

Abstract
Targeting glutamine catabolism has been attracting more research attention on the development of successful cancer therapy. Catalytic enzymes such as glutaminase (GLS) in glutaminolysis, a series of biochemical reactions by which glutamine is converted to glutamate and then alpha-ketoglutarate, an intermediate of the tricarboxylic acid (TCA) cycle, can be targeted by small molecule inhibitors, some of which are undergoing early phase clinical trials and exhibiting promising safety profiles. However, resistance to glutaminolysis targeting treatments has been observed, necessitating the development of treatments to combat this resistance. One option is to use synergy drug combinations, which improve tumor chemotherapy’s effectiveness and diminish drug resistance and side effects. This review will focus on studies involving the glutaminolysis pathway and diverse combination therapies with therapeutic implications.
Keywords: Metabolic reprogramming, Glutaminolysis, Glutaminase, Therapeutic resistance, Combination therapy
Introduction
Glutamine, the most abundant free amino acid in plasma, is the quintessential substrate in several biosynthesis processes, such as the synthesis of nucleotides (purines and pyrimidines), fatty acids, antioxidants, and different nonessential amino acids (NEAAs), many of which are vital for rapidly proliferating cells such as cancer cells [1]. Forty years ago, scientists revealed that glutamine serves as the major energy source in cultured cervical cancer cells (HeLa cells) [2]. The dependence on glutamine has subsequently been seen in other cancer types, such as pancreatic cancer, ovarian cancer, breast cancer, and glioblastoma multi-forme (GBM) [3–7]. While glutamine is a nonessential amino acid in most cases, it is indispensable for glutamine-addicted cancer cells, and glutamine deprivation impairs cancer growth [1]. Glutamine can also activate the mammalian target of rapamycin complex 1 (mTORC1) pathway by being exchanged for leucine through the large neutral amino acid transporter (LAT1) or by contributing to the production of more α-ketoglutarate after the conversion to glutamate, which eventually leads to cell growth and suppression of autophagy [8–10]. Inhibiting glutaminolysis, coupled with chemo medications or other metabolic inhibitors, triggers synergy drug combinations in glutamine-dependent tumors. Specifically, GLS inactivation causes redox imbalance, suppresses the synthesis of nucleotides, and induces replication stress, making cancer cells more dependent on poly (ADP-ribose) polymerase (PARP) DNA repair and thus sensitizing them to the PARP inhibitor [11]. On the other hand, pancreatic tumors were found to escape GLS inhibition by elevating glutamate production, such as through the glutaminase II pathway, where glutamine is converted to glutamate via the intermediates α-ketoglutaramate (KGM) and α-ketoglutarate (α-KG) [12]. Knockdown of glutamine transaminase K (GTK), a key enzyme of the glutaminase II pathway, in combination with GLS inhibition, can impair both metabolic pathways [12]. Pancreatic tumors undergo metabolic compensation to rely more on glucose metabolism after GLS inhibition [13]. Cancer cells that survive GLS inhibition tend to be more susceptible to metformin, a drug used to treat diabetes [13]. Moreover, even if glutamate production from other sources is limited, N-acetylaspartylglutamate (NAAG) can serve as an essential reservoir to provide glutamate to cancer cells via carboxypeptidase II (GCPII), thereby making GCPII a viable target for cancer therapy, either alone or in combination with GLS inhibition [14]. As a result, a combination of GLS inhibitors and other treatments could be a potential avenue to successful cancer therapy.
The crucial role of glutamine metabolism in cancer growth and survival
In order to decipher the fate of glutamine in cancer cells, glutamine metabolism, including glutaminolysis, has been an important and vigorously researched topic. Cancer cells transport exogenous glutamine into the cytoplasm via alanine, serine, cysteine transporter 2 (ASCT2, also called SLC1A5), and glutamine can subsequently be converted to glutamate via glutaminase 1 (GLS1 or GLS) or glutaminase 2 (GLS2) (Figure 1). Glutamate then undergoes a deamidation through two different possible pathways to produce α-ketoglutarate (α-KG), which is incorporated into the TCA cycle. The first pathway is catalyzed by glutamate dehydrogenase (GDH/GLUD1), which converts glutamate into ammonium and α-KG. The second pathway is catalyzed by a number of transaminases, such as glutamate-pyruvate transaminase and glutamate-oxaloacetate transaminase.
Figure 1.

The glutaminolysis pathway and its inhibitors.
Glutamine is an important nitrogen and carbon source for actively proliferating cells, especially glutamine-addicted cancer cells, and is an essential building block for numerous biosynthesis processes, such as the synthesis of glutathione, NADPH, nucleotides, and fatty acids. Glutamine can be imported from the extracellular space via alanine, serine, cysteine transporter 2 (ASCT2). Glutamine is then converted to glutamate by either glutaminase 1 (GLS1) or glutaminase 2 (GLS2). Glutamate is then converted to α-ketoglutarate (α-KG). This reaction can be catalyzed by either glutamate dehydrogenase (GDH) generating one equivalent of ammonia or by transaminases in which the nitrogen is incorporated into other amino acids. α-KG can be incorporated into the TCA cycle and its carbon converted to malate, oxaloacetate (OAA), and citrate subsequently. Malate can be transported from the mitochondrion to the cytoplasm, where it is a substrate of the malic enzyme 1 (ME1), thereby assisting in the production of NADPH from NADP+. OAA can be converted to aspartate and transported to the cytoplasm to participate in nucleotide synthesis. α-KG can also be transported to the cytoplasm as a source of acetyl-CoA, which is an essential substrate of fatty acid synthesis. There are several drugs developed to inhibit different parts of the glutaminolysis pathway. The first type is exemplified by glutamine analogs, such as DON, acivicin, and azaserine. The second group is represented by the SLC1A5 inhibitors, including GPNA and V-9302. Last, GLS1/2 is also a promising target for glutaminolysis inhibition. Several noncompetitive allosteric inhibitors have been identified, such as CB-839, BPTES, and compound 968.
The transaminase reactions can contribute to the production of other nonessential amino acids, including alanine and aspartate. Apart from its incorporation into the TCA cycle for the subsequent production of energy, α-KG can be exported from the mitochondria, where the TCA cycle occurs, to the cytosol and be converted to isocitrate by isocitrate dehydrogenase 1 (IDH1), which is then converted to citrate for de novo fatty acid synthesis [9,15]. Other TCA cycle metabolites produced from glutamine can be released into the cytosol and be converted into other molecules that also assist proliferating cells. For example, pyruvate can be generated from malate, concomitant with the production of NADPH via malic enzyme 1 (ME1) to replenish cell reduction capacity. Oxaloacetate (OAA) and glutamate can undergo a transamination reaction and generate α-KG and aspartate, which can be transported to the cytosol and participate in nucleotide synthesis [16–18]. Furthermore, glutamine is also the precursor (via glutamate) of glutathione (GSH), which is an endogenous antioxidant that shields cancer cells from excessive oxidative stress and sustains their survival [1]. Glutamine provides nitrogen for nucleobase synthesis, carbon for the TCA cycle [19], and participates in lipid synthesis and redox hemostasis. These are necessary for the survival and proliferation of cancer cells, making glutaminolysis an attractive target for developing new small molecule anticancer drugs [20,21].
Glutamine-addicted cancers
While an accurate proportion of human cancers that display glutamine addiction is yet to be defined, it has been noted that particular cancer types or cancer cell lines may be more prone to glutamine addiction than others, such as pancreatic cancer, acute myelogenous leukemia, GBM, myeloma, clear cell renal cell carcinoma (ccRCC), lung cancer and triple-negative breast cancer (TNBC) [22–26]. The dependency on glutamine in glioblastoma was interpreted by the high concentration of glutamine in glioblastoma of patients injected with 13C6-glucose [27]. Lung cancer cells become addicted to glutamine as a result of aberrantly triggered oncogenes and the lack of tumor suppressors in contrast to normal counterparts [28]. Glutamine addiction may be specific to a particular breast cancer subtype; for instance, the TNBC MDA-MB-453 cell line is tamine dependent, while the luminal A subtype MCF-7 breast cancer cell line is not [29].
