

Abstract
A repressive chromatin state featuring trimethylated lysine 36 on histone H3 (H3K36me3) and DNA methylation suppresses cryptic transcription in embryonic stem cells. Cryptic transcription is elevated with age in yeast and nematodes, and reducing it extends yeast lifespan, though whether this occurs in mammals is unknown. We show that cryptic transcription is elevated in aged mammalian stem cells, including murine hematopoietic stem cells (mHSCs) and neural stem cells (NSCs) and human mesenchymal stem cells (hMSCs). Precise mapping allowed quantification of age-associated cryptic transcription in hMSCs aged in vitro. Regions with significant age-associated cryptic transcription have a unique chromatin signature: decreased H3K36me3 and increased H3K4me1, H3K4me3, and H3K27ac with age. Genomic regions undergoing such changes resemble known promoter sequences and are bound by TBP even in young cells. Hence, the more permissive chromatin state at intragenic cryptic promoters likely underlies increased cryptic transcription in aged mammalian stem cells.
Editor summary:
The authors show that aged mammalian stem cells produce aberrant transcripts due to profound yet characteristic changes to chromatin during aging, a phenomenon only previously known to occur in simple invertebrate models, limiting their lifespan.
Introduction
Aging, organismal degeneration over time, is a universal phenomenon. Although the specific manifestations of aging vary between different organisms, the molecular and cellular mechanisms that drive this process are broadly conserved among eukaryotes1. One such hallmark, epigenetic alteration, drives the opening of chromatin with age, which limits lifespan2. Chromatin structure can also have a profound effect on the regulation of transcription; there are numerous examples of age-associated transcriptional dysregulation that can impair cellular function and induce a partial loss of cell identity3–5. Recent work in yeast and worms has also revealed that an open chromatin state, characterized by loss of trimethylated lysine 36 of histone H3 (H3K36me3) within gene bodies, causes an increase in cryptic transcription during aging6. As reduction of H3K36me3 levels is a common age-associated phenotype in eukaryotes (reviewed in7,8), increased cryptic transcription may play an underappreciated role in aging.
Cryptic transcription is aberrant transcription initiation from non-promoter regions within the gene body. This phenomenon has been extensively studied in yeast (reviewed in ref.9) and more recently in mammals10–12. It is thought to result from the chromatin state changes that arise when RNA polymerase II (Pol II) transits the gene body, during which histones are removed and histone modifications are co-transcriptionally conferred (reviewed in ref.13). Following transcription, a closed chromatin state is restored by recruitment of the FACT complex, which promotes the re-incorporation of nucleosomes in yeast and mammals14,15; the histone deacetylase complex Rpd3S in yeast16; and the histone demethylase Kdm5b12 and the DNA methyltransferase Dnmt3b11 in mice. Loss of any of these proteins, which are recruited by H3K36me3, causes increased cryptic transcription.
Evidence from yeast and worms suggests that increased cryptic transcription has a negative impact on lifespan. Perturbations that decrease H3K36me3 increase cryptic transcription and decrease lifespan in yeast, while those that increase H3K36me3 have the opposite effect on both lifespan and cryptic transcription6. Similarly, in worms, knocking down met-1, which is thought to trimethylate H3K36, shortens lifespan, while RNAi against jmjd-2, an H3K36 demethylase, extends it17,18, though the impact of these treatments on cryptic transcription has not been assessed. Nevertheless, an increase in cryptic transcription, likely driven by chromatin structure dysregulation downstream of H3K36me3, is a conserved feature of aging in yeast and worms.
Given the broad conservation of molecular mechanisms that drive aging, we asked whether cryptic transcription increases with age in mammals and if chromatin dysregulation contributes to this process. In particular, we focused on stem cells, which may play an outsized role in aging (reviewed in ref.19). Analysis of transcriptome data, mapping of transcription start sites, and assessment of several transcription-associated histone modifications suggest that cryptic transcription increases with age in mammals, and that this is associated with an opening of the chromatin, just as seen in yeast and worms.
Results
Mammalian stem cell aging models
We focused on aging in two models: murine hematopoietic stem cells (mHSCs) and human mesenchymal stem cells (hMSCs). Transcriptional and chromatin-level changes were previously characterized in HSCs isolated from young (4mo) and aged (24mo) mice20; these data were reanalyzed to look for evidence of cryptic transcription. We used culture expanded hMSCs isolated from cord blood as an in vitro model of aging in a human stem cell population, as this recapitulates phenotypes observed in MSCs isolated from aged individuals21. Culture expanded hMSCs have decreased differentiation potential; an increased proportion of senescent cells, measured by β-galactosidase staining; and decreased growth rate (Extended Data Figure 1). Reduced differentiation potential occurs before decreased growth rate and increased β-galactosidase activity; for this study, we expanded the cells to the point that the mock-aged cells had decreased differentiation potential, but had not yet slowed growth or increased cellular senescence.
Increased cryptic transcription inferred from aging RNA-seq
Intragenic cryptic transcription is transcription initiation from non-promoter regions within gene bodies16, and should be reflected in RNA-seq data as an increase in mapped reads downstream of the cryptic initiation site relative to upstream thereof. We developed a method to detect cryptic transcription in RNA-seq samples by examining exon-based expression levels within transcripts. If downstream exons have a higher transcripts per kilobase million (TPM) than the first (or second) exon of a transcript, this indicates that cryptic transcription occurs in the cognate transcript. When comparing between samples, elevated cryptic transcription is inferred from an increased average exon-based TPM ratio between samples. Our method detected a significant increase in the average exon-based TPM ratio in RNA-seq data from murine embryonic stem cells (mESCs) that lack Setd2 and are known to have elevated cryptic transcription11 (Extended Data Figure 1). Analysis of mESCs undergoing Setd2 knockdown22 or knockout23 and Setd2 knockout murine oocytes24 confirmed elevated cryptic transcription in these systems (Extended Data Figure 1).
We applied this method to RNA-seq datasets from aged mHSCs20 and culture-expanded hMSCs. The average exon-based TPM ratio is increased in mHSCs isolated from old mice vs. young, indicating that cryptic transcription increases with age (Figure 1A, shown separately in Extended Data Figure 1). A similar phenomenon is observed in hMSCs (Figure 1B and Extended Data Figure 1). Transcripts with higher expression tend to have greater increases in exon-based TPM ratios with age in both mHSCs and hMSCs (Extended Data Figure 1), suggesting that highly expressed genes are more susceptible to age-associated increases in cryptic transcription. The exon-based TPM ratio increases deeper into the transcript, consistent with sustained transcription from cryptic sites, as the number of exons that have exon-based TPM ratios greater than 2 increases as exon number increases (Extended Data Figure 1). The RNA-seq data from mHSCs and hMSCs indicates that, globally, cryptic transcription is elevated with age in mammalian stem cells.
Figure 1. Age-associated increase in cryptic transcription detected in mHSC and hMSC RNA-seq data.
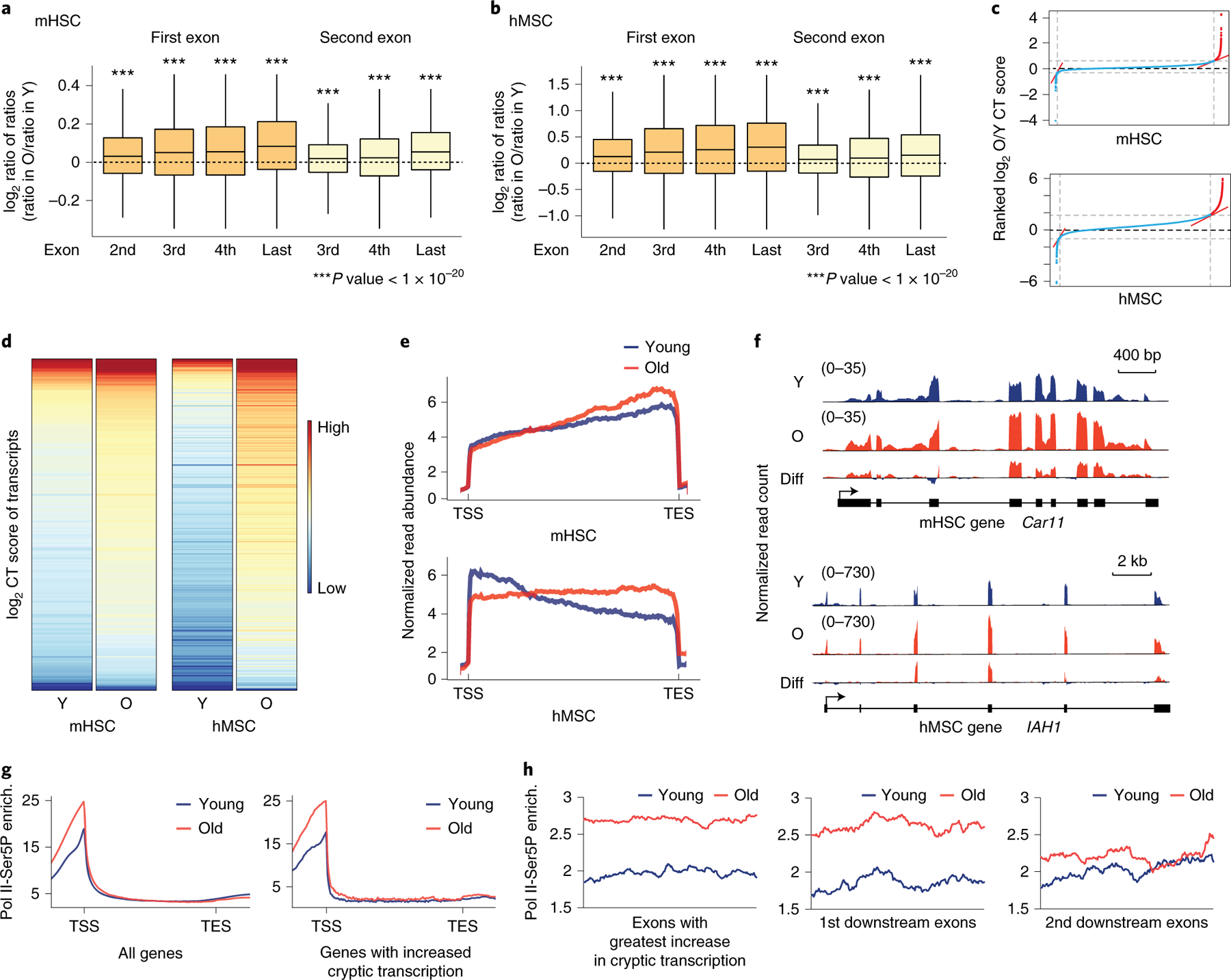
A and B) Boxplots of the log2-transformed ratio of reads mapping to the indicated exon vs. reads mapping to the first or second exon (dark or light orange, respectively) in old vs. young samples (ratio in old divided by ratio in young, ratio of ratios), in mHSCs (A) and hMSCs (B). Expressed major transcripts with at least 3 exons were included for mHSC (n=10068) and hMSCs (n=9230). Bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range. Significance was determined by a two-sided Wilcoxon signed-rank test with the null hypothesis that the log2-transformed ratios are equal to 0. p-values reported in Supplementary Table 1. C) Ranked plot of CT scores for mHSCs (top) and hMSCs (bottom). The red lines show inflection points with a tangent of 1; transcripts with significantly increased CT scores, i.e., those with an age-associated increase in cryptic transcription, are located to the right of the second inflection point. D) Heatmaps of the CT scores of transcripts with age-increased cryptic transcription in mHSCs (left) and hMSCs (right). E) Metagene plot of RNA-seq read density in old (red) and young (blue) samples. mHSCs are on top; hMSCs on the bottom. F) Age-associated cryptic transcription in mHSCs (left, Car11) and hMSCs (right, IAH1), depicting mapped reads from young samples (top, blue), old samples (middle, red), and the difference (old-young, in red where reads are higher in old samples, blue in young; bottom panel). G) Metagene plots of RNA Pol II-Ser5P ChIP-seq enrichment in young and old hMSCs. The plot on the left shows enrichment among all transcripts; the one on the right shows enrichment only in transcripts identified as having increased cryptic transcription with age. H) Enrichment plots of RNA Pol II Ser5P ChIP-seq in exons with the highest CT score of transcripts with age-increased cryptic transcript and the first and second downstream exons. In all panels, Y: young; O: old; Diff: difference; enrich.: enrichment.
