

Abstract
Infectious hematopoietic necrosis virus (IHNV) is the causative agent of IHN triggering a systemic syndrome in salmonid fish. Although IHNV has always been associated with low levels of mortality in Italian trout farming industries, in the last years trout farmers have experienced severe disease outbreaks. However, the observed increasing virulence of IHNV is still based on empirical evidence due to the poor and often confounding information from the field. Virulence characterization of a selection of sixteen Italian isolates was performed through in vivo challenge of juvenile rainbow trout to confirm field evidence. The virulence of each strain was firstly described in terms of cumulative mortality and survival probability estimated by Kaplan–Meier curves. Furthermore, parametric survival models were applied to analyze the mortality rate profiles. Hence, it was possible to characterize the strain-specific mortality peaks and to relate their topology to virulence and mortality. Indeed, a positive correlation between maximum mortality probability and virulence was observed for all the strains. Results also indicate that more virulent is the strain, the earliest and narrowest is the mortality peak. Additionally, intra-host viral quantification determined in dead animals showed a significant correlation between viral replication and virulence. Whole-genome phylogeny conducted to determine whether there was a relation between virulence phenotype and IHNV genetics evidenced no clear clustering according to phenotype. Moreover, a root-to-tip analysis based on genetic distances and sampling date of Italian IHNV isolates highlighted a relevant temporal signal indicating an evolving nature of the virus, over time, with the more virulent strains being the more recent ones. This study provides the first systematic characterization of Italian IHNV’s virulence. Overall results confirm field data and point out an abrupt increase in IHNV virulence, with strains from 2015–2019 showing moderate to high virulence in rainbow trout. Further investigations are needed in order to extensively clarify the relation between evolution and virulence of IHNV and investigate the genetic determinants of virulence of this viral species in rainbow trout.
Keywords: IHNV, virulence, rainbow trout
1. Introduction
Infectious hematopoietic necrosis (IHN) is an OIE/World Organization for Animal Health-listed infectious disease triggering a systemic syndrome in salmonid fish (OIE/World Organization for Animal Health 2019) and it is caused by a bullet-shaped, enveloped virus (infectious hematopoietic necrosis virus, IHNV) belonging to the species Salmonid Novirhabdovirus of the genus Novirhabdovirus (ICTV/International Committee on Taxonomy of Viruses 2020). Its negative-sense, single-stranded RNA genome architecture consists of six genes in the order 3ʹ-N-P-M-G-NV-L-5ʹ, which encode six structural and non-structural proteins: the nucleocapsid protein (N), the phosphoprotein (P), the matrix protein (M), the glycoprotein (G), the non-virion protein (NV), and the polymerase protein (L) (Kurath et al. 1985; Morzunov, Winton, and Nichol 1995; Schutze et al. 1995). The phylogenetic analysis of the glycoprotein gene of IHNV has enabled the identification of five genogroups named U (upper), M (middle), L (lower), E (European), and J (Japanese) (Kurath et al. 2016) presenting characteristic geographic patterns (Kurath et al. 2003; Enzmann et al. 2005, 2010; Nishizawa et al. 2006; Kuzmin et al. 2009; He et al. 2013).
Originally enzootic in North America, IHNV spread in Europe from its M ancestor forming the monophyletic genogroup E (Enzmann et al. 2010) that eventually established in rainbow trout (Oncorhynchus mykiss). Phylogenetic analyses support the hypothesis that IHNV introduction in Europe had occurred through the transportation of infected fish and contaminated eggs (Kurath et al. 2003; Enzmann et al. 2010). The first descriptions of IHN in Italy date back to 1987 (Bovo et al. 1987). Despite the prevention and surveillance programs that have been applied in fish farms since then and even though movements of live animals between member states are regulated within the European Union (Directive 2006/88/EC), IHN still represents a major threat to Italian and European trout farming, as well as to the Northern European salmon industry that still remains free from this listed virus.
A recent study carried out over a 30-year period focused on the molecular evolution and phylogeography of IHNV and viral hemorrhagic septicemia virus (VHSV) in Italy showed that all IHNV isolates fell within genogroup E and formed a number of clusters possibly representing independent introduction events or the parallel evolution of separate viral lineages (Abbadi et al. 2016). Notably, IHNV shows a higher substitution rate and is subject to stronger positive selective pressure than VHSV (mean rates of 11 and 7.3 × 10–4 nucleotide substitutions per site, per year, respectively) (Abbadi et al. 2016). Historically, viral hemorrhagic septicemia has been identified as the major issue for Italian and European trout farms, with a higher mortality rate than the one caused by IHN. Unexpectedly, in the last years, Italian trout farmers have experienced more and more severe disease outbreaks caused by IHNV, which appears to have gained virulence in rainbow trout (Toffan, personal observations). However, due to the confounding factors always present under field conditions (e.g. co-occurring infections, fish farming conditions, and medical treatments) and to the lack of record keeping on past IHN outbreaks, the increased virulence of IHNV remains just a mere empirical observation.
To confirm field evidence and investigate the hypothesis that IHNV has been gaining virulence in recent years, a selection of Italian isolates with different genetic features and covering a wide time span was used to challenge rainbow trout juveniles under controlled conditions. Their virulence was described in terms of cumulative mortality, rate and time of maximum mortality, duration of the mortality event, and intra-host viral replication. Data herein presented will help unravel the mechanisms orchestrating virulence of Italian IHNV strains and will provide useful information for future studies focused on understanding the evolution and virulence of this viral species.
2. Materials and methods
2.1. Viral strains selection
IHNV (n = 16) used for in vivo trials were isolated from different rainbow trout (O. mykiss) farms, located in northeastern Italy, during active and passive surveillance activities carried out by the National Reference Laboratory for fish diseases located at the Istituto Zooprofilattico Sperimentale delle Venezie (IZSVe). Viruses were selected taking into account the following criteria: (1) field information related to clinical signs and mortality at farm level, (2) collection date, and (3) viral genetic features. In particular, IHNV strains were selected to represent the genetic variability observed for such viruses in Italy, according to the molecular characterization presented in Abbadi et al. (2016), which highlighted the simultaneous circulation of different viral lineages (Supplementary Fig. S1). Detailed information related to each virus and abbreviations of names we are going to refer to throughout the manuscript are reported in Table 1.
Table 1.
Italian IHNV strains isolated from O. mykiss and used in the present study for experimental challenges.
| Strain | Abbreviation | Year | Genetic group within genogroup E | Coinfection in the field | Size/age of field infected fish | Reported virulence in the filed | Virulence in in vivo trial | Acc. number |
|---|---|---|---|---|---|---|---|---|
| IHNV/O.mykiss/I/TN/459/Sep97 | IHNV/459/97 | 1997 | non A | NO | n.a. | n.a. | Moderate | MN914166b |
| IHNV/O.mykiss/I/TN/139/Mar01 | IHNV/139/01 | 2001 | non A | NO | n.a. | n.a. | Low | MK829690c |
| IHNV/O.mykiss/I/TV/3/Dec02 | IHNV/3/02 | 2002 | non A | YES (VHSV) | n.a. | n.a. | Low | MK829692c |
| IHNV/O.mykiss/I/TV/310/May05 | IHNV/310/05 | 2005 | A | NO | 100–300 gr | Low | Low | MK829693c |
| IHNV/O.mykiss/I/VI/409/Nov06 | IHNV/409/06 | 2006 | A | NO | 1 year | Low | Low | MK829696c |
| IHNV/O.mykiss/I/TN/534/Dec11 | IHNV/534/11 | 2011 | non A | NO | 60–700 gr | Low | Low | MK829700c |
| IHNV/O.mykiss/I/VR/143/Apr14 | IHNV/143/14 | 2014 | A | NO | juveniles | Low | Low | MK829701c |
| IHNV/O.mykiss/I/TN/62/Feb15 | IHNV/62/15 | 2015 | non A | YES (VHSV) | adult trout | High | Moderate | MK829704c |
| IHNV/O.mykiss/I/TN/68/Feb15 | IHNV/68/15 | 2015 | A | YES (VHSV) | 30–40 gr | High | Moderate | MK829703c |
| IHNV/O.mykiss/I/TN/86/Feb15 | IHNV/86/15 | 2015 | non A | YES (VHSV) | 500–600 gr | High | Moderate | MK829702c |
| IHNV/O.mykiss/I/TN/509/Oct15 | IHNV/509/15 | 2015 | A | NO | 50–60 gr | High | High | MK829705c |
| IHNV/O.mykiss/I/TN/72/Feb16 | IHNV/72/16 | 2016 | non A | YES (VHSV) | 100 gr | High | High | MN914161b |
| IHNV/O.mykiss/I/TN/220/Mar18 | IHNV/220/18 | 2018 | A | YES (VHSV) | 60–80 gr | High | High | MN914163b |
| IHNV/O.mykiss/I/TN/223/Mar18 | IHNV/223/18 | 2018 | non A | YES (VHSV) | a | Low | High | MN914164b |
| IHNV/O.mykiss/I/TN/224/Mar18 | IHNV/224/18 | 2018 | non A | YES (VHSV) | adult trout | High | High | MN914165b |
| IHNV/O.mykiss/I/TN/91/Feb19 | IHNV/91/19 | 2019 | non A | YES (VHSV) | 60–80 gr | High | High | MN914162b |
2.2. Virus propagation and plaque purification
Viruses stored at −80°C at the IZSVe repository were propagated in epithelioma papulosum cyprini (EPC) cell monolayers according to standard procedures. Plaque purification of each viral isolate was executed according to Panzarin et al. (2020). Only discrete plaques (i.e. lytic foci of the cells monolayer) with defined edges were collected. Each plaque was diluted with 350 µl of minimum essential medium (MEM) Eagle (Sigma-Aldrich), and 200 µl of each diluted plaque were then used to infect 24-h-old EPC cells seeded in Falcon® 12-well polystyrene culture plates (Corning) for propagation at 15°C. This step was necessary to multiply the viruses collected after the first plaque-purification round and to obtain sufficient material for purity check by qualitative real-time RT-PCR (rRT-PCR) targeting VHSV, IHNV, and the infectious pancreatic necrosis virus (IPNV) (Overturf, Lapatra, and Powell 2001; Jonstrup et al. 2013, Orpetveit et al. 2010). Purity assessment was indeed a prerequisite for the following plaque-purification steps, since VHSV, IHNV, and IPNV can be found in coinfection as previously reported (Abbadi et al. 2016). Upon satisfactory rRT-PCR results, two additional plaque-purification and propagation rounds were performed as described above. For each strain, a final amplification was then performed in three 75 cm2 flasks (Falcon®) and the cell culture supernatant was collected at the appearance of complete cytopathic effect, pooled, clarified for 10 min at 2,800 g at 4°C, and checked again by rRT-PCR (Overturf, Lapatra, and Powell 2001; Orpetveit et al. 2010; Jonstrup et al. 2013) and sequencing (Abbadi et al. 2016). Viral titers were expressed as TCID50/ml (Fifty-percent tissue culture infectious dose/ml) according to Reed and Müench formula (Reed and Muench 1938). Viral batches were finally aliquoted and stored at −80°C until use.
