

Abstract
Human pluripotent stem cells (hPSCs) are a promising source of cells for cell replacement-based therapies as well as modeling human development and diseases in vitro. However, achieving fate control of hPSC with a high yield and specificity remains challenging. The fate specification of hPSCs is regulated by biochemical and biomechanical cues in their environment. Driven by this knowledge, recent exciting advances in micro/nanoengineering have been leveraged to develop a broad range of tools for the generation of extracellular biomechanical and biochemical signals that determine the behavior of hPSCs. In this review, we summarize such micro/nanoengineered technologies for controlling hPSC fate and highlight the role of biochemical and biomechanical cues such as substrate rigidity, surface topography, and cellular confinement in the hPSC-based technologies that are on the horizon.
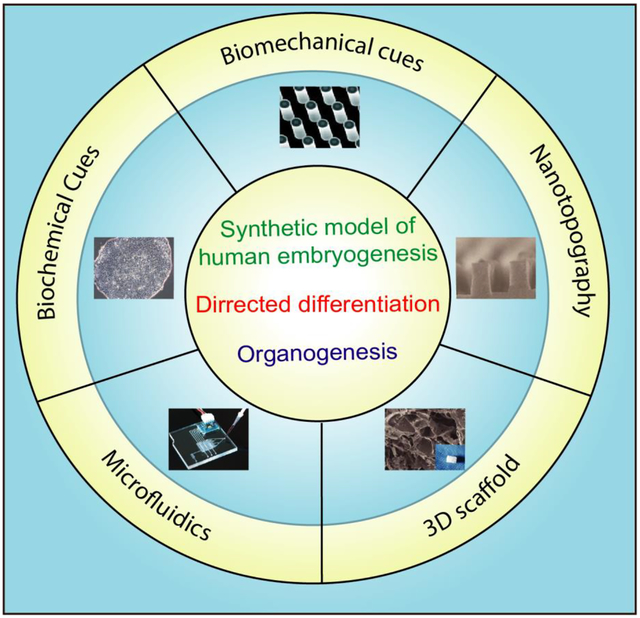
Graphical abstract
1. Introduction
Human pluripotent stem cells (hPSCs) have steadily risen to the forefront as a powerful tool for fundamental studies of human development as well as for translational research in drug development and regenerative medicine [1–4]. hPSCs, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can give rise to all three germ layers (i.e., endoderm, mesoderm, and ectoderm) and downstream somatic cell types. This differentiation potential, along with their ability to self-renew without losing stemness, makes them ideal for the generation of cells for biomedical applications, including disease modeling [3, 5] and high-throughput in vitro drug screening.
The development of the human embryo is arguably the most dynamic process that occurs in the human life cycle. It involves a plethora of events, including cell sorting, self-organization into 3D structures, patterning, migration, and specification. The complexity of embryonic development highlights the amazing capability of hPSCs to respond to a wide range of environmental parameters in very distinct and specific ways. This has prompted researchers to design different biomimetic and biological systems that allow for multiparametric microenvironmental control. Understanding how the niche microenvironment regulates their self-renewal and differentiation will be invaluable for both developmental studies and cell replacement therapy development.
Over the past few years, various micro/nanoengineering methods for hPSC fate and function control have been introduced and implemented for biomedical and biological research [1, 2]. These micro/nanoengineered technologies have shed light onto how cellular behaviors such as cell proliferation, migration, survival, and differentiation are controlled by biophysical signals of cell microenvironment, such as ECM topography and rigidity, cell shape and geometry, spatial organization of adhesive proteins, and extracellular forces. For example, substrate rigidity and dimensionality have been controlled with both hydrogels and the use of micro-post arrays and have been shown to affect hESC differentiation and self-organization [3]. Geometrical confinement is commonly achieved through micro-contact printing and has been shown to affect both cytoskeletal traction force [4] and morphogen distribution among cells [5]. Nanoscale ridge/groove patterns fabricated using UV-assisted capillary force lithography have been utilized to induce hESC differentiation into a neural lineage [6]. Knowing the importance of the microenvironment on hESC behavior, researchers have bioengineered a variety of platforms that can capture critical features of the in vivo environment, including microfluidic devices and microfabricated cell culture substrates. The robust control of parameters such as the biochemical and physical properties of the cell microenvironment in these bioengineered systems has allowed for studies on how exogenous factors influence embryonic development.
This review aims to present an overview of the state of the art of micro/nanoengineered technologies for controlling hPSC fate and function. First, we summarize diverse culture platforms and the biochemical cues used for maintaining pluripotency and self-renewal of hPSC. We then discuss the roles of biomechanical cues such as substrate rigidity, surface topographies, and cellular confinement in determining hPSC fate. Finally, we discuss the application of microfluidic devices for engineering hPSCs.
2. Biochemical approaches for in vitro hPSC maintenance
Research with hPSCs requires long-term cell culture without loss of pluripotency. Traditionally, hPSCs have been cultured on feeder cells, which are cells that secret multiple growth factors that support hPSC self-renewal (Fig. 1a) [7, 8]. For example, mitotically inactivated mouse embryonic fibroblast cells (MEFs), which have been utilized to maintain mouse ESC self-renewal, are commonly used in the maintenance of hPSCs. However, there is a risk of murine pathogens transferring from the MEFs to the hPSCs. These pathogens can cause zoonosis in cell transplantation recipients [9, 10]. Additionally, feeder-based cultures suffer from cytogenic aberrations due to the repeated enzymatic treatments, which poses a challenge for achieving controllable hPSC culture systems [11]. Murine feeder cells can be replaced by human feeders such as human adult marrow cells and human foreskin fibroblasts [12–14]. However, using feeder cells results in increasing the cost of hPSC production, limiting the scaling-up of hPSCs for clinical applications [9]. More recently, feeder-free systems have been introduced with the use of conditioned medium (CM) in conjunction with human serum and purified ECM proteins like Matrigel [15, 16] (Fig. 1b). In the case of ECM protein substrates, researchers found a twofold increase in the expansion of cells compared to hESCs grown in MEF-CM [10]. Batch-to-batch variation of biological materials and the need for costly tests to ensure the absence of pathogens have led researchers to develop synthetic substrates. Thus far, defined peptide and protein surfaces have been used as synthetic ECM for cell culture (Fig. 1c). Melkoumian et al. [17] developed synthetic peptide-acrylate surfaces (PAS) to create an appropriate environment for pluripotency maintenance of different hPSC lines in several commercially available media, including KnockOut SR-supplemented medium and the chemically defined medium mTeSR1 for more than ten passages. Their study showed that high functional peptide density on the plating substrate and uniform peptide distribution result in hESC expansion, phenotypic marker expression, and cell morphology similar to that on Matrigel.
Fig. 1.
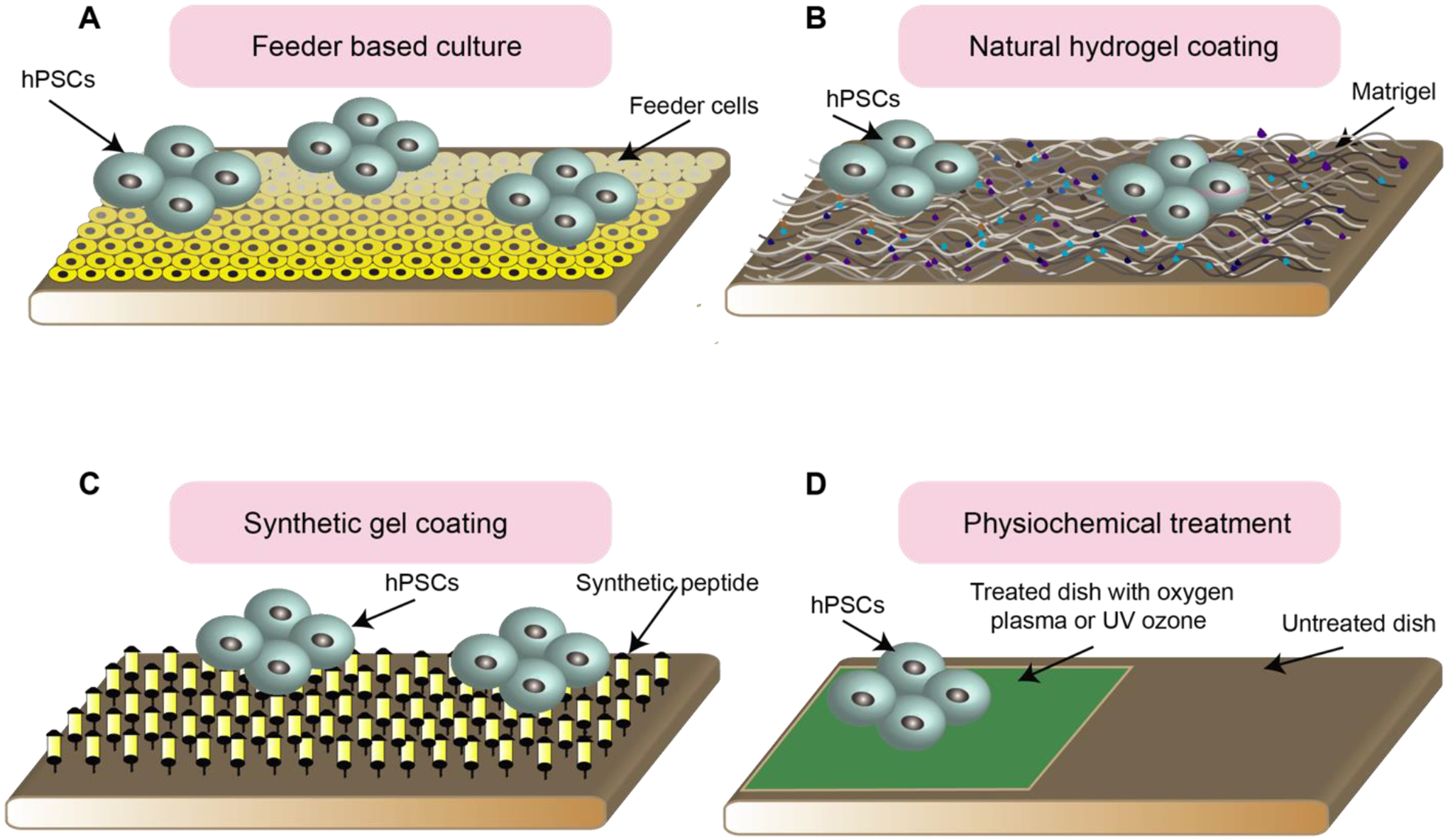
Culture platforms for hPSC maintenance and expansion. (A) Cartoon showing culturing hPSCs on the feeder culture. (B) Cartoon showing feeder free culture of hPSCs on the substrates coated with natural hydrogel. (C) Cartoon showing feeder free culture of hPSCs on the substrates coated with synthetic gel. (D) Cartoon showing feeder free culture of hPSCs on the dishes treated with UV ozone or oxygen plasma.
Similarly, Kolhar et al. [18] have generated a unique peptide-based surface using a high-affinity cyclic RGD peptide for the long time culture of hPSCs. This substrate provides a surface supporting integrin-mediated cell attachment, protecting hPSCs against apoptosis generated from loss of attachment to the extracellular matrix substrate, called anoikis. Several other studies are using synthetic polymers to provide a desirable environment for the long-term self-renewal of hPSCs. For informative discussions, readers are referred to these excellent papers [18–24].
Cells in culture respond to a plethora of biochemical and biomechanical signals. When using polymers as substrates, polymer features can be used to increase cell control and cultivation efficiency. Microarrays are a great tool for identifying appropriate polymer features [25, 26]. In this process, a large number of monomers with different ratios can be synthesized in nanoliter volumes. Brafman et al. [27], probed the use of an array-based high-throughput screening approach to identify a synthetic polymer, poly(methyl vinyl ether-alt-maleic anhydride) (PMVE-alt-MA), which could promote attachment, self-renewal, and proliferation of several hPSC lines over five passages. In another related study, Hansen et al. [28] reported a two-step approach for the rapid fabrication of 7,316 polymer features on a glass slide to discover the best substrate for cultivation and self-renewal of hESC. The process consists of generating a fluorous mask in which two monomers along with a photo-initiator and cross-linker are printed. This process is done to generate a large number of compositions with various chemical characteristics [29].
In addition to functionalizing the surface of the substrate as described above, physical methods can also be used to prepare new 2D surfaces for long-time self-renewal of hESCs. For example, oxygen plasma-etched tissue culture polystyrene (PE-TCP) surfaces can be generated by putting polystyrene substrates under radio frequency oxygen plasma (Fig. 1d). This treatment raises the oxygen content at the surface of the substrate by 1.6-fold, enabling attachment and proliferation of hESCs. Mahlstedt et al. [30] investigated the use of PE-TCP for long-term pluripotency maintenance of various hESC cell lines, including HUES7 and NOTT1. Furthermore, oxygen plasma etching can modify the surface chemistry of the standard tissue culture polystyrene (TCPS) to support hPSC growth and proliferation. Supporting this view, Saha et al. [31] developed culture conditions based on UV/ozone radiation modification of cell culture plates to provide a suitable substrate for hPSC culture. This attractive cell culture platform generates more than three times the number of cells generated by feeder-containing substrates. Surface analysis of the modified TCP showed an increased concentration of molecular species. This leads to a surface with a higher degree of hydrophilicity, which increases protein binding and cell attachment.