We identified transcripts with an age-associated increase in cryptic transcription, i.e., transcripts for which the exon-based TPM ratio is highest in the old vs. young samples (Figure 1C; see Methods); 199 transcripts were identified in mHSCs and 304 in hMSCs. Heatmaps of the exon-based TPM ratios confirm this increase (Figure 1D), and a scatterplot of the old ratio vs. the young ratio by transcript shows that the identified transcripts all lie above the line y=x (Extended Data Figure 1). RNA-seq metagene plots show that read density increases towards the 3’ end of the transcripts vs. the 5’ end in old samples (Figure 1E). Two examples of cryptic transcripts identified by this analysis are shown in Figure 1F. The transcripts identified by our analysis are longer than average (Extended Data Figure 1), suggesting that gene length is associated with elevated cryptic transcription with age.
We performed ChIP-seq for Pol II using a C-terminal domain serine 5 phospho-specific antibody (Pol II-Ser5P) in young and old hMSCs. Pol II-Ser5P is highly enriched in promoter regions (Figure 1G). While Pol II-Ser5P enrichment in gene bodies is similar in young and old cells, there is a slight increase in intragenic Pol II-Ser5P enrichment in old cells when only examining genes with elevated cryptic transcription with age (Figure 1G). Examination of the exons in which increased cryptic transcription is first detected, we observe higher enrichment of Pol II-Ser5P in old cells than young, which gradually decreases downstream from the presumptive cryptic initiation site (Figure 1H). This analysis supports the idea that the genes we identified exhibit increased cryptic transcription with age.
We asked whether cryptic transcription is elevated during aging in other adult stem cells. Neural stem cells (NSCs) are maintained in a resting state, but are activated under certain conditions25. We examined RNA-seq data from quiescent and activated NSCs (qNSCs and aNSCs, respectively) isolated from the subventricular zone of young and old mice. Principle component analysis showed that most gene expression changes correlate with activation state and sex, while age has less of an effect (Figure 2A). In both males and females, aNSCs show an increased exon-based TPM ratio with age, though qNSCs do not (Figure 2B). In males, this increase is only evident in the last exon, while in females, there is a gradual increase throughout the transcript. In aNSCs isolated from female mice, only transcripts in the fourth expression quartile show elevated cryptic transcription with age, suggesting a link between expression level and age-increased cryptic transcription. No such trend was observed in aNSCs from male mice (Extended Data Figure 2). We identified 237 transcripts in males and 266 in females that exhibit increased cryptic transcription with age, confirmed by heatmaps of the young and old ratios vs. the first exon (Figure 2C) and metagene plots of RNA-seq signal (Figure 2D); as in mHSCs and hMSCs, these transcripts tend to be longer (Extended Data Figure 2). Increased cryptic transcription is also a hallmark of NSC aging, but only in activated NSCs.
Figure 2. Analysis of RNA-seq in NSCs and other mammalian tissues suggests a widespread increase in cryptic transcription during mammalian aging.
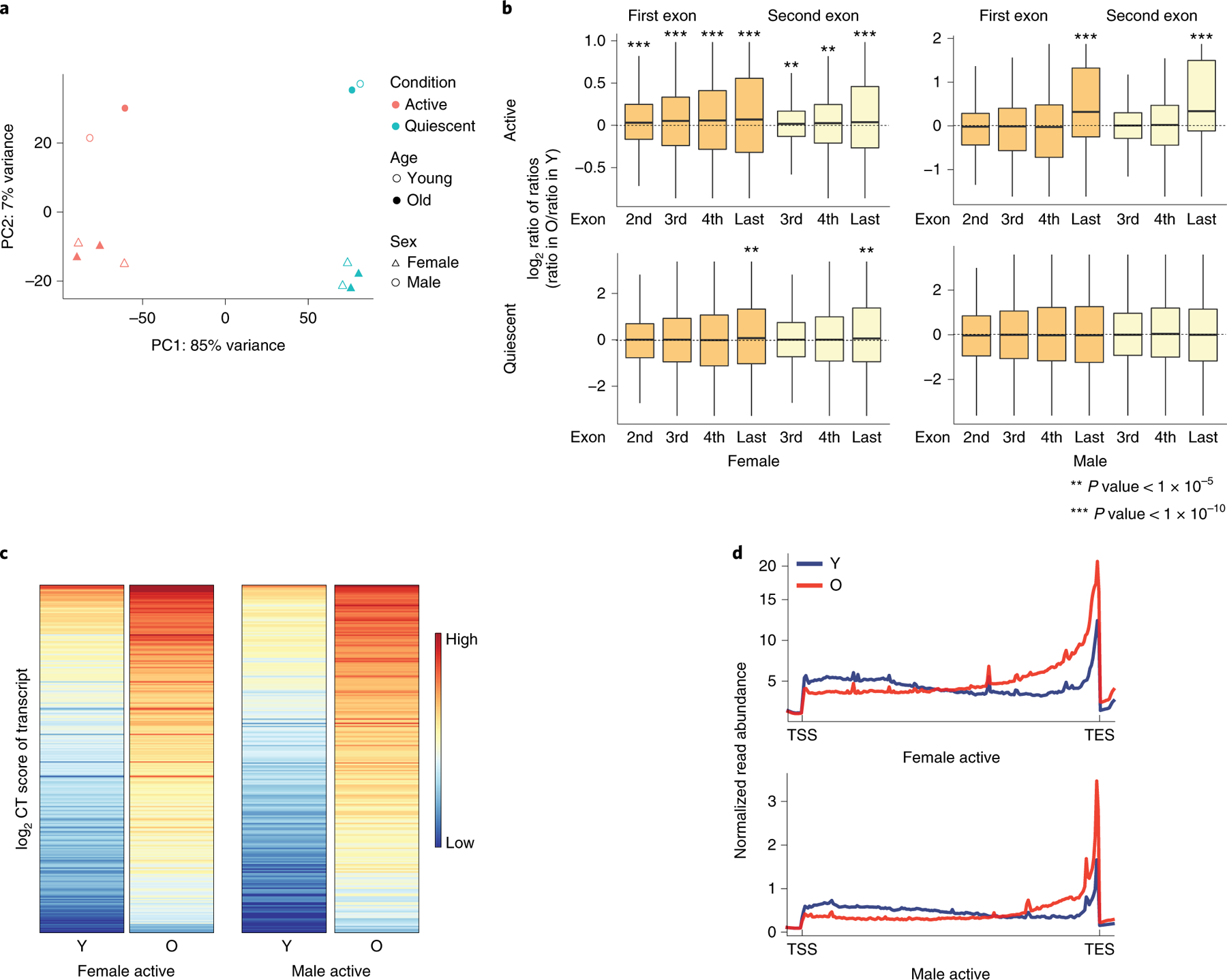
A) Principle component analysis (PCA) of transcriptomes in murine NSCs shows that the primary differences in transcription are based on sex and status (activated vs. quiescent). B) Boxplots of the log2-transformed ratio of reads mapping to the indicated exon vs. reads mapping to the first or second exon (dark and light orange, respectively) in old vs. young samples (ratio in old divided by ratio in young, or ratio of ratios), as indicated, in activated (top) or quiescent (bottom) NSCs in females (left) and males (right). Expressed major transcripts with at least 3 exons were included in the analysis for active female NSC (n=6110), quiescent female NSC (n=4985), active male NSC (n=4654), and quiescent male NSC (n=2956). Bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range. A two-sided Wilcoxon signed-rank test was used to determine significance with the null hypothesis that the calculated log2 ratios are equal to 0. p-values reported in Supplementary Table 1. C) Heatmaps of the CT score of cryptic transcripts shows an increase in old vs. young cells in aNSCs from both female (left) and male (right) mice. D) Metagene plot of RNA-seq read density in old (red) and young (blue) aNSC samples. Cells isolated from females are on the top; those from males on the bottom. In all panels, Y: young; O: old; PC: principle component.
We looked for signs of increased cryptic transcription with age in publicly available aging and senescence RNA-seq datasets (E-GEOD-59966; E-GEOD-46486; GSE53330; EMTAB-4879; and refs.26–35). In 21 out of 25 datasets spanning a range of tissues, cryptic transcription is elevated with age (Extended Data Figure 2), similar to what was observed in the stem cells. This suggests that increased cryptic transcription is a hallmark of aging across mammalian tissues. We could not detect increased cryptic transcription in samples from Rett and Werner syndromes36,37, premature aging models (Extended Data Figure 2), indicating that these models do not recapitulate this feature of aging. Taken together, our data indicate that elevated cryptic transcription is a hallmark of mammalian aging.
Identification of sites of cryptic transcription initiation
Our analysis of RNA-seq data cannot identify the sites from which cryptic transcripts initiate (cryptic transcription start sites, cTSSes). For this, we used a protocol called DECAP-seq in which the 5’ ends of transcripts are sequenced11. Sufficient RNA for this protocol could only be obtained from hMSCs. A modified peak-calling algorithm was used to identify DECAP-seq peaks in young and old hMSC samples. Most peaks cluster around annotated RefSeq TSSes (“endogenous TSSes;” Figure 3A). The DECAP-seq signal persists at a low level throughout gene bodies in young and old samples, suggesting that cryptic transcription occurs in young and old hMSCs. DECAP-seq peaks greater than 2000bp distant from an endogenous TSS were considered to indicate cryptic transcription, to ensure that no signal from annotated promoters is mis-called as cryptic and prevent chromatin signatures associated with such promoters from confounding assessment of chromatin state at cryptic promoters; subsequent analysis considers only these peaks. Fifty-eight percent of the total peaks were found in both samples, while nearly 12% were unique to the young sample and almost 30% were unique to the old sample (Figure 3B), which suggests that cryptic transcription increases with age.
Figure 3. Sequencing the 5’ ends of capped RNA shows increased cryptic transcription during hMSC aging.
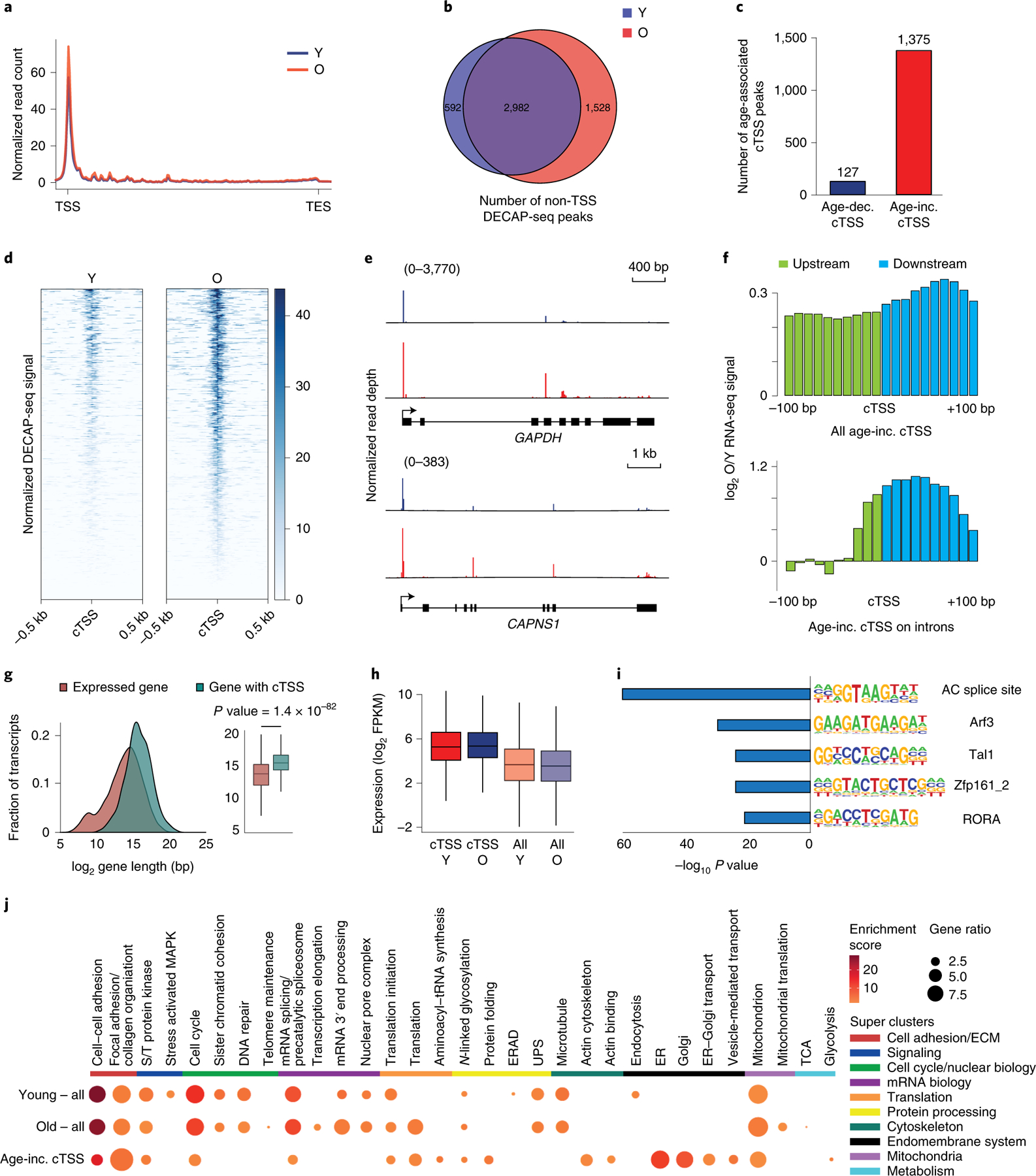
A) Metagene plot showing the distribution of normalized DECAP-seq reads along expressed genes. Young sample in blue; old in red. B) Venn diagram showing the overlap of DECAP-seq peaks (cTSSes) in young and old samples. C) Bar chart indicating the number of DECAP-seq peaks with significantly increased reads in young (blue, age-dec. cTSS) and old (red, age-inc. cTSS) samples. The latter group are as age-associated cTSSes. D) Read pile up of DECAP-seq signal surrounding age-associated cTSSes; young sample on the left and old on the right. E) Mapped DECAP-seq reads from young samples (top, blue) and old samples (bottom, red) along the GAPDH and CAPAS1 genes, which have age-associated cryptic transcription. F) Log2-transformed old to young ratio of averaged normalized RNA-seq reads mapping to the 200bp interval surrounding age-associated cTSSes. All sites are shown in the top plot; only those sites located in introns are shown in the bottom. The region upstream of the age-increased cTSS is green; downstream is blue. G) Length distribution of genes that undergo age-associated cryptic transcription (n=873, blue) and all genes (n=14062, red). p-values were calculated by a two-sided Wilcoxon signed-rank test with the null hypothesis that the average length is the same. H) Boxplot of the log2-transformed FPKM of genes that undergo age-associated cryptic transcription (n=873) compared to all expressed genes (n=13148). Genes with FPKM < 1 were filtered. I) HOMER analysis of the DNA sequence ±200bp from the identified age-associated cTSSes showing known motifs with significant p-values. J) Heatmap showing the results of DAVID GO cluster enrichment of genes containing DECAP-seq peaks in young hMSCs, old hMSCs, and those that increase with age. For boxplots, bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range. S/T protein kinase: serine/threonine protein kinase; ERAD: endoplasmic reticulum-associated degradation; ER: endoplasmic reticulum; UPS: ubiquitin-proteasome system. In all panels, Y: young; O: old, TSS: transcription start site; cTSS: cryptic transcription start site.
