

Abstract
PI-103 (7) is a potent dual phosphatidylinositol 3-kinase (PI3K)/mTOR inhibitor, but its rapid in vivo metabolism hinders its further clinical development. To improve the bioavailability of PI-103, we designed and synthesized a PI-103 bioisostere, PI-103BE (9) in which the phenolic hydroxyl group of PI-103 was replaced by a boronate, a structural modification known to enhance bioavailability of molecules containing phenolic hydroxyl moieties. In cell culture, PI-103BE is partially converted to its corresponding boronic acid (10) and to a lesser extent the active ingredient, PI-103. This mixture contributes to the in vitro activity of 9 that shows reduced potency compared to the parent compound. When administered to mice by oral gavage, 9 displays a significantly improved pharmacokinetic profile compared to PI-103, which shows no oral bioavailability at the same dose. Drug exposure of 9 as measured by the area under curve (AUC) value is 88.2 ng/mL*h for 7 and 8879.9 ng/mL*h for 10. When given by intraperitoneal injection (IP), the prodrug afforded an AUC of 32.3 ng/mL*h for 7 and 400.9 ng/mL*h for 10, compared to 9.7 ng/mL*h from PI-103 injection. In plasma from both pharmacokinetic studies, 9 is fully converted to 10 and 7, with the boronic acid metabolite (10) displaying antiproliferative activities comparable to 9, but weaker than 7. The boronic bioisostere of PI-103 thus offers an improved bioavailability that could be translated to in vivo efficacy of PI-103.
Keywords: PI-103 bioisosteres, Boron-containing compound, Synthesis, Pharmacokinetics, Bioavailability
Introduction
The PI3K/AKT/mTOR pathway is a validated therapeutic target for the treatment of several malignancies including chronic lymphocytic leukemia (CLL),1 follicular lymphoma,2 and breast cancer.3,4 As of January 2020, FDA has approved four PI3K inhibitors as oncology agents: idelalisib (July 2014, NDA 206545) for leukemia and two types of lymphoma5 copanlisib (September 2017, NDA 209936) for relapsed follicular lymphoma, duvelisib (September 2018, NDA 211155) for relapsed or refractory chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL)6 (Fig. 1), and alpelisib (BYL719), an alpha-specific PI3K inhibitor for use in combination with endocrine therapy fulvestrant for treatment of HR-positive and HER2/neu-negative breast cancer.7 More PI3K inhibitors are being developed in different stages of clinical trials.8-11 Pharmacological inhibition of PI3Ks downregulates the expression of programmed death ligand-1 (PD-L1), as recent data from clinical trials and preclinical mouse models indicate that the therapeutic inhibition of PI3Ks also enhances antitumor immune-intrinsic properties,12,13 suggesting additional clinical utility of PI3K inhibition. PI-103 is a potent dual PI3K and mTOR inhibitor.14 Although the efficacy and tolerability of PI-103 have been examined in preclinical models, PI-103 has not yet entered clinical trials mainly because of its poor pharmacokinetic performance.15,16 Recently, Zhu and Merino et al designed a PI-103 prodrug (Fig. 1) to improve metabolic stability of the compound. Their study showed the enhanced selectivity of the prodrug against Kasumi-1 AM cells over normal cells, but no bioavailability data were provided.17
Fig. 1.
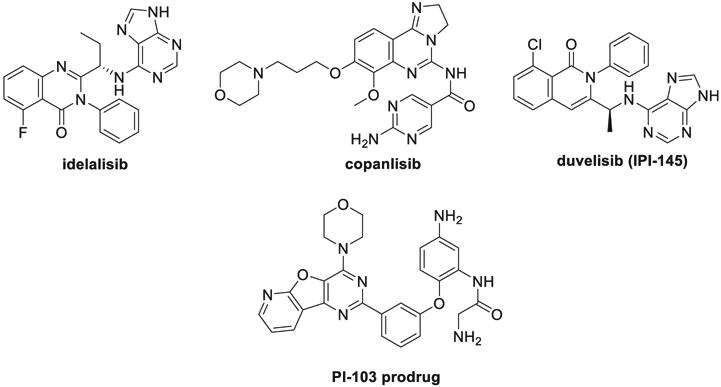
PI3K inhibitors in clinical use and PI-103 prodrug.