3. Biomechanical approaches for in vitro culture or differentiation of hPSC
3.1. Mechanical stiffness of the extracellular matrix
It has been demonstrated that hPSCs have mechano-sensitive and mechano-responsive properties that affect their self-renewal and differentiation [4, 32–34]. Substrate rigidity modulates hPSC behaviors partially through intracellular cytoskeleton and actomyosin contractility [32]. Engler et al. were the first to use tissue-mimicking matrix stiffness to observe how mechanical properties of the ECM affect hPSC differentiation [35]. They showed that culturing human mesenchymal stem cells (hMSCs) in polyactrylamide (PA) hydrogel substrates with brain-mimicking stiffness led to neurogenesis, while muscle- and bone-like stiff PA substrates promoted cardiogenesis and osteogenesis, respectively. Keung et al. also used PA hydrogels to modulate substrate rigidity and found that soft substrate stiffness in vitro promoted hPSC neural ectoderm differentiation [32].
In addition to PA hydrogel substrates, elastomeric micropost arrays can be used to modulate substrate rigidity and study its effect on cytoskeleton contractility and differentiation of hESCs [4, 33] (Fig. 2a). Top surfaces of the micropost array are functionalized with adhesive ECM proteins to promote hPSC attachment. The substrate rigidity of the array can be easily controlled by changing post height while leaving other substrate properties such as surface chemistry and adhesive ligand density unchanged. Moreover, each post functions as a cantilever to measure subcellular contractile force [34, 36]. The micropost array has been useful for the study of mechanotaxis [36], single-cell mechanical homeostasis [37], and stem cell differentiation [38]. This section will mainly discuss the use of polydimethylsiloxane (PDMS) microposts to modulate substrate rigidity and measure contractile forces to study the differentiation of hPSCs.
Fig. 2.
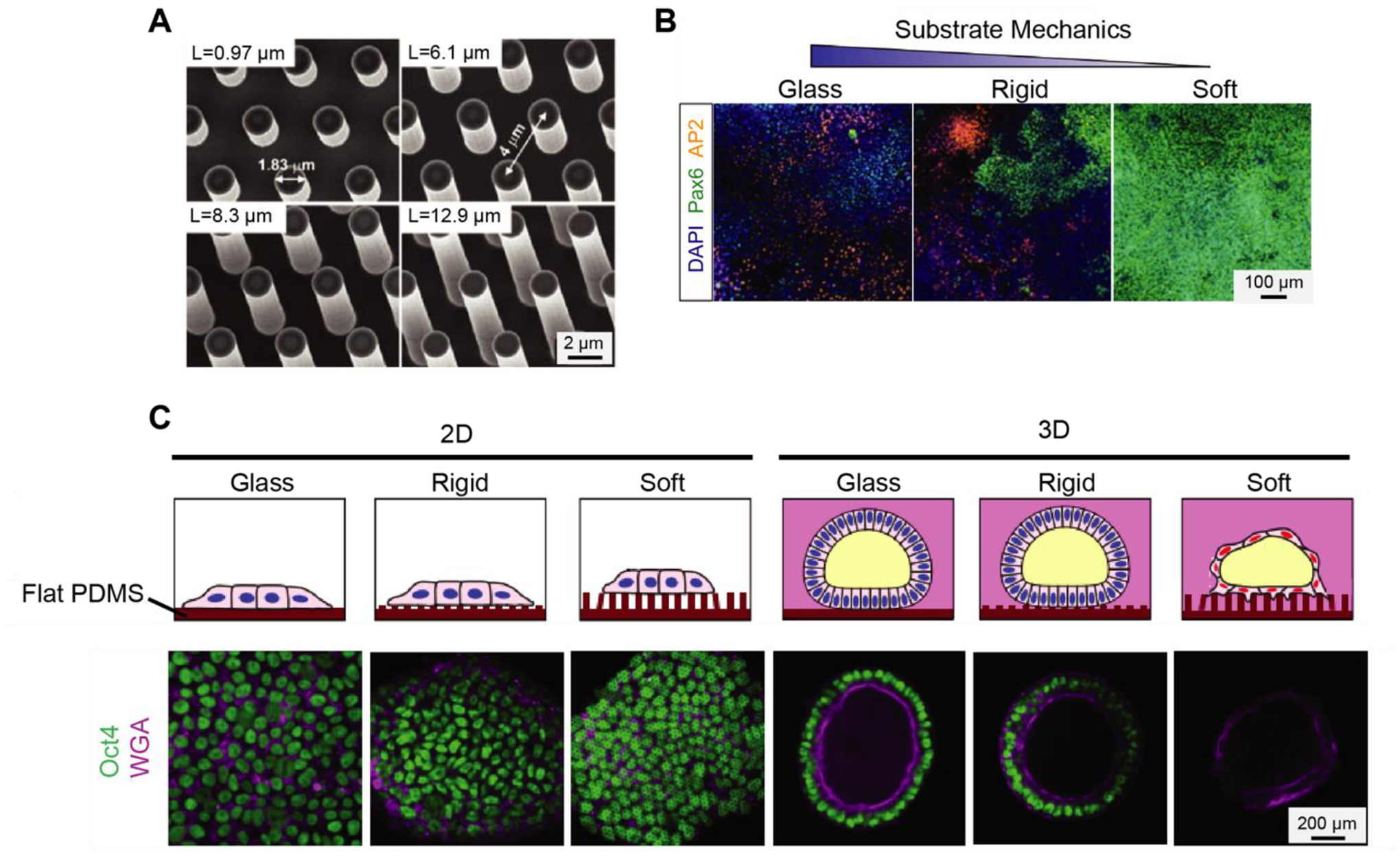
Microposts arrays are used to study the mechanoresponsive behaviors of hPSCs. (A) Scanning Electron Microscopy (SEM) images of microfabricated silicon micropost array masters with different post height. Adapted with permission from [33]. (B) Immunofluorescence and quantitative results are showing PAX6+ neural epithelial cells and AP2+ neural crest cells cultured on vitronectin-coated coverslips and rigid and soft PDMS micropost array. Adapted with permission from [33]. (C) Confocal micrographs showing staining of OCT4 and WGA for hPSCs cultured in the indicated conditions. Adapted with permission from [39].
Using a PDMS micropost array, Sun et al. [4] showed that hESCs are mechanosensitive, as their contractility was increased with increasing substrate rigidity. They also showed that rigid substrates support the pluripotency of hESCs, while soft substrates promote the differentiation of hESCs as reflected by the down-regulation of E-cadherin. Another work from Sun et al. [33] showed that neural induction of hPSCs could be expedited using a PDMS micropost array of low rigidity (Fig. 2b). Furthermore, the authors demonstrated that such mechanotransductive neuronal differentiation of hPSCs involved Smad phosphorylation and nucleocytoplasmic shuttling controlled by mechanosensitive Hippo-YAP activities. The micropost array has also been used to study hPSCs in mechanically controlled 3D culture environments. For example, Shao et al. [39] investigated the effect of substrate rigidity on hPSC self-organized amniogenesis using a PDMS micropost array (Fig. 2c). Interestingly, the authors found the development of squamous amniotic ectoderm-like cysts occurred only in hPSCs cultured on microposts with low rigidity. In addition, they discovered that hPSCs cultured on both soft and rigid microposts in 2D conditions without Geltrex overlay maintained pluripotency and did not form cysts. These results demonstrated that both 3D dimensionality of the ECM and low mechanical rigidity were needed to trigger the amniotic differentiation of hPSCs.
3.2. Nano topography controls hPSC fate
Within native tissues, cells interact with different nanoscale features of the surrounding extracellular matrix, varying from porous fibrous connective tissue to more tightly woven basement membranes [40]. These nanometers to micrometer topographical features possess a complicated mixture of ridges, grooves, fibers, and pores [41] which regulate cell-cell interaction, cell-soluble factor interaction, cell–ECM interaction, and cell-mechanical stimuli interactions [42–45]. Basement membrane, a common type of ECM, is an example of an in vivo substrate that presents a mixture of different surface topographies regulating the fate and function of different types of cells. The effects of surface topography on cell behavior have been under investigation for several years, and research has shown that mammalian cells respond to synthetic nanotopographies [46–48]. Different nanoengineering tools and synthesis approaches have been introduced to create different nanotopographical surfaces, nanopatterns, and scaffolds for in vitro stem cell research.
Nanotopographical features are used for both maintenance [49, 50] and differentiation [51–54] of many cell types. In particular, recent studies have demonstrated that some types of topographical features can provide regulatory signals for adhesion, proliferation, and self-renewal of hPSCs [49, 55–58]. For example, Bae et al. [49] cultured cells on a nanopillar topography to investigate its effect on colony formation and the expression of pluripotency markers in hESCs. Cell-nanopillar interaction leads to cytoskeletal reorganization by forming focal adhesions and restricted colony spreading, which increases E-cadherin mediated cell-cell adhesions in hESC colonies. It was demonstrated that the formation of a compact colony is indispensable for the retention of an undifferentiated hESC state. Further, culture in the nanopillar surface resulted in a higher expression of pluripotency markers as compared to culture on a flat substrate. In another study, Chen et al. [57] used nanorough glass coverslips with various roughness levels and reported an optimized level of nanoroughness that promotes proper cell function and enhances expression of pluripotency markers (Fig. 3a).
Fig. 3.
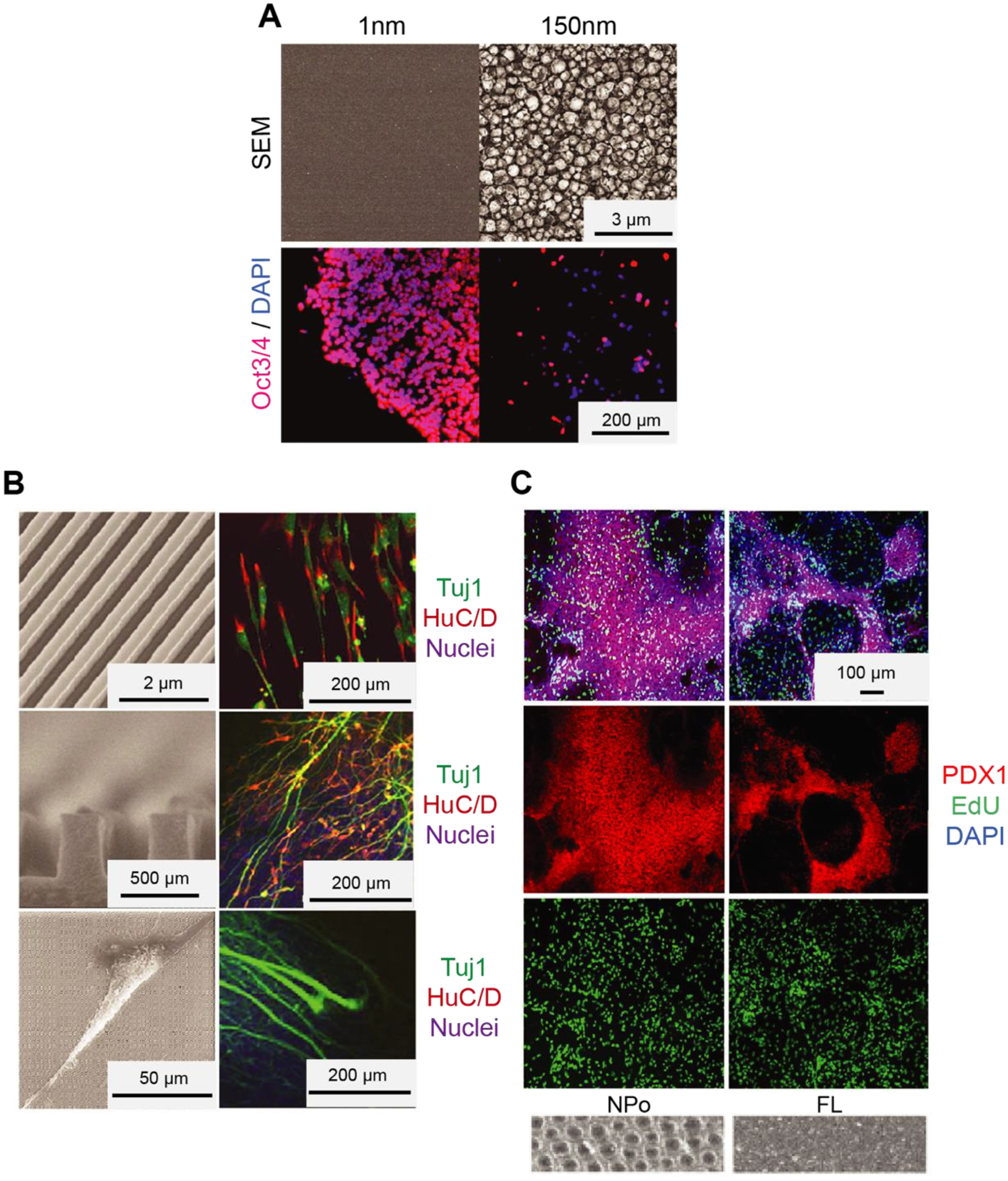
Nanotopography regulates hPSCs self-renewal and differentiation. (A) Culturing hESCs on platforms with surface topographies. SEM images of glass surfaces with surface topographies (top) and immunofluorescence images of hESCs (bottom) cultured on a glass surface with their indicated root-mean-square (RMS) nanoroughness Rq. The cells were stained for nuclei (DAPI; blue) and pluripotency marker (OCT3/4; red). Courtesy of the U.S. National Library of Medicine [57]. (B) Differentiation of hESCs into selective neurons on ridge/groove patterns. SEM images of a bird’s eyes view of 350-nm ridge/groove pattern arrays (height of 500 nm, the spacing of 350 nm) (left top), a cross-section (left middle), and a SEM image which shows hESCs on the ridge/groove pattern arrays (left bottom). Immunofluorescence images of hESCs stained with nuclei, neural and glial marker (HUC/D, TUJ1). Courtesy of the U.S. National Library of Medicine [6]. (C) Differentiation of hESC-derived endoderm to pancreatic progenitors on nanopores with 200 nm diameter. Immunofluorescence images of hESC-derived endoderm with nuclei (DAPI) and critical transcription factor for pancreatic development (PDX1) (If available from the original paper, add SEM images or cartoons in c, to show what the topography looks like) Adapted with permission from [64].
