

Abstract
BACKGROUND AND AIMS:
Hepatitis B virus (HBV) precore (PC) and dual basal core promoter (BCP) mutations halt and down-regulate hepatitis B e antigen (HBeAg) production respectively. PC mutation is rarely associated with HBV genotype A. We sought to examine the association of these variants with HBV genotypes, age, and HBeAg status in a racially diverse population in North America. Prospective study included 1,036 (808 adults, 228 children) participants in the Hepatitis B Research Network. PC and BCP variants were determined by Sanger sequencing, and dominant HBV species (>50%) were reported.
APPROACH AND RESULTS:
Median age was 36.3 years (range, 2–80), 44.6% HBeAg(+), 74.2% Asians, 13.3% black, and 9.7% white. The dominant PC variant was present in 29.4% participants, including 20 with subgenotype A1 or A2. Seventeen of 20 participants with genotype A and PC had a compensatory C1858T mutation. In the HBeAg(+) cohort, the prevalence of PC and/or BCP variants increased from 14.4% in the first two decades to 51% after 40 years of age. Among those aged 2–18, 52% and 83% with dominant PC and BCP variants were HBeAg(+) compared to 3.8% and 29% in the >40 years age group. HBeAg clearance rates were significantly higher for those with dominant PC or BCP variants: 24.4 and 15.0 per 100 person-years compared to 6.0 in wild-type HBV (P < 0.0001).
CONCLUSIONS:
PC variants can be present in HBV genotype A and are usually associated with C1858T, which preserves the pregenome encapsidation sequence. Selection of PC and BCP variants occurred at a young age, with increasing prevalence across age groups. HBeAg(+) participants with dominant PC and BCP variants progressed to the HBeAg(−) phase of chronic HBV infection significantly faster. This finding has potential clinical and therapeutic implications.
Hepatitis B virus (HBV) infection is a significant public health problem worldwide and a major cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma (HCC). According to the World Health Organization (WHO) Global Hepatitis Report in 2017, an estimated 257 million persons worldwide have chronic HBV infection, with the highest prevalence in Asia and Africa.
At least nine genotypes of HBV, designated A-I, have been described that vary by >8% in nucleotide sequence and differ greatly in geographical distribution.(1,2) Genotype A is the predominant cause of hepatitis B in Northern Europe and was formerly the most frequent genotype found in the United States. Genotype D is most commonly found in Mediterranean areas of Europe, Northern Africa, India, and the Middle East. Genotypes B and C are found most frequently in East Asia, with genotype C being predominant in Northern and genotype B in Southern Asia. Genotype E is found predominantly in Africa, where genotype A is also prevalent, but designated A1 because it differs slightly (4%) but distinctly from the European strains, which are designated A2.(3,4) In the United States, HBV genotype usually matches the country of origin (B and C among Asian Americans; E among blacks).
HBV genotypes also vary in clinical and virological features, risk of HCC, and response to therapy.(5–9) A prominent difference among genotypes is its association with a variant in the precore (PC) region where a G (Guanine) is replaced by an A (Adenine) at nucleotide (nt) position 1896 (codon 28), the “classical” PC stop codon variant: G1896A. This mutation abrogates the production of hepatitis B e antigen (HBeAg).(10) The PC G1896A variant is most frequently associated with genotypes B and D followed by genotype C. HBV genotype A is believed to be incompatible with the PC G1896A variant because it harbors 1858C (Cytosine), which is paired to nt1896 in the stem-loop structure of epsilon (ε) at the pregenome encapsidation sequence. A change from G to A at nt1896 would cause instability of the stem-loop structure.(11) In contrast, genotype non-A usually harbors 1858T. The G1896A mutation, in this case, actually strengthens the stability of the HBV pregenome encapsidation sequence.
The most common mutations in the basal core promoter (BCP) region (A1762T [Thymine], G1764A) can down-regulate, but not abolish, HBeAg production. Contrary to the PC G1896A variant, the BCP variant can be found across all genotypes.(12)
We determined the prevalence of the PC G1896A and BCP variants across the HBV genotypes among participants enrolled in the Hepatitis B Research Network (HBRN) Cohort Study and examined the association of these variants with virological parameters and age in this large, racially diverse cohort of children and adults with chronic HBV infection living in North America.
Patients and Methods
The HBRN is a cooperative network of 21 adult and seven pediatric clinical centers in the United States and in Toronto, Ontario, Canada, a data coordinating center, a virology testing laboratory, an immunology core, and a serum and tissue repository.(13) The HBRN Cohort Study enrolled persons with hepatitis B surface antigen (HBsAg) in serum who were without evidence of hepatic decompensation, HCC, organ transplant, human immunodeficiency virus (HIV) infection, and were not receiving antiviral therapy (AVT) at enrollment, as detailed in our publications.(13,14) All protocols were approved by the HBRN Steering Committee, the institutional review board, or research ethics board at each participating site and a central data safety and monitoring board selected by the National Institute of Diabetes and Digestive and Kidney Diseases, which was responsible for funding the Network. All participants provided written informed consent/assent before participation.
As of July 2018, a total of 2,112 (1,795 adults, 317 children) participants with chronic hepatitis B enrolled in the study had BCP and PC testing. Participants were excluded from this analysis if they had acute hepatitis B, or were coinfected with HIV, hepatitis C virus, or hepatitis D virus. We also excluded participants who were targeted for other HBRN studies (e.g., immunology and pregnancy projects) in order to keep “randomness” of the sample analyzed so that the prevalence of BCP/PC and other variables represent an unbiased estimate of those in the HBRN study. A total of 322 participants met the exclusion criteria. The resulting 1,790 participants were potentially eligible for this study. The final cohort was further reduced to 1,036 (808 adults, 228 children) for the following reasons: 699 in whom BCP/PC sequence could not be determined because of technical problems including sample condition and/or low HBV DNA as well as assay sensitivity, and 55 who received AVT ≤48 weeks before the BCP/PC sample collection (Supporting Fig. S1). Participant information at the initial evaluation, including sex, age at the time blood sample was collected for BCP/PC testing, race, continent of birth, and presumed mode of HBV transmission, was recorded.
Laboratory Tests
ROUTINE LABORATORY TESTING
Complete blood counts and chemistry panels, including serum aminotransferase levels, were tested at local sites and the results recorded in a central database maintained by the HBRN Data Coordinating Center. Additional serum samples were shipped to a central repository and laboratories for virological testing. HBV-DNA levels were determined using a real-time PCR assay (COBAS Ampliprep/COBAS TaqMan HBV Test, v2.0; Roche Molecular Diagnostics, Branchburg, NJ) with a lower limit of detection of 20 IU/mL. HBeAg status was determined by a quantitative HBeAg (qHBeAg) test (Roche Diagnostics). If the result was below the limit of quantification, qualitative enzyme immunoassays for HBeAg and hepatitis B e antibody (anti-HBe) at local sites were used to determine the status of HBeAg and anti-HBe. HBeAg(+) participants had repeat HBeAg and anti-HBe testing every 6 months to determine the time of HBeAg clearance.
QUANTITATIVE HBsAg AND qHBeAg
Both tests were made available by Roche Diagnostics for research purposes using their Elecsys platform and performed at the HBRN central virology laboratory at the University of Washington (Seattle, WA). The quantitative HBsAg (qHBsAg) assay had a measuring range of 0.05–130,000 IU/mL. Results for qHBsAg are presented as log10 IU/mL. qHBeAg testing was modified as described.(15,16) Initial testing was done by utilizing the Paul Ehrlich Institute (PEI) HBeAg standard material (PEI U/mL) and then converted to the International WHO HBeAg quantitative standard (1 PEI Unit × 1.1 = IU/mL). Utilizing standardized sample dilutions, the assay had a measuring range of 0.3–149,500 IU/mL. Samples with results <0.3 IU/mL are reported as “below the limit of detection” (BLD), and participants with a qHBeAg result BLD were considered to be HBeAg(−).
HBV GENOTYPING
Testing was performed by an automated mass spectrometry test using a 420-base-pair fragment of the S gene, including the a-determinant. Subtyping was done based on Sanger sequence of a larger fragment spanning nt311–1026 of the HBV genome, followed by phylogenetic clustering to reference sequences.(17) The sensitivity of the genotyping method is 50 genome equivalents per reaction.
BCP AND PC VARIANTS
The BCP fragment was amplified by first-round PCR with primers 1521sense GGGGCGC ACCTCTCTTTACG and 2324antisense CTGGA GGAGTGCGAATCCACAC at 55°C annealing temperature for 35 cycles, followed by 30 cycles of a nested PCR reaction with primers 1572sense GGACCGTGTGCACTTCGCTTCA and 2169anti-sense GGATCTTCC AAATTAITICC CAC CCA GG at 50°C annealing temperature. The sensitivity of this approach is estimated to be 500 genome equivalents per reaction. The presence of BCP mutations, A1762T and/or G1764A, and the PC mutations G1896A and C1858T, was determined by the majority call of the Sanger chromatograms of both strands of a fragment spanning nt1572–2187 of the HBV genome. BCP and PC results were presented using validated scoring system based on dominant viral species (>50%) of the Sanger chromatogram at nt1762, 1764, and 1896. Both HBV genotyping and BCP/PC variant testing were performed at the Molecular Epidemiology and Bioinformatics Laboratory in the Division of Viral Hepatitis at the Centers for Disease Control and Prevention.
