

Abstract
Via conversion to Katritzky pyridinium salts, alkyl amines can now be used as alkyl radical precursors for a range of deaminative functionalization reactions. The key step of all these methods is single electron reduction of the pyridinium ring, which triggers C–N bond cleavage. However, little has been done to understand how the precise nature of the pyridinium influences these events. Using a combination of synthesis, computation, and electrochemistry, this study delineates the steric and electronic effects that substituents have on the canonical steps and the overall process. Depending on the approach taken, consideration of both the reduction and the subsequent radical dissociation may be necessary. Whereas the electronic effects on these steps work in opposition to each other, the steric effects are synergistic, with larger substituents favoring both steps. This understanding provides a framework for future design of pyridinium salts to match the mode of catalysis or activation.
Keywords: Deamination, Pyridinium, Single-Electron Transfer, Electrochemical, Radical Fragmentation
TOC Graphic
INTRODUCTION
Amines have long been recognized as a useful substrate class for synthesis. In particular, the wide diversity of commercially available alkyl amines offers opportunities for the preparation of a variety of molecules in pharmaceutical discovery,12 and the ubiquity of alkyl amines as advanced intermediates, biomolecules, and drugs provides opportunities for late-stage derivatization.3 However, the use of alkyl amines has classically been limited to the synthesis of nitrogen-containing products. Until recently, deaminative methods were largely limited to alkyl amine derivatives with highly specific structural requirements surrounding the alkyl group or C–N bond (benzylic, allylic, α-carbonyl, or strained).4 However, the recognition that Katritzky pyridinium salts, or 2,4,6-triphenylpyridinium salts,5 can be utilized as alkyl radical precursors has opened new opportunities in synthesis via deamination. Indeed, there has been a growing interest in Katritzky alkylpyridinium salts since our 2017 publication6 demonstrating that they can be used in nickel-catalyzed cross-couplings.7 Excitingly, the development of these methods has demonstrated that the utility of pyridinium salts extends beyond serving as a pseudohalide; the pyridinium group can participate in activation modes unknown for halide counterparts. Methods to harness these largely untapped pyridinium reagents now utilize a variety of methods to activate the pyridinium ring via single-electron transfer (SET), including transition metal catalysis,8 photoredox catalysis,9 and photoactivation of electron donor-acceptor (EDA) pairs (Scheme 1).10 Via these methods, amino groups can now be transformed to aryl, vinyl, alkynyl, allyl, alkyl, boryl, and carbonyl substituents.
Scheme 1. Seminal methods using alkylpyridinium salts in cross-coupling.

However, limitations in this chemistry remain. The structure of the pyridinium moiety is largely dictated by the pyrylium precursors that are readily available, with 2,4,6-triphenylpyridinium salts nearly exclusively used. Although medicinal chemists have adopted these methods, the poor atom economy of the 2,4,6-triphenylpyridinium moiety hinders adoption in process chemistry and other large-scale synthesis. Further, the synthesis of 2,4,6-triphenylpyridinium salts is only possible for constrained tertiary alkyl groups with smaller steric footprints, such as cyclopropyls.8c, 11 Finally, notable differences in reactivity between primary and secondary alkylpyridinium salts have been observed in certain activation classes. This trend is most prevalent under photoredox catalysis, where often only secondary alkylpyridinium salts undergo activation by the photocatalyst.8h, 9a, 9b This trend is also found within transition metal catalysis, where different conditions are often needed to effect reactions with primary and secondary alkylpyridinium salts.8c, 8e–g These observations have precipitated some effort toward understanding the SET and C–N bond cleavage steps. To date, these studies have largely focused on the differences due to the alkyl substituent.5, 12 For example, in studying the requirement for secondary alkyl groups in their photoredox-catalyzed alkynylation, Gryko and coworkers observed complete reversibility in the electrochemical reduction of primary alkylpyridinium salts, while secondary alkyl and benzylic pyridinium salts exhibited quasireversible or irreversible cyclic voltammetry (CV) traces.9a This result is consistent with the increased difficulty associated with forming primary alkyl radicals.