We identified non-endogenous TSS-associated DECAP-seq peaks with more reads in the old sample vs. the young. While only 127 such peaks are found in the young sample, there are 1375 peaks with higher signal in the old sample (Figure 3C); these peaks are found in both young and old samples. This is consistent with increased cryptic transcription in old hMSCs, in line with our other analyses. A metagene plot shows that these sites have higher DECAP-seq signal in the old vs. young samples (Figure 3D), while those identified as having more signal in the young show the converse (Extended Data Figure 3). DECAP-seq signal along the whole gene is shown for GAPDH and CAPNS1. An enrichment of DECAP-seq signal is seen around the endogenous TSS of each gene, and several peaks can be found within the gene bodies; the intragenic peaks are higher in the old samples vs. the young (Figure 3E). We consider the 1375 loci identified by this analysis to be bona fide sites of age-associated cryptic transcription.
If the age-increased DECAP-seq peaks are sites where cryptic transcription increases with age, the RNA-seq data should reflect this. There is an increase in the ratio of RNA-seq reads mapping immediately up- and downstream from these sites (Figure 3F, top), which is more obvious when considering the 158 cTSSes located within introns (Figure 3F, bottom). The transcripts that have increased cryptic transcription with age by DECAP-seq are longer (Figure 3G) and tend to be more highly expressed (Figure 3H). There is a slight trend of higher gene expression with age in genes with an age-associated increase in cryptic transcription, though there is no clear trend in genes with decreased cryptic transcription with age (Extended Data Figure 3). We searched for enriched transcription factor binding sites (TFBSs) near age-associated cTSSes. HOMER38 analysis identified enrichment of five motifs within 200bp of these DECAP-seq peaks (Figure 3I and Extended Data Figure 3). Changes in the abundance or binding of transcription factors that recognize these sites with age could contribute to increased Pol II recruitment to the cTSSes and therefore increased cryptic transcription.
We explored the effects of cryptic transcription on hMSC biology. An ~30% reduction in SETD2 levels decreases the self-renewal of these cells (Extended Data Figure 3), consistent with increased cryptic transcription being deleterious in yeast6,39. To better understand the specific processes that cryptic transcription might impact, we performed DAVID clustering analysis of genes producing cryptic transcripts in young and in old hMSCs, as well as those genes that have increased cryptic transcription with age (Figure 3J). In young cells, clusters include cell adhesion, cell cycle, mRNA processing, protein processing, and mitochondria. Specific to young hMSCs are clusters related to stress-activated MAPK signaling, endoplasmic reticulum-associated protein degradation, and endocytosis. Many of the processes impacted by cryptic transcription in young cells continue to be impacted in old hMSCs. Unique to old cells, we observed clusters related to telomere maintenance; translation and protein folding; and endoplasmic reticulum and Golgi function. As cryptic transcription can interfere with normal transcription and aberrant proteins can be translated from cryptic transcripts11,39–41, we expect that cryptic transcription will disrupt these processes. When considering some of the transcription factors that may bind in the vicinity of age-increased cTSSes, we found that they may regulate many of these same processes (Extended Data Figure 3).
Chromatin state is maintained along expressed genes with age
Transcription is regulated at the level of chromatin, which becomes more open with age in many systems (reviewed in ref.2), and could contribute to increased cryptic transcription by promoting intragenic Pol II entry. We performed ChIP-seq to characterize how several transcription-associated histone modifications and heterochromatin-associated histone marks change with age in hMSCs. Additionally, we performed ChromHMM analysis42 to understand how the chromatin state changes. A 10 state model in ChromHMM identified four categories of chromatin states: heterochromatin (states 1–3), enhancers (states 4–6), transcribed genes (states 7–9), and chromatin with no enriched histone modifications (state 10) (Figure 4A). These designations are consistent with the genome-level distribution of states and their enrichment around TSSes and transcription end sites (Extended Data Figure 4).
Figure 4. Chromatin states associated with transcription are largely preserved during aging.
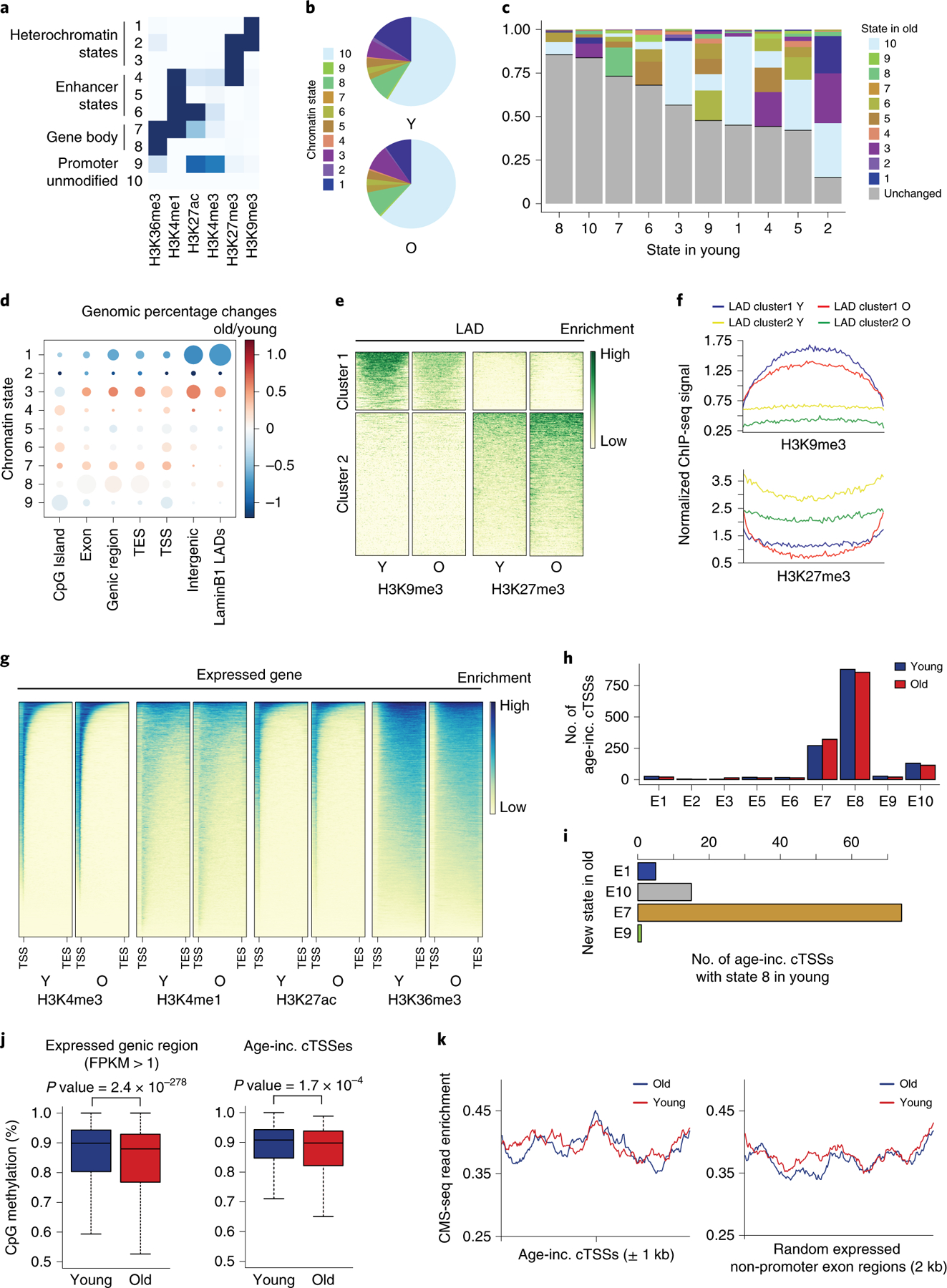
A) Chromatin states identified in a 10 state ChromHMM model. B) Proportion of the genome covered by each state in young (top) and old (bottom) samples. C) Stacked bar graph showing how the chromatin states change with age; each column represents the totality of the indicated state in the young sample and the shaded regions indicating its state in old. D) Proportional dot plot showing the change in chromatin state with age, segregated by genomic region. Each column represents the total genomic space of the indicated region; the dot size indicates the age-averaged proportion of that region that is covered by that state; and dot color represents its change with age in that region. E) Heatmaps depicting the changes in repressive histone modifications H3K9me3 and H3K27me3 in LADs with age. The maps on the left show the young sample. F) Metagene plot showing the same LAD regions as in (E). G) Read pile ups showing the enrichment of the indicated histone modifications along expressed genes (FPKM>1). The maps on the left show the young sample. H) Bar chart showing the distribution of age-increased cTSSes identified by DECAP-seq across chromatin states in young and old hMSCs. I) Bar chart showing the chromatin state in old hMSCs of age-increased cTSSes that were in state 8 in young cells and changed states with age. J) Boxplots showing % CpG methylation in young and old hMSCs across expressed genic regions (1kb window, n=60418; left) or surrounding age-increased cTSSes (±500bp, n=259; right). Bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range. p-values were calculated by a two-sided Wilcoxon signed-rank test with the null hypothesis that methylation levels are equal in young and old samples. K) Metagene plots showing 5-hydroxymethylcytosine (CMS-IP-seq) enrichment around age-increased cTSSes (left) or random expressed genic regions (right). In all panels, Y: young; O, old; TSS: transcription start site; TES: transcription end site; LADs: lamin-associated domains; inc.: increased.
Heterochromatin decreases with age in hMSCs; however, there are only small changes in the fraction of the genome associated with enhancer- and transcription-related chromatin states (Figure 4B and Extended Data Figure 4). Most changes occur between similar states, e.g., state 7 changing to state 8 by losing enrichment of several histone modifications (Figure 4C and Extended Data Figure 4). The most drastic changes in chromatin state are a loss of constitutive heterochromatin, a gain of facultative heterochromatin, and a decrease in the overlap between H3K4me1 and H3K27ac with H3K36me3.