STEM-LOOP STRUCTURE PREDICTION AND ENERGY CALCULATION
Only the stem-loop region of the core promoter sequences was selected for RNA structure calculations. Sequences that had large gaps in the stem-loop region and/or had more than one International Union of Pure and Applied Chemistry code assignment in any position of the stem-loop in the majority call sequence were removed from the analysis in order to avoid ambiguities in the structure predictions, leaving the number of available fragments at n = 605. Structure prediction and stem-loop energy (delta G) was calculated by the CLC Genomics Workbench v 11.0 Secondary RNA structure Tool. Stem-loops were rendered by the RNAfold webserver (The Vienna RNA web services).(18)
STATISTICAL ANALYSES
Demographic, clinical, and virological characteristics were summarized using frequencies and percentages for categorical variables. As appropriate, mean values with SD, or median values with 25th and 75th percentiles, were used to summarize continuous variables. Given that the distribution of clinical variables such as HBV DNA, qHBsAg, and qHBeAg levels were skewed, they were summarized using medians and 25th and 75th percentiles. We used Wilcoxon’s test to compare continuous characteristics and chi-square test to compare categorical characteristics between wild types (WTs) and variants. Fisher’s exact tests were used to compare the distribution of WTs and variants of interests across each age group and genotype, and to compare HBeAg positivity between WTs and variants of interests within each age group. Wilcoxon’s tests were used to compare quantitative HBeAg between WTs and variants of interests within each age group among the HBeAg-positive group. P values from these tests were adjusted for multiple comparisons using Holm’s step-down procedure. The statistical software package, SAS (version 9.4 (SAS Institute, Cary, NC), was used to perform statistical analyses.
Results
The demographic, clinical, and virological features of the 1,036 participants included in this study are shown in Table 1. The majority (74.2%) of the participants were Asians, followed by blacks (13.3%) and whites (9.7%). Roughly half (48.9%) of the cohort was male. In this cohort, 461 (44.6%) were HBeAg(+) and 228 (22.0%) were aged ≤18 years.
TABLE 1.
Demographic, Clinical, and Virological Features of the Participants
| Variable | Total n = 1,036 | WT n = 478 (46.3%) | PC G1896A only n = 190 (18.4%) | BCP only n = 250 (24.2%) | BCP and PC G1896A n = 114 (11.0%) |
|---|---|---|---|---|---|
| Sex | n = 1,036 | n = 478 | n = 190 | n = 250 | n = 114 |
| Male | 507 (48.9%) | 208 (41.1%) | 104 (20.6%) | 130 (25.7%) | 64 (12.6%) |
| Female | 529 (51.1%) | 270 (51.3%) | 86 (16.3%) | 120 (22.8%) | 50 (9.5%) |
| Race | n = 1,031 | n = 475 | n = 188 | n = 250 | n = 114 |
| White | 100 (9.7%) | 49 (49.0%) | 13 (13.0%) | 23 (23.0%) | 15 (15.0%) |
| Black | 137 (13.3%) | 64 (47.1%) | 24 (17.6%) | 34 (25.0%) | 14 (10.3%) |
| Asian | 765 (74.2%) | 353 (46.3%) | 143 (18.8%) | 185 (24.3%) | 81 (10.6%) |
| Other/mixed | 29 (2.8%) | 9 (31.0%) | 8 (27.6%) | 8 (27.6%) | 4 (13.8%) |
| Age (years) | n = 1,036 | n = 478 | n = 190 | n = 250 | n = 114 |
| 0–18 | 228 (22.0%) | 171 (75.0%) | 21 (9.2%) | 31 (13.6%) | 5 (2.2%) |
| >18–40 | 377 (36.4%) | 185 (49.1%) | 66 (17.5%) | 95 (25.2%) | 31 (8.2%) |
| >40 | 431 (41.6%) | 122 (28.6%) | 103 (24.1%) | 124 (29.0%) | 78 (18.3%) |
| HBeAg status | n = 1,033 | n = 476 | n = 190 | n = 249 | n = 114 |
| Negative | 572 (55.4%) | 164 (28.9%) | 165 (29.0%) | 134 (23.6%) | 105 (18.5%) |
| Positive | 461 (44.6%) | 312 (67.7%) | 25 (5.4%) | 115 (24.9%) | 9 (2.0%) |
| Genotype | n = 1,028 | n = 475 | n = 189 | n = 247 | n = 113 |
| A | 185 (18.0%) | 106 (57.6%) | 14 (7.6%) | 54 (29.3%) | 10 (5.4%) |
| B | 374 (36.4%) | 180 (48.3%) | 112 (30.0%) | 45 (12.1%) | 36 (9.7%) |
| C | 339 (33.0%) | 142 (41.9%) | 28 (8.3%) | 127 (37.5%) | 42 (12.4%) |
| D | 87 (8.5%) | 35 (41.2%) | 17 (20.0%) | 16 (18.8%) | 17 (20.0%) |
| E | 36 (3.5%) | 10 (27.8%) | 16 (44.4%) | 3 (8.3%) | 7 (19.4%) |
| Other (F, G, and multiple) | 7 (0.7%) | 2 (28.6%) | 2 (28.6%) | 2 (28.6%) | 1 (14.3%) |
| HBV DNA (log10 IU/mL) | n = 1,036 | n = 478 | n = 190 | n = 250 | n = 114 |
| <4 | 386 (37.3%) | 134 (35.0%) | 108 (28.2%) | 79 (20.6%) | 62 (16.2%) |
| 4-<6 | 207 (20.0%) | 42 (20.4%) | 55 (26.7%) | 71 (34.5%) | 38 (18.4%) |
| 6-<8 | 146 (14.1%) | 67 (45.9%) | 14 (9.6%) | 54 (37.0%) | 11 (7.5%) |
| >=8 | 297 (28.7%) | 235 (79.1%) | 13 (4.4%) | 46 (15.5%) | 3 (1.0%) |
Note: Bolded rows indicate the number of study subjects with available data.
Of the samples from these 1,036 participants, 478 (46.3%) had dominant WT sequences in both BCP and PC regions, 250 (24.2%) had dominant BCP variant only, 190 (18.4%) dominant PC variant only, and 114 (11.0%) dominant BCP and PC variants.
Among the HBeAg (+) participants, 67.7% had dominant WT HBV, 24.9% had dominant BCP only, 5.4% had dominant PC only, and 2% had dominant BCP + PC variants. In contrast, among HBeAg(−) participants, dominant WT, BCP only, PC only, and BCP + PC variants were present in 28.9%, 23.6%, 29%, and 18.5%, respectively (Fig. 1).
FIG. 1.
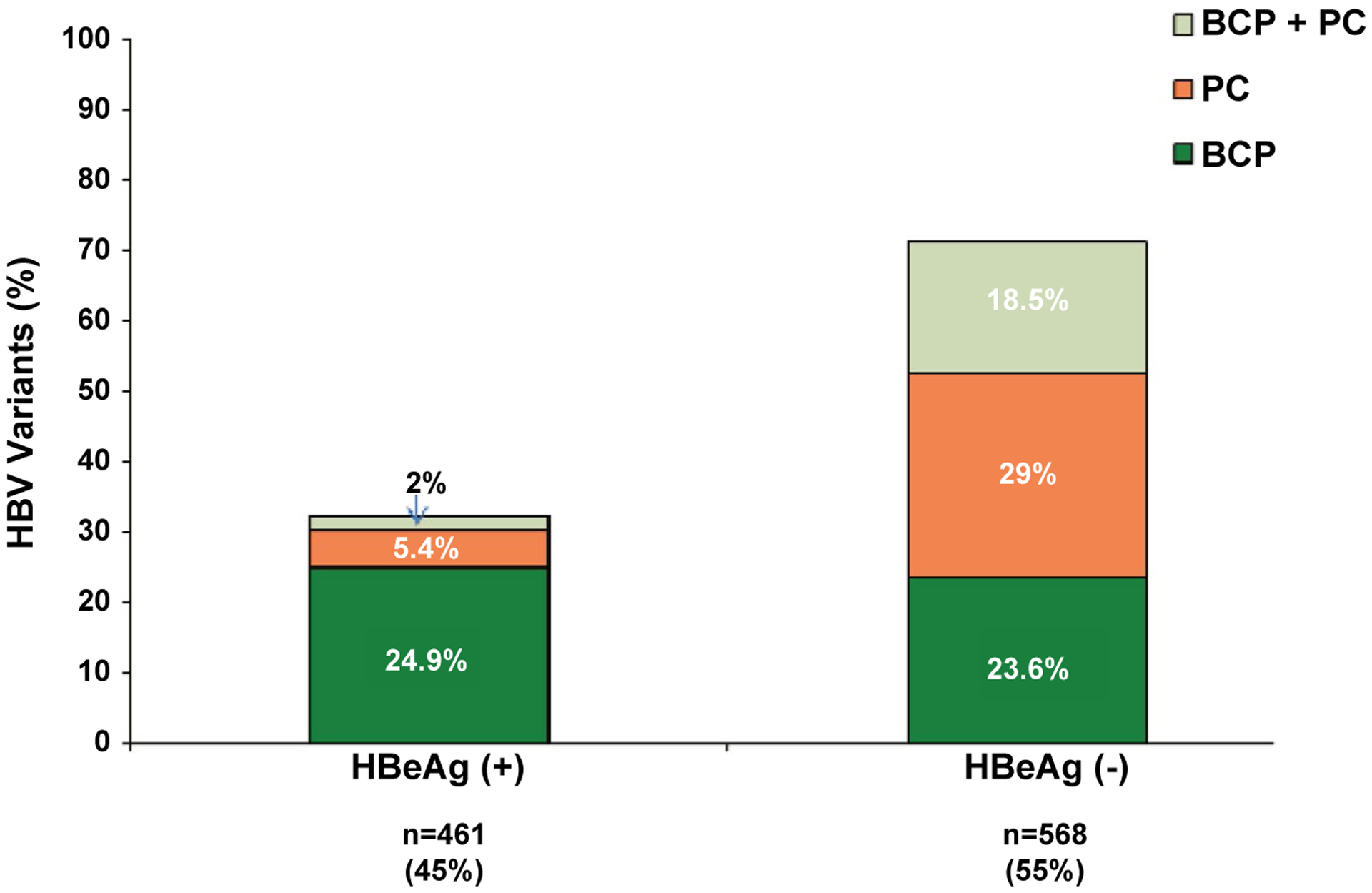
PC and BCP variants by HBeAg status.