We analyzed the fractions of cancer cell lines that are more dependent on the glutaminolysis pathway using data collected from DepMap (https://depmap.org/portal [30] Table 1). The data collected from DepMap are recorded using CERES dependency scores [32] that are based on data from cell depletion arrays. Glutamine-addicted cancer cells may express a higher level of the following genes: GLS1, GLS2, MYC, ASCT2, and GLUD1. Consequently, we calculated the dependent rate of cell lines toward certain genes by dividing the number of cell lines dependent on the genes (as the CERES score of 0 indicates that the gene is not essential for the cell line, and a lower score indicates that the gene is more likely to be crucial for the survival of the cell line, a cell line with a CERES score below 0 are taken as gene dependent [31,32]) by the total number of cell lines. Since glutamine synthetase (GLUL) catalyzes the synthesis of glutamine from glutamate and ammonia, GLS1/GLUL, GLUD1/GLUL, and (GLS1 + GLUD1)/GLUL were used to show the activity of glutamine catabolism as compared to glutamine anabolism [5]. Acute myeloid leukemia (AML), cervical squamous, esophagus adenocarcinoma, nonsmall cell lung cancer (NSCLC), non-Hodgkin’s lymphoma, ovary adenocarcinoma, thyroid carcinoma, and several types of soft tissue sarcoma manifest high ratios of GLS1/GLUL, GLUD1/GLUL, and (GLS1 + GLUD1)/GLUL suggesting a greater reliance on extracellular glutamine catabolism (Table 1).
Table 1.
Fraction of Cell Lines Dependent on Glutaminolysis: Data analysis is based on https://depmap.org/portal/.
| Primary Disease | Lineage | Gene Dependency Rate Formula = ((cell line counts that scores of 0 and −1 represent the median effects of nonessential genes and common core essential genes)/total cell line counts) * 100% |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GLS1 | GLS2 | MYC | SLC1A5 | GLUD1 | GLUL | GLS1/GLUL | GLUD1/GLUL | (GLS1+GLUD1)/GLUL | ||
|
| ||||||||||
| Bile | Cholangiocarcinoma (27) | 88.89 | 96.30 | 100.00 | 100.00 | 85.19 | 44.44 | 2.00 | 1.92 | 3.92 |
| Duct (30) | Gallbladder Adenocarcinoma (3) | 100.00 | 100.00 | 100.00 | 66.67 | 66.67 | 66.67 | 1.50 | 1.00 | 2.50 |
| Blood (44) | ALL (12) | 75.00 | 100.00 | 100.00 | 100.00 | 66.67 | 41.67 | 1.80 | 1.60 | 3.40 |
| AML (21) | 95.24 | 95.24 | 100.00 | 100.00 | 85.71 | 38.10 | 2.50 | 2.25 | 4.75 | |
| CLL (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| CML (7) | 100.00 | 85.71 | 100.00 | 100.00 | 85.71 | 57.14 | 1.75 | 1.50 | 3.25 | |
| Unspecified Leukemia (3) | 66.67 | 100.00 | 100.00 | 100.00 | 66.66 | 66.67 | 1.00 | 1.00 | 2.00 | |
| Bone (28) | Chondrosarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A |
| Choroma (3) | 100.00 | 100.00 | 100.00 | 100.00 (4 cell lines) | 100.00 | 50.00 (4 cell lines) | 2.00 | 2.00 | 4.00 | |
| Ewing Sarcoma (16) | 75.00 | 100.00 | 100.00 | 100.00 | 75.00 | 43.75 | 1.71 | 1.71 | 3.43 | |
| Osteosarcoma (8) | 62.50 | 100.00 | 100.00 | 100.00 | 62.50 | 75.00 | 0.83 | 0.83 | 1.67 | |
| Breast (34) | Breast Adenocarcinoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A |
| Breast Carcinoma (18) | 83.33 | 94.44 | 100.00 | 100.00 | 77.78 | 11.11 | 7.50 | 7.00 | 14.50 | |
| Breast Ductal Carcinoma (15) | 100.00 | 93.33 | 100.00 | 100.00 | 86.67 | 46.67 | 2.14 | 1.86 | 4.00 | |
| Central Nervous | Glioma (50) | 90.00 | 94.00 | 100.00 | 100.00 | 94.00 | 56.00 | 1.61 | 1.68 | 3.29 |
| System (61) | Medulloblastoma (9) | 66.67 | 100.00 | 100.00 | 100.00 | 77.78 | 44.44 | 1.50 | 1.75 | 3.25 |
| Meningioma (2) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| Cervix (11) | Cervical Adenocarcinoma (3) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 66.67 | 1.50 | 1.50 | 3.00 |
| Cervical Carcinoma (4) | 75.00 | 100.00 | 100.00 | 100.00 | 75.00 | 100.00 | 0.75 | 0.75 | 1.50 | |
| Cervical Squamous (4) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 50.00 | 2.00 | 2.00 | 4.00 | |
| Colorectal (37) | Colorectal Adenocarcinoma (37) | 75.68 | 81.08 | 100.00 | 100.00 | 91.89 | 67.57 | 1.12 | 1.36 | 2.48 |
| Embryo (1) | Teratoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 |
| Epidermoid Carcinoma (1) | Skin Squamous (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 |
| Esophagus (23) | Esophagus Adenocarcinoma (3) | 100.00 | 100.00 | 100.00 | 100.00 (5 cell lines) | 66.67 | 40.00 (5 cell lines) | 2.50 | 1.67 | 4.17 |
| Esophagus Squamous (20) | 90.00 | 100.00 | 100.00 | 100.00 | 95.00 | 55.00 | 1.64 | 1.73 | 3.36 | |
| Eye (6) | Retinoblastoma (1) | 0.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A |
| Uveal Melanoma (5) | 100.00 | 80.00 | 100.00 | 100.00 | 100.00 | 60.00 | 1.64 | 1.73 | 3.36 | |
| Fibroblast (1) | Fibroblast Soft Tissue (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 |
| Gastric (26) | Gastric Adenocarcinoma (26) | 80.77 | 88.46 | 100.00 | 100.00 | 80.77 | 38.46 | 2.10 | 2.10 | 4.20 |
| Kidney (23) | Malignant Rhabdoid Tumor (7) | 100.00 | 100.00 | 100.00 | 100.00 (8 cell lines) | 100.00 | 87.50 (8 cell lines) | 1.14 | 1.14 | 2.29 |
| Renal Cell Carcinoma (21) | 76.19 | 90.48 | 100.00 | 100.00 | 85.71 | 57.14 | 1.33 | 1.50 | 2.83 | |
| Liver (22) | Hepatoblastoma (1) | 0.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 1.00 | 1.00 |
| Hepatocellular Carcinoma (20) | 80.00 | 95.00 | 100.00 | 100.00 | 80.00 | 55.00 | 1.45 | 1.45 | 2.91 | |
| Lung (107) | Lung Carcinoid (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 |
| Mesothelioma (10) | 100.00 | 100.00 | 100.00 | 100.00 | 90.00 | 80.00 | 1.25 | 1.13 | 2.38 | |
| NSCLC (78) | 85.90 | 98.72 | 100.00 | 100.00 | 93.59 | 50.00 | 1.72 | 1.87 | 3.59 | |
| SCLC (18) | 77.78 | 100.00 | 94.44 | 100.00 | 83.33 | 55.56 | 1.40 | 1.50 | 2.90 | |
| Lymphocyte (19) | ATL (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 |
| Hodgkin Lymphoma (2) | 100.00 | 50.00 | 100.00 | 100.00 (3 cell lines) | 100.00 | 66.67 (3 cell lines) | 1.50 | 1.50 | 3.00 | |
| Lymphoma Unspecified (3) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 66.67 | 1.50 | 1.50 | 3.00 | |
| Non Hodgkin Lymphoma (13) | 100.00 | 100.00 | 100.00 | 100.00 (14 cell lines) | 69.23 | 28.57 (14 cell lines) | 3.50 | 2.42 | 5.92 | |
| Ovary (43) | Brenner Tumor (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A |
