

Summary
Toll-like receptors are essential for animal development and survival, with conserved roles in innate immunity, tissue patterning, and cell behavior. The mechanisms by which Toll receptors signal to the nucleus are well characterized, but how Toll receptors generate rapid, localized signals at the cell membrane to produce acute changes in cell polarity and behavior is not known. We show that Drosophila Toll receptors direct epithelial convergent extension by inducing planar polarized patterns of cortical Src and PI3-kinase (PI3K) activity. Toll receptors target Src activity to specific sites at the membrane, and Src recruits PI3K to the Toll-2 complex through tyrosine phosphorylation of the Toll-2 cytoplasmic domain. Reducing Src or PI3K activity disrupts planar polarized myosin assembly, cell intercalation, and convergent extension, whereas constitutive Src activity promotes ectopic PI3K and myosin cortical localization. These results demonstrate that Toll receptors direct cell polarity and behavior by locally mobilizing Src and PI3K activity.
eTOC blurb
Toll receptors provide critical spatial cues that drive cell movements during convergent extension. Tamada and Shi et al. show that Toll receptors organize cell movements by generating planar polarized patterns of Src and PI3K activity. Toll-2 tyrosine phosphorylation recruits PI3K to the Toll-2 complex, promoting localized myosin assembly and cell rearrangement.
Introduction
Cell-surface receptors convert extracellular signals into changes in cell behavior that are essential for the formation, remodeling, and repair of multicellular tissues. Toll-like receptors (TLRs) are a conserved family of receptors with roles in dorsal-ventral patterning (Morisato and Anderson, 1995) and cell-pathogen recognition in the innate immune system (Anderson, 2000; Leulier and Lemaitre, 2008). More recently, members of the Toll receptor family have also been shown to regulate cell behavior during the development of epithelial tissues and the nervous system (Anthoney et al., 2018; Paré and Zallen, 2020). Toll receptors organize cell movement (Paré et al., 2014; Benton et al., 2016; Sun et al., 2017), direct nervous system wiring (McIlroy et al., 2013; Ballard et al., 2014; Ward et al., 2015; Foldi et al., 2017), repair wounds (Carvalho et al., 2014; Capilla et al., 2017), and eliminate less fit cells through cell competition (Meyer et al., 2014), defining new functions for this receptor family. In the immune system, TLR signaling activates the expression of immune response genes by promoting the nuclear translocation of transcriptional regulators in the NF-κB, AP-1, and IRF families (Kawai and Akira, 2011; Fitzgerald and Kagan, 2020). However, how Toll receptors elicit fast-acting and localized responses in cells to generate spatially regulated changes in cell behavior is not understood.
Tyrosine phosphorylation is a rapid and reversible mechanism that transduces extracellular information into molecular changes within cells. Cell-surface receptors such as growth factor receptors initiate signaling cascades through their intrinsic tyrosine kinase activity, which is activated by ligand binding and receptor dimerization (Lemmon and Schlessinger, 2010; Kovacs et al., 2015). In addition, receptors that lack kinase activity can also participate in tyrosine kinase signaling by recruiting nonreceptor tyrosine kinases. Src family nonreceptor tyrosine kinases are essential for signaling by growth factor receptors, T cell and B cell receptors, integrins, and cadherins (Thomas and Brugge, 1993), and have diverse roles in cell-cell adhesion (Takahashi et al., 2005; McLachlan et al., 2007; Shindo et al., 2008; Forster and Luschnig, 2012, Hunter et al., 2018), cell-matrix adhesion (Huveneers and Danen, 2009), and cytoskeletal organization (Thomas and Wieschaus, 2004; Andreeva et al., 2014; Sun et al., 2017; Hunter et al., 2018). A critical event in receptor-initiated tyrosine kinase signaling pathways is often phosphorylation of the receptor itself, which generates binding sites that recruit downstream effector proteins (Pawson, 2004; Lemmon and Schlessinger, 2010). Although canonical TLR signaling involves a serine/threonine kinase cascade triggered by the recruitment of TLR-associated adaptor proteins, tyrosine kinases in the Src family have also been shown to facilitate the response to TLR signaling in cultured cells (Abram and Lowell, 2008; Page et al., 2009; Chattopadhyay and Sen, 2014). However, the effects of Src family kinases on Toll receptor signaling in vivo, and whether tyrosine kinase pathways mediate the rapid and localized effects of Toll receptors on cell behavior, are not known.
Convergent extension is a conserved morphogenetic process that shapes developing organs and elongates the body axis of multicellular organisms (Huebner and Wallingford, 2018). In Drosophila, three Toll family receptors expressed in distinct striped patterns provide critical spatial cues that orient cell rearrangements and elongate the embryo along the head-to-tail axis (Paré et al., 2014). Toll receptors guide cell movements by directing the planar polarized localization of proteins involved in actomyosin contractility and cell adhesion (Paré et al., 2014; Paré and Zallen, 2020). However, the signaling pathways that generate spatially regulated changes in the organization and activity of the force-generating machinery downstream of Toll receptors are unknown. Here we show that a Src-mediated tyrosine kinase pathway is essential for planar polarity and Toll receptor signaling during convergent extension in Drosophila. Toll receptors generate a planar polarized pattern of Src kinase activity, and Src in turn phosphorylates the Toll-2 C-terminal domain, promoting the association of Toll-2 with PI3-kinase (PI3K). Drosophila Src family kinases, the regulatory and catalytic subunits of the PI3K complex, and tyrosine phosphorylation of the Toll-2 cytoplasmic domain are all necessary for planar polarity, cell intercalation, and convergent extension. These results identify a localized signaling mechanism that allows Toll receptors to induce rapid changes in cell polarity and behavior during development.
Results
Localized Src activity promotes Toll receptor tyrosine phosphorylation
To understand how Toll receptors direct cell rearrangements during convergent extension, we sought to identify proteins that are required for receptor activity. The immediate effectors of Toll receptor signaling are predicted to physically associate with Toll receptors, act in a spatially localized fashion within cells, and regulate cell polarity and behavior during convergent extension. The three Toll family receptors required for convergent extension, Toll-2, Toll-6, and Toll-8, do not possess intrinsic tyrosine kinase activity, but we hypothesized that they could interact with nonreceptor tyrosine kinases to transmit polarity signals within cells. The two Drosophila Src-family kinases, Src42 and Src64, are both expressed and active during convergent extension (Takahashi et al., 2005; Sopko et al., 2014). Src42 and Src64 are uniformly distributed throughout the plasma membrane of embryonic epithelial cells (Figures 1A and S1A–C). By contrast, a phosphospecific antibody that binds to the active form of Src42 (Shindo et al., 2008) detected a planar polarized enrichment of active Src42 kinase at adherens junctions between anterior and posterior cells (Figures 1A and S1D, see Table S1 for full statistical information). To determine whether this pattern of Src42 activity requires Toll receptor signaling, we analyzed the localization of active Src42 in embryos that lack all three Toll receptors. The planar polarized pattern of active Src42 was severely disrupted in Toll-2,6,8 triple mutants (Figures 1B, 1C, and S1E), whereas total Src42 localization was unaffected (Figures S1J and S1K). Defects in active Src42 localization were detected in Toll-2 single mutants and Toll-6,8 double mutants, indicating that multiple Toll receptors contribute to this pattern (Figure S1F). Active Src42 localization was defective in a subset of cells in Toll-2 mutants (Figures S1G–S1I), consistent with the region-specific expression and function of Drosophila Toll receptors (Paré et al., 2014). These results demonstrate that Toll receptors generate a planar polarized pattern of Src42 activity during convergent extension.
Figure 1. Src42 activity is spatially regulated and promotes Toll-2 phosphorylation in vitro and in vivo.
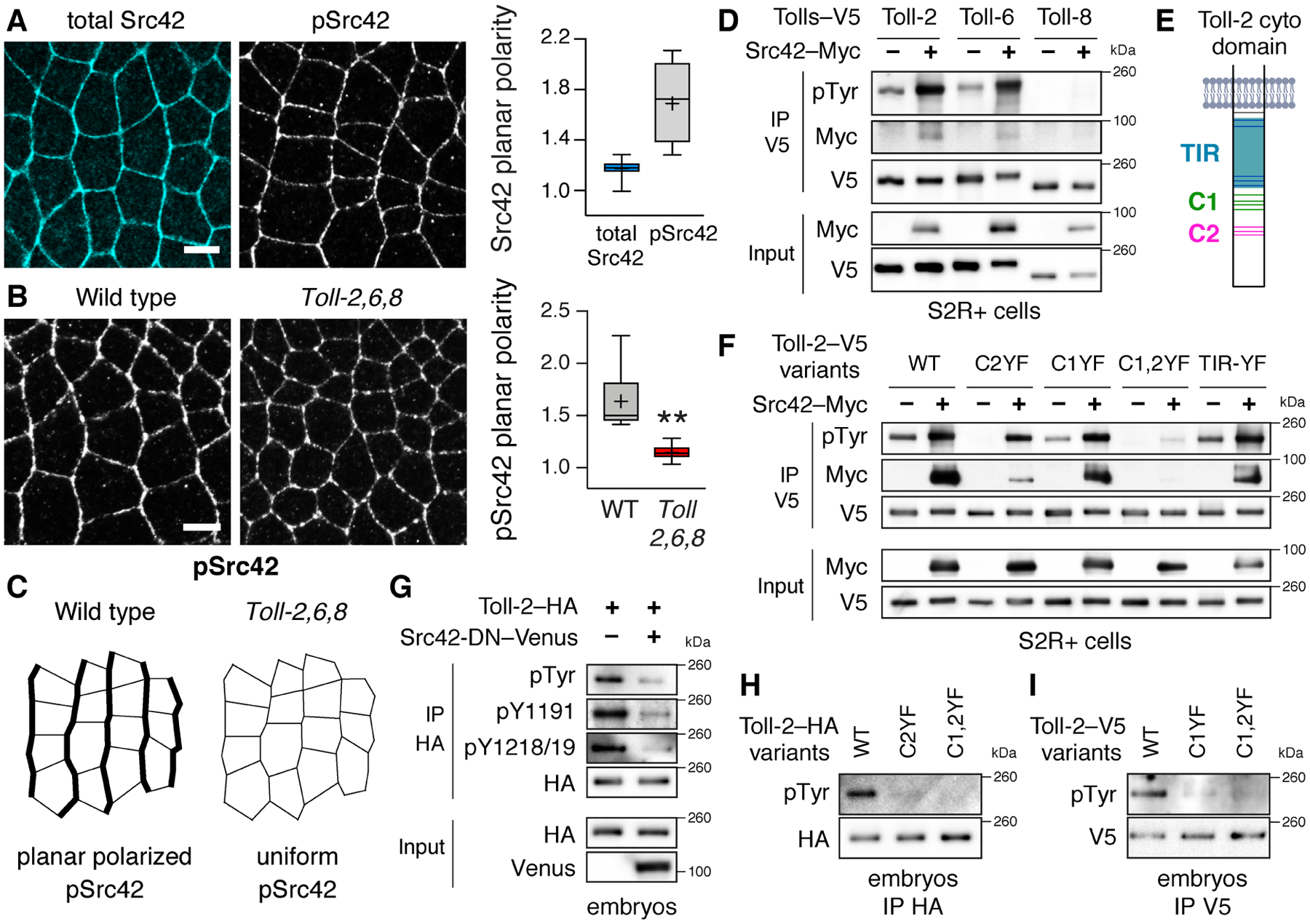
(A) Phosphorylated Src42 (pSrc42), but not total Src42, is enriched at vertical edges during convergent extension. Planar polarity, vertical-to-horizontal edge intensity ratio. (B,C) pSrc42 planar polarity is reduced in Toll-2,6,8 mutants. (D) Src42 promotes the tyrosine phosphorylation of Toll-2 and Toll-6, but not Toll-8, in Drosophila S2R+ cells. (E) Toll-2 cytoplasmic domain. Gray, membrane. Blue, TIR domain. Green and magenta, C-terminal tyrosine clusters C1 and C2. Tyrosines, horizontal lines. (F) Src42 promotes the tyrosine phosphorylation of wild-type Toll-2 (WT) and Toll-2 variants lacking tyrosine cluster C1 (Toll-2C1YF) or C2 (Toll-2C2YF), but not a variant lacking both clusters (Toll-2C1,2YF) in S2R+ cells. (G) Toll-2 tyrosine phosphorylation is reduced in embryos expressing dominant negative Src42 (Src42-DN–Venus), detected with a pan-phosphotyrosine (pTyr) antibody and phosphospecific antibodies to Toll-2 pY1191 or pY1218/1219. (H,I) Toll-2 tyrosine phosphorylation is reduced in embryos expressing Toll-2 phosphovariants from the Toll-2 locus. Toll-2 variants were immunoprecipitated (IP) from S2R+ cells (D,F) or lysates of staged embryos (G–I) with V5 or HA antibodies. Boxes, 2nd and 3rd quartiles; whiskers, min to max; horizontal line, median; +, mean, 3–9 fixed stage 7 embryos/genotype, **p=0.01, Welch’s t-test. Anterior left, ventral down. Bars, 5 μm. See also Figures S1 and S2 and Table S1 for full n and p values.
An early event in receptor-activated tyrosine kinase signaling pathways is often phosphorylation of the receptor itself (Pawson, 2004; Lemmon and Schlessinger, 2010). We therefore tested whether Src kinases promote Toll receptor phosphorylation. We found that Src42 and Src64 significantly enhanced the tyrosine phosphorylation of Toll-2 and Toll-6, but not Toll-8, in Drosophila S2R+ cells (Figures 1D, 1F, S2A and S2B). Toll-2 and Toll-6 also coimmunoprecipitated with both kinases in vitro. To identify the residues that are targeted by Src kinases, we mutated different sets of tyrosines in Toll-2. The Toll-2 cytoplasmic domain contains a conserved Toll/interleukin-1 receptor (TIR) domain and a unique C-terminal extension that is present in a subset of Drosophila Toll receptors (Figure 1E) (Leulier and Lemaitre, 2008). Mutating all 5 tyrosines in the Toll-2 TIR domain to unphosphorylatable phenylalanine residues (Toll-2TIR-YF) had no effect on Toll-2 tyrosine phosphorylation when coexpressed with Src42 or Src64 in S2R+ cells (Figures 1F and S2B). By contrast, mutating the 7 tyrosines in the Toll-2 C-terminal domain, which are present in two clusters, significantly reduced Toll-2 tyrosine phosphorylation and disrupted its interaction with both kinases (Figures 1E, 1F, S2B, and S2E). The more C-terminal tyrosine cluster (C2) was particularly important for the association between Toll-2 and Src42, whereas tyrosine cluster C1 was dispensable for this interaction (Figure 1F). These results demonstrate that Src family kinases promote the phosphorylation of a subset of Drosophila Toll receptors in vitro.
To examine whether Toll-2 is tyrosine phosphorylated in vivo, we used CRISPR approaches to generate embryos that express C-terminally tagged Toll-2 proteins from the Toll-2 locus (Zhang et al., 2014; Paré et al., 2019). Toll-2–HA and Toll-2–V5 expressed from the endogenous locus were present in the expected striped pattern (Figures S2F and S2G). Both proteins were tyrosine phosphorylated in embryos during convergent extension, detected with a pan-phosphotyrosine antibody and with phosphospecific antibodies to specific tyrosines in the Toll-2 C-terminal domain (Figures 1G, S2C, and S2D). By contrast, Toll-2 tyrosine phosphorylation was significantly reduced in embryos that lack either C-terminal tyrosine cluster of Toll-2 or that express a catalytically inactive variant of Src42, Src42K276R (Src DN), which is predicted to act as a dominant negative for both Src kinases (Figures 1G–1I). These results demonstrate that Toll-2 tyrosine phosphorylation requires Src family kinase activity in vivo.
Src kinases act combinatorially to generate planar polarity
The findings that Toll receptors generate planar polarized Src activity, and that Src family kinases promote Toll receptor phosphorylation, raised the possibility that Toll receptors could direct planar polarity through a Src-dependent signaling pathway. To investigate this possibility, we analyzed whether Src kinases are required for cell polarity and behavior during convergent extension. As Src42 and Src64 both enhance Toll-2 and Toll-6 tyrosine phosphorylation in vitro, and often function redundantly in vivo (Takahashi et al., 2005; Shindo et al., 2008), we wanted to examine the effects of disrupting both kinases simultaneously. However, disrupting the expression of both Src kinases in the embryo is technically challenging, as maternal Src42 expression cannot be eliminated using standard genetic methods due to the proximity of Src42 to the centromere (Chou and Perrimon, 1996). To circumvent this limitation, we generated embryos that were defective for both proteins using CRISPR/Cas9 to target the Src42 locus in the female germline (Port et al., 2014) and a maternally expressed shRNA to deplete maternal Src64 transcripts (Ni et al., 2011). Embryos with an >85% reduction in Src42 and Src64 levels by western blot or in cortical Src42 localization by immunofluorescence (Figures S3A–S3D) were selected for further analysis, and are referred to as Src KD embryos.