The oxidation of boronic acids and their esters to alcohols is a biorthogonal reaction that can occur inside living systems without interfering with native biochemical processes.18 This approach was first applied to the development of imaging probes19 and was later successfully used to design the bioisosteres or prodrugs of phenolic compounds including 4-hydroxytamoxifen,20 endoxifen,21 fulvestrant,22,23 GW7604,24 combretastatin A-4,25 metalloproteinase inhibitor,26 SN-38 (camptothecin derivative),27 and estrone.28,29 We have found the replacement of the phenolic hydroxyl group with a boronic acid or a boronate significantly improves the bioavailability of the drugs and led to targeted uptake and accumulation in solid tumors.21,24,30-33 Chang et al also reported that an imaging agent attached to aryl boronic acid moiety can spread throughout the body of living mice to reach deep tumor tissues due to the unique biocompatibility of boronic acid.19 It was also reported that the insulin derivatives modified with aliphatic phenylboronic acid conjugates could provide long-term glucose-mediated insulin activity.34 All of these results encouraged us to modify the hydroxyl group of PI-103 with a boron-containing moiety to enhance its bioavailability (Fig. 2). Herein, we report the synthesis and biological evaluation of PI-103BE (9), a PI-103 bioisostere.
Fig. 2.

Design and synthesis of PI-103BE (9).
Results and discussion
Synthesis of PI-103BE (9)
PI-103BE (9) was prepared as shown in Fig. 2. Condensation reaction of ethyl 3-aminofuro[2,3-b]pyridine-2-carboxylate (1) with 3-methoxybenzoyl chloride (2) provided the corresponding amide 3 in dichloromethane. The treatment of 3 with aqueous ammonium hydroxide gave 4 and subsequent cyclization of 4 with aqueous sodium hydroxide afforded 5. The demethylation of 5 followed by acetylation formed 6. The chlorination of 6 and subsequent substitution with morpholine resulted in the formation of PI-103 (7).35,36 7 was converted to the aryl triflates 8 as a colorless solid by trifluoromethanesulfonic anhydride. The formation of 9 (PI-103BE) was carried out via a boronation reaction using bis(pinacolato)diboron as the boron reagent, KOAc as the base, and PdCl2(dppf) as the catalyst in dioxane at 80° C.37
Reagents and conditions: (a)Et3N, DCM, 77%; (b)28% aq NH3, MeOH, 87%; (c)2N NaOH, MeOH, reflux, 88%; (d)(i) 48% HBr, AcOH, reflux, (ii) Ac2O, AcONa, reflux, 65% for two steps; (e) (i) POCl3, reflux, (ii) morpholine, reflux, 32% for two steps; (f) Tf2O, py, DCM, 84%; (g) PdCl2(dppf), Bis(pinacolato)diboron, KOAc, dioxane, 80°C, 76%.
In vitro antiproliferative properties
We first evaluated the antiproliferation activity of the bioisostere PI-103BE (9) against human lung carcinoma A549, human breast adenocarcinoma MDA-MB-231, human cervical cancer HeLa, human prostate carcinoma 22Rv1, human prostate adenocarcinoma PC-3, and human ovarian adenocarcinoma SKOV-3 cell lines. The results in Table 1 show that bioisostere 9 with IC50 values of 0.57 μM, 0.44 μM, 3.82 μM, 0.66 μM, 0.72 μM, and 1.33 μM is 3 ~ 24 times less potent than PI-103 (7) against these six cell lines, respectively. To test if the reduced activity of 9 is a result of partial conversion into the active ingredient (7), we tested the drug stability in cell culture by determination the concentrations of PI-103BE (9), PI-103BA (10), and PI-103 at various time points of treatment. It was found that PI-103BE underwent rapid hydrolysis in culture medium to become PI-103BA (10), the boronic acid of the bioisostere (> 95%) (Table 2). A small fraction of 7 at < 4% was also detected in culture medium (Table 2), indicating that the activity of the bioisostere in vitro may be accounted for by the combination of reduced potency of 9 and the incomplete conversion to PI-103 (7). Fig. 3 shows the conversion of 10 to 9 and 7 in aqueous media.
Table 1.
In vitro antiproliferative activity of PI-103BE (9) on a panel of cancer cell lines.
| Compounds | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| A549 | MDA-MB-231 | HeLa | 22Rv1 | PC-3 | SKOV-3 | |
| PI-103 (7) | 0.18 ± 0.04 | 0.14 ± 0.01 | 0.16 ± 0.05 | 0.10 ± 0.03 | 0.17 ± 0.03 | 0.11 ± 0.03 |
| PI-103BE (9) | 0.57 ± 0.06 | 0.44 ± 0.04 | 3.82 ± 0.18 | 0.66 ± 0.03 | 0.72 ± 0.02 | 1.33 ± 0.11 |
±: standard error (SEM) of 3 replicate experiments.
Table 2.