2.3. Ethics statement
The procedures for animal experiments were evaluated by the Animal Welfare Body and the Ethics Committee of the IZSVe, according to the Directive 2010/63/EU on the protection of animals used for scientific purposes implemented at a national level through Legislative Decree No. 26/2014. The experimental design was finally authorized by the Italian Ministry of Health (authorization no. 735/2016-PR 22/7/2016).
2.4. Experimental challenges
A batch of 935 rainbow trout (O. mykiss) was purchased from a commercial farm classified within Category I according to EU Directive 88/2006. Fish (average weight of 0.6 g) were acclimated for 10 days in a single 2,500-l tank at 10 ± 2°C and tested against main fish pathogens. Fish were fed with commercial feed for the entire duration of the trial. After acclimation, rainbow trout were divided into seventeen experimental groups of 55 fish each, housed in 80-l tanks, at 10 ± 2°C, in a natural light/dark cycle (14 h/10 h) and challenged with 104 TCID50/ml of each viral strain (Table 1). The challenge was performed by immersion for 3 h in static water reducing the level from 80 to 20 l and pouring the needed amount of virus directly into the tank. Additional aeration was supplied during the infection. At the end of the challenge, the water level was restored by adding clean freshwater (10 l/h). One group out of seventeen was mock infected with sterile MEM Eagle (Sigma-Aldrich) to act as negative control. Fish were monitored daily for a 4-week period. Daily mortality was recorded, and moribund and dead fish were collected. At the end of the observation period, survivor fish were euthanized by overdose of anesthetic (Tricaine, Pharmaq). Dead fish and survivors were stored at −80°C until use.
2.5. Assessment of IHNV quantitative real-time RT-PCR performances
2.5.1. Preparation of in vitro transcribed RNA standards
The reference isolate IT-217A (Johansson et al. 2009) was used as source of RNA and purified using the NucleoSpin RNA kit (Macherey-Nagel). Partial N gene of IHNV was amplified with the Qiagen® OneStep RT-PCR Kit (Qiagen) according to the manufacturer’s instructions. Oligonucleotides used for RT-PCR are reported in Supplementary Table S1. To allow the subsequent RNA in vitro transcription, the forward primer incorporated the T7 RNA polymerase promoter sequence. The RT-PCR was performed under the following thermal condition: 50°C for 30 min, 95°C for 15 min, and followed by 35 cycles of 30-s denaturation at 94°C, 1 min annealing at 58°C, and 1 min elongation at 72°C; the reactions were terminated with 10-min elongation at 72°C. PCR products were analyzed for purity and size by gel electrophoresis.
In vitro transcription reaction was performed at 37°C for 4 h using the MEGAshortscript™ T7 Transcription Kit (ThermoFisher Scientific) following the manufacturer’s instructions. The reaction was completed by incubation for 15 min at 37°C with 1 μl of TURBO DNase. IHNV synthetic RNA was then purified using the MEGAclear™ Transcription Clean-Up Kit (ThermoFisher Scientific) following the manufacturer’s recommendations. In vitro transcribed RNA was then quantitated with the Qubit® RNA High Sensitivity (HS) Assay kit (ThermoFisher Scientific), supplemented with 40 U of RNAsin Plus RNase Inhibitor (Promega), aliquoted, and stored at −80°C until use.
2.5.2. Determination of the limit of detection and limit of quantification of the qRT-PCR protocol
Ten-fold serial dilutions of IHNV in vitro transcribed RNA were prepared in order to develop standard curves to determine the limit of detection (LoD) and limit of quantification (LoQ) of the quantitative real-time RT-PCR (qRT-PCR) protocol adopted. The concentration of RNA used ranged between 10–1 and 108 copy number per µl. Each dilution was tested in triplicate, together with a no-template control. The employed primers and probe (Supplementary Table S1) targeted a conserved region of the nucleocapsid gene, according to Purcell et al. (2013). Quantitative qRT-PCR reactions were carried out in a final volume of 25 µl using the QuantiTect Multiplex RT-PCR Kit (Qiagen) in a reaction mix containing 1× RT-PCR master mix, 0.6 µM of each primer, 0.25 µM of probe, 0.2 µl of enzyme mix, and 5 µl of in vitro transcribed RNA template dilutions. Amplification reactions were performed in a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) with the following thermal profile: 20 min incubation at 50°C, 15 min of initial denaturation at 95°C, 45 cycles of 45-s denaturation at 94°C, and 45-s annealing at 60°C. Amplification data were analyzed with the Bio-Rad CFX Manager software V3.1. The slopes of the standard curves were used to calculate the percentage of reaction efficiency (E) of the qRT-PCR assays according to the equation E% = (10–1/slope − 1) × 100.
LoD was defined as the lowest sample dilution for which all the replicates tested positive. LoQ was determined by the lowest sample dilution for which all the replicates tested positive with a coefficient of variation (CV) of the estimated copy numbers < 40 per cent (Bustin et al. 2009). Such a threshold ensured that concentration values obtained for each dilution were contained within a range smaller than 1 log10 unit, with an error rate lower than 5 per cent.
2.6. Quantitation of the IHNV gene copy number in experimentally infected fish
Intra-host viral loads for each experimental group was assessed in five dead specimens collected at the peak of mortality predicted by each model (see Table 2 and Supplementary Fig. 2A, B and C) or in its proximity, assuming that all the dead animals were positive for the respective challenge virus. Viral replication was also estimated in all the survivor fish euthanized at the end of the observation period for each experimental group. Fish were analyzed singularly. After removal of the caudal fin and gut, the body trunk (approximately 1 g) was homogenized with sterile quartz powder and diluted with 5 ml of MEM Eagle (Sigma-Aldrich). Homogenates were subsequently clarified for 10 min at 2,800 g at 4°C. Total nucleic acids were purified from 160 µl of tissues homogenate supernatant using the automatic Microlab STARlet (Hamilton) and employing the LSI MagVet™ Universal Isolation Kit (ThermoFisher Scientific), according to the manufacturers’ instructions. Total RNA was quantitated with the Qubit® RNA HS Assay kit (ThermoFisher Scientific) and all the samples were normalized at a RNA concentration of 5 ng µl−1.
Table 2.
Mean value and CI of the mortality peak parameters max_rate, tmax_rate, and fwhm estimated for each IHNV strain by the parametric flexible survival model.
| Strain | max_rate (95% CI) | tmax_rate (dpi) (95% CI) | fwhm (days) (95% CI) | Virulence |
|---|---|---|---|---|
| IHNV/139/01 | 0.012 (0.007–0.022) | 12.39 (9.77–13.84) | 10.05 (n.d.) | Low |
| IHNV/3/02 | n.d. (n.d.) | n.d. (n.d.) | n.d. (n.d.) | Low |
| IHNV/310/05 | 0.009 (0.004–0.017) | 12.45 (10.03–14.17) | 9.99 (n.d.) | Low |
| IHNV/409/06 | 0.013 (0.008–0.022) | 13.49 (10.76–17.16) | 15.34 (n.d.) | Low |
| IHNV/534/11 | 0.003 (0.001–0.010) | 16.81 (11.58–30.00) | n.d. (n.d.) | Low |
| IHNV/143/14 | 0.005 (0.002–0.011) | 14.42 (11.17–30.00) | n.d. (n.d.) | Low |
| IHNV/459/97 | 0.029 (0.023–0.041) | 15.41 (12.56–18.90) | n.d. (n.d.) | Moderate |
| IHNV/62/15 | 0.045 (0.033–0.059) | 10.18 (8.79–11.52) | 9.07 (7.66–10.92) | Moderate |
| IHNV/86/15 | 0.056 (0.042–0.073) | 9.54 (8.38–10.94) | 8.54 (7.09–10.05) | Moderate |
| IHNV/68/15 | 0.061 (0.048–0.080) | 9.02 (8.03–10.30) | 8.13 (6.80–9.41) | Moderate |
| IHNV/509/15 | 0.078 (0.063–0.094) | 9.83 (8.79–10.99) | 8.48 (7.21–9.76) | High |
| IHNV/72/16 | 0.117 (0.100–0.141) | 9.48 (8.44–10.30) | 6.86 (5.93–7.85) | High |
| IHNV/220/18 | 0.115 (0.097–0.139) | 8.61 (7.80–9.54) | 6.74 (5.64–7.79) | High |
| IHNV/223/18 | 0.160 (0.135–0.191) | 8.26 (7.57–8.84) | 5.35 (4.53–6.28) | High |
| IHNV/224/18 | 0.193 (0.162–0.230) | 7.97 (7.39–8.44) | 4.59 (3.95–5.29) | High |
| IHNV/91/19 | 0.164 (0.138–0.191) | 8.26 (7.68–8.96) | 5.29 (4.53–6.22) | High |
n.d. = not determinable.
