

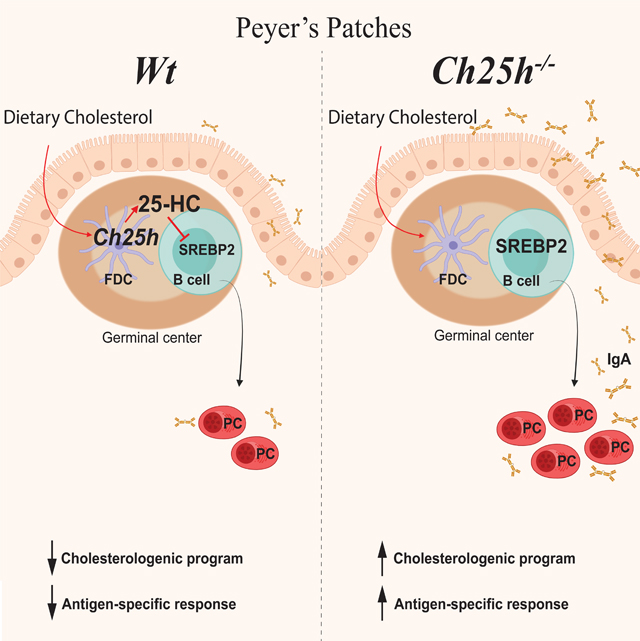
Summary
Diets high in cholesterol alter intestinal immunity. Here, we examined how the cholesterol metabolite 25-hydroxycholesterol (25-HC) impacts the intestinal B cell response. Mice lacking cholesterol 25-hydroxylase (CH25H), the enzyme generating 25-HC, had higher frequencies of IgA-secreting antigen-specific B cells upon immunization or infection. 25-HC did not affect class switch recombination, but rather restrained plasma cell (PC) differentiation. 25-HC was produced by follicular dendritic cells and increased in response to dietary cholesterol. Mechanistically, 25-HC restricted activation of the sterol sensing transcription factor SREBP2, thereby regulating B cell cholesterol biosynthesis. Ectopic expression of SREBP2 in germinal center B cells induced rapid PC differentiation, whereas SREBP2 deficiency reduced PC output in vitro and in vivo. High cholesterol diet impaired, whereas Ch25h deficiency enhanced, the IgA response against Salmonella and the resulting protection from systemic bacterial dissemination. Thus, a 25-HC-SREBP2 axis shapes the humoral response at the intestinal barrier, providing insight into the impact of high dietary cholesterol in intestinal immunity.
Graphical Abstract
eTOC Blurb
25-hydroxycholesterol (25-HC) is a metabolite of dietary cholesterol generated by the enzyme CH25H. Trindade et al. demonstrate that 25-HC produced by follicular dendritic cells inhibits the activity of the transcription factor SREBP2 in intestinal germinal center B cells and thereby reduces antigen-specific IgA response during enteric infection.
Introduction
In the small intestine, most of the T cell-dependent IgA response originates in Peyer’s patches, the lymphoid organ closest to the intestinal lumen (Hand and Reboldi, 2021). The anatomical position of Peyer’s patches places them at the primary site of absorption for both dietary and bacterial products, exposing them to a variety of metabolic cues. While metabolites absorbed from the intestinal lumen can shape Peyer’s patches organogenesis (van de Pavert et al., 2014) and migration of adaptive immune cells (Mora et al., 2006), precise mechanistic understanding of how diet-derived metabolites impact B cell differentiation and IgA production in Peyer’s patches is lacking.
In humans and mice, cholesterol absorption is restricted to the small intestine and cholesterol byproducts are known to modulate lymphocyte function (Cyster et al., 2014). Diets high in cholesterol impact adaptive immune responses, including B cell activity (Petta et al., 2018). Several cholesterol byproducts have immunomodulatory properties, including 25-hydroxycholesterol (25-HC). 25-HC, the oxysterol produced by the enzyme cholesterol 25-hydroxylase (CH25H), is the substrate necessary for the generation of 7α,25-HC, the ligand with highest affinity for the orphan G protein-coupled receptor 183 (GPR183, also known as EBI2). Despite 7α,25-HC and 25-HC differing only in the hydroxylated group at the 7α position, 25-HC does not bind to EBI2. EBI2 drives migration of several immune cell types (Kelly et al., 2011; Pereira et al., 2009; Yi et al., 2012; Baptista et al., 2019; Emgård et al., 2018; Chu et al., 2018; Melo-Gonzalez et al., 2019 ) and CH25H is required for this process. 25-HC can also have biological activity independent from its role as the intermediary for EBI2 ligand generation, primarily acting on innate myeloid cells such as macrophages (Reboldi et al., 2014; Blanc et al., 2012; Gold et al., 2014; Liu et al., 2012; Dang et al., 2017).
25-HC was originally identified as a potent inhibitor of sterol biosynthesis, with higher suppressive potency than cholesterol itself (Goldstein et al., 2006). 25-HC prevents sterol biosynthesis by maintaining inactive sterol response element binding proteins 2 (SREBP2) in the endoplasmic reticulum (ER) (Goldstein et al., 2006). When the intracellular level of 25-HC decreases, SREBP2 moves into the Golgi, where it is cleaved by proteases and subsequently activated as a transcription factor (Sakai et al., 1996). Despite its critical role in maintaining cellular cholesterol homeostasis, insight into the role of the SREBP2 pathway in immune cells has been limited (Kidani et al., 2013).
B cell responses in the Peyer’s patches, and the subsequent IgA production, are driven by continual exposure to microbial-derived molecules (Jung et al., 2010; Reboldi et al., 2016; Belkaid and Naik, 2013; Hooper and Macpherson, 2010) as evidenced by the presence of chronic germinal center (GC) composed of discrete clusters of GC B cells, T follicular helper cells (Tfh cells) and follicular dendritic cells (FDCs) in the center of B cell follicles. Peyer’s patches GC B cells give rise to plasma cells (PCs) and memory B cells. In this study, we sought to define how 25-HC controls intestinal IgA responses and to understand how dietary cholesterol regulates B cell differentiation in the GC. We found that IgA PC generation in intestinal lymphoid organs is controlled by 25-HC independently of EBI2. Mice lacking Ch25h had normal IgA class switch recombination in vivo but show increased antigen (Ag)-specific IgA response to toxin and enteric pathogens. In Peyer’s patches, FDCs generated 25-HC which suppressed the synthesis of cholesterol metabolism genes in GC B cells. Mechanistically, 25-HC inhibited SREBP2 activation, nuclear translocation and transcriptional activity: in the absence of 25-HC, expression of SREBP2 target genes increased in GC B cells and ectopic expression of Srebf2 in GC B cells increased PC differentiation. Conversely, GC B cells lacking SREBP2 maintained a GC phenotype and had reduced propensity to become PCs. Our results establish a role for a dietary-derived sterol in controlling B cell fate in the mucosal draining lymph node during T cell-dependent IgA responses.
Results
CH25H controls antigen-specific intestinal IgA response independently of EBI2.
To measure the impact of the 25-HC producing enzyme CH25H on mucosal B cell responses, we immunized Ch25h−/− mice and co-housed, littermate controls with the intestinal antigen cholera toxin and quantified the IgA response in the intestinal lamina propria. While the IgA+ PC fraction (Fig.1A) was identical between genotypes, cholera toxin-specific IgA-secreting cells (ASC) were present in higher numbers in the duodenum of mice lacking CH25H (Ch25h−/−) (Fig.S1A): the total number of IgA-ASC was not different between genotypes (Fig.S1B), leading to a higher frequency of cholera toxin-IgA-ASC in Ch25h−/− mice (Fig.1B). In line with the overall distribution of IgA-ASC in the small intestine (Fig.S1C), we only observed a consistent frequency of IgA-ASC in the duodenum, with very little IgA or cholera toxin-IgA-ASC in the ileum of Ch25h−/− mice (Fig.S1D,E). Since CH25H is required for the generation of 7α,25-HC, the most potent ligand for EBI2, a GPCR that is expressed on B cells and controls B cell positioning and differentiation, we tested whether CH25H deficiency might impact antigen-specific IgA response through EBI2 by immunizing Ebi2-deficient mice (Ebi2−/−) and littermate controls. Ebi2−/− mice showed unchanged number of cholera toxin-IgA-ASC, IgA-ASC (Fig.S1F,G), and overall frequency of cholera toxin-IgA-ASC (Fig.1C) suggesting that CH25H controls the intestinal humoral response independently of EBI2 migration. cholera toxin-specific IgA titers were increased in the small intestinal lavage (Fig.1D), but not in the serum (Fig.1E), highlighting how CH25H is predominantly affecting intestinal, but not systemic, antigen-specific IgA responses.
Figure 1. 25-HC, but not EBI2, shapes antigen-specific IgA response in duodenal lamina propria.

(A) Representative flow cytometric plot and compiled analysis of IgA+ plasma cells in duodenal lamina propria of Ch25h−/− and littermate control mice. (B) Representative ELISPOT and compiled percentage of cholera toxin-IgA secreting PCs from lamina propria of Ch25h−/− and littermate control immunized for 3 weeks with cholera toxin. (C) Percentage of cholera toxin-specific IgA secreting PCs in LP of Ebi2−/− mice immunized for 3 weeks with cholera toxin. (D,E) ELISA of cholera toxin-specific IgA in small intestine lavage and serum of Ch25h−/− and littermate control mice immunized for 3 weeks with cholera toxin. (F) Representative ELISPOT and compiled percentage of Salmonella-specific IgA PCs in intestinal lamina propria of Ch25h−/− and littermate control mice and two weeks post-infection. Data are representative of three independent experiment, ns=non-significant, *** p<0.005; ****p<0.001 (unpaired Student’s T test).
To further investigate the role of CH25H on intestinal IgA response, we infected Ch25h−/− mice and littermate controls with aroA-deficient (ΔAroA) Salmonella enterica Serovar Typhimurium, a defective strain of Salmonella that has an intact route of entry into Peyer’s patches but reduced infectivity (Martinoli et al., 2007), allowing for the measurement of IgA response in B6 mice (Monack et al., 2004). In absence of CH25H, we observed an increased number of Salmonella-IgA-ASC in the lamina propria (Fig.1F). Overall, our results show that mice lacking CH25H are characterized by an enhanced antigen-specific IgA response against toxin and enteric bacteria, suggesting that 25-HC can control T cell-dependent IgA responses.
PC-generation, not class switch recombination, is controlled by 25-HC in Peyer’s patches.
IgA-secreting PCs represent the terminal differentiation step of B cells stimulated by intestinal antigen in the Peyer’s patches. Upon Ag stimulation and GC entry, GC B cells differentiate into IgA PC that egress from the Peyer’s patches and migrate to the lamina propria (LP) using a combination of integrin and chemokine receptors. We therefore asked whether CH25H’s effect on antigen-specific IgA ASC generation was anatomically restricted and initiated in Peyer’s patches. To this end we took advantage of FTY720, a sphingosine 1-phosphate receptor (S1PR1) antagonist that can prevent IgA PC egress from Peyer’s patches (Gohda et al., 2008).
We immunized Ch25h−/− mice and littermate controls with cholera toxin and administered FTY720 to trap newly generated, antigen-specific PCs in Peyer’s patches. As expected, FTY720 treatment increased the number of IgA-ASC in the Peyer’s patches (Fig.2A, Fig.S1H), but only Ch25h–deficient mice showed an enhanced antigen-specific PC response (Fig.2B, Fig.S1I). In contrast, mice lacking EBI2 showed no increase in cholera toxin response, highlighting how CH25H, but not EBI2, is involved in controlling specific IgA response in Peyer’s patches (Fig.2C, Fig.S1J). In line with the data from LP, blocking of S1PR1-dependent ASC egress with FTY720 during Salmonella infection also enhanced IgA-ASC response in Peyer’s patches of Ch25h−/− mice (Fig.2D). While Ebi2−/− mice have shown a systemic decrease of PC response (Pereira et al., 2009), we did not observe an effect on PC generation in Peyer’s patches (Fig.S1K), and Ch25h−/− mice, but not Ebi2−/− mice, were able to generate more antigen-specific PCs. These results suggest the existence of a process dependent on CH25H, but independent of EBI2, that shapes IgA specific response in Peyer’s patches. Ch25h−/− mice showed no defect in GC formation (Fig.S2A,B), GC structure (Fig.S2C,D) or Tfh cell frequency (Fig.S2E) compared to littermate controls. While previous work has suggested that 25-HC could block IgA class switching (Bauman et al., 2009), ex-vivo FACS analysis of B cell subsets from Peyer’s patches and mesenteric lymph nodes in Ch25h+/− and Ch25h−/− showed that lack of 25-HC has no impact on the ability of B cells to undergo IgA class switch recombination (Fig.2E, Fig.S2F,G).