In contrast, little effort has been directed towards understanding the role of the pyridinium ring itself on the reactivity of these compounds, with the recent use of tris(p-methoxyphenyl)pyridinium salt by Martin and coworkers as the sole exception.8g In his CV studies of these “tuned” pyridinium salts, the reduction of primary alkylpyridinium salts is again reversible, and that of secondary alkylpyridiniums is quasireversible. However, at elevated temperatures, the reversibility of this reduction decreased slightly, suggesting that temperature may play a role in the improved reactivity of the tris(4-methoxyphenyl) pyridinium salt. Despite this work, it remains unknown what implications further permuations of the pyridinium salt would have.
Given the tunability of the pyridinium moiety by means of different substituents and its importance in the activation process, a greater understanding of its role in controlling reactivity would be beneficial. Herein, we describe our detailed study of the effect of substitution on the pyridinium ring of secondary alkylpyridinium salts. We show that electronic effects play a crucial role in modulation of the reduction potential, while steric bulk at the 2,6-positions facilitates cleavage of the C–N bond. This work lays the foundation for fundamental understanding of the SET and C–N bond cleavage steps to guide the development of future methods especially to enable the inclusion of problematic substrates.
RESULTS AND DISCUSSION
In consideration of the structure and reactivity of alkylpyridiniums, several canonical steps are important. Specifically, Figure 1 outlines these steps, comprised of: (1) reduction of the pyridinium 1 to give a neutral pyridyl radical 2, (2) possible oxidation of this neutral pyridyl radical (the reverse of the reduction), (3) cleavage of the C–N bond of the pyridyl radical 2 to release pyridine 4 and an alkyl radical, and (4) the potential recombination of pyridine 4 and alkyl radical to reconstitute the pyridyl radical 2. For these efforts, we deployed electrochemical measurements and DFT calculations to interrogate the factors controlling reactivity. In order to generate results that would advance the use of these reagents across multiple activation modes (metal catalysis, photoredox catalysis, and EDA photoactivation), electrochemical activation of the pyridiniums was examined. This mode eliminates variables associated with different catalytic conditions. This approach allowed us to dissect the thermodynamics of the first step (pyridinium salt to pyridyl radical; 1 → 2) as well as the thermodynamics (2 → 4) and kinetics of step two (2 → 3/3 → 2).
Figure 1.

Generic energy profile for C–N bond activation and parameters discussed in this study.
We considered systems with several different substitutions of the pyridinium fragment. One class we investigated is 2,4,6-triaryl pyridinium salts (Scheme 2B).13 These 2,4,6-triaryl pyridinium salts (1a-Cy–1e-Cy, 1g-Cy) are easily synthesized by refluxing cyclohexyl amine with pyrylium 7, which was generated from two equivalents of an acetophenone (5) and one equivalent of a benzaldehyde (6) (Scheme 2A). This protocol allows ready synthetic access such that comparison of experimental and computed reactivity data is possible. In our computational studies, we considered both cyclohexyl and isopropyl groups at R1 to confirm that cyclic and acyclic systems behaved similarly. We originally attempted to synthesize pyridinium 1f with 3,5-difluorophenyl substitution. However, insolubility of 1f prevented characterization and accurate measurement of its reduction potential. All further analysis of this compound was solely computational. Due to this difficulty, we prepared pyridinium salt 1g as an additional data point in correlating the experimental and computed reduction potentials. In addition, a complimentary set of hypothetical pyridinium salts were also subjected to computation (Scheme 2B). Substitution in these compounds is more diverse and includes groups that permit both electronic and steric differentiation that we could not easily synthesize. Because we saw good agreement between the cyclohexyl and isopropyl pyridinium salts for 1a–1g, we focused on the isopropyl pyridiniums for 1h–1m. The 4-aryl substituent was also removed from these hypothetical pyridinium salts to facilitate computation. Finally, pyridinium salt 1n-Cy used to determine the effect of the 2,6-aryl substituents on the reduction potential.