Participants born in Asia (n = 680) were infected predominantly with genotypes B (47.5%) or C (40.1%). Genotype A was identified in 5.4% (A1 4.1%, A2 1.2%, and other A 0.1%) of Asians. The majority of African-born persons (n = 106) had genotype A (54.7%; A1 51.9% and A2 2.8%) followed by genotype E (31.1%). Participants born in North America (n = 205) had more diverse genotypes, the majority having genotype A (39%: A2 34.1% and A1 4.9%) followed by C (28.8%), B (22.4%), D (7.3%), and E (0.5%).
DISTRIBUTION OF PC AND BCP VARIANTS ACROSS THE HBV GENOTYPES
The predominant classical PC G1896A variant was identified in 304 (29.3%) participants. Other PC variants, including mixtures of G1896A, G1896C, and G1896T, without a majority call were identified in only 13 (1.3%) participants. Therefore, the PC variant will hereafter be referred to as the G1896A variant, unless specified otherwise.
For HBeAg(+) participants, the dominant PC variant was infrequent and was mostly associated with genotype B (11.6%), followed by genotype C (4.5%) and genotype D (3%; Fig. 2A). Among the HBeAg(−) participants, the PC variant as the dominant viral species, either alone or in combination with BCP, was most frequently associated with genotype B (64%) and genotype D (63.5%), followed by genotype C (43.9%; Fig. 2B). There were only 10 HBeAg(+) and 26 HBeAg(−) participants with genotype E in this study; 5 (50%) of the HBeAg(+) participants had dominant PC, and 21 (81%) of the HBeAg(−) participants had the dominant PC variant either alone or in combination with BCP.
FIG. 2.
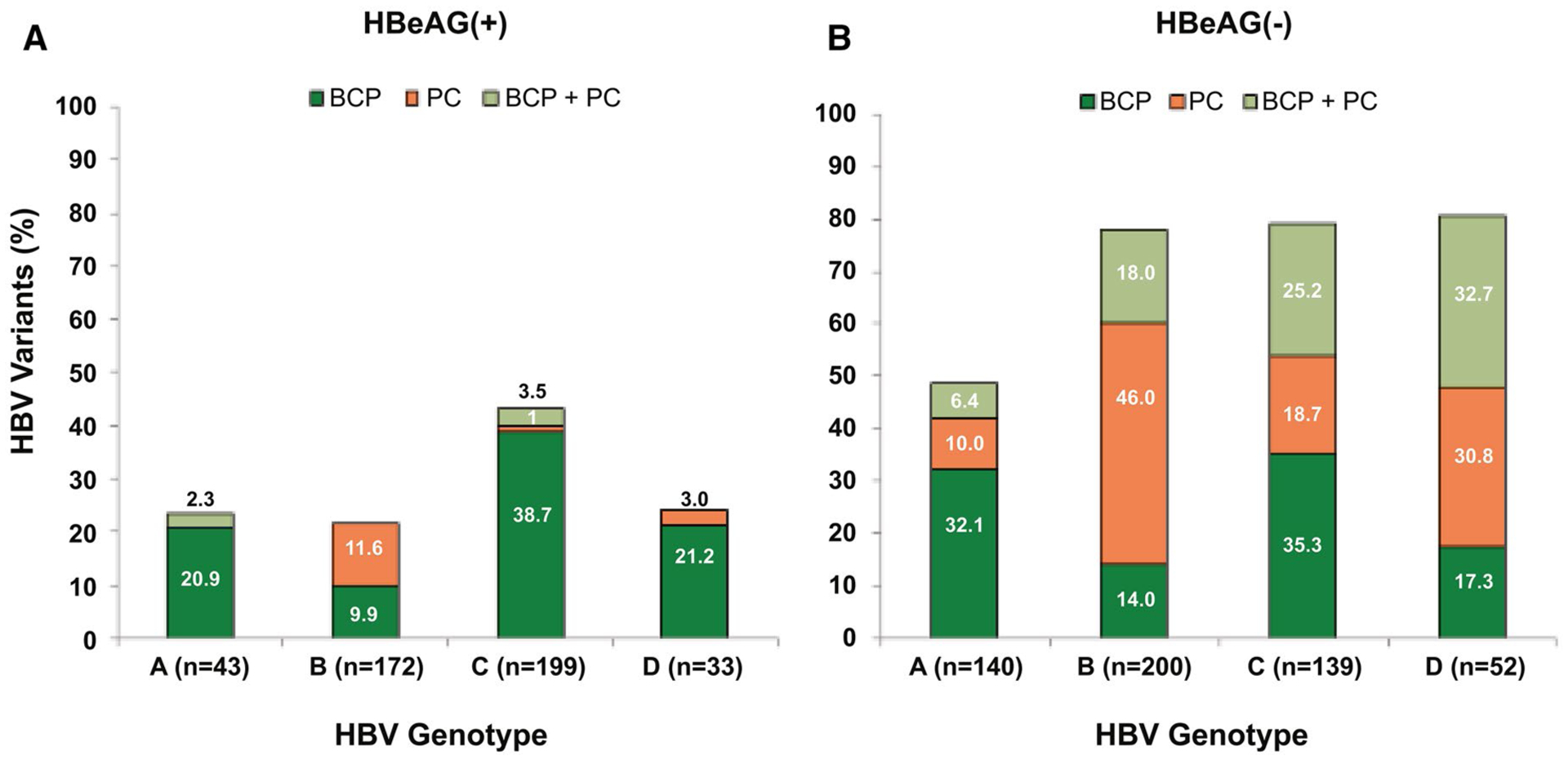
Prevalence of PC and BCP variants across HBV genotypes and HBeAg status. (A) HBeAg(+) participants; (B) HBeAg(−) participants.
The dominant PC variant was detected in 16.4% HBeAg(−) and 2.3% HBeAg(+) participants with genotype A infection. PC variants had similar prevalence in subgenotypes A1 and A2: 11 of 92 (12%) and 9 of 84 (11%) participants, respectively (Fig. 3A). All 20 participants with the dominant PC variant and genotype A were adults, and all but 1 were HBeAg(−). Among the 11 participants with subgenotype A1 and the dominant PC variant, 4 (36.4%) were born in Africa and 7 (63.6%) were born in Asia. In contrast, 8 of 9 (89%) participants with subgenotype A2 and the dominant PC variant were born in North America, and only 1 (11%) was born in Asia. The nucleotide at 1858 was T and not C in 18 participants with subgenotype A1 and in 17 with subgenotype A2 infection. The PC G1896A variant was the dominant species in 17 (49%) of these 35 with C1858T allowing base-pairing with Adenine at nt1896. Two participants with subgenotype A1, but none of those with subgenotype A2, and 1858C were noted to have the G1896A variant.
FIG. 3.
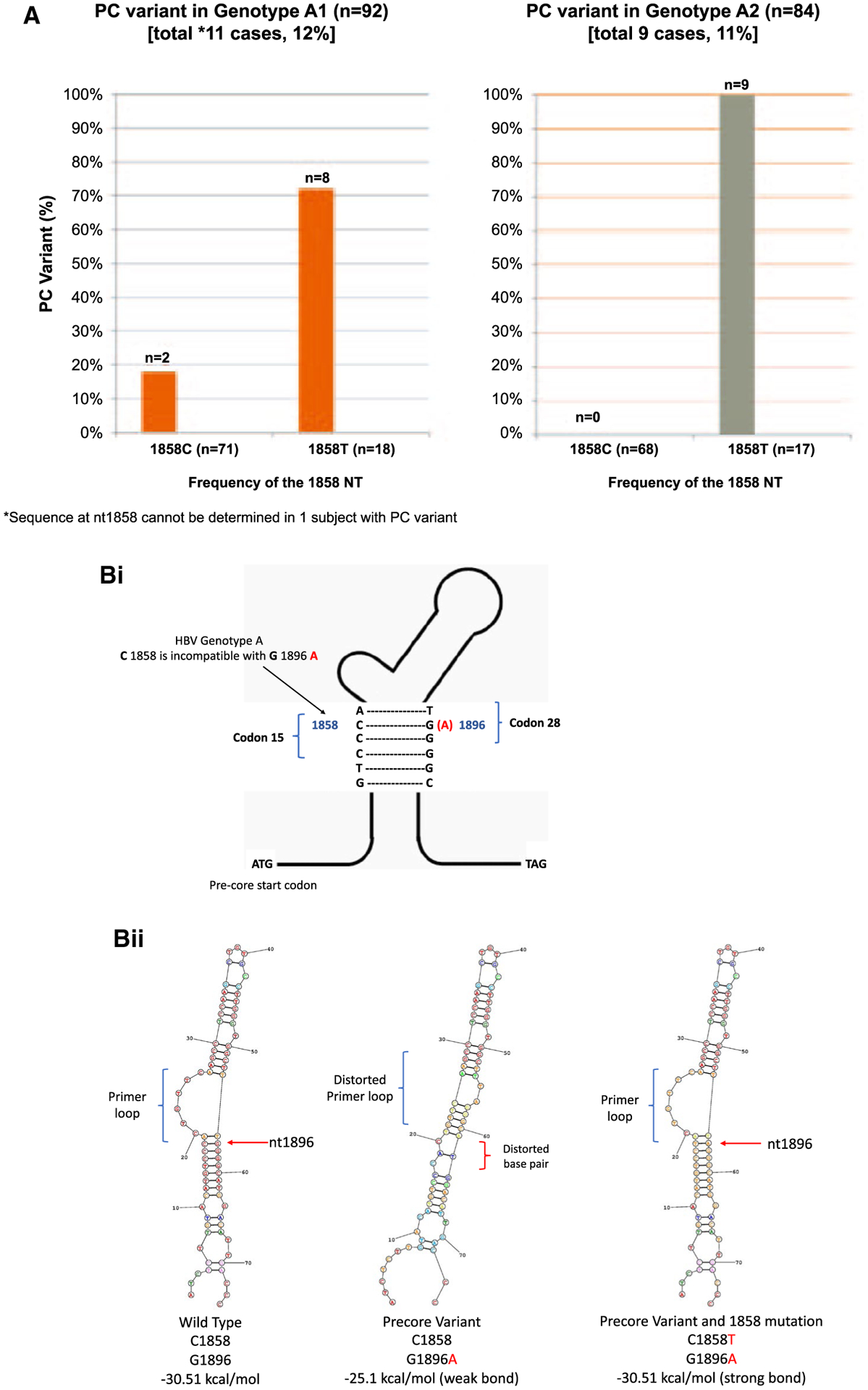
(A) Prevalence of PC G1896A variant in HBV genotype A. (Bi) HBV pregenome encapsidation sequence. (Bii) Stem-loop configurations of HBV genotype A with and without substitution at nt1986 (PC) and nt1858 positions. The stem-loop energy (kcal/mol) indicates the thermodynamic stability of the HBV pregenome encapsidation sequence. Lower energy numbers indicate stronger bonds between base pairs.
