial somatic mutations in the genes that encode the Giα and the A1AR in unique subtypes of RVOT tachycardia,2,3 as well as known constitutively active somatic mutations in GNAS that elevate basal levels cAMP in endocrine tumors,7 we hypothesized that cAMP-mediated RVOT tachycardia could be caused by a somatic mutation in GNAS.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Index Case
A 67-year-old man presented with repetitive monomorphic VT at rest. The longest spontaneous run of VT was 17 beats. Cardiac evaluation included a normal ECG and signal averaged ECG. Cardiac catheterization disclosed no significant coronary artery disease and left ventricular ejection fraction was 50%. There was no family history of VT or sudden cardiac death. During electrophysiologic study, sustained monomorphic VT was reproducibly induced with rapid ventricular pacing either in the presence or absence of concurrent isoproterenol infusion. The VT morphology was similar to the patient’s spontaneous arrhythmia, having a left bundle branch block (LBBB), inferior axis configuration with a precordial transition in lead V3. VT terminated with rapid ventricular pacing, adenosine, verapamil and edrophonium. The site of origin of tachycardia was localized to the septal RVOT by endocardial catheter activation mapping. VT was successfully ablated by application of radiofrequency energy (30 W for 60 seconds).
Genetic analysis
Prior to ablation and after informed consent was obtained, percutaneous endomyocardial biopsies were obtained with a bioptome from multiple right ventricle sites as part of a Cornell University Medical College Institutional Review Board approved protocol. A single myocardial biopsy sample was obtained from the site of origin of arrhythmia in the RVOT (guided by multiplane fluoroscopy) and from 2 sites in the right ventricular apex (remote site). The biopsy specimens were frozen rapidly and stored for molecular analysis. Genomic DNA was prepared using proteinase K digestion and phenol-chloroform extraction.8 Using 25 ng of the patient’s genomic DNA as a template, all exons corresponding to the coding region of Gsα and their flanking intronic sequences were PCR amplified [(94°C × 60s, 60°C × 60s, 72°C × 60s) × 35 cycles, 72°C × 7 min] using [α−32P]dCTP and rTth DNA Polymerase XL (Applied Biosystems). Novel and previously described9 primer sequences are listed in Supplemental Table 1. 32P-labeled PCR products were denatured and studied by single strand conformation polymorphism analysis (SSCP) using 6% polyacrylamide gels in the presence or absence 5% (v/v) glycerol as previously described,6,10 When a GNAS SSCP variant was identified, the PCR product containing the variant was subjected to PCR amplification and cloned into a sequencing vector. Approximately 10 clones from each sample were sequenced with bidirectional DNA sequence analysis using Big Dye terminators on an ABI 377 automated sequencer (Applied Biosystems).
Functional studies of the Gαs W234R mutation
Intracellular cAMP Accumulation
Mutated GNAS, identified in genomic samples, was introduced into full length GNAS cDNA via site-directed mutagenesis kit (QuikChange XL, Stratagene) with oligonucleotides 5΄-CGATGAACGCCGCAAGCGATCCAGTGCTTCAACG- 3΄ and 5΄- CGTTGAAGCACTGGATCCGCTTGCGGCGTTCATCG-3΄. Mutant and wild-type cDNAs were subcloned into pCEP4 plasmid and transiently transfected into Gsα -deficient S49 cyc− murine lymphoma cells (provided by Dr. Xin-Yun Huang, Cornell Medicine).11 After 48 hours, S49 cyc− expression of Gsα isoforms was analyzed by Western blot with anti-Gsα antibody (Santa Cruz Biotechnology). Bound antibody was detected via an ECL Advance Western Blotting Detection Kit (Amersham Biosciences). Parallel transfected cultures were treated with isoproterenol (100 μM) and intracellular cAMP levels were assayed by a cAMP-125I radioimmunoassay kit (DuPont NEN, Boston, MA).12
Expression and Purification of Recombinant Gsα in Sf9 Cells
Recombinant wild-type and mutant (W234R) Gsα proteins were produced using a baculovirus expression system. Full-length GNAS cDNA encoding wild-type or W234R was amplified by PCR and cloned into pFastBac donor plasmid (Invitrogen) along with an NH2-terminal hexahistidine tag using the following oligonucleotides: 5΄-ATGCATCACCATCACCATCACATGGGC-3΄ and 5΄-TTAGAGCAGCTCGTACTGACGAAGGTG-3΄. All inserts were verified by automated sequence analysis. Plasmids were transfected into DH10Bac cells to generate recombinant bacmid DNA per the instructions of the manufacturer (Invitrogen). Gsα baculovirus was harvested from Sf9 insect cells transfected with recombinant bacmid DNA and then used to infect additional Sf9 cells in order to prepare recombinant wild-type or W234R mutant Gsα protein. Recombinant proteins were purified from infected Sf9 cell lysates by Ni2+-NTA affinity chromatography (Invitrogen).13
Trypsin Protection Assay
Wild-type or mutant Gsα protein (1.0 mg/ml) was incubated for 60 minutes at room temperature with 100 μM GTP or GTPγS. Trypsin was added to a final concentration of 100 μg/ml. After incubation for an additional 10 minutes at room temperature, the digested protein was analyzed by SDS-PAGE.14
GTPγS Binding and GTP Hydrolysis
GTP binding (kapp) and hydrolysis (kcat) were determined as previously described.14 Values for kcat were calculated directly from a single round of GTP hydrolysis, performed in triplicate experiments, by allowing recombinant G-protein to bind GTP in the absence of Mg2+. Catalysis was initiated by addition of Mg2+ and rebinding of radiolabeled substrate was prevented by addition of excess unlabeled guanine nucleotide.
Structural Modeling
The effects of the W234R mutation on Gsα structure-function were probed by in silico modeling. A model for the mutant Gsα protein was generated from the crystal structure of bovine Gsα15 (Protein Data Bank accession code 1AZT) by substituting arginine for tryptophan at the conserved amino acid residue 234 and subsequent energy minimization was determined using the GROMOS96 force field in DeepView.16 Structural figures were generated with PyMol software (http://www.pymol.org). Predictive protein-protein interactions were assessed using Mutabind2 software (https://lilab.jysw.suda.edu.cn/research/mutabind2/).17
Whole cell patch clamp electrophysiology
We transiently transfected Chinese Hamster Ovary (CHO) cells (ATCC, Manassas, VA) with cDNA encoding human cardiac L-type Ca2+ channel subunits (α1, β2a and α2-δ) and wild-type or W234R human GNAS, using Superfect transfection reagent and CMV promoter-based expression vectors. The mutation was made using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA). cDNA encoding eGFP was co-transfected to facilitate identification of transfected cells using an IX50 inverted microscope equipped with fluorescence optics (Olympus, Cypress, CA).
CHO cell expression studies were conducted using 6 independent transfections per group, for 6 separate days of patch-clamp recording performed over several weeks. On each day, fresh transfection mixtures were prepared to avoid recurring pipetting errors. Wild-type recordings were matched with those of the mutant each day to minimize variation from day-to-day transfection efficiency. Plasmid sequences were identical except for the point mutation to avoid variation from different promoter efficiencies. Individual cells were chosen for recording based on visual inspection of robust GFP expression as a marker of efficient transfection.