Figure 2. 25-HC modulates antigen-specific PC generation in Peyer’s patches.

(A) Flow cytometric analysis of plasma cells in Peyer’s patches of Ch25h−/− and littermate control mice treated daily with FTY720 or saline for one week. (B) Number of cholera toxin -specific IgA secreting cells in Ch25h−/−, (C) Ebi2−/− and wild type mice, measured by ELISPOT. (D) Representative ELISPOT and compiled Salmonella-specific IgA secreting cells in Peyer’s patches of Ch25h−/− and littermate control mice treated with FTY720. Data indicate the frequency of Salmonella-specific-ASC among Peyer’s patch cells. (E) Analysis of Class switching recombination in Peyer’s patches by flow cytometry. (F) Quantification of secreted IgA by ELISA from GC B cells cultured with NB21 cells and incubated with the indicated sterols for 3.5 days. A,B,C,E are pooled from at least three independent experiments, D, F from two independent experiments. Statistical significance was measured by two-way ANOVA using Bonferroni’s corrections (B,C,F) **p<0.01, unpaired Student’s t test (D).
To assess whether 25-HC could directly restrain PC generation from GC B cells, we used NB21 culture assay, in which the feeder cell layer expresses CD40L, BAFF and IL-21 (Kuraoka et al., 2016). Sorted GC B cells were cocultured with NB21 and then treated with different oxysterols: 25-HC, but not 7α,25-HC or cholesterol, decreased the Ab-secreting cell generation (Fig.2F), while minimally impacting overall survival (Fig.S2H). Similar results were obtained with GC B cells stimulated with anti-CD40 and cultured with NB21 supernatant, ruling out a direct effect of 25-HC on NB21 cells (Fig.S2I), and suggests that 25-HC acts on GC B cells to restrain their ability to differentiate into PCs. Overall, our data suggest that intestinal and systemic antibody responses are differentially impacted by oxysterols, with 25-HC controlling local antigen-specific PC responses in an EBI2-independent manner during enteric response.
FDCs contribute to 25-HC Peyer’s patches levels and dietary cholesterol reduces IgA response via CH25H.
25-HC can be secreted (Reboldi et al., 2014) and it diffuses to the surrounding tissue (Cyster et al., 2014; Yi et al., 2012) to generate a 25-HC niche, a microanatomical area rich in 25-HC that can regulate nearby immune cells in a EBI2-independent way. Ch25h expression has been reported in several cell types of both hematopoietic and non-hematopoietic origin, mainly in the context of EBI2 ligand generation. However, the contribution of Ch25h-expressing cells to the 25-HC niche in vivo is less clear. While the relative quantity of EBI2 ligand 7α,25-HC can be determined in a transwell assay based on the EBI2-mediated migration, 25-HC is not specifically measured in that setting. To unambiguously establish functional 25-HC substrate amounts in tissue extracts, we designed a modified transwell assay and verified it using synthetic oxysterols (Fig.S3A). We first inactivated 7α,25-HC in samples by incubating it with 293T cells expressing HSD3B7, a 7α-oxidized cholesterol dehydrogenase enzyme that oxidizes all existing EBI2 ligand; subsequently we harvested and incubated 25-HC-containing extract with 293T cells expressing CYP7B1. This second incubation converted 25-HC to 7α,25-HC, which was then measured in the transwell using EBI2-expressing M12 cells (Fig.S3B). Analysis of intestine and intestinal secondary lymphoid organs showed that 25-HC can be measured in all of these tissues and required the activity of CH25H enzyme. Thus, intestinal tissues retain 25-HC that is not converted into EBI2 ligand in unmanipulated mice (Fig.S3C). Ch25h is induced by Toll-like receptor agonists (Park and Scott, 2010), and broad antibiotic treatment (ampicillin, vancomycin, neomycin, and metronidazole) sharply reduces the 25-HC amount in Peyer’s patches, with individual antibiotic showing different degrees of effectiveness, suggesting the existence of specific commensal-oxysterol generation circuits (Fig.S3D).
In Peyer’s patches, follicular and GC B cells did not express Ch25h, which was instead highly abundant in non-hematopoietic (CD45−) cells (Fig.S4A). PC differentiation largely takes place in the GC, therefore we reasoned that a 25-HC niche anatomically restricted to the GC could influence activated B cell fate decision. In GC, stromal follicular dendritic cells (FDCs) that display antigens in the light zone (LZ) for BCR selection, have high levels of Hsd3b7 transcripts (Yi et al., 2012), thus they are able to inactivate the EBI2 ligand. In this way EBI2-expressing follicular B cells remain outside the GC, while GC B cells, which don’t express EBI2, reach and reside inside the GC structure (Cyster et al., 2014). As 25-HC is not processed by HSD3B7, the GC environment is likely to contain higher concentrations of 25-HC compared to the follicles. Since EBI2 ligand regulates naïve B cell positioning, we posit that most of 25-HC in the follicles is not freely available as it is converted to 7α,25-HC by CYP7B1 (Fig.S4B). In line with this concept, Peyer’s patches CD45- cells express a high level of Hsd3b7 transcripts (Fig.S4C). FDCs express Ch25h (Rodda et al., 2018), but whether FDCs are critical to establish GC 25-HC production is unknown.
We employed two different strategies to assess whether FDCs were the primary source of 25-HC in Peyer’s patches. First, we used CD21-DTR mice, where Cd21-Cre mice are crossed with mice carrying Simian diphtheria toxin receptor (DTR) downstream of a floxed stop element in the ROSA26 locus. We generated bone marrow chimeras using CD21-DTR mice (or littermate controls) as hosts. In this model the DTR is expressed only in non-hematopoietic CD21-expressing cells, i.e. FDCs (Wang et al., 2011) and short term DT injection led to FDC ablation (Fig.S4D). While there was a small reduction in EBI2 ligand, 25-HC was largely reduced in Peyer’s patches upon FDC depletion (Fig.3A). This result supports FDCs as the primary generator of 25-HC in GC and suggests that the follicular production of EBI2 ligand (Rodda et al., 2018) can occur in cells other than FDCs. To confirm this result, we alternatively depleted FDCs by blocking lymphotoxin (LT) signaling through LTβ receptor (LTβR) on FDCs using soluble LTβR fused to human IgG Fc (LTβR-Fc). The blockade prevents the interaction between LT-α2β1-expressing GC B cells and LTβR-expressing FDCs, leading to FDC death (Mackay et al., 1997). This approach led to the same result as in CD21-DTR mice: FDC depletion (Fig.S4E,F) only marginally impacted EBI2 ligand generation but led to a dramatic reduction of 25-HC (Fig.3B). Thus, in Peyer’s patches FDCs are responsible for the 25-HC rich environment in the GC, while only minimally contributing to the overall EBI2 ligand tissue concentration, which is likely underpinned by other CH25H+ cells (Fig.S4G) (Pikor et al., 2021; Rodda et al., 2018).
Figure 3. FDCs and diet impact tissue 25-HC levels and control IgA response.

(A) CD21-DTR and littermate control mice were reconstituted with WT bone marrow and treated with diphtheria toxin for 16hrs: lipids were extracted from Peyer’s patches for 25-HC measurement using transwell migration assay with EBI2+ M12 cells. (B) Transwell migration assay of EBI2+ M12 cells in response to lipid extracted from Peyer’s patches of wild type mice treated with recombinant LTβR-FC or isotype control. (C) Relative migration of EBI2+ M12 cells exposed to Peyer’s patches lipid extracts of wild type mice treated for 1 weeks with 0.15 % High cholesterol diet (Western diet-HCF) or normal chow (NF). (D,E) Total number of cholera toxin-specific ASC detected by ELISPOT in mesenteric lymph nodes and Peyer’s patches from Ch25h−/− and littermate control fed with mice NF or 0.15% of HCF. Results are pooled from 3 or 4 independent experiments. ns=non-significant, *p<0.05, **p<0.01,*** p<0.005, ****p<0.001 (unpaired Student’s T test) in (A,B,C) and ns=non-significant, *p<0.05, **p<0.01,***p<0.005, ****p<0.001 (two-way ANOVA) in (D,E).
Since CH25H requires cholesterol to generate 25-HC we asked whether changes in dietary cholesterol content could shape the intestinal 25-HC niche. We fed mice with normal food (NF) or normal food with the exclusive addition of 0.15% cholesterol found in the Western diet (high cholesterol food, HCF) for 1 week and measured 25-HC in Peyer’s patches. We found that 25-HC was increased in animals fed HCF (Fig.3C), suggesting that altering the intestinal absorption of dietary cholesterol can shape the 25-HC niche. EBI2 ligand was also increased in Peyer’s patches, highlighting a link between dietary cholesterol and immunomodulatory sterols. We then asked whether diet-dependent changes in the Peyer’s patches 25-HC niche could influence B cell fate. For this, mice fed with different diets were immunized with cholera toxin and treated with FTY720. WT mice fed HCF showed decreased generation of cholera toxin-IgA-ASC compared to WT mice fed with NF. In contrast, Ch25h−/− mice showed only a modest reduction in cholera toxin-IgA-ASC (Fig.3D). While this effect was pronounced in Peyer’s patches, we observed a similar effect of HCF in mesenteric lymph nodes of WT, but not Ch25h−/−, mice (Fig.3E), suggesting that 25-HC dependent restraint on PC generation can occur in other intestinal lymphoid organs. Total IgA was largely unaffected in both Peyer’s patches and mesenteric lymph nodes (Fig.S4H,I).
SREBP2 activation is induced by BCR signaling and is controlled in vivo by 25-HC.
Given the role of 25-HC in controlling activation and nuclear translocation of sterol response element- binding proteins (SREBP2) (Goldstein et al., 2006), we analyzed the SREBP2 transcriptional activity in B cells. Since SREBP2 regulates its own transcription (Sato et al., 1996), we initially measured Srebf2 transcripts in Peyer’s patches B cell subsets (Fig.4A). GC B cells and PC showed the highest amounts of Srebf2 expression, followed by memory B cells, and follicular B cells with the lowest Srebf2 transcription. Similar dynamics of Srebf2 transcription was observed in B cell subsets from the mesenteric lymph nodes (Fig.S5A). Transcripts for Srebf1a, an isoform of SREBP1 protein than can control certain aspects of lipid metabolism, were detected at a considerably lower amounts in both Peyer’s patches and the mesenteric lymph nodes, with both GC B cells and PC transcribing Srebf1a (Fig.4B, Fig.S5B). To determine whether 25-HC can control SREBP2 amounts in vivo we analyzed Srebf2 transcripts in Ch25h−/− mice. Srebf2 expression was increased in GC B cells and PCs in both Peyer’s patches and mesenteric lymph nodes in the absence of 25-HC (Fig.4C, Fig.S5C), while no difference was observed in follicular B cells and a minimal increase was noted in memory B cells. These results demonstrate that in vivo 25-HC levels control SREBP2 activation in some antigen-experienced B cell subsets.
Figure 4. 25-HC regulates SREBP2 transcription and activation in stimulated B cells.