The stem-loop region contains the HBV pregenome encapsidation sequence (Ɛ) that is critical for HBV replication. The stem-loop energy (kcal/mol) indicates the thermodynamic stability of the structure of epsilon (Ɛ), with lower numbers indicating stronger bonds. The stem-loop energy levels across WT-HBV genotypes B to E were similar and ranged between −31.6 and −32.8 kcal/mol (data not shown). WT-HBV genotype A had lower stem-loop energy at −30.6 kcal/mol compared to other genotypes (P < 0.0001). The stem-loop structures and energy stabilities of HBV genotype A G1896A PC variant in association with T or C at the nt1858 position were examined (Fig. 3B). Compared to the WT sequence with G1896 and C1858, the bond in the stem-loop was significantly weakened (−25.1 kcal/mol) when G1896A was present in association with C1858. The bond was restored with a similar energy level (−30.5 kcal/mol) when G1986A was paired with C1858T.
The double BCP TA variant was the most dominant species (82.4%) among the BCP variants in all the genotypes. The single A1762T or G1764A mutation was identified in 57 (6.3%) and other variants in the BCP region without majority call in only 17 (1.9%) participants. Therefore, the BCP variant will hereafter be referred to the double BCP TA variant, unless specified otherwise. The distribution of BCP variants across the HBV genotypes for HBeAg(−) and HBeAg(+) participants is shown in Fig. 2A,B. The double BCP TA variant, alone or in combination with PC, was most frequently associated with genotype C, followed by genotype D and A, for both HBeAg(+) and HBeAg(−) participants.
DISTRIBUTION OF PC AND BCP VARIANTS FOR HBeAg(+) AND (−) PARTICIPANTS ACROSS THE AGE GROUPS
For the HBeAg(+) participants, the prevalence of dominant WT HBV progressively decreased with age from 79.9% in those aged 2–18 years to 44.2% in those aged >40 years (Fig. 4A). Dominant PC and BCP variants, either alone or in combination, were already present in 7.2% and 14.4%, respectively, in participants between 2 and 18 years and increased to 10.2% and 50.7% among those >40 years. Among the HBeAg(−) participants, dominant PC and BCP variants, either alone or in combination, were present in 42.8% and 25% of participants between age 2 and 18 years respectively, and increased to 49.8% and 46.7% among those >40 years (Fig. 4B).
FIG. 4.
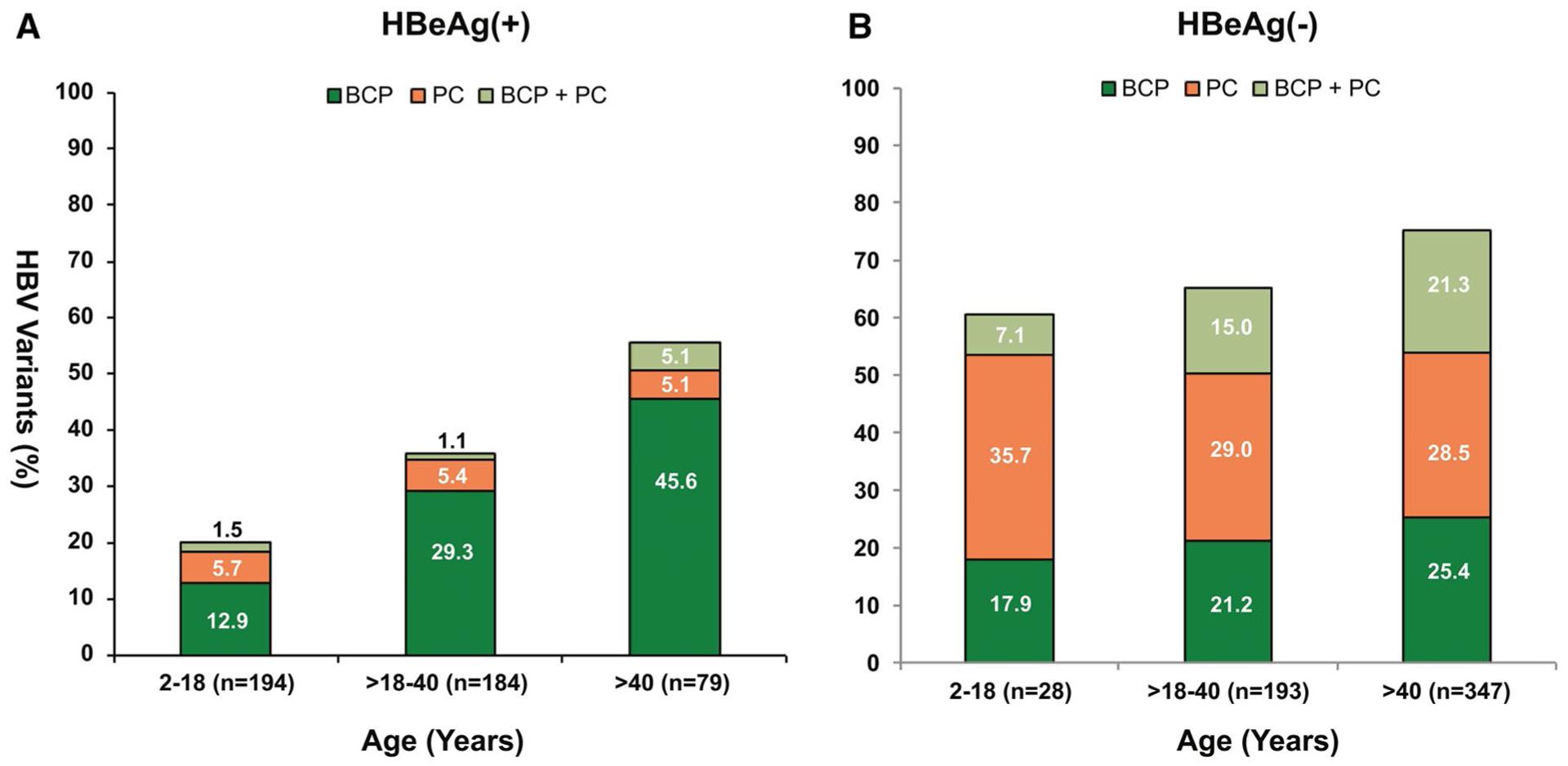
PC and BCP variants by age and HBeAg status. (A) HBeAg(+) participats; (B) HBeAg(−) participants.
COMPARISON OF HBeAg STATUS AND qHBeAg LEVELS IN PARTICIPANTS WITH WT, PC, AND BCP VARIANTS ACROSS THE AGE GROUPS
Given that the emergence of PC and BCP variants is associated with abolished or reduced production of HBeAg, the relationship between HBeAg status and HBV variants across the age groups was examined (Fig. 5A). Among participants with dominant WT HBV, prevalence of HBeAg was >90% in those between 2 and 18 years of age, decreasing steadily with age; however, 28.9% of those aged >40 years remained HBeAg positive. Prevalence of HBeAg across the age groups for those with dominant BCP variants was similar to those with WT HBV (P > 0.05). As expected, prevalence of HBeAg was lower among participants with the dominant PC variant compared to those with WT HBV in each age group (P < 0.001). However, 12 of 23 (52%) participants aged 2–18 years were HBeAg(+), decreasing to 15% by the fourth decade and only 4% in those >40 years. Participants with both dominant PC and BCP variants had similar prevalence of HBeAg across the age groups as those with the dominant PC variant only.
FIG. 5.