Toll receptors direct convergent extension by activating myosin contractility at interfaces between anterior and posterior cells (vertical edges) and targeting the adherens junction regulator Par-3 to complementary interfaces between dorsal and ventral cells (horizontal edges) (Bertet et al., 2004; Zallen and Wieschaus, 2004; Blankenship et al., 2006; Rauzi et al., 2008; Fernandez-Gonzalez et al., 2009; Simoes et al., 2010). Consistent with a role for Src kinases in Toll receptor signaling, reducing the expression of both kinases severely disrupted planar polarity. In contrast to wild-type embryos, myosin II cortical localization was significantly reduced and Par-3 localization expanded to occupy interfaces at all orientations in Src KD embryos (Figures 2A and 2B). Similar defects were observed in embryos that express dominant negative Src42 (Figures S3E–S3G). By contrast, loss of either Src42 or Src64 alone did not significantly affect planar polarity (Figures S3J–S3O). Expression of constitutively active Src42Y511F (Src42 CA) (Tateno et al., 2000) had the opposite effect, resulting in ectopic myosin II localization at the cortex and reduced levels of cortical Par-3, disrupting the planar polarized distributions of both proteins (Figures 2C and 2D). Despite these defects, the localization of adherens junction proteins to the apical junctional domain occurred normally in embryos with increased or decreased Src activity, indicating that apical-basal polarity was not affected (Figures S3H and S3I). These results demonstrate that Src activity is necessary and sufficient for the localized assembly of myosin at the cell cortex, consistent with a model in which Src plays an instructive role in directing planar polarity.
Figure 2. Src kinases regulate planar polarity and cell intercalation during convergent extension.
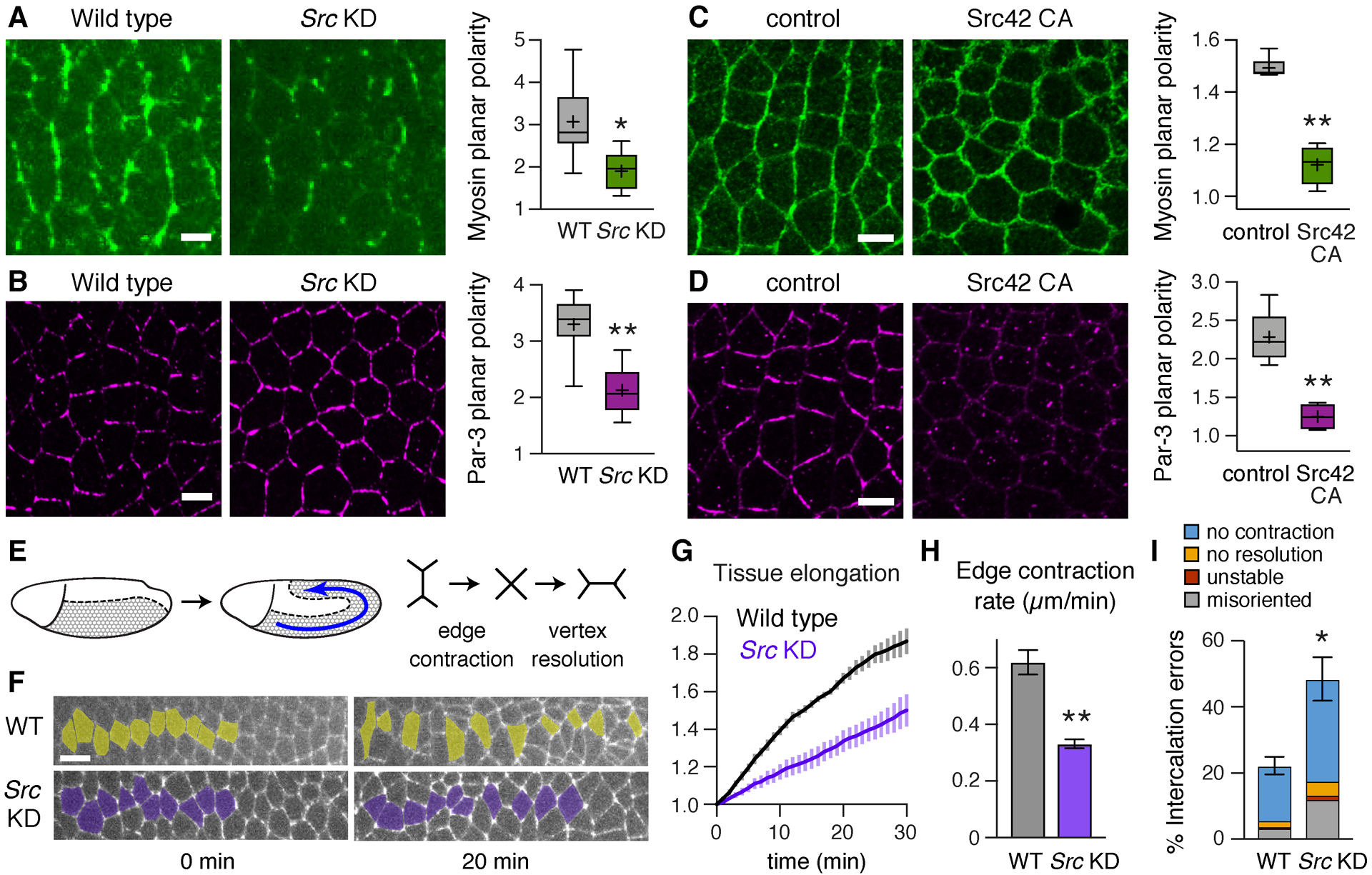
(A,B) Localization of myosin II (myosin–mCherry) (A) and Par-3 (B) in wild-type (WT) and Src KD embryos. (C,D) Localization of myosin II (myosin–GFP) (C) and Par-3 (D) in Src42 CA embryos and Gal4 controls. Both Src KD and Src42 CA disrupt myosin planar polarity (vertical-to-horizontal edge intensity ratio) and Par-3 planar polarity (horizontal-to-vertical edge intensity ratio). (E) The Drosophila germband elongates along the anterior-posterior (AP) axis (left) through planar polarized cell rearrangements (right). (F) Stills from movies of WT and Src KD embryos expressing E-cadherin–GFP (0 min, onset of axis elongation). Anterior and posterior cells become separated by intercalation in WT, but many cells fail to separate in Src KD. (G) Axis elongation (tissue AP length relative to t=0) is reduced in Src KD embryos (p<0.02 at t=30 min). (H) The rate of vertical edge contraction is reduced in Src KD embryos. (I) Intercalation errors are increased in Src KD embryos. (A–D) Boxes, 2nd and 3rd quartiles; whiskers, min to max; horizontal line, median; +, mean. (G–I) Mean±SEM. Living embryos are shown in (A and F), fixed embryos in (B–D), 4–6 stage 7 embryos/genotype in (A–D) and 4 stage 7–8 embryos/genotype in (G–I), *p≤0.02, **p≤0.004, Welch’s t-test. Anterior left, ventral down. Bars, 5 μm (A–D), 10 μm (F). See also Figure S3.
The critical outputs of Toll receptor signaling in the Drosophila embryo are planar polarized cell rearrangements that lead to body axis elongation (Irvine and Wieschaus, 1994; Paré et al., 2014). To determine whether Src activity is necessary for these cell- and tissue-level behaviors, we generated time-lapse movies of Src KD embryos that express E-cadherin–GFP to track cell behavior. Convergent extension is driven by cell intercalation, in which vertical edges contract to produce 4-cell vertices (Bertet et al., 2004) and rosettes where 5 or more cells meet (Blankenship et al., 2006). These structures resolve in a perpendicular direction, driving tissue elongation (Figure 2E). Src KD embryos displayed a significant reduction in axis elongation, elongating to less than half the extent of wild type (Figures 2F and 2G, Movies S1 and S2). This defect required the loss of both kinases, as the double mutant was more severe than loss of either kinase alone (Figures S3P and S3Q). Src KD embryos also displayed defects in cell intercalation, including slowed vertical edge contraction (Figure 2H) and an increase in the percentage of cells that failed to initiate rearrangement (no contraction), 4-cell vertices or rosettes that failed to resolve (no resolution), and new edges that only formed transiently (unstable) or in the wrong direction (misoriented) (Figure 2I). These results demonstrate that Src family kinases are necessary for planar polarity, cell intercalation, and axis elongation, indicating that Src activity is required for multiple outputs of Toll receptor signaling during convergent extension.
Toll-2 forms a complex with the regulatory subunit of PI3K
To determine how Src activity influences Toll receptor signaling, we looked for proteins that interact with tyrosine-phosphorylated Toll-2. PI3K was an intriguing candidate, as the regulatory subunit of the PI3K complex (p85 in mammals, PI3K21B or PI3K-reg in Drosophila) is often recruited to tyrosine-phosphorylated receptors at the plasma membrane (Wymann and Pirola, 1998; Vanhaesebroeck et al., 2010), and PI3K regulates front-rear polarity in migrating cells (Cain and Ridley, 2009) and apical-basal polarity in epithelia (Gassama-Diagne et al., 2006; Pinal et al., 2006; Martin-Belmonte et al., 2007; Chartier et al., 2011). Mammalian TLRs can interact with p85 in vitro, either directly (Arbibe et al., 2000; Sarkar et al., 2004) or through adaptor proteins (Ojaniemi et al., 2003; Gelman et al., 2006; Rhee et al., 2006; Santos-Sierra et al., 2009). However, the functional significance of the association between TLRs and PI3K is unclear, as PI3K potentiates the expression of immune response genes downstream of TLR activation in culture, but the loss of PI3K enhances immune responses in vivo, suggesting that PI3K activity dampens the immune response instead of being critical for its activation (Fukao et al., 2002; Troutman et al., 2012). Therefore, the relationship between TLR signaling and PI3K activity is poorly understood.
To test whether Drosophila Toll receptors interact with PI3K-reg, we examined their association in vitro. PI3K-reg strongly co-immunoprecipitated with Toll-2 and Toll-6, and displayed a weaker association with Toll-8, when expressed in S2R+ cells (Figure S4A). In contrast to the interaction between mammalian TLRs and PI3K, which requires the TIR domain (Arbibe et al., 2000; Sarkar et al., 2004), the TIR domain of Toll-2 was dispensable for this interaction (Figures 3A, 3B, and S4B). Instead, the association between Toll-2 and PI3K-reg required the Toll-2 C-terminal domain (Figures 3B and S4B), indicating that this interaction occurs through a distinct mechanism.
Figure 3. Toll-2 interacts with the regulatory subunit of PI3K and regulates its localization.
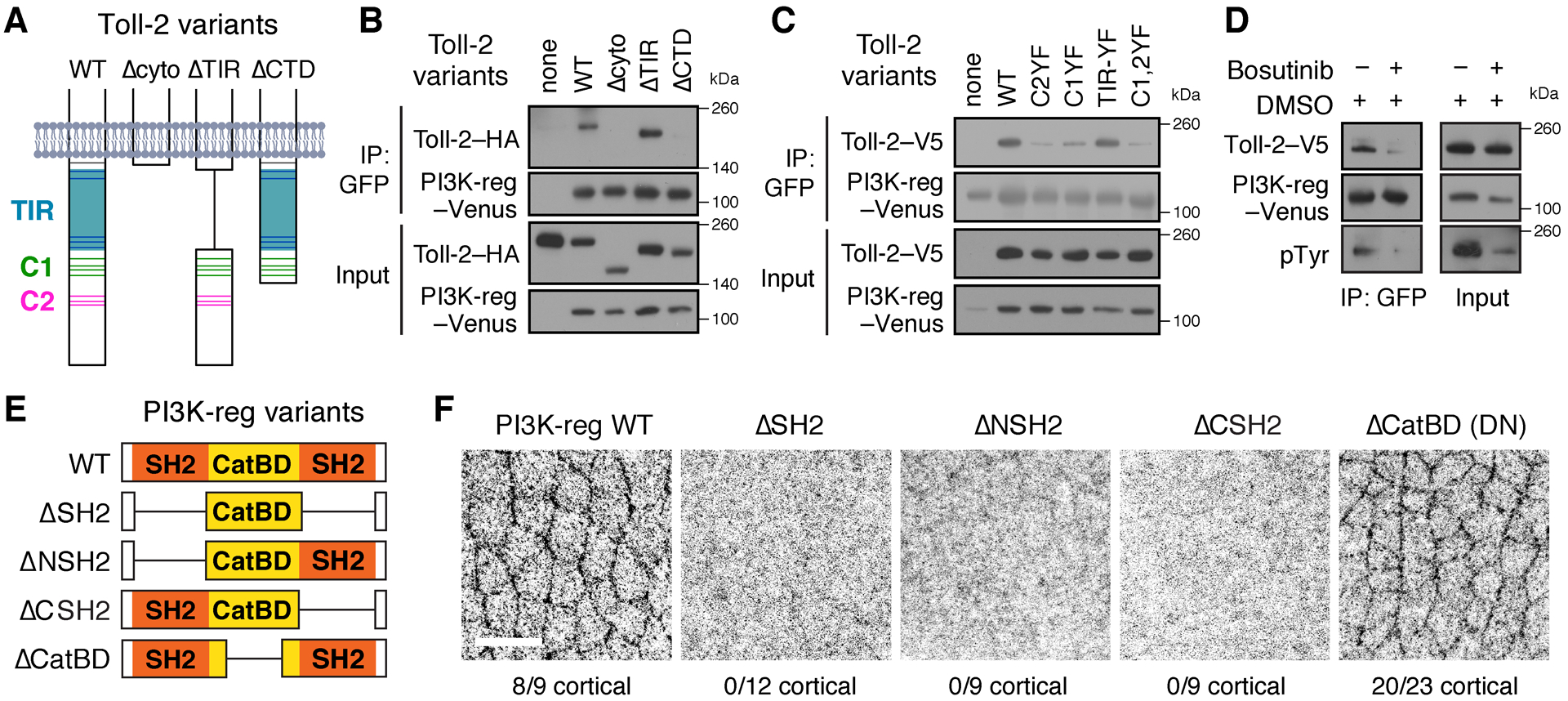
(A) Wild-type (WT) Toll-2 cytoplasmic domain and deletion variants. Gray, membrane. Blue, TIR domain. Green and magenta, C-terminal tyrosine clusters C1 and C2. Tyrosines, horizontal lines. (B,C) Immunoprecipitation (IP) of PI3K-reg–Venus pulled down Toll-2 variants C-terminally tagged with HA (B) or V5 (C) in Drosophila S2R+ cells. This interaction requires the Toll-2 C-terminal domain (CTD) and the tyrosine clusters C1 and C2. (D) Toll-2–V5 was not efficiently immunoprecipitated with PI3K-reg–Venus in the presence of Bosutinib (1 μM Bosutinib in 0.01% DMSO vs. 0.01% DMSO alone). (E) WT PI3K-reg and deletion variants. (F) Localization of WT PI3K-reg and deletion variants tagged with C-terminal HA, 9–23 fixed stage 7 embryos/genotype. Anterior left, ventral down. Bar, 10 μm. See also Figure S4.
To investigate the molecular basis of the interaction between Toll-2 and PI3K, we characterized the domains required for this interaction. The PI3K-reg subunit contains two Src-homology 2 (SH2) domains and a central domain that interacts with the PI3K catalytic subunit (p110 in mammals and PI3K92E or PI3K-cat in Drosophila (Figure 3E) (Wymann and Pirola, 1998). Deletion of either SH2 domain of PI3K-reg nearly eliminated its association with Toll-2 in S2R+ cells, whereas removing the central PI3K-cat-binding domain had no effect (Figure S4E). The finding that the SH2 domains of PI3K-reg mediate its interaction with Toll-2 raised the possibility that this interaction could require Toll-2 tyrosine phosphorylation. Consistent with this, inhibiting tyrosine phosphorylation using the Src/Abl tyrosine kinase inhibitor Bosutinib disrupted the association between Toll-2 and PI3K-reg in vitro (Figures 3D and S4D). Moreover, mutating either or both C-terminal tyrosine clusters of Toll-2 significantly reduced the interaction between Toll-2 and PI3K-reg, whereas mutating the tyrosines in the TIR domain had no effect (Figures 3C and S4C). These results demonstrate that Toll-2 binding to PI3K-reg requires the SH2 domains of PI3K-reg and two tyrosine clusters in the Toll-2 C-terminal domain, suggesting that this interaction requires Toll-2 tyrosine phosphorylation.
PI3K is necessary for planar polarity and convergent extension
If PI3K is an important effector of Toll-2 signaling during convergent extension, then PI3K localization or activity is predicted to be planar polarized. Consistent with this prediction, PI3K-reg tagged at its C-terminus with Venus or HA localized to vertical cell interfaces during convergent extension and was enriched in the apical junctional domain (Figures 4A, 4D, and S5A–S5C). The SH2 domains of PI3K-reg, but not the PI3K-cat-binding domain, were necessary and sufficient for PI3K-reg cortical localization and planar polarity in vivo, consistent with the requirements for Toll-2 binding in vitro (Figures 3F, S4E–S4G, and S5C). To test whether Toll receptors are required for PI3K-reg planar polarity, we examined PI3K-reg localization in embryos that were defective for Toll receptor activity. PI3K-reg planar polarity was significantly reduced in embryos injected with dsRNAs targeting Toll-2 and Toll-8 (Toll-2,8 KD embryos) (Figures 4A and 4D). Similar defects were observed in embryos injected with dsRNAs to Toll-2 and Toll-6 and in embryos depleted for all three receptors (Figure S6). Toll-2,8 KD embryos also had a significant decrease in the level of phosphorylated Akt, a conserved readout of PI3K activity (Figure S5D). These results demonstrate that Toll receptors regulate the localization and the activity of PI3K.