Nanotopographical features can also be utilized to direct the differentiation of hESCs into different cell types (Fig. 3b, c) such as neural [6, 59–62], cardiac [63], and pancreatic cells [64]. Lee et al. used nanoscale ridge/groove patterns to induce hESCs into a neural lineage [6]. They showed that hESCs seeded on gelatin-coated nanoscale pattern arrays in DMEM/FBS medium could rapidly and effectively differentiate into neuronal lineage without using differentiation-inducing agents. Elongation of the cytoskeleton guided by ridge/groove patterns led to a transfer of tensional force to the nuclei, which influenced signal transduction and gene expression. Similarly, in another study by Pan et al. [61], it was found that hPSCs cultured on the nanografted substrates efficiently differentiate into the neuronal lineage and show elongated and aligned nuclei in the direction of nano/microstructures with increased contact guidance [61, 65]. Another study from Lu et al. [66] showed that nanofibrous scaffolds could cause differentiation of hESCs into the neural lineage when combined with treating the cells with neural induction medium containing Noggin/retinoic acid. It was further proposed that topographical features might improve the cardiomyogenic differentiation of hESCs. Interestingly, Lee et al. [63] reported that hPSCs cultured with no exogenous chemicals for differentiation on a nanorough graphene substrate show enhanced cardiomyogenic differentiation compared to cells cultured on glass or Matrigel. hESCs cultured on the nanorough graphene showed enhanced cell adhesion, leading to the cardiomyogenic differentiation through the ERK signaling pathway. More recently, Kim et al. [64] demonstrated that nanopore-patterned surfaces could remarkably promote the pancreatic differentiation of hPSCs. This study showed that nanopores of 200 nm diameter lead to a 3-fold increase in the percentage of pancreatic cells compared to hESCs cultured on flat surfaces. TAZ was identified as a significant player in the nanopore-induced mechanotransduction facilitating the pancreatic differentiation of hPSCs.
3.3. Cellular confinement
Cells in vivo exist in limited spaces, either encapsulated in ECM or surrounded by other cells and exposed to different gradients of soluble factors and local adhesive motifs [74]. Geometrical confinement of the cells in ECM is crucial for controlling dynamic cellular behaviors, including asymmetrical cell division and cell migration. Coupled with manipulating culture conditions, 2D micropatterning provides important advantages as an assay technology because of its scalability, reproducibility, and high compatibility with live imaging. Conventional culturing systems, such as homogeneous plates and tissue culture dishes with uniform surface treatments, do not properly recapitulate the spatial cell confinement in vivo. In the past 15 years, a plethora of methods have been developed to generate micro/nanoscale patterns of ECM proteins in various shapes and sizes to get insights into the role of spatial confinement in tissue morphogenesis [75]. In particular, a handful of technologies such as microcontact printing (μCP) [76–78], microstencils [79], microwell culture [80, 81], and photopatterning [5] processes have been implemented to study hPSCs.
Micro-contact printing is the most common technique for generating micro/nanoscale adhesive ECM patterns on glass substrates and tissue culture dishes [78, 82, 83]. In this process, an elastomeric PDMS patterned stamp is coated with adhesive proteins or a solution of thiol-containing molecules that can be spontaneously absorbed by the stamp owning to hydrophobic interactions. When the stamp is dried, it is brought into conformal contact with a second substrate which effectively creates protein patterns (Fig. 4a). Since its invention, μCP has been widely adapted to generate micro/nanopatterns of ECM proteins on substrates despite the drawbacks of requiring a two-step coating process and specific humidity conditions [74]. μCP has also been used as an efficient method for generating patterning with hPSCs. Lee et al. [77] reported that by treating hESCs with BMP2 and activin A and by precisely controlling colony size, the cells could differentiate into either definitive endoderm or mesoderm lineages. BMP2 and Activin A act synergistically to activate endoderm-specific genes and mesoderm-specific genes in the system. However, colony size can selectively guide these primitive streak-like cells to either definitive endoderm or mesoderm lineages. In another related study, Hoof et al. achieved hESCs differentiation into pancreatic endoderm-like cells by seeding cells onto a patterned substrate [84].
Fig. 4.
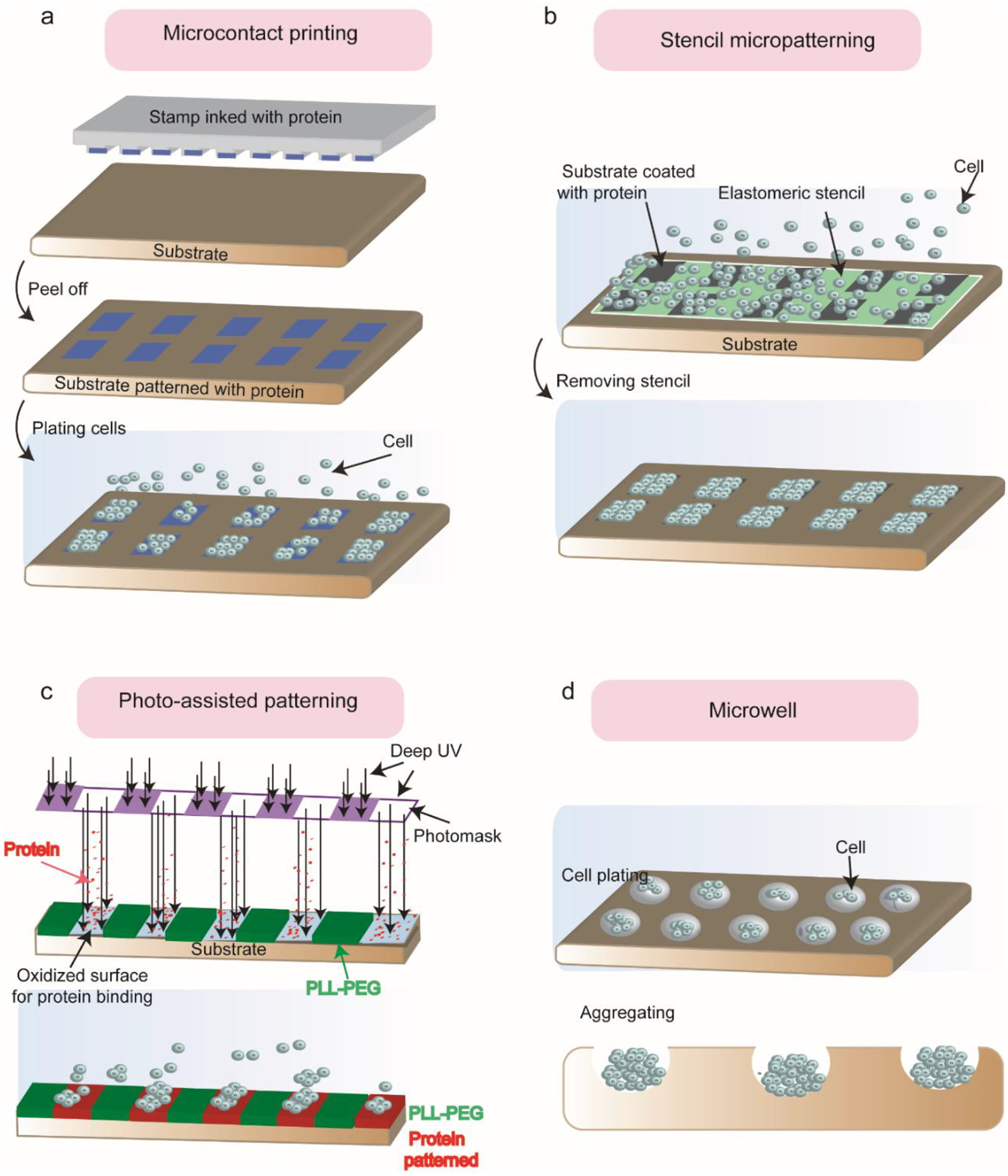
Micro/nano-patterned cell adhesive cues on the substrate. (A) Schematic of microcontact printing (μCP). The protein-coated PDMS stamp is brought into contact with the activated substrate. After peeling the stamp off, the patterned protein is transferred into the substrate. (B) Schematic of elastomeric stencil micropatterning. After coating the substrate with ECM protein, the stencil is applied onto the cell-culture substrate during the seeding process and peeled off after cell plating. (C) Schematic of deep UV-activated micropatterning. Deep UV removes the cell-repellent PLL-g-PEG coating and oxidized the surface underneath for proper binding to the soluble ECM protein (fibronectin) molecules. Therefore, micro-patterns will be transferred from the photomask to the substrate in the presence of cells. (D) Schematic of the microwell for patterning hPSCs. A specific number of cells are seeded into each microwell depending on the size of the well. Cells will aggregate and form the shape of the well.
Another technique to create regular micropatterns of hESCs is stencil-assisted micropatterning. The stencil is a thin sheet with an array of microscale through-holes that will self-seal against the target substrate. As early as 1967, nickel stencils [85] and stainless-steel stencils [86] were used to generate cellular micropatterns on non-adherent acetate. However, the metallic stencils cannot be completely sealed against the target substrate. Researchers have reported the fabrication of a rubber-like stencil that allows for the creation of cellular micropatterns of different cell types on a substrate. In this case, the stencil is applied onto the cell-culture substrate before the seeding process and peeled off after (Fig. 4b). Several studies describe the microstencil method as a robust and simple method for generating hPSC micropatterns capable of working with various ECM proteins and different culture substrates [87]. Yao et al. [79] used stencil micropatterning to generate multilayered hPSC-derived colonies and induce them toward hepatocyte-like cells by performing a multi-staged 17-day differentiation protocol on the cells. In multilayered colonies, cell-cell interaction was enhanced, leading to more mature hepatocyte-like cells with higher levels of Albumin (hepatic marker) than hepatocyte-like cells obtained through more conventional methods.
Both μCP and stencil-assisted micropatterning technologies need access to microfabrication tools, limiting their usage for laboratories that do not have access to microfabrication technologies [74]. A new approach that overcomes this limitation is deep UV-activated micropatterning. This method can create dynamic and stable ECM adhesion patterns on target substrates with a sub-micron resolution [88–91]. It consists of deep UV exposure that oxidizes a polymer coating (e.g., poly(L-lysine)- g-poly (ethylene glycol), PLL-PEG) on a cell culture substrate (e.g., glass or PS) covered by a photomask. Exposed surface areas will become hydrophilic and undergo covalent binding to ECM proteins (Fig. 4c). Using such micro-patterned substrates, Warmflash et al. [5] showed that confinement of hESCs to a disk-shaped pattern is a key factor for recapitulating germ layer patterning. It was shown that colonies with larger diameters resulted in differentiation of hESCs into spatially organized three germ layers. However, in smaller colonies, the inner layer (ectoderm layer) disappeared, and the two outer layers were extended into the center of the colony.Following this work, there are several other studies that use UV-activated micropatterning to generate human gastruloids and study the role of signaling pathways such as BMP, WNT, and ACTIVIN. For informative discussions, readers are referred to these excellent papers [92–96]. To investigate the combined effect of cellular confinement and the biochemical and mechanical properties of the environment, 2D patterning has been coupled with bioengineered tools that allow for the control of chemical signaling and force measurement. Integrative tools such as these allow for comprehensive studies on the interplay between tissue geometry, mechanical force, and cell signaling.
Geometrical confinement plays an essential role in the self-organization of hPSCs into in vivo- like structures. However, efficient generation of these structures also requires control over cell number and cell type composition in the initial cell cluster. Microwell arrays offer control over blastocyst-like structures [97–99]. Yu et al. [98] used a sequential differentiation protocol to generate human blastoids from naïve hPSCs cultured in AggreWell plates. These human blastoids resemble the human blastocyst both morphologically and transcriptionally. Further, they show that cells from the blastoid can give rise to downstream embryonic and extraembryonic cell types. Liu et al. [99] cultured induced PSCs in AggreWell plates to generate blastocyst-like structures coined iBlastoids. These iBlastoids consist of a trophectoderm-like cyst with an inner aggregate of epiblast- and primitive endoderm-like cells. Further, they show that the iBlastoids can recapitulate aspects of early implantation. Each of these human blastocyst models provides unique insights into the mechanisms driving early human embryogenesis. While the generation efficiency of these structures remains relatively low, future efforts could include a move towards automated, high throughput protocols like the one designed by Czerniecki et al. for the generation of human kidney organoids [100].
3.4. Local mechanical perturbation
Tissues and cells in human and animal bodies are continuously subjected to different mechanical stresses, including shear, tensile, and compressive stresses. Mechanical stimuli play essential roles in various biological actions, including proliferation, differentiation, migration, and contraction [102, 103]. Many techniques and tools have been generated to identify the role of mechanical forces in tissue engineering, cell biology, and regenerative medicine. These technologies can be used to study force-dependent dynamics or measure the local mechanical properties of some molecules in mechanotransduction. In this section, four different techniques used to study the mechanical properties of hPSCs, including an optical tweezer, atomic force microscopy, acoustic tweezer, and stretchable membrane, are discussed.