After 24–48 hours, we quantified the effects of the Gsα W234R mutation on basal channel function using whole-cell patch clamp at room temperature (~17–20 °C), and standard intracellular and bath solution except that BaCl2 (20 mM) was included in the bath solution as a surrogate charge carrier for CaCl2. Recordings were performed using a MultiClamp 700A amplifier, a Digidata 1300 Analogue/Digital converter, and pClamp9 software (Molecular Devices, San Jose, CA). Cells were held at −80 mV and then stepped to membrane potentials between −60 and +60 mV at 10 mV intervals, and then peak current density was measured and plotted against voltage. Currents were corrected for cell membrane area, quantified by measuring cell capacitance.
In silico modeling of GNAS mutation
For mathematical modeling, we used a human ventricular cell model.18 The input for the human ventricular cell model was parameter values. Output was transmembrane potential as a function of time. We used the parameter values to simulate normal (non-mutant) action potentials. This is the standard setup for which the model was developed and validated against a range of experimental data, including action potentials, calcium transients and ionic currents. The Gsα mutation was modeled by increasing the value of the parameter controlling permeability of the L-type Ca2+ current as determined in our experimental study. The mutation was modeled as a 50% and 100% increase in the permeability of the L-type calcium current. The protocol to induce delayed after depolarizations (DADs) consisted of 2 Hz pacing for 30 s, followed by a pacing pause.19
The type of model used in this study (a dynamic model representing a complex biological system) inevitably contains several simplifications and assumptions. For one, as is standard for this type of model, it contains four separate intracellular Ca2+ compartments that each have a uniform Ca2+ concentration, although Ca2+ almost certainly changes more continuously within the cell, especially within the bulk intracellular space. Other simplifications include omission of chloride currents and phosphorylation dynamics.
Statistical Methods
Continuous variables are presented as mean±SD. Based on normality of distribution, pairwise comparisons of continuous variables were made using Student’s t-test. Comparisons of more than two continuous variables were performed using ANOVA. Assumptions of normality of distribution and common variance were verified. Post-hoc pairwise comparisons of means were performed using the Scheffe test. P<0.05 was considered statistically significant. Statistics were performed using SPSS version 24 (IBM, Chicago, IL).
Results
Molecular Genetic Analysis
Percutaneous endomyocardial biopsies from the site of VT origin and remote right ventricular sites were obtained. Genomic DNA was extracted and all 13 exons corresponding to the coding region of GNAS and their flanking intronic sequences were PCR amplified. A single variant SSCP pattern in the amplicon of GNAS exon 9 was detected in our patient’s DNA sample obtained from the VT site of origin. The PCR product containing the SSCP variant was identified, and the original sample was PCR amplified and cloned into a sequencing vector. Bidirectional automated sequence analysis of the exon 9 amplicon revealed a T to C transition at nucleotide 700 that substitutes arginine for tryptophan at amino acid residue 234 (W234R) (Figure 1A). Residue W234 is conserved across several species, including mouse, rat, dog, cow and human, and across other human G proteins, including Gi, Gt and Gq (https://blast.ncbi.nlm.nih.gov) (Figure 1B). No other variants were observed in GNAS amplicons from the RVOT biopsy sample, and no sequence variants were found in any amplicon from the right ventricular apex biopsy samples.
Figure 1.
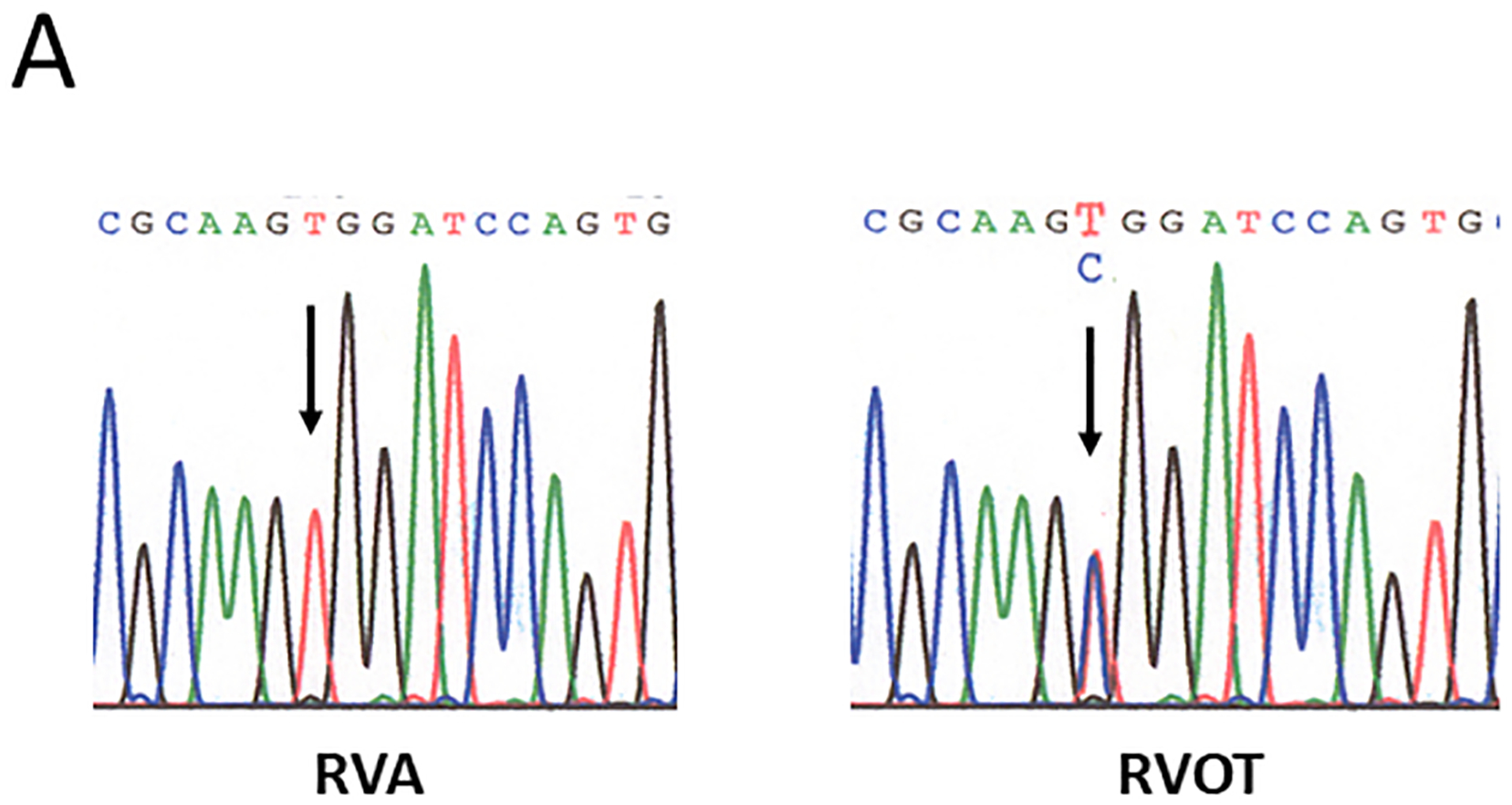

Sequence of polymerase chain reaction-amplified DNA from right ventricular apex (RVA) and right ventricular outflow tract (RVOT) biopsy samples. A, Novel point mutation (W234R) in the switch II region of Gsα was identified from a biopsy sample obtained from the site of origin of ventricular tachycardia in the RVOT, but not from the RVA. B, Gsα protein sequence alignment showing site of point mutation (W234R) within a conserved region across several species.