We examined how chromatin state changes with age in particular genomic regions (Figure 4D and Extended Data Figure 4). Intergenic regions and lamin-associated domains (LADs) are enriched for state 1, which is drastically reduced during aging; other LADs show increased enrichment for state 3 during aging (Figure 4E and F; Extended Data Figure 4). Within CpG islands and genic regions, there is increased enrichment of states 3 and 7, while states 5 and 9 are generally depleted with age. This suggests a decrease in transcription with age, consistent with a decline in TPMs of expressed genes with age (not shown). This analysis indicates that normally heterochromatic regions of the genome become more open with age, while genic regions become more open and more closed at different loci.
There are subtle age-associated changes in histone modification enrichment in expressed genes. H3K4me3 distribution does not change with age, but its enrichment around the promoter increases (Figure 4G). Conversely, the distribution of H3K4me1 and H3K27ac becomes wider with age (Figure 4C and D). H3K36me3 levels are depleted within gene bodies (Figure 4G). As H3K36me3 promotes the restoration of chromatin structure following Pol II transit16, this may have effects on the chromatin that are undetectable by ChromHMM analysis. Although chromatin structure at actively expressed genes is largely preserved, the chromatin at these loci may be more open in old hMSCs.
To assess whether changes in chromatin state impact cryptic transcription, we examined how chromatin state at age-increased cTSSes changes with age. These sites are largely located in states 7 and 8, while a smaller fraction of peaks are in state 10 in young cells (Figure 4H). Most cTSSes maintain their chromatin state with age (Figure 4C). However, there is a slight shift from state 8 (H3K36me3 only) to state 7 (H3K36me3/H3K4me1/H3K4me3/H3K27ac) among cTSSes with increased expression during aging (Figure 4I), which is not observed for cTSSes whose expression decreases with age (Extended Data Figure 4). Overall, these data suggest that while chromatin state is preserved during aging, the chromatin is more accessible, and there is an increase of transcription-permissive histone modifications near age-elevated cTSSes.
To determine whether increased cryptic transcription is associated with a more open chromatin structure, we assessed DNA methylation. Whole genome bisulfite sequencing (WGBS) revealed a global reduction of CpG methylation during hMSCs aging (Extended Data Figure 4), which was also observed for 5 hydroxymethylcytosine (5hmC), a product of DNA demethylation by TET enzymes (Extended Data Figure 4). This suggests that although chromatin state, as defined through histone modifications, is largely preserved during aging, globally, the chromatin becomes more open.
When considering genic regions, there is a significant reduction of DNA methylation with age (Figure 4J), which is both indicative of a more open chromatin state and known to contribute to elevated cryptic transcription in ESCs11. At age-increased cTSSes, we also observe lower levels of CpG methylation during aging (Figure 4J), indicating that more open chromatin is associated with increased cryptic transcription during hMSCs aging. These sites show higher levels of 5hmC than random genic regions (Figure 4K, Extended Data Figure 4), indicating that DNA methylation is actively regulated at age-increased cTSSes in young cells. 5hmC levels remain steady at these sites during aging (Figure 4K), despite their decreased DNA methylation, suggesting that the reduction in this is caused by decreased de novo DNA methylation. Loss of CpG methylation with age at age-increased cTSSes indicates that the more open chromatin structure in aged cells promotes elevated cryptic transcription.
Age-increased cTSSes gain a promoter-like chromatin state
The observed chromatin changes at age-increased cTSSes prompted us to more closely examine the histone modification milieu at these sites. We hypothesized that the chromatin structure around age-associated cTSSes might take on an active promoter-like state with age. H3K4me3 is specifically enriched near age-increased cTSSes in old, but not young, hMSCs (Figure 5A), though this is not observed at cTSSes with decreased expression with age (Extended Data Figure 5). The pattern of H3K4me3 enrichment around the age-associated cTSSes is comparable to what is seen at endogenous TSSes (Extended Data Figure 5). A similar trend is seen for H3K4me1 and H3K27ac (Extended Data Figure 5). H3K36me3 levels are decreased around age-increased cTSSes in the old samples compared to the young (Figure 5A and Extended Data Figure 5). This reduction in H3K36me3 enrichment could contribute to the observed reduction of DNA methylation at these sites11. H3K36me3 levels at age-associated cTSSes are much higher than at endogenous TSSes, likely contributing to the low levels of transcription observed at these sites. In hMSCs the chromatin state around age-increased cTSSes becomes more active promoter-like during aging, with a reduction in H3K36me3 and an accumulation of H3K4me3.
Figure 5. Chromatin near cTSSes takes on promoter-like characteristics with age.
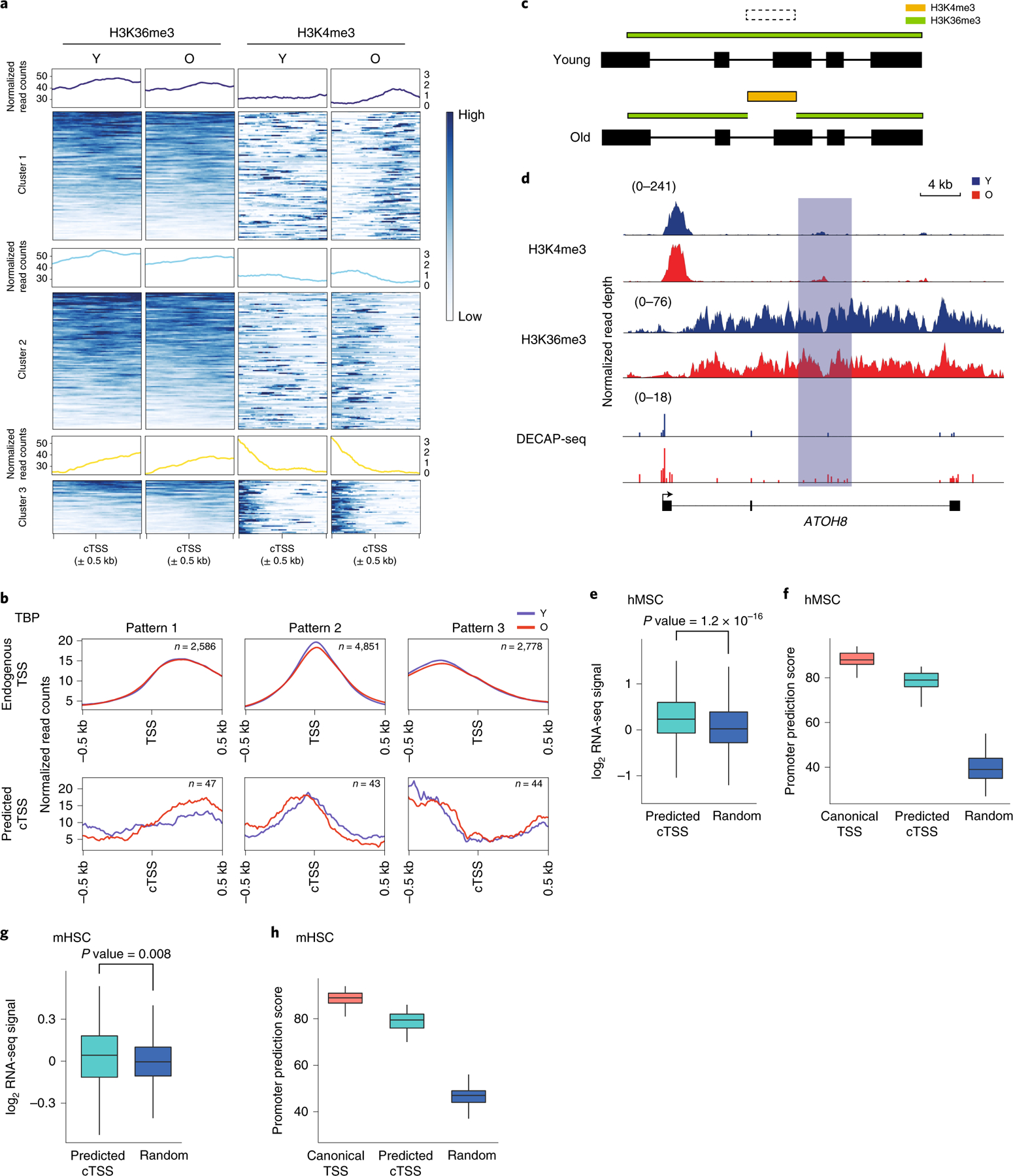
A) Read pile ups showing enrichment of H3K36me3 (left) and H3K4me3 (right) in old and young hMSCs surrounding the 1375 age-increased cTSSes identified by DECAP-seq. Loci are clustered by H3K4me3 enrichment pattern. B) Metagene plots showing TBP enrichment patterns of endogenous TSSes (n=10215, top) and the age-increased cTSSes (n=134, bottom) previously identified that are associated with TBP peaks. cTSSes without TBP signal were excluded from analysis. C) Depiction of the predicted change in chromatin state our algorithm uses to find additional putative cTSSes, based on the change in chromatin state observed at cTSSes in panel (A). H3K4me3 enrichment is represented in gold and H3K36me3 in green. D) An example of a putative age-increased cTSS identified by our chromatin state algorithm. The predicted cTSS in ATOH8 is highlighted in light blue. H3K4me3, H3K36me3, and DECAP-seq signal tracks are shown, with the signal for the young sample in blue and the old in red. E) Boxplot showing the log-normalized ratio of ratios of RNA-seq reads mapping to the exon downstream of the putative age-increased cTSSes identified by chromatin state vs. the first exon of the transcript in old vs. young hMSCs. Predicted age-increased cTSSes found within expressed major transcripts (n = 1056, teal) and randomly selected genic regions (n=1000, blue) were analyzed. F) Boxplot showing promoter prediction scores for endogenous TSSes (n=100, red), putative age-increased cTSSes (n=100, teal), and random genomic sequences (n=100, blue) in hMSCs. G and H) as (E) and (F), respectively, except these depict putative age-increased cTSSes identified in mHSCs (n=264). P-values for both hMSCs and mHSCs boxplots were calculated by a two-sided Wilcoxon rank-sum test against the null hypothesis that the RNA-seq ratios or promoter scores are equal in the two groups, as appropriate. In all panels, Y: young; O: old; TSS: transcription start site; cTSS: cryptic transcription start site; Random: random genomic region. For boxplots, bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range.
We asked whether age-associated cTSSes have other characteristics of promoters. ChIP-seq for TBP revealed that most peaks are located within 2000bp of an annotated promoter (Extended Data Figure 5). TBP is also bound near a subset of age-associated cTSSes to a similar extent in old and young hMSCs (Figure 5B). Clustering of TBP ChIP-seq signal showed that TBP enrichment is similar around endogenous and cryptic TSSes, suggesting that at least part of the transcriptional machinery recognizes select age-associated cTSSes in a manner that allows transcription. The pre-initiation complex may be actively recruited to age-increased cTSSes and, as the local chromatin state becomes more open and permissive with age, transcription initiation may increase at these loci.
If age-associated cTSSes gain an active promoter-like chromatin structure in aged hMSCs, this chromatin signature could be associated with age-increased cTSSes that were not identified by DECAP-seq. We searched for sites within gene bodies where H3K4me3 is gained and H3K36me3 enrichment decreases with age (Figure 5C). This allowed us to identify an additional 2118 putative age-associated cTSSes in hMSCs, exemplified by a site in ATOH8 (Figure 5D). The putative age-increased cTSS is enriched for H3K4me3 in both young and old cells, but there is a local depletion of H3K36me3 with age. The increase of DECAP-seq reads in this region in the old vs. young sample also suggests an age-associated increase in cryptic transcription, though it does not rise to the level of significance required by our differential peak calling algorithm.
To confirm that these sites are associated with elevated cryptic transcription, we analyzed RNA-seq and DECAP-seq data surrounding these loci. There is a significant increase in DECAP-seq signal in old hMSCs vs. young associated with these sites (Extended Data Figure 5). The ratio of RNA-seq signal in the exon downstream of the predicted cTSS vs. the first exon of the transcript is elevated in old vs. young hMSCs (Figure 5E). Putative age-increased cTSSes have higher scores in a promoter 2.0 analysis43 than random sequences, indicating that they have promoter-like characteristics (Figure 5F). We performed a similar analysis using ChIP-seq and RNA-seq data from aging mHSCs20 and identified 510 putative age-associated cTSSes. As in hMSCs, intragenic sites that lost H3K36me3 and gained H3K4me3 with age have an age-associated elevation of RNA-seq reads downstream of the predicted cTSS (Figure 5G). Likewise, these loci also have high promoter prediction scores (Figure 5H). Taken together, these analyses suggest that the sites identified by our chromatin state analysis are bona fide age-increased cTSSes.