Scheme 2. Pyridinium salts used for computational and electrochemical investigations.

Pyridinium Salt Reduction.
From the pyridinium, the first part of the amine activation is reduction of the pyridinium moiety. In assessing this reduction step, a relevant numerical measure would be the thermodynamic stability of the pyridyl radical 2 relative to the corresponding pyridinium cation 1. The relative Gibbs free energies (ΔG°calc) were computed using UM06/6–311+G(d,p), SMD: DMF //UB3LYP/6–31G(d) and the values listed are relative to the most readily reduced pyridinium salt 1f (Table 1).
Table 1.
| |||||
|---|---|---|---|---|---|
| ΔG°calcb | ΔG°expc | ||||
| (kcal/mol) | |||||
| Entry | Ar | σ a | R1 = i-Pr | R1 = Cy | R1=Cy |
| 1 | 4-MeOC6H4, 1a | –0.27 | 9.2 | 9.5 | 8.4 |
| 2 | 4-MeC6H4, 1b | –0.17 | 8.3 | 8.3 | 6.3 |
| 3 | Ph, 1c | 0.00 | 5.9 | 6.1 | 5.0 |
| 4 | 4-FC6H4, 1d | 0.06 | 5.4 | 5.7 | 4.4 |
| 5 | 4-CF3C6H4, 1e | 0.54 | 0.8 | 0.4 | 0.0 |
| 6 | 3,5-F2C6H3, 1f | 0.68d | 0.0 | 0.0 | nde |
| 7 | 3,5-Me2C6H3, 1g | – | nd | 7.6 | 6.8 |
Hammett σp parameters14 for substituents.
Relative Gibbs free energies computed using UM06/6–311+G(d,p), SMD: DMF // UB3LYP/6–31G(d).
ΔG0exp calculated using ΔG0exp =-FE°exp.
For 3,5-difluoro substitution, the σ parameter was estimated additively as a sum of two fluorine σm values of 0.34.
Insolubility of this pyridinium salt prevented accurate measurement of the reduction potential.
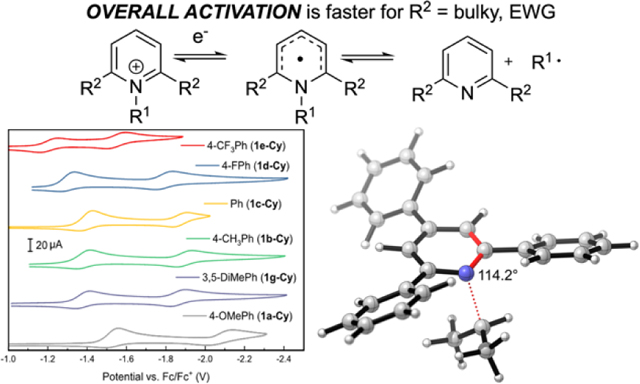
The reduction potentials of cyclohexylpyridinium salts were also measured experimentally by recording the cyclic voltammograms (CVs) shown in Figure 2A. Because the redox waves for the pyridinium derivatives surveyed in Figure 2A show varying levels of reversibility, we also employed differential pulse voltammetry (DPV) to provide an alternative measure of the potentials at which the cyclohexylpyridinium salts are reduced in anhydrous DMF containing 0.1 M TBAPF6 as supporting electrolyte. Relative redox potentials (E°rel) recorded by DPV (see Supporting Information) were calibrated against an internal ferrocene standard and were converted to the relative thermodynamics of the reduction using equation 1 (where n = 1).
Figure 2.