Functional studies of the W234R mutation in Gsα
To evaluate the functional effects of Gsα W234R, we generated GNAS cDNA corresponding to this mutant allele by site-directed mutagenesis of wild-type GNAS cDNA. Mutant and wild-type GNAS cDNA were transfected into Gsα deficient S49 cyc− lymphoma cells and synthesis of both recombinant proteins was confirmed by Western blot (Figure 2). To determine the physiologic activity of these G proteins, we assessed isoproterenol-mediated changes in cAMP levels in S49 cyc− lymphoma cells transfected with wild-type, W234R Gsα and equal ratios (1:1) of wild-type and mutant construct. Three separate experiments for each combination of the three constructs were carried out in triplicate.
Figure 2.
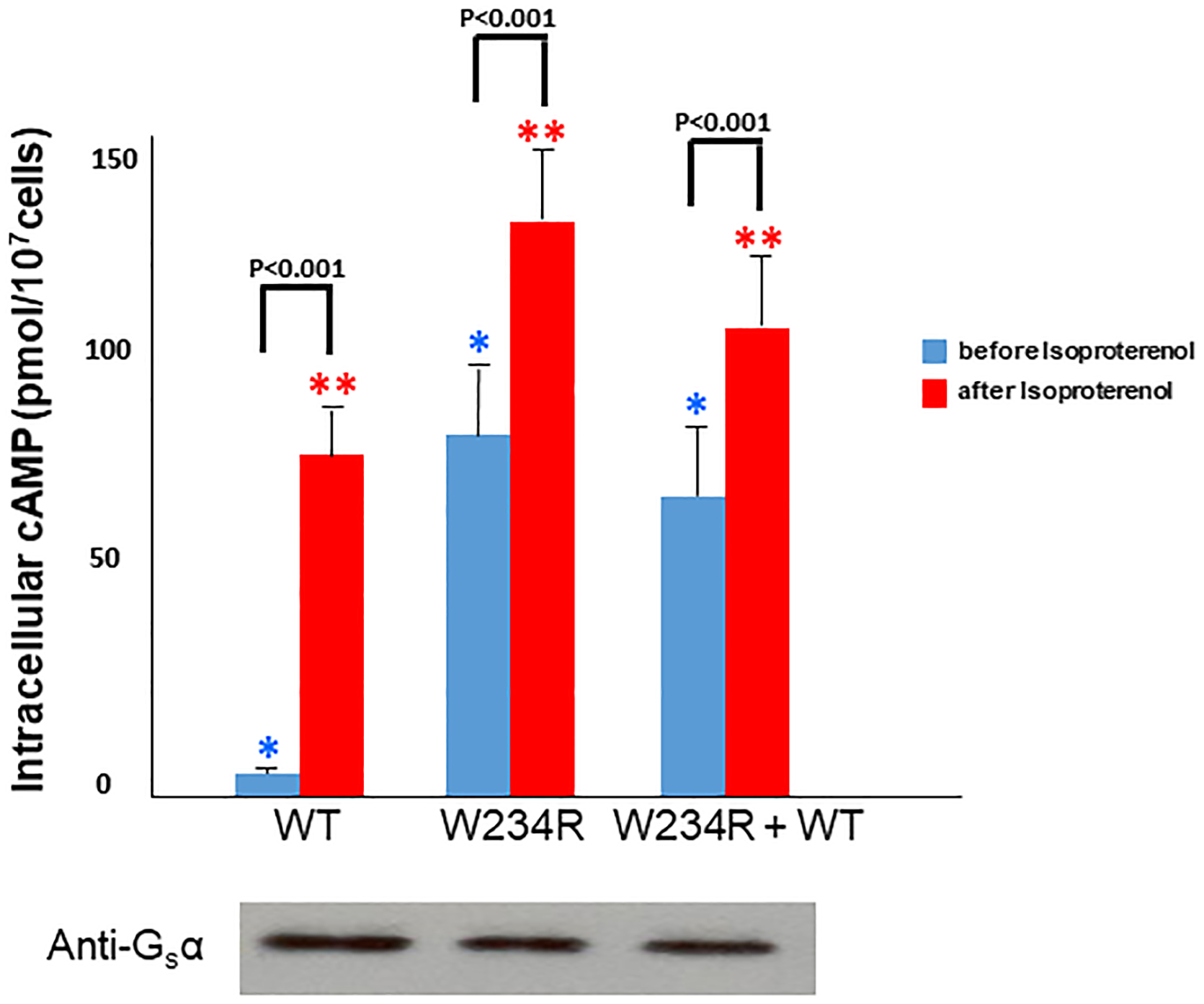
Effects of W234R mutation in Gsα (stimulatory G protein alpha- subunit) on cyclic AMP (cAMP) cellular content. S49 cycˉ Gsα deficient cells were transfected with either wild-type or W234R mutant Gsα cDNA. Western blot demonstrated similar expression levels of both wild-type and mutant protein. cAMP levels were assayed before and after treatment with isoproterenol (ISO). Three separate experiments for each construct were carried out in triplicate. Thus, there are 9 data points for each condition. Basal cAMP levels in cells transfected with wild-type Gsα were 5.0±0.7 pmol/107 cells, whereas basal cAMP levels in cells transfected with mutant Gsα were 79±12 pmol/107 cells, P<0.001. * P< 0.001 for comparison of basal cAMP levels among the three constructs. ** P<0.001 for comparison of cAMP levels after treatment with ISO across all three constructs.
S49 cyc− lymphoma cells transfected with W234R mutant GNAS demonstrated markedly elevated basal levels of cAMP (79±12 pmol/107 cells), which were 15.8-fold higher than that observed in basal wild-type GNAS transfected cells (5.0±0.7 pmol/107 cells), P<0.001. For cells transfected with equal ratios (1:1) of wild-type and mutant constructs, the intracellular levels of cAMP were 12-fold greater than control, 60±8 pmol/107 cells vs. 5.0±0.7 pmol/107 cells, P<0.001. Stimulated levels of cAMP increased nearly 2-fold in cells transfected with Gsα W234R, from 79±9 pmol/107 cells (basal) to 135±18 pmol/107 cells, P<0.001. Basal levels of cAMP in W234R Gsα transfected cells approximated the stimulated (isoproterenol) cAMP levels in wild-type transfection (79±9 pmol/107 cells) vs.79±12 pmol/107 cells, respectively, P=NS (Figure 2). These data are consistent with agonist-independent (constitutive) stimulation of mutant Gsα.