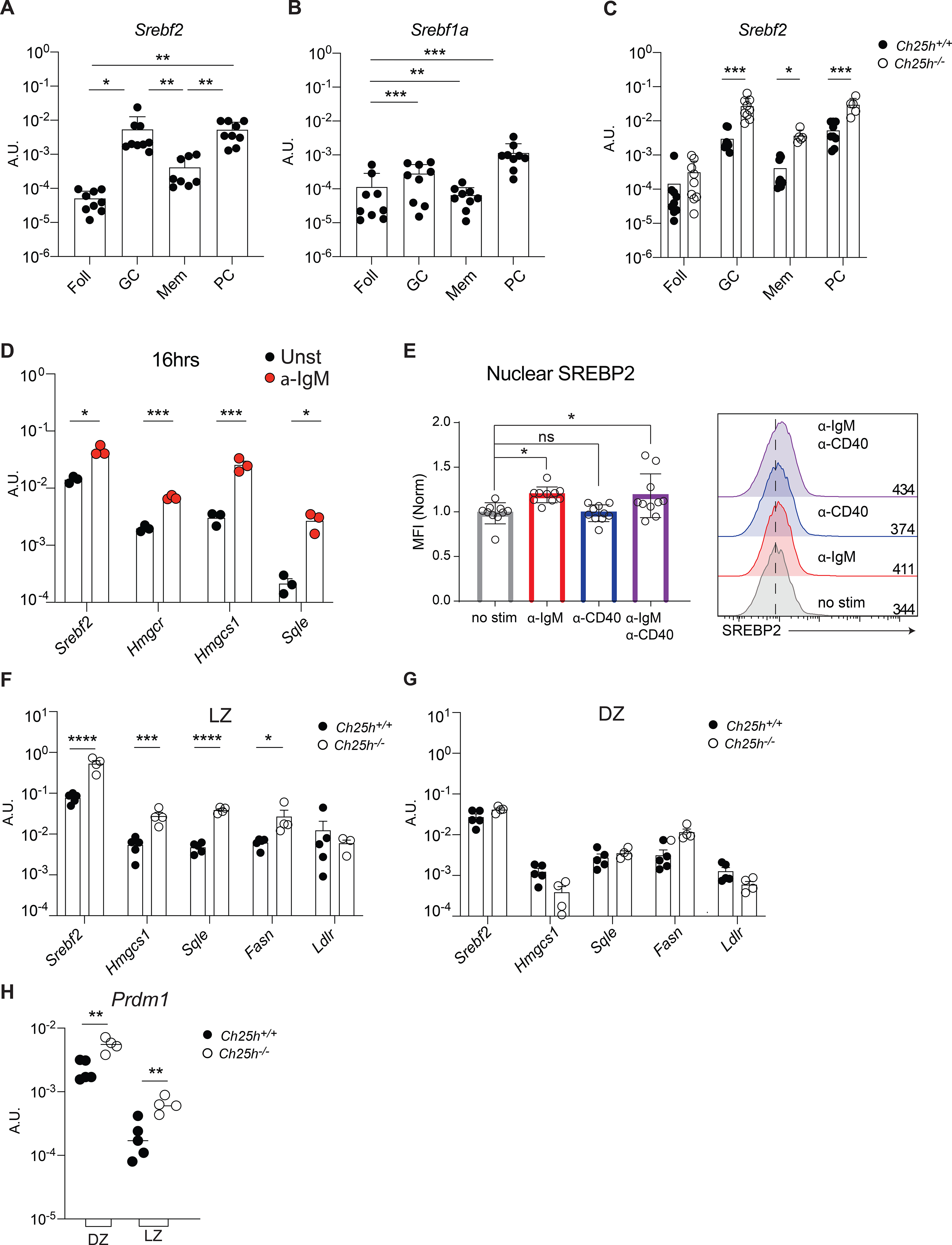
Transcript level of Srebf2 (A) and Srebf1a (B) in B cell subsets from Peyer’s patches of wild-type mice and (C) from Peyer’s patches of Ch25h−/− and littermate control mice measured by qPCR. (D) Analysis of gene expression of Srebf2 and SREBP2-target in follicular B cells stimulated with anti-IgM for 16h. (E) Cumulative and representative flow cytometry of SREBP2 nuclear staining in follicular B cells stimulated with the indicated stimuli. (F) qPCR results of Srebf2 and its target genes in sorted B cells of germinal center LZ and (G) DZ. (H) Blimp expression in sorted DZ (CXCR4+CD86−) and LZ (CXCR4−CD86+) GC B cells from Peyer’s patches of Ch25h−/− and littermate control mice. Each symbol represents one independent mouse from 9–10 (A,B,C, E) or 4 mice (F,G). Graphs show mean and SEM. *p<0.05,**p<0.01,***p<0.005, ****p<0.001(two-way ANOVA).
SREBP2 induction is generally driven by low levels of intracellular sterols, including select oxysterols, but whether other signals can control SREBP2 activation was unclear. All activated B cell subsets, but not naïve B cells, showed high Srebf2 transcripts to some extent, thus we reasoned that BCR stimulation and T cell help could possibly induce SREBP2 activation. Follicular B cells stimulated with anti-IgM Ab for 16h showed Srebf2 induction and increase in SREBP2 target genes Hmgcr, Hmgcs1 and Sqle (Fig.4D). In contrast, anti-CD40 stimulation alone, which mimics T cell help, had little effect on Hmgcr, Hmgcs1 transcription compared to stimulation with both anti-IgM and anti-CD40, but it was able to induce Sqle transcription (Fig.S5D). These results suggest that SREBP2 protein abundance is increased in BCR-stimulated B cells.
We first analyzed the amount of SREBP2 by staining stimulated B cells intracellularly with an Ab that recognizes a site in SREBP2 that is only present in the ER-resident, unprocessed SREBP2. In line with the qPCR data, BCR stimulation drove SREBP2 accumulation (Fig.S5E). SREBP2 function is mediated by its nuclear translocation. Therefore, we sought to investigate the cellular dynamics of SREBP2 in stimulated B cells by flow cytometric analysis of isolated cell nuclei (Gallagher et al., 2018). We validated the nuclear isolation by analyzing the B cell line WEHI-231 expressing cytosolic GFP, nuclear RFP (H2B-RFP) and stained with fluorescent ER tracker. This strategy allowed for the quantification of nuclear transcription factors at single cell level without ER contamination, which is critical since inactive SREBP2 is sequestered in the ER (Fig.S5F). We used an Ab directed against the N-terminus of SREBP2, which functions as the transcription factor: B cells stimulated through the BCR induced SREBP2 activation and its nuclear translocation (Fig.4E). IRF4, a transcription factor that is upregulated upon BCR stimulation and is critical for PC formation, also translocated to the nucleus in a similar fashion (Fig.S5G). Specificity of the staining was validated using B cells lacking SREBP2 (Cd21CreSrebf2floxflox) that showed no SREBP2 induction upon anti-IgM stimulation (Fig.S5H). SREBP2 nuclear translocation in response to BCR stimulation was mainly dependent on BCR signaling transducers as the Bruton’s Tyrosine Kinase (BTK) inhibitor Ibrutinib and Syk inhibitor R406 maintained SREBP2 sequestered in the cytoplasm (Fig.S5I). In contrast, AKT inhibitor API1 did not affect SREBP2 activation, while inhibitors for mTOR (Rapamycin) and HMGCR (Mevastatin) only partially prevented SREBP2 nuclear translocation.
Since SREBP2 induction is controlled by BCR stimulation, and the 25-HC niche requires FDCs that display antigens to GC B cells, we hypothesized that modulation of SREBP2 by 25-HC would be restricted to the GC in the LZ. Sorted LZ and dark zone (DZ) GC B cells (Fig.S6A) from Ch25h−/− and littermate controls indicated that both Srebf2 and SREBP2 target genes are induced to a greater extent in LZ compared to DZ, and their expression was restrained by 25-HCs (Fig.4F,G). Since Ch25h deficiency led to increased GC-derived antigen-specific PC (Fig.2B) we analyzed the expression of Prdm1, the gene that encodes for Blimp-1, the master transcription factor required for PC differentiation. We observed that LZ and DZ GC B cells from Peyer’s patches of Ch25h−/− mice showed increased Prdm1 transcription compared to littermate control GC B cells (Fig.4H). Together, our data demonstrate that in vivo, 25-HC restrains cholesterologenic gene transcription in anatomically distinct subsets of GC B cells and regulates the initiation of PC transcriptional program in GC B cells, possibly by impeding SREBP2 activation.
Ectopic SREBP2 activation is sufficient to drive PC differentiation.
To test whether SREBP2 activation could drive B cell differentiation into PCs per se, we sought to manipulate SREBP2 cellular localization, and therefore activity, in Peyer’s patches GC B cells. We took advantage of mice expressing Cre in the Aicda locus and the fluorescent protein tdTomato (tdTom) downstream a loxP-flanked STOP cassette in the ROSA26 locus (AicdaCre/+Rosa26 lox-stop-lox-tdTom). In these animals only GC B cells and post-GC B cells (memory B cells and PCs) express tdTom. We retrovirally transduced bone marrow from AicdaCre/+Rosa26 lox-stop-lox-tdTom with a retrovirus encoding constitutively active nuclear (n) form of SREBP2 downstream of the floxed-stop cassette (followed by an internal ribosomal entry site and a CD90.1 (Thy-1.1) reporter)(Green et al., 2011), which resulted in restricted expression of nSREBP2 to B cells that expressed AID (GC B cells, memory B cells and PCs) (Fig.5A). Overexpression of nSREBP1a, which is not controlled by 25-HC, was used as control. Since nSREBPs can translocate into nucleus despite high intracellular sterol concentration, this approach uncoupled intracellular metabolism and SREBP2 activation in the context of B cell differentiation. Active SREBP2 led to increased PC generation in Peyer’s patches, while active SREBP1a resulted in a minimal increase of PC fate compared to empty vector (EV) transduced cells (Fig.5B,C). SREBP1a overexpression reduced GC output, albeit not to the level observed with SREBP2 modulation, possibly through its impact on fatty acid biosynthesis. The enhanced PC generation from B cells with active nSREBP2 and the unaffected B cell fate in presence of nSREBP1a is in line with the notion that 25-HC specifically controls SREBP2, but not SREBP1 processing (Reboldi et al., 2014). Analysis of LP PCs, identified by the surface amino acid transporter CD98 (Fig.5D), (which specifically marks PCs (Tellier et al., 2016) and it is as susceptible to enzymatic cleavage during digestion as CD138) revealed that transduced B cells were able to reach the tissue (Fig.5E). However, comparison of transduced cell frequency in Peyer’s patches and LP revealed that SREBP-2 transduced cells were present in higher frequency compared to both EV and SREBP1a transduced cells, showing that increased SREBP2 activity was more efficient in generating LP-homing PCs (Fig.5F). These data establish that alteration of SREBP2 activity instructs B cells to assume distinct cell fates.
Figure 5. SREBP2 ectopic expression induces plasma cell development.
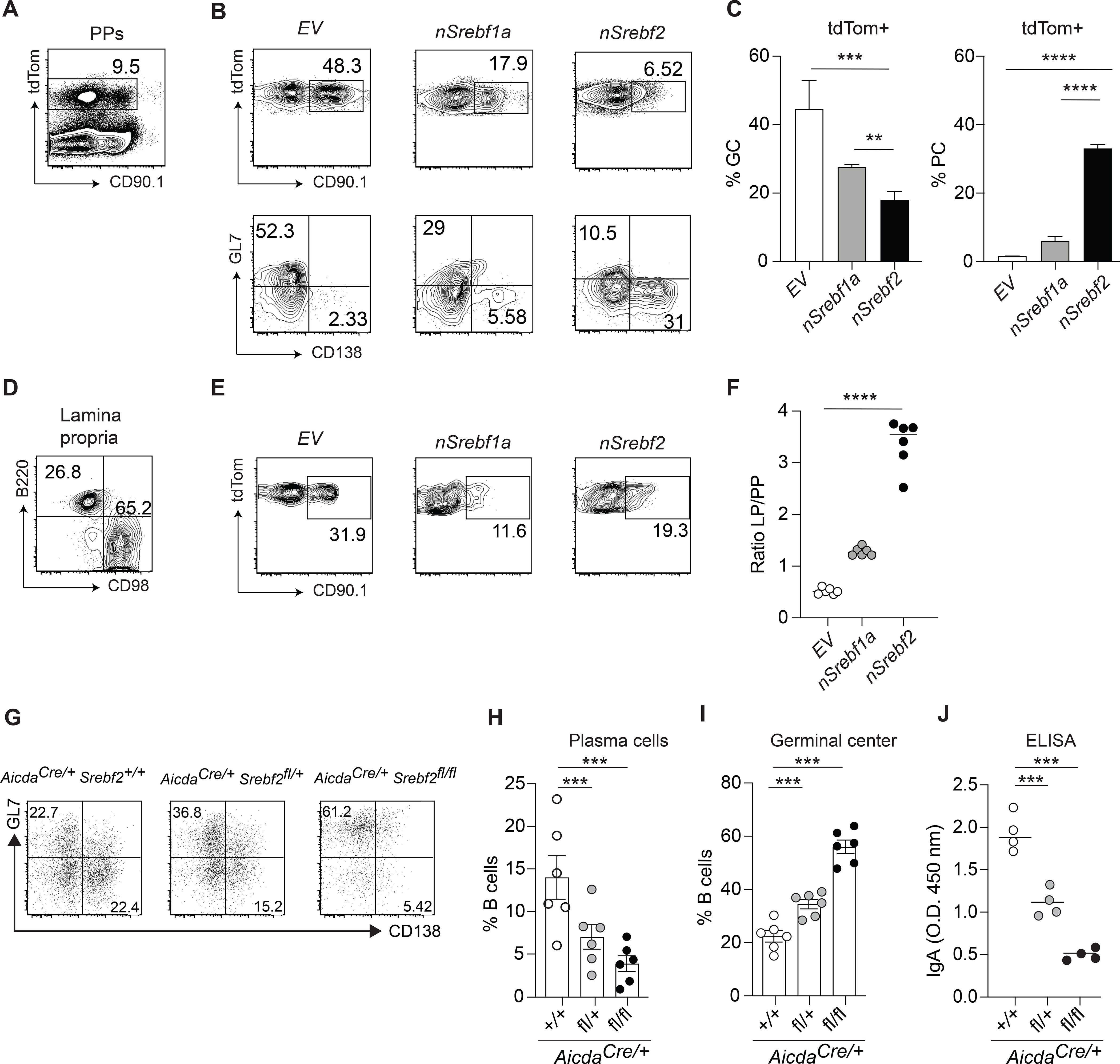
(A) Representative plot of AicdaCre/+Rosa26 tdTom/+ BM transduced with CD90.1 retroviral vector in Peyer’s patches.