Figure 2.
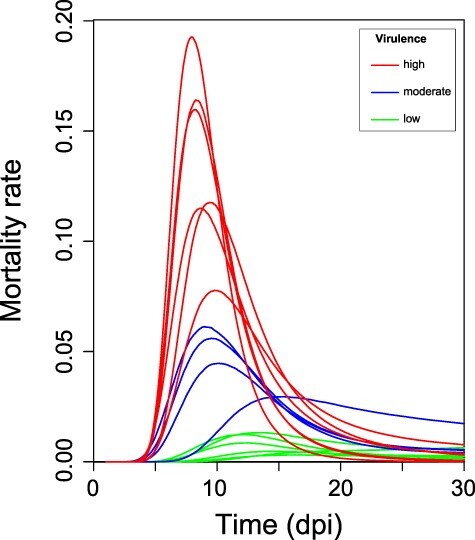
Mortality rate profiles of the IHNV isolates. The depicted curves represent the negative slope of the survival function of each isolate, inferred by the parametric survival model presented in this work. Lines were color-coded according to the virulence of the analyzed IHNV strain.
The detection and absolute quantification of IHNV gene copy number was carried out applying the qRT-PCR protocols described above (Section 2.5.2) and using 25 ng of total RNA as input template. Absolute quantitation of IHNV RNA, expressed as gene copy number/ng of total RNA (CN/ng), was performed by interpolation of the Ct values obtained with the standard curve developed with six dilutions of in vitro transcribed RNA tested in triplicate. Dilutions ranged between 5 × 101 and 5 × 106 copy number/reaction. For each run, linearity (R2), reaction efficiency (E), and slope were extrapolated from the standard curves.
2.7. Statistical analysis
2.7.1. Determination of sample size for experimental challenges
In order to discriminate between the different virulence categories defined in Section 2.7.2, the theoretical number of animals for each experimental challenge group was determined by conditional Fisher’s exact test for two proportions with Walters’ normal approximation, using the statistical software SAS, assuming an α error of 0.05 (one tail) and power 1 − β = 0.90.
2.7.2. Survival analysis and mortality kinetics modeling
The analyses described below were conducted under R environment (R Development Core Team 2018) using the packages survival (Therneau 2020), flexsurv (Jackson 2016), and betareg (Grün, Kosmidis, and Zeileis 2012).
Mortality data were first analyzed by plotting Kaplan–Meier survival curves for each strain. According to the cumulative per cent mortality (CPM) recorded for each IHNV field isolate, viruses were arbitrarily categorized into three classes of virulence: (1) CPM ≤ 40 per cent—low virulent, (2) 40 per cent < CPM < 70 per cent—moderate virulent, and (3) CPM ≥ 70 per cent—high virulent. Log-rank tests were conducted on strains belonging to the same virulence class in order to assess the homogeneity of each group. In addition, the mortality rate determined by each strain was characterized by analyzing the event density function that coincides with the negative slope of the survival function S(t). The event density function is better described by parametric models, in contrast to discrete assessments of S(t) from non-parametric Kaplan–Meier estimators or semi-parametric Cox proportional hazard models. For this reason, different models based on Weibull, exponential, gamma, generalized-gamma, log-normal, log-logistic, and Gompertz distributions were tested, but none showed an acceptable fit with the observed data. Therefore, flexible parametric survival models (Royston and Parmar 2002; Jackson 2016) were used, in which the baseline survival was modeled by means of restricted cubic splines that permitted obtaining smooth survivor curves and better fit to the data, as assessed by comparisons with Kaplan–Meier curves. The methodology and model building strategy are described in Supplementary Methods. For each viral strain, event density values were predicted over 500 individual time points in the observation period, by multiplying the hazard and survival values assessed by the model. Hence, maximum mortality rate (max_rate), time to maximum mortality rate (tmax_rate), and mortality peak width, expressed as full width at half maximum (fwhm), were calculated. Their confidence intervals (CIs) were obtained from 1,000 simulated mortality rate profiles produced by parametric bootstrap. In order to solely report data supported estimates, CIs were calculated only in case all simulated peaks could be described in terms of max_rate, tmax_rate, and fwhm over the experimental time slot. The relationships between these parameters and mortality (expressed in the range 0–1, with 1 corresponding to 100 per cent) or time were in addition analyzed by means of linear or generalized linear (Grün, Kosmidis, and Zeileis 2012) models. In particular, the trend lines describing mortality and time-dependent max_rate variation were inferred by generalized linear models assuming a beta distribution of the observed values and employing a logit link function.
2.7.3. Intra-host viral replication
Counts of viral gene copy numbers were analyzed by a generalized linear model, employing a left-truncated negative binomial distribution in order to account for the LoQ of the qRT-PCR (counts ≥ 100) and overdispersion of the data (R packages gamlss and gamlss.tr.; Rigby and Stasinopoulos (2005); Stasinopoulos and Rigby (2018); for further details see Supplementary Methods). Viral counts and their relative dispersion (sigma) were modeled via a logarithmic link function. The former were related to the explicative variables cumulative mortality (in the range 0–1), status (alive/dead), and their interaction. Differently, sigma was found to depend on virulence. The statistical significance of each variable was assessed by likelihood ratio test.
2.8. Whole-genome sequencing
A high-throughput sequencing approach was applied in order to obtain the complete genome of all IHNV strains used in this study. Isolates were sequenced using Illumina MiSeq and detailed procedures regarding samples processing, sequencing, and bioinformatics analyses are reported in Zamperin et al. (2019).
2.9. Phylogenetic analysis
A whole-genome phylogeny based on concatenated nucleotide sequences of the coding regions was performed. The consensus sequences obtained were aligned and compared to IHNV complete genomes available in GenBank using MEGA 7.0 (Kumar, Stecher, and Tamura 2016).
The phylogenetic relationship among isolates was inferred using the maximum likelihood (ML) method available in the PhyML program version 3.1 (Guindon et al. 2010). The trees incorporated a general time-reversible (GTR) model of nucleotide substitution with a gamma-distribution of among-site rate variation (with four rate categories, Γ4) and a Subtree pruning and regrafting (SPR) branch-swapping search procedure (Darriba et al. 2012). The robustness of nodes was assessed with 1,000 bootstrap replicates performed using the GTR model. Phylogenetic trees were visualized with the FigTree v1.4 software (http://tree.bio.ed.ac.uk/software/figtree/). A phylogenetic tree based on the complete G gene sequence, comprising a wider sequence dataset, was also performed (Supplementary Fig. S1).
Pairwise amino acid similarities (p-distance method) of Italian IHNV sequences were determined with the MEGA 7.0 software. Analyses were performed firstly among all sequences and then considering those within each class of virulence.
Moreover, we also attempted to explore the association between genetic divergence through time and sampling dates in order to observe whether there is a relevant temporal signal in the dataset. Thus, we performed a regression of root-to-tip genetic distances against sampling date using TempEst v1.5.1 (Rambaut et al. 2016). This analysis used Italian IHNV concatenated nucleotide sequences of the coding regions with the ML phylogenetic tree described above as input.
2.10. Analysis of selection pressures
To assess the selection pressure acting on the codons of each gene, the ratios of non-synonymous (dN) and synonymous (dS) nucleotide substitutions per site were estimated using the HyPhy open source software package available at the datamonkey web server (https://www.datamonkey.org/) (Delport et al. 2010). Posterior probability (PP) values ≥ 0.9 and P-values ≤ 0.1 were considered significant. These ratios were estimated using four different codon-based ML approaches (Yang 2000) including single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), mixed effects model of evolution (MEME), and fast unbiased Bayesian approximation (FUBAR). The analyses employed the best-fitting nucleotide substitution model identified for each dataset and neighbor-joining phylogenetic trees as input.
3. Results
3.1. Survivorship curves
To compare the virulence of different viral isolates in experimentally challenged rainbow trout, we developed Kaplan–Meier curves by plotting for each strain the survival rate over the observation period (Fig. 1). Mock infected fish suffered no mortality. Our data highlighted a great variation in terms of virulence in challenge groups, with cumulative mortality ranging between 5.7 and 100 per cent (Fig. 1; Supplementary Table S2). Based on the previously established virulence categorization, six strains were characterized as low virulent, four as moderately virulent, and six as highly virulent. Notably, the viruses classified as highly virulent (IHNV/509/15, IHNV/72/16, IHNV/220/18, IHNV/223/18, IHNV/224/18, and IHNV/91/19) were the most recent IHNV strains in our selection and determined a dramatic decrease in survival probability in the challenged trout over the first 10 days post infection (dpi), with the appearance of the first dead fish at 4 dpi. Differently and despite being assigned to the high virulence class, strain IHNV/509/15 was notably slower and had a lower final mortality compared to the rest of the high virulence isolates. Hence, isolates belonging to a highly virulent class were characterized by low homogeneity in terms of survival trend, as assessed by log-rank test (P = 2 × 10–15).