To further delineate the mechanism by which Gsα W234R increases basal intracellular cAMP levels, the structural and functional properties of recombinant wild-type and W234R Gsα proteins were assessed with a trypsin protection assay. When bound to GDP, wild-type Gsα subunits are digested by trypsin into small fragments. Such extensive proteolysis occurs when wild-type Gsα subunits are incubated with GTP since the latter is hydrolyzed to GDP by Gsα GTPase activity. In contrast, GTPγS is relatively resistant to hydrolysis, and thus, in the presence of GTPγS, wild-type Gsα is protected against trypsin proteolysis and 43 kDa products accumulate. Mutant Gsα W234R is also partially protected from proteolysis after incubation with GTP (Figure 3A), findings consistent with diminished GTPase activity and increased basal intracellular cAMP levels.
Figure 3.
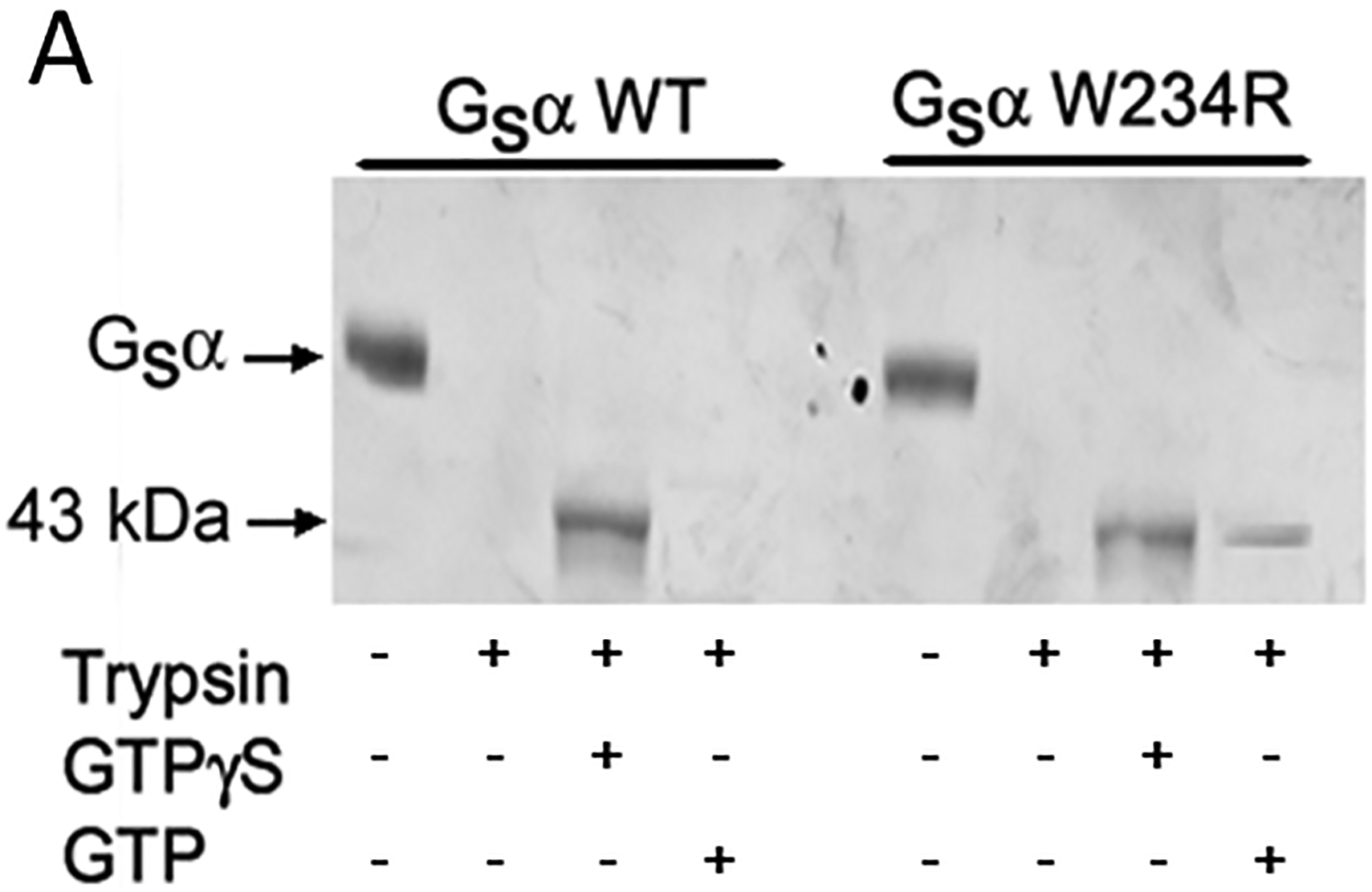
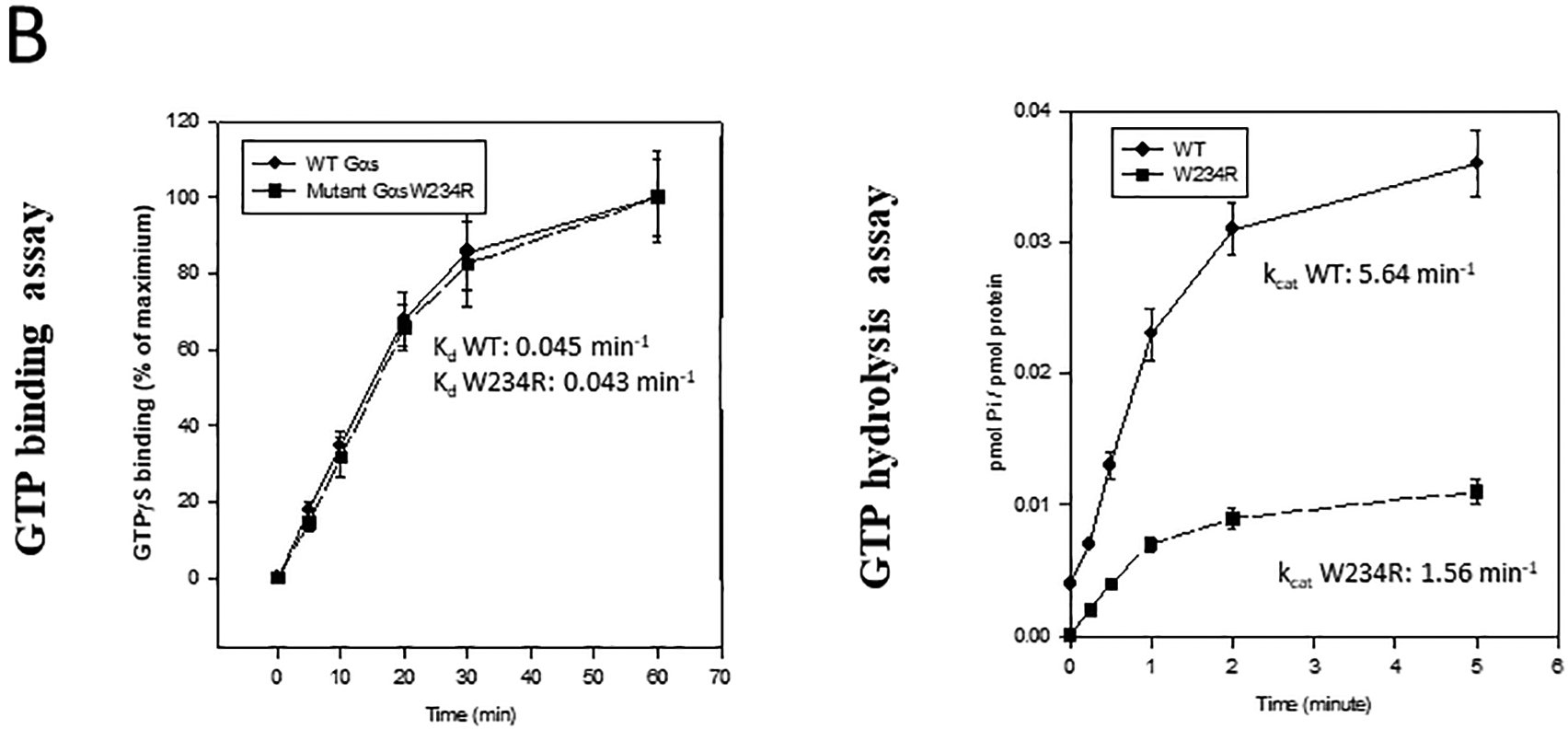
Structural and functional properties of wild-type and mutant W234R Gsα recombinant proteins. A, Wild-type Gsα is protected from trypsin proteolysis after incubation with GTPγS (43kDa proteolytic fragments accumulate) but not with GTP. B, Left panel. Recombinant wild-type and mutant Gsα proteins were incubated with 35[S]GTPγS and analyzed as described in Methods. No effect of mutation on GTP binding was observed and similar Kd values were calculated for both proteins (P=0.46). B, Right panel. Wild-type and mutant Gsα proteins were incubated with [γ−32P]GTP. Aliquots (50 μl) were analyzed for [32P]Pi at the indicted time. kcat was calculated by observation of a single round of GTP hydrolysis performed in triplicate experiments.