| Mixed Germ Cell (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| Ovary Adenocarcinoma (38) | 92.11 | 100.00 | 100.00 | 100.00 | 89.47 | 36.84 | 2.50 | 2.43 | 4.93 | |
| Ovary Carcinoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| SCCOHT (2) | 100.00 | 100.00 | 100.00 | 100.00 | 50.00 | 50.00 | 2.00 | 1.00 | 3.00 | |
| Pancreas (34) | Exocrine (34) | 88.24 | 100.00 | 100.00 | 100.00 | 88.24 | 52.94 | 1.67 | 1.67 | 3.33 |
| PNS (20) | Neuroblastoma (20) | 85.00 | 90.00 | 100.00 | 100.00 | 95.00 | 60.00 | 1.42 | 1.58 | 3.00 |
| Plasma (21) | Multiple Myeloma (21) | 100.00 | 95.24 | 100.00 | 100.00 | 76.19 | 71.43 | 1.40 | 1.07 | 2.47 |
| Prostate (1) | Prostate Adenocarcinoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A |
| Skin (52) | Melanoma (46) | 87.00 | 93.48 | 100.00 | 100.00 (47 cell lines) | 80.43 | 53.19 (47 cell lines) | 1.63 | 1.51 | 3.15 |
| Merkel Cell Carcinoma (2) | 100.00 | 100.00 | 100.00 | 100.00 (3 cell lines) | 100.00 | 33.33 (3 cell lines) | 3.00 | 3.00 | 6.00 | |
| Skin Squamous (4) | 100.00 | 100.00 | 100.00 | 100.00 | 50.00 | 25.00 | 4.00 | 2.00 | 6.00 | |
| Soft Tissue (39) | ATRT (3) | 66.67 | 100.00 | 100.00 | 100.00 (5 cell lines) | 100.00 | 20.00 (5 cell lines) | 3.33 | 5.00 | 8.33 |
| Epithelioid Sarcoma (2) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 50.00 | 2.00 | 2.00 | 4.00 | |
| Fibrosarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| Leiomyosarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 (2 cell lines) | 100.00 | 50.00 (2 cell lines) | 2.00 | 2.00 | 4.00 | |
| Liposarcoma (8) | 100.00 | 87.50 | 100.00 | 100.00 | 100.00 | 37.50 | 2.67 | 2.67 | 5.33 | |
| Malignant Rhabdoid Tumor (7) | 85.71 | 85.71 | 100.00 | 100.00 (8 cell lines) | 100.00 | 37.50 (8 cell lines) | 2.29 | 2.67 | 4.95 | |
| Pleomorphic Sarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| Rhabdomyosarcoma (11) | 81.82 | 90.91 | 100.00 | 100.00 | 54.55 | 36.36 | 2.25 | 1.50 | 3.75 | |
| Synovial Sarcoma (5) | 80.00 | 100.00 | 100.00 | 100.00 | 80.00 | 60.00 | 1.33 | 1.33 | 2.67 | |
| Thyroid Sarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| Undifferentiated Sarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 0.00 | N/A | N/A | N/A | |
| Thyroid (6) | Thyroid Carcinoma (5) | 80.00 | 80.00 | 100.00 | 100.00 | 80.00 | 40.00 | 2.00 | 2.00 | 4.00 |
| Thyroid Squamous (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| Upper Aerodigestive (32) | Upper Aerodigestive Squamous (32) | 87.50 | 90.63 | 100.00 | 100.00 (33 cell lines) | 90.63 | 45.45 (33 cell lines) | 1.93 | 1/99 | 3.92 |
| Urinary Tract (29) | Bladder Carcinoma (29) | 75.86 | 93.10 | 96.55 | 100.00 | 96.55 | 55.17 | 1.38 | 1.75 | 3.13 |
| Uterus (26) | Choriocarcinoma (2) | 0.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | 1.00 | 1.00 |
| Clear Cell Carcinoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| Endometrial Adenocarcinoma (15) | 60.00 | 100.00 | 100.00 | 100.00 | 73.33 | 73.33 | 0.82 | 1.00 | 1.82 | |
| Endometrial Adenosquamous (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 1.00 | 1.00 | 2.00 | |
| Endometrial Squamous (3) | 66.67 | 100.00 | 100.00 | 100.00 | 100.00 | 66.67 | 1.00 | 1.50 | 2.50 | |
| Endometrial Stromal Sarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| MMMT (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| Mullerian Carcinoma (1) | 0.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
| Uterine Carcinosarcoma (1) | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 0.00 | N/A | N/A | N/A | |
Biomarkers determining glutamine addiction in cancer
In order to select patients for future clinical trials evaluating the efficacy of glutaminolysis inhibitors, certain biomarkers need to be identified to determine the cancers that are glutamine addicted. The study by Yuneva et al. showed that MYC expression results in a strong dependence on glutamine in cancer cells, so much so that upon removal of glutamine, these cells undergo apoptosis [33]. This is because, largely glutaminolysis is stimulated by an MYC-regulated transcriptional program. Thus, MYC causes the mitochondrial TCA cycle to become reliant on glutamine [34]. MYC is also responsible for activating SLC38A5 (SN2), a major transporter of glutamine, and SLC1A5 (ASCT2), an amino acid transporter that is involved in glutamine-dependent mTORC1 activation. mTORC1, a protein translation regulator, has been shown to be responsive to the levels of glutamine in the cell [22]. Furthermore, in cells that are dependent on glutamine, higher than average levels of SLC1A5 are often present [35]. For example, SLC1A5 is highly expressed in TNBC patients, and knocking it down improved survival in tumor-bearing mice [26]. Additionally, it has been discovered that in cells displaying an overexpression of MYC, glutaminase levels are higher than those in cells with low MYC expression levels [11,36]. Thus, all of these—MYC, SLC1A5, and mTORC1 levels in addition to levels of glutaminase—can be used in future clinical trials as biomarkers to select patients when evaluating the potential efficacy of glutaminolysis inhibitors as they all are present at greater levels in glutamine-dependent cells.
Targeting glutaminolysis in combination with other therapies for cancer treatment
Decades ago, several glutamine antagonists, such as DON (6-diazo-5-oxo-L-norleucine), acivicin, and azaserine, were developed by researchers to inhibit glutamine metabolism in cancer cells (Table 2) [37– 41]. Although such glutamine analogs demonstrate high efficiency in obstructing cancer cell growth, they are not ideal cancer drugs because of their high toxicity [1]. To alleviate gastrointestinal (GI) toxicities, scientists developed DON prodrugs that optimized the delivery of DON to tumors with limited exposure to GI tissues and suggested administrating these prodrugs in a low dosage [38,42]. Moreover, treating MC38 colon cancer cells with JHU083, a prodrug of DON developed by Leone et al., not only decreased glycolysis and oxidative phosphorylation but also promoted oxidative metabolism of effector T cells [43]. Taken together, prodrugs of the glutamine antagonist DON together with optimized delivery strategies and dosages may show promising outcomes in patients with glutamine-addicted tumors in clinical trials in the future.
Table 2.
Small molecule inhibitors targeting glutaminolysis pathway.