A) Cyclic voltammograms recorded for the set of cyclohexylpyridinium salts 1a-g listed in Table 1. CVs were recorded under a N2 atmosphere using a glassy carbon working electrode at a scan rate of 100 mV/s. Potentials are reported versus an internal Fc/Fc+ reference. B) Experimentally determined relative reduction potentials of pyridinium salts vs corresponding DFT-computed values from Table 1. C) Relative Gibbs free energy of single-electron reduction for a set of pyridinium cations vs Hammett parameters from Table 1. Black and white markers represent N–i-Pr and N-Cy respectively.
| (1) |
The computationally determined reduction potentials of the N-Cy pyridinium salts were compared to the values determined experimentally by DPV (Table 1). Strong agreement between the computational and experimental data indicates that we appropriately capture the reactivity of the salts in the reduction step, confirming the suitability of the chosen level of theory (Figure 2B). The computational and experimental data indicates that more electron-rich systems are more difficult to reduce. A strong Hammett correlation is observed for both N-Cy and N–i-Pr systems upon plotting ΔG0red vs σ (Figure 2C).14 Both N-Cy and N–i-Pr systems exhibit near identical behavior and are equally sensitive to the electronic character of the aryl groups as reflected in the similar values of the linear fitting coefficients.
The relative free energies of the reduction were computed for a series with differing 2,6-substitution (Table 2). Notably, these calculations reproduce the trend in Figure 3A with 2,6-diphenyl substitution facilitating reduction (entries 3 vs 4). Similar to our experimental results, electron-withdrawing groups show a shift to more positive reduction potentials (entries 4 vs 5 and 6). An unexpected steric effect was also observed. When groups with similar Hammett parameters were compared, a lower reduction potential was observed with larger 2,6-substituents (entries 1 vs 2). These trends suggest that steric strain facilitate SET by destabilizing the pyridinium salt. These substituent effects work synergistically; groups that possess both steric bulk and electron-withdrawing properties minimize the reduction potential (entry 6).
Table 2.
Single-electron reduction of hypothetical pyridinium cations.
| ||||
|---|---|---|---|---|
| Entry | R2 | σpa | νa | ΔG0redb (kcal/mol) |
| 1 | t-Bu, 1h | –0.20 | 1.24 | 19.2 |
| 2 | Me, 1i | –0.17 | 0.52 | 34.2 |
| 3 | Ph, 1j | –0.01 | 0.57 | 24.5 |
| 4 | H, 1k | 0.00 | 0.00 | 30.4 |
| 5 | Cl, 1l | 0.23 | 0.55 | 15.6 |
| 6 | CF3, 1m | 0.54 | 0.91 | 0.0 |
Hammett sigma para (σp) and Charton steric (ν) parameter15 for the R groups.
Relative Gibbs free energy are computed with UM06/6–311+G(d,p), SMD: 1,4-dioxane // UB3LYP/6–31G(d).
Figure 3.


(a) CVs recorded for 2,4,6-triphenylcyclohexyl pyridinium salt 1c-Cy and 4-phenyl-cyclohexyl pyridinium salt 1n-Cy under a N2 atmosphere using a glassy carbon working electrode at a scan rate of 100 mV/s. Potentials are reported versus an internal Fc/Fc+ reference. (b) Molecular diagram of 1c-Cy with ellipsoids at 50% probability. H-atoms and BF4– omitted for clarity. (c) Molecular diagram of 1c-iPr with ellipsoids at 50% probability. H-atoms, BF4–, and disorder omitted for clarity.
As we were examining the electrochemical reductions of these alkylpyridinium salts, we observed that compound 1n-Cy, lacking the 2,6-diphenyl groups, was harder to reduce than the parent 2,4,6-triphenylpyridinium 1c-Cy (Figure 3A). Because the crystal structure of 1c-Cy and i-Pr variant 1c-iPr showed little conjugation between the 2,6-aryl groups and the pyridinium ring (Figure 3B, C), we set out to interrogate whether this difference was due primarily to steric or electronic effects.