Discussion
The impact of changes to the chromatin landscape with age on cells and organisms has not been exhaustively characterized, though they are known to contribute to reduced transcriptional fidelity2. Here we show that intragenic cryptic transcription is elevated with age in mammals and link this increase to an altered chromatin state that is more permissive for transcription activation. Cryptic promoters are sites from which Pol II can aberrantly initiate transcription. These sites have sequence features similar to those in endogenous TSSes and cTSSes can be bound by TBP in young and old cells (Extended Data Figure 3; Figure 5), suggesting that they are functioning promoters that are routinely silenced, likely due to a repressive chromatin state downstream of H3K36me310–12 (Figure 6). This histone modification is reduced within gene bodies with age, especially around age-increased cTSSes (Figures 4 and 5). A concomitant increase in H3K4me1, H3K4me3, and H3K27ac in these regions with age results in an active promoter-like chromatin state (Figure 5). Age-associated cTSSes tend to lose DNA methylation during aging (Figure 4), further opening the chromatin. As the intragenic chromatin state becomes more permissive for transcription initiation during aging, levels of cryptic transcription increase.
Figure 6. Model of the mechanisms driving elevated cryptic transcription in mammalian aging.
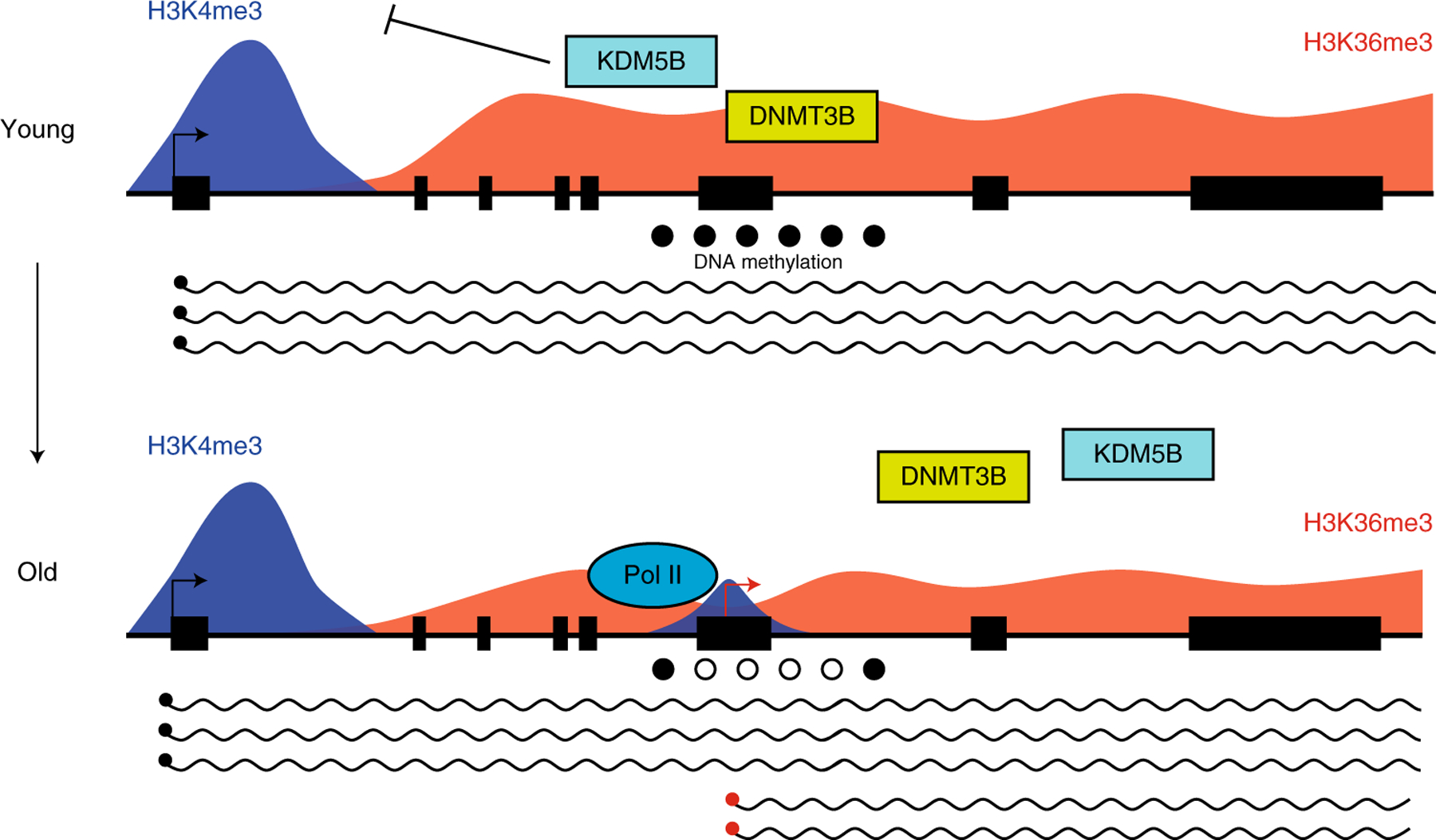
In young cells, H3K36me3 (red peaks) within the gene body of actively transcribed genes serves as a scaffold for the chromatin modifiers KDM5B and DNMT3B (top panel). These enzymes remove intragenic H3K4me3 (blue peaks) and confer DNA methylation (solid black circles) at CpG residues, respectively, generating a repressive chromatin environment that maintains cTSSes in an inactive state10–12. During aging, H3K36me3 levels are reduced within gene bodies, which inhibits the recruitment of KDM5B and DNMT3B to these regions, resulting in accumulation of H3K4me3 and reduced DNA methylation (bottom panel). This generates in a chromatin state that is more permissive for transcription initiation at cTSSes (red arrow), causing an elevation of cryptic transcription with age.
Cryptic transcription increases in a wide range of tissues during aging (Figures 1 and 2; Extended Data Figures 1 and 2), suggesting it may contribute to aging pathologies throughout the body. In murine aNSCs, age-associated cryptic transcription has distinct characteristics in males and females (Figure 2). A similar trend is observed in the human dermal fibroblasts dataset analyzed in Extended Data Figure 2 (ref.30): when segregated by sex, only males show an elevation of cryptic transcription (not shown). The functional consequences and mechanisms responsible for this difference remain unclear. Sex differences in adult neurogenesis have been reported in rodents, including in cell proliferation (aNSCs), and stress affects neurogenesis in a sex-specific manner in rodents (reviewed in44). Little is known about the underlying mechanisms or consequences for healthy cognitive aging in males and females. Our findings raise the possibility that sex-specific changes in cTSS usage may be a novel mechanism responsible for sex differences in neurogenesis during aging.
How elevated cryptic transcription contributes to aging phenotypes remains unclear; however, evidence is accumulating that increased cryptic transcription is deleterious in mammals as it is in yeast6,39. Mild reduction in SETD2 levels reduces the self-renewal of hMSCs (Extended Data Figure 3). Similarly, loss of Dnmt3a, Dnmt3b, Kdm5b, and Setd2, which have characterized roles in repressing cryptic transcription, limits the self-renewal and/or differentiation capacity of stem cells12,23,45–47. In hMSCs, cryptic transcription increases with age in genes whose products contribute to telomere maintenance, proteostasis, and the endomembrane system (Figure 3). We expect that increased cryptic transcription in genes involved in these processes will impact these processes during aging, either at the transcriptional level39,40 or by generating aberrant proteins that directly disrupt them11,41. As cryptic transcription increases with age throughout the body, and is epigenetically encoded, interventions that target this phenomenon may be approachable pro-longevity treatments.
Methods
hMSC cell culture and growth curve
Human cord blood mesenchymal stem cells (hMSCs) were isolated from Wharton’s jelly of a newborn Caucasian male (ATCC #PCS-500–010, lot #63216949). Cells were grown in low glucose DMEM (Life Technologies 11885–084) with 10% FBS (Life Technologies 16000–044, lot #1314735) and 1× penicillin/streptomycin (Life Technologies 15140–122) (“hMSC growth medium”) at 37°C with 5% CO2 and 3% O2. Medium was replaced every 4 days. Cells were grown to ~70% confluence and split 1:4. Cell density was too low for hemocytometer measurement, so growth was estimated as 2 population doublings per passage.
hMSC senescence-associated β-galactosidase assay
Approximately 5×104 hMSCs were plated in one well of a 6 well plate and incubated at 37°C with 5% CO2 and 3% O2 for 4–6 hours. Staining was performed as described in ref.48
hMSC differentiation assays
Approximately 105 hMSCs were plated in one well of a 12 well plate and grown to confluence under conditions described above. Differentiation was performed using the Adipocyte Differentiation tool (ATCC, PCS500050) or Osteocyte Differentiation tool (ATCC, PCS500052) following manufacturer’s instructions. Adipogenic differentiation was assessed after 14 days by oil red O staining and osteogenic differentiation after 21 days by Alizarin Red S staining following standard protocols49,50.
SETD2 knockdown in hMSCs
Lentiviruses expressing an shRNA against SETD2 (MISSION shRNA #TRCN0000003029, Sigma) were used to knock down expression of SETD2. Lentiviruses were produced by co-transfection of the SETD2 shRNA or the MISSION non-targeting control shRNA (NT; #SHC002, Sigma) plasmid and plasmids encoding VSVG and ΔR8.9 (from Xhixun Dou) using Lipofectamine 2000 (11668030, Thermo Fisher) into HEK 293T cells following manufacturer’s protocols.
PD8 hMSCs were infected with SETD2 or NT shRNAs at 3 MOI in hMSC growth medium with 6μg/mL polybrene for 6 hours, then cultured for 2 days in hMSC growth medium at 37°C, 5% CO2, 3% O2. Cells underwent selection in hMSC growth medium with 1μg/mL puromycin for 6 days, after which they were returned to hMSC growth medium.
RT-qPCR to assess SETD2 knockdown in hMSCs
Total RNA was extracted from ~5.5×105 PD12 hMSCs expressing either an NT shRNA or shRNA against SETD2 using the RNEasy Mini Kit (Qiagen 74104) following the manufacturer’s protocol. 1μg of DNase-treated RNA was used for cDNA synthesis or mock (-RT) reactions using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher, #4368814). cDNA and -RT samples were diluted 1:30; mixed cDNA from NT and SETD2 shRNA samples was used as standards, diluted 1:10, 1:100, and 1:1000. Reactions were assembled in triplicate, with 1× Fast SYBR Green Master Mix (Thermo Fisher, #4385612), 0.1μM forward and reverse primers, and 3.2μL template per 10μL reaction. Reactions were run on a Viia7 machine using the fast program. Data were analyzed using the quantity calculated by the QuantStudio v1.3 software package (Thermo Fisher). Primer sequences: SETD2-F: AAACCAGGTGCTCAGCTTATCC, SETD2-R: CCCATGACGTTCCAGAAAGG; GAPDH-F: CAGCCTCAAGATCATCAGCA, GAPDH-R: TGTGGTCATGAGTCCTTCCA.
hMSC RNA-seq library preparation
Total RNA was extracted from PD12 and PD32 hMSCs using the RNEasy Mini Kit (Qiagen 74104) following the manufacturer’s protocol. 100ng of DNase-treated total RNA was depleted of rRNA (NEB, E6310) and underwent stranded RNA-seq library synthesis using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, E7760).
hMSC ChIP-seq library preparation
hMSCs were fixed for ChIP according to standard protocols51. Samples were sonicated to release chromatin with an average fragment length of 400bp in an EpiShear Multi-Sample sonicator (Active Motif 53062).
For each ChIP, 10μL of clarified lysate was added to 90μL of HBSS and used as input into the True MicroChIP Kit (Diagenode, C01010130); following manufacturer’s instructions. Antibodies used: histone H3: Active Motif #61475, lot #17316003 and Millipore #05–928, lot #2884434; H3K4me1: AbCam #ab8895, lot #GR1278894; H3K4me3: Diagenode #C15410030, lot #002; H3K9me3: Active Motif #39765, lot #16513004; H3K27ac: Active Motif #39133, lot #01613007; H3K27me3: Active Motif #39155, lot #23813016; H3K36me3: Active Motif #61101, lot #32412003; RNA polymerase II: Active Motif #39097, lot #29613012; and TBP: Cell Signaling Technologies #44059S, lot #1. 2μL of clarified lysate was used as input and was treated as a sample starting from the reverse crosslinking step. Sequencing libraries were prepared using the MicroPlex Library Preparation Kit v2 (Diagenode, C05010014) following manufacturer’s instructions.
hMSC DECAP-seq library preparation
DECAP-seq was performed as described in ref.11 with modifications. The starting material was poly-A+ RNA was isolated from 100μg of total RNA using the Dynabeads mRNA Purification Kit (Life Technologies, 61006). 5’ phosphate groups were removed using CIP (NEB, M0290), and Cap-Clip acid pyrophosphatase (CellScript, C-CC15011H) was used to decap the mRNAs. Size selection of products was performed after 5’ adapter ligation. A negative control library was generated as in ref.18.