Stability of PI-103BE (9) in cell culture medium.
| Compound | Concentration (μM) and fraction of total drug content (%) in cell culture media | ||||
|---|---|---|---|---|---|
| 0.5 h | 1 h | 3 h | 8 h | 24 h | |
| PI-103BE (9) | 0.04 (1.61%) | 0.01 (0.32%) | 0.00 (0%) | 0.00 (0%) | 0.00 (0%) |
| PI-103BA (10) | 2.39 (97.95%) | 3.08 (97.47%) | 3.52 (96.70%) | 3.55 (96.73%) | 3.53 (96.19%) |
| PI-103 (7) | 0.05 (2.02%) | 0.08 (2.52%) | 0.12 (3.30%) | 0.12 (3.27%) | 0.14 (3.81%) |
Fig. 3.

Metabolism of PI-103BE (9) to active metabolite PI-103 (7).
Pharmacokinetics and in vivo metabolites of PI-103BE (9) in mice plasma
To determine the bioavailability of PI-103BE (9), we conducted pharmacokinetic studies of 9 in mice with PI-103 (7) as a control. Mice were administered a single dose of 10 mg/kg of 9 or 7 by intraperitoneal injection and blood samples were collected at 0.5, 1, 3, 8, and 24 h post drug administration. Measured plasma concentrations of the administered drug and metabolites are plotted over a period of 24 h in Fig. 4 and summarized in Table 3. Peak concentration of 7 in the blood of 9-treated mice reached 14.9 ng/mL at 0.5 h after IP administration, then decreased over time, and at 24 h, maintained a concentration of PI-103 at 0.4 ng/mL (Fig. 4A). The major form of the drug was PI-103BA, the boronic acid while no PI-103BE was detected in plasma, consistent with the stability profile of the bioisostere in culture media. In comparison, the peak concentration of PI-103 in the blood of 7-treated mice, reached at 1 h, was at 1.4 ng/mL, 10 times lower than achieved by the i.p. administration of PI-103BE (Fig. 4B).
Fig. 4.

Single dose pharmacokinetics of PI-103BE (A) and PI-103 (B) in mice.
Table 3.
Pharmacokinetics parameters of PI-103BE (9) and PI-103 (7) in mice after intraperitoneal injection (IP).
| Time Point (h) | PI-103BE (9) | PI-103 (7) | |
|---|---|---|---|
| PI-103 (ng/mL) | PI-103BA (10) (ng/mL) | PI-103 (ng/mL) | |
| 0.5 | 14.9 ± 0.5 | 30.6 ± 1.4 | 0.1 ± 0.0 |
| 1 | 5.4 ± 0.1 | 44.5 ± 0.9 | 1.4 ± 0.1 |
| 3 | 1.3 ± 0.1 | 28.0 ± 1.0 | 1.0 ± 0.1 |
| 8 | 1.0 ± 0.0 | 17.2 ± 0.3 | 0.3 ± 0.0 |
| 24 | 0.4 ± 0.0 | 6.5 ± 0.2 | NF |
| Cmax | 14.9 ng/mL | 44.5 ng/mL | 1.4 ng/mL |
| T max | 0.5 hr | 1.0 hr | 3.0 hr |
| t 1/2 | 12.4 hr | 10.2 hr | 2.0 hr |
| AUC | 32.3 ng/mL*h | 400.9 ng/mL*h | 9.7 ng/mL*h |
We next examined the pharmacokinetic profile of PI-103BE via oral administration, again in comparison with PI-103. After a single dose of 10 mg/kg by oral gavage of PI-103BE or PI-103, blood samples were collected from mice and analyzed for concentration of PI-103 and 9 at 0.5, 1, 3, 8, and 24 h time points after drug administration. Table 4 shows the pharmacokinetics parameters of PI-103BE (9) and PI-103 (7) in mice after oral administration. In 9-treated mice, the peak plasma concentration of PI-103 was 8.2 ng/mL, reached at 1 h after oral administration. In contrast, no PI-103 was found in mice given a single oral dose of PI-103, indicating no oral bioavailability. Importantly, the predominant form in 9-treated mice plasma is PI-103BA (10), the corresponding free boronic acid of 9. The maximum concentration of 10 reached 709.2 ng/mL at 1 h, and its plasma level remained above 100 ng/mL at 24 h. These results confirm that PI-103 is poorly bioavailable in vivo, especially by oral administration, whereas PI-103BE (9) has significantly enhanced bioavailability in both its boronic acid form and the active ingredient form of PI-103 as a metabolite of the bioisostere.
Table 4.