In studies that directly assess guanine nucleotide binding and hydrolysis, we observed no difference between GTPγS binding to wild-type or to W234R Gsα (Kd 0.045±0.006 versus Kd = 0.043±0.007, P=0.46) (Figure 3B). However, mutant Gsα exhibited reduced GTP hydrolysis. kcat for wild-type Gsα was 5.64±1.0 min−1 and for Gsα W234R was 1.56±0.7min−1, P<0.001. These data suggest that Gsα W234R is trapped in the GTP bound active state, allowing Gsα W234R to increase basal intracellular cAMP levels in the absence of an agonist.
Models of Mutant Gsα Structure
The crystallographic structure of Gsα in a complex with GTPγS has been previously reported and provides a model to determine the conformational consequences of Gsα mutations15 (Figure 4). GTP binding to Gsα produces conformational changes in three switch regions (I-III), that in turn determine the interactions of Gsα with the G protein coupled receptor, βδ dimers and its effector, adenylyl cyclase. GTP hydrolysis is thought to occur by deprotonation of an attacking water molecule and transfer of a proton to an oxygen of the γ-phosphate.16 This process is facilitated by two catalytic residues in Gsα: R201 and Q227. Both sites have been associated with constitutively activating endocrine mutations.20–25
Figure 4.

Ribbon diagram for the Ras-like domain of bovine Gsα (PDB ID 1AZT), with W234 mutated to R (red sticks, label underlined). The bound GTP analog is colored according to atom types, and surrounding secondary structure elements are labeled according to Sunahara et al.15 Catalytically important residues are shown in yellow, and the switch II helix α2 in cyan.
W234 is located at the center of an evolutionarily conserved hydrophobic core that stabilizes the position of switch II including Q227. Mutation of W234 substitutes an arginine and introduces a polar side chain into this hydrophobic environment. As a result, the region is predicted to destabilize the orientation of Q227 for GTP hydrolysis. Based on modeling,17 the ΔΔGbind conferred by the W234R mutation would be 2.36 kcal/mol, which is consistent with a destabilizing and deleterious mutation (ΔΔGbind >1.5 kcal/mol). In addition, the W234R mutation places the positively charged arginine next to K53, a conserved positively charged residue of the P-loop motif that binds the substrate phosphates, which would lead to repulsion between the two sites. The net result of these effects is that GTP binding occurs in a conformation unsuitable for hydrolysis (Figure 4).
Whole cell patch clamp electrophysiology
We quantified whole-cell Ba2+ currents using whole-cell patch clamp in CHO cells co-expressing cardiac L-type Ca2+ channel subunits with wild-type or W234R mutant Gsα. Compared to wild-type Gsα, co-expression with W234R mutant Gsα increased mean peak L-type calcium channel current density by 48±15 % (P<0.05; n =24 – 27 cells per group) (Figure 5).
Figure 5.

Whole-cell patch clamp. A, Exemplar traces measured using whole-cell patch clamp from CHO cells expressing cardiac L-type calcium channel subunits (α1, β2a and α2-δ) and wild-type or W234R human Gsα as indicated. Zero current level indicated by dashed line. Voltage protocol shown in lower inset. Arrow, approximate time point at which peak current was measured. B. Mean current density-voltage relationships for cells as in panel A; n = 27 (wild-type) and n = 24 (W234R). *P < 0.05 comparing wild-type versus W234R currents.
In silico model of increased calcium channel current density due to W234R Gsα mutation
Simulations with wild-type Gsα did not elicit DADs or triggered activity. Simulating the effects of W234R Gsα mutation by increasing L-type calcium current permeability by 50% resulted in prolonged action potential duration but no DADs. However, an increase of 100% in L-type calcium current permeability caused multiple DADs and triggered a spontaneous action potential following cessation of pacing, secondary to calcium-induced calcium release from the ryanodine receptor (Figure 6).
Figure 6.