Whole genome bisulfite (WGBS) library preparation
WGBS was performed on genomic DNA isolated from approximately 1×106 PD12 and PD 32 hMSCs as described52.
hMSC CMS-IP-seq library preparation
CMS-IP-seq was performed on approximately 1×106 PD12 and PD32 hMSCs as described, using an in-house anti-CMS antibody53,54.
Murine NSC isolation
Activated and quiescent NSCs (aNSC and qNSC) were freshly isolated from the subventricular zone of adult (~7 month) and aged (19–21 month) hGFAP-GFP transgenic mice (FVB/N background) according to established methods55,56. NSCs were isolated by FACS using the surface markers prominin-1 (CD133) and the EGF receptor, and GFAP-GFP fluorescence (Supplementary Figure 1). Cells were stained with 1:300 EGF-Alexa 647 (Thermo Fisher, E-35351), and 1:400 Prominin-1-biotin (Thermo Fisher, 13-1331-80). Dead cells were excluded using propidium iodide. Cells were sorted on a BD FACS Aria into PBS. aNSC are prominin-1+; GFP+; EGFR+ and qNSC are prominin-1+; GFP+; EGFR-. FACS plots are available upon request. This was performed in biological triplicate, generating each RNA-seq library from a single animal, using ~400 cells per library. Mice were maintained and used according to protocols approved by Stanford’s Administrative Panel on Laboratory Animal Care (APLAC) and in alignment with institutional and national guidelines.
Murine NSC RNA-seq library preparation
RNA-seq libraries were generated by GENEWIZ LLC. using the SMART-Seq v4 Ultra Low Input RNA Kit for cDNA synthesis and the Nextera XT DNA Library Preparation Kit.
High throughput sequencing
Murine NSC RNA-seq libraries were sequenced on the Illumina HiSeq 2500 platform at GENEWIZ LLC. All other libraries were sequenced on the Illumina HiSeq 2500 platform at the Human Genome Sequencing Center at Baylor College of Medicine. Sequencing data were deposited in the GEO database (#GSE156409).
Accessing public datasets
Publicly-available datasets for aging mHSC20 and various non-stem cell aging and senescence datasets (E-GEOD-59966; E-GEOD-46486; GSE53330; E-MTAB-4879; and refs.26–37) were downloaded from the GEO database using fastq-dump version 2.8.0 (https://ncbi.github.io/sra-tools/fastq-dump.html).
Data quality control, read trimming, and read mapping
Sequencing adaptors were trimmed and low quality reads removed using Trim Galore version 0.4.4 with default parameters (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). RNA-seq reads were mapped to the genome using HISAT2 version 2.1.0 (ref.57); DECAP-seq reads using STAR version 2.7 (ref.58); ChIP-seq reads using bowtie2 version 2.2.4 (ref.59); and WGBS and CMSIP-seq reads using Bismark version 0.22.3 (ref.60). Human genome assembly hg19 and mouse genome assembly mm10 were used as reference genomes. Reads mapped to the ENCODE blacklist regions61,62 were filtered. Multi-aligned reads were removed.
RNA-seq and global cryptic transcription event assessment
Transcript-level read count and FPKM and TPM measurements were performed using salmon version 1.0.0 (ref.63) with the following parameters: -l A –validateMappings.
Cryptic transcription was assessed by exon. The exon-level read count was performed using featureCounts version 1.5.3 (ref.64) with the following parameters: -t exon -g transcript_id -f -O -T 8 -p -C. Cryptic transcript identification was performed on the transcript with the highest TPM as determined in the transcript-level analysis (the “major transcript”). Major transcripts with length >3 kb and TPM ≥1 were used for further analysis.
The length-normalized TPM of each exon was calculated, and the relative expression value (Ei) was determined:
Where i is the i th exon of a transcript; L is the length of the exon; and b = 1 or 2.
Fold change of relative expression was then calculated:
Where i is the i th exon of a transcript, O is the data of the old sample, and Y is the data of the young sample.
To assess age-associated cryptic transcription, the log2-transformed FCi for the second, third, fourth, and last exons of major transcripts relative to their first and second exons was calculated in old vs. young samples; FCi >0 indicates increased cryptic transcription with age. The same analysis was performed on major transcripts separated into expression quantiles. We compared the averaged Ei values (exons 2 through the second to last exon) in old vs. young cells, using a two-tailed Wilcoxon signed rank test to determine significance vs. the null hypothesis that the average FCi was the same in both samples. To identify transcripts with age-increased cryptic transcription, we used the CT score, i.e., the Ei of the exon with the maximum FCi relative to the first exon. Transcripts with significantly large ratio of ratios were identified using the rank ordering algorithm in ROSE65,66. These were further filtered : 1) all exons downstream of the identified exon have a higher Ei in the old sample than in the young; 2) transcripts in which the first exon of a transcript has the highest FCi were excluded; and 3) the expression of the second exon in the old sample cannot be lower than in the young.
DECAP-seq data analysis
Strand-specific reads were analyzed independently. cTSSes were identified in 10bp bins across the genome by comparing sample data to the negative control. Read count assessment was performed using the csaw package version 1.20 (ref.67) and the count data were normalized using TMM in edgeR version 3.12 (ref.68). Bins were tested for significant differences using a Poisson test with the null hypothesis that the signal in the sample is equal to the signal in the negative control.
Where p is the output p-value, x is the number of reads in a 10bp bin of the sample, and λ is the number of reads in the negative control.
Bins were merged into 100bp windows, with the most significant bin representing the window. FDR was calculated using the Benjamini & Hochberg method69. cTSSes were defined as windows with FDR < 0.05 and fold change > 1.2 in the sample vs. the negative control. Windows overlapping with annotated TSSes and 3’ UTRs were filtered. A common cTSS was defined as a peak identified in young and old datasets; otherwise, the cTSS was considered unique to the young or old sample.
Age-associated cTSS peaks were defined as windows in the old sample with significantly more reads than negative control (FDR < 0.05) and significantly more reads than the young sample (FDR < 0.05 and fold change > 1.2).
We performed DAVID clustering (version 6.8 with default parameters)70 on 3 separate gene lists: the first comprised transcripts containing 3574 DECAP-seq peaks found in young cells; second list the transcripts with the 4510 peaks in old cells; the third contained transcripts with the 1375 age-increased DECAP-seq peaks. Gene lists and full DAVID results provided in Supplementary Tables 2–5.
Analysis of age-increased cryptic transcription
The distribution of length and expression levels of transcripts with age-increased cryptic transcription was compared to the major transcripts of expressed genes, as defined above. Significance was determined using a two-tailed Wilcoxon rank sum test vs. the null hypothesis that they are equal.
Motifs within 200bp of age-increased cTSSes were identified by HOMER version 4.8 findMotif.pl with default parameters38. We chose thirteen TFs that bind the identified motifs, have the largest changes in expression with age in hMSCs, and have ENCODE ChIP-seq datasets. Target genes of called peaks downloaded from the ENCODE website (www.encodeprojects.org) were identified with ChIPseeker version 1.8.6 (ref.71) using default parameters. The following datasets were used: ENCFF207AVV, ENCFF753WNT, ENCFF983STO, ENCFF719PKP, ENCFF777ZEH, ENCFF341NJI, ENCFF687AQV, ENCFF088XQT, ENCFF764OZD, ENCFF905PYM, ENCFF333FZO, ENCFF650QJC, ENCFF889AKD, ENCFF815HWK, ENCFF031ZWH, ENCFF004QBE, ENCFF449PID, ENCFF169TCW, ENCFF429BQL, ENCFF440PZY, ENCFF400JCO, and ENCFF116OUV. GO-term enrichment analysis was performed using the enrichGO function from clusterProfiler R package version 3.0.4 (ref.72). GO terms were grouped into groups and larger GO clusters. GO cluster enrichment was calculated as follows. A single list of GO terms was compiled for each TF that included all GO terms significantly enriched any dataset for that TF. GO terms that were broadly represented among the various datasets were grouped and clustered. Gene lists for the GO clusters were compiled by merging all genes in the GO terms that fell into the selected GO groups in that GO cluster. GO cluster enrichment was determined for each TF using the one-tailed hypergeometric test73 with the assumption that more genes are regulated by the TF in question than expected by chance. Results of the GO analysis and clustering provided in Supplementary Table 6.
FPKM fold changes (old/young) of all the major transcripts were calculated and ranked. The rank distributions of transcripts with age-increased and age-decreased cTSSes were compared to the distribution of the rank list of all the genes.
RNA-seq read coverage 100bp up- and downstream of age-associated cTSSes was counted using deeptools version 3.2.0 (ref.74) with the computeMatrix function and a 10bp bin size. The old to young ratio of summed read counts for each bin was calculated. A two-sided Wilcoxon rank sum test vs. the null hypothesis that the ratio was equal in young and old samples was used to determine significance.
Heatmaps and metagenes plot were made using deeptools version 3.2.0 (ref.74) using plotHeatmap and computeMatrix.
hMSC WGBS data analysis
Duplicated reads were removed by the deduplicate_bismark function from Bismark version 0.22.3 (ref.60). DNA methylation information was extracted using the bismark_methylation_extractor function. Statistical analysis of DNA methylation and annotation was performed using methylKit R package version 1.16.1 (ref.75). Methylated CpG sites with read coverage of at least 10 were included in the analysis. Sites in the top 0.1% of read coverage were excluded. Coverage values between samples were normalized using a scaling factor derived from differences between median of coverage distribution. Percent CpG methylation on expressed genic regions (FPKM>1) and age-increased cTSS regions (up and downstream 500bp) was calculated.
hMSC CMS-IP-seq data analysis
5hmC peaks were called using macs2 version 2.1.0 (ref.76) callpeak function with input as control, q-value < 0.01. Signal tracks were generated using the macs2 version 2.1.0 bdgcmp function with -m ppois parameter. Enrichment analysis of 5hmC peak at age-increased cTSSes was performed using the regioneR R package version 1.22 (ref.77) permTest function with the universe/background limited to non-promoter genic regions.
hMSC ChIP-seq data analysis
Duplicated reads were removed using Picard (http://broadinstitute.github.io/picard). Peak calling was performed using MUSIC78. Differentially bound regions were identified by the csaw version 1.20 package67 with default parameters, using get_motimal_broad_ERs model for H3K9me3, H3K36me3 and H3K27me3 and get_optimal_punctate_ERs model for the remaining datasets. Differentially bound regions were identified using the csaw version 1.20 package67 with H3 total and input as internal control for the histone markers and non-histones (TBP and RNA Pol II-Ser5P), respectively.
Heatmaps of read abundance of histone markers in genic regions and around TSSes/cTSSes were generated by deeptools version 3.2.0 (ref.74).
TBP read abundance within 500bp of age-increased cTSSes or endogenous TSSes was calculated in 10bp bins using cTSSes or TSSes as reference and processed TBP bigwig files as input; regions without TBP signal were excluded. Three clusters were made using the k-means method.
H3K36me3 coverage around age-increased cTSSes and endogenous TSSes was calculated using deeptools version 3.2.0 computeMatrix function74 in 10bp bins. Endogenous TSSes of major transcripts were used in this analysis (defined above). Changes in total read depth around cTSSes and TSSes in both young and old samples was assessed using a two-sided Wilcoxon rank sum test vs. the null hypothesis that the normalized read depth is equal.
Chromatin state analysis
Chromatin states were identified using a 10-state model in chromHMM version 1.17 (ref.42) with pre-defined peaks as input. In addition to pre-defined genomic regions in the package, we assessed the enrichment of chromatin states 1kb up- and downstream of TSSes and transcription end sites (TESes), using genomic coordinates from hg19.
Heatmaps of the LADs (downloaded from the USCS genome browser using human assembly hg19) were made with the deeptools version 3.20 plotHeatmap function74 using histone modification signal tracks as sample and H3 total signal tracks as control. LADs were separated into two groups by k-means clustering based on H3K9me3 enrichment.
Integration of chromHMM results with DECAP-seq defined age-increased cTSSes was performed by counting the number of age-increased cTSSes in each chromatin state in both young and old samples. Chromatin state changes at age-increased cTSS regions were analyzed by tracing the transition of chromatin states from young to old in regions 1kb up- and downstream of cTSSes.
cTSS region prediction from histone modification patterns
Gene body regions at least 1kb away from any endogenous TSSes, were considered putative age-increased cTSS regions if they: 1) contained significantly more H3K4me3 reads in the old sample than the young and the regions did not overlap with H3K36me3 peak in the old sample or 2) contained significantly fewer H3K36me3 reads in the old sample than the young and the region was within 2kb of a H3K4me3 peak in the old sample. Significance was determined with a Poisson test, using signal abundance of the young sample as , vs. the null hypothesis that signal abundance is the same in both samples.