| Small Molecule Inhibitors Targeting Glutaminolysis Pathway | |||||
|---|---|---|---|---|---|
|
| |||||
| Small Molecule Inhibitors | Chemical Structure | Inhibition Type | Enzyme Target | Inhibitory Activity | Reference |
|
| |||||
| 6-diazo-5-oxo-l-norleucine (DON) |
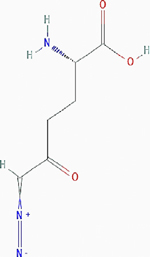
|
Irreversible inhibition | Nonselective. Glutamine utilizing enzymes, including glutamine amidotransferases | Inhibits the enzymes by covalently modifying the cysteine residues in the glutamine active sites irreversibly. | PubChem CID: 9087 URL: https://pubchem.ncbi.nlm.nih.gov/compound/9087#section=2D-Structure [38,40,90] |
| Acivicin |
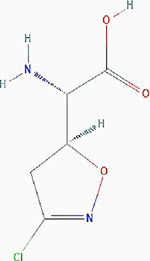
|
Irreversible inhibition | Nonselective. Glutamine utilizing enzymes, including glutamine amidotransferases, γ-glutamyltranspeptidase (GGT) | Inhibits the enzymes by forming an imine–thioether adduct at the cysteine residue of the active site as a result of nucleophilic substitution of the chlorine atom. Inhibits GGT by covalently modifying the Thr391 residue at the active site. |
PubChem CID: 294641 URL: https://pubchem.ncbi.nlm.nih.gov/compound/294641#section=2D-Structure [40,91,92] |
| Azaserine |
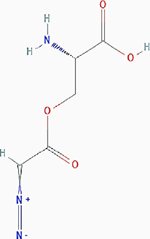
|
Irreversible inhibition | Nonselective. Glutamine utilizing enzymes, including glutamine amidotransferases, γ-glutamyltranspeptidase (GGT) | Inhibits GGT by covalently modifying the Thr391 residue at the active site. | PubChem CID: 460129 URL: https://pubchem.ncbi.nlm.nih.gov/compound/460129#section=2D-Structure [40,92] |
| l-γ-Glutamyl-p-nitroanilide (GPNA) |
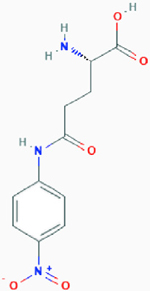
|
Competitive inhibition | Glutamine transporter SLC1A5, sodium-dependent and independent amino acid transporters | Proposed model: Inhibits SLC1A5 by binding to the binding site with high affinity (the ligand part binds to the lipophilic pocket of SLC1A5 and forms a hydrogen bond). | PubChem CID: 81732 URL: https://pubchem.ncbi.nlm.nih.gov/compound/81732#section=2D-Structure [47,48] |
| V-9302 |
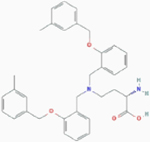
|
Competitive inhibition | Glutamine transporter SLC1A5 | In silicon model: V-9302 docks into the orthosteric binding site situated at the transmembrane region of SLC1A5. | PubChem CID: 127035871 URL: https://pubchem.ncbi.nlm.nih.gov/compound/127035871#section=2D-Structure [49] |
| Compound 968 |
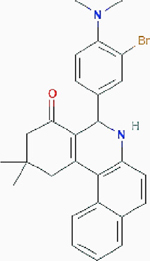
|
Allosteric inhibition | Glutaminase (GLS): kidney-type glutaminase: kidney glutaminase (KGA) and glutaminase C (GAC), and liver-type glutaminase (GLS2) | Inhibits glutaminases by binding to GAC monomers and preventing the formation of activated GAC tetramers. | PubChem CID: 3099980 URL: https://pubchem.ncbi.nlm.nih.gov/compound/3099980#section=2D-Structure [50–52] |
| bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) |
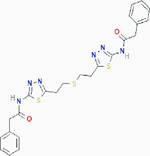
|
Allosteric inhibition | Selectively inhibits kidney-type glutaminase (GLS): kidney glutaminase (KGA) and glutaminase C (GAC) | Inhibits KGA and GAC by trapping their tetramers in a conformation that is nonfunctioning. | PubChem CID: 3372016 URL: https://pubchem.ncbi.nlm.nih.gov/compound/3372016#section=2D-Structure [93–95] |
| CB-839 (Telaglenastat) |
|
Allosteric inhibition | Selectively inhibits kidney-type glutaminase (GLS): kidney glutaminase (KGA) and glutaminase C (GAC) | Inhibits KGA and GAC by forming hydrogen bonds with cKGA (catalytic domain Ile221-Leu533 of KGA) tetramer. | PubChem CID: 71577426 URL: https://pubchem.ncbi.nlm.nih.gov/compound/71577426#section=2D-Structure [58,59] |
On the other hand, glutaminolysis has been an attractive cancer therapy target for the past decade (Figure 1). Researchers have developed multiple medications blocking various steps of glutamine catabolism pathways to impede glutamine-addicted cancer cells’ growth. Targeting amino acid transporter SLC1A5 plays a vital role in importing extracellular glutamine. High SLC1A5 expression level has been documented to be correlated to poor overall survival in various types of cancers, including nonsmall cell lung cancer, liver cancer, and breast cancer [44–46]. The SLC1A5 inhibitor L-γ-glutamyl-p-nitroanilide (GPNA) is an analog of glutamine that blocks SLC1A5 [47,48]. V-9302, a small molecule antagonist, is another SLC1A5 inhibitor. In a previous study, blocking SLC1A5 with V-9032 diminished cancer growth, disturbed redox homeostasis, and inflicted cell death in preclinical models [49]. Small-molecule allosteric inhibitors of GLS have been identified, including compound 968, BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide), and CB-839 (Telaglenastat). Compound 968 can interact with kidney glutaminase (KGA or GLS1) and its slicing variant, glutaminase C (GAC) [50]. It inactivates GAC by inhibiting nonactive monomeric GAC from forming enzymatically active GAC tetramer [51]. Compound 968 also inhibits the liver-type glutaminase (GLS2 or LGA) and its slicing variant, glutaminase B (GAB), in luminal-subtype breast cancer [52]. While the inhibitory potency of BPTES is substantial, its low solubility makes it difficult to deliver in vivo [1,53]. Hence, scientists are now vigorously engaged in concerted efforts to refine BPTES and create BPTES analogs in the hope of boosting its efficacy for cancer therapy [53,54]. Although these small molecule drugs showed profound efficiency in inhibiting glutaminolysis and are capable of hampering cancer cell growth in preclinical models, the emergence of therapeutic resistance has been a refractory issue. It is common in real-world scenarios to find heterogeneity within a single tumor relying on different metabolic pathways [55–57]. Solely inhibiting glutaminolysis is not enough to cure patients whose tumors exhibit high intratumoral heterogeneity. Therefore, scientists now aim to target additional pathways besides glutaminolysis to optimize cancer treatments.
Targeting GLS1 by CB-839 synergizes with other clinical drugs
CB-839, another important GLS inhibitor, selectively inhibits both KGA and GAC [58]. CB-839 has undergone many clinical trials for many cancer types, including clear cell renal cell carcinoma (ccRCC), colorectal cancer, ovarian cancer (NCT03875313, NCT02861300, NCT03944902). CB-839 is also currently undergoing a phase I clinical trial to treat patients with relapsed and/or treatment-refractory leukemia (NCT02071927).
Over the past decade, CB-839 has been used in various preclinical studies and clinical trials in combination with various clinical drugs, and some of them have shown a promising synergistic effect (Table 3). In a patient-derived TNBC xenograft model and a basal HER2+ cell line xenograft model, CB-839 resulted in significant reductions in tumor growth, either alone or in combination with paclitaxel, which showed strong synergy to suppress the regrowth of the tumors [59]. A phase II combination trial of CB-839 and paclitaxel in patients with advanced TNBC was also performed (NCT03057600). In addition, the combined use of CB-839 and olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, resulted in potently synergistic cell death presumably due to extreme oxidative stress and DNA replication stress in tumors, reinforcing the rationale for a clinical trial setting to incorporate GLS inhibitors with PARP inhibitors (Figure 2) [11,60]. Furthermore, CB-839 could act synergistically with the epidermal growth factor receptor (EGFR) inhibitor erlotinib or the EGFR-targeted monoclonal antibody cetuximab for cancer therapy [61,62]. Specifically, CB-839, in combination with erlotinib, synergistically triggered in vitro apoptosis and reduced in vivo tumor growth in EGFR mutant nonsmall cell lung cancer [61]. A combination of CB-839 and cetuximab also showed a synergistic effect and decreased cell viability in colorectal cancer cells [62]. CB-839 is currently undergoing clinical trials, both alone or in conjunction with everolimus (NCT03163667), cabozantinib (NCT03428217), or nivolumab (NCT02771626), for various cancers [63]. The phase I or phase II clinical trials showed positive results and demonstrated that all the drug combinations could be tolerated [63]. CB-839 has a synergistic effect of promoting ER stress and apoptosis caused by carfilzomib in treating myeloma cell lines with the proteasome inhibitor-resistant phenotype [64]. By activating mitochondrial-mediated apoptosis, CB-839 displayed good synergism with ABT-199, a BCL-2 inhibitor, killing acute myeloid leukemia blasts [65]. In the tumor microenvironment [66], tumor cells outcompete T-cells for scarce nutrients such as glutamine. By diverting glutamine from cancer cells to active T-cells, CB-839 is proposed to have synergistic effects with nivolumab, an immunoglobulin G4 PD-1 immune checkpoint inhibitor (Figure 2) [63]. Inhibition of glutamine metabolism can hamper oxidative and glycolytic metabolism in cancer cells while stimulating oxidative metabolism in effector T cells and rendering these Tcells highly activated and long-lived [67]. These findings signify that targeting glutamine addiction through GLS1 inhibition in combination with other drugs provides a promising therapeutic strategy for several types of cancers.