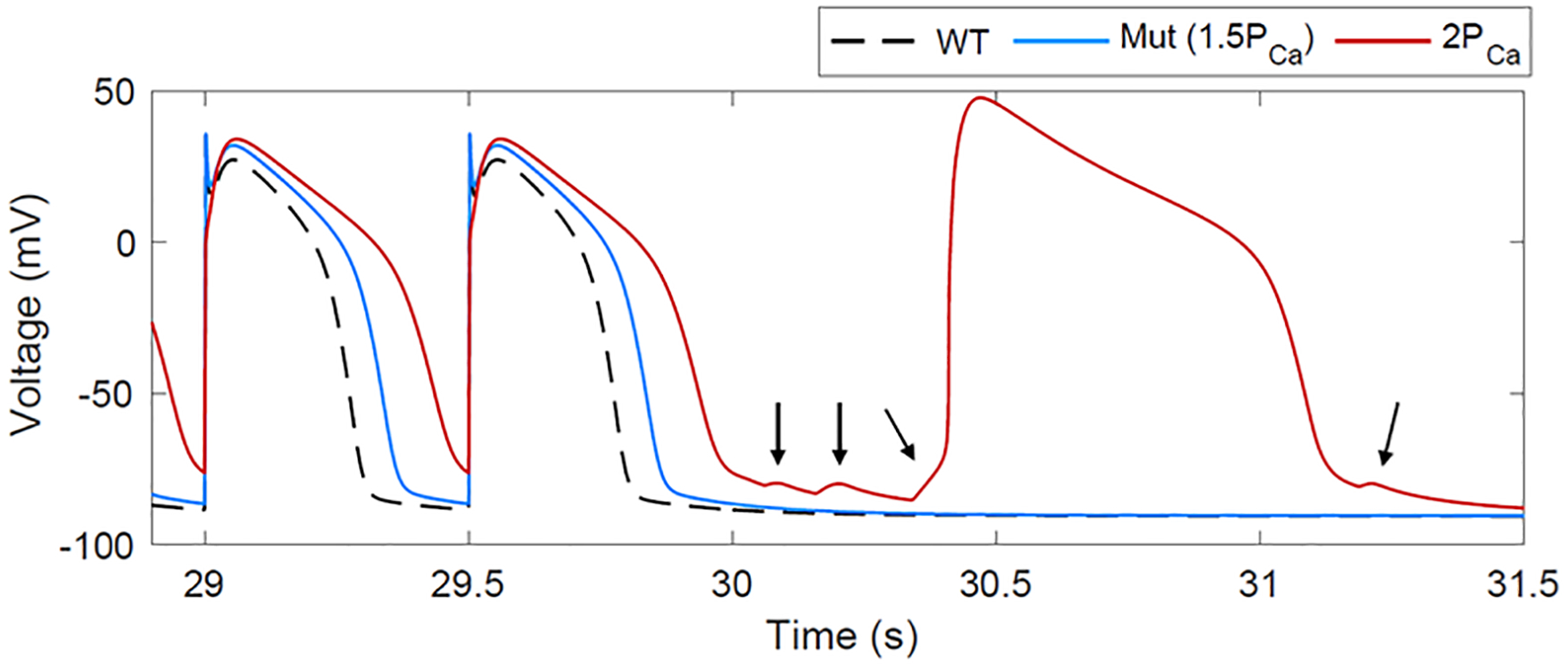
Model prediction of proarrhythmic electrical activity due to increased L-type calcium current secondary to W234R Gsα mutation. Simulations of wild-type ionic behavior in response to 2 Hz pacing for 30 s followed by a pacing pause results in normal action potentials (black trace). Simulating the W234R Gsα mutation causes action potential prolongation (50% increase in the permeability of the L-type calcium current: blue trace). Challenging the cell by increasing the calcium current further leads to multiple DADs (arrows) and a spontaneously triggered action potential (100% increase in the permeability of the L-type calcium current; red trace).
Discussion
We identified a constitutively activating somatic GNAS mutation from the arrhythmogenic site in the RVOT from a patient with repetitive monomorphic VT. W234R is a nonconservative charge-changing Gsα mutation, which impairs GTP hydrolysis and elevates basal cAMP levels. In addition, transfection studies demonstrated an increase in calcium current due to the W234R Gsα mutation and in silico studies were consistent with the presence of DADs and triggered beats. Along with our previous observation that somatic mutations in the genes encoding Gi2α and A1AR can cause atypical forms of RVOT tachycardia,2,3 the present findings further strengthen the proposal that RVOT tachycardia can be linked to somatic mutations in signal transduction proteins that elevate cAMP, leading to DADs and triggered activity
Repetitive monomorphic VT is characterized by brief paroxysms of nonsustained VT at rest, occurring in the absence of structural heart disease.6,26 These arrhythmias may become sustained under conditions associated with enhanced catecholamine stimulation and/or programmed stimulation, as demonstrated in our patient. Mapping studies confirm a discrete, focal outflow tract source. The tachycardia has features consistent with calcium-dependent, cAMP-mediated VT, i.e., termination in response to adenosine, vagal maneuvers, and blockade of the slow-inward calcium current. Our patient’s tachycardia demonstrated these characteristics, suggesting a pathogenic mechanism that is Gsα-mediated and dependent upon increased intracellular calcium.
Heterotrimeric G proteins, consisting of α, β, and γ subunits, play a central role in a variety of intracellular signal transduction systems (Figure 7).27 G proteins are activated by binding of agonists (e.g., catecholamines) to G protein coupled receptors, resulting in release of GDP and binding of guanosine triphosphate (GTP). Binding of GTP results in conformational changes in three switch regions, causing the GTP-α complex to dissociate from both the receptor and noncovalently bound β and γ subunits. The activated α subunit and the βγ dimer regulate the activity of downstream effectors, such as AC, phospholipases and ion channels. This activity is terminated by intrinsic GTPase activity in the α subunit, which hydrolyzes GTP to GDP, reconstituting the inactive heterotrimeric complex. In the case of cardiac myocytes, the beta-adrenergic receptor is associated with the heterotrimeric G protein Gsα; the M2 muscarinic receptor and A1AR are associated with Giα.
Figure 7.
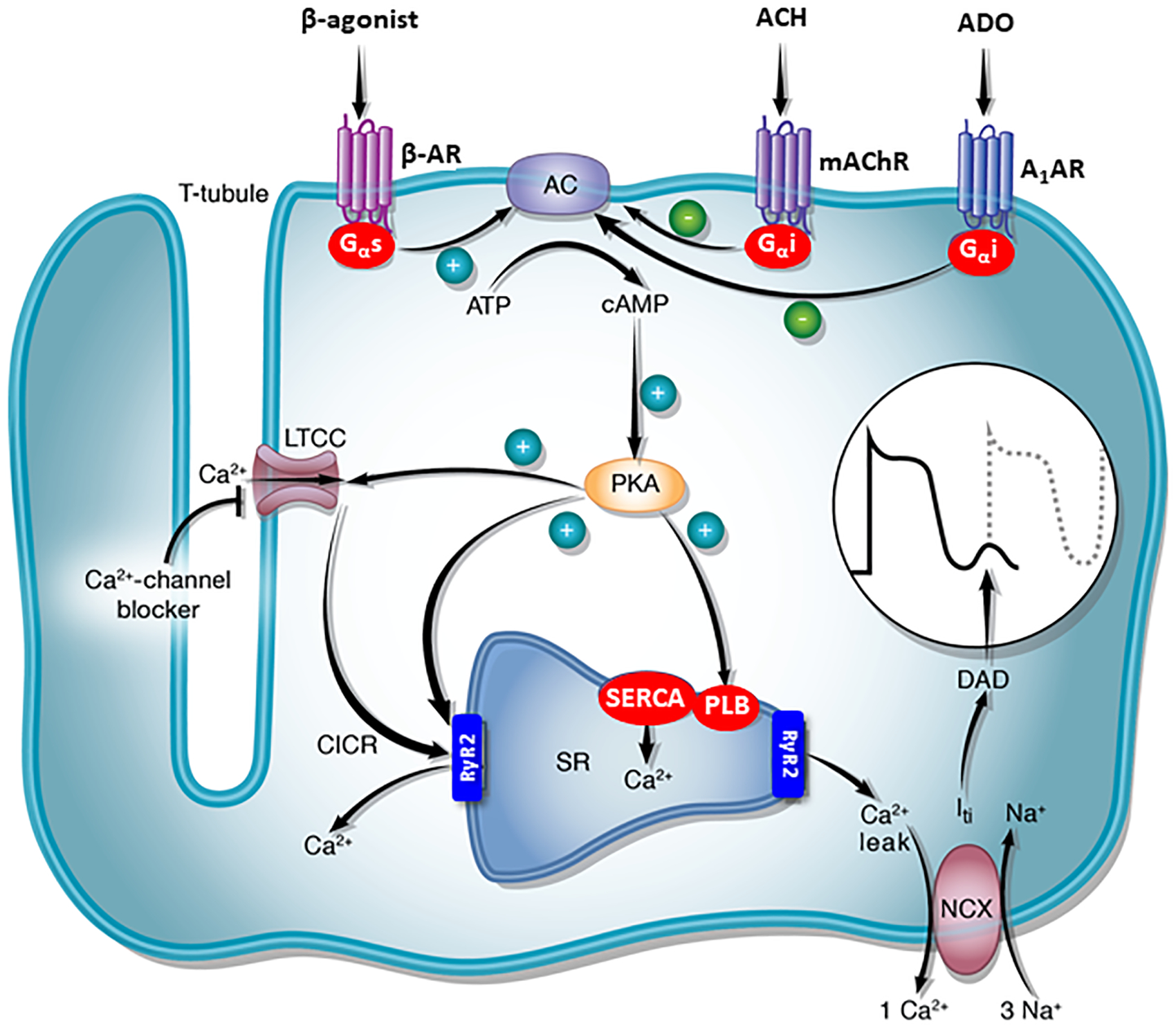
Schematic showing the signal transduction cascade that mediates ventricular tachycardia due to cAMP-mediated triggered activity. Clinical right ventricular outflow tract tachycardia is usually adrenergically-mediated, and is frequently triggered by exercise or stress. In contrast, the somatic mutation W234R Gsα doesn’t require an agonist for activation since it is constitutively active, accounting for resting ventricular tachycardia. Abbreviations: AC, adenylyl cyclase; A1AR, adenosine receptor A1; ACH; acetylcholine; ADO, adenosine; β-AR, β-adrenergic receptor; CICR, calcium-induced calcium release; DAD, delayed afterdepolarization; Gi2α, inhibitory G-protein; Gsα, stimulatory G-protein; Iti, transient inward current; LTCC, L-type calcium channel; mAChR, muscarinic acetylcholine receptor; NCX, sodium–calcium exchanger; PKA, cAMP-dependent protein kinase (protein kinase A); PLB, phospholamban; RyR2, ryanodine receptor; SERCA, sarco/endoplamic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
Functional studies of mutant W234R Gsα transfected into Gsα -deficient S49 cyc− murine lymphoma cells showed a marked increase (approximately 16-fold) in basal levels of cAMP compared to cells transfected with wild-type Gsα (Figure 2). Moreover, basal levels of cAMP of mutant Gsα approximated adrenergically-stimulated (via isoproterenol) levels of cAMP in wild-type Gsα transfected cells. These findings suggest that the somatic mutation obtained from the site of VT origin caused constitutive activation of Gsα. This was further supported by detection of reduced GTPase hydrolysis of mutant W234R Gsα (Figure 3). Furthermore, structural modeling predicted that the W234R mutation causes GTP binding in a conformation unsuitable for hydrolysis (Figure 4). Finally, whole cell patch clamp electrophysiology of mutant W234R Gsα increased L-type calcium channel current density by approximately 50% (Figure 5), and when further augmented, as would occur with adrenergic stimulation, DADs and triggered activity were simulated in an in silico model of human ventricular myocytes (Figure 6).