Figure 4. The PI3K complex is necessary for planar polarity during convergent extension.
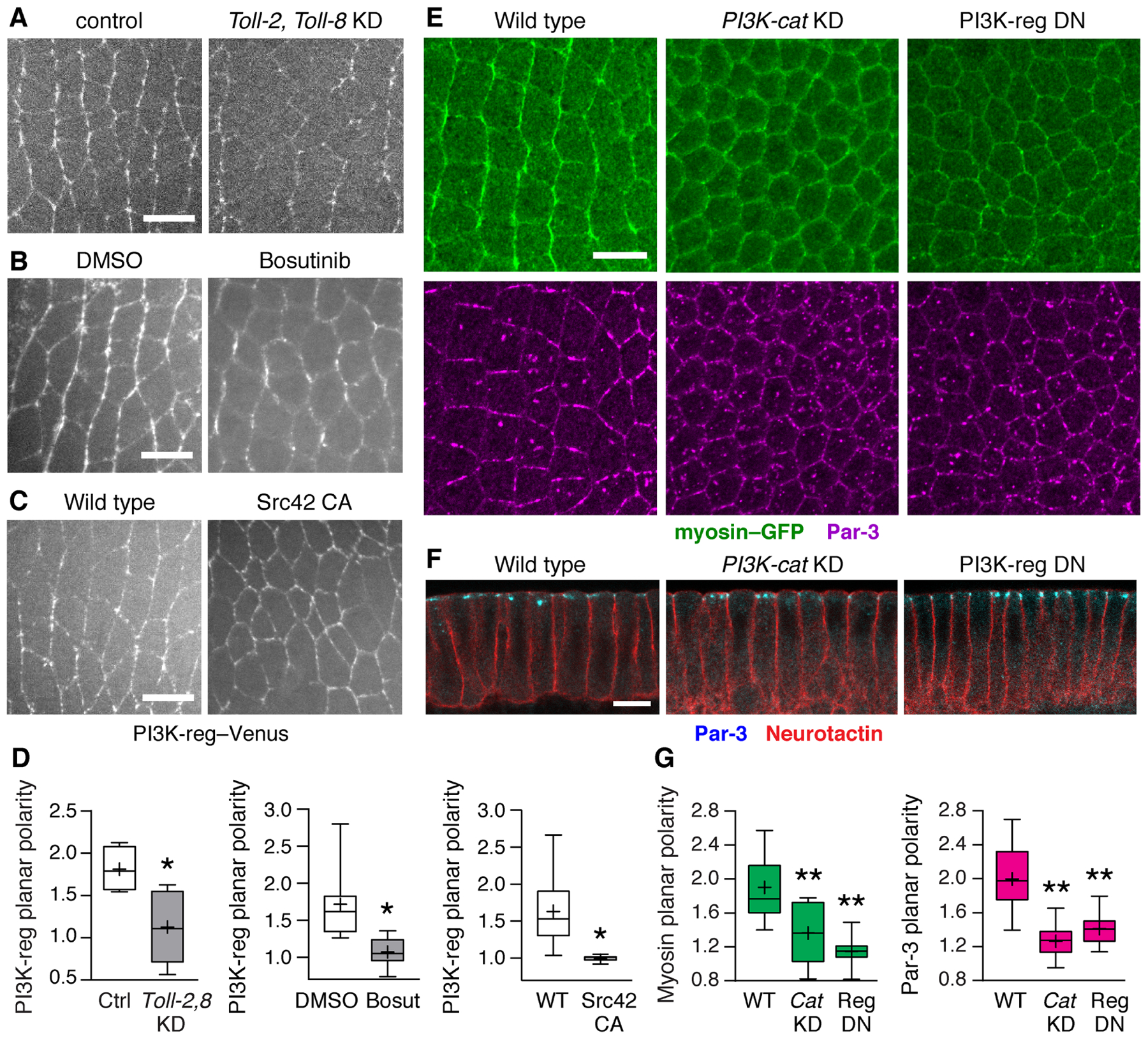
(A–C) PI3K-reg–Venus localization in (A) embryos injected with dsRNAs to Toll-2 and Toll-8 (Toll-2,Toll-8 KD) and control embryos injected with Toll-3 dsRNA, (B) embryos injected with Bosutinib (100 μM Bosutinib in 0.6% DMSO) and DMSO controls (0.6% DMSO) (final concentrations), and (C) embryos expressing constitutively active Src42 (Src42 CA) and wild-type Gal4 controls. (D) PI3K-reg–Venus planar polarity (vertical-to-horizontal edge intensity ratio). (E) Localization of myosin II (myosin–GFP, top panels) and Par-3 (bottom panels) in wild-type (WT), PI3K-cat KD, and PI3K-reg DN embryos. (F) Apical-basal polarity occurs normally in PI3K-cat KD and PI3K-reg DN embryos. (G) Myosin–GFP planar polarity (vertical-to-horizontal edge intensity ratio) and Par-3 planar polarity (horizontal-to-vertical edge intensity ratio). Boxes, 2nd and 3rd quartiles; whiskers, min to max; horizontal line, median; +, mean. Living stage 7 embryos are shown in (A–C), fixed stage 7 embryos in (E and F), 3–14 embryos/genotype. *p<0.03, **p<0.001, Welch’s t-test. Anterior left, ventral down (A, B, C, and E). Cross sections, apical up (F). Bars, 10 μm. See also Figures S5 and S6.
The findings that Toll receptors are required for PI3K-reg planar polarity, and that Toll-2 tyrosine phosphorylation is necessary for binding to PI3K-reg, suggested that Src activity could be important for PI3K planar polarity. To investigate this possibility, we examined whether PI3K-reg localization is sensitive to increased or decreased Src activity. We found that PI3K-reg cortical localization was significantly reduced in embryos injected with the Src/Abl inhibitor Bosutinib (Figure 4B). Conversely, expression of constitutively active Src42 recruited PI3K-reg to ectopic sites at the cell cortex (Figure 4C). Both perturbations strongly disrupted PI3K-reg planar polarity (Figure 4D). These results demonstrate that regulated Src activity is necessary for the planar polarized distribution of PI3K during convergent extension.
To determine if PI3K-reg is an important effector of Toll receptor signaling, we analyzed the effects of PI3K on planar polarity and cell behavior during convergent extension. As the complete loss of the catalytic or regulatory subunits of PI3K leads to early developmental arrest (Jagut et al., 2013), we used three approaches to reduce PI3K activity during convergent extension, including RNA interference to PI3K-cat (Ni et al., 2011), injection of the PI3K inhibitor wortmannin (Wymann and Pirola, 1998), and expression of dominant negative PI3K-reg lacking the PI3K-cat-binding domain (PI3K DN) (Dhand et al., 1994). All three methods effectively reduced PI3K expression or activity during convergent extension, detected by qRT-PCR or by a significant decrease in phosphorylated Akt levels (Figures S5E–S5G). Embryos expressing an shRNA targeting the catalytic subunit of PI3K (PI3K-cat KD) displayed significantly reduced myosin II and Par-3 planar polarity (Figures 4E and 4G). Similar defects were observed in embryos expressing PI3K-reg DN (Figures 4E and 4G) and in embryos injected with PI3K-cat dsRNA (Figure S5H) or wortmannin (Figures S5I). PI3K-cat KD and PI3K-reg DN embryos also had significantly reduced levels of the phosphorylated myosin regulatory light chain, which was also observed in Toll-2,8 KD embryos, supporting the idea of shared functions of Toll receptors and PI3K in regulating myosin activation (Figures S5D, S5E, and S5G). The apical localization of Par-3 and the basolateral localization of the transmembrane protein Neurotactin were unaffected in PI3K-defective embryos, indicating that apical-basal polarity occurred normally (Figure 4F). These results demonstrate that PI3K is required for myosin II and Par-3 planar polarity.
To determine whether PI3K is required for cell behavior during convergent extension, we generated time-lapse movies of PI3K-defective embryos (Movies S3–S7). PI3K-cat KD, PI3K-cat dsRNA-injected, and PI3K-reg DN embryos displayed cell intercalation defects that resembled Src KD embryos, including slowed edge contraction, an increased percentage of vertical edges that failed to contract, and delayed vertex resolution (Figure 5). The defects in cell intercalation that result from disrupted PI3K expression or activity demonstrate that PI3K is required for rapid and oriented cell rearrangements. Together with the findings that Src and Toll receptors regulate PI3K-reg planar polarity, these results are consistent with a model in which Toll/Src signaling directs planar polarity and cell behavior by generating spatially localized PI3K activity.
Figure 5. PI3K is required for cell intercalation and convergent extension.
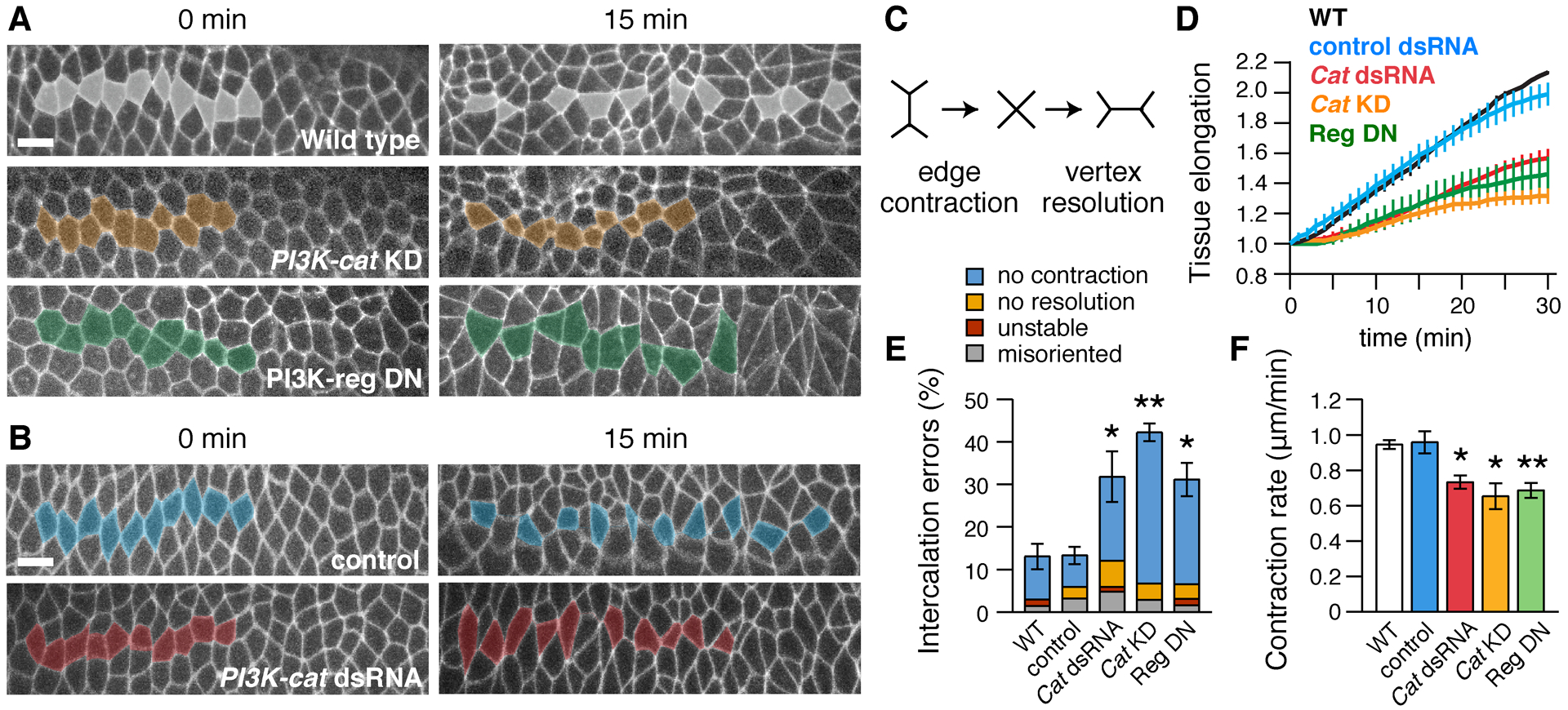
(A,B) Stills from movies of (A) wild-type (WT), PI3K-cat KD, and PI3K-reg DN embryos expressing Spider–GFP and (B) control (Toll-3 dsRNA-injected) and PI3K-cat dsRNA-injected embryos expressing Resille–GFP (0 min, onset of elongation). Anterior and posterior cells become separated by intercalation in WT, but many cells fail to separate in PI3K-defective embryos. (C) Schematic of cell intercalation. (D) Axis elongation (tissue AP length relative to t=0) is reduced in PI3K-cat KD (p=0.003 at t=30 min), PI3K-reg DN (p=0.03), and PI3K-cat dsRNA embryos (p=0.01). (E) Intercalation errors are increased in PI3K-cat KD, PI3K-reg DN, and PI3K-cat dsRNA embryos. (F) The rate of vertical edge contraction is reduced in PI3K-cat KD, PI3K-reg DN, and PI3K-cat dsRNA embryos. (D–F) Mean±SEM, 3–5 living stage 7–8 embryos/genotype. *p<0.05, **p≤0.01, Welch’s t-test. Anterior left, ventral down. Bars, 10 μm.
Toll-2 tyrosine phosphorylation is necessary for planar polarity and convergent extension
Thus far, our data show that Src and PI3K activity are necessary for convergent extension and that Toll-2 tyrosine phosphorylation is necessary for its association with PI3K. These results suggest that the phosphorylation of Toll-2 by Src family kinases could direct the localization of active PI3K complexes in the cell. If tyrosine phosphorylation is necessary for Toll-2 activity, then preventing Toll-2 phosphorylation should interfere with its function. To test this, we engineered flies that express wild-type Toll-2 (Toll-2WT) or Toll-2 variants that cannot be phosphorylated on tyrosines targeted by Src (Toll-2C2YF or Toll-2C1,2YF) from the endogenous Toll-2 locus (Figure 6D). As a readout of Toll-2 signaling, we analyzed the borders of the Toll-2 stripes, where Toll-2 activity is necessary for planar polarity (Figures 6A–6C). In contrast to control embryos expressing Toll-2WT, embryos expressing Toll-2C2YF or Toll-2C1,2YF displayed a significant reduction in myosin accumulation at Toll-2 stripe borders, resembling Toll-2 null mutants (Figures 6E, 6H, and 6I). Moreover, embryos expressing Toll-2C2YF or Toll-2C1,2YF failed to recruit PI3K-reg and deplete Par-3 from the Toll-2 stripe borders, demonstrating that tyrosine phosphorylation is required for multiple aspects of Toll-2 activity (Figures 6F, 6G, and 6J–6L). Toll-2 stripe borders were more jagged in Toll-2C2YF mutants, consistent with a failure to achieve normal levels of myosin contractility (Figure S6E). These results indicate that the C-terminal tyrosines in Toll-2, which are targeted by Src and mediate its interaction with PI3K, are necessary for Toll-2 to generate planar polarity.
Figure 6. Toll-2 tyrosine phosphorylation is required for planar polarity.