Table 3.
Compounds that synergize with glutamine catabolism inhibitors.
| Small Molecule Inhibitors Targeting Glutaminolysis | Compounds that Synergize with Glutamine Catabolism Inhibitors | Cell Line | Results and Cellular Target/Mechanism | Reference |
|---|---|---|---|---|
|
| ||||
| BPTES [bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide] | shRNA silencing glutamine transaminase K (GTK) | Pancreatic cancer | Result: Decreased intracellular glutamate level and inhibited cell growth. Target: GTK. |
[14] |
2-(Phosphonomethyl)-pentanedioic acid 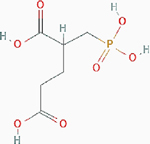 [2-PMPA] [2-PMPA] |
Patient-derived ovarian and pancreatic orthotopic tumors | Result: Reduced tumor growth. Target: GCPII. |
PubChem CID: 10130754 URL: https://pubchem.ncbi.nlm.nih.gov/compound/10130754#section=2D-Structure [14] | |
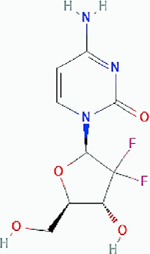 Gemcitabine Gemcitabine |
Pancreatic ductal adenocarcinoma (PDAC) | Result: Combination therapy did not improve single treatment efficacy. Target: DNA replication. |
PubChem CID: 60750 URL: https://pubchem.ncbi.nlm.nih.gov/compound/60750#section=2D-Structure [13,96] | |
Metformin 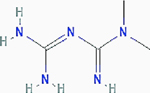
|
Pancreatic and prostate cancer | Results: Greater decrease in cancer cell number and decreased levels of lactate, glucose-6-phosphate, glucose-1-phosphate, and UDP-glucose (pancreatic). Target: glucose metabolism |
PubChem CID: 4091 URL: https://pubchem.ncbi.nlm.nih.gov/compound/4091#section=2D-Structure [13,85] | |
| CB-839 (Telaglenastat) |
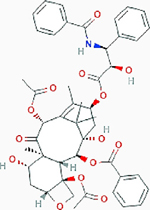 Paclitaxel Paclitaxel |
TNBC and a basal HER2+ cell line | Result: Reductions in tumor growth and suppressed regrowth of tumor. Mechanism: Inducing ER stress which promotes degradation of SLC1A5 by ubiquitin ligase RNF5 |
PubChem CID: 36314 URL: https://pubchem.ncbi.nlm.nih.gov/compound/36314#section=2D-Structure [97,98] |
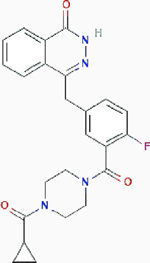 Olaparib Olaparib |
Ovarian cancer and renal cell carcinoma | Result: Potently synergistic cell death. Target: poly (ADP-ribose) polymerase (PARP) |
PubChem CID: 23725625 URL: https://pubchem.ncbi.nlm.nih.gov/compound/23725625#section=2D-Structure [11,60] | |
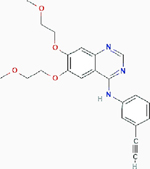 Erlotinib Erlotinib |
EGFR mutant nonsmall cell lung cancer | Result: In vitro apoptosis and reduced in vivo tumor growth. Target: EGFR tyrosine kinase |
PubChem CID: 176870 URL: https://pubchem.ncbi.nlm.nih.gov/compound/176870#section=2D-Structure [61] | |
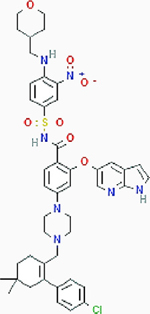 ABT-199 ABT-199 |
Myeloid leukemia | Result: Killing acute myeloid leukemia blasts. Target: BCL-2 |
PubChem CID: 49846579 URL: https://pubchem.ncbi.nlm.nih.gov/compound/49846579#section=2D-Structure [65] | |
| Nivolumab | Clear cell renal cell carcinoma | Result: Activation of T cells and proposed synergistic effects. Target: G4 PD-1 immune checkpoint |
[63] | |
Figure 2.

Emerging combination therapy: Combined blockage of glutaminolysis with inhibition of other pathways.
CB-839 or BPTES, small molecule inhibitors of GLS, combined with other drugs, is an emerging cancer treatment strategy. (1) CB-839 combinations: CB-839 combined with cabozantinib, a small molecule inhibitor of growth factor signaling, or with everolimus, an mTOR inhibitor, are currently undergoing clinical trials. In addition, CB-839 synergizes with nivolumab, a human IgG4 monoclonal antibody blocking PD-1, and this combination is also undergoing a Phase II clinical trial. Poly (ADP-ribose) polymerase (PARP) is important for cancer cells to reduce ROS stress and sustain genome integrity. GLS inhibition by, e.g. CB-839 treatment, suppresses the generation of nucleotides and causes replication stress, which may activate PARP-dependent DNA repairs. Treating cancer cells with a combination of CB-839 and PARP inhibitor, such as olaparib, has been shown to achieve synergistic antitumor activity. NAAG has been identified as a glutamine reservoir in cancer cells and is hydrolyzed to generate glutamate and NAA by carboxypeptidase II (GCPII), which can be inhibited by 2-PMPA. Combining 2-PMPA with CB-839 showed a more significant decrease in tumor size compared to treating the tumor with either of these drugs alone. (2) BPTES combination: BPTES combined with metformin, a glucose metabolism inhibitor, results in greater inhibition of tumor growth. The glutaminase II pathway provides cancer cells with a mechanism for bypassing GLS inhibition. Inhibition of both GLS1/2 and the glutaminase II pathway is a novel strategy for treating glutamine-addicted cancers. The effect of each drug or shRNA (highlighted in yellow) is shown in red, metabolites are shown in black, and enzymes are shown in blue boxes.
Dual inhibition of glutaminase I and glutaminase II pathways
A study recently discovered that the inhibition of GLS1 could only partially reduce tumor growth and not entirely hinder cancer cell proliferation [12]. By following glutamine metabolism in mice bearing patient-derived pancreatic cancer using (m+7) -labeled glutamine [68], the authors observed an increase in (m+5) glutamate production upon GLS1 inhibition using BPTES-NP (nanoparticles encapsulated BPTES). As glutamate and 13C5 glutamate are the only two possible (m+5) glutamate produced from -labeled glutamine, and glutamate is highly improbable, GLS1 inhibition should lead to a reduction in (m+5) glutamate production if it is the only pathway for the generation of glutamate. This is because the formation of 13C5 glutamate requires a transamination reaction from 13C5 α-KG, which requires the production of glutamate via GLS1. The result obtained thus indicates the presence of a different glutamate generation pathway other than glutaminolysis [12]. The study indicated that the 13C5 glutamate production occurs by the transamination of 13C5 α-KG derived from an alternative pathway: the glutaminase II pathway [12]. The glutaminase II pathway begins with a glutamine transaminase (most notably glutamine transaminase K; GTK) that catalyzes the transamination of glutamine to α-ketoglutaramate (KGM) in the presence of a suitable α-keto acceptor. In the second step, the enzyme ω-amidodicarboxylate amidohydrolase then catalyzes the conversion of KGM to α-KG (Figure 2). Glutamate can be further derived from α-KG by a transamination reaction or by GDH [12]. The carbon backbone for glutamate is derived from ⍺-KG in the transamination reaction. This sequence of reactions thereby helps to demonstrate how both 15N-labels are lost.
Glutaminase II Pathway
| (1) |
| (2) |
Transamination Reaction
| (3) |
KGM, the glutaminase II pathway intermediate, was studied to further validate that the glutaminase II pathway is responsible for the increased (m+5) glutamate production. Since ω-amidase only catalyzes reactions involving the open form of KGM (note that at physiological pH, 99.7% of KGM is cyclized to a lactam and only 0.3% is in the open-chain (substrate) form) [69], mass spectrometry and 1H NMR were employed to quantify the levels of KGM in the tumors, where the KGM structure could only be adequately identified in 1H NMR. Elevated KGM levels were found in tumors treated with BPTES-NP as compared to tumors treated with blank-NP. For further verification, GTK, the primary enzyme for the glutaminase II pathway that is responsible for generating KGM, was then analyzed. In eight pancreatic cancer cell lines derived from patients, the highest expression of GTK was found in P198 cells. shRNA knockdown of GTK in P198 resulted in reduced cell numbers, indicating that GTK is essential for the proliferation of cancer cells. Moreover, knockdown of GTK led to the complete suppression of in vivo pancreatic tumorigenesis [70]. With the role of the glutaminase II pathway in cancer now uncovered, a novel class of specific small molecule inhibitors with dual targeting of both the GLS1 and glutaminase II pathways could be a potential therapeutic strategy for cancer therapy.