Figure 1.
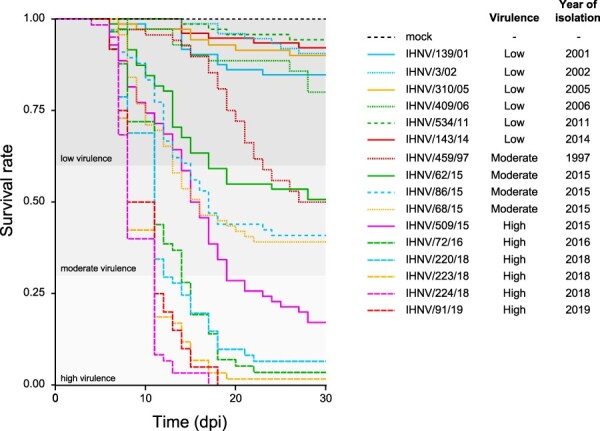
Kaplan–Meier curves of the sixteen IHNV isolates tested for their virulence in rainbow trout. The y-axis reports the survival rate, while the x-axis reports the observation period expressed as days post infection (dpi). The thresholds that distinguish the high (CPM ≥ 70 per cent), moderate (40 per cent < CPM < 70 per cent), and low (CPM ≤ 40 per cent) virulent classes as assessed in this study are also reported.
On the other hand, low-virulent viruses caused mortality with a chronic trend clearly homogeneous among strains, as confirmed also by the log-rank test (P = 0.0746). Over the first half of the observation period, strain IHNV/459/97 showed mortality kinetics comparable to those of low-virulent viruses. However, at 17 dpi we observed a moderate decrease in survival probability, resulting in a cumulative mortality of 50 per cent. Similarly, strains IHNV/86/15 and IHNV/68/15 showed an initial mortality pattern similar to that of the high-virulent strain IHNV/509/15. However, by Day 17 the survival curve of the latter split from that of the former ones, determining the assignment of the three strains to two different virulence categories. The different survival trend curves observed among the moderately virulent strains impeded the designation of this class as homogeneous (P = 0.0454).
3.2. Mortality modeling
In order to further characterize the observed mortality kinetics, we estimated the mortality rate, defined as the death probability per unit of time (day) and described by the event density function. A flexible parametric survival model (Royston and Parmar 2002) was constructed and validated by direct comparison with the relative Kaplan–Meier curves (see Supplementary Methods and Fig. S2A, B and C). For each IHNV strain we then derived the mortality rate profile as well as its 95 per cent CI over the observation period (Fig. 2 and Supplementary Fig S3A, B and C). At a first visual inspection, most of the profiles appeared as a right-skewed peak whose maximum identified the model predicted time of highest death probability (tmax_rate). The uncertainty in the profile shape was clearly higher for IHNV low-virulent strains, because few death events were recorded from fish infected with these isolates. As shown in Fig. 2 and according to Table 2, peak height (maximum mortality rate, max_rate) was positively correlated with virulence. In detail, in low-virulent strains, max_rate ranged between 0.003 and 0.013; increased to 0.029–0.061 for moderately virulent strains; and max_rate ranged between 0.078 and 0.193 for high-virulent strains. In order to better evaluate how virulence could influence peak topology, in addition to height (max_rate), peak position (tmax_rate) and width (fwhm) were also estimated for all the strains (Table 2 and Supplementary Fig. S3A, B and C). Such parameters could not be inferred for strain IHNV/3/02 given the trend in mortality observed for this virus. For the highly and moderately virulent stains, virulence increase appeared to be inversely related with peak width and time of display of maximum mortality rate (tmax_rate), meaning that the more virulent the virus was, the shorter and the earlier the mortality event appeared. Besides, the higher uncertainty in predictions of mortality peaks related to low-virulent strains resulted in wider or non-estimable CIs, in particular for tmax_rate and fwhm (Table 2).
The relationship between max_rate, tmax_rate and fwhm with respect to cumulative mortality was then analyzed. As previously observed, IHNV-mediated death rate was clearly correlated with virulence class (Fig. 3A), as shown by the exponential relationship between max_rate and mortality. The analysis of tmax_rate allowed us to better assess the differences between the three classes of virulence according to the events of maximum mortality. Precisely, isolates belonging to low, moderate, and high class of virulence elicited mortality peaking in mean times ranging between 12.39 and 16.81, 9.02 and 15.41, and 7.97 and 9.83 dpi, respectively. Generally, tmax_rate and cumulative mortality values were inversely related to the classes of virulence as suggested by the slope value (−6.41) (Fig. 3B). Thus, the higher the mortality rate induced by the isolates, the earlier the event of maximum mortality occurred. Similar to tmax_rate, peak widths further contributed to distinguish between the three classes of virulence in terms of duration of the event of mortality. Notably, fwhm median values for the low, moderate, and high class of virulence were 10.05, 8.54, and 6.04 days, respectively. As for tmax_rate, peak widths showed an inverse relation to mortality (slope −6.85) (Fig. 3C).
Figure 3.
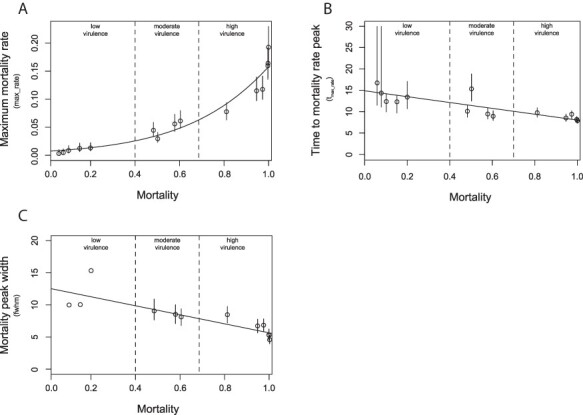
Graphical representation of the relation between IHNV-elicited cumulative mortality and the mortality peak topographic parameters max_rate (A), tmax_rate (B), and fwhm (C). For each viral strain the model predicted means and 95 per cent CIs of the mortality peak parameter are represented as circles and vertical lines. Cumulative mortality values are reported in the 0–1 scale. The vertical dashed lines correspond to the mortality threshold values that determine the different virulence categories.
Further, by relating max_rate, tmax_rate, and fwhm with respect to the year of isolation of the strains, we observed a progressive increase in IHNV virulence over time, with the most recent strains showing a moderate to high virulence in rainbow trout. Indeed, max_rate and year of isolation of the strains were positively correlated (Fig. 4A). Conversely, a negative correlation was observable when comparing the year of isolations of the strain to the tmax_rate and fwhm with slope values of (−0.30) and (−0.37), respectively (Fig. 4B and C). Besides, it is observable that all the calculated parameters related to max_rate, tmax_rate, and fwhm remained almost unchanged between 1996 and 2014, while the trend remarkably changed from 2015 (Fig. 4). Hence, the more recent the strains were, the more severe the induced mortality was.
Figure 4.
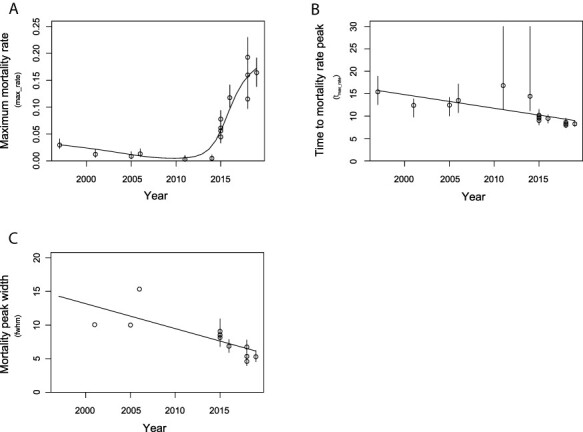
Graphical representation of the relation between IHNV year of isolation and the mortality peak topographic parameters max_rate (A), tmax_rate (B), and fwhm (C). For each viral strain the model predicted means and 95 per cent CIs of the mortality peak parameter are represented as circles and vertical lines, respectively.
3.3. Intra-host viral replication
Upon performing IHNV qRT-PCR protocol characterization by defining LoD and LoQ of 101 and 102 copy number µl−1, respectively, challenged fish were analyzed. The frequency of positive animals among dead specimens was assumed to be 100 per cent, thus viral load was estimated by qRT-PCR on five animals randomly selected at the peak of mortality (tmax_rate) or in its proximity. Differently, as frequency of positive animals was unknown in survivor fish, all specimens euthanized and collected at the end of the experiment were subject to molecular testing in order to estimate their viral load.