(B) Representative plot of frequency and fate of tdTom+ B cells transduced with indicated vectors. (C) Summary of the data in (B). (D) Representative gating of tdTom+ PCs in lamina propria. (E) Representative plot of frequency and fate of tdTom+ B cells transduced with indicated vectors. (F) Ratio of transduced B cells in lamina propria and Peyer’s patches from data in (B) and (E). (G) Representative flow cytometry of GC B cells from mice of the indicated genotype cultured with NB21 cells for 2.5 days. (H,I) Frequency of PCs and GC B cell from G. (J) Quantification of secreted total IgA in A by ELISA. Data represents 3 independent experiments. Graphs show mean and SEM.**p<0.01,***p<0.005, ****p<0.001(two-way ANOVA).
SREBP2 is required for the generation of PC in vitro and in vivo
To assess whether SREBP2 is necessary for PC differentiation, we generated Aicdacre Srebf2 flox/flox mice. In these animals Srebf2 was virtually undetectable in GC B cells and SREBP2-target genes were also transcribed at a lower level compared to littermate controls (Fig.S6B). We sorted GC B cells from Aicdacre Srebf2 flox/flox, Aicdacre Srebf2 flox/+ and Aicdacre Srebf2+/+ and assessed their potential for PC differentiation by co-culturing them with NB21 cells: Srebf2-deficient GC B cells were largely unable to differentiate into PCs and maintained their GC B cell identity (Fig.5G,H,I). In line with previous reports indicating that Srebf2 gene dosage can affect metabolic gene levels (Rong et al., 2017) we observed an intermediate PC output from GC B cells from Aicdacre Srebf2 flox/+ mice compared to Aicdacre Srebf2 flox/flox and Aicdacre Srebf2+/+ GC B cells (Fig.5G,H,I). Overall cellular recovery was reduced in cultures containing Aicdacre Srebf2 flox/flox GC B cells suggesting that SREBP2 deficiency is not conducive for transition to PCs and GC B cell survival (Fig.S6C). IgA secretion was also reduced from Aicdacre Srebf2 flox/flox GC B cell cultures (Fig.5J). Since IgA class switch was unaffected in GC B cells lacking SREBP2 (Fig.S6D), this result also indicates that SREBP2 is required for efficient GC B cell differentiation into PCs. While Aicdacre Srebf2 flox/flox mice did not show altered overall IgA-ASC in LP (Fig.S6E) or increased GC B cell numbers in Peyer’s patches (Fig.S6F) in steady state, oral immunization with cholera toxin led to a drastic decrease of antigen-specific-IgA ASC and titer (Fig.6A,B).
Figure 6. SREBP2 controls germinal center differentiation potential in vitro and in vivo.
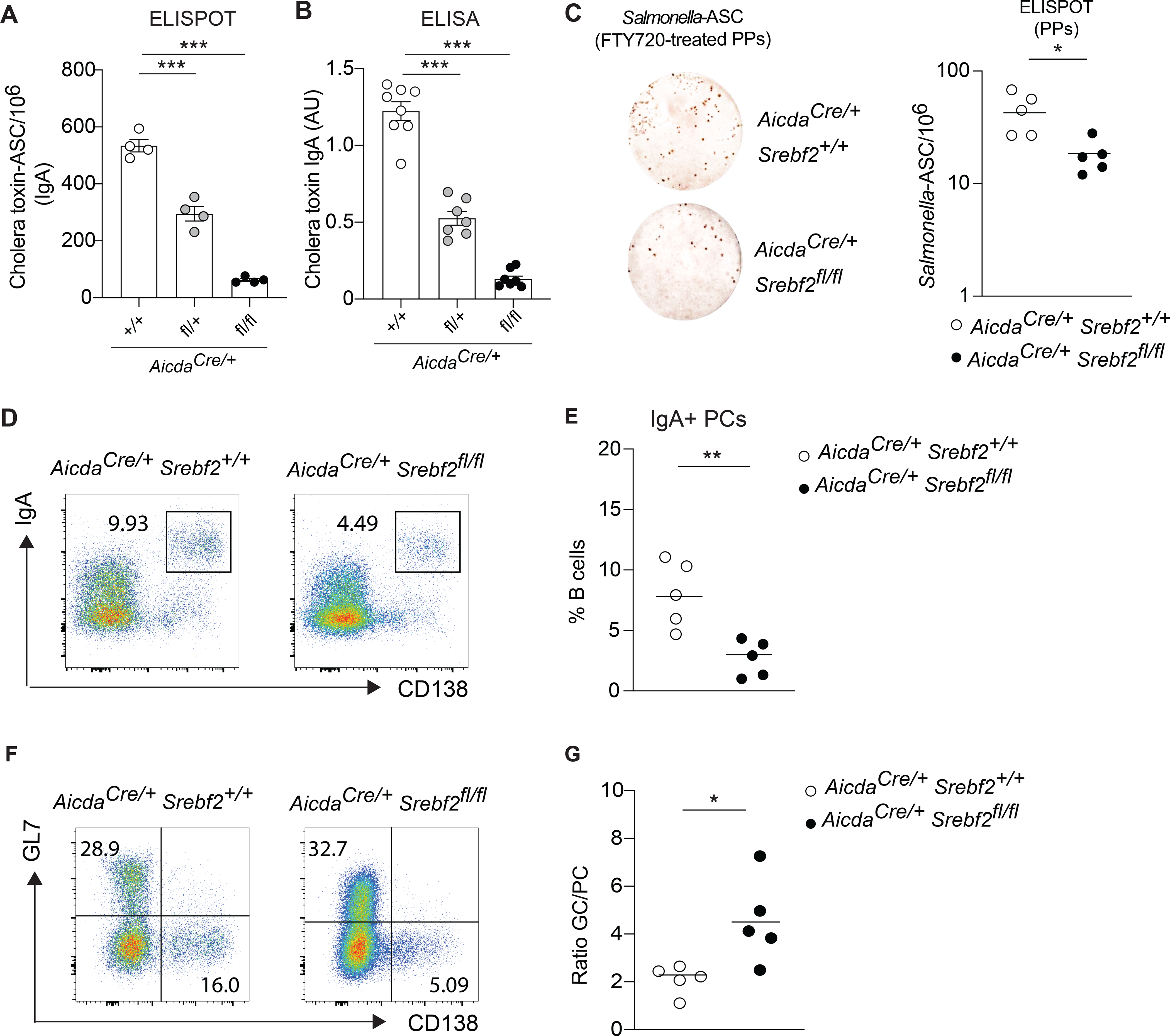
(A) Number of cholera toxin-IgA ASC cells in lamina propria of AicdaCre/+Srebf2 flox/flox and littermate control mice, quantified by ELISPOT. (B) Quantification of cholera toxin-IgA in small intestine lavage of mice treated 3 weeks with cholera toxin. (C) Representative ELISPOT and compiled data showing Salmonella-specific IgA secreting cells in Peyer’s patches of AicdaCre/+Srebf2 +/+ and AicdaCre/+Srebf2 flox/flox mice infected with Salmonella and treated with FTY720. (D) Representative flow cytometry and (E) compiled frequency of IgA+ PCs in Peyer’s patches of AicdaCre/+Srebf2 +/+ and AicdaCre/+Srebf2 flox/flox mice infected with Salmonella and treated with FTY720 as in (C). (F) Representative flow cytometry of GC B cell and PCs from Peyer’s patches of AicdaCre/+Srebf2 +/+ and AicdaCre/+Srebf2 flox/flox mice infected with Salmonella and treated with FTY720 as in (C, D). (G) Ratio of GC B cell and PCs in Peyer’s patches from (F). Each symbol represents one mouse from 3–4 independent experiments. *p<0.05, **p<0.01,***p<0.005 (two-way ANOVA).
Finally, to assess the role of SREBP2 in GCs during intestinal response, we infected Aicdacre Srebf2 +/+ and Aicdacre Srebf2 flox/flox mice with ΔAroA Salmonella and administered FTY720. We observed a decreased number of Salmonella-IgA-ASC in both Peyer’s patches and mesenteric lymph nodes in the absence of SREBP2 in GC B cells (Fig.6C, S7A) and FACS analysis revealed a clear reduction in IgA+ PCs (Fig.6D,E S7B,C). In line with in vitro experiments described above, removal of Srebf2 in GC B cells interfered with normal B cell output, leading to an overrepresentation of GC B cells at the expense of PCs (Fig.6F,G; S7D,E). Reduced generation of PCs in Aicdacre Srebf2 flox/flox mice was linked to the inability of GC B cells to sense cholesterol-derived metabolite, as HCF treatment decreased PC output in AicdacreSrebf2 +/+, but had no effect on AicdacreSrebf2 flox/flox mice in both Peyer’s patches (Fig.S7F) and mesenteric lymph node (Fig.S7G). Taken together, our findings reveal a role for the sterol sensor SREBP2 in controlling B cell fate during intestinal immune response.
25-HC and dietary cholesterol control susceptibility to intestinal pathogen
To test whether 25-HC-dependent control of IgA PC differentiation could impact protection during enteric infection, we initially infected Ch25h−/− mice and littermate controls with ΔAroA Salmonella. 14 days after infection, mice were orally gavaged with invasive WT Salmonella and pathogen dissemination was assessed 48 hours later (Fig.7A). Mice lacking Ch25h showed reduced Salmonella colony-forming units (CFU) in mesenteric lymph nodes and spleen compared to littermate controls, while Salmonella was present at high concentration in the fecal lavage (Fig.7B), suggesting that Ch25h−/− mice mounted a more efficient mucosal response to restrain Salmonella entry from the lumen. In line with this notion, Salmonella-specific IgA amounts, measured by flow cytometry (Moor et al., 2016) (Fig.7C), and by ELISA (Fig.7D), were higher in intestinal lavage of Ch25h−/− mice compared to controls, highlighting how 25-HC restrains IgA response and impacts immune exclusion mechanisms at the mucosal interface (Moor et al., 2017).
Figure 7. Susceptibility to Salmonella systemic dissemination is shaped by 25-HC and dietary cholesterol.