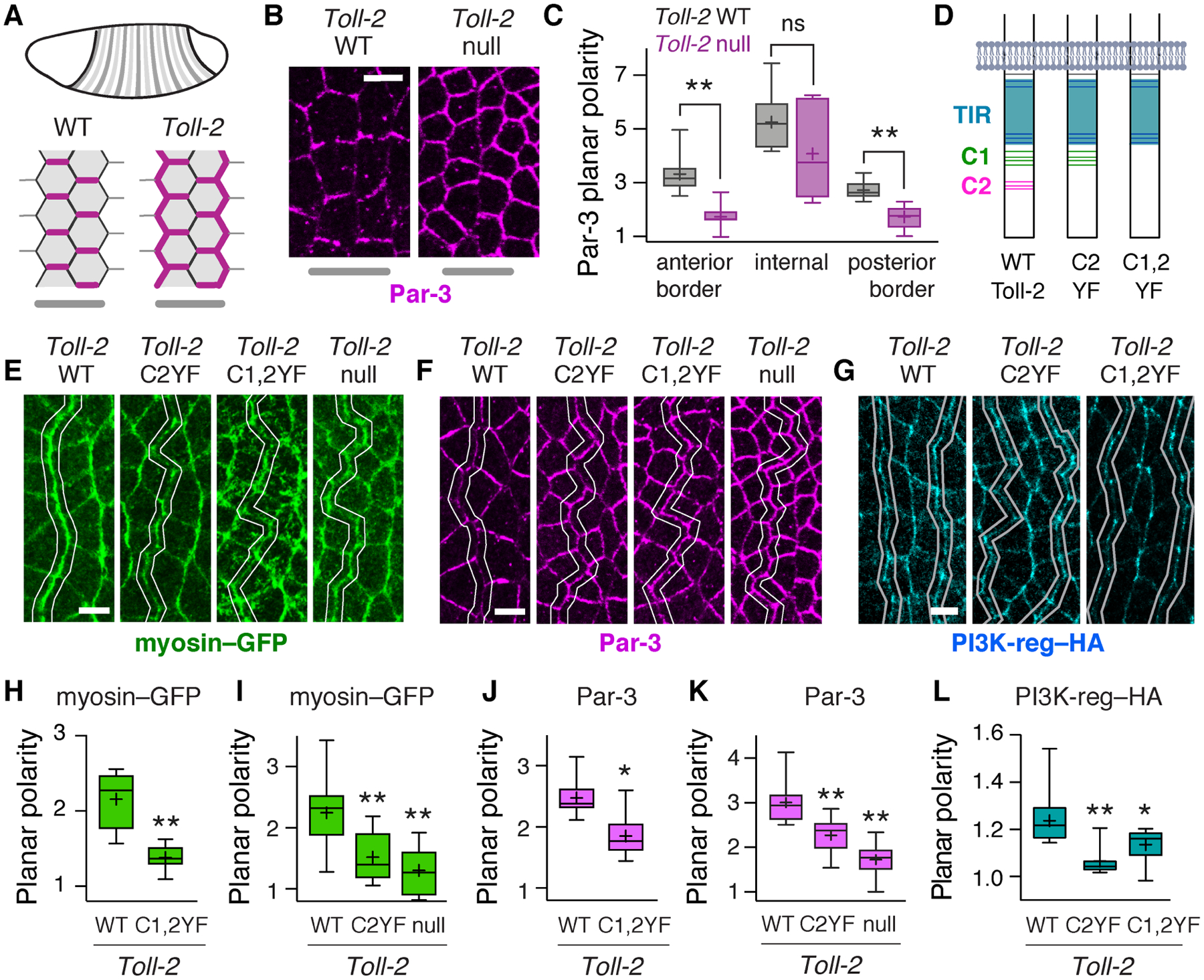
(A) Toll-2 is required for planar polarity at Toll-2 stripe borders. Par-3 (magenta), Toll-2 stripes (gray). (B,C) Par-3 planar polarity is defective at Toll-2 stripe borders in Toll-2 null mutants, identified by Wingless staining. (D) Toll-2 cytoplasmic domain. Gray, membrane. Blue, TIR domain. Green and magenta, C-terminal tyrosine clusters C1 and C2. Tyrosines, horizontal lines. (E–G) Localization of myosin–GFP (E), Par-3 (F), and PI3K-reg–HA (G) at Toll-2 stripe borders (white lines) identified by Wingless staining in embryos expressing the indicated Toll-2 variants from the Toll-2 locus. (H–L) Myosin–GFP planar polarity (vertical-to-horizontal edge intensity ratio) (H,I), Par-3 planar polarity (horizontal-to-vertical edge intensity ratio) (J,K), and PI3K–HA planar polarity (vertical-to-horizontal edge intensity ratio) (L) at Toll-2 stripe borders. Toll-2C1,2YF images in E and F are from the same embryo. Toll-2 null mutant images in B and F are from the same embryo. Boxes, 2nd and 3rd quartiles; whiskers, min to max; horizontal line, median; +, mean, 6–16 fixed stage 7 embryos/genotype, *p<0.03, **p<0.01, Welch’s t-test. Anterior left, ventral down. Bars, 5 μm. See also Figure S6.
Because Toll-2, Toll-6, and Toll-8 function additively to regulate convergent extension (Paré et al., 2014), local defects in cell polarity can be observed in Toll-2 single mutants. However, at the tissue level, the remaining receptors are capable of driving convergent extension in the absence of any single receptor (Paré et al., 2014). Therefore, to investigate the effects of Toll-2 tyrosine phosphorylation on convergent extension, we analyzed the effects of Toll-2 phosphovariants in time-lapse movies of embryos that lack the other two receptors. As expected, wild-type Toll-2 was sufficient to support axis elongation in embryos mutant for Toll-6 and Toll-8 (Figures 7A and 7B). By contrast, embryos expressing Toll-2C2YF, which shows reduced tyrosine phosphorylation in vivo and does not bind to PI3K-reg in vitro, displayed significantly reduced axis elongation and more frequent errors in edge contraction and vertex resolution in a Toll-6,8 mutant background, similar to the defects in Toll-2,6,8 triple mutants (Figures 7A–7C). These results demonstrate that the C-terminal tyrosines of Toll-2, which are targeted by Src kinases and mediate its association with PI3K, are essential for the effects of Toll-2 on planar polarity, cell intercalation, and axis elongation, suggesting that Toll-2/PI3K complex formation is required for all aspects of Toll-2 signaling during convergent extension.
Figure 7. Toll-2 tyrosine phosphorylation is required for cell intercalation and convergent extension.
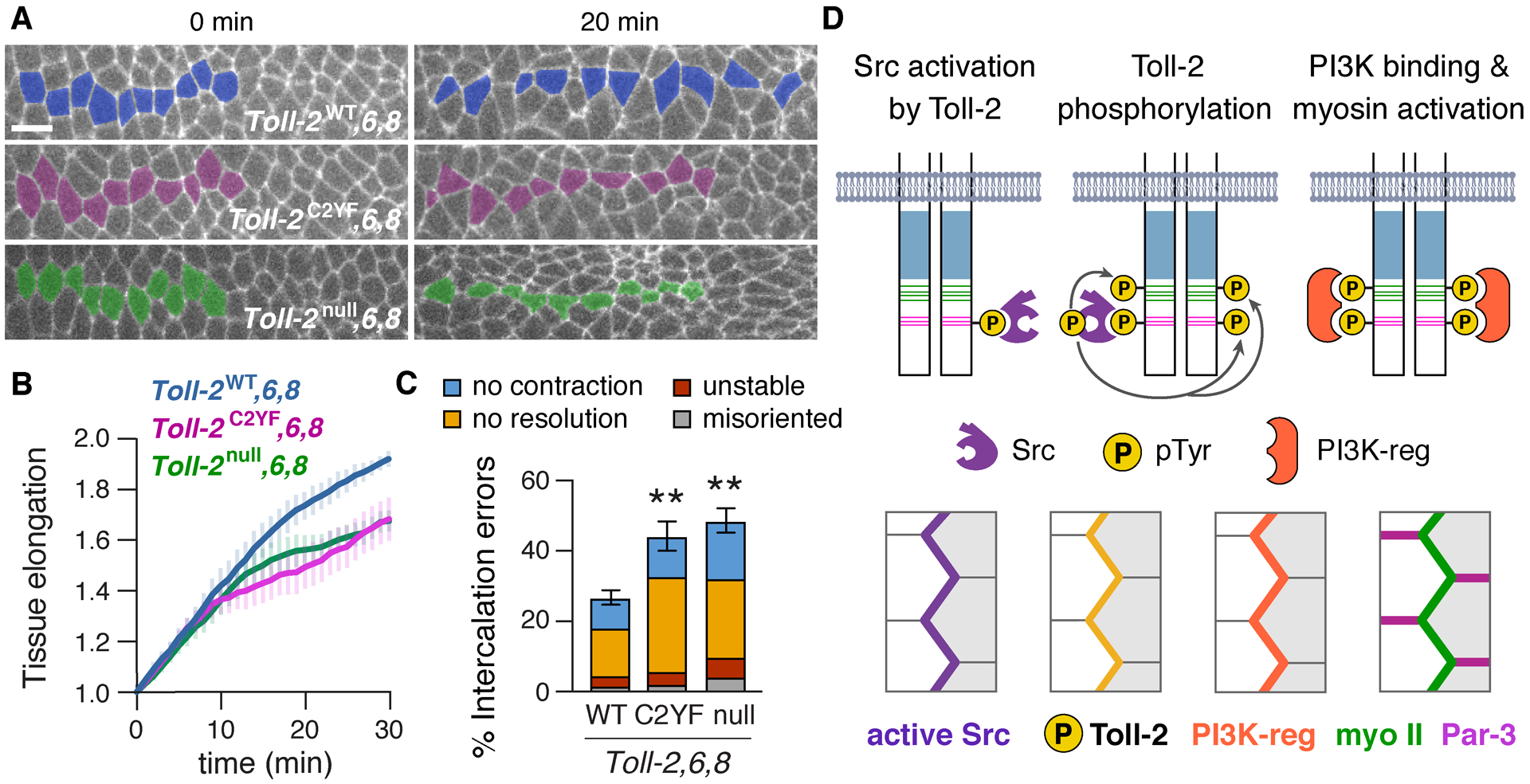
(A) Stills from movies of Toll-6,8 mutant embryos that express Toll-2WT, Toll-2C2YF, or no Toll-2 from the endogenous locus and Spider–GFP (0 min, onset of axis elongation). Cell intercalation is reduced in Toll-2C2YF,6,8 and Toll-2null,6,8 embryos. (B) Axis elongation (tissue AP length relative to t=0) is reduced in Toll-2C2YF,6,8 and Toll-2null,6,8 embryos compared with Toll-2WT,6,8 (p<0.03 at t=30 min). (C) Intercalation errors are increased in Toll-2C2YF,6,8 and Toll-2null,6,8 embryos compared with Toll-2WT,6,8. (D) Model of Toll receptor signaling. Mean±SEM, 4–9 living stage 7–8 embryos/genotype, **p≤0.007, Welch’s t-test. Anterior left, ventral down. Bar, 10 μm.
Discussion
Toll family receptors are best known for their roles in signaling to the nucleus to activate the expression of immune response genes. Here we show that Toll receptors also direct cell polarity and behavior during convergent extension by promoting the localized activities of Src and PI3K at the cell membrane. Toll receptors recruit active Src kinases to planar polarized membrane domains, and localized Src activity generates spatially regulated actomyosin contractility by promoting the phosphorylation of two clusters of tyrosines in the Toll-2 cytoplasmic domain, creating docking sites for PI3K-reg (Figure 7D). Src and PI3K are critical effectors of Toll receptor signaling, as loss of Src or PI3K recapitulates the effects of Toll receptor mutants on planar polarity, cell intercalation, and convergent extension. Moreover, disruption of a single cluster of tyrosines in the Toll-2 C-terminal domain, which is phosphorylated in a Src-dependent fashion, inhibits Toll-2/PI3K complex formation and abolishes Toll-2 function during convergent extension. Taken together, these results identify Src and PI3K-reg as critical effectors of Toll receptor signaling during convergent extension, and reveal a noncanonical mechanism by which Toll receptors induce spatially regulated cell behavior by mobilizing two potent signaling proteins to specific subcellular domains.
These results demonstrate that a critical symmetry-breaking event during convergent extension in Drosophila is the formation of localized Toll-2/PI3K complexes that generate tissue-wide patterns of actomyosin contractility. The upstream signals that activate Toll receptors in specific cellular domains are not known, but could involve a localized extracellular ligand that increases Toll-2 binding to Src or enhances Src activity. Alternatively, mechanisms that promote Toll receptor clustering could amplify the activity of associated Src kinases by enhancing their proximity to multiple substrates (Cooper and Qian, 2008). Toll-2 and Toll-6 both interact with PI3K-reg and are tyrosine phosphorylated by Src kinases in vitro, suggesting that a Src/PI3K signaling mechanism could link both Toll-2 and Toll-6 to PI3K. By contrast, Toll-8 is not phosphorylated by Src kinases, although it does associate weakly with PI3K-reg. Toll-8 could signal through a distinct mechanism, or it could communicate with Src and PI3K indirectly through heterodimerization with receptors such as Toll-6, which is expressed in many of the same cells (Paré et al, 2014). The defects in embryos lacking Src or PI3K appear to be more severe than the defects in Toll-2,6,8 mutants, suggesting that Src and PI3K may represent a common mechanism that mediates signaling downstream of multiple receptors during convergent extension.
Tyrosine kinase signaling pathways are widely used in multicellular organisms, with a particular enrichment of tyrosine phosphorylation at sites of contact between cells and with the extracellular matrix (Giannone and Sheetz, 2006; McLachlan and Yap, 2007). Identifying the substrates required for the diverse roles of tyrosine kinase signaling in vivo remains a formidable challenge. Here we demonstrate that Src-dependent phosphorylation of a single cluster of tyrosines in the Toll-2 C-terminal domain is sufficient to eliminate Toll-2 function during convergent extension. In addition to Toll-2, Src kinase may also promote the phosphorylation of other proteins involved in Toll receptor signaling. For example, Src family kinases can phosphorylate Rho-kinase and recruit Rho-kinase and myosin II to focal adhesions and adherens junctions in fibroblasts and epithelial cells (Lee et al., 2010; Andreeva et al., 2014; Gomez et al., 2015), and can phosphorylate and activate the Abl tyrosine kinase, which regulates planar polarized junctional dynamics during convergent extension (Tamada et al., 2012). In addition, Src kinases can stabilize cell adhesion (Pang et al., 2005; Takahashi et al., 2005; McLachlan et al., 2007), enhance E-cadherin dynamics (Shindo et al., 2008; Forster and Luschnig, 2012; Hunter et al., 2018; Chen et al., 2019), influence cytoskeletal organization (Thomas and Wieschaus, 2004; Sun et al., 2017; Andreeva et al., 2014; Hunter et al., 2018), and modulate cell polarity (Nelson et al., 2012; Olivares-Castiñeira and Llimargas, 2018). The recruitment of active Src kinases to Toll receptor complexes in Drosophila demonstrates an essential role for tyrosine kinase pathways in Toll receptor signaling, and suggests a mechanism by which Toll receptors could trigger a wide range of cellular events that contribute to epithelial remodeling.
PI3K is a conserved lipid kinase with widespread roles in cell growth, proliferation, survival, and polarity (Katso et al., 2001; Cain and Ridley, 2009; Vanhaesebroeck et al., 2010). Here we demonstrate that PI3K directs planar polarity in the Drosophila embryo by promoting the localized assembly of contractile myosin networks in epithelial cells. These results are consistent with altered cell topology and deregulated myosin activity observed in epithelial cells lacking the PI3K-antagonizing enzyme PTEN (Ma et al., 2008; Bardet et al., 2013; Grego-Bessa et al., 2015). The mechanisms by which PI3K influences tissue structure are not well understood. PI3K promotes the activity of the Rac GTPase in a number of cell types (Cain and Ridley, 2009; Chartier et al., 2011), but PI3K can also influence epithelial organization by activating PDK1 (Grego-Bessa et al., 2015), and PI3K regulates the localization and activity of RhoA and Rho-kinase in fibroblasts (Yoneda et al., 2005) and endothelial cells (Graupera et al., 2008). PI3K could influence planar polarity by regulating Rho GTPase signaling, which has been linked to Toll-2 (Kolesnikov and Beckendorf, 2007) and is essential for convergent extension (Simoes et al., 2010; Levayer et al., 2011; Kasza et al., 2014; Simoes et al., 2014; Munjal et al., 2015). The effectors that link Toll-2/Src/PI3K signaling to changes in actomyosin organization remain to be identified. These could include classical PI3K effectors that signal to the actin cytoskeleton (Pinner and Sahai, 2008; Xue and Hemmings, 2013) or other proteins such as RhoGEFs that interact with the phospholipid product of PI3K, phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (Shewan et al., 2011).
TLRs initiate signaling by recruiting adaptor proteins that assemble large multiprotein signaling complexes (Kawai and Akira, 2011; Luo et al., 2019). The Toll receptor signaling complexes involved in morphogenesis have not been defined. Here we demonstrate that the C-terminal-most tyrosine cluster of Toll-2 is targeted by Src kinases and recruits Src and PI3K to the Toll-2 complex. This tyrosine cluster bears homology to immunoreceptor tyrosine-based activation motifs (ITAMs and hemITAMs). These motifs are present in several classes of immunoreceptors and their associated adaptor proteins and, when phosphorylated, create binding sites for downstream effector proteins (Hamerman and Lanier, 2006; Abram and Lowell, 2007; Bauer and Steinle, 2017). Toll-2 therefore displays features of multiple receptors in the immune system: an extracellular domain and intracellular TIR domain characteristic of TLRs, and a C-terminal extension that couples to effectors through Src family kinases and PI3K, reminiscent of other immunoreceptors such as C-type lectin-like receptors and the T cell receptor/CD3 complex (Clements et al., 1999; Bauer and Steinle, 2017). Although mammalian TLRs do not display this bipartite structure, tyrosine-rich adaptor proteins that associate with mammalian TLRs, such as BCAP and SCIMP, could create docking sites for the assembly of downstream signaling components (Troutman et al., 2012; Luo et al., 2019). Toll-family receptors have conserved roles in regulating spatially regulated cell behaviors in flies and mammals, including axon growth (Ma et al., 2006; Cameron et al., 2007), synapse formation (McIlroy et al., 2013; Ballard et al., 2014; Ward et al., 2015; Foldi et al., 2017), epithelial remodeling (Eldon et al., 1995; Kleve et al., 2006; Kolesnikov and Beckendorf, 2007), and wound healing (Huebener and Schwabe, 2013; Carvalho et al., 2014). An intriguing possibility is that Src and PI3K could represent a core module of a spatially regulated branch of Toll receptor signaling that promotes the rapid and directional mobilization of cells during tissue development and immune defense.