Optical tweezers [104, 105] and magnetic tweezers [106–108] are techniques commonly used to provide force and displacement on the surface of the cell or within a defined region of a cell (Fig. 5a). In this technique, microbeads are functionalized with an antibody or adhesive ligand to bind to specific receptors on the surface of the cells. The tweezers apply forces to the microbeads to balance forces transferred from cells to the beads. This force can be calculated with parameters of the microbeads and optical/magnetic fields [109]. Optical tweezers have been utilized to compare the mechanical properties of undifferentiated hPSCs with the mechanical properties of differentiated hPSCs [110, 111]. Tan et al. [110] used optical tweezers to explore how dynamic and static micromechanical properties of hESCs vary when differentiating toward cardiac cells. It was shown that pluripotent stem cell-derived cardiomyocytes (hPSC-CM) have a higher stiffness than undifferentiated hESC due to increased organized myofibrillar assembly. Similarly, atomic force microscopy (AFM) can probe cell components by applying force in the resolution of 10−12 N and displacement with a resolution of 1 nm. In this technique, an electronic controller moves an elastic cantilever beam over the cell, which causes mechanical perturbations (Fig. 5b) [103, 112]. A nano-microscopic tip at the end of the cantilever beam is functionalized with an adhesive ligand that binds to cell receptors. Cantilever movement caused by the electronic controller generates a local stretch or indentation to the cell that can be calculated by measuring the deflection of the cantilever beam. It was found that AFM can quantify the beat force of either a cluster or a single cardiomyocyte cell. Liu et al. [113] used AFM to measure the mechanobiological properties of hPSC-CM, including cellular elasticity, contraction rate, beat force, and duration. As well as measuring the cellular elasticity, AFM enables the measurement of viscoelastic properties at the cellular level. Li et al. [114] proposed a new method to extract viscoelastic properties of living cells exposed to specific types of drugs. For informative discussions about measuring viscoelastic properties of cells using AFM, readers are referred to these excellent papers [115, 116].
Fig. 5.
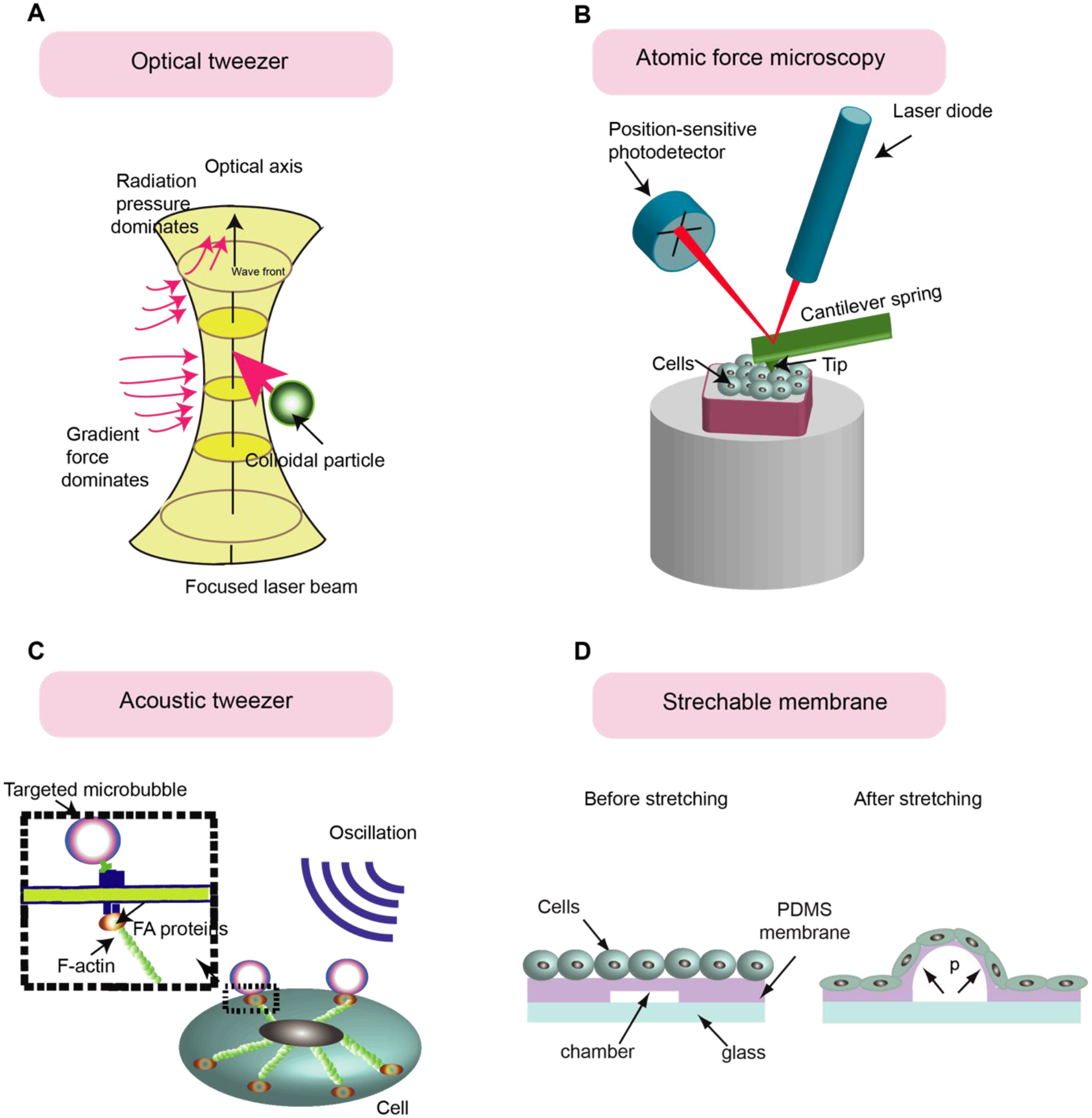
Techniques to apply mechanical perturbations to hPSCs. (A) Optical tweezer provides force and displacement on the cell’s surface or within a defined region of a cell by controlling the displacement of microbeads. (B) Atomic Force Microscopy to apply mechanical perturbations to the cells. An electronic controller moves the cantilever beam, functionalized with the adhesive ligand, to bind the cell surface via adhesive ligand-receptor binding and provide mechanical perturbations to the cells. (C) In the acoustic tweezers cytometry (ATC) method, Acoustic wave vibrating the lipid microbubbles covalently attached to the cell’s surface and applied force to the cell. (D) Stretchable substrate technique to apply mechanical strain to the cells. The cells are plated on the PDMS membranes containing some chambers. By connecting chambers to the vacuum, the PDMS membranes and cells attached to the membrane will be stretched.
Acoustic tweezer cytometry (ATC) applies a local mechanical load to the cells [117, 118]. In this method, lipid microbubbles functionalized with specific ligands can covalently attach to the surfaces of the cells via adhesive ligand-receptor binding (Fig. 5c). An acoustic wave is utilized to vibrate the lipid microbubbles and apply force to the cells. The parameters of the exerted force include frequency, magnitude, period, and duration and are determined by the ultrasound parameters. To improve the cloning efficiency and survival rate of hESCs, Chen et al. [118] used ATC to provide mechanical stimulation to disassociated single hESCs. Integrin-mediated adhesion formation and strengthening by ATC stimulations facilitated the spread of the disassociated hESC, which rescued the cells from hyperactivated actomyosin activities that prompt downstream apoptotic signaling pathways.
Cell stretching devices are utilized to carry out stretching of single cells, colonies, and tissue samples in a way that captures the patterns of deformation experienced by different cell types in the body, including vascular cells, cardiomyocytes, fibroblast, and skeleton muscles. Several studies have reported that mechanical strain can direct the differentiation of hPSCs. Li et al.[119, 120] investigated how uniaxial mechanical strain in parallel to the signaling pathways regulated by TGF-β can modulate the differentiation of neural crest stem cells (NCSCs) into smooth muscle cells (SMCs). In another study, Teramura et al. [120] showed that cyclic strain alters the alignment of actin fibers in hiPSC and the expression of pluripotency markers. Xue et al. [121] reported a micropatterned hPSC-based neuroectoderm developmental model in which pre-patterned geometrical confinement induces emergent neuroepithelial and neural plate border cells. To see the effect of mechanical force on cell differentiation, a custom-designed cell stretching device (Fig. 5d) was generated and applied to stretch central regions of micropatterned cell colonies. They showed that stretching leads to the activation of the BMP signaling pathway and differentiation into neuro plate border cells in the central region of the pattern.
4. 3D biodegradable scaffolds
3D tissue scaffolds are often used for biological applications such as tissue engineering [122]. Porous biodegradable scaffolds can provide a desirable environment to host cell proliferation and adhesion. Furthermore, they can provide a complex 3D matrix for cell maintenance and differentiation. The application of scaffold biomaterials to mimic ECM requires that the biomaterial have high biocompatibility, proper chemistry to induce cell proliferation and adhesion, and the mechanical properties and degradation rate of the ECM of interest. The level of porosity, pore distribution, and the exposed surface area also play a significant role in the architecture of the ECM and penetration of cells into the scaffold volume [123]. Various natural and synthetic biomaterials have been utilized to generate scaffolds for in vitro stem cell research. Scaffolds that have been implemented for hPSC culture are classified into three groups: bioactive hydrogel scaffolds, synthetic biodegradable polymers, and micro/nanofibrous scaffolds.
4.1. Natural scaffolds
Naturally derived hydrogels, including collagen, alginates, and chitosan extracted from animals, plants, and human tissues, exhibit promising biocompatibility and low toxicity for cell culture [124, 125]. Collagen is a widely used natural material for making scaffolds composed of fibrous proteins with a stiff helical structure that provides a suitable structure for cell distribution and capillary formation [124, 126–128]. Chen et al. [129] incorporated hESC-MSCs within a silk-collagen sponge scaffold that provided mechanical strength in conjunction with neo-ligament tissue regeneration to induce tendon-like cells. hESC-MSCs positively expressed tendon-related gene markers including Epha4, Scleraxis, and Collagen type I & III. They also exhibited tenocyte-like morphology when exposed to mechanical stimuli.
Alginate, present in the cell walls of brown alginate, is another naturally derived polysaccharide that is a proper candidate for making 3D scaffolds [130]. Gerecht-Nir et al. [131] used alginate as a scaffold to direct the differentiation of hESCs. They reported the generation of human embryoids (hEBs) and induced vasculogenesis in the forming hEBs within three-dimensional porous alginate scaffolds. They showed that the environment provided by the alginate scaffold pores enables the formation of round, small-sized hEBs and subsequent vasculogenesis. It was concluded that, in addition to chemical cues, physical constraints could also induce and direct differentiation of hESCs. Future use of natural hydrogels, however, might be limited by batch-to-batch variability, low competence for biochemical modifications, and their poor potential in translational research due to the risk of immunogenicity and disease transfer.
4.2. Synthetic biodegradable polymers
Recent studies have focused on the use of different types of polymeric biomaterials including poly(lactic-co-glycolic acid) [132–134], poly(- glycerol sebacate) [135, 136], poly(methyl methacrylate) [137], and poly(caprolactone) [138–140] as a supportive structure for hPSC viability, attachment, and differentiation. Synthetic hydrogels like these are preferable to natural hydrogels because they have a fully defined chemistry and can be engineered to have specific biophysical (e.g., 3D architecture, mechanical stiffness) and biochemical (e.g., growth factors) properties. Further, they have a lower risk for immunogenic reactions [124, 133]. Levenberg et al. [141] explored the neuronal differentiation of hPSCs on 3D polymeric scaffolds made from poly(lactic-co-glycolic acid) and poly(L-lactic acid). In this study, neural rosette-like structures generated throughout the scaffolds in the presence of differentiation factors in the medium, including neurotrophin 3 [NT-3], retinoic acid [RA], and nerve growth factor [NGF]. Synthetic scaffolds can also be used for the 3D cultivation in vivo. Bai et al. [142] generated embryoid bodies and then seeded on the alginate/ poly(lactic-co-glycolic acid) scaffolds and then transplanted into the nude mice for several weeks. The results indicate hESCs differentiate into chondrocytes and further to cartilage like tissue.
A remarkable achievement was accomplished by developing polymer grafted carbon nanotubes (CNTs) scaffolds for directing the differentiation of hPSCs toward neuron cells. CNTs are of high strength but flexible. Furthermore, they are conductive, and their conductivity remains unchanged during harsh situations [143]. These characteristics make polymer grafted CNTs a promising scaffold material for inducing neuronal lineage from hESCs. Supporting this view, Chao et al. [144] generated a thin film scaffold comprising of biocompatible polymer Poly(acrylic acid) (PAA) grafted CNTs which can promote differentiation of hESCs into the neuron cells. According to the observations, PAA is a weak acid by nature that has a negative effect on neuron differentiation. However, the nanoscale fiber morphology of CNTs can enhance both cell adhesion and protein adsorption, making PAA grafted onto CNTs a proper substrate for neuron differentiation and neuron cell attachment. In addition to the neural differentiation of hESCs, studies have used CNTs to investigate the effect of matrix properties on hESC differentiation into other cell types. Sridharan et al. [145] reported the differentiation of hESCs into the ectodermal lineage on the collagen-carbon nanotube (collagen/CNT) composite material.