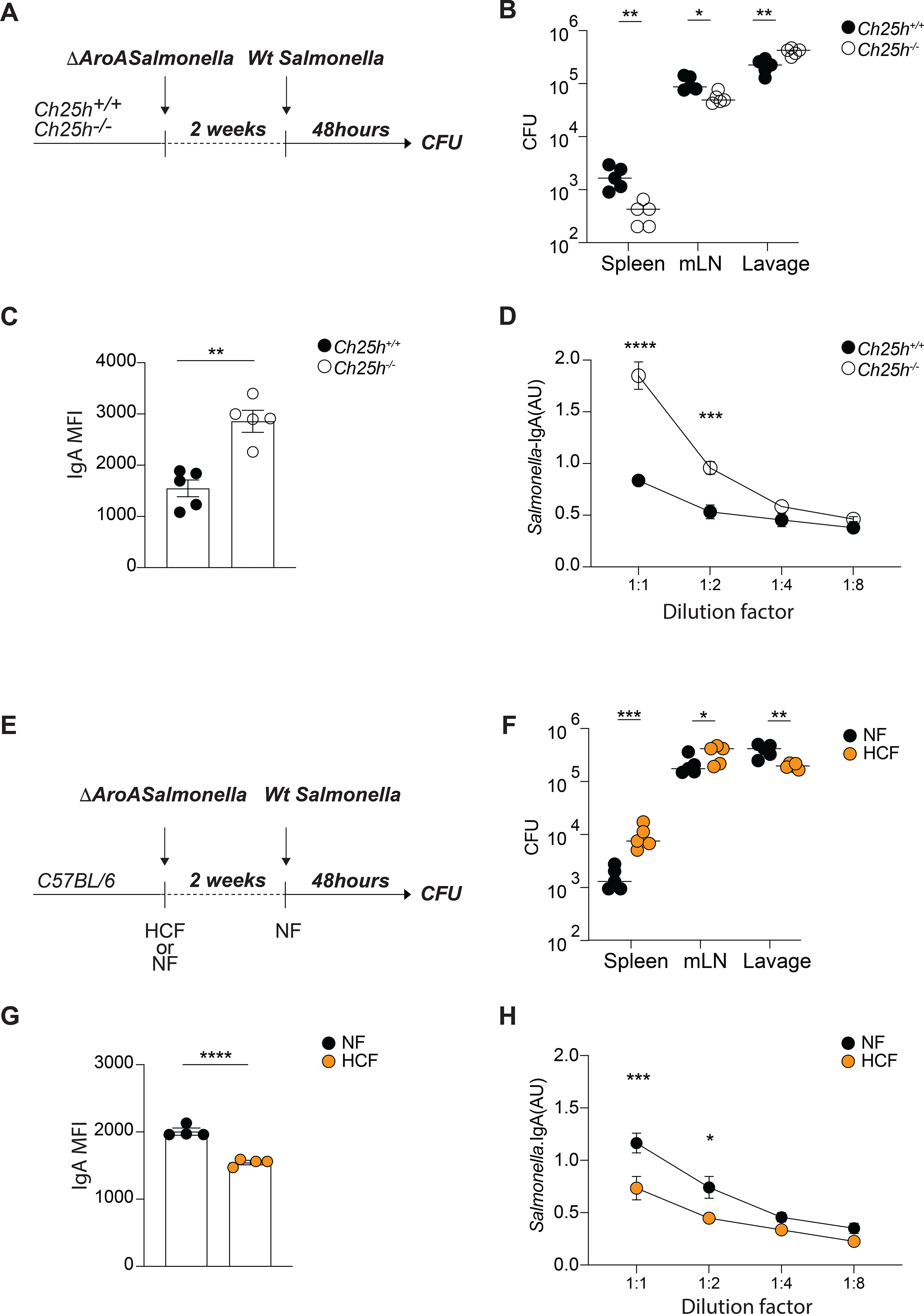
(A) Experimental setup of ΔAroA Salmonella and Wt Salmonella sequential infection using Ch25h+/+ and Ch25h−/− mice. (B) Salmonella CFU quantification in spleen, mesenteric lymph nodes and intestinal lavage from mice infected as in (A). (C) Analysis of mean fluorescence intensity (MFI) of IgA binding to Salmonella by flow cytometry in intestinal lavages of Ch25h+/+ and Ch25h−/− mice infected as in (A). (D) Salmonella-specific IgA titer measured by ELISA in intestinal lavages of Ch25h+/+ and Ch25h−/− mice infected as in (A). (E) Experimental setup of ΔAroA Salmonella and Wt Salmonella sequential infection in C56Bl6 mice during dietary perturbation. (F) Salmonella CFU quantification in spleen, mesenteric lymph nodes and intestinal lavage from mice infected as in (E). (G) Analysis of mean fluorescence intensity (MFI) of IgA binding to Salmonella by flow cytometry in intestinal lavages from mice infected as in (E). (H) Salmonella-specific IgA titer measured by ELISA in intestinal lavages of mice infected as in (E). Each dot represents an individual mouse from two independent experiments. Data are presented as mean + SEM and statistical differences were measured as multiple t-test in (B,C,F,G) or one-way ANOVA in (D and H) with *p<0.05, **p<0.01,***p<0.005, ****p<0.001.
To test whether dietary cholesterol could shape susceptibility to enteric infection, WT mice were either fed NF or HCF during primary infection with ΔAroA Salmonella, and subsequently kept on, or switched to, NF during infection with invasive WT Salmonella (Fig.7E). High dietary cholesterol contents permitted increased Salmonella entry and dissemination, as seen by CFU measured in lumen, mesenteric lymph nodes and spleen (Fig.7F). Accordingly, Salmonella specific-IgA response was blunted by high cholesterol diet (Fig.7G,H). This finding suggests that inhibition of pathogen-specific B cell responses by cholesterol metabolites contributes to the increased risk of infection associated with obesity (Falagas and Kompoti, 2006).
Discussion
We found that oxysterol 25-hydroxycholesterol (25-HC) acts on GC B cells in Peyer’s patches to restrain PC differentiation by inhibiting SREBP2 activation. Our in vitro experiments suggest that 25-HC acts directly on GC B cells to undergo PC differentiation, supporting an effect of 25-HC on adaptive immune cells independent of its role in the generation of 7α,25-dihydroxycholesterol (7α,25-HC), the ligand for EBI2. In the absence of 25-HC-producing enzyme CH25H, the antigen-specific IgA PC response is enhanced during oral immunization and enteric infections. CH25H was required in Peyer’s patches to control PC output during T cell-dependent responses, without any measurable effect on IgA CSR. Moreover, increasing cholesterol in the diet was sufficient to modulate 25-HC tissue concentration in Peyer’s patches and to reduce antigen-specific IgA response.
Our findings identify FDCs as the dominant cell type required to establish the 25-HC niche in the GCs. FDCs have a well-established role in maintaining the anatomical organization of follicular and GC response by enzymatic degradation of metabolites acting on GPCR as described for the EBI2 ligand 7α,25-HC (Yi et al., 2012) and the P2YR8 ligand S-geranylgeranyl-L-glutathione (Lu et al., 2019; Muppidi et al., 2015). Our data show for the first time that an FDC-derived metabolite can be locally maintained and act directly on GC B cells. Other cells have been reported to express Ch25h, but anatomy places FDCs at a prime location to directly control GC B cells via 25-HC in an EBI2 independent fashion, without requiring long range tissue diffusion. Our results do not exclude the possibility that in conditions of increased concentrations of tissue cholesterol, cells other than FDCs could produce 25-HC outside of the GC, and therefore contribute to PC inhibition at different stages of B cell activation.
25-HC’s ability to block activation, and subsequent transcriptional activity of SREBP2, an ER protein that senses intracellular sterol concentration, is well documented, but its regulation remains largely unexplored in adaptive immune cells. B cells activated in vitro by IgM BCR stimulation induce SREBP2. Similarly, LZ GC B cells, which continuously test their BCRs against Ag displayed by FDCs, are also characterized by increased SREBP2 induction and transcriptional activity. We suggest a model in which SREBP2 activation is tuned by 25-HC in the LZ to reduce PC differentiation. Alterations of SREBP2 levels sharply affect GC B cell fate, suggesting that environmental cues able to control SREBP2 activation might play a role in GC output in addition to the widely studied BCR affinity and T cell help. Although our over-expression results suggested a striking effect for SREBP2 activation in PC differentiation, the in vivo phenotype in mice lacking the SREBP2 inhibitor 25-HC was only evident when antigen-specific response was tracked. While our data suggest a dominant role for CH25H in mediating dietary cholesterol inhibition of PC response, we cannot exclude the possibility that other oxysterols or cholesterol itself could play a compensatory role in the absence of 25-HC. In line with the negative feedback exerted by high concentrations of cholesterol and selected oxysterols on cholesterol metabolism via SREBP2 inhibition, CH25H-independent PC inhibition could take place during prolonged cholesterologenic diet. Moreover, other external cues such as those derived from commensals, could modulate the strictness of 25-HC/SREBP2 crosstalk in GC B cells. The original report describing Ch25h−/− mice reports increased IgA production (Gold et al., 2014), which is attributed to aberrant CSR based on in vitro experiments. We did not observe 25-HC inhibition of IgA CSR in Peyer’s patches or mesenteric lymph nodes in our mouse colony. However, the initial IgA phenotype in Ch25h−/− mice (Gold et al., 2014) could be easily reconciled with our data by invoking an overt SREBP2 activation and increase in PC differentiation mediated by a stronger T cell dependent commensal response in their colony.
SREBP2 has a well-established role in controlling cholesterol metabolism in non-hematopoietic cells: our findings here establish a critical role for SREBP2 in controlling GC B cell fate. While in vitro differentiation into PCs was inhibited in absence of SREBP2, the nature of the genetic model we used prevented us from uncovering the intrinsic role of SREBP2 in PC survival. Our in vitro data suggest that BCR stimulation is the dominant signal for SREBP2 expression and activation, and it is likely to shape GC B cell fate in the LZ; however, we cannot rule out a role for T cells in maintaining SREBP2 activation and promoting differentiation in PCs. The SREBP2 signature can be clearly observed in LZ GCs of mouse (Radtke and Bannard, 2019) and human (Holmes et al., 2020), suggesting an evolutionary conserved mechanism to turn on cholesterologenic genes upon selection in the LZ.
We propose a model centered on the ability of FDCs to convert dietary cholesterol into 25-HC and to reduce antigen-specific PC responses in Peyer’s patches. Evolutionary, dietary cholesterol metabolites- GC B cell axis might have evolved to allow dynamic B cell responses adaptation to the rapidly changing intestinal environment. Intestinal cholesterol absorption is dependent on bile acid. Gut commensals have profound effects on bile acid: selected members of the microbiome are able to degrade bile acid (Buffie et al., 2014) and germ-free mice show increased bile acid concentration in the small intestine (Sayin et al., 2013). Thus, we posit that early dysbiosis will result in decreased dietary sterol absorption. As consequence, decreased 25-HC levels will be sensed by GC B cells and will induce a shift in B cell differentiation controlled by SREBP2 transcriptional activity. Since multiple antigens simultaneously drive chronic intestinal GCs, a metabolite-based mechanism that shapes GC B cell fate would provide a rapid mode of antibody regulation in response to fluctuations in the luminal commensal composition.
In addition to microbial alterations, metabolic requirements for controlling PC differentiation could be altered by changes in cholesterol absorption driven by diet. Thus, our findings may be of broader relevance to understand humoral response in human populations exposed to high cholesterol diet. Obesity, which is associated with high plasma cholesterol, is rapidly increasing worldwide and it has profound effects on the immune homeostasis, especially in the gut. Enhanced proinflammatory T cell responses (Garidou et al., 2015; Luck et al., 2015), changes in microbiome (Cani et al., 2008; Ding et al., 2010) and altered IgA responses (Luck et al., 2019; Muhomah et al., 2019) are reported in animal models of obesity. While the exact mechanisms underlying immune modulation associated with diet variations remain largely unknown due to technical limitations, we speculate that 25-HC/SREBP2 axis contributes to altered B cells response observed in animal and human exposed to high fat diet. Long term cholesterol uptake will lead to blunted IgA plasma cell response in the gut, possibly shifting IgA coating of commensals and microbiome composition. Future studies need to assess the role of oxysterol-sensing proteins in other intestinal immune cells (dendritic cells, macrophages, T cells) and non-immune cells (epithelial, endothelial, neuronal) using cell type-specific genetic modifications to map with increased granularity the dietary cholesterol-centered responses at the mucosal interface. Moreover, specific diet formulations administered at different time points during development will be essential to dissect the relative contribution of cholesterol- and non-cholesterol-derived lipids in shaping distinct stages of intestinal immune reactions.
Limitation of the study
In this study we found that 25-HC production in Peyer’s patches is controlled by FDCs via dietary cholesterol and that 25-HC suppression of SREBP2 in GC B cells controls plasma cell differentiation.
However, because additional cells, including other stromal cell subsets, express Ch25h, we could not exclude the possibility that these cells might also produce 25-HC and contribute to SREBP2 restriction in germinal center B cells during antigen-specific IgA response. In addition, intestinal immune response is very sensitive to commensal composition and our data show that 25-HC production can be tuned by antibiotic treatment. Thus, it is possible that the antigen-specific IgA response might be amplified or reduced according to microbiome niche present in animal facilities.
Finally, mice consume most of their food in frequent, small meals during the night while human consume their food in three-four meals during the day. Therefore, such different feeding behavior might impact oxysterol control of B cell response in Peyer’s patches in human and mice.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Lead contact Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrea Reboldi (andrea.reboldi@umassmed.edu).
Materials Availability
This study did not generate new unique reagents.
Data and code availability
Data and code availability This study did not generate any unique datasets or code.
EXPERIMENTAL MODELS
Mice
C57BL/6J (CD45.2) (Stock No: 000664), Ly5.2 (CD45.1) congenic B6.SJL-Ptprca Pepcb/BoyJ (Stock No 002014), Rosa26-flox-stop-flox-DTR (Stock No: 007900), Srebf2 flox/flox (Stock No: 031792), Ch25h−/− (Stock No: 016263), Gpr183−/− (Pereira et al., 2009) Aicda-cre ( Stock No: 007770), Cd21-Cre (Stock No: 006368), Rosa26-flox-stop-flox-tdTomato (Stock No: 007914) were purchased from the Jackson Laboratory. All mice were bred and maintained under standard 12:12 hours light/dark conditions and housed in specific pathogen-free (SPF) conditions at the University of Massachusetts Medical School. Female and male mice were analyzed at 8–12 weeks of age and littermates of the same sex were cohoused and randomly assigned to assigned groups. Mice were either fed a standard chow diet (ISO-PRO 3000 sterile rodent diet #5P76 (LabDiet)) for the duration of the experiment or a high cholesterol diet where 0.15% cholesterol is added to the ProLab RMH 3000 5P76 diet. (0.15% HCD Envigo TD.180381customized diet). All procedures were conformed to ethical principles and guidelines approved by the UMMS Institutional Animal Care and Use Committee.