Pharmacokinetics parameters of PI-103BE (9) and PI-103 (7) in mice after oral administration.
| Time Point (hrs) |
PI-103BE (9) | PI-103 (7) | |
|---|---|---|---|
| PI-103 (7) (ng/mL) | PI-103BA (10) (ng/mL) | PI-103 (7) (ng/mL) | |
| 0.5 | ND | 402.8 ± 22.4 | ND |
| 1 | 8.2 ± 0.7 | 709.2 ± 11.9 | ND |
| 3 | 6.5 ± 0.4 | 516.2 ± 13.9 | ND |
| 8 | 4.1 ± 0.3 | 354. 2 ± 3.2 | ND |
| 24 | 0.7 ± 0.1 | 106.4 ± 2.2 | ND |
| Cmax | 8.2 ng/mL | 709.2 ng/mL | 0 |
| T max | 1 hr | 1 hr | ND |
| t 1/2 | 6.5 hr | 9.2 hr | ND |
| AUC0-24h | 88.2 ng/mL*h | 8879.9 ng/mL*h | 0 |
±: standard error (SEM) of 5 mice; ND: not detected.
Activity of the major metabolite PI-103BA (10)
Both in vitro stability and in vivo pharmacokinetic studies confirm that the boronic acid PI-103BA (10) is the major metabolite of prodrug 9, which may contribute to the overall therapeutic efficacy of the prodrug. We therefore sought to determine the anticancer activity of the boronic acid metabolite of 9. To prepare the compound 10, we performed the oxidation of 9 by sodium periodate, which led to the hydrolysis of 9 to yield the boronic acid 10 (PI-103BA) in THF-H2O. The antiproliferative activity of 10 was then evaluated in four cancer cell lines, A549, MDA-MB-231, HeLa, and 22RV1. As shown in Fig. 5, compound 10 displays dose dependent antiproliferative activities against all four cancer cell lines with potency comparable to that of the prodrug 9, but less than PI-103 (7).
Fig. 5.

Antiproliferative activity of PI-103BA (10) in A549, MDA-MB-231, HeLa, and 22RV1 human carcinoma cell lines.
Conclusion
Based on the unique properties of the boronic acid and its ester as a bioavailability-enhancing molecular functional group, we replaced the hydroxyl group of the PI3k inhibitor PI-103 (7) with the boronic acid pinacol ester moiety to construct a PI-103 bioisostere, PI-103BE (9). PI-103BE (9) was synthesized from ethyl 3-aminofuro[2,3-b]pyridine-2-carboxylate (1) and 3-methoxybenzoyl chloride (2) in seven steps. In vitro, the bioisostere 9 displays an antiproliferative activity against a panel of cancer cell lines with reduced potency compared to PI-103. In vivo pharmacokinetics studies in mice demonstrate that PI-103BE (9) affords significantly improved bioavailability over PI-103, which may be translatable to efficacy in vivo by virtue of enhanced drug exposure that has not been achieved by PI-103.
Experimental section
General information
All reactions were carried out under air atmosphere, unless otherwise mentioned. Commercially available materials were used as received without further purification. Reactions were monitored by thinlayer chromatography (TLC) carried out on commercial silica gel plates using UV light as a visualizing agent. Commercial silica gel was used for column chromatography. 1H and 13C NMR spectra were recorded on 400 MHz or 600 MHz spectrometer. 1H NMR spectra were referenced to Chloroform-d (7.26 ppm) or DMSO-d6 (2.50 ppm), and reported as follows; chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet). Chemical shifts of the 13C NMR spectra were measured relative to Chloroform-d (77.23 ppm) or DMSO-d6 (39.51 ppm). Mass spectral data were obtained from Bruker Daltonics Data analysis 3.2 mass spectrometer and Thermo LTQ Orbitrap-XL mass spectrometer in positive ion modes. Unless specified otherwise, all tested compounds were confirmed to be > 95% pure by HPLC.
Chemical synthesis of PI-103 bioisostere
Ethyl 3-(3-methoxybenzamido)furo[2,3-b]pyridine-2-carboxylate (3):
To a mixture of ethyl 3-aminofuro[2,3-b]pyridine-2-carboxylate (1) (2.06 g, 10 mmol) and Et3N (2.6 mL, 20 mmol) in dichloromethane (30 mL) was added dropwise 3-methoxybenzoyl chloride (2) at 0°C. After stirred at room temperature for 2.5 h, the reaction mixture was diluted with dichloromethane and washed with saturated NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude was recrystallized from ethyl acetate to give 3 (2.4 g, 77% yield) as a colorless solid: 1H NMR (400 MHz, Chloroform-d) δ 10.55 (s, 1H), 9.08 (dd, J = 8.1, 1.7 Hz, 1H), 8.54 (dd, J = 4.7, 1.8 Hz, 1H), 7.61 – 7.55 (m, 2H), 7.45 (t, J = 7.9 Hz, 1H), 7.36 (dd, J = 8.1, 4.7 Hz, 1H), 7.15 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 4.51 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H).