4.3. Micro/Nanofibrous scaffolds
Nanomaterials have emerged as a great candidate for making scaffolds because of their resemblance to natural ECM, which provides a fitting environment for cell adhesion, proliferation, and differentiation [146, 147]. Furthermore, they are biodegradable and have suitable surface chemistry, appropriate mechanical properties, and the capability to be formed into various sizes and shapes. It has been demonstrated that nanofibrous scaffolds can support the self-renewal of hESCs. Gauthaman et al. [148] cultured hESCs on a scaffold made from Polycaprolactone /gelatin (PCL/gelatin) nanofibrous and PCL/collagen. It was observed that hESCs could proliferate on both scaffolds, showing the capability of nanofibrous scaffolds for long-term maintenance of stemness characteristics of hESCs (Fig. 6b). One possible reason is that the porous nature of the scaffold and large surface to volume ratio offer proper cell and matrix interaction for MEFs attachment and prevent the direct contact of hESCs and MEFs due to the fibroblast-like cell growth of MEFs and colony formation of hESCs in vitro. Supporting this view, Lu et al. [149] reported using an engineered 3D microfiber system supporting long-term hPSCs self-renewal under defined conditions. The unique ability to form microscale fibrous matrices allowed cells to be encapsulated in the scaffold with excellent viability. One advantage of the micro-fibrous system is its ability to support cell culture and differentiation within the same 3D system by manipulating specific medium components. Another study from the same group [66] indicated that nanofibrous scaffolds could also differentiate hESCs into the neural lineage by treating the cells with a neural induction medium containing Noggin/retinoic acid.
Fig. 6.

Scaffold regulates hPSCs self-renewal and differentiation. (A) Derivation of cartilage-like tissue from hESCs on alginate/PLGA (Synthetic polymer) scaffolds, SEM examinations of the PLGA scaffold (left), and the cells/alginate/PLGA complex. (B) The nanofibrous scaffold supports colony formation and maintains the stemness of hESCs. SEM examination of the electrospun nanofibrous scaffold (PCL/gelatin (1:9%w/v)) (left). hESCs cultured on PCL/gelatin nanofibrous scaffolds and MEFs. Courtesy of the U.S. National Library of Medicine [148].
5. Controlling hPSC fate by microfluidic devices
The microenvironment, including extracellular matrix, soluble factors, and mechanical cues, is important for controlling hPSC behavior. Microfluidic systems allow researchers to precisely modulate the microenvironment to control hPSCs maintenance and differentiation through microscale biochemical [150–154] and mechanical stimulation [155–157]. Microfluidic platforms have also been widely used in cell sorting [158–161] and high-throughput single cell analysis [162–167].
There are several recent studies using microfluidic devices to precisely control the hPSC microenvironment and study its effect on hPSC maintenance and differentiation [150, 168–171]. For example, a cell culture platform named inverting microwell array chip was developed to generate hiPSC aggregates with controlled size and geometry [168] (Fig. 7a). The microwells have a depth of 0.8 mm and each wall has a slope angle of 45°C. The cell aggregates were first formed on the bottom of the PEG-based microwells. After the cellular aggregates formed, the chip was inverted to plate the aggregates onto the polystyrene surface. This platform has the potential to study autocrine and paracrine signaling by modulating aggregate size and spacing. Additionally, Sikorski et al. generated a microfluidic device to support the robust generation of clonally-derived colonies to study heterogeneity of hESCs [169]. The single ESCs were cultured in individually addressable chambers to track cell proliferation, morphology, and OCT4 expression. They revealed that low OCT4 expression was correlated with a low growth rate and a less compact morphology. Microfluidic devices were also used to identify the optimal culture conditions of hESCs and hiPSCs [170, 171]. Matsumura et al. [170] found that laminin promoted hiPSC proliferation better than Matrigel. In another study, Yoshimitsu et al. [171] found laminin and fibronectin to be better than collagen and gelatin in terms of attachment and growth rate in hiPSC maintenance.
Fig. 7.
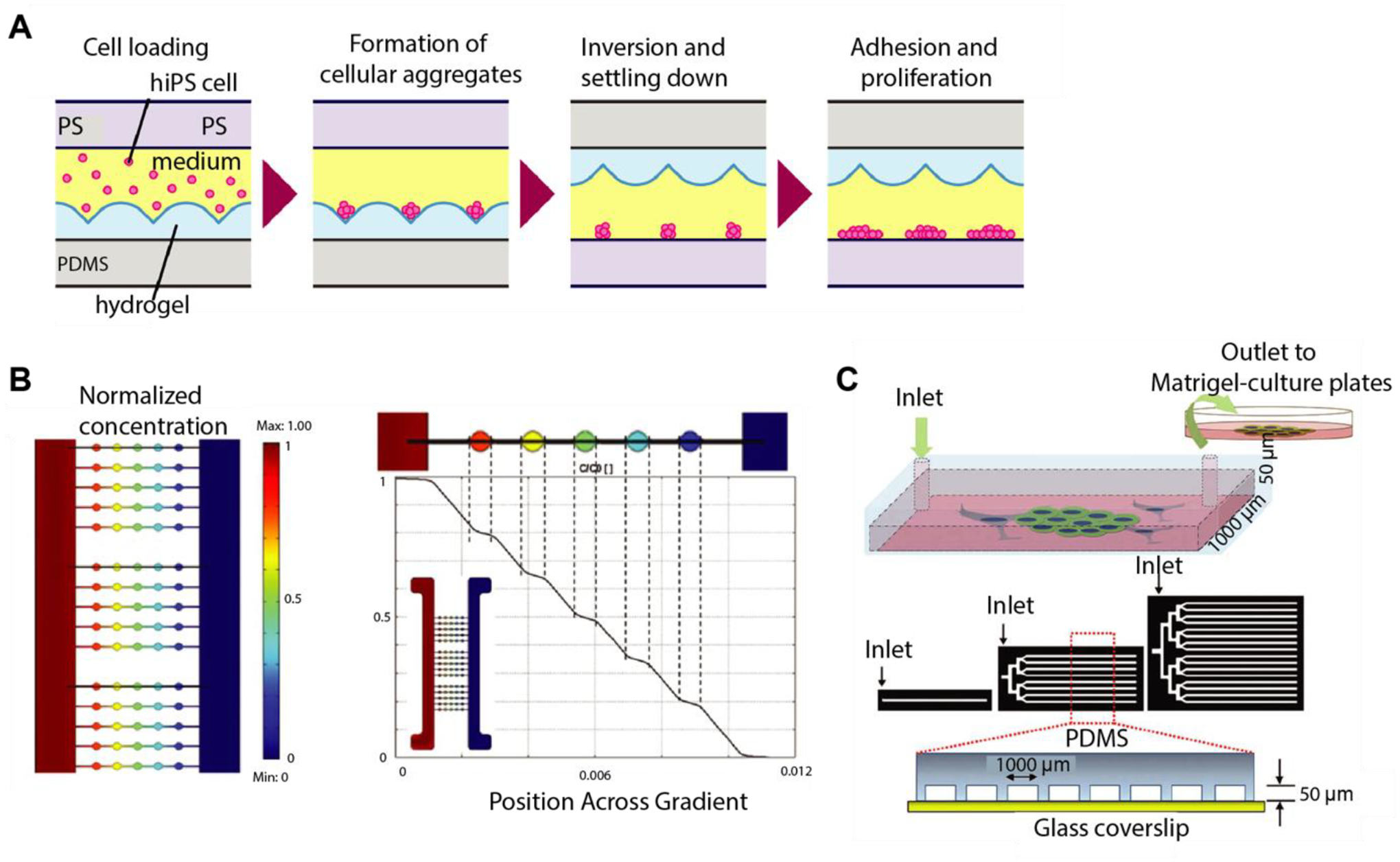
Different applications of microfluidics in hPSC culture. (A) Schematics showing the culture process of hiPSCs in the inverting microwell chip. Courtesy of the U.S. National Library of Medicine [168]. (B) Computational modeling of mass transport within the microbioreactor. Adapted with permission from [152]. (C) Schematics of adhesion strength-based isolation of hPSCs in microfluidic devices. Adapted with permission from [161].
Microfluidic devices can generate chemical gradients to precisely assess the phenotype of hPSCs or model early development [152–154]. Park et al. [151] cultured hESC-derived neural progenitor cells in microfluidic chambers for eight days under gradients of different growth factors including Shh, FGF8, and BMP4. They observed the opposite effect of BMP4 and Shh on differentiation and proliferation of hESC-derived neurons; BMP4 inhibited the SHH mediated proliferation of neural projector cells. A microfluidic device was also used to provide a temporal and spatial gradient of multiple morphogens (Wnt3a, Activin A, BMP4, and their inhibitors) on embryoid bodies (EB) to study the effect of these molecular factors on the fate specification and mesoderm differentiation of hESCs [152] (Fig. 7b). This study showed that a linear concentration of morphogen gradients resulted in non-linear EB differentiation responses. More recently, Kamei et al. developed PDMS devices using soft lithography and 3D printing. They exposed hESCs in a micro-channel to 3D gradients of chemicals created by differences in molecular weight [153]. They showed that the concentration of growth factors in the culturing medium is critical for the formation of hESC spheres. In another study from Zheng et al. [172], a PDMS-based microfluidic device was used to generate chemical gradients to achieve a controllable model system showing developmental events in the post-implantation human embryo. Manfrin et al. [173] used a microfluidic device to expose hPSC colonies to spatiotemporally controlled morphogen gradients generated from artificial signaling centers. They showed exposing hPSC colonies to a localized source of BMP4 resulted in a shift from radial symmetry to axially arranged differentiation domains.
Label-free, microfluidic cell sorting platforms have been widely investigated because of the minimal sample preparation required, the ability to apply precise forces, and their greater compatibility with downstream analysis as compared to traditional cell sorting techniques such as fluorescence-activated cell sorting (FACS) [158–161]. For example, Wang et al. integrated optical tweezers with microfluidic technologies to handle small cell population sorting [158].
They isolated OCT4-GFP+ hESCs from OCT4-GFP− differentiated cells with a 90% recovery rate and 90% purity. Choudhury et al. developed a microfluidic platform to separate hESCs from differentiated cells based on the difference in their cytoskeletal elasticity [159]. The elastic cells were more likely to flow along narrow separation channels than the inelastic ones. In another study, undifferentiated hESCs were isolated from a heterogeneous population based on the hESC surface marker SSEA-4 using an antibody-functionalized PDMS channel [reference]. Singh et al. [160] utilized the differential adhesive strength between hPSCs and somatic cells to rapidly isolate fully reprogramed hiPSCs from heterogeneous reprogramming culture with 95%-99% purity and >80% survival [161] (Fig. 7c). Future work could involve the integration of microfluidic sorting platforms with imaging technologies and downstream biochemical and genomic analysis.
One notable advantage of microfluidics is the potential to integrate lab-based testing in a single chip to perform high-throughput single cell analysis such as on-chip immunoassays [162, 163] and single cell real-time PCR [164–167]. Recently, such technologies have been utilized in hPSC research to study the heterogeneity of hPSCs. Kamei et al. [162] demonstrated the culture and analysis of hESC colonies in an integrated microfluidic platform termed hESC-μChip. hESC-μChip is capable of culturing hESCs in addressable chambers and running phenotypical and functional analyses, including live-cell imaging and immunocytochemistry. In another study, Kamei et al. performed single-cell profiling of protein expression (OCT4 and SSEA-1) with a similar device [163]. In this device, every single chamber could run immunocytochemistry under different hPSC culture conditions. They found that culture in different conditions resulted in the generation of hPSC lines of different phenotypes in which growth rate, morphology, and pluripotency, and differentiation markers all varied. High-throughput single cell analysis methods are essential to study how heterogeneity in hPSC populations can lead to different fate determinations. Microfluidic devices are a uniquely suited tool for single-cell gene expression measurements with a low sample population, reduced cost, and high sensitivity [164–167]. White et al. [166] generated a fully integrated microfluidic device to perform RT-PCR from hundreds of single cells per run. All steps, including cell capture, cell lysis, reverse transcription, and quantitative PCR, were processed in the chip. They observed coregulation of miR-145 and OCT4 in the single cells, which is not apparent from population level measurements. Another study used microfluidic-based single cell gene expression analysis and showed that hiPSCs were more heterogeneous in gene expression than hESCs [167].
6. Conclusion and future perspective
HPSC systems constitute a promising tool for the generation of human tissues, screening for patient-specific therapeutic and drug responses, and in vitro modeling of human diseases. Over the years, progression in the study of hPSC has revealed the significance of the stem cell niche in stem cell development. Rapid advances in the fields of micro/nanoengineering have allowed the engineering of platforms that offer robust control of the cell microenvironment. This controllability has permitted a more comprehensive exploration of parameter space as it concerns the effect of microenvironmental cues on hPSC behaviors such as self-organization and differentiation. Researchers have uncovered and leveraged a wide range of mechano-sensitive and -responsive cellular processes for enhanced control of cell fate and behavior. As in vitro niches become more able to capture the in vivo environment, researchers will be able to access a wider range of developmental events in a laboratory setting.
Advances in in vitro niche technologies could lead to advances in cell replacement therapies and drug development. Of particular interest is the development of platforms that facilitate a high-throughput generation of organoids. By providing the proper biophysical and biochemical factors, PSCs can be made to differentiate and self-organize to form tissue-specific organoids including the optic cup[174], brain [175, 176], intestine [176], liver [177], and kidney [178]. Additionally, with the advent of gene editing processes such as CRISPR/Cas9, mutation correction and personalized medicine are now possible in patient-specific iPSC-derived organoids [179]. In the future, patient-specific iPSC-derived organoids could be used to predict individualized drug efficacy and epithelial response, as has recently been shown for patients with cystic fibrosis using adult tissue-derived organoids [180]. Recently, an increasing number of studies have demonstrated that, in addition to iPSC transdifferentiation ability, iPSC-derived cells can also display therapeutic effects via the paracrine mechanism. Exosomes have emerged as an important paracrine factor for iPSCs to repair injured cells through the delivery of bioactive components. Reports on the use of iPSC-derived exosomes on animal disease models are increasing and to date include heart, liver, limb, skin, eye, bone, and neurological disease models [181–186]. While this approach presents a promising alternative for the safe and reliable delivery of bioactive molecules to target cells, further analysis of the bioactive molecules inside exosomes needs to be carried out for future translational applications.