Generally, IHNV viral load in dead specimens was highly variable, ranging between 20 and 2.58 × 104 gene CN/ng. Such variability seemed to depend on the virulence class of the isolate under analysis, as it proved smaller, with higher viral loads, among fish infected with highly virulent strains compared with fish infected with medium–low-virulent strains (Supplementary Tables S4 and S5). When looking at replication as a function of cumulative mortality, a positive correlation was observed in dead IHNV-challenged fish (slope = 4.72, 95 per cent CI = 2.78–6.66) (Fig. 5). In contrast, we did not observe any mortality-dependent variation in viral replication in survived fish (slope = 1.38, 95 per cent CI = −1.04–3.80) (Fig. 5, Supplementary Table S4). In addition, no correlation was found between the frequency of observation of the IHNV-positive survivors (Supplementary Table S2) and the virulence class of the challenging viral strain (data not shown).
Figure 5.
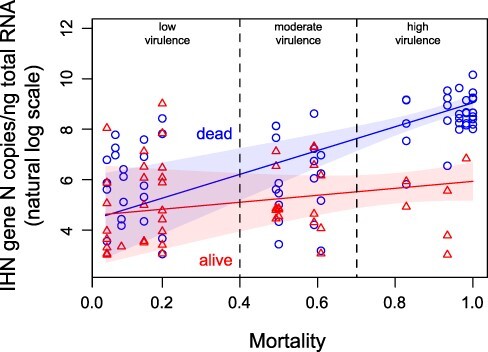
Graphical representation of the relation between IHNV-elicited cumulative mortality and viral titer. Viral titer values, expressed as gene copy number/ng of total RNA (natural log scale), assessed in dead and live animals are depicted as blue circles and red triangles, respectively. With the same color code, model predicted means and 95 per cent CIs are represented by solid lines and shaded regions. The vertical dashed lines correspond to the mortality threshold values determining the different virulence categories.
3.4. Molecular characterization of Italian IHNV
A ML phylogenetic analysis, based on concatenated nucleotide sequences of the complete genomes, confirmed that all the Italian IHNV strains fell within the European genogroup and six isolates (IHNV/220/18, IHNV/509/15, IHNV/68/15, IHNV/409/06, IHNV/310/05, and IHNV/143/14) were found within genetic group A. Two main monophyletic groups were identified in the phylogeny and include the sixteen Italian viruses showing no specific clustering with respect to their virulence phenotype. However, root-to-tip analysis performed on concatenated nucleotide sequences revealed a strong association between genetic distances and sampling dates of the Italian IHNV isolates (correlation coefficient, R2 = 0.8199). Such a result shows the presence of a sufficient temporal signal, which can plausibly be interpreted as the diverging over time of the Italian IHN viruses (Fig. 6).
Figure 6.
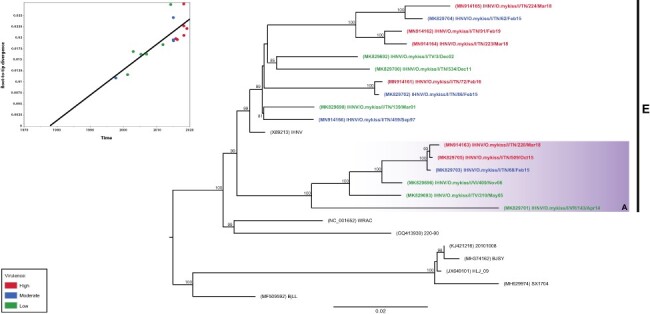
ML phylogenetic tree based on whole-genome concatenated nucleotide sequences of the coding regions. The Italian IHNV isolates are color-coded according to the assigned class of virulence, where red is high (CPM ≥ 70 per cent), blue is moderate (40 per cent < CPM < 70 per cent), and green is low (CPM ≤ 40 per cent). Reference strains sequences are shown in black. IHNV genogroup E is designated by a vertical bar. The colored box represents the genetic group (A) within genogroup E. The numbers at nodes show bootstrap values (only values ≥ 70 per cent are reported). Branch lengths are scaled according to the number of nucleotide substitutions per site. The tree is midpoint rooted for clarity only. The upper-left panel represents the root-to-tip regression of the genetic distance against sampling dates. Dots representing Italian IHNV strains are color-coded as in the phylogeny.
A high variability among strains was corroborated by a low percentage of amino acid similarity calculated among all the Italian IHNV (95.27–99.90 per cent). Such a variability was observed also when calculated within the three classes of virulence (low: 96.01–98.83 per cent; moderate: 96.07–98.17 per cent; and high: 95.87–99.55 per cent) (Table 3).
Table 3.
Percentages of amino acid similarities calculated among all Italian IHNV sequences and within each class of virulence.
| Group | Amino acid sequences similarity (%) |
|---|---|
| All IHNV | 95.27–99.90% |
| Highly virulent | 95.87–99.55% |
| Moderately virulent | 96.07–98.17% |
| Low virulent | 96.01–98.83% |
An association study was also attempted in order to identify the virulence markers correlated with different virulence phenotypes. This approach used a statistical association analysis that combined genetic and mortality data in order to find specific single amino acid polymorphisms in all the viral coding regions presumably involved in IHNV virulence. However, no signature associated with virulence phenotype was detected. The amino acid signatures under positive pressure in the two monophyletic groups were also different, which suggests that their acquisition had occurred as independent selection events (data not shown).
3.5. Selection pressures in IHNV
The non-synonymous (dN) to synonymous (dS) substitutions rates estimated for the N, P, M, G, NV, and L proteins were 0.1028, 0.2258, 0.152, 0.3319, 0.314, and 0.0842, respectively. The lower mean dN/dS ratio suggests that the sequences are undergoing purifying selection over the sampling period. Despite the action of the negative selection on viral coding sequences, a number of codons in the G and L genes were under positive selection (P-value ≤ 0.1; PP ≥ 0.9). Results in Table 4 show the amino acid positions identified by at least two models among SLAC, FEL, MEME, and FUBAR. More precisely, of the four amino acid sites found in the glycoprotein under positive selection, two (247 and 376) were confirmed by three methods (FEL, MEME, and FUBAR). The other two sites (91 and 257) were identified by two methods (FEL and FUBAR). For the polymerase protein, the only site under positive selection (1,524) was confirmed by FEL, MEME, and FUBAR methods.
Table 4.
Amino acid sites under putative positive selection (PSS, positive selected sites) detected using different analytical models (SLAC, FEL, MEME, and FUBAR).
| SLAC | FEL | MEME | FUBAR | ||
|---|---|---|---|---|---|
| Gene | PSS | P-value ≤ 0.1 | PP ≥ 0.9 | ||
| 91 | – | 0.0874 | – | 0.961 | |
| Ga | 247 | – | 0.0298 | 0.0743 | 0.987 |
| 257 | – | 0.0639 | – | 0.967 | |
| 376 | – | 0.0870 | 0.0575 | 0.965 | |
| L | 1,524 | – | 0.056 | 0.0977 | 0.976 |
4. Discussion
The first description of IHNV in Italy dates back to 1987 (Bovo et al. 1987) and since then fish farmers and institutions have made great efforts to control and prevent the spread of the disease. In recent years, IHNV has progressively gained virulence to the point of severely affecting the profitability of trout industry (Abbadi et al. 2016; Kibenge 2018). In the present study, experimental trials were conducted under controlled conditions by challenging juveniles rainbow trout with IHNV isolates differing in terms of genetic features and covering a wide time span. We confirmed the hypothesis of the increasing virulence of Italian IHNV isolates over time, with the most recent strains being more pathogenic. Moreover, we highlighted the suitability of combining in vivo challenges data together with the proposed statistical models and molecular analysis to characterize the virulence of IHNV.
It is worth mentioning that the virus isolates tested here were subjected to three plaque purifications prior to amplification as challenge stocks. This was necessary mainly to separate coinfection of IHNV from VHSV. The plaque-purification assay is able to eliminate the natural quasi-species variation within the original virus population. This means that the virulence of the original virus population may not be fully represented in the challenge stocks. However, the significant variations in virulence suggest that the plaque-purification procedure in EPC cells did not lead to uniform virulence levels, very likely reflecting the variation in the original isolates.
To compare the virulence of the different isolates, we initially developed Kaplan–Meier curves that showed a pronounced heterogeneity of viral phenotypes that led to the arbitrarily classification of viruses into virulence classes based on cumulative mortality, namely low, moderate, and highly virulent. The analyses of homogeneity of each class of virulence through the log-rank test highlighted that low-virulent strains were homogeneous supposing a chronic trend in mortality, while the highly virulent isolates resulted as an inhomogeneous group in terms of survival probability. Interestingly, this latter group included the most recent Italian IHNV strains, from Year 2015 to 2019. Most likely, the variation in virulence of recent strains acted as a driver of epidemiological success among other co-circulating isolates in the same geographic area and population. Similar observations have been broadly reported by other authors. Indeed, Breyta et al. (2016) studied the virulence of different IHNV isolates, involved in displacement events overtime on trout populations and under controlled conditions. Their results suggested that the viral increasing virulence might have potentially influenced both the IHNV emergence and the displacement events in trout populations.