To probe the relative importance of these effects, the calculated reduction potentials were subjected to a multilinear regression analysis using Hammett (σp) and Charton (ν)15 parameters of the 2,6-substituents (R2) as independent variables. For this system, the following correlation in equation 2 was obtained. The statistical model is in excellent agreement with DFT-calculated values across the entire range of free energies (Figure 4). The negative values of the coefficients indicate that electron-withdrawing groups and large steric groups facilitate reduction. The larger absolute value of the coefficient for σp vs ν highlights that electronic effects are more pronounced in this series.16
| (2) |
Figure 4.

Relative Gibbs free energy of single-electron reduction for a set of the hypothetical N–i-Pr pyridinium cations 1h-m. DFT computed values vs those calculated via multilinear regression (eq 2).
The electronic effect is consistent with electron-withdrawing groups facilitating uptake of an electron, but the steric impact on the reduction potential was not obvious. To interrogate the origin of this steric effect, the computed structures of pyridinium cations and their corresponding radicals were analyzed (Figure 5).
Figure 5.

A) Distortion of the unsubstituted pyridinium cation 1k-iPr upon reduction to 2k-iPr. B) Distortion of the diphenyl substituted pyridinium cation 1j-iPr upon reduction to 2j-iPr. Values of the dihedral angle (degrees) for the bonds highlighted in red are shown. Optimizations were performed using UB3LYP/6–31G(d). Optimizations using explicit and implicit solvation did not produce significantly different results.
To maintain aromaticity, the starting pyridinium systems tend to be as flat as possible; however, with the 2,6-diphenyl groups, the isopropyl is forced out of the pyridinium plane by approximately 15° (180° in 1k-iPr vs. 165.5° in 1j-iPr). On the other hand, the pyridyl radical contains 7 electrons in the 6-membered ring and is no longer fully aromatic. As a consequence, the nitrogen adopts a configuration intermediate between sp2 and sp3 (dihedral angle = 162.7° in 2k-iPr vs. 180° in 1k-iPr). Notably, 2,6-disubstitution amplifies this effect (122.2° in 2j-iPr vs. 162.7° in 2k-iPr), allowing the steric clash between the isopropyl and phenyl groups to be alleviated. Pyramidalization values (see SI; sum of bond angles around nitrogen atom; planar = 360°) also indicate deviation from planarity in both systems as well as more significant distortion in 2,6-disibstituted systems (347.8° for 2j-iPr vs 356.5° for 2k-iPr). Importantly, this increased sp3 character on nitrogen weakens the C–N bond and preorganizes the pyridyl radical for C–N bond cleavage as the overlap between C-N σ* and the pyridyl π orbital gets larger.
C–N Bond Cleavage.
With the stage set by reduction to form the alkyl radical by dissociation, we turned to examination of the substituent effects on the thermodynamic and kinetic aspects of this homolytic bond cleavage process. In addition to the changes in reduction potential discussed above, we also observed that varying the substitution of the pyridinium salt correlates with variable extents of redox reversibility, as can be noted by inspection of the CV traces in Figure 3A. This variability suggested to us that slow chemical steps may be linked to the electrochemical reduction steps revealed through the voltammetry (i.e., EC reaction pathways).
To understand these effects on the thermodynamics of this dissociation step, we turned to a computational approach. With respect to electronic effects on the thermodynamics of radical dissociation, we observed that electron-donating substituents substantially favored radical dissociation (Table 3). Indeed, a plot of calculated Gibbs free energies of dissociation against the Hammett parameters of substituents indicates a moderately strong (ρ = 4.3, 4.7) positive Hammett correlation (Figure 6). For both N–i-Pr and N-Cy systems, the magnitude of the electronic effect is smaller than in the preceding SET step. As shown in Figure 5B, the nitrogen lone pair in the radical is still partially conjugated with the π-system. There is a net decrease in the π electron density to only 6 electrons upon radical dissociation. Thus, electron donors would be expected to favor dissociation due to a formal decrease in π-electron density.
Table 3.