Analysis of putative age-increased cTSS regions
Putative age-associated cTSS regions were analyzed by Promoter – 2.0 (ref.43). To generate promoter scores, 100 iterations were performed in which 100 randomly-selected promoters, putative age-increased cryptic promoters, and genomic sequences (2kb each) were analyzed. Promoter score was defined as the number of sequences identified as containing promoter features. The sample sequences analyzed each time were distinct and samples were randomly selected without replacement.
The number of RNA-seq reads were compared between the exon downstream of the identified region and the first exon of the transcript, following normalization by mapped read count and exon length. A two-sided Wilcoxon rank sum test was used to determine significance. The DECAP-seq signal within 1kb of the midpoint of the identified region in young and old samples was compared. A two-sided Wilcoxon rank sum test was used to determine significance with the null hypothesis that the normalized DECAP-seq signal is equal in the young and old samples.
Statistics & Reproducibility
In this study, we 1) reanalyzed publicly available aging RNA-seq and ChIP-seq datasets and 2) generated RNA-seq, ChIP-seq, WGBS, and CMS-IP-seq datasets from NSCs isolated from young and old mice and from culture-expanded hMSCs, which were validated has having young (low passage) or old (high passage) phenotypes. Mice were randomly assigned to be sacrificed as adults or as aged adults. No statistical method was used to predetermine number of mice sacrificed at each timepoint. For hMSCs, cells were randomly allocated to be processed or continue growing. During hMSC assessment, experimenters were not blinded to whether the cells were low vs. high passage. Results were consistent across three independent culture expansions. For sequencing data analysis, only reads that failed standard QC assessments (detailed above) were excluded. Experimenters were not blinded to the identity of the datasets. The statistical methods used to distinguish differences between samples are detailed in the corresponding sections of the Methods.
Extended Data
Extended Data Fig. 1. Additional analysis of aging RNA-seq from mHSCs and hMSCs.
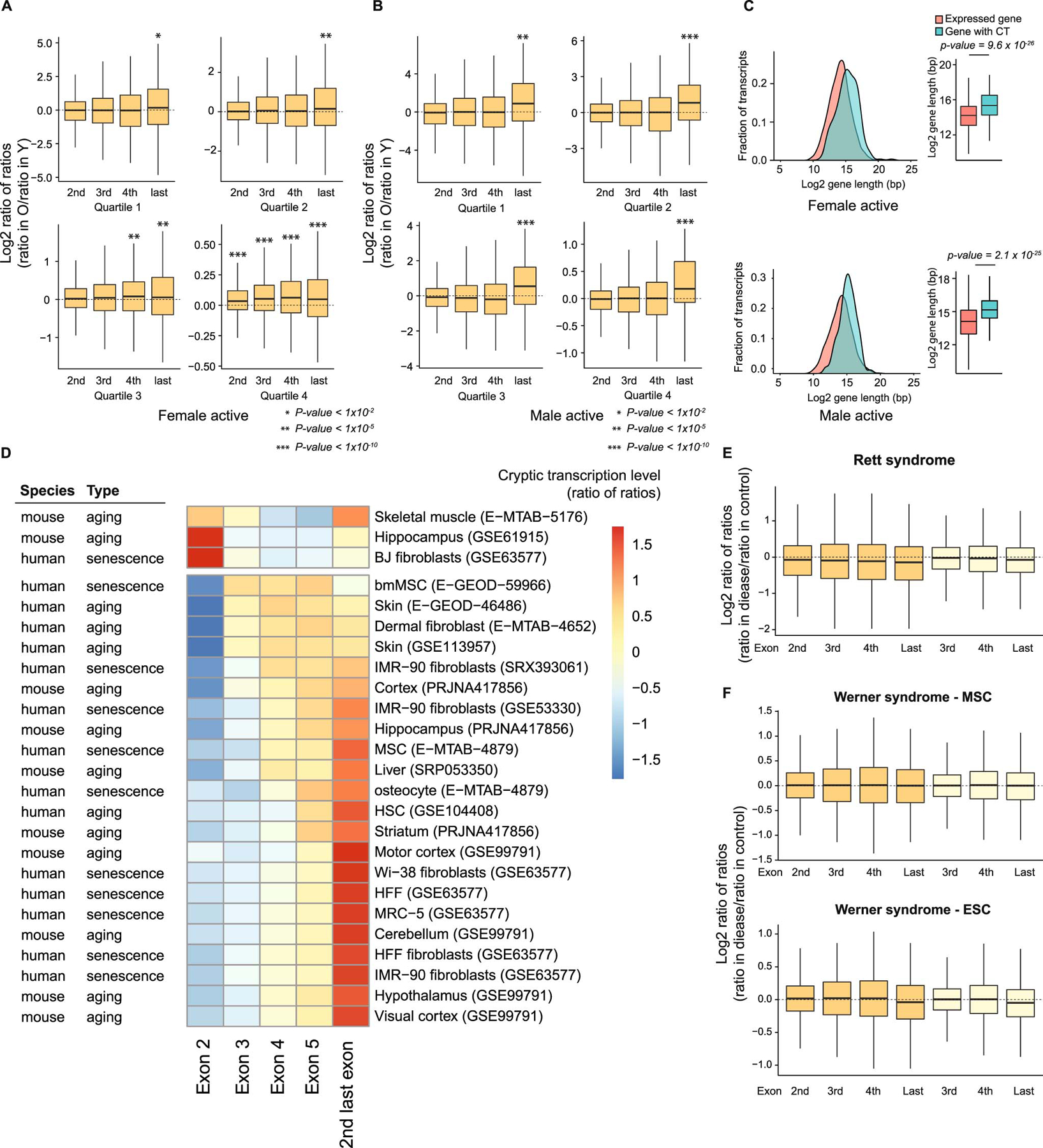
A) Growth curve of culture-expanded hMSCs; PD: population doubling. B) Proportion of senescence-associated β-galatosidase stained hMSCs at the indicated PDs, showing standard error of the mean. In total, 1629, 1641, and 293 cells were analyzed in PD 12, PD 32 and PD 42, respectively. C) Adipogenic and osteogenic differentiation of hMSCs is shown by Oil Red O (ORO) and Alizarin Red S (ARS) staining. Experiments were performed 3 times. D) Boxplots of the log2-transformed ratio of reads mapping to the indicated exon vs. reads mapping to the first or second exon (dark and light orange, respectively) in Setd2-perturbed vs. control samples (ratio in Setd2-perturbed divided by ratio in control, or ratio of ratios). Samples used: Setd2 knockout (n=6869, GSE 72855; n=7821, E-GEOD-54932)11,23 or knockdown (n=6606, E-GEOD-51006)22 in murine embryonic stem cells and knockout in murine oocytes (n=7143, GSE112832)24.E and F) Boxplots showing the log2-transformed ratio of RNA-seq reads mapping to the indicated exons vs. the first exon of genes in mHSCs (E) and hMSCs (F). Young samples are in blue and old in red; Y: young, O: old. G and H) Boxplots of the log2-transformed ratio of reads mapping to the indicated exons vs. reads mapping to the first exon in old vs. young samples (ratio in old divided by ratio in young, or ratio of ratios) divided by expression quartile in mHSCs (G) and hMSCs (H). I and J) Bar charts of transcripts in which the indicated exon has a 2-fold increase (red) or decrease (blue) in TPM vs. the first exon; mHSCs in (I) and hMSCs in (J). K and L) Histograms of the CT scores of major transcripts; mHSCs in (K) and hMSCs in (L). M and N) Scatterplots showing the log2-transformed CT scores in old vs. young samples; mHSCs in (M) and hMSCs in (N). Blue indicates an age-associated increase in cryptic transcription. O and P) Length distribution of transcripts with increased cryptic transcription (n=210 for mHSCs and n=305 for hMSCs) with age vs. expressed major transcripts with at least 3 exons. mHSCs are in (O) and hMSCs in (P). For boxplots, bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and the whiskers show 1.5-fold of the interquartile range. p-values were calculated using a two-sided Wilcoxon signed-rank test vs. the null hypothesis that the samples have the same average value or the log2-transformed ratio of ratios equals 1. Exact p-values for panels D, G, and H are provided in Supplementary Table 1. Expressed major transcripts with at least 3 exons were included in the CT analyses for mHSCs (n=10068, panels E, G, and O) and hMSCs (n=9230, panels F, H, and P).
Extended Data Fig. 2. Analysis of aging RNA-seq in NSCs and other tissues.
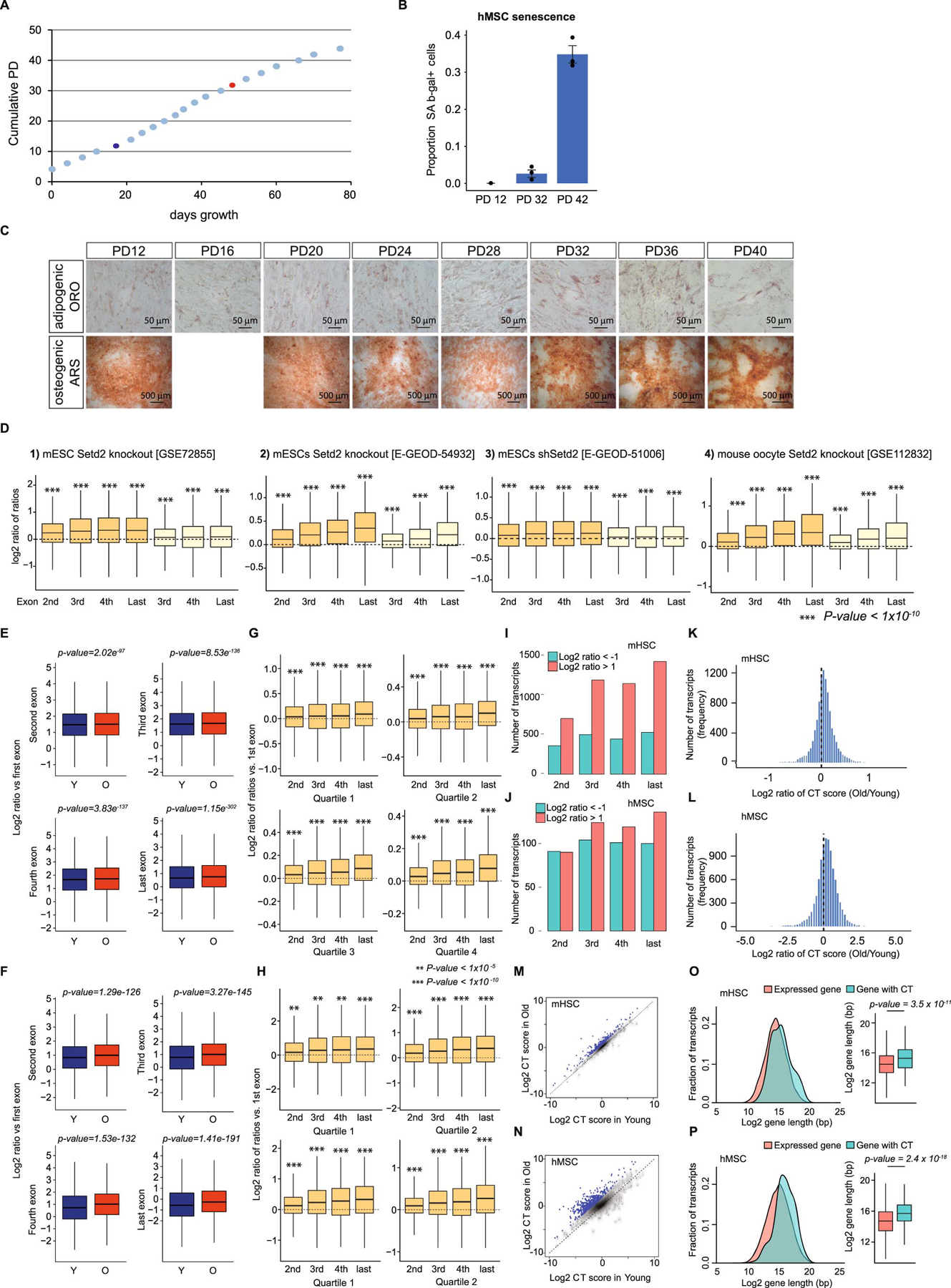
A and B) Boxplots showing the log2-transformed ratio of ratios (indicated exon vs. first exon) for transcripts in aNSCs, separated into quartiles by expression levels. aNSCs isolated from female mice are shown in (A) and from males in (B); Y indicates young and O old. Expressed major transcripts with at least 3 exons were included in the analyses for females (n=6110) and males (n=4654). P-values were calculated using a two-sided Wilcoxon signed-rank test with the null hypothesis that the calculated log2 ratios are equal to 0; exact p-values are provided in Supplementary Table 1. C) Comparison of the distribution of the length of transcripts with an increase in cryptic transcription with age vs. expressed major transcripts with at least 3 exons in aNSCs, shown as a histogram and boxplot. aNSCs isolated from females on top (n=266 for genes with CT and n=6110 for all major transcripts) and from males on the bottom (n=237 for genes with CT and n=4654 for all major transcripts). P-values were calculated using a two-sided Wilcoxon signed-rank test. D) Heatmap depicting the log2-transformed ratio of ratios (indicated exon vs. the first exon) from a variety of mammalian aging or senescence RNA-seq datasets, identified in the figure (E-GEOD-59966; E-GEOD-46486; GSE53330; E-MTAB-4879; and refs.26–35). E and F) Boxplots showing the log2-transformed ratio of ratios (indicated exon vs. first or second exon) for transcripts in fibroblasts from Rett syndrome patients vs. controls36 (E) and cells engineered to carry a mutation in LMNA that causes Werner syndrome37 (F). Expressed major transcripts with at least 3 exons were included in the analysis (Rett syndrome, n=7302; Werner syndrome MSCs, n=8934, Werner syndrome ESCs, n=10185). No significant result founds were in (E) and (F) using a two-sided Wilcoxon signed-rank test. For boxplots, bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range.