Cell culture
Freshly isolated germinal center B cells from Peyer’s patches and mesenteric lymph nodes were FACS sorted and cocultured with NB-21 feeder cells as previously described and with adjustments (Kuraoka et al., 2016; Stewart 2018). Briefly, 103 NB-21.2D9 cells/well in 100 uL of B cell media (RPMI supplemented with 10% heat-inactivated FBS, 10 mM HEPES, 1mM sodium pyruvate, 100 units/mL penicillin, 100 mg/mL streptomycin, MEM nonessential amino acid and 55 mM 2-Mercaptoethanol) were seeded into 96-well plates and growth at 37°C and 5% of CO2, one day before B cell co-culturing. On the next day, 104 B cells in 100 uL of B cell media was added to each well. 100uL of media was replaced on day 2. Cholesterol, 7α,25-HC or 25-HC were added to the B cell/NB-21 co-culture at days 0 and 2. Culture plates were centrifuged at day 3.5 and supernatant was collected for IgA detection.
PlatE cells were cultured in DMEM supplemented with 10% FCS and 1% of Penicillin-streptomycin at 37°C and use as system for retroviral packaging. PLAT-E cells were transfected with murine stem cell virus (MSCV) retroviral constructs encoding nuclear portion of murine Srebf2 or Srebf1a with Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. For transduction of BM-derived cells, BM cells from AicdaCre/+ Rosa26 lox-stop-lox-tdTom were harvested 4 days after 5-flurouracil (Sigma) i.p. injection and cultured in the presence of recombinant IL-3, IL-6, and mouse stem cell factor (SCF) (100 ng/ml, Peprotech). BM cells were spin-infected twice with a retroviral construct expressing nuclear portion of murine Srebf2 or Srebf1a and an internal ribosomal entry site (IRES)–Thy1.1 cassette as a reporter. One day after the last spin infection, the cells were injected into lethally irradiated C57BL/6 recipients.
EBI2-transduced M12 B cell line and Mock control were grown to confluency in T75 flasks in RPMI containing 5% FBS and 1% of Penicillin-Streptomycin, washed three times in migration media (RPMI + 0.5% fatty acid free BSA), and desensitized for 20 min at 37°C in migration media prior to use for chemotaxis assays.
HEK293T cell line was cultured in 24 well tissue culture plates in DMEM containing with 10% FCS and 1% of Penicillin-streptomycin and 10mM of HEPES, at 37°C. After reaching approximately 90% of confluence, cells were transfected with pENTR-Hsd3b7 or pENTR-Cyp7b1 vectors. 16–18 hours after transfection, medium was replaced with migration media containing 10% of tissue lipid extracts and incubated for 24 hours before the chemotaxis assay.
Bacterial strains
The following S. typhimurium strains on SL1344: metabolically defective (aroA) S. typhimurium strain and wildtype (Wt) invasive S. typhimurium were provided by Milena Bogunovic lab (UMMS) and grown at 37°C in Luria broth supplemented with appropriate antibiotics to preserve mutation and plasmid.
METHOD DETAILS
In vivo treatment, Immunization and Infection Models
For cholera toxin responses, mice were immunized with 10 ug of cholera toxin (List Biological Laboratories) in PBS by oral gavage. Animals received cholera toxin every 7 days for three consecutive weeks. Mice were analyzed 7 days after last immunization. For Salmonella infection mice were orally gavage three times, unless differently specified in the figure legend, on alternate days with 109 CFUs of aroA-Salmonella in 200 ul 5% sodium bicarbonate. Serial dilutions of bacterial preparations were plated onto LB-agar plates to confirm administered dose. 109 CFU (Wt) Salmonella were injected once orally, and mice were harvested 48 hours later. To prevent lymphocyte egress from Peyer’s patches, the S1PR1 agonist FTY-720 (Fingolimod-HCl., Selleck Chemicals ) was dissolved in saline solution and administered to mice daily i.p. at 1 mg/kg for 3–7 days. For antibiotics treatment mice were gavage with 200ml of antibiotic cocktail containing metronidazole 1mg/ml, ampicillin 0.5 mg/ml, neomycin 0.5 mg/ml, vancomycin 0.5 mg/ml or single antibiotic at the same concentration per day for 1 week. For LTbR-blocking experiments, animals were treated with 100 μg of LTβR-Fc or hIgG-FC on days −3 and −1 before sacrifice. For Diphtheria toxin treatment mice received 100 ng of DT (EMD Bioscience) i.p 18 h before sacrifice.
Bone marrow chimeras
Cd21cre/Rosa26DTR or control mice were lethally irradiated twice with 550 rads gamma-irradiation, 3 hours apart, then intravenously injected with 1–3 × 106 bone marrow cells. Bone marrow was harvested by flushing both tibia and femurs of donor mice. Mice were analyzed 8–10 weeks after cell transfer and 18h after diphtheria toxin treatment.
Immunofluorescence and Immunohistochemistry
Tissues were frozen in OCT. Cryosections of 7μm thickness were dried for 1 hour at RT, fixed in 4°C acetone for 10 min, and dried for 1 hour at RT or overnight at −20°C. For FDC and GC quantification slides were stained ON at 4°C with the following primary antibodies: Goat purified anti-mouse IgD (), biotin-conjugated anti mouse CD35 and AF647-GL7 followed by Cy3 anti-goat, AF488 streptavidin and 4′,6-diamidino-2-phenylindole (DAPI). For immunohistochemistry, acetone fixed cryosections were stained with the following antibodies: goat purified anti-mouse IgD and biotin anti mouse GL7. Then sections were stained with the secondary antibodies anti-goat HRP and streptavidin Alkaline phosphatase (AP). The total FDC area was calculated as ratio between CD35+ area and total PP area.
Isolation of lymphocytes
Peyer’s patches and mesenteric lymph nodes were digested in digestion media (RPMI, 5% heat inactivated FBS, 10 mM HEPES, 1% P/S, 50 ug/mL DNase I (Sigma DN25), and 0.5 mg/mL Collagenase IV (Worthington Biochemical LS004189)) rotating at 37°C for 15 min. The digested tissue was then smashed through 70μm cell strainer. Spleens were harvested and smashed through 70μm cell strainer. Isolated cells were then washed with FACS buffer (1X DPBS, 2% heat inactivated FBS, 2mM EDTA) and counted for further analysis. For isolation of lamina propria (LP) lymphocytes, small intestine was dissected and flushed with cold PBS and Peyer’s Patches were removed. The small intestine was divided into 3 equal parts, the proximal (duodenum) and distal (ileum) were opened longitudinally and vortexed in a 50ml conical tube containing HBSS supplemented with 5% heat-inactivated FBS and 10mM Hepes. Epithelial cells were removed by rotating the small intestine tissue in pre-digestion media (RPMI medium, 5% heat-inactivated FBS, 10mM Hepes, 10mM EDTA) for 30 minutes at 37°C. The intestinal pieces were then washed with complete media (10% heat inactivated FBS, 10 mM HEPES, 1% P/S), chopped with scissors, and digested at 37°C for 30 minutes in digestion media. Digested tissue was passed through 70 um cell strainer and isolated cells were resuspended in 40% Percoll-RPMI and layered with 80% Percoll-RPMI and subsequently centrifuged for 20min at 2200 rpm without break. The isolated LP cells were enumerated on a BD LSRII using AccuCheck Counting Beads (Invitrogen) as per manufacturer recommendations.
Flow cytometry and cell sorting
Mesenteric lymph node, Peyer’s patches, and spleen were collected as described above. Cell suspensions were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen) or Fixable Viability Dye eFluor780 (Invitrogen 65086514) in FACS buffer, and Fc receptors blocked with anti-mouse CD16/32 (2.4G2). BD Cytofix/Cytoperm kit (BD BDB554714) was used for fixation and intracellular staining. Cells were incubated for 20 min on ice with antibodies to B220, IgA, IgD, IgM, IgG1, IgG2b, IgG3, GL7, CD138, CD38, CD45.1, CD90.2, CXCR4 and CD86 (the antibodies used are described in table S1). Data were collected on a BD LSR II and analyzed in FlowJo v10.7 software. For cell sorting, cells were stained as described above and sorted on a BD FACSAria II with a 70- or 84-micron nozzle. Cells were maintained at 4°C until sorting. Sorted plasma cells from PPs and mLN were used for either ELISPOT assay or analysis. Germinal center, Follicular B cells, Memory and plasma cells were sorted as live IgD− GL7+ CD38− CD138−, live IgD+ GL7− CD138−, live IgD− GL7+ CD38+ CD138−, IgD− GL7− CD38− CD138+ respectively into TRIZOL (Invitrogen) for RNA extraction or into tissue culture plates containing complete media for further analysis. Light zone and dark zone germinal center B cells were sorted based on expression of CXCR4 and CD86.
RNA extraction and Real Time PCR
Total RNA was isolated with 0.5 mL of TRIZOL reagent (Invitrogen) following the manufacturer’s protocol. Reverse transcription was performed using Superscript III reagent kit from Invitrogen. All samples were checked for quality (A260/A280 ratio of 1.8–2.0). Real Time PCR was performed by using iQ SYBR Green superMix (BioRad). Gene expression levels were determined using a comparative method (DCq) normalizing the target mRNA to bactin as endogenous internal control. Forward and reverse primers sequences are listed in table S1.
Nuclei Preparation
Nuclei preparations were carried out as previously described (Gallagher, M. P. Immunohorizons, 2018). Briefly, follicular B cells were isolated from spleens of wild type or Cd21creSrebf2fl/fl mice using EasySep mouse B cell isolation kit and protocol (StemCell). Isolated cells were then cultured in vitro in the presence or absence of 5 mg/ml anti-IgM F(ab)2 (Jackson Immunoresearch) and/or 5 mg/ml anti-CD40 (BioXCell) for 16 or 48 hours. Inhibition experiments were carried out by culturing isolated follicular B cells with 5 ug/ml anti-IgM in the presence or absences of 10 nM Ibrutinib, 1 mM R406 (Invitrogen), 1 mM API1(Sigma), 50 nM Rapamycin (Selleck Chemical) or 1 mM Mevastatin (Selleck Chemical) for 16 hours. Following the culture, the cells were harvested and lysed in Sucrose Buffer A (10 mM HEPES (Gibco), 8 mM MgCl2, 320 mM Sucrose, 0.1 % (v/v) Triton-X 100 (Sigma), and 1X complete, EDTA-free Protease Inhibitor Cocktail (Roche) on ice for 15 minutes. The cells were then centrifuged at 2000g for 10 minutes and washed twice with Sucrose Buffer B (Sucrose Buffer A without Triton-X 100). Nuclei were fixed in 4% Paraformaldehyde (electron microscopy grade, Electron Microscopy Sciences), diluted in Sucrose Buffer B, at room temperature for 25 minutes. Nuclei were stained one ice for 30 minutes with primary purified SREBP2 antibody and directly conjugated IRF4 antibody, washed twice, and then stained with secondary anti-rabbit antibody on ice for 30 minutes, before flow cytometry analysis.
Analysis of specific IgA binding to Salmonella
Anti-Salmonella IgA response was analyzed by flow cytometry. Briefly, peracetic acid killed Salmonella was prepared as previously described (Moor et al. 2017). Briefly an overnight culture of GFP+Salmonella was resuspended with 1% of peracetic acid (Sigma) and incubated for 60 min at room temperature. Bacteria were washed once in 40 ml of sterile PBS and subsequently three times in 50 ml sterile 1× PBS. The final pellet was resuspended to yield a density of 1010 particles per ml in sterile PBS (determined by optical density (OD600)) and stored at 4 °C. Fecal lavages of mice previously vaccinated with Salmonella were cleaned from commensal bacteria by centrifugation at 16000g for 5 minutes. Serial dilutions of supernatant were used as source of IgA to stain 105 GFP+ Salmonella and incubated for 1 hour at 4°C. Next, detection of IgA binding Salmonella was performed by staining the bacteria with anti-mouse IgA and fixable dye for 30 minutes at 4°C. Finally, bacteria were fixed in 2% of PFA solution and IgA-specific Salmonella were analyzed by flow cytometry.
Evaluation of Salmonella CFU
MLNs, and Spleen from single mice infected with aroA and Wt Salmonella were harvested after 48hours of Wt Salmonella infection, weighted, and dissolved in sterile PBS at a concentration of 100mg/mL. Serial dilutions of the homogenized tissues were plated on MacConkey agar (Teknova) plates containing 50 μg/ml streptomycin. The next day grown colonies were counted and the total number was calculated as colony forming unit (CFU). Fecal lavage was collected with 3 mL of PBS and plated and counted as above.