Limitations of the study
This study demonstrates a functional connection between Toll receptors and localized Src and PI3K activity during convergent extension in Drosophila. However, several limitations remain. First, we were unable to generate phosphospecific antibodies to the C2 tyrosine cluster of Toll-2, and the other phosphospecific Toll-2 antibodies that we generated and used in this study worked only for western blots, but not for immunostaining. Therefore, we could not analyze the spatiotemporal pattern of Toll-2 tyrosine phosphorylation in the embryo. Second, we analyzed embryos that only had a partial loss of Src42, PI3K-reg, and PI3K-cat function, due to essential roles of these proteins at earlier stages of development. Therefore, these proteins may have additional functions in this tissue that were not detected in this study. Third, we focused on protein localization and cell behavior in the adherens junction domain, and our results do not exclude the possibility that defects in basolateral regions of these cells or in other parts of the embryo could contribute to the phenotypes observed.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jennifer Zallen (zallenj@mskcc.org).
Materials Availability
Materials generated in this study are available upon request.
Data and Code Availability
No large-scale datasets or new code were generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila Stocks and Genetics
Wild-type embryos were Oregon R and control genotypes are indicated for each experiment. Fly stocks were grown on molasses/cornmeal/yeast food and embryos were collected on apple juice agar plates at 25°C (Figures 3–5, 6G, 6L, S4, and S5) or room temperature (Figures 1, 2, 6A–F, 6H–K, 7, S1–S3, S6). The sex of the embryos was not considered relevant to this study and was not determined. The following knock-in lines were generated in this study using the Toll-2attP landing site (Paré et al., 2019): Toll-2–HA, Toll-2C2YF–HA, Toll-2C1,2YF–HA, Toll-2–V5, Toll-2C1YF–V5, Toll-2C1,2YF–V5, Toll-2WT, Toll-2C2YF, and Toll-2C1,2YF. The following transgenic lines were generated in this study: pUASp-Src42K276R–Venus (attP2) (Src DN), pCFD3-Src42-gRNA (attP2), pUASp-PI3K-reg–Venus (VK37), pUASp-PI3K-reg–HA (VK37), pUASp-PI3K-reg-ΔNSH2–HA (VK37), pUASp-PI3K-reg-ΔCSH2–HA (VK37), pUASp-PI3K-reg-ΔSH2–HA (VK37), and pUASp-PI3K-reg-ΔCatBD–HA (VK37). The following published alleles and transgenic lines were used: Toll-2attP (Toll-2null) (Toll-2 coding region replaced with attP-3xP3-DsRed-attP (Paré et al., 2019), Toll-65A (frameshift mutation after amino acid 35) (Paré et al., 2014), Toll-859 (deletion of the Toll-8 open reading frame) (Yagi et al., 2010), and Toll-8145 (a 1.2 kb deletion in the Toll-8 5’ UTR) (Kim et al., 2006), ubi-E-cadherin–GFP (Oda and Tsukita, 2001), spider–GFP and resille–GFP (gifts of Alain Debec, Institut Jacques Monod, Paris, France), myosin–mCherry (Martin et al., 2009) and myosin–GFP (Royou et al., 2004) (mCherry or GFP fused to the sqh myosin regulatory light chain and expressed from the sqh promoter), sqh-gap43–mCherry (Martin et al., 2010), Src64–YFP (CPTI000567) (Lye et al., 2014), pUASt-Src42Y511F (Src42 CA) (Tateno et al., 2000), eve–YFP (Ludwig et al., 2011) and matαtub67 Gal4 (gift of Daniel St Johnston, University of Cambridge, Cambridge, UK). To analyze total Src42 and pSrc42 localization in Toll-2,6,8 triple mutants in Figure 1B and Figure S1, the F1 progeny of Toll-2attP/CyO,hb-lacZ; Toll-65A, Toll-859/+ males and females were analyzed. Toll-2attP, Toll-65A, Toll-859 triple mutants were identified by the absence of lacZ and Toll-8 immunostaining, Toll-2attP single mutants were identified by the absence of lacZ and the presence of Toll-8 immunostaining, and Toll-65A, Toll-859 double mutants were identified by the presence of lacZ and the absence of Toll-8 immunostaining.
To disrupt maternal Src42 expression, males bearing a pCFD3-Src42-gRNA transgene expressing a gRNA targeting the first splice acceptor site of Src42 (Src42 gRNA) were crossed to females that harbor a nos-Cas9 transgene expressed in the female germline (Port et al., 2014) and the F2 progeny were analyzed. To disrupt maternal Src64 expression, males bearing the Src64 shRNA transgene P{TRiP.GL00297}attP2 targeting the 5’ UTR of Src64 (Ni et al., 2011) were crossed to matαtub67 Gal4 females and the F2 progeny were analyzed. To generate embryos with reduced maternal expression of both Src42 and Src64 (Src KD embryos), matαtub67 Gal4; pCFD3-Src42-gRNA females were crossed to nanos-Cas9; P{TRiP.GL00297}attP2 males and the F2 progeny were analyzed. This method consistently reduced Src64 expression, but Src42 expression was more variable. Src KD embryos with an >85% reduction in Src42 and Src64 levels by western blot, or in Src42 fluorescence intensity by immunostaining with the Src42 antibody (1:2,000; gift of K. Saigo and T. Kojima, University of Tokyo, Tokyo, Japan) (Takahashi et al., 2005), were selected for analysis.
To generate embryos that express dominant negative Src42, males bearing a pUASp-Src42K276R–Venus (Src DN) transgene were crossed to matαtub67 Gal4; sqh-sqh–mCherry females and the F2 progeny were analyzed. The F1 progeny of matαtub67 Gal4/+; sqh-sqh–mCherry/+ females were analyzed as a control. Similar defects were observed in Src KD and Src DN embryos, suggesting that Src DN inhibits both Src42 and Src64. To generate embryos that express constitutively active Src42, males bearing a pUASt-Src42Y511F (Src42 CA) transgene (Tateno et al., 2010) were crossed to matαtub67 Gal4; sqh-sqh–GFP females and the F1 progeny were analyzed. The F1 progeny of matαtub67 Gal4; sqh-sqh–GFP females crossed to Oregon R males were analyzed as a control.
To generate PI3K-cat KD embryos, males bearing the PI3K-cat shRNA transgene P{TRiP.HMC05152}attP40 (Ni et al., 2011) targeting exon 4 of PI3K-cat were crossed to matαtub67 Gal4; sqh-sqh–GFP females and the F2 progeny were analyzed. To generate PI3K-reg DN embryos, males bearing a pUASp-PI3K-reg-ΔCatBD–HA transgene were crossed to matαtub67 Gal4 females and the F2 progeny were analyzed. Embryos that exhibited severe morphological defects prior to the onset of axis elongation were not included in the analysis. The F1 progeny of matαtub67 Gal4/+; sqh-sqh–GFP/+ flies were analyzed as a control.
To visualize PI3K-reg–Venus localization in wild type, males bearing a pUASp-PI3K-reg–Venus (VK37) transgene were crossed to matαtub67 Gal4; sqh-gap43–mCherry females and the F2 progeny were analyzed live. Embryos with intermediate levels of PI3K-reg–Venus expression were selected for analysis. Embryos that expressed high levels of PI3K-reg–Venus exhibited defects and were not analyzed. Gap43–mCherry was included to identify edges for quantification. To visualize PI3K-reg–Venus localization in embryos expressing Src42 CA, the progeny of matαtub67 Gal4/pUASp-PI3K-reg–Venus females crossed to pUASt-Src42Y511F males were analyzed live. The F1 progeny of matαtub67 Gal4/pUASp-PI3K-reg–Venus females crossed to Oregon R males were analyzed as a control.
To visualize PI3K-reg–HA localization in wild type, males bearing pUASp-PI3K-reg–HA (VK37), pUASp-PI3K-reg-ΔNSH2–HA (VK37), pUASp-PI3K-reg-ΔCSH2–HA (VK37), pUASp-PI3K-reg-ΔSH2–HA (VK37), or pUASp-PI3K-reg-ΔCatBD–HA (VK37) were crossed to matαtub67 Gal4 females and the F2 progeny were fixed and stained for analysis. To visualize PI3K-reg–HA localization in Toll-2 knock-in mutants, in vitro synthesized PI3K-reg–HA transcripts were injected into early embryos, which were then fixed and stained for analysis.
Cell Culture
Drosophila S2R+ cells (Drosophila Genomics Resource Center, stock number 150) were cultured at room temperature or 25°C in Schneider’s medium supplemented with 10% fetal bovine serum (F4135 Millipore Sigma).
METHOD DETAILS
Cloning
See Table S2 for primer sequences. The pUASp-W-attB vector was a gift of Michael Buszczak (University of Texas Southwestern Medical Center, Dallas TX). pUASp-W-msVenus was generated by inserting an msVenus cDNA encoding monomeric superfolder Venus (msVenus) (gift of Ben Glick, University of Chicago, Chicago, IL) preceded by a 4xGSS linker into pUASp-W-attB. pUASp-W-HA was generated by cloning a PCR fragment encoding a 1xHA tag (TACCCATACGATGTTCCAGATTACGCT) into pUASp-W-attB. pUASp-V5 was generated by inserting complementary oligonucleotides encoding the V5 tag (GGTAAGCCTATCCCTAACCCTCTCCTCGGTCTC-GATTCTACG) into pUASp-W-attB linearized with BamHI and MluI. Linearized backbones for Gibson cloning were generated using the restriction enzymes BamHI (for C-terminal msVenus and V5 fusions), MluI (for C-terminal HA fusions), or both (when cloned directly into pUASp-W-attB) in CutSmart Buffer for 1 h at 37°C (NEB) to excise the CAT/CmR-ccdB lethal genes from the pUASp-W-attB backbone. All PCR products and linearized backbones were gel purified (Illustra GFX Kit, GE) and Gibson assembled using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, NEB).
To generate pUASp-PI3K-reg–Venus, the PI3K-reg open reading frame was amplified from the LD42724 cDNA plasmid (generated by the Berkeley Drosophila Genome Project and obtained through the Drosophila Genome Resource Center). The PCR fragment was cloned into pUASp-W-msVenus digested with BamHI by Gibson assembly. To generate pUASp-PI3K-reg–HA, pUASp-PI3K-reg-ΔNSH2–HA (deletion of amino acids 20–120), pUASp-PI3K-reg-ΔCSH2–HA (deletion of amino acids 325–406), pUASp-PI3K-reg-ΔSH2–HA (deletion of amino acids 20–120 and 325–406), and pUASp-PI3K-reg-ΔCatBD–HA (PI3K-reg DN) (amino acids 185–277 were replaced with a 4xGSS linker), 2–5 PI3K-reg fragments were PCR amplified from the LD42724 cDNA plasmid using primers to add a C-terminal HA tag and cloned into the linearized pUASp vector. All PI3K-reg plasmids were inserted into the VK37 landing site on chromosome II. To generate pCS2-PI3K-reg–HA, PI3K-reg–HA was amplified from pUASp-PI3K-reg–HA and cloned into the pCS2+ vector (Turner and Weintraub, 1994) for in vitro transcription of PI3K-reg–HA mRNA from the SP6 promoter.
To generate pUASp-Toll-2–V5, pUASp-Toll-6–V5, and pUASp-Toll-8–V5, the open reading frames of wild-type Toll-2, Toll-6, and Toll-8 were amplified with the indicated primers and cloned into linearized pUASp–V5 by Gibson assembly. The pUASp-Toll-2C1,2YF–V5 construct was generated by 4-part Gibson assembly with the linearized pUASp-V5 backbone, synthesized dsDNA fragment GB004, and two fragments amplified from pUASp-Toll-2–V5 using KSB047 with KSB014 and KSB048 with KSB006. The pUASp-Toll-2C1YF–V5 construct was generated by 3-part Gibson assembly with the linearized pUASp backbone and two fragments amplified from pUASp-Toll-2–V5 and pUASp-Toll-2C1,2YF–V5 using KSB047 with KSB056 and KSB048 with KSB055. The pUASp-Toll-2C2YF–HA construct was generated by 3-part Gibson assembly with the linearized pUASp backbone and two fragments amplified from pUASp-Toll-2–V5 and pUASp-Toll-2C1,2YF–V5 using MT001 with MT002 and MT003 with MT004. The pUASp-Toll-2TIRYF–V5 construct contained the following mutations: Y1045, Y1056, Y1156, Y1171, and Y1178 and was generated by 4-part Gibson assembly with the linearized pUASp-V5 backbone, synthesized dsDNA fragment GB010 (IDT), and two fragments amplified from pUASp-Toll-2–V5 using KSB033 with KSB035 and KSB036 with UASp-Toll-2-V5_r.
The pUASp-Toll-2–HA construct was previously described (Paré et al., 2014). To generate pUASp-Toll-2-Δcyto–HA (deletion of amino acids 1040–1385), pUASp-Toll-2-ΔCTD–HA (deletion of amino acids 1239–1385), and pUASp-Toll-2-ΔTIR–HA (deletion of amino acids 1044–1181), full-length Toll-2 and Toll-2 fragments were PCR amplified from pUASp-Toll-2 and cloned into the linearized pUASp-W-HA backbone by Gibson assembly. The pUASp-Toll-2Δcyto–HA construct was generated by 2-part Gibson assembly with the linearized pUASp-W-HA backbone and a fragment amplified from pUASp-Toll-2–HA using Toll2:HA 1xHA Fwd with Toll2:HADEL Cyto Rev. The pUASp-Toll-2ΔCTD–HA construct was generated by 2-part Gibson assembly with the linearized pUASp-W-HA backbone and a fragment amplified from pUASp-Toll-2–HA using Toll2:HA 1xHA Fwd with Toll2:HADEL C-Rev. The pUASp-Toll-2ΔTIR–HA construct was generated by 3-part Gibson assembly with the linearized pUASp-W-HA backbone and two fragments amplified from pUASp-Toll-2–HA using Toll2:HA 1xHA Fwd with Toll2:HADEL TIR R1 and Toll2:HADEL TIR F2 with Toll2:HADEL TIR R2. The pUASp-Toll-2Y1056F–HA construct was generated by 3-part Gibson assembly with the linearized pUASp backbone and two fragments amplified from pUASp-Toll-2–HA using KSB047 with KSB039 and KSB038 with KSB048. The pUASp-Toll-2Y1191F–HA construct was generated by 3-part Gibson assembly with the linearized pUASp backbone and two fragments amplified from pUASp-Toll-2–HA using KSB047 with KSB042 and KSB041 with KSB048.
To generate pCFD3-Src42-gRNA, the oligos 5′-GTCGAAACCTACCAATAGGGTGAC-3′ and 5′-AAACGTCACCCTATTGGTAGGTTT-3′ were annealed and cloned into the pCFD3 vector digested with BbsI (Port et al., 2014) (Addgene 49410). pCFD3-Src42-gRNA was inserted into the attP2 site on chromosome III. To generate pUASp-Src42–Myc and pUASp-Src64–Myc, the Src42 open reading frame (PA isoform) or the Src64 open reading frame (PE isoform) were PCR amplified and cloned into the pENTR/D-TOPO vector (Invitrogen), followed by recombination into pPWM (gift of Terence Murphy, Carnegie Institute, Baltimore, MD) using the Gateway system (Invitrogen).
To generate pEntr-Src42-DN–Venus, a fragment of the Src42 open reading frame was PCR amplified from flies bearing a Src42K276R transgene (Takahashi et al., 2005) and cloned into pENTR-Src42 digested with MfeI and HindIII. A DNA fragment encoding msVenus was cloned into the AscI site of pEntr-Src42-DN and Src42-DN–Venus was recombined into pUASp-W-attB using the Gateway system to generate pUASp-Src42-DN–Venus. The pUASp-Src42-DN–Venus transgene was inserted into the attP40 site on chromosome II.
Toll-2 knock-in lines
Toll-2 donor constructs were generated in a backbone containing inverted attB sites and introduced into the Toll-2attP landing site (Paré et al., 2019) with transgenically expressed phiC31 integrase or a coinjected vasa-phiC31 plasmid (p3xP3-EGFP.vas-int.NLS, Addgene 60948), following the strategy described in (Zhang et al., 2014). Donor constructs were diluted in H2O and injected at 150 ng/μl with 200 ng/μl vasa-PhiC31 or at 100 ng/μl with 130 ng/μl vasa-PhiC31. Injected embryos were incubated at 18°C for 48–72 h until they reached the larval stage, transferred to extra-wet food vials, and grown to the adult stage at 25°C. Adult injected males were crossed individually to y, w; Sp/CyO; Dr/TM3 females at 25°C for 3 days, insertion lines were screened for the loss of 3xP3-DsRed in the larval CNS and adult eye, and the presence and correct orientation of the insertion were confirmed by PCR amplification and sequencing.
The pattB-Toll-2-WT plasmid was generated by 3-part Gibson assembly with the following fragments: the left and right fragments of the pBS-attB backbone (Addgene 61255) were amplified using primers ZKM018 with ZKM019 and ZKM018 with ZKM020. The Toll-2 genomic region from cut to cut was amplified using primers ZKM085 and ZKM086, which added 20 bp overlaps to the attB sequence. The pattB-Toll-2-YF constructs were generated by 3-part Gibson assembly with the following fragments: the left and right backbone fragments were amplified from pattB:Toll-2-WT using KSB005 with ZKM019 and KSB006 with ZKM020. The Toll-2 coding region was amplified from pUASp-Toll-2–V5 constructs that contained the corresponding mutations using KSB007 with KSB008.