Extended Data Fig. 3. Additional analysis of cryptic transcription in aging hMSCs.
DECAP-seq read pile ups around cTSSes that were identified as having higher DECAP-seq peaks in the young hMSC sample vs. the old, i.e., sites where cryptic transcription decreases with age. B) Genes were ranked by the ratio of their FPKM in young cells vs. FPKM in old. Histograms showing the ranked distribution of genes in the following categories: all genes (top); genes with sites that have an age-associated increase in cryptic transcription (middle); and genes with sites that have a decrease in cryptic transcription with age (bottom). FC indicates fold change. C) RT-qPCR results showing a mild decrease (~30%) in SETD2 RNA levels upon SETD2 knockdown in hMSCs. D) Growth curve showing growth of hMSCs expressing a control, non-targeting (NT) shRNA vs. those expressing SETD2 shRNA. E) Complete HOMER de novo motif results of the significant motifs found from age-increased cTSSes flanking regions (±200bp). Known promoter elements are highlighted in green. The at-AC splice acceptor sequence is shown in blue. Transcription factors that bind motifs similar to the ones identified by HOMER are shown in grey if they are not expressed in hMSCs (FPKM<1); ones listed in black are expressed in hMSCs. Transcription factors highlighted in bold and indicated with an asterisk show the highest age-associated changes in expression and were included in a GO analysis. The p-value was directly calculated by HOMER Motif Analysis38. F) GO analysis of putative targets of the indicated transcription factors in ENCODE datasets. Gene ratio indicates the proportion of genes in the dataset that fall in the GO cluster. In all panels, cTSS: cryptic transcription start site. Enrichment p-values were generated by a one-sided hypergeometric test to determine if the list contain more genes for the GO cluster than expected by chance.
Extended Data Fig. 4. Genome-wide analysis of chromatin states.
A) Emission parameters of the 10-state ChromHMM model in hMSCs. B) Enrichment of the ChromHMM states in the indicated genomic regions in young hMSCs. C) As (B), except in old hMSCs. D) Enrichment of the ChromHMM states around annotated TSSes and TESes in young and old hMSCs. E) Transition map of ChromHMM states in old vs. young hMSCs. State in old is along the x-axis and state in young along the y-axis. F) Two examples of a decline in H3K9me3 (ChromHMM state 1) enrichment at LADs with age. Normalized mapped reads are shown in blue for young hMSCs and in red for old. G) Distribution of the chromatin states of age-decreased cTSSes determined by DECAP-seq in young and old hMSCs. H) Methylated CpG distributions in young (left) and old (right) hMSCs. I) Number of CMSIP-seq (5-hydroxymethylcytosine) peaks in young and old hMSCs. J) Graphical representation of a one-sided permutation test with the null hypothesis that the number of CMS-IP-seq peaks that overlap with age-increased DECAP-seq peaks is equal to the background level of CMS-IP-peaks. This shows a significant overlap of CMS-IP-seq peaks with age-increased cTSSes. In all panels, Y: young; O: old; TSS: transcription start site; TES: transcription end site; LAD: lamin-associated domain.
Extended Data Fig. 5. Chromatin state changes around age-increased cTSSes.
A) Read pile ups of H3K4me3 around annotated TSSes (left), age-increased cTSSes (middle), and age-decreased cTSSes (right), independently clustered into 3 groups. B) As in (A), except H3K4me1 enrichment is shown. C) As in (A), except depicting H3K27ac enrichment. D) Boxplots showing H3K36me3 enrichment around age-increased cTSSes (n=1373) and endogenous TSSes (n=13802) in young and old hMSCs. p-values were calculated using a two-sided Wilcoxon signed-rank test with the null hypothesis that enrichment was equal in the young and old samples. E) Bar chart showing the proportion of TBP ChIP-seq peaks around endogenous TSSes in young and old hMSCs. F) Metagene plot of TBP enrichment around annotated TSSes in hMSCs. G) DECAP-seq signal around putative age-associated cTSSes predicted in hMSCs by the chromatin state model. Averaged read depth of putative age-associated promoter regions (±1kb of the midpoint of the identified region) in young (blue) and old (red) is shown on the left at 100bp resolution; a boxplot of the log2-transformed ratio of signal in old vs. signal in young shown on the right (n=166). Distinct random genic non-promoter regions (length =2kb) were used as control (n=2774). p-values were calculated using a two-sided Wilcoxon signed-rank test vs. the hypothesis that the RNA-seq ratios were equal in the putative age-increased cTSSes vs. control regions, as appropriate. Regions without DECAP-seq signal were excluded from analysis. In all panels, Y: young; O: old; TSS: transcription start site; cTSS: cryptic transcription start site. For boxplots, bounds of box show the 25th and 75th percentiles; the central lines in the box plots represent the median value; and whiskers show 1.5-fold of the interquartile range.
Supplementary Material
Acknowledgements
We thank Dr. Rui Chen and the Human Genome Sequence Center at Baylor College of Medicine for performing the Illumina sequencing reported here. This work was funded by NIH grants R01AG052507 to WD and R01AG053268 to AEW; R01HL134780 and R01HL146852 to YH; CPRIT award R1306, to WD; and a Ted Nash Long Life Foundation research grant to WD. BSM was supported by NIH training grant T32AG000183.
Footnotes
Competing Interests
The authors declare no competing interests.
Code Availability
All code for implementing the analyses described in this paper is available at Github: https://github.com/NyxSLY/ASCT.
Data Availability
All RNA-seq, ChIP-seq, WGBS, and CMS-IP-seq data has been deposited in the GEO database at NCBI (#GSE156409).
References
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Booth LN & Brunet A The Aging Epigenome. Mol. Cell 62, 728–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhan M et al. Temporal and spatial transcriptional profiles of aging in Drosophila melanogaster. Genome Res. 17, 1236–1243 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai RW et al. Multi-level remodeling of transcriptional landscapes in aging and longevity. BMB Rep. 52, 86–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Son HG, Altintas O, Kim EJE, Kwon S & Lee SJV Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 18, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sen P et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 29, 1362–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCauley BS & Dang W Histone methylation and aging: Lessons learned from model systems. Biochim. Biophys. Acta - Gene Regul. Mech 1839, 1454–1462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen P, Shah PP, Nativio R & Berger SL Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hennig BP & Fischer T Chromatin and cryptic transcription. Transcription 4, 97–101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho S et al. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 41, 2881–2893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neri F et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543, 72–77 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Xie L et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30, 1473–1484 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venkatesh S & Workman JL Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol 16, 178–189 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Belotserkovskaya R et al. FACT facilitates transcription-dependent nucleosome alteration. Science (80-.) 301, 1090–1093 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Kaplan CD, Laprade L & Winston F Transcription elongation factors repress transcription initiation from cryptic sites. Science (80-.). 301, 1096–1099 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Carrozza MJ et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Pu M et al. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 29, 718–731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ni Z, Ebata A, Alipanahiramandi E & Lee SS Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11, 315–325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodell MA & Rando TA Stem cells and healthy aging. Science (80-.). 350, 1199–1204 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Sun D et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner W et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One 4, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrari KJ et al. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 53, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y et al. H3K36 histone methyltransferase Setd2 is required for murine embryonic stem cell differentiation toward endoderm. Cell Rep. 8, 1989–2002 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Xu Q et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat. Genet 51, 844–856 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Urbán N, Blomfield IM & Guillemot F Quiescence of Adult Mammalian Neural Stem Cells: A Highly Regulated Rest. Neuron 104, 834–848 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Adelman ER et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 9, 1080–1101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boisvert MM, Erikson GA, Shokhirev MN & Allen NJ The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 22, 269–285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke LE et al. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U. S. A 115, E1896–E1905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleischer JG et al. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 19, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaisers W et al. Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PLoS One 12, 1–21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacRae SL et al. DNA repair in species with extreme lifespan differences. Aging (Albany. NY). 7, 1171–1184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marthandan S et al. Conserved senescence associated genes and pathways in primary human fibroblasts detected by RNA-seq. PLoS One 11, 1–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marthandan S et al. Similarities in Gene Expression Profiles during In Vitro Aging of Primary Human Embryonic Lung and Foreskin Fibroblasts. Biomed Res. Int 2015, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rai TS et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of Neoplasia. Genes Dev. 28, 2712–2725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stilling RM et al. De-regulation of gene expression and alternative splicing affects distinct cellular pathways in the aging hippocampus. Front. Cell. Neurosci 8, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson BS et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat. Med 23, 1203–1214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science (80-.). 348, 1160–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinz S et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDaniel SL et al. H3K36 Methylation Regulates Nutrient Stress Response in Saccharomyces cerevisiae by Enforcing Transcriptional Fidelity. Cell Rep. 19, 2371–2382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haupt S, Söntgerath VSA, Leipe J, Schulze-Koops H & Skapenko A Methylation of an intragenic alternative promoter regulates transcription of GARP. Biochim. Biophys. Acta - Gene Regul. Mech 1859, 223–234 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Cheung V et al. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 6, 2550–2562 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ernst J & Kellis M ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knudsen S Promoter2.0: For the recognition of PolII promoter sequences. Bioinformatics 15, 356–361 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Yagi S & Galea LAM Sex differences in hippocampal cognition and neurogenesis. Neuropsychopharmacology 44, 200–213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Challen GA et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15, 350–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziller MJ et al. Dissecting the Functional Consequences of De Novo DNA Methylation Dynamics in Human Motor Neuron Differentiation and Physiology. Cell Stem Cell 22, 559–574.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart MH et al. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal in mice. Blood 125, 2075–2078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mori E et al. Impaired adipogenic capacity in induced pluripotent stem cells from lipodystrophic patients with BSCL2 mutations. Metabolism. 65, 543–556 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Liu B et al. A protocol for isolation and identification and comparative characterization of primary osteoblasts from mouse and rat calvaria. Cell Tissue Bank. 20, 173–182 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dang W et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lister R et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pastor WA et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y, Pastor WA, Zepeda-Martínez JA & Rao A The anti-CMS technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat. Protoc 7, 1897–1908 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Codega P et al. Prospective Identification and Purification of Quiescent Adult Neural Stem Cells from Their In Vivo Niche. Neuron 82, 545–559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leeman DS et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science (80-.). 359, 1277–1283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krueger F & Andrews SR Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amemiya HM, Kundaje A & Boyle AP The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep 9, 1–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunham I et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y, Smyth GK & Shi W FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Lovén J et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lun ATL & Smyth GK Csaw: A Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 44, e45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benjamini Y & Hochberg Y Controlling the False Discovery Rate : A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. . Ser. B (Methodol.) 57, 289–300 (2016). [Google Scholar]
- 70.Dennis G et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, (2003). [PubMed] [Google Scholar]
- 71.Yu G, Wang LG & He QY ChIP seeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson NL, Kotz S & Kemp AW Univariate Discrete Distrubtions, Second Edition. (John Wiley and Sons, Inc., 1992). [Google Scholar]
- 74.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akalin A et al. MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gel B et al. RegioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harmanci A, Rozowsky J & Gerstein M MUSIC: identification of enriched regions in ChIP-Seq experiments using a mappability-corrected multiscale signal processing framework. Genome Biol. 15, 474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq, ChIP-seq, WGBS, and CMS-IP-seq data has been deposited in the GEO database at NCBI (#GSE156409).