The findings above are consistent with the arrhythmic phenotype in our patient. The presenting arrhythmia, repetitive monomorphic VT, occurs at rest, but yet is cAMP-mediated.6 Agonist-independent G protein activation due to reduced GTPase activity may explain how cAMP- mediated tachycardia can occur in the absence of an apparent increase in endogenous or exogeneous catecholamine stimulation. Enhanced catecholamine stimulation, under appropriate conditions, may further augment elevated basal cAMP levels and facilitate initiation of sustained monomorphic VT. It is possible that compensatory mechanisms (i.e., enhanced phosphodiesterase activity or reduced adenylyl cyclase) may also be activated and prevent continuous VT despite persistent cAMP stimulation.
Although germline transmission of activating GNAS mutations is generally presumed to be lethal, somatic mutations, especially with restricted tissue expression and receptor coupling can lead to more limited manifestations. Constitutively activating mutations of Gsα (R201 and Q227) have been described in acromegaly, thyroid adenomas, the McCune-Albright syndrome, and less commonly in ovarian, testicular stromal Leydig cell tumors and Cushing’s syndrome.20–25 These somatic mutations have biochemical consequences similar to W234R and are characterized by loss of GTPase activity, impaired hydrolysis of GTP, sustained stimulation of adenylyl cyclase and persistent elevation of cAMP. Based on these data, we hypothesize that similar gain-of-function mutations in the stimulatory G protein Gsα may be responsible for some forms of cAMP mediated, adenosine-sensitive RVOT tachycardia.
The mutation identified in GNAS as well as previously reported arrhythmogenic somatic mutations in GNAI3 (Gi2α) and ADORA1, identified by sequencing DNA from a 1–2 mm3 biopsy sample from the arrhythmogenic focus, is consistent with there being more than one single cardiac cell affected by a somatic G protein mutation.2,3 However, it remains unknown whether these somatic mutations occur during embryonic heart development in a single cell that clonally expands or whether they possibly occur during postnatal life in multipotent cardiac cells that retain the capacity to proliferate in the adult heart.28 The significance and prevalence of somatic gene mutations in individuals with RVOT tachycardia merits further investigation.
Other arrhythmogenic cardiac somatic mutations, although rare, have also been identified in the gene encoding connexin 40 (GJA5) in 3 patients with atrial fibrillation.29 Additionally, somatic mutations in genes encoding the transcriptional factors GATA4, GATA5 and GATA6 are increasingly recognized as being associated with some forms of congenital heart disease, including ventricular septal defects and tetralogy of Fallot.30
Study Limitations
We performed targeted sequencing of extracted genomic DNA of all exons corresponding to the coding region of GNAS and their flanking intronic sequences to identify the W234R variant in this study. Although we cannot rule out the presence of modifying polymorphisms that could have been implicated in our patient’s RVOT tachycardia, in vitro functional studies (i.e., cAMP assay, trypsin protection assay, whole cell patch clamping), structural modeling, and in silico modeling all suggest that the identified mutation causes phenotypic changes consistent with of our patient’s VT. Moreover, the mutation was only found at the site of arrhythmia origin and not at remote right ventricular biopsy sites, indicating the focal and somatic nature of the mutation. The generalizability of our findings to other patients with RVOT tachycardia is unknown since myocardial biopsies are rarely performed in these patients and the focal origin of the mutation makes exact sampling of the critical tissue often fortuitous.
Supplementary Material
What Is Known?
Right ventricular outflow tract (RVOT) tachycardia occurs in the absence of structural heart disease and is due to a non-reentrant mechanism.
This form of tachycardia is usually sustained and induced with catecholamine stimulation and burst pacing. However, a variant manifests as repetitive monomorphic ventricular tachycardia and occurs independently of catecholamine stimulation.
What the Study Adds?
A cardiac somatic mutation in GNAS was identified from an endomyocardial biopsy obtained from the site of origin of the tachycardia. The mutation increases basal levels of cAMP by approximately 16-fold compared with wild-type GNAS. Further characterization of the mutation showed diminished GTPase activity and impaired GTP hydrolysis. Transfection experiments showed that the mutant protein increases basal inward calcium current by ~50% and in silico modeling demonstrated delayed afterdepolarizations and triggered activity.