Glutaminase 1 (GLS1) and glutaminase 2 (GLS2)
There are two isoenzymes of glutaminase, glutaminase 1 (GLS1 or GLS) and glutaminase 2 (GLS2 or LGA), which were initially found in the kidney and the liver, respectively [71]. Both are essential enzymes required in the regulation of glutamine metabolism that catalyzes the hydrolysis of glutamine to glutamate [72,73]. However, in tumorigenesis, GLS1 and GLS2 lead to opposite effects [74,75]. Studies have shown the significant role of GLS2 as a tumor suppressor in cancers such as gastric cancer [76,77]. The idea that GLS2 functions as a tumor suppressor while GLS1 promotes cancer growth has led to the assumption that inhibiting the GLS1 pathway is a sufficient anticancer strategy [74]. However, this is not completely true, as emerging evidence illustrates the importance of GLS2 in cancer cell proliferation, and a study on luminal-subtype breast cancer suggests a dual-GLS1/GLS2 inhibition to be more effective than GLS1 inhibition alone [52]. GLS2 is also expressed, though downregulated, in pancreatic cancer cells [78]; hence, it is possible that the resistance to BPTES-NP in pancreatic cancer could come from the compensation of GLS2. Although compound 968 inhibits both GLS1 and GLS2, this inhibition will not prevent the generation of glutamate via the glutaminase II pathway, as discussed in section 2.2. Therefore, dual glutaminase I and II pathway inhibition would be a better option.
Combined treatment of GLS1 inhibitor with blockage of glutamate release from its reservoir
N-Acetyl-aspartyl-glutamate (NAAG), the third most abundant neurotransmitter in the human body, has been studied thoroughly with respect to its function in several neurological disorders [79]. However, its role in cancer metabolism, its feasibility as a cancer therapy target, and its capacity as a noninvasive cancer progression indicator have only recently been discovered. A study by Nguyen et al. revealed that NAAG is a quintessential glutamate reservoir and that GCPII is responsible for the hydrolysis of NAAG to N-acetyl-aspartate (NAA) and glutamate (Figure 2) [14]. Knocking down the GCPII gene in pancreatic cancer P198 cells, using both shRNA lenti-virus and CRISPR, and treating them with BTPES, showed accentuation of decreased intracellular glutamate level and an inhibitory effect on cell proliferation compared to the control cells [14]. The possibility of targeting GCPII as a means of cancer therapy was then further tested using 2-(phosphonomethyl)-pentanedioic acid (2-PMPA), which was found to be a selective competitive inhibitor of GCPII, to treat patient-derived ovarian and orthotopic pancreatic tumors, and this treatment resulted in a significant reduction in tumor sizes [14]. A combination therapy using CB-839 and 2-PMPA was then tested, and, importantly, the decrease in tumor growth was found to be greater than when either treatment was used alone [14]. This is because these two compounds target two different glutamate metabolic pathways, which reduces drug resistance. Nguyen et al. also found that the concentrations of NAAG in MYC- transformed lymphoma B cells [80–82], high-grade ovarian tumors, and brain tumors are higher than those in low-grade cancers of the same type [14]. This is corroborated with other studies showing that GCPII exists at higher levels and exhibits greater activity in metastatic prostate cancer cells than in either benign or normal cells by at least a factor of ten [83]. NAAG also has the potential to be used as a noninvasive marker of predicting tumor growth. The study done by Nguyen et al. found that increases and decreases in NAAG concentrations in plasma took place before corresponding increases and decreases in tumor growth, respectively [14,84]. This correlation may allow physicians to monitor and stay one step ahead of tumor development and should have translational implications by introducing a new strategy for future clinical testing.
In short, NAAG’s role in glutamine metabolism and its correlation with cancer progression make this compound and the proteins involved in its metabolism likely candidates for future research and potential therapeutic targets. However, due to the propensity of many cancers for drug resistance, any future therapies targeting NAAG metabolism will likely be more effective when combined with other metabolic inhibitors, especially glutaminase inhibitors.
Combination therapy with GLS inhibitor and other metabolic inhibitors
BPTES-nanoparticles, where nanoparticles made up of poly (ethylene glycol) (PEG) and poly (lactic-coglycolic acid) (PLGA) encapsulate the drug to increase blood circulation time and tumor uptake, was an attempt to overcome the solubility limitation of BPTES. Indeed, it was shown to have better pharmacokinetics than unencapsulated BPTES [13]. The combined treatment of BPTES-NPs and gemcitabine, the current standard post-PDAC surgery adjuvant chemotherapy, did not result in improved tumor reduction [13]. Upon BPTES-NPs treatment, the residual tumor relies on glycolysis and glycogen synthesis, as evidenced by increases in the levels of lactate, glycogen, glucose-6-phosphate, glucose-1-phosphate, and UDP-glucose. Elgogary et al. then tested the combination of BPTES-NP and metformin, a diabetic drug currently in phase III clinical trials for cancer therapy (NCT01101438). Metformin has the ability to reduce glucose metabolism [13,85]. It has been previously shown that by inhibiting hexokinase-II, which catalyzes glucose phosphorylation, metformin reduces the supply of cellular energy and glucose metabolism [86]. With the combined therapy of BPTES-NPs and metformin, a greater decrease tumor growth as compared to either drug acting alone, as well as a decreased level of lactate, glucose-6-phosphate, glucose-1-phosphate, and UDP-glucose was detected.
Combination therapy has the ability to combat intratumoral metabolic heterogeneity and has important implications for the design of more effective treatment strategies. The rationale for combination therapy is to use drugs that work via different mechanisms, thereby decreasing the likelihood that resistant cancer cells will develop. With many types of combination therapies investigated, the combination of BPTES and metformin was selected by tracking the metabolic pathways utilized in the tumor cells remaining after GLS inhibition and identifying the best-suited drugs for combined synergistic therapy.
Conclusion
Resistance to therapy is a major impediment in the treatment of cancer, especially in heterogeneous cancers. In comparison, malignant cells rely more on glucose, glutamine, and other substrates than their noncancer counterparts. Given that glutamine metabolism underpins human malignancies, and GLS expression in cancer cells can determine their response toward GLS inhibiting drugs, biomarkers such as GLS can be used to select patients who are most likely to respond to and benefit from the anti-GLS therapy [87]. The utilization of L-[11C]glutamine positron emission tomography (PET) imaging demonstrates the value of noninvasive biomarkers that may be paired with therapies targeting tumor metabolism [88]. Future analysis of the molecular-based metabolic vulnerabilities of cancer cells will allow for the stratification of additional resistant patient populations who may benefit from GLS inhibitor treatment in combination with other anti-cancer drugs. Since combination therapy could decrease the likelihood of the development of resistant cancer cells through the inhibition of several different pathways, it is by far the most effective cancer treatment [89]. Future investigations that will provide more mechanistic insights into the metabolic adaptation of glutamine-dependent tumors will further highlight and potentiate novel strategies for cancer therapy.