In this review, we have summarized the state of the art micro/nanoengineered approaches for accurate regulation of various aspects of the cell microenvironment to control hPSC fate and function. These approaches incorporate a variety of engineering technologies, including biomaterials, microfabricated systems, and microfluidics. We anticipate that in the future, researchers will be able to better address issues in fundamental hPSC studies and biomedical applications, including toxicity and drug screening and regenerative medicine.
Table 1.
Different studies using nanotopography for hPSC research.
| Application | Feature size | Fabrication Technique | Topography | Material | Remarks | Ref |
|---|---|---|---|---|---|---|
| HPSC maintenance | 1–150 nm | Lithography and replica-molding | Nano roughness | Silica-based Glass wafer | Alter cell morphology, adhesion, and proliferation. | [57] |
| HPSC maintenance | 1–150 nm | Photolithography and reactive ion etching (RIE) | Nano roughness | Silica-based Glass wafer | Mediate hESCs function, including attachment, morphology, proliferation, and differentiation. | [67] |
| HPSC maintenance | 30 nm | Chemical vapor deposition (CVD) | Multi-walled carbon nanotube–graphene hybrid | Carbon nanotubes-graphene | Maintain attachment, proliferation, and stemness of hESCs. | [55] |
| Neuronal lineages differentiation | 360 nm | Laser inference lithography (NIL) and replica-molding | Ridge/groov e-patterned surface | PDMS | Ridge/groove nanotopography directs differentiation of hiPSCs towards the neuronal lineage. | [61] |
| Neuronal lineages differentiation | - | Nanoimprinting | Ridge/groov e-patterned surface | Glass coverslip | Nanoscale ridge/groove pattern arrays directs differentiation of hESCs into selective neuron cells. | [6] |
| Neuronal lineages differentiation | 80–250 nm | Nanoimprinting | Grating-pillar | Thermoplastic polycarbonate | Substrate topography, with optimal dimension and geometry, modulates the neural fate of hPSCs. | [59] |
| Neuronal lineages differentiation | 250 nm | Soft lithography | Nano-grating | PDMS | Nano-grating substrates direct neural differentiation of hPSCs through actomyosin contractility. | [65] |
| Neuronal lineages differentiation | 1–200 nm | Reactive-ion etching (RIE) | Random nanoscale | Glass coverslip | Nanotopographic substrates enhance hPSC motor neuron progenitor cell differentiation. | [62] |
| Cardiomyogenic lineages differentiation | - | Chemical vapor deposition (CVD) | Nanorough graphene | Graphene | Improving cardiomyogenic differentiation of hESCs on the nanorough graphene. | [63] |
| Endothelial lineages differentiation | - | Salt leaching process | Porous sponges | Poly-(l-lactic acid) (PLLA) and polylactic-glycolic acid (PLGA) | Endothelial cells are derived from hESCs on the porous sponge PLGA. | [68] |
| Pancreatic lineages differentiation | - | Electrospinning | Nanofibrous | Poly-L-lactic acid and polyvinyl alcohol (PLLA/PVA) | Synthetic scaffolds lead to the differentiation of hiPSC to pancreatic cells. | [69] |
| Pancreatic lineages differentiation | 100–400 nm | Electrochemical method | Nanopillar/nanopore | Oxalic AAO (O-AAO) and phosphoric AAO (P-AAO | Nanotopographical surface improves 3-dimensional differentiation of pancreatic cells from hPSCs. | [70] |
| Chondrogenic lineages differentiation | - | Electrospinning | Nanofibrous | Polyethersulfone (PES) | Nanofiber-based polyethersulfone scaffold directs differentiation of hiPSCs to chondrogenic. | [71] |
| Retinal lineages differentiation | 150–190 μm | - | Porous structure | Gelatin, chondroitin sulfate, and hyaluronic acid (GCH) | Biodegradable scaffold improves differentiation of hPSC into the retinal cells. | [72] |
| Hepatogenic lineages differentiation | - | Electrospinning | Nanofibrous | Polyethersulfone/collagen | Enhancing hepatogenic differentiation of hPSC on the aligned polyethersulfone. | [73] |
Table 2.
Summary of different studies using cellular confinement for hPSC research.
| Application | Feature size | Method | Remarks | Ref |
|---|---|---|---|---|
| Amniotic tissue array generation | 40–100 μm | Microcontact printing | The amnion microtissue array was generated and used to screen clinically relevant drugs and their effects in amniogenesis. | [101] |
| Germ layers differentiation | 200–1200 μm | Microcontact printing | Colony size can selectively guide primitive streak-like cells to either definitive endoderm or mesoderm lineages. | [77] |
| Pancreatic endoderm-like cell differentiation | 100–120 μm | Microcontact printing | HESCs differentiate into pancreatic endoderm-like cells by seeding cells onto a patterned substrate. | [84] |
| Hepatocyte-like cell differentiation | 200–1200 μm | Stencil-assisted micropatterning | A multi-staged 17-day differentiation protocol was applied on the multilayered hPSC-derived colonies to induce hepatocyte-like cells. | [79] |
| Generation of human gastruloid | 500–1000 μm | UV patterning | Colonies with larger diameters resulted in differentiation of hESCs into spatially organized three germ layers. | [5] |
| Generation of human gastruloid | 500–1000 μm | UV patterning | Wnt signaling is required to generate primitive streak cells from hPSCs in the micropattern colony, and stimulation with Wnt and Activin is necessary to induce an organizer. | [96] |
| Generation of human gastruloid | 500–1000 μm | UV patterning | A balance between edge sensing and secreted inhibitors controls the self-organization of hPSCs to gastruloid. | [95] |
| Generation of human gastruloid | 500–1000 μm | UV patterning | WNT signaling memory is essential for ACTIVIN to function as a morphogen in human gastruloids. | [94] |
| Generation of human gastruloid | 700–800 μm | UV patterning | The timing of signaling events in the WNT, NODAL, and BMP cascade controls self-organized patterning in human gastruloids. | [93] |
| Generation of human gastruloid | 700–800 μm | UV patterning | The dynamics of signaling events in the NODAL, WNT, and BMP cascade during self-organized fate patterning in human gastruloids were studied. | [92] |
| Generation of blastocyst-like structure | - | Microwell | HPSCs have been used to generate blastocyst-like structures resembling human blastocysts in terms of their size, cell number, morphology, and composition, and allocation of different cell lineages. | [98] |
| Generation of blastocycst-like structure | - | Microwell | Generation of human blastocysts by reprogramming fibroblasts into iBlastoids. | [99] |
| Generation of kidney organoid | - | Microwell | High-throughput screening improves kidney organoid differentiation from hPSCs and enables automated multidimensional phenotyping. | [100] |
Highlights:
This review summarized state of the art of micro/nanoengineered technologies for controlling human pluripotent stem cell fate and function.
The role of biochemical and biomechanical cues, 3D biodegradable scaffolds, and microfluidic systems in determining hPSC fate is discussed.
The challenges and perspectives faced by human pluripotent stem cell biologists and bioengineers are discussed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
All authors (below) certify that they have taken part in the revised version of the manuscript titled “Micro/nanoengineered technologies for human pluripotent stem cells maintenance and differentiation: A review”. All authors take responsibility for the content.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Kim D-H, Wong PK, Park J, Levchenko A, Sun Y, Annual review of biomedical engineering, 11 (2009) 203–233. [DOI] [PubMed] [Google Scholar]
- [2].Park J, Kim P, Helen W, Engler AJ, Levchenko A, Kim D-H, Integrative biology, 4 (2012) 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shao Y, Taniguchi K, Gurdziel K, Townshend RF, Xue X, Yong KMA, Sang J, Spence JR, Gumucio DL, Fu J, Nature Materials, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sun Y, Villa-Diaz LG, Lam RH, Chen W, Krebsbach PH, Fu J, PLoS One, 7 (2012) e37178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Warmflash A, Sorre B, Etoc F, Siggia ED, Brivanlou AH, Nature methods, 11 (2014) 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee MR, Kwon KW, Jung H, Kim HN, Suh KY, Kim K, Kim K-S, Biomaterials, 31 (2010) 4360–4366. [DOI] [PubMed] [Google Scholar]
- [7].Lim JWE, Bodnar A, Proteomics, 2 (2002) 1187–1203. [DOI] [PubMed] [Google Scholar]
- [8].Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM, science, 282 (1998) 1145–1147. [DOI] [PubMed] [Google Scholar]
- [9].Yang K, Lee J, Cho S-W, International journal of stem cells, 5 (2012) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li Y, Powell S, Brunette E, Lebkowski J, Mandalam R, Biotechnology and bioengineering, 91 (2005) 688–698. [DOI] [PubMed] [Google Scholar]
- [11].Shao Y, Sang J, Fu J, Biomaterials, 52 (2015) 26–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hovatta O, Mikkola M, Gertow K, Strömberg AM, Inzunza J, Hreinsson J, Rozell B, Blennow E, AndaÈng M, Ährlund‐ Richter L, Human Reproduction, 18 (2003) 1404–1409. [DOI] [PubMed] [Google Scholar]
- [13].Richards M, Fong C-Y, Chan W-K, Wong P-C, Bongso A, Nature biotechnology, 20 (2002) 933–936. [DOI] [PubMed] [Google Scholar]
- [14].Amit M, Margulets V, Segev H, Shariki K, Laevsky I, Coleman R, Itskovitz-Eldor J, Biology of reproduction, 68 (2003) 2150–2156. [DOI] [PubMed] [Google Scholar]
- [15].Carpenter MK, Rosler E, Rao M, Cloning & Stem Cells, 5 (2003) 79–88. [DOI] [PubMed] [Google Scholar]
- [16].Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK, Nature biotechnology, 19 (2001) 971–974. [DOI] [PubMed] [Google Scholar]
- [17].Melkoumian Z, Weber JL, Weber DM, Fadeev AG, Zhou Y, Dolley-Sonneville P, Yang J, Qiu L, Priest CA, Shogbon C, Nature biotechnology, 28 (2010) 606–610. [DOI] [PubMed] [Google Scholar]
- [18].Kolhar P, Kotamraju VR, Hikita ST, Clegg DO, Ruoslahti E, Journal of biotechnology, 146 (2010) 143–146. [DOI] [PubMed] [Google Scholar]
- [19].Derda R, Li L, Orner BP, Lewis RL, Thomson JA, Kiessling LL, ACS chemical biology, 2 (2007) 347–355. [DOI] [PubMed] [Google Scholar]
- [20].Nandivada H, Villa-Diaz LG, O’Shea KS, Smith GD, Krebsbach PH, Lahann J, Nature protocols, 6 (2011) 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Villa-Diaz LG, Nandivada H, Ding J, Nogueira-de-Souza NC, Krebsbach PH, O’shea KS, Lahann J, Smith GD, Nature biotechnology, 28 (2010) 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ross AM, Nandivada H, Ryan AL, Lahann J, Polymer, 53 (2012) 2533–2539. [Google Scholar]
- [23].Irwin EF, Gupta R, Dashti DC, Healy KE, Biomaterials, 32 (2011) 6912–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li L, Klim JR, Derda R, Courtney AH, Kiessling LL, Proceedings of the National Academy of Sciences, 108 (2011) 11745–11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anderson DG, Levenberg S, Langer R, Nature biotechnology, 22 (2004) 863–866. [DOI] [PubMed] [Google Scholar]
- [26].Mei Y, Gerecht S, Taylor M, Urquhart AJ, Bogatyrev SR, Cho SW, Davies MC, Alexander MR, Langer RS, Anderson DG, Advanced Materials, 21 (2009) 2781–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brafman DA, Chang CW, Fernandez A, Willert K, Varghese S, Chien S, Biomaterials, 31 (2010) 9135–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hansen A, Mjoseng HK, Zhang R, Kalloudis M, Koutsos V, de Sousa PA, Bradley M, Advanced healthcare materials, 3 (2014) 848–853. [DOI] [PubMed] [Google Scholar]