3-(3-Methoxybenzamido)furo[2,3-b]pyridine-2-carboxamide (4):
To a solution of 3 (2.1 g, 6.7 mmol) in MeOH (150 mL) was added 28% aqueous NH3 (175 mL). After stirred at room temperature for 12 h, the reaction mixture was concentrated to give the product 4 (1.81 g, 87% yield): 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 8.80 (dd, J = 8.0, 1.8 Hz, 1H), 8.53 (dd, J = 4.7, 1.8 Hz, 1H), 8.46 (s, 1H), 8.09 (s, 1H), 7.59 – 7.48 (m, 4H), 7.26 (ddd, J = 7.7, 2.8, 1.5 Hz, 1H), 3.86 (s, 3H).
2-(3-Methoxyphenyl)pyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-4(3H)-one (5):
To a mixture of 4 (1.8 g, 5.8 mmol) in i-PrOH (70 mL) was added 2 N NaOH (27 mL). After refluxed for 3 h, the reaction mixture was neutralized with concentrated HCl and the resulting solid was collected to give 5 (1.5 g, 88% yield) as a canary yellow solid: 1H NMR (400 MHz, DMSO-d6) δ 13.25 (s, 1H), 8.67 (dd, J = 4.9, 1.8 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.75 (t, J = 2.1 Hz, 1H), 7.64 (dd, J = 7.7, 4.8 Hz, 1H), 7.49 (t, J = 8.0 Hz, 2H), 7.18 (dd, J = 8.2, 2.6 Hz, 1H), 3.88 (s, 3H).
3-(4-Oxo-3,4-dihydropyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenyl acetate (6):
A mixture of 5 (770 mg, 2.7 mmol), 48% HBr (50 mL), and AcOH (50 mL) was refluxed for 48 h. The reaction mixture was concentrated in vacuo. Acetic anhydride (30 mL) and NaOAc (100 mg) were added and the reaction mixture was refluxed for 0.5 h. After concentration, the solid obtained was washed with methanol and Et2O to give 6 (0.50 g, 65% yield) as a gray solid: 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 8.68 – 8.62 (m, 2H), 8.10 – 8.05 (m, 1H), 7.97 (t, J = 2.1 Hz, 1H), 7.66 – 7.60 (m, 2H), 7.40 (dd, J = 8.0, 2.3 Hz, 1H), 2.34 (s, 3H).
3-(4-Morpholinopyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenol (7):
Compound 6 (1.0 g, 3.6 mmol) in phosphorus oxychloride (10 mL) was refluxed for 3 h and concentrated in vacuo. The residue was dissolved in THF (15 mL) and morpholine (15 mL) was added to it. After refluxed for 1.5 h, the reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous MgSO4. After concentration, the solid obtained was washed with CHCl3/MeOH (96:4) to give 7 (0.40 g, 32% yield) as a colorless solid: 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H), 8.66 (dd, J = 11.9, 6.2 Hz, 2H), 7.93 – 7.85 (m, 2H), 7.62 (dd, J = 7.7, 4.8 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 6.88 (dd, J = 7.9, 2.5 Hz, 1H), 4.11 (t, J = 4.6 Hz, 4H), 3.85 (t, J = 4.7 Hz, 4H). 13C NMR (101 MHz, DMSO-d6): δ 162.7, 158.9, 158.0, 150.4, 148.9, 147.0, 139.5, 133.3, 132.2, 129.8, 121.3, 119.2, 117.6, 115.1, 115.0, 66.5, 45.8. HRMS (ESI) calcd C19H17N4O3 [M + H]+ 349.1300, found 349.1294.
3-(4-Morpholinopyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenyltrifluoromethane sulfonate (8):
To the phenol 7 (174 mg, 0.5 mmol) in anhydrous CH2C12 was added pyridine (0.1 mL, 1 mmol) and the solution was cooled to 0 °C. Trifluoromethanesulfonic anhydride (0.13 mL, 0.6 mmol) was added dropwise and the mixture was warmed to r.t. The reaction was complete within 10 min. The reaction mixture was diluted with dichloromethane and washed successively with saturated NaHCO3 and brine. After drying over MgSO4, the solution was concentrated and the residue was purified by column chromatography on silica gel with hexane–ethyl acetate (4:1 to 2:1) as eluent to give the triflates 8 (0.20 g, 84% yield) as pale yellow: 1H NMR (400 MHz, DMSO-d6) δ 8.72 – 8.67 (m, 2H), 8.55 (dt, J = 7.8, 1.4 Hz, 1H), 8.39 (t, J = 2.0 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.67 – 7.62 (m, 2H), 4.13 (t, J = 4.7 Hz, 4H), 3.85 (t, J = 4.7 Hz, 4H).