Acknowledgments
This review has been prepared on the basis of the research works carried out in the laboratory of A Le supported by the National Institutes of Health (NIH) Grants R01-CA193895, R01-CA112314 (to AL). The work in the laboratory of YA Shen was supported by a TMU grant 108-5403-003-112 and a grant from the Ministry of Science and Technology, Taiwan (MOST 109-2314-B-038-021-MY3). Special thanks to Dr. Arthur Cooper for his helpful suggestions.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
* * of outstanding interest
- 1.Li T, Le A: Glutamine metabolism in cancer. Adv Exp Med Biol 2018, 1063:13–32. [DOI] [PubMed] [Google Scholar]
- 2.Reitzer LJ, Wice BM, Kennell D: Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979, 254:2669–2676. [PubMed] [Google Scholar]
- 3.Camelo F, Le A: The intricate metabolism of pancreatic cancers. Adv Exp Med Biol 2018, 1063:73–81. [DOI] [PubMed] [Google Scholar]
- 4.Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD: Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 2013, 9:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, et al. : Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol 2014, 10:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun X, Wang M, Wang M, Yu X, Guo J, Sun T, Li X, Yao L, Dong H, Xu Y: Metabolic reprogramming in triple-negative breast cancer. Front Oncol 2020, 10:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinones A, Le A: The multifaceted metabolism of glioblastoma. Adv Exp Med Biol 2018, 1063:59–72. [DOI] [PubMed] [Google Scholar]
- 8.Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN: Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012, 47:349–358. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Venneti S, Nagrath D: Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng 2017, 19:163–194. [DOI] [PubMed] [Google Scholar]
- 10.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. : Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136:521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen Y-A, Hong J, Asaka R, Asaka S, Hsu F-C, Suryo Rahmanto Y, Jung J-G, Chen Y-W, Yen T-T, Tomaszewski A, et al. : Inhibition of the MYC-regulated glutaminase metabolic Axis is an effective synthetic lethal approach for treating chemoresistant ovarian cancers. Canc Res 2020, 80: 4514–4526. ** The authors showed how targeting GLS1 with CB-839 synergized with olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, and resulted in cancer cell death. This study is a great example of how inhibiting GLS1 using CB-839 can synergize with other clinical drugs.
- 12. Udupa S, Nguyen S, Hoang G, Nguyen T, Quinones A, Pham K, Asaka R, Nguyen K, Zhang C, Elgogary A, et al. : Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics 2019, 19, e1800451. ** The authors discovered that pancreatic cancer cells utilize the Glutaminsae II pathway when GLS1 is inhibited, and knocking down GTK, an important enzyme of the Glutaminsae II pathway, inhibited pancreatic tumorigenesis in vivo. This study demonstrates the potential of dual inhibition of GLS and the Glutaminase II pathway for cancer treatment.
- 13.Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, Fu J, Chen B, Xia S, Liu Y, et al. : Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A 2016, 113:E5328–E5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen T, Kirsch BJ, Asaka R, Nabi K, Quinones A, Tan J, Antonio MJ, Camelo F, Li T, Nguyen S, et al. : Uncovering the role of N-Acetyl-Aspartyl-Glutamate as a glutamate reservoir in cancer. Cell Rep 2019, 27:491–501. e496. ** The authors discovered that NAAG is a reservoir for glutamate in cancer cells, and inhibition of the conversion of NAAG to glutamate using 2-PMPA coupled with inhibition of GLS1 using CB-839 resulted in more pronounced reduction in tumor growth. This study demonstrates that inhibiting GLS1 together with blocking the release of glutamate from its reservoir is a promising strategy for cancer treatment.
- 15.Park JK, Coffey NJ, Limoges A, Le A: The heterogeneity of lipid metabolism in cancer. Adv Exp Med Biol 2018, 1063:33–55. [DOI] [PubMed] [Google Scholar]
- 16.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. : Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB: Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007, 104:19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukhopadhyay S, Saqcena M, Foster DA: Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2:807–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. : Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabol 2012, 15: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang CV, Hamaker M, Sun P, Le A, Gao P: Therapeutic targeting of cancer cell metabolism. J Mol Med (Berl) 2011, 89: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA, et al. : Dysregulated metabolism contributes to oncogenesis. Semin Canc Biol 2015, 35(Suppl):S129–S150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wise DR, Thompson CB: Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010, 35:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolzoni M, Chiu M, Accardi F, Vescovini R, Airoldi I, Storti P, Todoerti K, Agnelli L, Missale G, Andreoli R, et al. : Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood 2016, 128: 667–679. [DOI] [PubMed] [Google Scholar]
- 24.Abu Aboud O, Habib SL, Trott J, Stewart B, Liang S, Chaudhari AJ, Sutcliffe J, Weiss RH: Glutamine addiction in kidney cancer suppresses oxidative stress and can Be exploited for real-time imaging. Canc Res 2017, 77: 6746–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanhove K, Derveaux E, Graulus GJ, Mesotten L, Thomeer M, Noben JP, Guedens W, Adriaensens P: Glutamine addiction and therapeutic strategies in lung cancer. Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, et al. : ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35:3201–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marquez J, Alonso FJ, Mates JM, Segura JA, Martin-Rufian M, Campos-Sandoval JA: Glutamine addiction in gliomas. Neurochem Res 2017, 42:1735–1746. [DOI] [PubMed] [Google Scholar]
- 28.Mohamed A, Deng X, Khuri FR, Owonikoko TK: Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin Lung Canc 2014, 15:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang CV: MYC, microRNAs and glutamine addiction in cancers. Cell Cycle 2009, 8:3243–3245. [DOI] [PubMed] [Google Scholar]
- 30.DepMap Broad: DepMap 20Q4 Public. figshare. Dataset 2020, 10.6084/m9.figshare.13237076.v2. [DOI]
- 31.Dempster JM, Rossen J, K M, Pan J, Kugener G, Root DE, Tsherniak A: Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. BioRxiv 2019. [Google Scholar]
- 32.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. : Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017, 49:1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y: Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007, 178: 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. : Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008, 105:18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhutia YD, Ganapathy V: Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta 2016, 1863:2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. : c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458:762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffey GL, Ehrlich J, Fisher MW, Hillegas AB, Kohberger DL, Machamer HE, Rightsel WA, Roegner FR: 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. I. Biologic studies. Antibiot Chemother (Northfield) 1956, 6:487–497. [PubMed] [Google Scholar]
- 38.Lemberg KM, Vornov JJ, Rais R, Slusher BS: We’re not “DON” yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol Canc Therapeut 2018, 17:1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hidalgo M, Rodriguez G, Kuhn JG, Brown T, Weiss G, MacGovren JP, Von Hoff DD, Rowinsky EK: A Phase I and pharmacological study of the glutamine antagonist acivicin with the amino acid solution aminosyn in patients with advanced solid malignancies. Clin Canc Res 1998, 4: 2763–2770. [PubMed] [Google Scholar]
- 40.Lyons SD, Sant ME, Christopherson RI: Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. J Biol Chem 1990, 265:11377–11381. [PubMed] [Google Scholar]
- 41.Rais R, Jancarik A, Tenora L, Nedelcovych M, Alt J, Englert J, Rojas C, Le A, Elgogary A, Tan J, et al. : Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem 2016, 59:8621–8633. [DOI] [PubMed] [Google Scholar]
- 42.Tenora L, Alt J, Dash RP, Gadiano AJ, Novotna K, Veeravalli V, Lam J, Kirkpatrick QR, Lemberg KM, Majer P, et al. : Tumor-targeted delivery of 6-Diazo-5-oxo-l-norleucine (DON) using substituted acetylated lysine prodrugs. J Med Chem 2019, 62: 3524–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. : Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu K, Kaira K, Tomizawa Y, Sunaga N, Kawashima O, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Yamada M, et al. : ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br J Canc 2014, 110: 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P, Ge M, Hu J, Li X, Che L, Sun K, Cheng L, Huang Y, Pilo MG, Cigliano A, et al. : A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66:167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernhardt S, Bayerlová M, Vetter M, Wachter A, Mitra D, Hanf V, Lantzsch T, Uleer C, Peschel S, John J: Proteomic profiling of breast cancer metabolism identifies SHMT2 and ASCT2 as prognostic factors. Breast Canc Res 2017, 19:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Esslinger CS, Cybulski KA, Rhoderick JF: Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem 2005, 13: 1111–1118. [DOI] [PubMed] [Google Scholar]
- 48.Chiu M, Sabino C, Taurino G, Bianchi MG, Andreoli R, Giuliani N, Bussolati O: GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino Acids 2017, 49: 1365–1372. [DOI] [PubMed] [Google Scholar]
- 49. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, et al. : Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 2018, 24: 194–202. * The authors demonstrated that blocking ASCT2 using V-9032 can reduce cancer growth and lead to cancer cell death. This study shows how glutamine uptake via transporters can be targeted for cancer therapy.