Thermodynamics of dissociation of the pyridyl radical.
| ||||
|---|---|---|---|---|
| ΔG0 rd (kcal/mol)b | ||||
| Entry | Ar | σa | R1 = Cy | R1 = i-Pr |
| 1 | 4-MeOC6H4, 2a | –0.27 | –4.1 | –5.2 |
| 2 | 4-MeC6H4, 2b | –0.17 | –3.6 | –4.7 |
| 3 | Ph, 2c | 0.00 | –2.4 | –3.4 |
| 4 | 4-FC6H4, 2d | 0.06 | –2.8 | –3.8 |
| 5 | 4-CF3C6H4, 2e | 0.54 | 0.5 | –0.9 |
| 6 | 3,5-F2C6H3, 2f | 0.68 | –0.1 | –1.5 |
Hammett σp parameters for the aryl substituents.
Gibbs free energy for dissociation are computed using UM06/6–311+G(d,p), SMD: DMF //UB3LYP/6–31G(d).
Figure 6.

Gibbs free energies for radical dissociation of the pyridyl radical vs Hammett parameters. Data from Table 3. Black and white markers represent N–i-Pr and N-Cy, respectively.
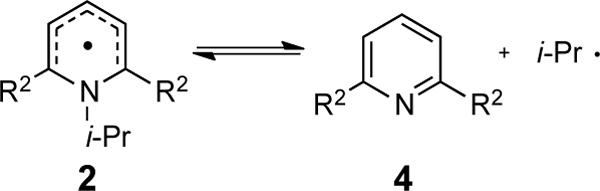
In the above series, the phenyl groups at the 2,6-positions have similar steric features. To estimate to what degree changing the steric bulk at these positions would change the overall thermodynamics, the free energy of radical dissociation was computed for a series of hypothetical 2,6-substituted pyridyl radicals (Table 4). A pronounced dependence between the thermodynamics and the size of the R2 groups is observed. Indeed, there is a good linear relationship between the radical dissociation free energies and the Charton values of the R2 groups (Figure 7B). This steric effect is readily rationalized; as the R group dissociates, unfavorable steric interactions with the R2 substituents are eliminated. On the other hand, a plot of σ vs ΔG° suggests that there is no apparent correlation in this series with the R2 electronic character (σ, Figure 7A). However, a multilinear regression reveals that both factors contribute to the variance (Figure 7C, eq 3) with normalized values showing that the electronic parameter contributes to ~25% of the variance and the steric parameter accounts for the majority of the variance at ~75% (see SI):
| (3) |
Table 4.
Free energies for the radical dissociation of hypothetical systems.
| ||||
|---|---|---|---|---|
| Entry | R2 | σa | νa | ΔG°rd (kcal/mol)b |
| 1 | t-Bu, 2h | –0.20 | 1.24 | -23.1 |
| 2 | Me, 2i | –0.17 | 0.52 | -11.4 |
| 3 | Ph, 2j | –0.01 | 0.57 | -5.1 |
| 4 | H, 2k | 0.00 | 0.00 | 1.2 |
| 5 | Cl, 2l | 0.23 | 0.55 | -8.0 |
| 6 | CF3, 2m | 0.54 | 0.91 | -9.0 |
Hammett sigma para (σp) and Charton steric (ν) parameters3 for the R groups.
Values are obtained using UM06/6–311+G(d,p),SMD: 1,4-dioxane // UB3LYP/6–31G(d).
Figure 7.

Gibbs free energies for radical dissociation of a set of the hypothetical N–i-Pr pyridyl radicals 2h-m. Data from Table 4. A) Vs Hammett parameters. B) Vs Charton parameters. C) Vs Hammett and Charton parameters.