These findings highlight the role of somatic cardiac mutations in genes that regulate cAMP and its downstream effects on intracellular calcium as a contributing mechanism for some forms of RVOT tachycardia.
Sources of Funding:
The work was supported in part by grants from National Institutes of Health (RO1 HL-56139, R01 HL66214), and The Rosenfeld Heart Foundation.
Nonstandard Abbreviations and Acronyms:
- A1AR
A1 adenosine receptor
- cAMP
cyclic adenosine monophosphate
- DAD
delayed afterdepolarization
- Gsα
stimulatory G protein alpha-subunit
- Giα
inhibitory G protein alpha subunit
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- PCR
polymerase chain reaction
- RVOT
right ventricular outflow tract
- SDS-PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- SSCP
single strand conformation polymorphism analysis
- VT
ventricular tachycardia
Footnotes
Disclosures: None
Supplemental Materials:
References:
- 1.Lerman BB, Belardinelli L, West GA, Berne RM, DiMarco JP. Adenosine–sensitive ventricular tachycardia: evidence suggesting cyclic AMP–mediated triggered activity. Circulation. 1986;74:270–280. [DOI] [PubMed] [Google Scholar]
- 2.Lerman BB, Dong B, Stein KM, Markowitz SM, Linden J, Catanzaro DF. Right ventricular outflow tract tachycardia due to a somatic cell mutation in G protein subunitαi2. J Clin Invest. 1998;101:2862–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung JW, Ip JE, Yarlagadda RK, Liu CF, Thomas G, Xu L, Wilkes D, Markowitz SM, Lerman BB. Adenosine-insensitive right ventricular tachycardia: novel variant of idiopathic outflow tract tachycardia. Heart Rhythm. 2014;11:1770–1778. [DOI] [PubMed] [Google Scholar]
- 4.Lerman BB, Wesley RC, DiMarco JP, Haines DE, Belardinelli L. Antiadrenergic effects of adenosine on His–Purkinje automaticity: evidence for accentuated antagonism. J Clin Invest. 1998;82:2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song Y, Thedford S, Lerman BB, Belardinelli L. Adenosine-sensitive afterdepolarizations and triggered activity in guinea pig ventricular myocytes. Circ Res. 1992;70:743–753. [DOI] [PubMed] [Google Scholar]
- 6.Lerman BB, Stein K. Engelstein ED, Battleman D, Lippman N, Bei D, Catanzaro. Mechanism of repetitive monomorphic ventricular tachycardia. Circulation. 1995;92:421–429. [DOI] [PubMed] [Google Scholar]
- 7.Weinstein LS, Chen M, Xie T, Liu J. Genetic diseases associated with heterotrimeric G proteins. Trends Pharmacol Sci. 2006;27:260–266. [DOI] [PubMed] [Google Scholar]
- 8.Strauss WM. Preparation of genomic DNA from mammalian tissue. Current Protocols in Molecular Biology. 1998;42:2.2.1–2.2.3. [DOI] [PubMed] [Google Scholar]
- 9.Ahrens W, Hiort O, Staedt P, Kirschner T, Marschke C, Kruse K. Analysis of the GNAS1 gene in Albright’s Hereditary Osteodystrophy. J Clin Endocrinol Metab. 2001;86:4630–4634. [DOI] [PubMed] [Google Scholar]
- 10.Borresen AL. Mismatch detection using heteroduplex analysis. Current Protocols in Human Genetics. 2002;33:7.3.1–7.3.3. [DOI] [PubMed] [Google Scholar]
- 11.Berlot CH. Expression and functional analysis of G protein alpha subunits in S49 lymphoma cells. Methods Enzymol. 2002;344:261–277. [DOI] [PubMed] [Google Scholar]
- 12.Zhao AZ, Bornfeldt KE, Beavo JA. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J Clin Invest. 1998;102:869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozasa T, Gilman AG. Purification of recombinant G proteins from Sf9 cells of hexahistidine tagging of associated subunits. Characterization of alpha 12 and inhibition of adenylyl cyclase by alpha z. J Biol Chem. 1995;270:1734–1741. [DOI] [PubMed] [Google Scholar]
- 14.Kleuss C, Raw AS, Lee E, Sprang SR, Gilman AG. Mechanism of GTP hydrolysis by G-protein α subunits. Proc Natl Acad Sci. 1994;91:9828–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunahara RK, Tesmer JJG, Gilman AG, Sprang SR. Crystal structure of the adenlylyl cyclase activator Gsα. Science. 1997;278:1943–1947. [DOI] [PubMed] [Google Scholar]
- 16.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 17.Zhang N, Chen Y, Lu H, Zhao F, Alvarez RV, Goncearenco A, Panchenko AR, Li M. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience. 2020;23:100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyer V, Mazhari R, Winslow RL. A computational model of the human left-ventricular epicardial myocyte. Biophys J. 2004;87:1507–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink M, Noble PJ, Noble D. Ca2+-induced delayed afterdepolarizations are triggered by dyadic subspace Ca22+ affirming that increasing SERCA reduces aftercontractions. Am J Physiol Heart Circ Physiol. 2011;301:H921–H935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330:566–568. [DOI] [PubMed] [Google Scholar]
- 21.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696. [DOI] [PubMed] [Google Scholar]
- 22.Lyons J, Landis CA, Harsh G, Vallar L, Grunewald K, Feichtinger H, Duh QY, Clark OH, Kawasaki E, Bourne HR, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659. [DOI] [PubMed] [Google Scholar]
- 23.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Freidman E, Spiegel AM. Activating mutations in the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695. [DOI] [PubMed] [Google Scholar]
- 24.Fragoso V, Latronico AC, Carvalho FM, Zerbini MC, Marcondes JA, Araujo LM, Lando VS, Frazzatto ET, Mendonca BB, Villares SM. Activating mutation of the stimulatory G protein (gsp) as a putative cause of ovarian and testicular human stromal Leydig cell tumors. J Clin Endocrinol Metab. 1998;83:2074–2078. [DOI] [PubMed] [Google Scholar]
- 25.Boston BA, Mandel S, LaFranchi S, Bliziotes M. Activating mutation in the stimulatory guanine nucleotide-binding protein in and infant with Cushing’s syndrome and nodular adrenal hyperplasia. J Clin Endocrinol Metab. 1994;79:890–893. [DOI] [PubMed] [Google Scholar]
- 26.Lerman BB. Mechanism, diagnosis, and treatment of outflow tract tachycardia. Nat Rev Cardiol. 2015;12:597–608. [DOI] [PubMed] [Google Scholar]
- 27.Farfel Z, Bourne HR, Iiri T. The expanding spectrum of G protein diseases. New Engl J Med. 1999;340:1012–1020. [DOI] [PubMed] [Google Scholar]
- 28.Maclellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. [DOI] [PubMed] [Google Scholar]
- 29.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang ASL, Stewart AFR, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–2688. [DOI] [PubMed] [Google Scholar]
- 30.Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J Mol Med. 2014;33:1227–1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.