4-Morpholino-2-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)pyrido[3′,2′:4,5] furo[3,2-d]pyrimidine
(PI-103BE, 9): A nitrogen-flushed 25 mL round-bottomed flask was charged with PdCl2(dppf) (8 mg, 0.01 mmol), KOAc (15 mg 0.15 mmol), bis(pinacolato)diboron (34 mg, 0.12 mmol), and the aryl triflate 8 (48 mg, 0.1 mmol), then anhydrous dioxane (3 mL) was added and the solution was stirred for 3 h at 80 °C. The reaction mixture was extracted with EtOAc, the organic layer was washed with brine, and dried over anhydrous MgSO4. The organic solvent was removed under reduced pressure and the crude was purified by column chromatography with hexane–ethyl acetate (5:1 to 3:1) as eluent to afford the product 9 (36 mg, 76% yield): 1H NMR (400 MHz, Chloroform-d) δ 8.89 (s, 1H), 8.62 (dd, J = 4.8, 1.5 Hz, 1H), 8.58 (d, J = 7.7 Hz, 1H), 7.93 (d, J = 7.2 Hz, 1H), 7.56 – 7.45 (m, 3H), 4.26 (t, J = 4.7 Hz, 4H), 3.94 (t, J = 4.8 Hz, 4H), 1.40 (s, 12H). 1H NMR (300 MHz, DMSO-d6): δ 8.76 (s, 1H), 8.73 (dd, J = 7.8, 1.8 Hz, 1H), 8.67 (dd, J = 5.1, 1.5 Hz, 1H), 8.53 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 7.2 Hz, 1H), 7.61 (m, 1H), 7.53 (t, J = 7.8 Hz, 1H), 4.11 (t, J = 4.7 Hz, 4H), 3.84 (t, J = 4.8 Hz, 4H), 1.34 (s, 12H). 13C NMR (75 MHz, DMSO-d6): 163.18, 159.34, 151.01, 149.54, 147.62, 138.16, 137.20, 134.65, 133.95, 133.05, 132.14, 129.17, 121.95, 115.47, 84.91, 67.08, 46.40, 25.80. HRMS (ESI) calcd C19H18BN4O4 [the free boronic acid of 9 + H]+ 377.1421, found 377.1416.
(3-(4-Morpholinopyrido[3′,2′:4,5]furo[3,2-d]pyrimidin-2-yl)phenyl)boronic acid (PI-103BA, 10):
9 (20 mg, 0.04 mmol) and sodium periodate (33 mg, 0.07 mmol) were dissolved in THF-H2O (5 mL, 4:1), to which 1 N HCl (0,5 mL) was added. The mixture was stirred at room temperature for 1 h, the reaction was quenched with brine and ethyl acetate (15 mL, 1;1). The precipitate was collected by filtration and washed with water and ethyl acetate 2–3 time, respectively to afford PI-103BA (10) (13 mg, 82% yield). The purity is > 98% based on HPLC. 1H NMR (300 MHz, DMSO-d6): δ 8.85 (s, 1H), 8.72–8.66 (m, 2H), 8.47 (d, J = 7.8 Hz, 1H), 7.91 (d, J = 7.2 Hz, 1H), 7.63 (m, 1H), 7.48 (t, J = 7.5 Hz, 1H), 4.14 (t, J = 5.1 Hz, 4H), 3.85 (t, J = 4.8 Hz, 4H). 13C NMR (75 MHz, DMSO-d6): 163.17, 159.90, 150.93, 149.49, 147.60, 137.73, 136.90, 134.67, 133.85, 132.88, 130.69, 128.51, 121.91, 115.53, 67.09, 46.35. HRMS (ESI) calcd C19H18BN4O4 [M + H]+ 377.1421, found 377.1419.
In vitro antiproliferative assay of PI-103 bioisostere toward cancer cell lines
For growth assay in the presence of PI-103 bioisosteres 9, the cancer cells were plated in plates at a density of 50,000 each well in 5% FBS DMEM medium. The cells were then treated with PI-103BE (9) separately at 6 different doses ranging from for 4 days, while equal treatment volume of DMSO and PI-103 (7) were used as vehicle control and positive control, respectively. Cell numbers were counted with a Coulter instrument (Beckman-Coulter). The ratio of drug-treated cell numbers to vehicle-treated cell numbers was defined as survival ratio. IC50 values were obtained from dose-response curves for each bioisostere. Experiments were conducted in triplicate and data represented as mean ± SD.