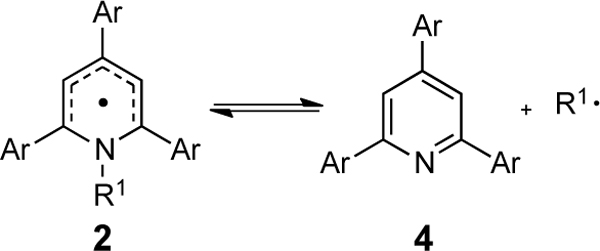
Having evaluated the thermodynamics, the kinetics of the dissociation step were next considered. The lack of full reversibility in the reduction of the pyridinium salts provides the opportunity to extract the rate of the pyridyl radical decomposition through analysis of the pyridinium CVs. To quantitate how pyridinium structure and substitution patterns influence this reversibility, CVs for each of the pyridinium salts of Table 1 were recorded at varying scan rates (5–1000 mV/s) to assess how electrode dynamics and the kinetics of possible chemical steps may influence the shape of CV traces (results obtained for 1c-Cy are shown as an example in Figure 8A). Through simulations of an EC reaction scheme involving an initial electrochemical step (i.e., pyridinium reduction) followed by a chemical step (i.e., pyridyl radical dissociation/C–N bond scission), we were able to reproduce the CVs with high fidelity (Figure 8B) and extract the amalgamated rate constants. Computation of the radical dissociation of pyridyl radicals derived from 1a–g gave rise to activation free energies that could be converted to krd values. These values correlated strongly to those determine through CV simulations (See Figure 8C, Table 5).
Figure 8.

A) Scan rate studies of the first reduction potential for pyridinium salt 1c-Cy. B) Experimental (solid) vs Simulated (dashed) of 1c-Cy first reduction at different scan rates. Simulation parameters: E0 = −1.34V vs Fc+/Fc, ks = 0.05 cm s−1, kf = 0.01 s-1. C) Experimentally determined rate constants of radical dissociation of 2 vs DFT-computed values.
Table 5.
Activation free energies for the radical dissociation of pyridyl radical 2 to pyridine 4 (Figures1 and2)
| |||||
|---|---|---|---|---|---|
| calc ΔG‡rdb | Expt ΔG‡rdc | ||||
| (kcal/mol) | |||||
| Entry | Ar | σ a | R1 = i-Pr | R1 = Cy | R1 = Cy |
| 1 | 4-MeOC6H4, 2a | –0.27 | 15.9 | 17.7 | 18.0 |
| 2 | 4-MeC6H4, 2b | –0.17 | 16.5 | 18.2 | 18.4 |
| 3 | Ph, 2c | 0.00 | 17.3 | 18.5 | 18.8 |
| 4 | 4-FC6H4, 2d | 0.06 | 16.9 | 18.3 | 18.5 |
| 5 | 4-CF3C6H4, 2e | 0.54 | 17.5 | 19.0 | 19.0 |
| 6 | 3,5-F2C6H3, 2f | 0.68d | 17.4 | 19.1 | nde |
| 7 | 3,5-Me2C6H3, 12g | – | nd | nd | 18.7 |
Hammett σp parameters for substituents.
Gibbs free energies of activation computed using UM06/6–311+G(d,p), SMD: DMF // UB3LYP/6–31G(d).
Experimental ΔG‡rd obtained from rate constants using the Eyring equation.
For 3,5-difluoro substitution, the σ parameter was estimated additively as a sum of two fluorine σm values of 0.34.
Insolubility of this pyridinium salt prevented accurate measurement.
The activation energy of the radical dissociation is much less affected by the electronics of pyridinium system (ρ = +1.3, +1.3; Figure 9) compared to the thermodynamics of radical dissociation (ρ = +4.2, +4.7; Figure 6).
Figure 9.

Activation free energy of radical dissociation for a set of pyridinium cations vs Hammett parameters. Data from Table 5. Black and white markers represent N–i-Pr and N-Cy, respectively.