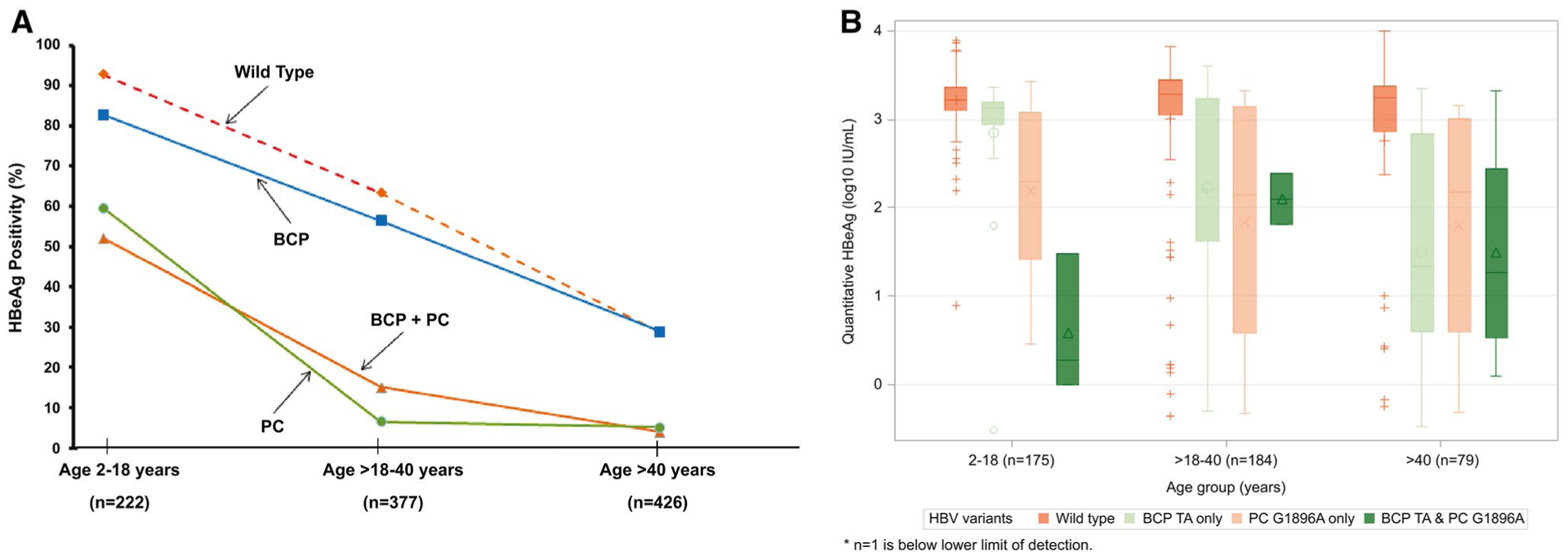
Distribution of WT, BCP, PC, and combination of BCP and PC variants across age groups. (A) WT indicates dominant WT at both BCP and PC regions. Overall P value < 0.001 for the prevalence of BCP or PC variants across age groups. Distribution of qHBeAg by age among HBeAg(+) participants with WT, BCP, PC, and combination of BCP and PC variants. (B) Distribution of qHBeAg by age WT indicates dominant WT at both BCP and PC regions.
Of the 461 HBeAg(+) participants included in this study, 410 (89%) had follow-up data on HBeAg status. Rates of HBeAg clearance among these HBeAg(+) participants, with dominant WT HBV (n = 272), BCP (n = 109), and dominant PC with or without BCP variants (n = 29) were further examined. After a median follow-up of 3.2 years, 22% participants with WT HBV achieved HBeAg clearance compared to 62% of those with dominant PC and 45% of those with dominant BCP variants. Rates of HBeAg clearance per 100 person-years were significantly higher for participants with dominant PC or BCP variants, 24.4 (95% confidence interval [CI], 15.4, 38.8) and 15.0 (95% CI, 11.4, 19.9), respectively, compared to those with WT HBV, 6.0 (95% CI, 4.7, 7.7; P < 0.0001; Supporting Table S1). This finding was true for both adults and children. Specifically, among the 167 HBeAg(+) children aged 2–18, incidence rates of HBeAg loss per 100 person-years were significantly higher for those with dominant PC variants, 25.1 (95% CI, 12.6, 50.2; P < 0.0001), and those with BCP variants, 9.9 (95% CI, 4.4, 22.0; P = 0.03), compared to those with WT HBV, 3.4 (95% CI, 2.1, 5.5).
Among HBeAg(+) participants, median qHBeAg levels for those with predominant WT HBV were consistently >3 log10 IU/mL across the age groups (Fig. 6B). The qHBeAg levels were more variable for those with dominant BCP or PC variants, but tended to be lower compared to WT HBV, especially for those aged >40 years.
FIG. 6.
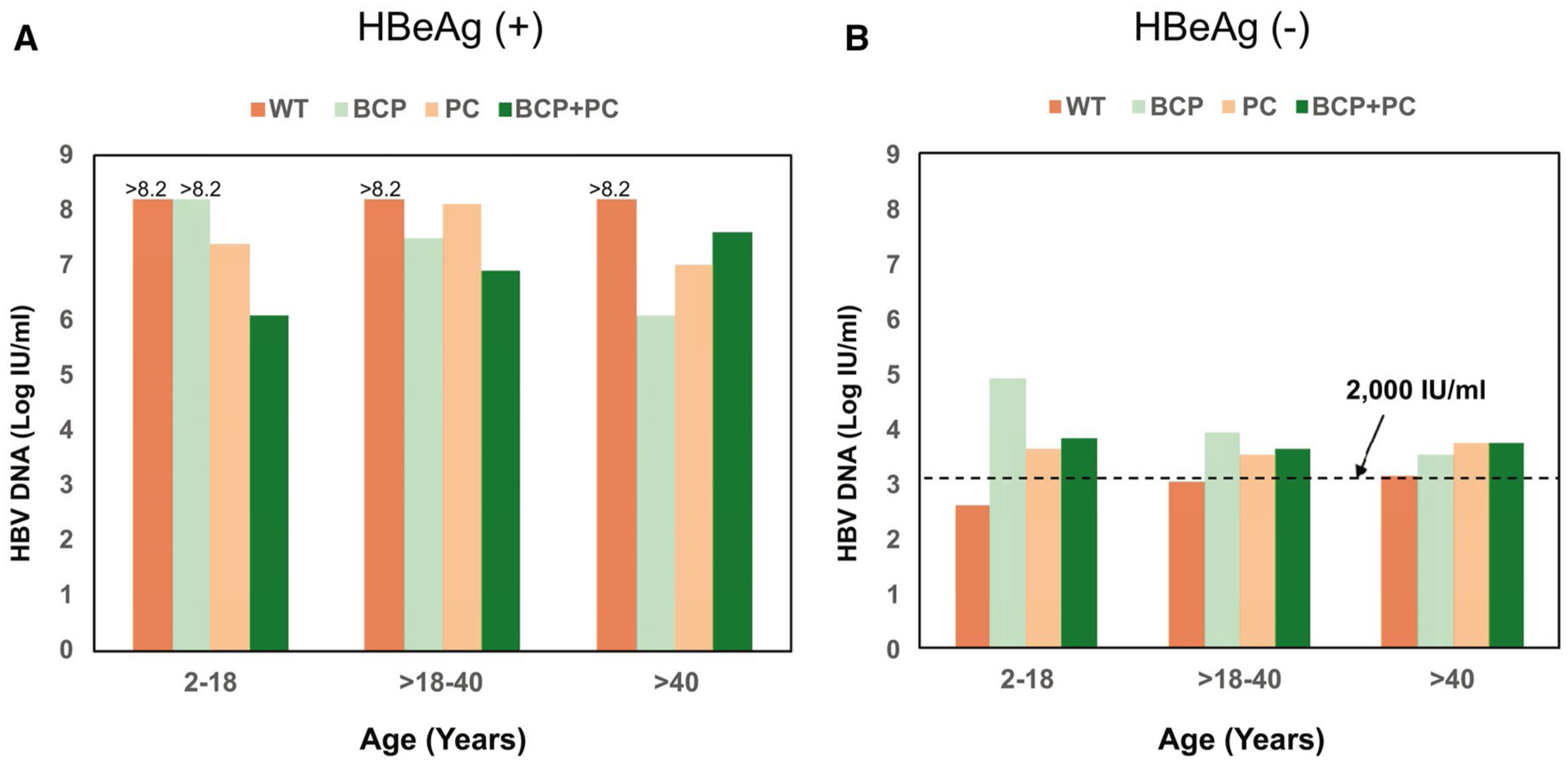
Distribution of HBV DNA by HBV variants, age, and HBeAg status. (A) HBeAg(+) participats; (B) HBeAg(−) participants.
COMPARISON OF HBV-DNA AND qHBsAg LEVELS IN PARTICIPANTS WITH WT-HBV, BCP, OR PC VARIANTS ACROSS THE AGE GROUPS
HBV-DNA levels were generally higher in HBeAg(+) than in HBeAg(−) participants (Fig. 6). Among the HBeAg(+) participants, median HBV-DNA levels were >8.2 log IU/mL for those with WT HBV across the different age groups. Those with dominant PC and BCP variants also had relatively high HBV-DNA levels, >6 log IU/mL (Fig. 6A). HBeAg(−) participants with WT HBV had low-median HBV-DNA levels (2.6–3.4 log10 IU/mL), consistent with an inactive phase of hepatitis B across the age groups; these levels tended to be lower than those with dominant PC (3.4–4.0 log10 IU/mL) or BCP (3.0–4.9 log10 IU/mL) variants (Fig. 6B).
The qHBsAg levels were higher in HBeAg(+) than HBeAg(−) participants (Data not shown). There were no differences in qHBsAg levels regardless of the presence or absence of dominant BCP or PC variants in both HBeAg(+) and HBeAg(−) groups.