However, solely observing the survival probability was not sufficient to fully describe the virulence of each strain. Consequently, we further characterized the mortality kinetics by estimating the mortality rate defined as the death probability per unit of time (day). Profiles obtained for each strain resulted as right-skewed peak whose maximum identified the predicted time of highest death probability. From these profiles it was also observable that more virulent was the strain, the earliest and narrowest was the mortality peak. Contrarily, a shift to later timing and an increase in duration of the morality peak were characteristics of low-virulent strains. Furthermore, to better evaluate how virulence could influence the peak topology, parameters such as maximum mortality rate (max_rate), time to maximum mortality rate (tmax_rate), and mortality peak width (fwhm) were assessed for each strain. Interestingly, virulence increase, in terms of cumulative mortality, appeared negatively correlated to time to maximum mortality rate (tmax_rate) and peak width (fwhm), while positively related to maximum mortality rate (max_rate). The mortality peak width strongly depends on how scattered over time the death events elicited by the viral strain under analysis are. As reported in Materials and Methods, all fish from the same batch were synchronously exposed to virus. However, such synchrony did not fully reflect into the time of death of each animal, most probably because of intra-population variability in virus–host interactions that determined survival versus death and, in the latter case, the different times of death. In addition, peak width may have been influenced by additional infection rounds caused by shedded virions. However, it is interesting to observe how such dependence of time to death on individual variability weakened with highly virulent viruses, as shown by the inverse relationship between fwhm and cumulative mortality. Thus, in case of highly virulent viruses, mortality seems to depend much more on virus than on host characteristics, therefore narrowing the mortality period and reestablishing the original infection synchrony. In addition, when relating max_rate, tmax_rate, and fwhm with respect to the year of isolation of the strains, similar trends were observed. Hence, overall these data confirmed an abrupt increase of IHNV virulence, with strains from 2015 to 2019 showing moderate to high virulence for rainbow trout. Given these results we then assumed that highly virulent strains could have an advantage over low-virulent strains in terms of viral replication. Notably, Kurath and Wargo (2015) described the virulence as a beneficial viral fitness trait; thus, the higher is the amount of viral loads and the greater is the cell damage and consequently higher are the morbidity and mortality.
To further examine our hypothesis aimed at linking viral replication to virulence, we first performed an intra-host viral quantification on IHNV-challenged fish. Analyses were performed individually on all survivors, while for dead animals we assumed that viral replication was equal to 100 per cent; therefore, we decided to analyze five animals at the time to maximum mortality rate (tmax_rate). Interestingly, dead specimens highlighted a considerable variability in terms of IHNV viral loads (20–2.58 × 104 gene CN/ng) and showed to be dependent on the virulence class of the analyzed samples. Hence, we observed a high variability among fish infected with low-virulent strains and a smaller one in those infected with high-virulent strains. Secondly, we attempted to look at viral replication as a function of virulence, expressed as cumulative mortality. As a result, a significant positive correlation was observed in dead specimens, while no mortality dependency was noticeable in the survivors. Our data are in accordance with those obtained in different in vivo experiments conducted using high- and low-virulent IHNV strains, demonstrating that differences in virulence correlated with significant variation in intra-host viral replication (Wargo, Garver, and Kurath 2010; Wargo and Kurath 2011). Similar evidence has been reported in literature on several virus–host systems, such as for avian influenza, vesicular stomatitis, and swine influenza (Suzuki et al. 2009; Furió et al. 2012; Pascua et al. 2012). It can be hypothesized that replication cycles of viruses with a high fitness result in a massive and faster viral replication within a narrowest time frame, while viruses with low fitness are subject to more frequent abortive replication cycles and/or with less efficiency resulting in asynchronous and time-lapse replication events. Another hypothesis is that highly virulent viruses gained the capacity of an early and rapid in vivo replication, taking advantage from the lag time required for the host innate immune system to be activated, in order to achieve a threshold level and withstand the host immune response (Peñaranda, Purcell, and Kurath 2009; Purcell et al. 2009b).
Additionally, to expand our understanding of the correlation between virulence phenotype and Italian IHNV genetics, a full-genome phylogenetic analysis was also conducted. The purpose of this analysis aimed to clarify the relationship between evolution and virulence for IHNV. Our results demonstrated no evident clustering of the isolates based on the assigned class of virulence. All the strains are resolved into two major well-supported sub-clusters within genotype E, one of which was previously designated as cluster A (Abbadi et al. 2016). Interestingly, both clusters include viruses of high, moderate, and low virulence, indicating a high phenotypic variability within clade. Notably, all the highly virulent strains appeared to originate from low/moderate virulence ancestors, suggesting a step-wise acquisition of virulence signatures in a time fashion manner. Indeed, an interesting result emerged when we investigated whether there was an association between genetic divergence through time and sampling dates. A relevant temporal signal was found within Italian IHNV dataset, suggesting that our strains are probably evolving over time with the more virulent strains being the more recent ones.
A more in-depth analysis of clade-specific signatures highlighted that these are not conserved among the two genetic clusters (data not shown). Besides, our study highlighted that no virulence markers are significantly associated to virulence. Taken together, these data corroborate the hypothesis that acquisition of virulence markers might have occurred through independent evolution mechanisms This might be explained by the fact that virulence markers are spread across the entire viral genome at different positions and that such markers might be acquired by the action of independent selection events. A more extensive dataset of sequences and viral phenotypes is certainly required to further clarify these assumptions. Besides, a high variability was observed in terms of amino acid percentage of similarity, calculated among all Italian IHNV isolates and within isolates of each class of virulence. A similar approach was adopted by Mochizuki et al. (2009) who wanted to evaluate the influence of evolutional divergence to virulence of Japanese IHNV isolates. Their results confirmed the increase in virulence of IHNV over time and also highlighted that nucleotide diversity of Japanese IHNV still continued by changing its virulence in rainbow trout.
We also estimated the non-synonymous (dN) to synonymous (dS) substitutions rate for each gene and results suggested that they are all undergoing purifying events (dN/dS < 1). Generally, purifying selection occurs in a biological mechanism in order to decrease the accumulation of deleterious mutations in the coding sequence, which in RNA viruses repeatedly occurs due to the error-prone nature of their RNA-dependent RNA polymerase (Elena and Sanjuán 2005). Despite the action of purifying selection, five codons were found to be strongly under positive selection (P ≤ 0.1; PP ≥ 0.9) and were confirmed by at least two of the applied analytical methods. It was striking to notice how the majority of these codons were located within the G gene (i.e. 91, 247, 257, and 376) that encodes the only structural protein capable of eliciting innate (Lorenzen et al. 2002) and adaptive (Engelking and Leong 1989; Boudinot et al. 1998) immune responses in host cells. Indeed, the IHNV glycoprotein has a higher genetic diversity compared to the other viral proteins and has been intensely studied either for the identification of antigenic constituent of interest in vaccines development or for phylogenetic analyses (LaPatra, Evilia, and Winston 2008; Bellec et al. 2017). Interestingly, the amino acids detected within the G gene were also reported as being positively selected in other studies (LaPatra, Evilia, and Winston 2008; He et al. 2013; Abbadi et al. 2016; Jalali et al. 2019). Notably, Jalali et al. (2019) through a 3D protein structural analysis evidenced that almost all the identified positively selected sites were mapped to a conserved structure on the surface of the protein. They also highlighted that amino acid variations at Positions 91 and 130 resulted in structural changes in the IHNV glycoprotein, which could be beneficial in terms of fitness advantage. However, other genes have been intensely investigated in order to understand the mechanisms governing IHNV virulence and pathogenicity. These studies generally applied the reverse genetics approach to exert single or multiple mutations on viral proteins involved in the different stages of the infection. For instance, Chen et al. (2019) focused on the viral assembly and budding by exploring the effect of mutations in the L-domains on the M and G of IHNV proteins implicated in specific interactions with host proteins.
Our study confirmed field data and evidenced an increased virulence of recent IHNV strains through experimental challenges and molecular and statistical analysis. However, virulence is the outcome of the continuous host–pathogen interaction during the infection cycle. Hence, it is important to consider each stage in the infection cycle (i.e. host entry, replication in target organs, and shedding) which represents a barrier that the pathogen must overcome to successfully infect the host (Peñaranda, Purcell, and Kurath 2009). Besides, aspects such as competition among strains, transmission routes, host adaptation, environmental conditions, as well as anthropogenic interventions (e.g. trade and sanitary practices) may influence the evolutionary dynamics and the virulence of IHNV (Abbadi et al. 2016; Panzarin et al. 2018).
In the last decades many efforts have been made to better understand the correlation of such aspects with IHNV pathogenicity (Peñaranda, Purcell, and Kurath 2009; Wargo, Garver, and Kurath 2010; Wargo and Kurath 2011; Kell, Wargo, and Kurath 2014; Breyta et al. 2016; Wargo et al. 2017), including the exploration of the transcriptomic response to viral infection (Purcell et al. 2011; Louboutin et al. 2021), thus providing a more complete picture of the pathways involved in host antiviral response to infection. Hence, to better comprehend and characterize Italian IHNVs virulence and evolution in-depth researches are required based, for instance, on transcriptome analyses that provide data useful to explore the effects of virus–host interactions, or else on reverse genetics, used to improve our knowledge of genetic determinants of IHNV virulence in rainbow trout.
Furthermore, an association study combining genetic and phenotype data should be performed for IHNV, as described by Panzarin et al. (2020). This would allow us to investigate the molecular basis of IHNV virulence in rainbow trout. Hence, more efforts toward IHNV whole-genome sequencing should be pursued, as the scarce availability of sequences could affect the statistical power when performing association studies. In addition, such studies combine in vivo virulence records with complete viral genomes, while the correlation of phenotype and genetic data using statistical methods leads to identify candidate virulence markers useful to predict the phenotype of unknown viral sequences.