The pattB-Toll-2-WT–HA plasmid was generated by 3-part Gibson assembly with the following fragments: the left and right backbone fragments were amplified from pattB-Toll-2-WT using ZKM083 with ZKM019 and KSB001 with ZKM020. The Toll-2 coding region was amplified from pattB:Toll2WT using ZKM096 with KSB002 to add the C-terminal HA tag. The pattB-Toll-2C1,2YF–HA and pattB-Toll-2C2YF–HA constructs were generated by 3-part Gibson assembly with the following fragments: the left and right backbone fragments were amplified from pattB-Toll-2-WT–HA using KSB006 with ZKM020 and ZKM019 with SS001. The Toll-2 coding region was amplified from pattB-Toll-2 containing the corresponding mutations using SS002 with KSB008.
The pattB-Toll-2-WT–V5 and pattB-Toll2-YF–V5 constructs were generated by 3-part Gibson assembly with the following fragments: the left and right backbone fragments were amplified from pattB-Toll-2-WT using KSB005 with ZKM019 and KSB009 with KSB010. The Toll-2 coding region was amplified from pUASp-Toll-2–V5 constructs containing the corresponding mutations using KSB007 with KSB011.
Antibody generation
Phosphospecific antibodies to the Toll-2 sites pY1218/1219 and pY1191 were generated by immunizing guinea pigs (Cocalico Biologicals) with the following synthetic phosphopeptides (Genscript): APPPPA{pTYR}{pTYR}{ABU}TEEMEANYSC (Toll-2 pY1218/1219) and CPIELSPRGNN{pTYR}TLDHH (Toll-2 pY1191). An antibody to Src64 was generated by immunizing guinea pigs with the CDFGLARVIADDE{pTYR}{SER}PKQG peptide. N- or C-terminal cysteines were added for conjugation to KLH, internal cysteines were mutated to {ABU} or {SER}. Toll-2 pY1191 antibody was affinity purified against the original, unconjugated phosphopeptide. The Toll-2 pY1218/1219 antibody bound specifically to phosphorylated Toll-2 without purification. The unpurified Src64 Y434 antibody was not phosphospecific and was used to identify total Src64 protein by western blotting.
Embryo injection
To analyze PI3K-reg–Venus localization in Toll receptor-deficient embryos, the Toll-2, Toll-6, Toll-8, and Toll-3 (control) dsRNAs were generated as described (Paré et al., 2014). In Figure 4A, embryos expressing PI3K-reg–Venus (the F2 progeny of pUASp-PI3K-reg–Venus males and matαtub67 Gal4; gap43–mCherry females) were collected for 1 h at 25°C and injected dorsally with Toll-2 and Toll-8 dsRNAs at 2 μg/μl each or control Toll-3 dsRNA at 4 μg/μl. After aging for 2.5 h at room temperature, embryos were mounted for live imaging, or 10 stage 7 embryos/sample were selected and subjected to western blot. To analyze PI3K-reg–Venus localization in Figure S6, the F2 progeny of pUASp-PI3K-reg–Venus males and matαtub67 Gal4 females were injected with water (control) or with Toll-2 and Toll-8 dsRNAs at 2 μg/μl each or with Toll-2 and Toll-6 dsRNAs at 2 μg/μl each. To analyze PI3K-reg–Venus localization in Toll-2,6 KD, ΔToll-8 embryos, the progeny of Toll-859/145 females that expressed pUASp-PI3K-reg–Venus from the matαtub67 Gal4 driver were injected with dsRNAs targeting Toll-2 and Toll-6 at 2 μg/μl each. PI3K-reg–Venus localization was analyzed in living embryos, where its planar polarity was most evident. A maximum of two dsRNAs were injected, as injecting more than two dsRNAs reduced the efficiency of knockdown.
For PI3K-cat dsRNA injection, dsRNA targeting exon 7 was generated with primers 5’-taatacgactcactatagggGCATCCGACCAGAACCTTT-3’ and 5’-taatacgactcactataggg-GCACGCGTCTTGTCAAAGT-3’ using the T7 MEGAscript Kit (Ambion). PI3K-cat dsRNA or control Toll-3 dsRNA was injected dorsally at 4 μg/μl into 0–1 h embryos expressing myosin–GFP or Resille–GFP. After aging for 2.5 h at room temperature, embryos were mounted for live imaging or fixed in a 1:1 mixture of 3.7% formaldehyde in PBS and heptane for 1 h, manually devitellinized, and Toll-3 and PI3K-cat dsRNA-injected embryos were stained in the same tube after prestaining the Toll-3 embryos for 1 h with Alexa 546-conjugated phalloidin (1:1,000; Invitrogen).
To analyze PI3K-reg localization in Toll-2YF mutants, PI3K-reg–HA mRNA was generated by in vitro transcription (mMESSAGE mMACHINE SP6 Transcription Kit) using pCS2-PI3K-reg–HA as a template. Purified mRNA was diluted to 2 μg/μl in H2O and injected ventrally into 1–2 h embryos that were the progeny of Toll-2WT, Toll-2C2YF, or Toll-2C1,2YF/CyO, hb-lacZ knock-in flies. Injected embryos were then incubated for 1 h at room temperature, fixed in a 1:1 mixture of 3.7% formaldehyde in PBS and heptane for 1 h, manually devitellinized, and stained. Mutant embryos were identified by the absence of lacZ signal from the CyO balancer.
For pharmacological inhibition of PI3K-cat, 50 μM wortmannin (Cell Signaling) in 2.5% DMSO, or 2.5% DMSO alone, were injected ventrally into the perivitelline space of early to mid-stage 6 embryos expressing myosin–GFP. The wortmannin concentration is predicted to be diluted 50-fold in the embryo, producing a final concentration of 1 μM wortmannin and 0.05% DMSO. Injected embryos were aged 5–10 min at room temperature and fixed 1:1 mixture of 18% formaldehyde in PBS and heptane for 10 min, manually devitellinized, and wortmannin- and control-injected embryos were stained in the same tube after prestaining the Toll-3 embryos with Alexa 546-conjugated phalloidin (1:1,000; Invitrogen). After mounting, stage 7 embryos were selected for imaging.
For pharmacological inhibition of Src/Abl kinases, 5 mM Bosutinib (Selleck Chemicals) in 30% DMSO, or 30% DMSO alone, were injected ventrally into the perivitelline space of early to mid-stage 6 embryos that were the F2 progeny of pUASp-PI3K-reg–Venus males and matαtub67 Gal4; gap43–mCherry females. The Bosutinib concentration is predicted to be diluted 50-fold in the embryo, producing a final concentration of 100 μM Bosutinib and 0.6% DMSO. Injected embryos were aged 5–10 min at room temperature to early stage 7 and mounted for live imaging.
Immunofluorescence
Embryos were fixed 12 min in a 1:1 mixture of 18% formaldehyde in 0.1 M phosphate buffer pH 7.2 and heptane and manually devitellinized. Primary antibodies used for immunofluorescence were guinea pig anti-Par-3 (1:500) (Blankenship et al., 2006), rabbit anti-Src42 (1:2,000; gift of K. Saigo and T. Kojima, University of Tokyo, Tokyo, Japan) (Takahashi et al., 2005), rabbit anti-phospho-Src42-Y400 (1:250; gift of S. Hayashi, RIKEN Center for Developmental Biology, Kobe, Japan) (Shindo et al., 2008), mouse anti-wingless (1:10, Developmental Studies Hybridoma Bank (DSHB), 4D4) (Brook and Cohen, 1993), mouse anti-V5 (1:100, Invitrogen), mouse anti-Neurotactin (1:200, DSHB, BP106) (Hortsch et al., 1990), mouse anti-β-gal (1:25, DSHB, 40–1a) (Ghattas et al., 1991), rabbit anti-GFP (1:100, Torrey Pines), chicken anti-GFP (1:500, Abcam), rat anti-HA (1:500, Roche), and guinea pig anti-runt (1:1,000) (Kosman et al., 1998). Primary antibodies were detected with Alexa Fluor secondary antibodies (1:500, Invitrogen). F-actin was detected with Alexa 546- or Alexa 647-conjugated phalloidin (1:1,000, Invitrogen). Embryos were mounted in ProLong gold (Invitrogen) and imaged on a Zeiss LSM700 laser-scanning confocal microscope with a PlanNeofluor 40×/1.3 NA oil-immersion objective; 1.0 μm z-slices were acquired at 0.5 μm z-steps. Maximum intensity projections of 1.5–2.0 μm in the apical junctional domain were generated for analysis.
Time-lapse imaging
Embryos were dechorionated for 2 min in 50% bleach, washed in water, and mounted in a 1:1 mixture of halocarbon 27 and 700 oil (Sigma) on a gas-permeable membrane (YSI). Time-lapse imaging was acquired on Ultraview Vox spinning disk confocal with a Zeiss Plan Neofluor 40×/1.3-NA oil-immersion objective and a Hamamatsu Orca-R2 camera using Volocity software. For time-lapse movies of WT (matαtub67 Gal4/E-cadherin–GFP, sqh-sqh–mCherry; pCFD3-Src42-gRNA/+) and Src KD (nanos-Cas9/+ or Y; matαtub67 Gal4/E-cadherin–GFP, sqh-sqh–mCherry; pCFD3-Src42-gRNA/P{TRiP.GL00297}) embryos, image stacks of embryos expressing E-cadherin–GFP and myo–mCherry were acquired at 0.5 μm z-steps and 15 s intervals. After imaging, each embryo was lysed and analyzed by single embryo western blot using Src42 and Src64 antibodies to confirm protein depletion. For time-lapse imaging of Toll-2WT,6,8, Toll-2C2YF,6,8, and Toll-2null,6,8 embryos, z-stacks of embryonic progeny from Toll-2WT, Toll-2C2YF, or Toll-2null/CyO, twi-Gal4, UAS-GFP; Toll-65A, Toll-859, Spider–GFP/eve–YFP were acquired at 15 s intervals and 1 μm z-steps. Triple mutant embryos were identified by the absence of GFP signal from the CyO balancer and the eve–YFP transgene. For time-lapse movies of PI3K-cat KD, PI3K-cat dsRNA-injected, and PI3K-reg DN embryos, image stacks of embryos expressing Spider–GFP (PI3K-cat KD and PI3K-reg DN embryos) or Resille–GFP (PI3K-cat dsRNA-injected embryos) were acquired at 1.0-μm z-steps and 15 s intervals. Live imaging of PI3K-reg–Venus was acquired using PlanApo 63×/1.4NA oil-immersion objective at 1.0 μm z-steps and 1 min intervals. Maximum intensity projections of an apical 2–3 μm region were generated for analysis.
Cell culture experiments
Drosophila S2R+ cells were cultured in Schneider’s medium with 10% fetal bovine serum at room temperature or 25°C. Transfections were performed with FuGENE HD (Promega) using the manufacturer’s protocol. The pUASp-Toll-2–V5 wild-type and Toll-2YF–V5 variants, or pUASp-Toll-2–HA wild-type and deletion variants, were cotransfected with pAc5.1/V5-HisB-GAL4 (Simoes et al., 2006) and pUASp-Src42–Myc, pUASp-Src64–Myc, pUASp-PI3K-reg–Venus, or pUASp-PI3K-reg–HA variants. Plasmids were transfected at 300 ng/μl each and incubated at 25°C for 36–48 h. For Src/Abl inhibition, a stock solution of 10 mM Bosutinib in DMSO, or DMSO alone, was diluted in Schneider’s Insect Media and added to cells at a final concentration of 1 μM Bosutinib, 0.01% DMSO and incubated for 48 h. After incubation, cells were lysed and subjected to immunoprecipitation.
Immunoprecipitation
Transfected S2R+ cells were lysed in lysis buffer [50 mM Tris-HCl (pH=7.5), 2 mM EDTA, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40, 1 mM phenylmethylsulphonyl fluoride, 2 mM Sodium Orthovanadate, Protease/Phosphatase Inhibitor Cocktail (Cell Signaling, 1:100)]. To analyze the binding between Toll-2WT or Toll-2YF variant and Src42, 0.5% TritonX-100 was used instead of Nonidet P-40. Lysates were centrifuged (13,000xg) 20 min at 4°C and supernatants were incubated with 10 μl anti-GFP nanobody conjugated to magnetic agarose beads (Chromotek) for 2 h at 4°C or anti-HA (1:400, Roche) or anti-V5 antibody (1:400, Invitrogen) for 30 min and then with Protein G Sepharose 4B (GE Healthcare) for 2 h at 4°C. Immunocomplexes were washed 3x in lysis buffer, eluted with SDS sample buffer, and analyzed by SDS-PAGE and western blot. For Toll2–V5 or Toll-2–HA immunoprecipitation in vivo, staged embryos (3.5–4.5 h) were collected at room temperature and lysed in lysis buffer. Toll-2 was immunoprecipitated and analyzed as described above.
Western blots
Immunoprecipitates or lysates of single or 10 embryos (hand-selected by stage, punctured with a glass needle, and boiled in SDS sample buffer) were applied to a Bis-Tris 4–12% acrylamide NuPage gel (Invitrogen), transferred to a PVDF membrane (Millipore), and immunoblotted with the following primary antibodies: mouse anti-V5 (1:2,500, Invitrogen), mouse anti-GFP (1:1000, Roche), rat anti-HA (1:2,000, Roche), mouse phosphotyrosine (1:4,000, 4G10 Millipore Sigma), mouse anti-Myc (1:1,500, DSHB) (Evan et al., 1985), rabbit anti-phosphomyosin (Ser19) (1:250, Cell Signaling), rabbit anti-phospho-Akt (Ser505) (1:250, Cell Signaling), rabbit anti-phospho-(Tyr) p85 PI3K binding motif (1:1000, Cell Signaling) (for Figure 3D only), mouse anti-β-actin (1:1,000, Abcam), mouse anti-β-catenin (1:500, DSHB) (Riggleman et al., 1990), guinea pig anti-Src64 (1:1,000, this study), or rabbit anti-Src42 (1:20,000) (Takahashi et al., 2005), purified anti-Toll-2-pY1191 (1:125, this study), anti-Toll-2-pY1218/1219 (1:500, this study), and HRP-conjugated secondary antibodies (1:10,000, Jackson Laboratory). Western blots are representative of 3–5 experiments.
Quantitative RT-PCR
Total RNA was extracted from ~60 staged embryos/sample using the TRIzol reagent (Invitrogen). RNA was purified according to the manufacturer protocol. 2 μg total RNA per sample was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). 50 ng cDNA was amplified per qRT-PCR reaction using TaqMan gene expression assays for PI3K-cat (Dm02142679_g1), PI3K-reg (Dm01842882_m1), and RpL32 (Dm02151827_s1) (Applied Biosystems). Reactions were carried out using a 7900HT Fast Real-Time PCR system (Applied Biosystems). The results are representative of at least two biological replicates and expression was normalized to RpL32 within each sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each result is representative of at least two independent experiments. Mean values were compared using Welch’s t-test (Glantz, 2002). Information about the n values, p values, and statistical tests used can be found in the figure legends and in Table S1.
To analyze planar polarity in fixed embryos, maximum intensity projections of 1.5–2.0 μm in the apical junctional domain of stage 7 embryos were analyzed for planar polarity as described (Simoes et al., 2010). The mean pixel intensity for vertical edges (oriented at 75–90° relative to the AP axis) and horizontal edges (oriented at 0–15° relative to the AP axis) were measured using SIESTA, a tool for Scientific ImagE SegmenTation and Analysis (Fernandez-Gonzalez and Zallen, 2011). Planar polarity was calculated as the ratio of the mean intensity at edges oriented at 75–90° relative to the mean intensity of edges oriented at 0–15° edges for myosin–GFP, PI3K-reg–Venus, total Src42, and pSrc42 after subtracting the cytoplasmic background signal calculated in SIESTA (the mean intensity of all pixels located at least 1 μm from a cell edge). The reciprocal ratio was calculated for Par-3. Planar polarity in Figure S1I was calculated as above using Toll-8 antibody staining to identify Toll-8 and non-Toll-8 cells. Planar polarity at Toll-2 stripe boundaries was calculated as the ratio of the mean intensity at edges that were oriented at 75–90° and were at the Toll-2 stripe boundary (identified by wingless staining for Par-3 and myosin–GFP and runt staining for PI3K-reg–HA) to the mean intensity of edges oriented at 0–15° that were within the Toll-2 stripe, after subtracting the cytoplasmic background signal. Image J was used to calculate the cytoplasmic background signal by measuring the average mean cytoplasmic pixel intensity of circles 3–8 μm2 in area within all of the visible Toll-2-expressing cells. For planar polarity measurements, the ratio of the mean intensity at edges oriented at 75–90° relative to the mean intensity of edges oriented at 0–15° or the reciprocal ratio was compared. A single mean value was obtained for each image, 1–2 images were acquired for each embryo, and the mean±SEM between images is shown.
To analyze cell intercalation in time-lapse movies, at least 50 edges that were vertically oriented (oriented at 75–90° relative to the AP axis) at the beginning of germband extension in stage 7 (t=0) were tracked for 30 min. The edge contraction rate was measured as the edge length at t=0 divided by the time required after t=0 to contract to a vertex. Only edges that successfully contracted to a 4-cell vertex were analyzed for the contraction rate measurements. This method underestimates the severity of the defects in mutant embryos, in which the most defective vertical edges never progressed to a vertex and were not included in the analysis. For edges that contracted to a 4-cell vertex or rosette, errors in vertex resolution were analyzed, including no resolution (no new edge formed after 10 min), unstable (the vertex initially resolved in the correct direction, but the edge contracted back to a vertex and remained a vertex at t=30 min), or misoriented (the vertex resolved incorrectly to form a vertical edge). To analyze tissue elongation in time-lapse movies, two cells that were located at least 30 cells apart along the AP axis and 2–3 cell diameters away from the ventral furrow at end of the fast phase of germband extension in late stage 8 were manually tracked back to the start of germband extension at the beginning of stage 7. Tissue elongation was calculated as the fold-change in distance between these cells along the AP axis. Tissue elongation values at t=30 min were compared for statistical analysis. For contraction rate, intercalation error, and tissue elongation measurements, a single value was obtained for each embryo and the mean±SEM between embryos is shown.