Pharmacokinetics study of PI-103BE (9) in mice
Sample collection of plasma:
Four to six weeks old female ovariectomized Nu/Nu mice were purchased from Charles River
Laboratories (Wilmington, MA). The above mice were used in the pharmacokinetic study of PI-103BE (9) and PI-103 (7). In oral pharmacokinetic study, mice (n = 5) were given a single dose of 10 mg/kg of PI-103BE (9) or PI-103 (7) dissolved in the oral gavage containing 5% dimethyl sulfoxide (DMSO), 40% polyethylene glycol 400, 55% saline. In the pharmacokinetic study by i.p. injection, mice were given a single dose of 10 mg/kg PI-103BE (9) or PI-103 (7) solution in ethanol by i.p. injection. After drug administration, blood samples were collected from the orbital sinus of the mice at 0.5, 1, 3, 8, and 24 h time points with each group of mice subjected to only one sampling. Blood samples were collected with a sterile capillary into 1.5 mL microcentrifuge tubes containing 0.1 mL of 10% EDTA anticoagulant. Plasma was then separated from cell pellets by centrifugation 3000 rpm, 5 min in a refrigerated centrifuge at 4 °C and transferred to a separate tube. Plasma samples were frozen at −80 °C until analysis.
Analysis of metabolites on TSQ Mass spectrometer:
Plasma samples were processed following a similar protocol.32 These samples were injected on the Hypersil GOLD C18 column (1.8 μm, 2.1mm × 50 mm) with UHPLC ultimate 3000 from Dionex coupled with a TSQ Vantage mass spectrometer coupled with a UHPLC ultimate 3000 from Dionex to quantitate the concentration of the main metabolites of PI-103BE (9) and PI-103 (7). The 10 μL samples were run with the gradient starting at 0.6 mL/min from 10% mobile phase B (Acetonitrile with 0.05% formic acid) and 90% mobile phase A (water with 0.05% formic acid) until 0.5 min, up to 100% B at 3 min, and until 6 min, then came back to 10% B until equilibration. The TSQ Vantage was set at spray vantage at 3200 V, vaporizer temperature at 365 °C, Sheath gas pressure at 42 PSI, Aug Gas pressure at 12 psi, Capillary temperature at 350 °C.
All procedures involving the animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by Xavier University Animal Care and Use Committee. The facilities and laboratory animals program of Xavier University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Supplementary Material
Acknowledgements
This work was funded by Sichuan University-Lu Zhou Strategic Cooperation Projects (2017 CDLZ-S34) (L. He) and supported by NIMHD at Xavier University of Louisiana through Grant (U54MD007595) (G. Wang).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2020.127258.
References
- 1.“FDA approves Zydelig for three types of blood cancers”. US Food and Drug Administration. July 23, 2014. [Google Scholar]
- 2.“FDA approves new treatment for adults with relapsed follicular lymphoma”. US Food and Drug Administration. September 14, 2017. [Google Scholar]
- 3.“FDA Approval for duvelisib (COPIKTRA, Verastem, Inc.) for adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL)”. US Food and Drug Administration September 24, 2018. September 24, 2018. [Google Scholar]
- 4.“FDA approves first PI3K inhibitor for breast cancer”. US Food and Drug Administration 2019-May-22019-05-24. [Google Scholar]
- 5.Raedler LA. Zydelig (Idelalisib): First-in-Class PI3 Kinase Inhibitor Approved for the Treatment of 3 Hematologic Malignancies. American health & drug benefits. 2015;8(Spec Feature): 157–162. [PMC free article] [PubMed] [Google Scholar]
- 6.Blair HA. Duvelisib: First Global Approval. Drugs. 2018;78(17):1847–1853. [DOI] [PubMed] [Google Scholar]
- 7.Juric D, Janku F, Rodon J, et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2019;5(2):e184475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurtz J-E, Ray-Coquard I. PI3 Kinase Inhibitors in the Clinic: An Update. Anticancer Research. 2012;32(7): 2463–2470. [PubMed] [Google Scholar]
- 9.Janku F Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: from laboratory to patients. Cancer Treat Rev. 2017;59:93–101. [DOI] [PubMed] [Google Scholar]
- 10.Zhao W, Qiu Y, Kong D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharmaceutica Sinica B. 2017;7(1):27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elmenier FM, Lasheen DS, Abouzid KAM. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur J Med Chem. 2019;183:111718. [DOI] [PubMed] [Google Scholar]
- 12.Oh T, Ivan ME, Sun MZ, et al. PI3K pathway inhibitors: potential prospects as adjuncts to vaccine immunotherapy for glioblastoma. Immunotherapy. 2014;6(6):737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103. [DOI] [PubMed] [Google Scholar]