Discussion
We examined the prevalence and patterns of HBV PC G1896A and BCP A1762T and G1764A variants among the racially diverse pediatric and adult participants enrolled in the HBRN Cohort Study. Our study is unique in its size (>1,000 participants), wide age range (2–80 years), and robust representation of the four most common HBV genotypes (A-D). The majority (67%) of our participants were born in Asia, with a substantial proportion born in North America (19%) and Africa (10%), allowing us to examine HBV PC and BCP variants in persons who acquired HBV infection from different continents. One of our major findings was the presence of the PC G1896A variant among participants with HBV genotype A and a T, but not a C, at nt1858. In addition, we observed that PC and BCP variants were detected at a young age in both HBeAg(+) and HBeAg(−) participants. We also noted that prevalence of HBeAg remained high in young persons with dominant PC or BCP variants, with a more rapid decline with age in those with dominant PC than those with dominant BCP variants.
In this study, which included 22% participants aged <18 years, we found that PC G1896A and BCP variants were present at a very young age, being detected as dominant species in 7.2% and 14.4% of the HBeAg(+) participants aged 2–18 years. Of note, whereas these variants block or decrease HBeAg production, some participants with these variants as dominant HBV sequences remained HBeAg(+), suggesting that these variants can be selected and become dominant even before HBeAg seroconversion. In fact, >50% of our participants aged <18 years who had dominant PC or BCP variants were HBeAg(+). In contrast, a recent study on African children living in Australia reported that 25 of 30 (83.3%) children infected with BCP or PC variants were HBeAg(−).(19) This could be explained by the difference in genotypes; all the African children with HBeAg(−) hepatitis in the Australian study had HBV genotype D or E, whereas 38 of 57 (67%) children with BCP or PC variants in our study had HBV genotype B or C.
Findings of mixed viral populations of WT and PC and/or BCP variants in HBeAg(+) patients had been previously reported, and this observation was more common in patients who cleared HBeAg during short-term follow-up.(20) In our study, we observed that prevalence of HBeAg among participants with dominant PC variants markedly declined between subjects aged 2–18 and those aged >18–40 years whereas the decline was much smaller in those with dominant BCP variants, attesting to the difference in impact of these two variants on HBeAg production. Rate of HBeAg clearance was highest for participants with dominant PC variants, followed by those with dominant BCP variants and lowest for those with dominant WT HBV. As expected, we found that qHBeAg levels were lower in participants with PC or BCP variants compared to those with WT HBV. This difference was more striking in those aged >40 years.
Among the HBeAg(+) participants, those with WT HBV had higher HBV-DNA levels compared to those with dominant PC or BCP variants. In contrast, HBeAg(−) participants with dominant PC or BCP variants tended to have higher serum HBV-DNA levels than those with WT HBV across age groups. These observations might suggest that patients with PC and BCP variants likely evolve to immune-active hepatitis B, whereas those with WT HBV are more likely to remain in the inactive phase after HBeAg clearance. Similar findings were observed in an earlier study conducted in 17 sites in the United States, but that study included adults only.(21) It has been postulated that selection of PC and BCP variants may facilitate escape of immune control. Prospective studies from Taiwan reported that the annual rates of active hepatitis B after spontaneous HBeAg seroconversion ranged from 1.5% to 3.3%.(22,23) Chu et al. reported on 37 patients with reactivation of hepatitis B (HBV DNA ≥104 copies/mL) after HBeAg seroconversion. Of these, 21 (56.8%) had a solely PC variant, 4 (10.8%) had a solely BCP variant, 10 (27.0%) had both variants, and only 2 (5%) had WT HBV.(23) Follow-up of our participants will be important to determine whether HBeAg(+) participants with dominant PC or BCP variants are more likely to evolve to active hepatitis B after HBeAg clearance.
A striking finding in our study is the detection of the dominant PC G1896A variant in 13% and 10% of participants with subgenotypes A1 and A2. Maintenance of the stem-loop structure is critical for the function of epsilon (Ɛ), which plays a pivotal role in pregenome encapsidation and HBV replication.(11) Nucleotides 1858 and 1896 are located opposite each other at the base of the stem-loop structure. In genotype A, the nt1858 is usually a C; a G to A change at nt1896 would weaken the pairing between these two nucleotides and the Ɛ signal structure. The PC G1896A variant is thus rarely detected in patients with genotype A infection. We found that 17 of 20 participants with genotype A and the dominant G1896A variant actually had C1858T, which allows base-pairing with A at nt1896 in the G1896A variant.
There is a paucity of reports on the prevalence of PC mutations among patients with genotype A infection. Publications to date have focused on Africans with predominantly subgenotype A1. In a study in South Africa,(24) variations of the PC sequence in HBV subgenotype A1 were examined in 25 patients (20 with HIV coinfection) with HBeAg-negative chronic hepatitis B. Six (24%) had start codon mutations: A1814C/T and G1816T, but none had a G1896A stop codon mutation, and all the samples had 1858C. Similar start-codon mutations at nt1814 were described in another study in Africa on patients with HBV genotype A.(25) In another report focused on HIV- and HBV-coinfected persons before antiretroviral therapy in South Africa, 44 sequences derived from HBeAg-negative samples were analyzed for mutations that are known to down-regulate or abolish HBeAg production.(26) All had subgenotype A1, and the PC G1896A variant was identified in 5 of the 39 samples that were successfully sequenced. Similar to our findings, in 4 cases the G1896A variant occurred together with C1858T.
We found that the PC G1896A variant was detected not only in subgenotype A1, but also in subgenotype A2, which is usually found in Europe and North America. In fact, the prevalence of the PC G1896A variant was almost identical in subgenotypes A1 and A2, 12% and 11%, respectively. Furthermore, whereas 4 of 11 participants with subgenotype A1 and the G1896A variant were born in Africa and the remaining 7 in Asia, 8 of 9 participants with subgenotype A2 and the G1896A variant were born in North America whereas the remaining participant was born in Asia. Surprisingly, 2 of 11 participants (1 born in Asia and 1 in Africa) with subgenotype A1 and the dominant PC G1896A variant had C, but not T, at nt1858. The predicted stem-loop energy would indicate a very unstable structure. There could be other compensatory mutations in these 2 participants that we have not identified. Unfortunately, we do not have sequences of follow-up samples from these 2 participants to determine whether changes at nt1858 or nt1896 occurred over time.
Prevalence of PC 1896A variants in this study (29.3%) is comparable to a previous study conducted on 694 patients in 17 centers in the United States (27%).(21) As has been reported by others, we found that prevalence of PC 1896A variants is related to HBV genotype, with the highest prevalence in genotypes B and D. Similarly, prevalence of BCP variants in this study (35.1%) is similar to the earlier study in the United States cited above (44%), with a higher prevalence in genotypes C and A.
The strengths of our study were the large cohort of racially diverse participants with chronic HBV infection, with ages spanning from 2 to 80 years and the follow-up data allowing us to compare the rates of HBeAg clearance between WT and PC and BCP HBV variants. However, there are several limitations. Given that HBRN only enrolled participants who were not receiving AVT, their liver disease tended to be less active and less advanced. Only samples with detectable HBV DNA could be applied to characterize the various HBV species that could skew the distribution of virological parameters of the cohort. Our study was also limited by the lack of sequencing data of serial samples to determine the temporal relationship of the changes in the PC and BCP regions and HBV-DNA levels during the course of infection.
This study is notable for a number of reasons. First, we reported the presence of the PC G1896A variant in 11.4% of participants with genotype A, with similar prevalence in subgenotypes A1 and A2. In most but not all cases, the G1896A variant was present in association with a compensatory C1858T mutation preserving base-pairing in the stem-loop structure of the pregenome encapsidation sequence. This finding has potential clinical significance; the selection of PC (G1896A) mutation may be important for genotype A HBV that harbors C1858T to persist by escaping immune surveillance.
Second, the study is worthwhile because we were able to evaluate the pediatric and adult populations in a continuum. We found that PC and BCP variants were present and could be dominant during the first two decades of life with increasing prevalence over time. Whereas most adults with the PC variant were HBeAg(−), the majority of children between 2 and 18 years old with the dominant PC variant were HBeAg(+). Our study demonstrates that the selection of HBV variants, especially PC, results in more-rapid HBeAg clearance compared to dominant WT HBV. These findings have clinical and therapeutic implications in understanding variations in rates of spontaneous HBeAg clearance and, potentially, response to AVT, especially for children and young adults. Longer-term observations will be critical to determine whether HBeAg(+) participants with PC and BCP variants are more likely to evolve to an HBeAg(−) immune-active state than those with WT HBV after HBeAg clearance.
Supplementary Material
Acknowledgment:
In addition to the authors, the HBRN would like to acknowledge the contributions of the following: Jay Hoofnagle, M.D., for critical review and data interpretation of the manuscript.