5. Conclusions
This study is the first systematic characterization of Italian IHNV virulence. Results confirm field data of an abrupt increase in IHNV virulence in a rather short time. Our data suggest that viral replication provides an important contribution in determining IHNV virulence. In addition, the present research highlights the distinctive statistical approach adopted, which made the proposed models suitable to characterize the virulence of IHNV and could also be exploited to study other viruses. Data shown in the present work provide a starting point for further research aiming to comprehend the mechanisms of IHNV pathogenesis and evolution.
Supplementary Material
Acknowledgements
The present work was developed within the framework of the Novimark Project (G88F13000660001). The project is funded under the ERA-Net. The content of this article reflects only the authors’ views and the ERA-Net Consortium is not liable for any use that may be made of the information contained therein. The authors wish to thank Andrea Marsella for his contribution in the experimental trials and Francesca Ellero for manuscript editing.
Contributor Information
Miriam Abbadi, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Michele Gastaldelli, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Francesco Pascoli, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Gianpiero Zamperin, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Alessandra Buratin, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Giulia Bedendo, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Anna Toffan, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Valentina Panzarin, Istituto Zooprofilattico Sperimentale delle Venezie, Viale dell’Università 10, 35020, Legnaro, Padova, Italy.
Data availability
IHNV complete genome sequences generated in this study have been deposited in GenBank under the accession numbers MN914161–MN914166. MiSeq raw data were submitted to the National Center for Biotechnology Information Sequence Read Archive under the accession numbers SRR10973039–SRR10973043 and SRR8943890.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
The present work was developed within the framework of the Novimark Project (G88F13000660001) funded by the Animal Health and Welfare (Anihwa) ERA-Net Consortium and the Current Research project RC IZSVE 13/19 funded by the Ministry of Health. Open access publishing costs were fully covered by the Italian Ministry of Health, Current Research project RC IZSVE 13/19.
Conflict of interest:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author contributions statement
V.P. and A.T. conceived the study and coordinated the work described. M.A., V.P., and M.G. wrote the manuscript. A.B. plaque-purified and propagated viral strains. F.P. and A.T. executed in vivo experimental challenges. G.Z. performed bioinformatics analysis. V.P. and M.A. carried out phylogenetic analysis. Statistical analyses were performed by M.G. M.A. and G.B. carried out biomolecular tests. V.P., M.G., A.T., M.A., and F.P. were involved in the interpretation of the results. All authors read and approved the final manuscript.
References
- Abbadi M. et al. (2016) ‘Molecular Evolution and Phylogeography of Co-circulating IHNV and VHSV in Italy’, Frontiers in Microbiology, 7: 1–13.doi: 10.3389/fmicb.2016.01306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellec L. et al. (2017) ‘Molecular Evolution and Phylogeography of Infectious Hematopoietic Necrosis Virus with a Focus on Its Presence in France over the Last 30 Years’, Journal of General Virology, 98: 2438–46.doi: 10.1099/jgv.0.000894. [DOI] [PubMed] [Google Scholar]
- Boudinot P. et al. (1998) ‘Combined DNA Immunization with the Glycoprotein Gene of Viral Hemorrhagic Septicemia Virus and Infectious Hematopoietic Necrosis Virus Induces Double-Specific Protective Immunity and Nonspecific Response in Rainbow Trout’, Virology, 249: 297–306.doi: 10.1006/viro.1998.9322. [DOI] [PubMed] [Google Scholar]
- Bovo G. et al. (1987) ‘Infectious Hematopoietic Necrosis: First Detection in Italy’, Bulletin of the European Association of Fish Pathologists, 7: 124. [Google Scholar]
- Breyta R. et al. (2016) ‘Increasing Virulence, but Not Infectivity, Associated with Serially Emergent Virus Strains of a Fish Rhabdovirus’, VirusEvolution, 2: vev018.doi: 10.1093/ve/vev018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin S. A. et al. (2009) ‘The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-time PCR Experiments’, Clinical Chemistry, 55: 611–22.doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Chen Y. et al. (2019) ‘The Role of Infectious Hematopoietic Necrosis Virus (IHNV) Proteins in Recruiting the ESCRT Pathway through Three Ways in the Host Cells of Fish during IHNV Budding’, Fish andShellfishImmunology, 92: 833–41.doi: 10.1016/j.fsi.2019.07.011. [DOI] [PubMed] [Google Scholar]
- Darriba D. et al. (2012) ‘jModelTest 2: More Models, New Heuristics and Parallel Computing’, NatureMethods, 9: 772.doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W. et al. (2010) ‘Datamonkey 2010: A Suite of Phylogenetic Analysis Tools for Evolutionary Biology’, Bioinformatics, 26: 2455–7.doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena S. F., and Sanjuán R. (2005) ‘Adaptive Value of High Mutation Rates of RNA Viruses: Separating Causes from Consequences’, Journal of Virology, 79: 11555–8.doi: 10.1128/jvi.79.18.11555-11558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelking H. M., and Leong J. C. (1989) ‘The Glycoprotein of Infectious Hematopoietic Necrosis Virus Elicits Neutralizing Antibody and Protective Responses’, VirusResearch, 13: 213–30.doi: 10.1016/0168-1702(89)90017-8. [DOI] [PubMed] [Google Scholar]
- Enzmann P. et al. (2010) ‘Evolution of Infectious Hematopoietic Necrosis Virus (IHNV), a Fish Rhabdovirus, in Europe over 20 Years: Implications for Control’, Diseases of Aquatic Organisms, 89: 9–15.doi: 10.3354/dao02182. [DOI] [PubMed] [Google Scholar]
- ——— (2005) ‘Infectious Hematopoietic Necrosis Virus: Monophyletic Origin of European Isolates from North American Genogroup M’, Diseases of Aquatic Organisms, 66: 187–95.doi: 10.3354/dao066187. [DOI] [PubMed] [Google Scholar]
- Furió V. et al. (2012) ‘Relationship between Within-host Fitness and Virulence in the Vesicular Stomatitis Virus: Correlation with Partial Decoupling’, Journal of Virology, 86: 12228–36.doi: 10.1128/JVI.00755-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün B., Kosmidis I., and Zeileis A. (2012) ‘Extended Beta Regression in {R}: Shaken, Stirred, Mixed, and Partitioned’, Journal of Statistical Software, 48: 1–25.doi: jstatsoft.org/v48/i11/. [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21.doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- He M. et al. (2013) ‘Dating the Divergence of the Infectious Hematopoietic Necrosis Virus’, Infection, Genetics and Evolution, 18: 145–50.doi: 10.1016/j.meegid.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Available online: ICTV/International Committee on Taxonomy of Viruses . <https://talk.ictvonline.org/taxonomy/> Accessed 30 Jul 2020.
- Jackson C. (2016) ‘{flexsurv}: A Platform for Parametric Survival Modeling in {R}’, Journal of Statistical Software, 70: 1–33.doi: 10.18637/jss.v070.i08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali S. A. H. et al. (2019) ‘Molecular Evolution and Selection Pressure Analysis of Infectious Hematopoietic Necrosis Virus (IHNV) Revealed the Origin and Phylogenetic Relationship of Iranian Isolates in Recent Epidemics in Iran’, Virology, 535: 45–58.doi: 10.1016/j.virol.2019.06.012. [DOI] [PubMed] [Google Scholar]
- Johansson T. et al. (2009) ‘Genetic and Serological Typing of European Infectious Haematopoietic Necrosis Virus (IHNV) Isolates’, Diseases of Aquatic Organisms, 86: 213–21.doi: 10.3354/dao02108. [DOI] [PubMed] [Google Scholar]
- Jonstrup S. P. et al. (2013) ‘Development and Validation of a Novel Taqman-Based Real-time RT-PCR Assay Suitable for Demonstrating Freedom from Viral Haemorrhagic Septicaemia Virus’, Journal ofFishDiseases, 36: 9–23.doi: 10.1111/j.1365-2761.2012.01416.x. [DOI] [PubMed] [Google Scholar]
- Kell A. M., Wargo A. R., and Kurath G. (2014) ‘Viral Fitness Does Not Correlate with Three Genotype Displacement Events Involving Infectious Hematopoietic Necrosis Virus’, Virology, 464–465: 146–55.doi: 10.1016/j.virol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibenge F. S. B. (2018) ‘Emerging Viruses in Aquaculture’, Current Opinion in Virology, 34: 97–103.doi: 10.1016/j.coviro.2018.12.008. [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., and Tamura K. (2016) ‘MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets’, Molecular Biology and Evolution, 33: 1870–4.doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurath G. et al. (1985) ‘Molecular Cloning of the Six mRNA Species of Infectious Hematopoietic Necrosis Virus, a Fish Rhabdovirus, and Gene Order Determination by R-loop Mapping’, Journal of Virology, 53: 469–76.doi: 0022-538X/85/020469-08$02.00/0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2003) ‘Phylogeography of Infectious Haematopoietic Necrosis Virus in North America’, Journal of General Virology, 84: 803–14.doi: 10.1099/vir.0.18771-0. [DOI] [PubMed] [Google Scholar]
- Kurath G., and Wargo A. (2015) ‘Evolution of Viral Virulence: Empirical Studies’. In: Weaver, S. C. et al. eds, Virus Evolution Current Research and Future Directions, pp. 155–213. Portland, OR: Caister Academic Press. [Google Scholar]
- Kurath G. et al. (2016) ‘Atlantic Salmon, Salmo salar L. Are Broadly Susceptible to Isolates Representing the North American Genogroups of Infectious Hematopoietic Necrosis Virus’, Journal ofFishDiseases, 39: 55–67.doi: 10.1111/jfd.12323. [DOI] [PubMed] [Google Scholar]
- Kuzmin I. V. et al. (2009) ‘The Rhabdoviruses: Biodiversity, Phylogenetics, and Evolution’, Infection, Genetics and Evolution, 9: 541–53.doi: 10.1016/J.MEEGID.2009.02.005. [DOI] [PubMed] [Google Scholar]
- LaPatra S. E., Evilia C., and Winston V. (2008) ‘Positively Selected Sites on the Surface Glycoprotein (G) of Infectious Hematopoietic Necrosis Virus’, Journal of General Virology, 89: 703–8.doi: 10.1099/vir.0.83451-0. [DOI] [PubMed] [Google Scholar]
- Lorenzen N. et al. (2002) ‘Immunity Induced Shortly after DNA Vaccination of Rainbow Trout against Rhabdoviruses Protects against Heterologous Virus but Not against Bacterial Pathogens’, Developmental and Comparative Immunology, 26: 173–9.doi: 10.1016/s0145-305x(01)00059-3. [DOI] [PubMed] [Google Scholar]
- Louboutin L. et al. (2021) ‘Comparative Analysis of the Course of Infection and the Immune Response in Rainbow Trout (Oncorhynchus mykiss) Infected with the 5 Genotypes of Infectious Hematopoietic Necrosis Virus’, Virology, 552: 20–31.doi: 10.1016/j.virol.2020.09.003. [DOI] [PubMed] [Google Scholar]
- Mochizuki M. et al. (2009) ‘Virulence Change of Infectious Hematopoietic Necrosis Virus against Rainbow Trout Oncorhynchus mykiss with Viral Molecular Evolution’, FishPathology, 44: 159–65.doi: 10.3147/jsfp.44.159. [DOI] [Google Scholar]
- Morzunov S. P., Winton J. R., and Nichol S. T. (1995) ‘The Complete Genome Structure and Phylogenetic Relationship of Infectious Hematopoietic Necrosis Virus’, VirusResearch, 38: 175–92.doi: 10.1016/0168-1702(95)00056-V. [DOI] [PubMed] [Google Scholar]
- Nishizawa T. et al. (2006) ‘Nucleotide Diversity of Japanese Isolates of Infectious Hematopoietic Necrosis Virus (IHNV) Based on the Glycoprotein Gene’, Diseases of Aquatic Organisms, 71: 267–72.doi: 10.3354/dao071267. [DOI] [PubMed] [Google Scholar]
- OIE/World Organization for Animal Health . (2019) On-line Update of Manual of Diagnostic Tests for Aquatic Animals; Chapter 2.3.4.- Infection with Infectious Haematopoietic Necrosis Virus. <http://www.oie.int/international-standard-setting/aquatic-manual/access-online/> Accessed 30 Jul 2020.