Supplementary Material
Movie S1. Related to Figure 2. Time-lapse video of convergent extension in a wild type embryo expressing E-cadherin–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S2. Related to Figure 2. Time-lapse video of convergent extension in a Src KD embryo expressing E-cadherin–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S3. Related to Figure 5. Time-lapse video of convergent extension in a wild-type embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S4. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-cat KD embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S5. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-reg DN embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S6. Related to Figure 5. Time-lapse video of convergent extension in a Toll-3 dsRNA-injected embryo expressing Resille–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S7. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-cat dsRNA-injected embryo expressing Resille–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-β-actin | Abcam | ab8224 |
| mouse anti-β-catenin | Developmental Studies Hybridoma Bank (DSHB) (Riggleman et al., 1990) | N2 7A1 |
| mouse anti-β-galactosidase | DSHB (Ghattas et al.,1991) | 40–1a |
| mouse anti-GFP | Roche | 11814460001 |
| mouse Living Colors mCherry | Takara | 632543 |
| mouse anti-Myc | DSHB (Evan et al., 1985) | 9E10 |
| mouse anti-Neurotactin | DSHB (Hortsch et al.,1990) | BP106 |
| mouse anti-phosphotyrosine | Millipore Sigma | 4G10 |
| mouse anti-V5 | Invitrogen | R96025 |
| mouse anti-Wingless | DSHB (Brook and Cohen, 1996) | 4D4 |
| rabbit anti-GFP | Torrey Pines | NC9589665 |
| rabbit anti-HA | Cell Signaling | C29F4 |
| rabbit phospho-Drosophila Akt (Ser505) | Cell Signaling | 4054S |
| rabbit phospho-Myosin Light Chain 2 (Ser19) | Cell Signaling | 3671S |
| rabbit anti-phospho-Src42-Y400 | Shindo et al., 2008 | N/A |
| rabbit-anti-Src42 | Takahashi et al., 2005 | N/A |
| guinea pig anti-Par-3 (Baz) | Blankenship et al., 2006 | N/A |
| guinea pig anti-Runt | Kosman et al., 1998 | N/A |
| guinea pig anti-Src64 (unpurified) | This paper | N/A |
| guinea pig anti-phospho-Toll-2 (pY1191) | This paper | N/A |
| guinea pig anti-phospho-Toll-2 (pY1218/1219) | This paper | N/A |
| rat anti-HA | Roche | 3F10 |
| chicken anti-GFP | Abcam | ab13970 |
| AlexaFluor-546 phalloidin | Invitrogen | A22283 |
| AlexaFluor-647 phalloidin | Invitrogen | A22287 |
| AlexaFluor-488 goat anti-mouse secondary | Invitrogen | A11001 |
| AlexaFluor-488 goat anti-rabbit secondary | Invitrogen | A11008 |
| AlexaFluor-488 goat anti-guinea pig secondary | Invitrogen | A11073 |
| AlexaFluor-546 goat anti-mouse secondary | Invitrogen | A11030 |
| AlexaFluor-546 goat anti-rabbit secondary | Invitrogen | A11035 |
| AlexaFluor-647 goat anti-guinea pig secondary | Invitrogen | A21450 |
| AlexaFluor-488 goat anti-chicken IgY | Abcam | 150173 |
| GFP-Trap-MA | Chromotek | gtma-20 |
| Protein G Sepharose 4B | GE Healthcare | 17061801 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bosutinib (SKI-606) | Selleck Chemicals | S1014 |
| Wortmannin | Cell Signaling | 9951S |
| Critical Commercial Assays | ||
| Taqman qRTPCR probe PI3K-cat | Applied Biosystems | Dm02142679_g1 |
| Taqman qRTPCR probe PI3K-reg | Applied Biosystems | Dm01842882_m1 |
| Taqman qRTPCR probe RpL32 | Applied Biosystems | Dm02151827_s1 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| D. melanogaster S2R+ cells | Drosophila Genomics Resource Center | Stock number 150 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster. Wild-type Oregon R strain | Bloomington Drosophila Stock Center | N/A |
| D. melanogaster. Toll-2 attP | Paré et al., 2019 | N/A |
| D. melanogaster. Toll-8 59 | Yagi et al., 2010 | N/A |
| D. melanogaster. Toll-8 145 | Kolesnikov and Beckendorf, 2007 | N/A |
| D. melanogaster. Toll-859, Toll-65A | Paré et al., 2014 | N/A |
| D. melanogaster. Toll-8 59 , Toll-6 5A , spider–GFP | Paré et al., 2014 | N/A |
| D. melanogaster. Toll-2 WT –HA | This paper | N/A |
| D. melanogaster. Toll-2 C2YF –HA | This paper | N/A |
| D. melanogaster. Toll-2 C1,2YF –HA | This paper | N/A |
| D. melanogaster. Toll-2 WT –V5 | This paper | N/A |
| D. melanogaster. Toll-2 C1YF –V5 | This paper | N/A |
| D. melanogaster. Toll-2 C1,2YF –V5 | This paper | N/A |
| D. melanogaster. Toll-2 WT | This paper | N/A |
| D. melanogaster. Toll-2 C2YF | This paper | N/A |
| D. melanogaster. Toll-2 C1,2YF | This paper | N/A |
| D. melanogaster. pUASp-Src42K276R–Venus (attP2) (Src DN) | This paper | N/A |
| D. melanogaster. pCFD3-Src42-gRNA (attP2) | This paper | N/A |
| D. melanogaster. pUASt-Src42Y511F (Src42 CA) | Tateno et al., 2000 | N/A |
| D. melanogaster. P{TRiP.GL00297}attP2 (Src64 shRNA) | Ni et al., 2011 | N/A |
| D. melanogaster. Src64–YFP (CPTI000567) | Lye et al., 2014 | N/A |
| D. melanogaster. P{TRiP.HMC05152}attP40 (PI3K-cat shRNA) | Ni et al., 2011 | N/A |
| D. melanogaster. pUASp-PI3K-reg–Venus (VK37) | This paper | N/A |
| D. melanogaster. pUASp-PI3K-reg–HA (VK37) | This paper | N/A |
| D. melanogaster. pUASp-PI3K-reg-ΔNSH2–HA (VK37) | This paper | N/A |
| D. melanogaster. pUASp-PI3K-reg-ΔCSH2–HA (VK37) | This paper | N/A |
| D. melanogaster. pUASp-PI3K-reg-ΔSH2–HA (VK37) | This paper | N/A |
| D. melanogaster. pUASp-PI3K-reg-ΔCatBD–HA (VK37) (PI3K-reg DN) | This paper | N/A |
| D. melanogaster. ubi-E-cadherin–GFP (II) | Oda and Tsukita, 2001 | N/A |
| D. melanogaster. eve–YFP BAC (attP2) | Ludwig et al., 2011 | N/A |
| D. melanogaster. myosin–GFP (III) | Royou et al., 2004 | N/A |
| D. melanogaster. myosin–mCherry (II) | Martin et al., 2009 | N/A |
| D. melanogaster. myosin–mCherry (III) | Martin et al., 2009 | N/A |
| D. melanogaster. resille–GFP (III) | gift of Alain Debec (Institut Jacques Monod) | N/A |
| D. melanogaster. spider-GFP (III) | gift of Alain Debec | N/A |
| D. melanogaster. sqh-gap43–mCherry (III) | Martin et al., 2010 | N/A |
| D. melanogaster. nos-Cas9 (X) | Port et al., 2014 | N/A |
| D. melanogaster. matαtub67 Gal4 | gift of Daniel St Johnston (University of Cambridge) | N/A |
| Oligonucleotides | ||
| See Table S2 | N/A | N/A |
| Recombinant DNA | ||
| Plasmid. pUASp-W-attB | gift of Michael Buszczak (UTSW Medical Center) | N/A |
| Plasmid. pUASp-W-msVenus | This paper | N/A |
| Plasmid. pUASp-W-V5 | This paper | N/A |
| Plasmid. pUASp-W-HA | This paper | N/A |
| Plasmid. LD42724 cDNA (PI3K-reg) | Berkeley Drosophila Genome Project (BDGP) Gold Collection | Drosophila Genomics Resource Center |
| Plasmid. pUASp-PI3K-reg–Venus | This paper | N/A |
| Plasmid. pUASp-PI3K-reg–HA | This paper | N/A |
| Plasmid. pUASp-PI3K-reg-ΔNSH2–HA | This paper | N/A |
| Plasmid. pUASp-PI3K-reg-ΔCSH2–HA | This paper | N/A |
| Plasmid. pUASp-PI3K-reg-ΔSH2–HA | This paper | N/A |
| Plasmid. pUASp-PI3K-reg-ΔCatBD–HA (PI3K-reg DN) | This paper | N/A |
| Plasmid. pCS2+ | Turner and Weintraub, 1994 | N/A |
| Plasmid. pCS2-PI3K-reg–HA | This paper | N/A |
| Plasmid. pUASp-Toll-2–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-2C1YF–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-2C2YF–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-2C1,2YF–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-2TIR-YF–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-2–HA | Paré et al., 2014 | N/A |
| Plasmid. pUASp-Toll-2-Δcyto–HA | This paper | N/A |
| Plasmid. pUASp-Toll-2-ΔTIR–HA | This paper | N/A |
| Plasmid. pUASp-Toll-2-ΔCTD–HA | This paper | N/A |
| Plasmid. pUASp-Toll-2-Y1056F–HA | This paper | N/A |
| Plasmid. pUASp-Toll-2-Y1191F–HA | This paper | N/A |
| Plasmid. pUASp-Toll-6–V5 | This paper | N/A |
| Plasmid. pUASp-Toll-8–V5 | This paper | N/A |
| Plasmid. pCFD3 | Addgene | 49410 |
| Plasmid. pCFD3-Src42-gRNA | This paper | N/A |
| Plasmid. pENTR/D-TOPO | Invitrogen | K240020 |
| Plasmid. pPWM | gift of Terence Murphy (Carnegie Institute) | N/A |
| Plasmid. pEntr-Src42–Myc | This paper | N/A |
| Plasmid. pUASp-Src42–Myc | This paper | N/A |
| Plasmid. pEntr-Src64–Myc | This paper | N/A |
| Plasmid. pUASp-Src64–Myc | This paper | N/A |
| Plasmid. pEntr-Src42-DN–Venus (Src42K276R) | This paper | N/A |
| Plasmid. pUASp-Src42-DN–Venus (Src42K276R) | This paper | N/A |
| Plasmid. p3xP3-EGFP.vas-int.NLS (vasa-PhiC31) | Addgene (Zhang et al., 2014) | 60948 |
| Plasmid. pBS-attB | Addgene | 61255 |
| Plasmid. pattB-Toll-2WT–HA | This paper | N/A |
| Plasmid. pattB-Toll-2C2YF–HA | This paper | N/A |
| Plasmid. pattB-Toll-2C1,2YF–HA | This paper | N/A |
| Plasmid. pattB-Toll-2WT–V5 | This paper | N/A |
| Plasmid. pattB-Toll-2C1YF–V5 | This paper | N/A |
| Plasmid. pattB-Toll-2C1,2YF–V5 | This paper | N/A |
| Plasmid. pattB-Toll-2WT | This paper | N/A |
| Plasmid. pattB-Toll-2C2YF | This paper | N/A |
| Plasmid. pattB-Toll-2C1,2YF | This paper | N/A |
| Software and Algorithms | ||
| SIESTA | Fernandez-Gonzalez and Zallen, 2011 | N/A |
| ImageJ and Fiji | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Prism 7 | Graphpad | N/A |
| Matlab | Mathworks | N/A |
| Other | ||
Highlights.