Harvard Consortium: Jianghe Niu, Ph.D., Karen Joanie Camoverde Reyes, M.D., Saad Choudhry, M.B.B.S., Pir Ahmad Shah, M.B.B.S., Imad Nasser, M.D. (Beth Israel Deaconess Medical Center, Boston, MA), Arley Donovan, Nifasha Rusibamayila, Cara Foley (Massachusetts General Hospital, Boston, MA). Minnesota Alliance for Research in Chronic Hepatitis B: Alisha C. Stahler, Linda Stadheim, R.N. (Mayo Clinic Rochester, Rochester, MN), John Lake, M.D., Philip Lacher (University of Minnesota, Minneapolis, MN), Shannon M. Riggs, L.P.N., A.S. (Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN). Midwest Hepatitis B Consortium: Kathryn Rushing, R.N., Rosemary A. Nagy, R.D.N., L.D., M.B.A., Jacki Cerkoski, R.N., M.S.N. (Saint Louis University School of Medicine, St. Louis, MO), Debra DeMarco Shaw, R.N., B.S.N., Lisa Kessels, R.N., Michael K. Klebert, Ph.D., R.N., A.N.P.-B.C. (Washington University School of Medicine, St. Louis, MO). University of Toronto Consortium: Seham Noureldin, Ph.D., Danie La, R.N., Lucie Liu, M.Sc., C.C.R.P., Diana Kaznowski, R.N., Jiayun Chen, Fengfei Huang, Doinita Vladutu, and Orlando Cerocchi (Toronto General Hospital, Toronto, Ontario, Canada), Athena Hau, B.Sc. (Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada). HBV CRN North Texas Consortium: Debra Rowan, L.V.N. (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Sheila Bass (University of Texas Southwestern, Dallas, TX), Barbara Lilly, B.S. (Baylor University Medical Center, Dallas, TX), Laurie A. Rodgers-Augustyniak, R.N., Shirley Montanye, R.N. (Department of Pediatrics, UTSW, Dallas, TX). Los Angeles Hepatitis B Consortium: Samuel French, M.D., Velma Peacock, R.N. (David Geffen School of Medicine, UCLA, Los Angeles, CA). San Francisco Hepatitis B Research Group Consortium: Marion Peters, M.D., Ashley Shobe, M.S., Rayshawnda Davis, Romuald Kuras, Claudia Ayala, M.S., Ivy Lau, B.S. (University of California-San Francisco, San Francisco, CA), Veronika Podolskaya, B.S., N.C.P.T., Anna von Bakonyi, L.V.N., C.C.R.C., Nata DeVole, R.N. (California Pacific Medical Center Research Institute, San Francisco, CA), Natasha Feier, M.S., Joel Feier, B.S., Camille Langlois, M.S. (Department of Pediatrics, UCSF, San Francisco, CA). Michigan Hawaii Consortium: Barbara McKenna, M.D., Karen Choi, M.D., Kelly Oberhelman, P.A.C., Sravanthi Kaza, B.pharm., Isabel Moran (University of Michigan, Ann Arbor, MI), Leslie Huddleston, N.P., Richmond Wong (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: A. Sidney Barritt, M.D., Tiffany Marsh, B.A., Vikki Metheny, A.N.P., Danielle Cardona, P.A.-C. (University of North Carolina at Chapel Hill, Chapel Hill, NC). Virginia Commonwealth University Medical Center: Paula G. Smith, R.N., B.S.N., Charlotte Hofmann, R.N. (Virginia Commonwealth University Health System, Richmond, VA). PNW/Alaska Clinical Center Consortium: Alycia Wolfstone, R.N., M.N. (University of Washington Medical Center, Seattle, WA), Jody Mooney, Lupita Cardona-Gonzalez (Virginia Mason Medical Center, Seattle, WA), Kara L. Cooper (Center for Clinical and Translational Research, Seattle Children’s Institute, Seattle, WA). Johns Hopkins University: Hongxia Li, M.B.B.S., M.S., Robert Anders, M.D., Ph.D., Hejab Imteyaz, Peter Lee, M.D., Kiyoko Oshima, M.D., Kim Kafka, R.N., Naureen Islam, B.S. (Department of Pediatrics, Johns Hopkins Medical Institutions, Baltimore, MD). Liver Diseases Branch, NIDDK, NIH: Nancy Fryzek, R.N., B.S.N., Elenita Rivera, B.S.N., Nevitt Morris, R.N., B.S.N., Vanessa Haynes-Williams, Amy Huang, R.N., Catherine Nadal, R.N., M.S., Jaha Norman-Wheeler, R.N., B.A. (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK, NIH: Jay H. Hoofnagle, M.D., Averell H. Sherker, M.D., Edward Doo, M.D., Rebecca J. Torrance, R.N., M.S., Sherry R. Hall, M.S. (National Institutes of Health, Bethesda, MD). Immunology Center: Mary E. Valiga, R.N., Keith Torrey, B.S., Danielle Levine, B.S., James Keith, B.S., Michael Betts, Ph.D. (University of Pennsylvania, Philadelphia, PA), Luis J. Montaner, D.V.M., D.Phil. (Wistar Institute, Philadelphia, PA). Data Coordinating Center: Frani Averbach, M.P.H., Tamara Haller, Regina Hardison, M.S., Stephanie Kelley, M.S., Christina Lalama, R.N., B.S.N., Sharon Lawlor, M.B.A., Hsing-Hua S. Lin, M.S., Ph.D., Manuel Lombardero, M.S., Andrew Pelesko, B.S., Donna Stoliker, Melissa Weiner, M.P.H., Ella Zadorozny, M.S., Qian Zhao, Ph.D. (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA).
The HBRN:
Minnesota Alliance for Research in Chronic Hepatitis B Consortium: Lewis R. Roberts, M.B., Ch.B., Ph.D. (Mayo Clinic Rochester, Rochester, MN), Mohamed A. Hassan, M.D. (University of Minnesota, Minneapolis, MN), Sarah Jane Schwarzenberg, M.D. (Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN). Midwest Hepatitis B Consortium: Adrian M. Di Bisceglie, M.D. (Saint Louis University School of Medicine, St. Louis, MO), Jeffrey Teckman, M.D. (Department of Pediatrics, Cardinal Glennon Children’s Medical Center, Saint Louis University, St. Louis, MO). University of Toronto Consortium: David K. Wong, M.D. (Toronto General Hospital, Toronto, Ontario, Canada), Joshua Juan, M.D. (Toronto General Hospital, Toronto, Ontario, Canada), Jordan Feld, M.D., M.P.H. (Toronto General Hospital, Toronto, Ontario, Canada), Colina Yim, N.P., M.N. (Toronto General Hospital, Toronto, Ontario, Canada), Keyur Patel, M.D. (Toronto General Hospital, Toronto, Ontario, Canada), Simon C. Ling, M.B.Ch.B. (Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada). HBV CRN North Texas Consortium: William M. Lee, M.D. (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Carol S. Murakami, M.D. (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Robert Perrillo, M.D. (Baylor University Medical Center, Dallas, TX), Son Do, M.D. (University of Texas Southwestern, Dallas, TX), Norberto Rodriguez-Baez, M.D. (Department of Pediatrics, University of Texas Southwestern, Dallas, TX). Los Angeles Hepatitis B Consortium: Steven-Huy B. Han, M.D. (David Geffen School of Medicine, UCLA, Los Angeles, CA), Tram T. Tran, M.D. (Cedars Sinai Medical Center, Los Angeles, CA). San Francisco Hepatitis B Research Group Consortium: Norah A. Terrault, M.D., M.P.H. (University of California-San Francisco, San Francisco, CA and Keck Medicine at the University of Southern California, Los Angeles, CA), Mandana Khalili, M.D., M.A.S. (Department of Medicine, University of California-San Francisco, San Francisco, CA), Stewart L. Cooper, M.D. (Division of General and Transplant Hepatology, California Pacific Medical Center, San Francisco, CA), Philip Rosenthal, M.D. (Department of Pediatrics, University of California-San Francisco, San Francisco, CA). Michigan Hawaii Consortium: Robert J. Fontana, M.D. (University of Michigan, Ann Arbor, MI), Naoky Tsai, M.D. (The Queen’s Medical Center, University of Hawaii, Honolulu, HI), Barak Younoszai, D.O. (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: Michael W. Fried, M.D. (University of North Carolina at Chapel Hill, Chapel Hill, NC), Andrew Muir, M.D. (Duke University Medical Center, Durham, NC), Donna Evon, Ph.D. (University of North Carolina at Chapel Hill, Chapel Hill, NC), Jama M. Darling, M.D. (University of North Carolina at Chapel Hill, NC). PNW/Alaska Clinical Center Consortium: Robert C. Carithers, M.D. (University of Washington Medical Center, Seattle, WA), Margaret Shuhart, M.D. (Harborview Medical Center, Seattle, WA), Kris V. Kowdley, M.D. (Virginia Mason Medical Center, Seattle, WA), Chia C. Wang, M.D. (Virginia Mason Medical Center, Seattle, WA), Karen F. Murray, M.D. (Department of Pediatrics, University of Washington, Seattle, WA). Virginia Commonwealth University Medical Center: Richard K. Sterling, M.D., M.Sc. (Virginia Commonwealth University Health System, Richmond, VA), Velimir A. Luketic, M.D. (Virginia Commonwealth University Health System, Richmond, VA). Johns Hopkins University: Kathleen B. Schwarz, M.D. (Department of Pediatrics, Johns Hopkins Medical Institutions, Baltimore, MD). Liver Diseases Branch, NIDDK: Marc G. Ghany, M.D., M.Hsc. (National Institutes of Health, Bethesda, MD), T. Jake Liang, M.D. (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK: Jay H. Hoofnagle, M.D. (National Institutes of Health, Bethesda, MD), Edward Doo, M.D. (National Institutes of Health, Bethesda, MD). Immunology Center: Jang-June Park, Ph.D. (University of Pennsylvania Perelman School of Medicine, Philadelphia, PA). Data Coordinating Center: Steven H. Belle, Ph.D., M.Sc.Hyg. (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA), Wendy C. King, Ph.D. (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA). Central Pathology: David Kleiner, M.D., Ph.D. (Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD).