- [29].Mei Y, Saha K, Bogatyrev SR, Yang J, Hook AL, Kalcioglu ZI, Cho S-W, Mitalipova M, Pyzocha N, Rojas F, Nature materials, 9 (2010) 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mahlstedt MM, Anderson D, Sharp JS, McGilvray R, Barbadillo Muñoz MD, Buttery LD, Alexander MR, Rose FR, Denning C, Biotechnology and Bioengineering, 105 (2010) 130–140. [DOI] [PubMed] [Google Scholar]
- [31].Saha K, Mei Y, Reisterer CM, Pyzocha NK, Yang J, Muffat J, Davies MC, Alexander MR, Langer R, Anderson DG, Proceedings of the National Academy of Sciences, 108 (2011) 18714–18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Keung AJ, Asuri P, Kumar S, Schaffer DV, Integrative Biology, 4 (2012) 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sun Y, Yong KMA, Villa-Diaz LG, Zhang X, Chen W, Philson R, Weng S, Xu H, Krebsbach PH, Fu J, Nature materials, 13 (2014) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS, Proceedings of the National Academy of Sciences, 100 (2003) 1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell, 126 (2006) 677–689. [DOI] [PubMed] [Google Scholar]
- [36].Saez A, Buguin A, Silberzan P, Ladoux B, Biophysical journal, 89 (2005) L52–L54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weng S, Shao Y, Chen W, Fu J, Nature materials, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fu J, Wang Y-K, Yang MT, Desai RA, Yu X, Liu Z, Chen CS, Nature methods, 7 (2010) 733–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shao Y, Taniguchi K, Gurdziel K, Townshend RF, Xue X, Yong KMA, Sang J, Spence JR, Gumucio DL, Fu J, Nature materials, 16 (2017) 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abrams G, Goodman S, Nealey P, Franco M, Murphy CJ, Cell and tissue research, 299 (2000) 39–46. [DOI] [PubMed] [Google Scholar]
- [41].Park S, Im GI, Journal of Biomedical Materials Research Part A, 103 (2015) 1238–1245. [DOI] [PubMed] [Google Scholar]
- [42].Gerecht S, Bettinger CJ, Zhang Z, Borenstein JT, Vunjak-Novakovic G, Langer R, Biomaterials, 28 (2007) 4068–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Diehl K, Foley J, Nealey P, Murphy C, Journal of Biomedical Materials Research Part A, 75 (2005) 603–611. [DOI] [PubMed] [Google Scholar]
- [44].Biela SA, Su Y, Spatz JP, Kemkemer R, Acta Biomaterialia, 5 (2009) 2460–2466. [DOI] [PubMed] [Google Scholar]
- [45].Wang J, Chen A, Lieu DK, Karakikes I, Chen G, Keung W, Chan CW, Hajjar RJ, Costa KD, Khine M, Biomaterials, 34 (2013) 8878–8886. [DOI] [PubMed] [Google Scholar]
- [46].Kolind K, Leong KW, Besenbacher F, Foss M, Biomaterials, 33 (2012) 6626–6633. [DOI] [PubMed] [Google Scholar]
- [47].Dalby MJ, Gadegaard N, Curtis G, Adam S, Oreffo C, Richard O, Current stem cell research & therapy, 2 (2007) 129–138. [DOI] [PubMed] [Google Scholar]
- [48].Kim D-H, Provenzano PP, Smith CL, Levchenko A, The Journal of cell biology, 197 (2012) 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bae D, Moon S-H, Park BG, Park S-J, Jung T, Kim JS, Lee KB, Chung H-M, Biomaterials, 35 (2014) 916–928. [DOI] [PubMed] [Google Scholar]
- [50].McMurray RJ, Gadegaard N, Tsimbouri PM, Burgess KV, McNamara LE, Tare R, Murawski K, Kingham E, Oreffo RO, Dalby MJ, Nature materials, 10 (2011) 637–644. [DOI] [PubMed] [Google Scholar]
- [51].Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CD, Oreffo RO, Nature materials, 6 (2007) 997–1003. [DOI] [PubMed] [Google Scholar]
- [52].McNamara LE, McMurray RJ, Biggs MJ, Kantawong F, Oreffo RO, Dalby MJ, Journal of tissue engineering, 1 (2010) 120623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yin Z, Chen X, Chen JL, Shen WL, Nguyen TMH, Gao L, Ouyang HW, Biomaterials, 31 (2010) 2163–2175. [DOI] [PubMed] [Google Scholar]
- [54].Yim EK, Pang SW, Leong KW, Experimental cell research, 313 (2007) 1820–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sebaa M, Nguyen TY, Paul RK, Mulchandani A, Liu H, Materials Letters, 92 (2013) 122–125. [Google Scholar]
- [56].Chen W, Villa-Diaz LG, Sun Y, Weng S, Kim JK, Krebsbach PH, Fu J, ASME 2012 Summer Bioengineering Conference, American Society of Mechanical Engineers; 2012, pp. 1293–1294. [Google Scholar]
- [57].Chen W, Villa-Diaz LG, Sun Y, Weng S, Kim JK, Lam RH, Han L, Fan R, Krebsbach PH, Fu J, ACS nano, 6 (2012) 4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen W, Sun Y, Fu J, 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences 2011, MicroTAS; 20112011. [Google Scholar]
- [59].Ankam S, Suryana M, Chan LY, Moe AAK, Teo BK, Law JB, Sheetz MP, Low HY, Yim EK, Acta biomaterialia, 9 (2013) 4535–4545. [DOI] [PubMed] [Google Scholar]
- [60].Ireland RG, Simmons CA, Stem Cells, 33 (2015) 3187–3196. [DOI] [PubMed] [Google Scholar]
- [61].Pan F, Zhang M, Wu G, Lai Y, Greber B, Schöler HR, Chi L, Biomaterials, 34 (2013) 8131–8139. [DOI] [PubMed] [Google Scholar]
- [62].Chen W, Han S, Qian W, Weng S, Yang H, Sun Y, Villa-Diaz LG, Krebsbach PH, Fu J, Nanoscale, 10 (2018) 3556–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lee T-J, Park S, Bhang SH, Yoon J-K, Jo I, Jeong G-J, Hong BH, Kim B-S, Biochemical and biophysical research communications, 452 (2014) 174–180. [DOI] [PubMed] [Google Scholar]
- [64].Kim JH, Kim HW, Cha KJ, Han J, Jang YJ, Kim DS, Kim J-H, ACS nano, 10 (2016) 3342–3355. [DOI] [PubMed] [Google Scholar]
- [65].Ankam S, Lim CK, Yim EK, Biomaterials, 47 (2015) 20–28. [DOI] [PubMed] [Google Scholar]
- [66].Lu HF, Lim S-X, Leong MF, Narayanan K, Toh RP, Gao S, Wan AC, Biomaterials, 33 (2012) 9179–9187. [DOI] [PubMed] [Google Scholar]
- [67].Chen W, Sun Y, Fu J, 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences 2011, MicroTAS; 20112011, pp. 36–38. [Google Scholar]
- [68].Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R, Proceedings of the national Academy of Sciences, 99 (2002) 4391–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Enderami SE, Kehtari M, Abazari MF, Ghoraeian P, Nouri Aleagha M, Soleimanifar F, Soleimani M, Mortazavi Y, Nadri S, Mostafavi H, Artificial cells, nanomedicine, and biotechnology, 46 (2018) 1062–1069. [DOI] [PubMed] [Google Scholar]
- [70].Kim JH, Park BG, Kim S-K, Lee D-H, Lee GG, Kim D-H, Choi B-O, Lee KB, Kim J-H, Acta biomaterialia, (2018). [Google Scholar]
- [71].Mahboudi H, Soleimani M, Enderami SE, Kehtari M, Hanaee-Ahvaz H, Ghanbarian H, Bandehpour M, Nojehdehi S, Mirzaei S, Kazemi B, Artificial cells, nanomedicine, and biotechnology, 46 (2018) 1948–1956. [DOI] [PubMed] [Google Scholar]
- [72].Singh D, Wang S-B, Xia T, Tainsh L, Ghiassi-Nejad M, Xu T, Peng S, Adelman RA, Rizzolo LJ, Biomaterials, 154 (2018) 158–168. [DOI] [PubMed] [Google Scholar]
- [73].Mahmoodinia Maymand M, Soleimanpour-Lichaei HR, Ardeshirylajimi A, Soleimani M, Enderami SE, Nojehdehi S, Behjati F, Kabir Salmani M, Artificial cells, nanomedicine, and biotechnology, 46 (2018) 853–860. [DOI] [PubMed] [Google Scholar]
- [74].Shao Y, Fu J, Advanced Materials, 26 (2014) 1494–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Falconnet D, Csucs G, Grandin HM, Textor M, Biomaterials, 27 (2006) 3044–3063. [DOI] [PubMed] [Google Scholar]
- [76].Peerani R, Rao BM, Bauwens C, Yin T, Wood GA, Nagy A, Kumacheva E, Zandstra PW, The EMBO journal, 26 (2007) 4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lee LH, Peerani R, Ungrin M, Joshi C, Kumacheva E, Zandstra P, Stem cell research, 2 (2009) 155–162. [DOI] [PubMed] [Google Scholar]
- [78].Bauwens CL, Peerani R, Niebruegge S, Woodhouse KA, Kumacheva E, Husain M, Zandstra PW, Stem cells, 26 (2008) 2300–2310. [DOI] [PubMed] [Google Scholar]
- [79].Yao R, Wang J, Li X, Jung Jung D, Qi H, Kee KK, Du Y, Small, 10 (2014) 4311–4323. [DOI] [PubMed] [Google Scholar]
- [80].Khademhosseini A, Ferreira L, Blumling J, Yeh J, Karp JM, Fukuda J, Langer R, Biomaterials, 27 (2006) 5968–5977. [DOI] [PubMed] [Google Scholar]
- [81].Mohr JC, de Pablo JJ, Palecek SP, Biomaterials, 27 (2006) 6032–6042. [DOI] [PubMed] [Google Scholar]
- [82].Kumar A, Whitesides GM, Applied Physics Letters, 63 (1993) 2002–2004. [Google Scholar]
- [83].Jackman RJ, Wilbur JL, Whitesides GM, Science, 269 (1995) 664. [DOI] [PubMed] [Google Scholar]
- [84].Van Hoof D, Mendelsohn AD, Seerke R, Desai TA, German MS, Stem cell research, 6 (2011) 276–285. [DOI] [PubMed] [Google Scholar]
- [85].Carter SB, Experimental cell research, 48 (1967) 189–193. [DOI] [PubMed] [Google Scholar]
- [86].Jimbo Y, Robinson HP, Kawana A, IEEE transactions on biomedical engineering, 40 (1993) 804–810. [DOI] [PubMed] [Google Scholar]
- [87].Paik I, Scurr DJ, Morris B, Hall G, Denning C, Alexander MR, Shakesheff KM, Dixon JE, Biotechnology and bioengineering, 109 (2012) 2630–2641. [DOI] [PubMed] [Google Scholar]
- [88].Théry M, J Cell Sci, 123 (2010) 4201–4213. [DOI] [PubMed] [Google Scholar]
- [89].Azioune A, Storch M, Bornens M, Théry M, Piel M, Lab on a chip, 9 (2009) 1640–1642. [DOI] [PubMed] [Google Scholar]
- [90].Vignaud T, Galland R, Tseng Q, Blanchoin L, Colombelli J, Théry M, J Cell Sci, 125 (2012) 2134–2140. [DOI] [PubMed] [Google Scholar]
- [91].Fink J, Théry M, Azioune A, Dupont R, Chatelain F, Bornens M, Piel M, Lab on a Chip, 7 (2007) 672–680. [DOI] [PubMed] [Google Scholar]
- [92].Chhabra S, Liu L, Goh R, Kong X, Warmflash A, PLoS biology, 17 (2019) e3000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Chhabra S, Liu L, Goh R, Warmflash A, BioRxiv, (2018) 440164. [Google Scholar]
- [94].Yoney A, Etoc F, Ruzo A, Carroll T, Metzger JJ, Martyn I, Li S, Kirst C, Siggia ED, Brivanlou AH, Elife, 7 (2018) e38279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Etoc F, Metzger J, Ruzo A, Kirst C, Yoney A, Ozair MZ, Brivanlou AH, Siggia ED, Developmental cell, 39 (2016) 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Martyn I, Kanno T, Ruzo A, Siggia E, Brivanlou A, Nature, 558 (2018) 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rivron NC, Frias-Aldeguer J, Vrij EJ, Boisset J-C, Korving J, Vivié J, Truckenmüller RK, Van Oudenaarden A, Van Blitterswijk CA, Geijsen N, Nature, 557 (2018) 106–111. [DOI] [PubMed] [Google Scholar]
- [98].Yu L, Wei Y, Duan J, Schmitz DA, Sakurai M, Wang L, Wang K, Zhao S, Hon GC, Wu J, Nature, 591 (2021) 620–626. [DOI] [PubMed] [Google Scholar]
- [99].Liu X, Tan JP, Schröder J, Aberkane A, Ouyang JF, Mohenska M, Lim SM, Sun YB, Chen J, Sun G, Nature, 591 (2021) 627–632. [DOI] [PubMed] [Google Scholar]
- [100].Czerniecki SM, Cruz NM, Harder JL, Menon R, Annis J, Otto EA, Gulieva RE, Islas LV, Kim YK, Tran LM, Cell stem cell, 22 (2018) 929–940. e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Esfahani SN, Shao Y, Irizarry AMR, Li Z, Xue X, Gumucio DL, Fu J, Biomaterials, 216 (2019) 119244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Chen S, Wang X, Cheng J, Kong C.-w., Cheng SH, Li RA, Sun D, 2013 13th IEEE International Conference on Nanotechnology (IEEE-NANO 2013), IEEE; 2013, pp. 333–336. [Google Scholar]
- [103].Baumgartner W, Hinterdorfer P, Ness W, Raab A, Vestweber D, Schindler H, Drenckhahn D, Proceedings of the National Academy of Sciences, 97 (2000) 4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bornschlögl T, Romero S, Vestergaard CL, Joanny J-F, Van Nhieu GT, Bassereau P, Proceedings of the National Academy of Sciences, 110 (2013) 18928–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Honarmandi P, Lee H, Lang MJ, Kamm RD, Lab on a chip, 11 (2011) 684–694. [DOI] [PubMed] [Google Scholar]
- [106].Garzon-Coral C, Fantana HA, Howard J, Science, 352 (2016) 1124–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP, Science, 323 (2009) 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Tanase M, Biais N, Sheetz M, Methods in cell biology, 83 (2007) 473–493. [DOI] [PubMed] [Google Scholar]