- 50.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et al. : Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Canc Cell 2010, 18:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stalnecker CA, Ulrich SM, Li Y, Ramachandran S, McBrayer MK, DeBerardinis RJ, Cerione RA, Erickson JW: Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci U S A 2015, 112:394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukey MJ, Cluntun AA, Katt WP, Lin MJ, Druso JE, Ramachandran S, Erickson JW, Le HH, Wang ZE, Blank B, et al. : Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep 2019, 29:76–88 e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Duvall B, Zimmermann SC, Gao R-D, Thomas AG, Kalčic F, Veeravalli V, Elgogary A, Rais R, Rojas C, Le A, et al. : Allosteric kidney-type glutaminase (GLS) inhibitors with a mercaptoethyl linker. Bioorg Med Chem 2020, 28:115698. * The authors synthesized and demonstrated the therapeutic potentials of new GLS inhibitors, which are BPTES derivatives containing mercaptoethyl linker. This study demonstrates the constant development and advancement of GLS targeting drugs.
- 54.Zimmermann SC, Wolf EF, Luu A, Thomas AG, Stathis M, Poore B, Nguyen C, Le A, Rojas C, Slusher BS, et al. : Allosteric glutaminase inhibitors based on a 1,4-Di(5-amino-1,3,4-thiadiazol-2-yl)butane scaffold. ACS Med Chem Lett 2016, 7: 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le A, Stine ZE, Nguyen C, Afzal J, Sun P, Hamaker M, Siegel NM, Gouw AM, Kang BH, Yu SH, et al. : Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia-cell cycle dual reporter. Proc Natl Acad Sci U S A 2014, 111: 12486–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nabi K, Le A: The intratumoral heterogeneity of cancer metabolism. Adv Exp Med Biol 2018, 1063:131–145. [DOI] [PubMed] [Google Scholar]
- 57.Dagogo-Jack I Shaw ATJNrCo: Tumour heterogeneity and resistance to cancer therapies, vol. 15; 2018:81. [DOI] [PubMed] [Google Scholar]
- 58.Ramachandran S, Pan CQ, Zimmermann SC, Duvall B, Tsukamoto T, Low BC, Sivaraman J: Structural basis for exploring the allosteric inhibition of human kidney type glutaminase. Oncotarget 2016, 7:57943–57954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. : Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Canc Therapeut 2014, 13:890–901. [DOI] [PubMed] [Google Scholar]
- 60.Okazaki A, Gameiro PA, Christodoulou D, Laviollette L, Schneider M, Chaves F, Stemmer-Rachamimov A, Yazinski SA, Lee R, Stephanopoulos G, et al. : Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J Clin Invest 2017, 127: 1631–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D, Graeber T, Jackson NJ, Czernin J, Emberley E, et al. : Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep 2017, 18: 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cohen AS, Geng L, Zhao P, Fu A, Schulte ML, Graves-Deal R, Washington MK, Berlin J, Coffey RJ, Manning HC: Combined blockade of EGFR and glutamine metabolism in preclinical models of colorectal cancer. Transl Oncol 2020, 13:100828. * The authors demonstrated that combining GLS1 targeting using CB-839 and the EGFR-targeted monoclonal antibody cetuximab decreased colorectal cancer cell viability. This study provides another example of the synergistic effect of the combination of CB-839 and other clinical drugs.
- 63.Raczka AM, Reynolds PA: Glutaminase inhibition in renal cell carcinoma therapy. Canc Drug Resist 2019, 2:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thompson RM, Dytfeld D, Reyes L, Robinson RM, Smith B, Manevich Y, Jakubowiak A, Komarnicki M, Przybylowicz-Chalecka A, Szczepaniak T, et al. : Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8:35863–35876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, et al. : Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126:1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Antonio MJ, Le A: Different tumor microenvironments lead to different metabolic phenotypes. Adv Exp Med Biol 2018, 1063: 119–129. [DOI] [PubMed] [Google Scholar]
- 67.Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. : Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, eaav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoang G, Udupa S, Le A: Application of metabolomics technologies toward cancer prognosis and therapy. Int Rev Cell Mol Biol 2019, 347:191–223. [DOI] [PubMed] [Google Scholar]
- 69.Hersh LB: Rat liver omega-amidase. Purification and properties. Biochemistry 1971, 10:2884–2891. [DOI] [PubMed] [Google Scholar]
- 70.Le A, Udupa S, Zhang C: The metabolic interplay between cancer and other diseases. Trends Canc 2019, 5:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katt WP, Lukey MJ, Cerione RA: A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med Chem 2017, 9:223–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML: The role of glutaminase in cancer. Histopathology 2020, 76: 498–508. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. : Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 2010, 107: 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang C, Liu J, Zhao Y, Yue X, Zhu Y, Wang X, Wu H, Blanco F, Li S, Bhanot G, et al. : Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 2016, 5, e10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z: Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010, 107: 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu L, Zhou D, Li F, Ji L: Glutaminase 2 functions as a tumor suppressor gene in gastric cancer. Transl Cancer Res 2020, 9: 4906–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matés JM, Campos-Sandoval JA, Márquez J: Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys Acta Rev Canc 2018, 1870:158–164. [DOI] [PubMed] [Google Scholar]
- 78.Saha SK, Islam SMR, Abdullah-Al-Wadud M, Islam S, Ali F, Park KS: Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J Clin Med 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neale JH, Yamamoto T: N-acetylaspartylglutamate (NAAG) and glutamate carboxypeptidase II: an abundant peptide neurotransmitter-enzyme system with multiple clinical applications. Prog Neurobiol 2020, 184:101722. [DOI] [PubMed] [Google Scholar]
- 80.Dang CV, Le A, Gao P: MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Canc Res 2009, 15:6479–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le A, Dang CV: Studying Myc’s role in metabolism regulation. Methods Mol Biol 2013, 1012:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kirsch BJ, Chang SJ, Le A: Non-hodgkin lymphoma metabolism. Adv Exp Med Biol 2018, 1063:95–106. [DOI] [PubMed] [Google Scholar]
- 83.Lapidus RG, Tiffany CW, Isaacs JT, Slusher BS: Prostate-specific membrane antigen (PSMA) enzyme activity is elevated in prostate cancer cells. Prostate 2000, 45:350–354. [DOI] [PubMed] [Google Scholar]
- 84.Asaka R, Le A: Dual role of N-acetyl-aspartyl-glutamate metabolism in cancer monitor and therapy. Mol Cell Oncol 2019, 6, e1627273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, et al. : Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Canc Res 2013, 73:4429–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salani B, Rio AD, Marini C, Sambuceti G, Cordera R, Maggi D: Metformin, cancer and glucose metabolism. Endocr Relat Canc 2014, 21:R461. [DOI] [PubMed] [Google Scholar]
- 87.Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, Semenza GL, Xie G: Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis 2019, 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Padakanti PK, Li S, Schmitz A, Mankoff D, Mach RH, Lee HS: Automated synthesis of [11C]L-glutamine on Synthra HCN plus synthesis module. EJNMMI Radiopharm Chem 2019, 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H: Combination therapy in combating cancer. Oncotarget 2017, 8:38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ahluwalia GS, Grem JL, Hao Z, Cooney DA: Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 1990, 46:243–271. [DOI] [PubMed] [Google Scholar]
- 91.Chittur SV, Klem TJ, Shafer CM, Davisson VJ: Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry 2001, 40:876–887. [DOI] [PubMed] [Google Scholar]
- 92.Wada K, Hiratake J, Irie M, Okada T, Yamada C, Kumagai H, Suzuki H, Fukuyama K: Crystal structures of Escherichia coli gamma-glutamyltranspeptidase in complex with azaserine and acivicin: novel mechanistic implication for inhibition by glutamine antagonists. J Mol Biol 2008, 380:361–372. [DOI] [PubMed] [Google Scholar]
- 93.Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, Hansen JC, Curthoys NP: Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 2007, 406:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, Wei W, Hurov JB: Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50: 10764–10770. [DOI] [PubMed] [Google Scholar]
- 95.Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, et al. : Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 2015, 125: 2293–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V: Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995, 22:3–10. [PubMed] [Google Scholar]
- 97.Cha YJ, Kim ES, Koo JS: Amino acid transporters and glutamine metabolism in breast cancer. Int J Mol Sci 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, et al. : Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Canc Cell 2015, 27: 354–369. [DOI] [PMC free article] [PubMed] [Google Scholar]