- Orpetveit I. et al. (2010) ‘Detection of Infectious Pancreatic Necrosis Virus in Subclinically Infected Atlantic Salmon by Virus Isolation in Cell Culture or Real-time Reverse Transcription Polymerase Chain Reaction: Influence of Sample Preservation and Storage’, Journal of Veterinary Diagnostic Investigation, 22: 886–95.doi: 10.1177/104063871002200606. [DOI] [PubMed] [Google Scholar]
- Overturf K., Lapatra S., and Powell M. (2001) ‘Real-time PCR for the Detection and Quantitative Analysis of IHNV in Salmonids’, Journal ofFishDiseases, 24: 325–33.doi: 10.1046/j.1365-2761.2001.00296.x. [DOI] [Google Scholar]
- Panzarin V. et al. (2020) ‘VHSV Single Amino Acid Polymorphisms (Saps) Associated with Virulence in Rainbow Trout’, Frontiers in Microbiology, 11: 1984.doi: 10.3389/fmicb.2020.01984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2018) ‘Low Evolutionary Rate of Infectious Pancreatic Necrosis Virus (IPNV) in Italy Is Associated with Reduced Virulence in Trout’, VirusEvolution, 4: vey019.doi: 10.1093/ve/vey019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascua P. N. et al. (2012) ‘Virulence and Transmissibility of H1N2 Influenza Virus in Ferrets Imply the Continuing Threat of Triple-Reassortant Swine Viruses’, Proceedings of the National Academy of Sciences of the United states of America, 109: 15900–5.doi: 10.1073/pnas.1205576109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñaranda M. M. D., Purcell M. K., and Kurath G. (2009) ‘Differential Virulence Mechanisms of Infectious Hematopoietic Necrosis Virus in Rainbow Trout (Oncorhynchus mykiss) Include Host Entry and Virus Replication Kinetics’, Journal of General Virology, 90: 2172–82.doi: 10.1099/vir.0.012286-0. [DOI] [PubMed] [Google Scholar]
- Purcell M. K. et al. (2009b) ‘Infectious Haematopoietic Necrosis Virus Genogroup‐Specific Virulence Mechanisms in Sockeye Salmon, Oncorhynchus nerka (Walbaum), from Redfish Lake, Idaho’, Journal ofFishDiseases, 32: 619–31.doi: 10.1111/j.1365-2761.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- ——— (2011) ‘Transcriptome Analysis of Rainbow Trout Infected with High and Low Virulence Strains of Infectious Hematopoietic Necrosis Virus’, Fish and ShellfishImmunology, 30: 84–93.doi: 10.1016/j.fsi.2010.09.007. [DOI] [PubMed] [Google Scholar]
- ——— (2013) ‘Universal Reverse-transcriptase Real-time PCR for Infectious Hematopoietic Necrosis Virus (IHNV)’, Diseases of Aquatic Organisms, 106: 103–15.doi: 10.3354/dao02644. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . (2018) ‘R: A Language and Environment for Statistical Computing’, R Foundation for Statistical Computing; Vienna, Austria: 2019. <https://www.R-project.org> accessed on 21 June 2021.doi: 10.1007/978-3-540-74686-7. [DOI] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, VirusEvolution, 2: vew007.doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L. J., and Muench H. (1938) ‘A Simple Method of Estimating Fifty per Cent Endpoints’, American Journal of Epidemiology, 27: 493–7.doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- Rigby R. A., and Stasinopoulos D. M. (2005) ‘Generalized Additive Models for Location, Scale and Shape (with Discussion)’, Applied Statistics, 54: 507–54. [Google Scholar]
- Royston P., and Parmar M. K. B. (2002) ‘Flexible Parametric Proportional-Hazards and Proportional-Odds Models for Censored Survival Data, with Application to Prognostic Modelling and Estimation of Treatment Effects’, Statistics in Medicine, 21: 2175–97.doi: 10.1002/sim.1203. [DOI] [PubMed] [Google Scholar]
- Schutze H. et al. (1995) ‘Complete Genomic Sequence of the Fish Rhabdovirus Infectious Haematopoietic Necrosis Virus’, Journal of General Virology, 76: 2519–27.doi: 10.1099/0022-1317-76-10-2519. [DOI] [PubMed] [Google Scholar]
- Stasinopoulos M., and Rigby B. (2018) Gamlss.tr: Generating and Fitting Truncated ‘gamlss.family’ Distributions. R package version 5.1-7. Available online: <https://cran.r-project.org/package=gamlss.tr> Accessed 30 Jul 2020.
- Suzuki K. et al. (2009) ‘Association of Increased Pathogenicity of Asian H5N1 Highly Pathogenic Avian Influenza Viruses in Chickens with Highly Efficient Viral Replication Accompanied by Early Destruction of Innate Immune Responses’, Journal of Virology, 83: 7475–86.doi: 10.1128/JVI.01434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau T. M. (2020) A package for survival analysis in R. R package version 3.2-7. Available online: https://CRAN.R-project.org/package=survival Accessed 30 Jul 2020.
- Wargo A. R., Garver K. A., and Kurath G. (2010) ‘Virulence Correlates with Fitness in Vivo for Two M Group Genotypes of Infectious Hematopoietic Necrosis Virus (IHNV)’, Virology, 404: 51–58.doi: 10.1016/j.virol.2010.04.023. [DOI] [PubMed] [Google Scholar]
- Wargo A. R., and Kurath G. (2011) ‘In Vivo Fitness Associated with High Virulence in a Vertebrate Virus Is a Complex Trait Regulated by Host Entry, Replication, and Shedding’, Journal of Virology, 85: 3959–67.doi: 10.1128/JVI.01891-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wargo A. R. et al. (2017) ‘Replication and Shedding Kinetics of Infectious Hematopoietic Necrosis Virus in Juvenile Rainbow Trout’, VirusResearch, 227: 200–11.doi: 10.1016/j.virusres.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. (2000) ‘Maximum Likelihood Estimation on Large Phylogenies and Analysis of Adaptive Evolution in Human Influenza Virus A’, Journal of Molecular Evolution, 51: 423–32.doi: 10.1007/s002390010105. [DOI] [PubMed] [Google Scholar]
- Zamperin G. et al. (2019) ‘Sequencing of Animal Viruses: Quality Data Assurance for NGS Bioinformatics’, Virology Journal, 16: 140.doi: 10.1186/s12985-019-1223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
IHNV complete genome sequences generated in this study have been deposited in GenBank under the accession numbers MN914161–MN914166. MiSeq raw data were submitted to the National Center for Biotechnology Information Sequence Read Archive under the accession numbers SRR10973039–SRR10973043 and SRR8943890.