- 14.Fan Q-W, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9(5):341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiarini F, Falà F, Tazzari PL, et al. Dual Inhibition of Class IA Phosphatidylinositol 3-Kinase and Mammalian Target of Rapamycin as a New Therapeutic Option for T-Cell Acute Lymphoblastic Leukemia. Cancer Res. 2009;69(8):3520–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raynaud FI, Eccles S, Clarke PA, et al. Pharmacologic Characterization of a Potent Inhibitor of Class I Phosphatidylinositide 3-Kinases. Cancer Res. 2007;67(12):5840–5850. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Mishra R, Yuan L, et al. Oxidative cyclization-induced activation of a phosphoinositide 3-kinase inhibitor for enhanced selectivity of cancer chemotherapeutics. ChemMedChem. 2019;14(22):1933–1939. [DOI] [PubMed] [Google Scholar]
- 18.Lippert AR, Van de Bittner GC, Chang CJ. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc Chem Res. 2011;44(9):793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van de Bittner GC, Dubikovskaya EA, Bertozzi CR, Chang CJ. In vivo imaging of hydrogen peroxide production in a murine tumor model with a chemoselective bioluminescent reporter. Proc Natl Acad Sci. 2010;107(50):21316–21321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Q, Zhong Q, Zhang Q, Zheng S, Wang G. Boron-Based 4-Hydroxytamoxifen Bioisosteres for Treatment of de Novo Tamoxifen Resistant Breast Cancer. ACS Med Chem Lett. 2012;3(5):392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Zhong Q, Zhang Q, Zheng S, Miele L, Wang G. Boronic prodrug of endoxifen as an effective hormone therapy for breast cancer. Breast Cancer Res Treat. 2015;152(2):283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo S, Zhang C, Bratton M, et al. ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models. Oncotarget. 2018;9(6):6924–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Zheng S, Akerstrom VL, et al. Fulvestrant-3 Boronic Acid (ZB716): An Orally Bioavailable Selective Estrogen Receptor Downregulator (SERD). J Med Chem. 2016;59(17):8134–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Zheng S, Guo S, et al. Rational Design of a Boron-Modified Triphenylethylene (GLL398) as an Oral Selective Estrogen Receptor Downregulator. ACS Med Chem Lett. 2017;8(1):102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong Y, Grembecka J, Edler MC, et al. Structure-Based Discovery of a Boronic Acid Bioisostere of Combretastatin A-4. Chem Biol. 2005;12(9):1007–1014. [DOI] [PubMed] [Google Scholar]
- 26.Jourden JLM, Daniel KB, Cohen SM. Investigation of self-immolative linkers in the design of hydrogen peroxide activated metalloprotein inhibitors. Chem Commun. 2011;47(28):7968–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Xie S, Ma L, Chen Y, Lu W. 10-Boronic acid substituted camptothecin as prodrug of SN-38. Eur J Med Chem. 2016;116:84–89. [DOI] [PubMed] [Google Scholar]
- 28.Govan JM, McIver AL, Riggsbee C, Deiters A. Hydrogen peroxide induced activation of gene expression in mammalian cells using boronate estrone derivatives. Angew Chem Int Ed. 2012;51 (36):9066–9070. [DOI] [PubMed] [Google Scholar]
- 29.Park H, McEachon JD, Pollock JA. Synthesis and characterization of hydrogen peroxide activated estrogen receptor beta ligands. Bioorg Med Chem. 2019;27(10):2075–2082. [DOI] [PubMed] [Google Scholar]
- 30.Zheng S, Guo S, Zhong Q, et al. Biocompatible boron-containing prodrugs of belinostat for the potential treatment of solid tumors. ACS Med Chem Lett. 2018;9(2):149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Q, Zhang C, Zhang Q, Miele L, Zheng S, Wang G. Boronic prodrug of 4-hydroxytamoxifen is more efficacious than tamoxifen with enhanced bioavailability independent of CYP2D6 status. BMC Cancer. 2015;15(1):625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Guo S, Yang L, et al. Metabolism, pharmacokinetics, and bioavailability of ZB716, a Steroidal Selective Estrogen Receptor Downregulator (SERD). Oncotarget. 2017;8(61):103874–103889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Guo S, Zhong Q, et al. Metabolism and Pharmacokinetic Study of the Boron-Containing Prodrug of Belinostat (ZL277), a Pan HDAC Inhibitor with Enhanced Bioavailability. Pharmaceuticals. 2019;12(4):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou DH-C, Webber MJ, Tang BC, Lin AB, Thapa LS, Deng D, et al. Glucose-responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc Natl Acad Sci USA. 2015;112(8):2401–2406. 10.1073/pnas.1424684112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayakawa M, Kaizawa H, Moritomo H, et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem. 2006;14(20):6847–6858. [DOI] [PubMed] [Google Scholar]
- 36.Hayakawa M, Kaizawa H, Moritomo H, et al. Synthesis and biological evaluation of pyrido[3′,2′:4,5]furo[3,2-d] pyrimidine derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem Lett. 2007;17(9):2438–2442. [DOI] [PubMed] [Google Scholar]
- 37.Thompson ALS, Kabalka GW, Akula MR, Huffman JW. The conversion of phenols to the corresponding aryl halides under mild conditions. Synthesis. 2005;2005(04):547–550. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.