Enzyme-linked ImmunoSorbent Assay (ELISA)
Ninety-six–well half area high binding flat bottom plates (Corning) were coated overnight at 4°C with 25ul of 2 μg/ml purified anti-IgA (RMA-1, BD), 2 ug/ml CT (List biological laboratories) or with lysate of 4×106 CFU/mL of Salmonella. Plates were washed and blocked with PBS–5% BSA before diluted intestinal wash or fecal samples were added and threefold serial dilutions were made. Samples were processed as described previously by Reboldi et al. and incubated overnight at 4°C. Bound antibodies were detected by anti-IgA-conjugated horseradish peroxidase (Southern Biotech) and visualized by the addition of Substrate Reagent Pack (Biolegend). Color development was stopped with 3 M H2SO4 stop solution (Biolegend). Purified mouse IgA (Southern Biotech) served as standard. Absorbances at 450 nm were measured on a tunable microplate reader (VersaMax, Molecular Devices) using SoftMax Pro 5 software. OD for antigen-specific IgA response was plotted as arbitrary unit (AU) and normalized to response from a pool (8–10) of immunized C57Bl6.
ELISpot
ELISpot plates (Millipore) were coated with 100μl of 2 μg/ml purified anti-IgA or lysate of heat-killed Salmonella in PBS overnight at 4°C. Plates were washed three times with PBS then blocked for 2 h at 37°C with 10% FCS-RPMI. Cells were isolated from Peyer’s patches, mesenteric lymph nodes and lamina propria and counted as described above then diluted in blocked ELISpot plates and incubated overnight in a 37°C 5% CO2 tissue culture incubator. The next day, plates were washed three times with PBS-0.1% Tween then PBS.
Detection of IgA spots was achieved by using anti-IgA-conjugated horseradish peroxidase and developed with 3-amino-9-ethylcarbazole (Sigma-Aldrich). Color development was stopped by washing several times with water. Once dried, plates were scanned and the spots counted using the CTL ELISPOT reader system (Cellular Technology). Measurement of total IgA and antigen-specific IgA secreting cells was plotted as total counted number or, when indicated, as percentage of cells.
Lipid extraction and 25-HC/7α,25-HC quantification
Lipids from 100 mg of Peyer’s patches and mesenteric lymph nodes were extracted using the Folch method (Folch et al., 1957). Lyophilized lipid was dissolved at 100 mg/ml in ethanol. 25-HC was measured by using HEK293T cells supernatant from cells transfected sequentially with pENTR-Hsd3b7 and pENTR-Cyp7b1 vectors. The 7α,25-HC activity was evaluated by transwell chemotaxis assay. Lipid extracts were diluted in 10 volumes of sterile migration media and tested for EBI2 dependent bioactivity by transwell chemotaxis assays of 50:50 mixed M12 B cell line transduced with an EBI2-IRES-GFP retroviral construct and mock M12 cells. The three-hour migration assay was performed at 37°C and migrated cells were stained for DAPI and analyzed by flow cytometry. The relative migration of EBI2−GFP+ M12 cells over M12 cells was normalized to migration toward lipid free migration media. The purified 7α,25-HC was used as positive control for chemotaxis.
Data analyses
Flow cytometry was performed using a LSRII and sort-purification was performed on a BD FACSAria III and FACSAria Fusion. All data were analyzed using FlowJo 10 (BD). All imagines were captured at the same magnification by using a florescence microscopy Axio Observer.Z1 (Carl Zeiss) and were analyzed by imaging processing software ImageJ (NIH).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism v8.0 using two-way ANOVA with Bonferroni’s multiple comparisons test or unpaired t-test Data are presented as means +/− SEM. Differences between group means were considered significant at indicated p value. The exact significance values are stated in all graphs and the number of biological replicates (n) is stated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD16/32, purified, Clone 93 | Biolegend | Cat#101330;RRID:AB_2783037 |
| Anti-mouse/human B220, BV711, Clone RA3-6B2 | Biolegend | Cat#103225;RRID: AB_492874 |
| Anti-mouse IgD, BV510, Clone 11-26c.2a | Biolegend | Cat#405723;RRID:AB_2562742 |
| Anti-mouse CD138, BV421, Clone 281–2 | Biolegend | Cat#142508;RRID:AB_11204257 |
| Anti-mouse CD38, PE-Cy7, Clone 90 | Biolegend | Cat#102718;RRID: AB_2072892 |
| Anti-mouse/human GL7, APC, Clone GL7 | Biolegend | Cat#144606;RRID:AB_2562184 |
| Anti-mouse/human GL7, PerCPCy5.5, Clone GL7 | Biolegend | Cat#144610;RRID: AB_2562978 |
| Anti-mouse/human GL7, AF488, Clone GL7 | Biolegend | Cat#144612;RRID: AB_2563284 |
| Anti-mouse IgG1, FITC, Clone RMG1-1 | Biolegend | Cat#406606;RRID: AB_493292 |
| Anti-mouse IgG2b, FITC, Clone RMG2b-1 | Biolegend | Cat#406706;RRID: AB_493296 |
| Anti-mouse CD98, AF647, Clone RL388 | Biolegend | Cat#128210;RRID: AB_2254922 |
| Anti-mouse CD90.1, APC, Clone OX-7 | Biolegend | Cat#202526;RRID: AB_1595470 |
| Anti-mouse CD184(CXCR4), PE, Clone 2B11 | BD Biosciences | Cat#551966;RRID: AB_394305 |
| Anti-mouse CD86, BV605, Clone GL-1 | Biolegend | Cat#105037;RRID:AB_11204429 |
| Anti-mouse IgA, FITC, polyclonal | Southern Biotech | Cat#1040-02;RRID:AB_2794370 |
| Anti-mouse IgA, purified, Clone C10-3 | BD Biosciences | Cat#556969;RRID:AB_396541 |
| Mouse IgA, unlabeled, Clone S107 | Southern Biotech | Cat#0106-01;RRID:AB_2714214 |
| Anti-mouse IgA-HRP, polyclonal | Southern Biotech | Cat#1040-05;RRID:AB_2714213 |
| Anti-mouse IgD, purified, polyclonal | Nordic Immunology | Cat#GAM/IgD(Fc)/7S |
| Anti-mouse/human GL7, biotin, Clone GL7 | Biolegend | Cat#144616;RRID: AB_2721505 |
| Anti-mouse CD21/CD35, AF647, Clone 7E9 | Biolegend | Cat#123424;RRID: AB_2629577 |
| Anti-mouse CD35, biotin, Clone 8C12 | BD Biosciences | Cat#553816;RRID:AB_395068 |
| Anti-goat Donkey-HRP, polyclonal | Jackson Immunoresearch | Cat#705-035-147;RRID: AB_2313587 |
| Anti-biotin-AP, 3D6.6 | Jackson Immunoresearch | Cat#200-052-211;RRID: AB_2339015 |
| Anti-goat Cy3, polyclonal | Jackson Immunoresearch | Cat#705-166-147;RRID: AB_2340413 |
| Anti-rabbit AF488, polyclonal | Jackson Immunoresearch | Cat#711-546-152; RRID:AB_2340619 |
| Anti-mouse CXCR5, BV421, Clone L138D7 | Biolegend | Cat#145512;RRID: AB_2562127 |
| Anti-mouse PD-1, PE-Cy7, Clone 29F.1A12 | Biolegend | Cat#135215;RRID:AB_10696422 |
| Anti-mouse CD4, FITC, Clone RM4-4 | Biolegend | Cat#116004;RRID: AB_313688 |
| Anti-mouse TCRbeta, BV605, Clone H57-597 | Biolegend | Cat#109241;RRID: AB_2629563 |
| Anti-mouse/human SREBP2 (a.a. 843–857), purified, polyclonal | Invitrogen | Cat#PA1-338;RRID:AB_2194237 |
| Anti-SREPB2, purified, Clone 22D5 | Millipore Sigma | Cat#MABS-1988 |
| Anti-mouse/human SREBP2 (a.a. 1–220), purified, polyclonal | Invitrogen | Cat#PA5-88943;RRID: AB_2805240 |
| Anti-mouse/human IRF4, EF660, Clone 3E4 | Invitrogen | Cat#50-9858-82;RRID: AB_2574393 |
| Bacterial and virus strains | ||
| S. enterica strain SL1344 | Gift from M. Bogunovic | N/A |
| S. enterica strain SL1344 aroA | Gift from M. Bogunovic | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Anti-IgM | Jackson ImmunoResearch Laboratories Inc. | Cat# 115-006-075 |
| Anti-CD40 | Bio X Cell | Cat# BE0016-2 |
| DAPI | Thermo Fisher | Cat#1306 |
| ER-Tracker | Invitrogen | Cat# E12353 |
| FTY-720 | Selleck Chemicals | Cat# S5002 |
| Cholera toxin | List Biological Laboratories | Cat#100B |
| Diphtheria toxin | Sigma | Cat#D0564 |
| 25-hydroxycholesterol | Avanti Polar Lipids | Cat# 700019P |
| 7a,25-dihydroxycholesterol | Avanti Polar Lipids | Cat# 700080P |
| Ampicillin | Corning | Cat# 61-238-RH |
| Metronidazole | MP Biomedicals | Cat#155710 |
| Neomycin | SIGMA | Cat#N1876 |
| Vancomycin | Fisher Bioreagents | Cat#BP2958-1 |
| Ibrutinib | Selleckchem | Cat# S2680 |
| R406 | Invivogen | Cat# inhr406 |
| API1 | Sigma | Cat# SML1342 |
| Rapamycin | LC Laboratories | Cat#R-5000 |
| Mevastatin | Cayman Chemical | Cat# 73573-88-3 |
| LTbR-hIgG-Fc | This paper | N/A |
| hIgG-Fc | This paper | N/A |
| 5-Fluorouracil | Sigma | Cat# F6627 |
| Collagenase IV | Worthington | Cat# LS004189 |
| DNase I | Sigma | Cat# DN25 |
| Percoll | Ge Healthcare | Cat# 17089101 |
| Trizol | Invitrogen | Cat#10296028 |
| Critical commercial assays | ||
| BD Cytofix/Cytoperm solution | BD Biosciences | Cat# 554714 |
| SYBR green master mix | Biorad | Cat# 1725270 |
| Script First-Strand kit | Invitrogen | Cat# 11904-018 |
| Elisa/Elispot diluent solution | Invitrogen | Cat#004202 |
| Fluoromount-G | SouthernBiotech | Cat#0100-01 |
| EasySep mouse B cell isolation kit | Stemcell | Cat#19854A |
| Experimental models: Cell lines | ||
| Human Embryonic Kidney Cells (HEK293T) | ATCC | Cat# CRL-3216 |
| M12-EBI2-GFP | Gift from J. Cyster | Kelly et al. 2011 |
| WEHI-231 | ATCC | Cat# CRL-1702 |
| Platinum-E (Plat-E) Retroviral Packaging Cell Line | Gift from S. Schwab | N/A |
| NB-21 | Gift from G. Kelsoe | Kuraoka M.et al.2016 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | 002014 |
| Mouse: Rosa26-flox-stop-flox-DTR | The Jackson Laboratory | 007900 |
| Mouse: Srebf2 flox/flox | The Jackson Laboratory | 031792 |
| Mouse: Ch25h−/− | The Jackson Laboratory | 016263 |
| Mouse: Gpr183−/− | Pereira et al., 2009 | N/A |
| Mouse: Aicda-cre | The Jackson Laboratory | 007770 |
| Mouse: Cd21-Cre | The Jackson Laboratory | 006368 |
| Mouse: Rosa26-flox-stop-flox-tdTomato | The Jackson Laboratory | 007914 |
| Oligonucleotides | ||
| See table S1 for list of quantitative RT-PCR primers | N/A | N/A |
| Recombinant DNA | ||
| MSCV-IRES-Thy1.1 | Addgene | Plasmid ID: 17442 |
| MSCV-Flox-STOP-Flox-IRES-Thy1.1 | Gift from J. Cyster | Green et al. 2011 |
| MSCV-IRES-H2B-RFP | Gift from J. Cyster | N/A |
| Software and algorithms | ||
| FlowJo V 10.6 software | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| FACSDiva V 7.0 software | BD | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| ZEN 3.1 | Carl Zeiss Microscopy | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Adobe Illustrator 2019 | Adobe Systems | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 9 | GraphPad Software | http://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| standard chow diet | LabDiet | Cat#5P76 |
| 0.15% high cholesterol diet | Envigo TD | Cat#180381 |
Highlights.