Toll receptors generate planar polarized patterns of Src and PI3K activity
Src and PI3K are required for localized myosin contractility and convergent extension
Src kinases promote Toll-2/PI3K-reg complex formation through Toll-2 phosphorylation
Toll-2 phosphorylation is necessary for planar polarity and convergent extension
Acknowledgments
We thank Zachary Mirman for generating the Toll-2WT line and Adam Paré and Zachary Mirman for initial observations about PI3K, Wilaysha Evans for help with genetic crosses, Matthew Laurie for the UASp-W-msVenus vector and Src42K276R–Venus transgene, Ben Glick for the monomeric superfolder Venus, Michael Buszczak for pUASp-W-attB, Shigeo Hayashi, Kaoru Saigo, and Tetsuya Kojima for antibodies, Frances Weis-Garcia for advice on antibody generation, and Eric Brooks and Marissa Gredler for comments on the manuscript. Stocks generated by the Transgenic RNAi Project at Harvard Medical School and obtained from the Bloomington Drosophila Stock Center (NIH P4OODO18537) and the KYOTO Stock Center at the Kyoto Institute of Technology were used in this study. This work was funded by NIH R01 grant GM079340 to JAZ. JS is supported by NIH/NCI F30 grant CA236441 and is a student of the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD program, which is supported by NIH/NIGMS T32 grant GM007739. JAZ is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abram CL, and Lowell CA (2007). The expanding role for ITAM-based signaling pathways in immune cells. Sci STKE 2007, re2. [DOI] [PubMed] [Google Scholar]
- Abram CL and Lowell CA (2008). The diverse functions of Src family kinases in macrophages. Front Biosci. 13, 4426–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KV (2000). Toll signaling pathways in the innate immune response. Curr. Opin. Immunol 12, 13–19. [DOI] [PubMed] [Google Scholar]
- Andreeva A, Lee J, Lohia M, Wu X, Macara IG, and Lu X (2014). PTK7-Src signaling at epithelial cell contacts mediates spatial organization of actomyosin and planar cell polarity. Dev. Cell 29, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthoney N, Foldi I, and Hidalgo A (2018). Toll and Toll-like receptor signalling in development. Development 145, dev156018. [DOI] [PubMed] [Google Scholar]
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, and Knaus UG (2000). Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol 1, 533–540. [DOI] [PubMed] [Google Scholar]
- Ballard SL, Miller DL, and Ganetzky B (2014). Retrograde neurotrophin signaling through Tollo regulates synaptic growth in Drosophila. J. Cell Biol 204, 1157–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardet PL, Guirao B, Paoletti C, Serman F, Leopold V, Bosveld F, Goya Y, Mirouse V, Graner F, and Bellaiche Y (2013). PTEN controls junction lengthening and stability during cell rearrangement in epithelial tissue. Dev. Cell 25, 534–546. [DOI] [PubMed] [Google Scholar]
- Bauer B, and Steinle A (2017). HemITAM: A single tyrosine motif that packs a punch. Sci. Signal 10, eaan3676. [DOI] [PubMed] [Google Scholar]
- Benton MA, Pechmann M, Frey N, Stappert D, Conrads KH, Chen YT, Stamataki E, Pavlopoulos A, and Roth S (2016). Toll Genes Have an Ancestral Role in Axis Elongation. Curr. Biol 26, 1609–1615. [DOI] [PubMed] [Google Scholar]
- Bertet C, Sulak L, and Lecuit T (2004). Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature 429, 667–671. [DOI] [PubMed] [Google Scholar]
- Blankenship J, Backovic S, Sanny J, Weitz O, and Zallen J (2006). Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev. Cell 11, 459–470. [DOI] [PubMed] [Google Scholar]
- Brook WJ, and Cohen SM (1996). Antagonistic interactions between wingless and decapentaplegic responsible for dorsal-ventral pattern in the Drosophila leg. Science 273, 1373–1377. [DOI] [PubMed] [Google Scholar]
- Cain RJ, and Ridley AJ (2009). Phosphoinositide 3-kinases in cell migration. Biol. Cell 101, 13–29. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, et al. (2007). Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci 27, 13033–13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capilla A, Karachentsev D, Patterson RA, Hermann A, Juarez MT, and McGinnis W (2017). Toll pathway is required for wound-induced expression of barrier repair genes in the Drosophila epidermis. Proc. Natl. Acad. Sci. U S A 114, E2682–E2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho L, Jacinto A, and Matova N (2014). The Toll/NF-kappaB signaling pathway is required for epidermal wound repair in Drosophila. Proc. Natl. Acad. Sc. U S A 111, E5373–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier FJM, Hardy EJL, and Laprise P (2011). Crumbs controls epithelial integrity by inhibiting Rac1 and PI3K. J. Cell Sci 124, 3393–3398. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S, and Sen GC (2014). Tyrosine phosphorylation in Toll-like receptor signaling. Cytokine Growth Factor Rev. 25, 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Crest J, Streichan SJ, and Bilder D (2019). Extracellular matrix stiffness cues junctional remodeling for 3D tissue elongation. Nat. Commun 10, 3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TB, and Perrimon N (1996). The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics 144, 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JL, Boerth NJ, Lee JR, and Koretzky GA (1999). Integration of T cell receptor-dependent signaling pathways by adapter proteins. Annu. Rev. Immunol 17, 89–108. [DOI] [PubMed] [Google Scholar]
- Cooper JA, and Qian H (2008). A mechanism for SRC kinase-dependent signaling by noncatalytic receptors. Biochemistry 47, 5681–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, and Waterfield MD (1994). PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J 13, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon E, Kooyer S, D’Evelyn D, Duman M, Lawinger P, Botas J, and Bellen H (1994). The Drosophila 18 wheeler is required for morphogenesis and has striking similarities to Toll. Development 120, 885–899. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, and Bishop JM (1985). Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell Biol 5, 3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Simoes S, Roper JC, Eaton S, and Zallen JA (2009). Myosin II dynamics are regulated by tension in intercalating cells. Dev. Cell 17, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, and Zallen JA (2011). Oscillatory behaviors and hierarchical assembly of contractile structures in intercalating cells. Phys Biol 8, 045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, and Kagan JC (2020). Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldi I, Anthoney N, Harrison N, Gangloff M, Verstak B, Nallasivan MP, AlAhmed S, Zhu B, Phizacklea M, Losada-Perez M, et al. (2017). Three-tier regulation of cell number plasticity by neurotrophins and Tolls in Drosophila. J. Cell Biol 216, 1421–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster D, and Luschnig S (2012). Src42A-dependent polarized cell shape changes mediate epithelial tube elongation in Drosophila. Nat. Cell Biol 14, 526–534. [DOI] [PubMed] [Google Scholar]
- Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, and Koyasu S (2002). PI3K-mediated negative feedback regulation of IL-12 production in DC.s Nat. Immunol. 3, 875–881. [DOI] [PubMed] [Google Scholar]
- Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, and Mostov K (2006). Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat. Cell Biol 8, 963–970. [DOI] [PubMed] [Google Scholar]
- Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, and Turka LA (2006). The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 25, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghattas IR, Sanes JR, and Majors JE (1991). The encephalomyocarditis virus internal ribosome entry site allows efficient coexpression of two genes from a recombinant provirus in cultured cells and in embryos. Mol. Cell Biol 11, 5848–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, and Sheetz MP (2006). Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol. 16, 213–223. [DOI] [PubMed] [Google Scholar]
- Glantz S (2002). Primer of Biostatistics (McGraw-Hill; ). [Google Scholar]
- Gomez GA, McLachlan RW, Wu SK, Caldwell BJ, Moussa E, Verma S, Bastiani M, Priya R, Parton RG, Gaus K, Sap J, and Yap AS (2015). An RPTPalpha/Src family kinase/Rap1 signaling module recruits myosin IIB to support contractile tension at apical E-cadherin junctions. Mol. Biol. Cell 26, 1249–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJH, Ridley AJ, Rurhberg C, Gerhardt H, and Vanhaesebroeck B (2008). Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453, 662–666. [DOI] [PubMed] [Google Scholar]
- Grego-Bessa J, Bloomekatz J, Castel P, Omelchenko T, Baselga J, and Anderson KV (2015). The tumor suppressor PTEN and the PDK1 kinase regulate formation of the columnar neural epithelium. eLife 5, e12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, and Lanier LL (2006). Inhibition of immune responses by ITAM-bearing receptors. Sci STKE 2006, re1. [DOI] [PubMed] [Google Scholar]
- Hortsch M, Patel NH, Bieber AJ, Traquina ZR, and Goodman CS (1990). Drosophila neurotactin, a surface glycoprotein with homology to serine esterases, is dynamically expressed during embryogenesis. (1990). Development 110, 1327–1340. [DOI] [PubMed] [Google Scholar]
- Huebener P, and Schwabe RF (2013). Regulation of wound healing and organ fibrosis by toll-like receptors. Biochimica et Biophysica Acta 1832, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, and Wallingford JB (2018). Coming to consensus: A unifying model emerges for convergent extension. Dev. Cell 46, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MV, Willoughby PM, Bruce AEE, and Fernandez-Gonzalez R (2018). Oxidative stress orchestrates cell polarity to promote embryonic wound healing. Dev. Cell 47, 377–387 e4. [DOI] [PubMed] [Google Scholar]
- Huveneers S, Danen EH (2009). Adhesion signaling-crosstalk between integrins, Src, and Rho. J. Cell Sci 122, 1059–1069. [DOI] [PubMed] [Google Scholar]
- Irvine K, and Wieschaus E (1994). Cell intercalation during Drosophila germband extension and its regulation by pair-rule segmentation genes. Development 120, 827–841. [DOI] [PubMed] [Google Scholar]
- Jagut M, Mihaila-Bodart L, Molla-Herman A, Alin MF, Lepesant JA, and Huynh JR (2013). A mosaic genetic screen for genes involved in the early steps of Drosophila oogenesis. G3 (Bethesda) 3, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza KE, Farrell DL, and Zallen JA (2014). Spatiotemporal control of epithelial remodeling by regulated myosin phosphorylation. Proc. Natl. Acad. Sci. U S A 111, 11732–11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, and Waterfield MD (2001). Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol 17, 615–675. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. [DOI] [PubMed] [Google Scholar]
- Kim S, Chung S, Yoon J, Choi KW, and Yim J (2006). Ectopic expression of Tollo/Toll-8 antagonizes Dpp signaling and induces cell sorting in the Drosophila wing. Genesis 44, 541–549. [DOI] [PubMed] [Google Scholar]
- Kleve CD, Siler DA, Syed SK, and Eldon ED (2006). Expression of 18-wheeler in the follicle cell epithelium affects cell migration and egg morphology in Drosophila. Dev. Dyn 235, 1953–1961. [DOI] [PubMed] [Google Scholar]
- Kolesnikov T, and Beckendorf SK (2007). 18 wheeler regulates apical constriction of salivary gland cells via the Rho-GTPase-signaling pathway. Dev. Biol 307, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman D, Small S, and Reinitz J (1998). Rapid preparation of a panel of polyclonal antibodies to Drosophila segmentation proteins. Dev. Genes Evol 208, 290–294. [DOI] [PubMed] [Google Scholar]
- Kovacs E, Zorn JA, Huang Y, Barros T, and Kuriyan J (2015). A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem 84, 739–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Tien SC, Jou TS, Chang YC, Jhong HG, and Chang ZF (2010). Src-dependent phosphorylation of ROCK participates in regulation of focal adhesion dynamics. J. Cell Sci 123, 3368–3377. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, and Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulier F, and Lemaitre B (2008). Toll-like receptors--taking an evolutionary approach. Nat. Rev. Genet 9, 165–178. [DOI] [PubMed] [Google Scholar]
- Levayer R, Pelissier-Monier A, and Lecuit T (2011). Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat. Cell Biol 13, 529–540. [DOI] [PubMed] [Google Scholar]
- Ludwig MZ, Manu Kittler R., White KP, and Kreitman M (2011). Consequences of eukaryotic enhancer architecture for gene expression dynamics, development, and fitness. PLoS Genet. 7, e1002364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Lucas RM, Liu L, and Stow JL (2019). Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell Sci 133, jcs239194. [DOI] [PubMed] [Google Scholar]
- Lye CM, Naylor HW, and Sanson B (2014). Subcellular localisations of the CPTI collection of YFP-tagged proteins in Drosophila embryos. Development 141, 4006–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Amonlirdviman K, Raffard RL, Abate A, Tomlin CJ, and Axelrod JD (2008). Cell packing influences planar cell polarity signaling. Proc. Natl. Acad. Sci. U S A 105, 18800–18805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, and Vartanian T (2006). Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J. Cell Biol 175, 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Gelbart M, Fernandez-Gonzalez R, Kaschube M, and Wieschaus E (2010). Integration of contractile forces during tissue invagination. J. Cell Biol 188, 735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Kaschube M, and Wieschaus E (2009). Pulsed contractions of an actin-myosin network drive apical constriction. Nature 457, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, and Mostov K (2007). PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy G, Foldi I, Aurikko J, Wentzell JS, Lim MA, Fenton JC, Gay NJ, and Hidalgo A (2013). Toll-6 and Toll-7 function as neurotrophin receptors in the Drosophila melanogaster CNS. Nat. Neurosci 16, 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, Kraemer A, Helwani FM, Kovacs EM, and Yap AS (2007). E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol. Biol. Cell 18, 3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, and Yap AS (2007). Not so simple: the complexity of phosphotyrosine signaling and cadherin adhesive contacts. J. Mol. Med 85, 545–554. [DOI] [PubMed] [Google Scholar]
- Meyer SN, Amoyel M, Bergantinos C, de la Cova C, Schertel C, Basler K, and Johnston LA (2014). An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 346, 1258236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisato D, and Anderson KV (1995). Signaling pathways that establish the dorsal-ventral pattern of the Drosophila embryo. Annu. Rev. Genet 29, 371–399. [DOI] [PubMed] [Google Scholar]
- Munjal A, Philippe JM, Munro E, and Lecuit T (2015). A self-organized biomechanical network drives shape changes during tissue morphogenesis. Nature 524, 351–355. [DOI] [PubMed] [Google Scholar]
- Nelson KS, Khan Z, Molnar I, Mihaly J, Kaschube M, and Beitel GJ (2012). Drosophila Src regulates anisotropic apical surface growth to control epithelial tube size. Nat. Cell Biol 14, 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D, Shim HS, Tao R, Handler D, Karpowicz P, et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, and Tsukita S (2001). Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J. Cell Sci 114, 493–501. [DOI] [PubMed] [Google Scholar]
- Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, and Hallman M (2003). Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur. J. Immunol 33, 597–605. [DOI] [PubMed] [Google Scholar]
- Olivares-Castineira I, and Llimargas M (2018). Anisotropic Crb accumulation, modulated by Src42A, is coupled to polarised epithelial tube growth in Drosophila. PLoS Genet. 14, e1007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page TH, Smolinska M, Gillespie J, Urbaniak AM, and Foxwell BMJ (2009). Curr. Molec. Med 9, 69–85. [DOI] [PubMed] [Google Scholar]
- Pang JH, Kraemer A, Stehbens SJ, Frame MC, and Yap AS (2005). Recruitment of phosphoinositide 3-kinase defines a positive contribution of tyrosine kinase signaling to E-cadherin function. J. Biol. Chem 280, 3043–3050. [DOI] [PubMed] [Google Scholar]
- Paré AC, Naik P, Shi J, Mirman Z, Palmquist KH, and Zallen JA (2019). An LRR Receptor-Teneurin System Directs Planar Polarity at Compartment Boundaries. Dev. Cell 51, 208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré AC, Vichas A, Fincher CT, Mirman Z, Farrell DL, Mainieri A, and Zallen JA (2014). A positional Toll receptor code directs convergent extension in Drosophila. Nature 515, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré AC, and Zallen JA (2020). Cellular, molecular, and biophysical control of epithelial cell intercalation. Curr. Topics in Dev. Biol 136, 167–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T (2004). Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203. [DOI] [PubMed] [Google Scholar]
- Pinal N, Goberdhan DC, Collinson L, Fujita Y, Cox IM, Wilson C, and Pichaud F (2006). Regulated and polarized PtdIns(3,4,5)P3 accumulation is essential for apical membrane morphogenesis in photoreceptor epithelial cells. Curr. Biol 16, 140–149. [DOI] [PubMed] [Google Scholar]
- Pinner S, and Sahai E (2008). PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10, 127–137. [DOI] [PubMed] [Google Scholar]
- Port F, Chen HM, Lee T, and Bullock SL (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. U S A 111, E2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauzi M, Verant P, Lecuit T, and Lenne PF (2008). Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nat. Cell Biol 10, 1401–1410. [DOI] [PubMed] [Google Scholar]
- Rhee SH, Kim H, Moyer MP, and Pothoulakis C (2006). Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J. Biol. Chem 281, 18560–18568. [DOI] [PubMed] [Google Scholar]
- Riggleman B, Schedl P, and Wieschaus E (1990). Spatial expression of the Drosophila segment polarity gene armadillo is posttranscriptionally regulated by wingless. Cell 63, 549–560. [DOI] [PubMed] [Google Scholar]
- Royou A, Field C, Sisson J, Sullivan W, and Karess R (2004). Reassessing the role and dynamics of nonmuscle myosin II during furrow formation in early Drosophila embryos. Mol. Biol. Cell 15, 838–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Sierra S, Deshmukh SD, Kalnitski J, Kuenzi P, Wymann MP, Golenbock DT, and Henneke P (2009). Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J. 28, 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, and Sen GC (2004). Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol 11, 1060–1067. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image anaysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan A, Eastburn DJ, and Mostov K (2011). Phosphoinositides in cell architecture. Cold Spring Harb. Perspect. Biol 3, a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo M, Wada H, Kaido M, Tateno M, Aigaki T, Tsuda L, and Hayashi S (2008). Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Development 135, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Simoes S, Blankenship JT, Weitz O, Farrell DL, Tamada M, Fernandez-Gonzalez R, and Zallen JA (2010). Rho-kinase directs Bazooka/Par-3 planar polarity during Drosophila axis elongation. Dev. Cell 19, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes S, Denholm B, Azevedo D, Sotillos S, Martin P, Skaer H, Hombria JC, and Jacinto A (2006). Compartmentalisation of Rho regulators directs cell invagination during tissue morphogenesis. Development 133, 4257–4267. [DOI] [PubMed] [Google Scholar]
- Simoes S, Mainieri A, and Zallen JA (2014). Rho GTPase and Shroom direct planar polarized actomyosin contractility during convergent extension. J. Cell Biol 204, 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopko R, Foos M, Vinayagam A, Zhai B, Binari R, Hu Y, Randklev S, Perkins LA, Gygi SP, and Perrimon N (2014). Combining genetic perturbations and proteomics to examine kinase-phosphatase networks in Drosophila embryos. Dev. Cell 31, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Amourda C, Shagirov M, Hara Y, Saunders TE, and Toyama Y (2017). Basolateral protrusion and apical contraction cooperatively drive Drosophila germ-band extension. Nat. Cell Biol 19, 375–383. [DOI] [PubMed] [Google Scholar]
- Schwartz D, Gygi SP (2005). An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnology 23, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Takahashi F, Ui-Tei K, Kojima T, and Saigo K (2005). Requirements of genetic interactions between Src42A, armadillo and shotgun, a gene encoding E-cadherin, for normal development in Drosophila. Development 132, 2547–2559. [DOI] [PubMed] [Google Scholar]
- Tamada M, Farrell DL, and Zallen JA (2012). Abl regulates planar polarized junctional dynamics through β-catenin tyrosine phosphorylation. Dev. Cell 22, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno M, Nishida Y, and Adachi-Yamada T (2000). Regulation of JNK by Src during Drosophila development. Science 287, 324–327. [DOI] [PubMed] [Google Scholar]
- Thomas JH, and Wieschaus E (2004). src64 and tec29 are required for microfilament contraction during Drosophila cellularization. Development 131, 863–871. [DOI] [PubMed] [Google Scholar]
- Thomas SM, and Brugge JS (1997). Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol 13, 513–609. [DOI] [PubMed] [Google Scholar]
- Troutman TD, Baza JF, and Pasare C (2012). Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 11, 3559–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, and Weintraub H (1994). Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev 8, 1434–1447. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Vogt PK, and Rommel C (2010). PI3K: from the bench to the clinic and back. Curr Top Microbiol Immunol 347, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Hong W, Favaloro V, and Luo L (2015). Toll receptors instruct axon and dendrite targeting and participate in synaptic partner matching in a Drosophila olfactory circuit. Neuron 85, 1013–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, and Pirola L (1998). Structure and function of phosphoinositide 3-kinases. Biochimica et Biophysica Acta 1436, 127–150. [DOI] [PubMed] [Google Scholar]
- Xue G, and Hemmings BA (2013). PKB/Akt-dependent regulation of cell motility. J Natl Cancer Inst 105, 393–404. [DOI] [PubMed] [Google Scholar]
- Yagi Y, Nishida Y, and Ip YT (2010). Functional analysis of Toll-related genes in Drosophila. Dev. Growth Differ 52, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HA, and Couchman JR (2005). The Rho kinases I and II regulate different aspects of myosin II activity. J. Cell Biol 170, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen J, and Wieschaus E (2004). Patterned gene expression directs bipolar planar polarity in Drosophila. Dev. Cell 6, 343–355. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koolhaas WH, and Schnorrer F (2014). A versatile two-step CRISPR- and RMCE-based strategy for efficient genome engineering in Drosophila. G3 (Bethesda) 4, 2409–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 2. Time-lapse video of convergent extension in a wild type embryo expressing E-cadherin–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S2. Related to Figure 2. Time-lapse video of convergent extension in a Src KD embryo expressing E-cadherin–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S3. Related to Figure 5. Time-lapse video of convergent extension in a wild-type embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S4. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-cat KD embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S5. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-reg DN embryo expressing Spider–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S6. Related to Figure 5. Time-lapse video of convergent extension in a Toll-3 dsRNA-injected embryo expressing Resille–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Movie S7. Related to Figure 5. Time-lapse video of convergent extension in a PI3K-cat dsRNA-injected embryo expressing Resille–GFP. Images acquired every 15 s. Anterior left, ventral down. Time in min:sec.
Data Availability Statement
No large-scale datasets or new code were generated in this study.