Potential conflict of interest:
Dr. Lau consults for and received grants from Abbott. She advises for and received grants from Gilead. She advises for Dicerna. Dr. Lok, M.D. advises for and received grants from Gilead and Target. She received grants from Bristol-Myers Squibb. Dr. Chung received grants from Gilead, AbbVie, Bristol-Myers Squibb, Merck, Boehringer Ingelheim, Roche, and Janssen. Dr. Lisker-Melman is on the speakers’ bureau for Gilead and AbbVie. Dr. Janssen consults for and received grants from Arbutus, Gilead, Janssen, Medimmune, Merck, and Roche. He consults for Arena, Enyo, GlaxoSmithKline, Vir, and Viroclinics. He received grants from AbbVie and Bristol-Myers Squibb.
The HBRN was funded as a Cooperative Agreement between the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the following investigators: Lewis R. Roberts, M.B., Ch.B., Ph.D. (U01-DK082843), Anna Suk-Fong Lok, M.D. (U01-DK082863), Steven H. Belle, Ph.D., M.Sc.Hyg. (U01-DK082864), Kyong-Mi Chang, M.D. (U01-DK082866), Michael W. Fried, M.D. (U01-DK082867), Adrian M. Di Bisceglie, M.D. (U01-DK082871), William M. Lee, M.D. (U01-DK082872), Harry L.A. Janssen, M.D., Ph.D. (U01-DK082874), Daryl T.-Y. Lau, M.D., M.P.H. (U01-DK082919), Richard K. Sterling, M.D., M.Sc. (U01-DK082923), Steven-Huy B. Han, M.D. (U01-DK082927), Robert C. Carithers, M.D. (U01-DK082943), Mandana Khalili, M.D. (U01-DK082944), Kathleen B. Schwarz, M.D. (U01-DK082916), an interagency agreement with the NIDDK: Lilia M. Ganova-Raeva, Ph.D. (A-DK-3002–001), and support from the intramural program, NIDDK, NIH: Marc G. Ghany, M.D. Additional funding to support this study was provided to Kyong-Mi Chang, M.D., the Immunology Center, (NIH/NIDDK Center of Molecular Studies in Digestive and Liver Diseases P30DK50306, NIH Public Health Service Research Grant M01-RR00040), Richard K. Sterling, M.D., M.Sc. (UL1TR000058, NCATS (National Center for Advancing Translational Sciences, NIH), Norah A. Terrault, M.D., M.P.H. (CTSA Grant No. UL1TR000004), Michael W. Fried, M.D. (CTSA Grant No. UL1TR001111), Anna Suk-Fong Lok (CTSA Grant No. UL1RR024986, U54TR001959), and Kathleen B. Schwarz, M.D. (CTSA Grant No. UL1TR000423). Additional support was provided by Gilead Sciences, Inc. and Roche Molecular Systems by a CRADA through the NIDDK.
Abbreviations:
- anti-HBe
hepatitis B e antibody
- AVT
antiviral therapy
- BCP
basal core promoter
- CI
confidence interval
- HBeAg
hepatitis B e antigen
- HBRN
Hepatitis B Research Network
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HIV
human immunodeficiency virus
- nt
nucleotide
- PC
precore
- qHBeAg
quantitative HBeAg
- qHBsAg
quantitative HBsAg
- WT
wild type
Footnotes
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.31506/suppinfo.
REFERENCES
- 1).Lin CL, Kao JH. Hepatitis B virus genotypes and variants. Cold Spring Harb Perspect Med 2015;5:a021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Genotypes Kramvis A. and genetic variability of hepatitis B virus. Intervirology 2014;57:141–150. [DOI] [PubMed] [Google Scholar]
- 3).Lago BV, Mello FC, Kramvis A, Niel C, Gomes SA. Hepatitis B virus subgenotype A1: evolutionary relationships between Brazilian, African and Asian isolates. PLoS One 2014;9:e105317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Kramvis A Molecular characteristics and clinical relevance of African genotypes and subgenotypes of hepatitis B virus. S Afr Med J 2018;108:17–21. [DOI] [PubMed] [Google Scholar]
- 5).Ahmed CS, Wang ZH, Bin Z, Chen J, Kamal M, Hou J. Hepatitis B virus genotypes, subgenotypes, precore, and basal core promoter mutations in the two largest provinces of Pakistan. J Gastroenterol Hepatol 2009;24:569–573. [DOI] [PubMed] [Google Scholar]
- 6).Ben-Ari Z, Ashur Y, Daudi N, Shmilovitz-Wiess H, Brown M, Sulkes J, et al. Genotype prevalence, viral load and outcome of hepatitis B virus precore mutant infection in stable patients and in patients after liver transplantation. Clin Transplant 2004;18:415–422. [DOI] [PubMed] [Google Scholar]
- 7).Blackberg J, Kidd-Ljunggren K. Genotypic differences in the hepatitis B virus core promoter and precore sequences during seroconversion from HBeAg to anti-HBe. J Med Virol 2000;60:107–112. [DOI] [PubMed] [Google Scholar]
- 8).De Mitri MS, Cassini R, Morsica G, Bagaglio S, Andreone P, Loggi E, et al. Virological analysis, genotypes and mutational patterns of the HBV precore/core gene in HBV/HCV-related hepatocellular carcinoma. J Viral Hepat 2006;13:574–581. [DOI] [PubMed] [Google Scholar]
- 9).Du H, Li T, Zhang HY, He ZP, Dong QM, Duan XZ, Zhuang H. Correlation of hepatitis B virus (HBV) genotypes and mutations in basal core promoter/precore with clinical features of chronic HBV infection. Liver Int 2007;27:240–246. [DOI] [PubMed] [Google Scholar]
- 10).Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 1989;2:588–591. [DOI] [PubMed] [Google Scholar]
- 11).Lok AS, Akarca U, Greene S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc Natl Acad Sci U S A 1994;91:4077–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen-negative chronic hepatitis B. Hepatology 2001;34:617–624. [DOI] [PubMed] [Google Scholar]
- 13).Ghany MG, Perrillo R, Li R, Belle SH, Janssen HLA, Terrault NA, et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin Gastroenterol Hepatol 2015;13:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Rosenthal P, Ling SC, Belle SH, Murray KF, Rodriguez-Baez N, Schwarzenberg SJ, et al. Combination of entecavir/peginterferon alfa-2a in children with hepatitis B e antigen-positive immune tolerant chronic hepatitis B virus infection. Hepatology 2019;69: 2326–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Wursthorn K, Zacher BJ, Jaroszewicz J, Darnedde M, Manns M, Wedemeyer H. Development of a protocol for the quantitative determination of HBeAg using the Elecsys(R) HBeAg immunoassay. J Viral Hepat 2011;18:e179–e183. [DOI] [PubMed] [Google Scholar]
- 16).Di Bisceglie AM, King WC, Lisker-Melman M, Khalili M, Belle SH, Feld JJ, et al. Age, race and viral genotype are associated with the prevalence of hepatitis B e antigen in children and adults with chronic hepatitis B. J Viral Hepat 2019;26:856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Ganova-Raeva L, Ramachandran S, Honisch C, Forbi JC, Zhai X, Khudyakov Y. Robust hepatitis B virus genotyping by mass spectrometry. J Clin Microbiol 2010;48:4161–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Gruber AR, Bernhart SH, Lorenz R. The ViennaRNA web services. Methods Mol Biol 2015;1269:307–326. [DOI] [PubMed] [Google Scholar]
- 19).Bannister EG, Yuen L, Littlejohn M, Edwards R, Sozzi V, Colledge D, et al. Molecular characterization of hepatitis B virus (HBV) in African children living in Australia identifies genotypes and variants associated with poor clinical outcome. J Gen Virol 2018;99:1103–1114. [DOI] [PubMed] [Google Scholar]
- 20).Chan HL, Hussain M, Lok AS. Different hepatitis B virus genotypes are associated with different mutations in the core promoter and precore regions during hepatitis B e antigen seroconversion. Hepatology 1999;29:976–984. [DOI] [PubMed] [Google Scholar]
- 21).Chu CJ, Keeffe EB, Han SH, Perrillo RP, Min AD, Soldevila-Pico C, et al. Prevalence of HBV precore/core promoter variants in the United States. Hepatology 2003;38:619–628. [DOI] [PubMed] [Google Scholar]
- 22).Hsu YS, Chien RN, Yeh CT, Sheen IS, Chiou HY, Chu CM, Liaw YF. Long-term outcome after spontaneous HBeAg seroconversion in patients with chronic hepatitis B. Hepatology 2002;35:1522–1527. [DOI] [PubMed] [Google Scholar]
- 23).Chu CM, Liaw YF. Predictive factors for reactivation of hepatitis B following hepatitis B e antigen seroconversion in chronic hepatitis B. Gastroenterology 2007;133:1458–1465. [DOI] [PubMed] [Google Scholar]
- 24).Mayaphi SH, Martin DJ, Mphahlele MJ, Blackard JT, Bowyer SM. Variability of the preC/C region of hepatitis B virus genotype A from a South African cohort predominantly infected with HIV. J Med Virol 2013;85:1883–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Olinger CM, Venard V, Njayou M, Oyefolu AO, Maïga I, Kemp AJ, et al. Phylogenetic analysis of the precore/core gene of hepatitis B virus genotypes E and A in West Africa: new subtypes, mixed infections and recombinations. J Gen Virol 2006;87: 1163–1173. [DOI] [PubMed] [Google Scholar]
- 26).Makondo E, Bell TG, Kramvis A. Genotyping and molecular characterization of hepatitis B virus from human immunodeficiency virus-infected individuals in Southern Africa. PLoS One 2012;7:e46345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.