- [109].Agrawal R, Smart T, Nobre-Cardoso J, Richards C, Bhatnagar R, Tufail A, Shima D, Jones PH, Pavesio C, Scientific reports, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Tan Y, Kong C.-w., Chen S, Cheng SH, Li RA, Sun D, Journal of biomechanics, 45 (2012) 123–128. [DOI] [PubMed] [Google Scholar]
- [111].Tan Y, Sun D, Cheng SH, Li RA, Robotics and Automation (ICRA), 2011 IEEE International Conference on, IEEE; 2011, pp. 4104–4109. [Google Scholar]
- [112].Matzke R, Jacobson K, Radmacher M, Nature Cell Biology, 3 (2001) 607–610. [DOI] [PubMed] [Google Scholar]
- [113].Liu J, Sun N, Bruce MA, Wu JC, Butte MJ, PloS one, 7 (2012) e37559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Li M, Liu L, Xiao X, Xi N, Wang Y, Journal of biological physics, 42 (2016) 551–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Chen J, Lu G, Journal of biomechanics, 45 (2012) 2810–2816. [DOI] [PubMed] [Google Scholar]
- [116].Chen J, Interface focus, 4 (2014) 20130055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Xue X, Hong X, Li Z, Deng CX, Fu J, Biomaterials, 134 (2017) 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chen D, Sun Y, Gudur MS, Hsiao Y-S, Wu Z, Fu J, Deng CX, Biophysical journal, 108 (2015) 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li X, Chu J, Wang A, Zhu Y, Chu WK, Yang L, Li S, PloS one, 6 (2011) e26029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Teramura T, Takehara T, Onodera Y, Nakagawa K, Hamanishi C, Fukuda K, Biochemical and biophysical research communications, 417 (2012) 836–841. [DOI] [PubMed] [Google Scholar]
- [121].Xue X, Sun Y, Resto-Irizarry AM, Yuan Y, Yong KMA, Zheng Y, Weng S, Shao Y, Chai Y, Studer L, Nature materials, (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mooney DJ, Sano K, Kaufmann PM, Majahod K, Schloo B, Vacanti JP, Langer R, Journal of biomedical materials research, 37 (1997) 413–420. [DOI] [PubMed] [Google Scholar]
- [123].Haycock JW, 3D cell culture, Springer; 2011. [Google Scholar]
- [124].Lee CH, Singla A, Lee Y, International journal of pharmaceutics, 221 (2001) 1–22. [DOI] [PubMed] [Google Scholar]
- [125].Bensaıd W, Triffitt J, Blanchat C, Oudina K, Sedel L, Petite H, Biomaterials, 24 (2003) 2497–2502. [DOI] [PubMed] [Google Scholar]
- [126].O’Brien FJ, Harley B, Yannas IV, Gibson LJ, Biomaterials, 26 (2005) 433–441. [DOI] [PubMed] [Google Scholar]
- [127].Pitaru S, Tal H, Soldinger M, Grosskopf A, Noff M, Journal of Periodontology, 59 (1988) 380–386. [DOI] [PubMed] [Google Scholar]
- [128].Flanagan TC, Wilkins B, Black A, Jockenhoevel S, Smith TJ, Pandit AS, Biomaterials, 27 (2006) 2233–2246. [DOI] [PubMed] [Google Scholar]
- [129].Chen JL, Yin Z, Shen WL, Chen X, Heng BC, Zou XH, Ouyang HW, Biomaterials, 31 (2010) 9438–9451. [DOI] [PubMed] [Google Scholar]
- [130].Hunt NC, Hallam D, Karimi A, Mellough CB, Chen J, Steel DH, Lako M, Acta biomaterialia, 49 (2017) 329–343. [DOI] [PubMed] [Google Scholar]
- [131].Gerecht‐ Nir S, Cohen S, Ziskind A, Itskovitz‐ Eldor J, Biotechnology and bioengineering, 88 (2004) 313–320. [DOI] [PubMed] [Google Scholar]
- [132].Lavik E, Klassen H, Warfvinge K, Langer R, Young M, Biomaterials, 26 (2005) 3187–3196. [DOI] [PubMed] [Google Scholar]
- [133].Tomita M, Lavik E, Klassen H, Zahir T, Langer R, Young MJ, Stem Cells, 23 (2005) 1579–1588. [DOI] [PubMed] [Google Scholar]
- [134].Kullenberg J, Rosatini F, Vozzi G, Bianchi F, Ahluwalia A, Domenici C, Tissue Engineering Part A, 14 (2008) 1017–1023. [DOI] [PubMed] [Google Scholar]
- [135].Neeley WL, Redenti S, Klassen H, Tao S, Desai T, Young MJ, Langer R, Biomaterials, 29 (2008) 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Redenti S, Neeley WL, Rompani S, Saigal S, Yang J, Klassen H, Langer R, Young MJ, Biomaterials, 30 (2009) 3405–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Tao S, Young C, Redenti S, Zhang Y, Klassen H, Desai T, Young MJ, Lab on a Chip, 7 (2007) 695–701. [DOI] [PubMed] [Google Scholar]
- [138].Sodha S, Wall K, Redenti S, Klassen H, Young MJ, Tao SL, Journal of Biomaterials Science, Polymer Edition, 22 (2011) 443–456. [DOI] [PubMed] [Google Scholar]
- [139].Steedman MR, Tao SL, Klassen H, Desai TA, Biomedical microdevices, 12 (2010) 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Redenti S, Tao S, Yang J, Gu P, Klassen H, Saigal S, Desai T, Young MJ, Journal of ocular biology, diseases, and informatics, 1 (2008) 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Levenberg S, Burdick JA, Kraehenbuehl T, Langer R, Tissue engineering, 11 (2005) 506–512. [DOI] [PubMed] [Google Scholar]
- [142].Bai HY, Chen GA, Mao GH, Song TR, Wang YX, Journal of biomedical materials research Part A, 94 (2010) 539–546. [DOI] [PubMed] [Google Scholar]
- [143].Chao T-I, Xiang S, Chen C-S, Chin W-C, Nelson A, Wang C, Lu J, Biochemical and biophysical research communications, 384 (2009) 426–430. [DOI] [PubMed] [Google Scholar]
- [144].Chao TI, Xiang S, Lipstate JF, Wang C, Lu J, Advanced Materials, 22 (2010) 3542–3547. [DOI] [PubMed] [Google Scholar]
- [145].Sridharan I, Kim T, Wang R, Biochemical and biophysical research communications, 381 (2009) 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Zhao C, Tan A, Pastorin G, Ho HK, Biotechnology advances, 31 (2013) 654–668. [DOI] [PubMed] [Google Scholar]
- [147].Gelain F, Bottai D, Vescovi A, Zhang S, PloS one, 1 (2006) e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Gauthaman K, Venugopal JR, Yee FC, Peh GSL, Ramakrishna S, Bongso A, Journal of cellular and molecular medicine, 13 (2009) 3475–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Lu HF, Narayanan K, Lim S-X, Gao S, Leong MF, Wan AC, Biomaterials, 33 (2012) 2419–2430. [DOI] [PubMed] [Google Scholar]
- [150].Khoury M, Bransky A, Korin N, Konak LC, Enikolopov G, Tzchori I, Levenberg S, Biomedical microdevices, 12 (2010) 1001–1008. [DOI] [PubMed] [Google Scholar]
- [151].Park JY, Kim SK, Woo DH, Lee EJ, Kim JH, Lee SH, Stem cells, 27 (2009) 2646–2654. [DOI] [PubMed] [Google Scholar]
- [152].Cimetta E, Sirabella D, Yeager K, Davidson K, Simon J, Moon RT, Vunjak-Novakovic G, Lab on a chip, 13 (2013) 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kamei K.-i., Mashimo Y, Koyama Y, Fockenberg C, Nakashima M, Nakajima M, Li J, Chen Y, Biomedical microdevices, 17 (2015) 36. [DOI] [PubMed] [Google Scholar]
- [154].Huang NF, Dewi RE, Okogbaa J, Lee JC, JalilRufaihah A, Heilshorn SC, Cooke JP, Am. J. Transl. Res, 5 (2013) 510–U596. [PMC free article] [PubMed] [Google Scholar]
- [155].Belair DG, Whisler JA, Valdez J, Velazquez J, Molenda JA, Vickerman V, Lewis R, Daigh C, Hansen TD, Mann DA, Stem Cell Reviews and Reports, 11 (2015) 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Korin N, Bransky A, Dinnar U, Levenberg S, Biomedical microdevices, 11 (2009) 87–94. [DOI] [PubMed] [Google Scholar]
- [157].Villa-Diaz LG, Torisawa Y.-s., Uchida T, Ding J, Nogueira-de-Souza NC, O’Shea KS, Takayama S, Smith GD, Lab on a Chip, 9 (2009) 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Wang X, Chen S, Kong M, Wang Z, Costa KD, Li RA, Sun D, Lab on a Chip, 11 (2011) 3656–3662. [DOI] [PubMed] [Google Scholar]
- [159].Choudhury D, Ramsay W, Brown G, Psaila N, Beecher S, Thomson R, Kiss R, Pells S, Willoughby N, Paterson L, SPIE MOEMS-MEMS, International Society for Optics and Photonics 2011, pp. 79290P-79290P-79296. [Google Scholar]
- [160].Jabart E, Rangarajan S, Lieu C, Hack J, Conboy I, Sohn L, Microfluidics and Nanofluidics, 18 (2015) 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Singh A, Suri S, Lee T, Chilton JM, Cooke MT, Chen W, Fu J, Stice SL, Lu H, McDevitt TC, Nature methods, 10 (2013) 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].ForáYu ZT, Lab on a Chip, 9 (2009) 555–563. [DOI] [PubMed] [Google Scholar]
- [163].Kamei K.-i., Ohashi M, Gschweng E, Ho Q, Suh J, Tang J, Clark AT, Pyle AD, Teitell MA, Lee K-B, Lab on a Chip, 10 (2010) 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zhong JF, Chen Y, Marcus JS, Scherer A, Quake SR, Taylor CR, Weiner LP, Lab on a Chip, 8 (2008) 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhong JF, Feng Y, Taylor CR, Current medicinal chemistry, 15 (2008) 2897–2900. [DOI] [PubMed] [Google Scholar]
- [166].White AK, VanInsberghe M, Petriv I, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL, Proceedings of the National Academy of Sciences, 108 (2011) 13999–14004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Narsinh KH, Sun N, Sanchez-Freire V, Lee AS, Almeida P, Hu S, Jan T, Wilson KD, Leong D, Rosenberg J, The Journal of clinical investigation, 121 (2011) 1217–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Satoh T, Sugiura S, Sumaru K, Ozaki S, Gomi S, Kurakazu T, Oshima Y, Kanamori T, Biomicrofluidics, 8 (2014) 024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Sikorski DJ, Caron NJ, VanInsberghe M, Zahn H, Eaves CJ, Piret JM, Hansen CL, Biotechnology journal, 10 (2015) 1546–1554. [DOI] [PubMed] [Google Scholar]
- [170].Matsumura T, Tatsumi K, Noda Y, Nakanishi N, Okonogi A, Hirano K, Li L, Osumi T, Tada T, Kotera H, Biochemical and biophysical research communications, 453 (2014) 131–137. [DOI] [PubMed] [Google Scholar]
- [171].Yoshimitsu R, Hattori K, Sugiura S, Kondo Y, Yamada R, Tachikawa S, Satoh T, Kurisaki A, Ohnuma K, Asashima M, Biotechnology and bioengineering, 111 (2014) 937–947. [DOI] [PubMed] [Google Scholar]
- [172].Zheng Y, Xue X, Shao Y, Wang S, Esfahani SN, Li Z, Muncie JM, Lakins JN, Weaver VM, Gumucio DL, Nature, 573 (2019) 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Manfrin A, Tabata Y, Paquet ER, Vuaridel AR, Rivest FR, Naef F, Lutolf MP, Nature methods, 16 (2019) 640–648. [DOI] [PubMed] [Google Scholar]
- [174].Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y, Nature, 472 (2011) 51. [DOI] [PubMed] [Google Scholar]
- [175].Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, Nature, 501 (2013) 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Nature, 470 (2011) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang R-R, Ueno Y, Zheng Y-W, Koike N, Nature, 499 (2013) 481. [DOI] [PubMed] [Google Scholar]
- [178].Takasato M, Er P, Becroft M, Vanslambrouck J, Stanley E, Elefanty A, Little M, Nature cell biology, 16 (2014) 118. [DOI] [PubMed] [Google Scholar]
- [179].Hockemeyer D, Jaenisch R, Cell stem cell, 18 (2016) 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Saini A, Cell Stem Cell, 19 (2016) 425–427. [Google Scholar]
- [181].Gao L, Wang L, Wei Y, Krishnamurthy P, Walcott GP, Menasché P, Zhang J, Science Translational Medicine, 12 (2020). [DOI] [PubMed] [Google Scholar]
- [182].Ye M, Ni Q, Qi H, Qian X, Chen J, Guo X, Li M, Zhao Y, Xue G, Deng H, International journal of biological sciences, 15 (2019) 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Kobayashi H, Ebisawa K, Kambe M, Kasai T, Suga H, Nakamura K, Narita Y, Ogata A, Kamei Y, Nagoya journal of medical science, 80 (2018) 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Winston CN, Aulston B, Rockenstein EM, Adame A, Prikhodko O, Dave KN, Mishra P, Rissman RA, Yuan SH, Journal of Alzheimer’s Disease, 67 (2019) 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Wang S, Hou Y, Li X, Song Z, Sun B, Li X, Zhang H, Aging (Albany NY), 12 (2020) 19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Wang AYL, International Journal of Molecular Sciences, 22 (2021) 1769.33578948 [Google Scholar]