CH25H deficiency enhances antigen-specific IgA plasma cell differentiation in PPs
25-HC suppresses SREBP2 transcriptional activity in activated B cells
Srebf2−/− germinal center B cells fail to differentiate into IgA plasma cells
Dietary cholesterol increases 25-HC level and reduces IgA against enteric pathogen
ACKNOWLEDGMENTS
We thank Michael Gallagher and Leslie Berg for the help with the nuclear isolation and staining, Grant Weaver and Lawrence Stern for the help with the generation of LTβR-Fc, Garnett Kelsoe for generously allowing use of NB21 cells and Joonsoo Kang for the critical reading of the manuscript. This work was supported by the Kenneth Rainin Foundation, Innovator Award (to A.R.), Charles H. Hood Foundation Child Health Research Awards Program (to A.R.), the NIH training grants T32 AI132152, T32 AI007349 (to F.R.). J.R.M. is supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute. We acknowledge the University of Massachusetts Flow Cytometry Core Facility for access to sorting services.
Footnotes
Declaration of interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baptista AP, Gola A, Huang Y, Milanez-Almeida P, Torabi-Parizi P, Urban JF Jr., Shapiro VS, Gerner MY, Germain RN, 2019. The Chemoattractant Receptor Ebi2 Drives Intranodal Naive CD4+ T Cell Peripheralization to Promote Effective Adaptive Immunity. Immunity 50, 1188–1201.e6. doi: 10.1016/j.immuni.2019.04.001 [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Naik S, 2013. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol 14, 646–653. doi: 10.1038/ni.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, Craigon M, Ruzsics Z, Haas J, Angulo A, Griffiths WJ, Glass CK, Wang Y, Ghazal P, 2012. The Transcription Factor STAT-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity 1–13. doi: 10.1016/j.immuni.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MRM, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG, 2014. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. doi: 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R, 2008. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481. doi: 10.2337/db07-1403 [DOI] [PubMed] [Google Scholar]
- Chu C, Moriyama S, Li Z, Zhou L, Flamar A-L, Klose CSN, Moeller JB, Putzel GG, Withers DR, Sonnenberg GF, Artis D, 2018. Anti-microbial Functions of Group 3 Innate Lymphoid Cells in Gut-Associated Lymphoid Tissues Are Regulated by G-Protein-Coupled Receptor 183. CellReports 23, 3750–3758. doi: 10.1016/j.celrep.2018.05.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Dang EV, Reboldi A, Yi T, 2014. 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol 14, 731–743. doi: 10.1038/nri3755 [DOI] [PubMed] [Google Scholar]
- Dang EV, McDonald JG, Russell DW, Cyster JG, 2017. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 1–27. doi: 10.1016/j.cell.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NMJ, Magness S, Jobin C, Lund PK, 2010. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 5, e12191. doi: 10.1371/journal.pone.0012191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emgård J, Kammoun H, García-Cassani B, Chesné J, Parigi SM, Jacob J-M, Cheng H-W, Evren E, Das S, Czarnewski P, Sleiers N, Melo-Gonzalez F, Kvedaraite E, Svensson M, Scandella E, Hepworth MR, Huber S, Ludewig B, Peduto L, Villablanca EJ, Veiga-Fernandes H, Pereira JP, Flavell RA, Willinger T, 2018. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 48, 120–132.e8. doi: 10.1016/j.immuni.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas ME, Kompoti M, 2006. Obesity and infection. The Lancet Infectious Diseases 6, 438–446. doi: 10.1016/S1473-3099(06)70523-0 [DOI] [PubMed] [Google Scholar]
- Gallagher MP, Conley JM, Berg LJ, 2018. Peptide Antigen Concentration Modulates Digital NFAT1 Activation in Primary Mouse Naive CD8+ T Cells as Measured by Flow Cytometry of Isolated Cell Nuclei. IH 2, 208–215. doi: 10.4049/immunohorizons.1800032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S, Dray C, Iacovoni JS, Courtney M, Collet X, Amar J, Servant F, Lelouvier B, Valet P, Eberl G, Fazilleau N, Douin-Echinard V, Heymes C, Burcelin R, 2015. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORγt and Controls Metabolic Disease. Cell Metabolism 22, 100–112. doi: 10.1016/j.cmet.2015.06.001 [DOI] [PubMed] [Google Scholar]
- Gohda M, Kunisawa J, Miura F, Kagiyama Y, Kurashima Y, Higuchi M, Ishikawa I, Ogahara I, Kiyono H, 2008. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J Immunol 180, 5335–5343. doi: 10.4049/jimmunol.180.8.5335 [DOI] [PubMed] [Google Scholar]
- Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, Aderem A, 2014. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proceedings of the National Academy of Sciences 111, 10666–10671. doi: 10.1073/pnas.1404271111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS, 2006. Protein Sensors for Membrane Sterols. Cell 124, 35–46. doi: 10.1016/j.cell.2005.12.022 [DOI] [PubMed] [Google Scholar]
- Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CDC, Schmidt TH, Xu Y, Proia RL, Coughlin SR, Cyster JG, 2011. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol 12, 672–680. doi: 10.1038/ni.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Reboldi A, 2021. Production and Function of Immunoglobulin A. Annu. Rev. Immunol 39, 695–718. doi: 10.1146/annurev-immunol-102119-074236 [DOI] [PubMed] [Google Scholar]
- Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, Nitzan M, Grunstein E, Pasqualucci L, Dalla-Favera R, Basso K, 2020. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med 217, 259. doi: 10.1084/jem.20200483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Macpherson AJ, 2010. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol 10, 159–169. doi: 10.1038/nri2710 [DOI] [PubMed] [Google Scholar]
- Jung C, Hugot J-P, Barreau F, 2010. Peyer’s Patches: The Immune Sensors of the Intestine. International Journal of Inflammation 2010, 1–12. doi: 10.4061/2010/823710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG, 2011. EBI2 Guides Serial Movements of Activated B Cells and Ligand Activity Is Detectable in Lymphoid and Nonlymphoid Tissues. J Immunol 187, 3026–3032. doi: 10.4049/jimmunol.1101262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, Graeber TG, Reue K, Brooks DG, Bensinger SJ, 2013. Sterol regulatory element–binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14, 489–499. doi: 10.1038/ni.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, Kelsoe G, 2016. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity 1–30. doi: 10.1016/j.immuni.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S-Y, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G, 2012. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity 1–14. doi: 10.1016/j.immuni.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E, Wolfreys FD, Muppidi JR, Xu Y, Cyster JG, 2019. S-Geranylgeranyl-l-glutathione is a ligand for human B cell-confinement receptor P2RY8. Nature 1–22. doi: 10.1038/s41586-019-1003-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, Chakraborty M, Cheng K, Tao Chan Y, Nøhr MK, Clemente-Casares X, Perry M-C, Ghazarian M, Lei H, Lin Y-H, Coburn B, Okrainec A, Jackson T, Poutanen S, Gaisano H, Allard JP, Guttman DS, Conner ME, Winer S, Winer DA, 2019. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nature Communications 10, 3650–17. doi: 10.1038/s41467-019-11370-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, Lei H, Luk CT, Shi SY, Surendra A, Copeland JK, Ahn J, Prescott D, Rasmussen BA, Chng MHY, Engleman EG, Girardin SE, Lam TKT, Croitoru K, Dunn S, Philpott DJ, Guttman DS, Woo M, Winer S, Winer DA, 2015. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metabolism 21, 527–542. doi: 10.1016/j.cmet.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL, 1997. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur. J. Immunol 27, 2033–2042. doi: 10.1002/eji.1830270830 [DOI] [PubMed] [Google Scholar]
- Martinoli C, Chiavelli A, Rescigno M, 2007. Entry Route of Salmonella typhimurium Directs the Type of Induced Immune Response. Immunity 27, 975–984. doi: 10.1016/j.immuni.2007.10.011 [DOI] [PubMed] [Google Scholar]
- Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, Tanes C, Fardus-Reid F, Swann JR, Bittinger K, Mabbott NA, Vallance BA, Willinger T, Withers DR, Hepworth MR, 2019. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J Exp Med 216, 728–742. doi: 10.1084/jem.20180871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Bouley DM, Falkow S, 2004. Salmonella typhimurium Persists within Macrophages in the Mesenteric Lymph Nodes of Chronically Infected Nramp1+/+ Mice and Can Be Reactivated by IFNγ Neutralization. Journal of Experimental Medicine 199, 231–241. doi: 10.1084/jem.20031319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor K, Diard M, Sellin ME, Felmy B, Wotzka SY, Toska A, Bakkeren E, Arnoldini M, Bansept F, Co AD, Völler T, Minola A, Fernandez-Rodriguez B, Agatic G, Barbieri S, Piccoli L, Casiraghi C, Corti D, Lanzavecchia A, Regoes RR, Loverdo C, Stocker R, Brumley DR, Hardt W-D, Slack E, 2017. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544, 498–502. doi: 10.1038/nature22058 [DOI] [PubMed] [Google Scholar]
- Moor K, Fadlallah J, Toska A, Sterlin D, Balmer ML, Macpherson AJ, Gorochov G, Larsen M, Slack E, 2016. Analysis of bacterial-surface-specific antibodies in body fluids using bacterial flow cytometry. Nat Protoc 11, 1531–1553. doi: 10.1038/nprot.2016.091 [DOI] [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, Andrian, von, U.H., 2006. Generation of Gut-Homing IgA-Secreting B Cells by Intestinal Dendritic Cells. Science 314, 1157–1160. doi: 10.1126/science.1132742 [DOI] [PubMed] [Google Scholar]
- Muhomah TA, Nishino N, Katsumata E, Haoming W, Tsuruta T, 2019. High-fat diet reduces the level of secretory immunoglobulin A coating of commensal gut microbiota. Biosci Microbiota Food Health 38, 55–64. doi: 10.12938/bmfh.18-027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi JR, Lu E, Cyster JG, 2015. The G protein–coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. Journal of Experimental Medicine 212, 2213–2222. doi: 10.1084/jem.20151250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Scott AL, 2010. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. Journal of Leukocyte Biology 88, 1081–1087. doi: 10.1189/jlb.0610318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JP, Kelly LM, Xu Y, Cyster JG, 2009. EBI2 mediates B cell segregation between the outer and centre follicle. Nature 460, 1122–1126. doi: 10.1038/nature08226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I, Fraussen J, Somers V, Kleinewietfeld M, 2018. Interrelation of Diet, Gut Microbiome, and Autoantibody Production. Front. Immun. 9, 642–9. doi: 10.3389/fimmu.2018.00439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikor NB, Cheng H-W, Onder L, Ludewig B, 2021. Development and Immunological Function of Lymph Node Stromal Cells. J Immunol 206, 257–263. doi: 10.4049/jimmunol.2000914 [DOI] [PubMed] [Google Scholar]
- Radtke D, Bannard O, 2019. Expression of the Plasma Cell Transcriptional Regulator Blimp-1 by Dark Zone Germinal Center B Cells During Periods of Proliferation. Front. Immun 9, 429–16. doi: 10.3389/fimmu.2018.03106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG, 2016. IgA production requires B cell interaction with subepithelial dendritic cells in Peyers patches. Science 352, aaf4822–aaf4822. doi: 10.1126/science.aaf4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG, 2014. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345, 679–684. doi: 10.1126/science.1254790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda LB, Lu E, Bennett ML, Sokol CL, Wang X, Luther SA, Barres Ben A, Luster AD, Ye CJ, Cyster JG, 2018. Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity 48, 1014–1028.e6. doi: 10.1016/j.immuni.2018.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong S, McDonald JG, Engelking LJ, 2017. Cholesterol auxotrophy and intolerance to ezetimibe in mice with SREBP-2 deficiency in the intestine. J. Lipid Res. 58, 1988–1998. doi: 10.1194/jlr.M077610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL, 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046. doi: 10.1016/s0092-8674(00)81304-5 [DOI] [PubMed] [Google Scholar]
- Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, Maeda M, 1996. Sterol-dependent Transcriptional Regulation of Sterol Regulatory Element-binding Protein-2. J. Biol. Chem 271, 26461–26464. doi: 10.1074/jbc.271.43.26461 [DOI] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F, 2013. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metabolism 17, 225–235. doi: 10.1016/j.cmet.2013.01.003 [DOI] [PubMed] [Google Scholar]
- Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, Nutt SL, 2016. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 17, 323–330. doi: 10.1038/ni.3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, Almeida FF, Ibiza S, Barbosa I, Goverse G, Labão-Almeida C, Godinho-Silva C, Konijn T, Schooneman D, O’Toole T, Mizee MR, Habani Y, Haak E, Santori FR, Littman DR, Schulte-Merker S, Dzierzak E, Simas JP, Mebius RE, Veiga-Fernandes H, 2014. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 1–17. doi: 10.1038/nature13158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG, 2011. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. Journal of Experimental Medicine 208, 2497–2510. doi: 10.1084/jem.20111449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson J-Å, Russell DW, Cyster JG, 2012. Oxysterol Gradient Generation by Lymphoid Stromal Cells Guides Activated B Cell Movement during Humoral Responses. Immunity 37, 535–548. doi: 10.1016/j.immuni.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and code availability This study did not generate any unique datasets or code.