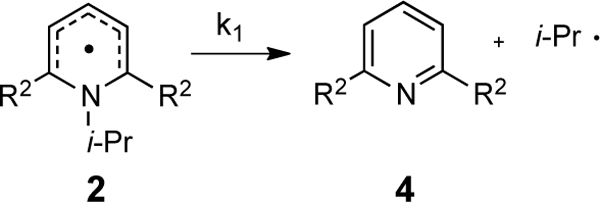
Since all of these substrates had aryl substituents of very similar steric bulk flanking the alkyl group that departs as a radical, we turned to computation to better understand steric nuances of these systems. In doing so, it appears that the weak electronic effects of substituents can be completely overridden by steric effects as shown by analysis of activation energies for the model systems (Table 6). Namely, larger R2 groups lower the dissociation barrier. For these model systems, there is no pronounced dependence of the dissociation activation energy on the electronics of the R2 groups (Figure 10). As with the thermodynamics, multilinear regression incorporating both the Hammett and Charton parameters reveals the strongest correlation (Figure 11). The majority of the variance is found in the Charton steric parameter as indicated by the larger coefficient in eq 4.17
| (4) |
Table 6.
Activation free energies for the radical dissociation of hypothetical systems.
| ||||
|---|---|---|---|---|
| Entry | R2 | σa | νa | ΔG‡ (kcal/mol)b |
| 1 | t-Bu, 2h | –0.20 | 1.24 | 11.4 |
| 2 | Me, 2i | –0.17 | 0.52 | 15.0 |
| 3 | Ph, 2j | –0.01 | 0.57 | 15.5 |
| 4 | H, 2k | 0.00 | 0.00 | 22.3 |
| 5 | Cl, 2l | 0.23 | 0.55 | 12.7 |
| 6 | CF3, 2m | 0.54 | 0.91 | 13.6 |
Hammett sigma para (σp) and Charton steric (ν) parameters3 for the R groups.
Values are obtained using UM06/6–311+G(d,p),4SMD: 1,4-dioxane // UB3LYP/6–31G(d).
Figure 10.

Activation free energy of radical dissociation for a set of model pyridyl radicals 2h-m vs Hammett parameters. Data from Table 6.
Figure 11.

Activation free energy of radical dissociation for a set of the hypothetical pyridyl radicals 2h-m vs those calculated via multilinear regression (eq 4. Data from Table 6.
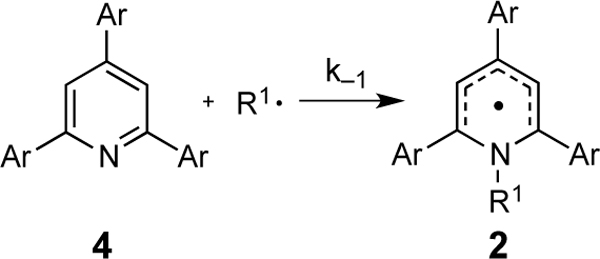
To gain insight into the orbital basis for these substituent effects, it is instructive to examine the kinetics of the reverse reaction, specifically the radical association with the neutral pyridine (Table 7). As would be expected from the relationship:
| (5) |
the electronic effects in radical association can be approximated by those from radical dissociation barriers (ρ = +1.3, +1.3; Figure 9) subtracted by those from the thermodynamics (ρ = +4.2, +4.7; Figure 6). Indeed, this relationship largely holds as shown in Figure 12 (ρ = –2.9, –3.4). Specifically, electron-withdrawing groups accelerate radical association while electron-donating groups strongly favor the dissociation thermodynamically and weakly accelerate dissociation kinetically.
Table 7.
Activation free energies for the attack of alkyl radicals on neutral pyridines.
| ||||
|---|---|---|---|---|
| ΔG≠ ra (kcal/mol)b | ||||
| Entry | Ar | σa | R1 = Cy | R1 = i-Pr |
| 1 | 4-MeOC6H4, 4a | –0.27 | 21.8 | 21.1 |
| 2 | 4-MeC6H4, 4b | –0.17 | 21.8 | 21.3 |
| 3 | Ph, 4c | 0.00 | 20.9 | 20.6 |
| 4 | 4-FC6H4, 4d | 0.06 | 21.1 | 20.7 |
| 5 | 4-CF3C6H4, 4e | 0.54 | 18.6 | 18.5 |
| 6 | 3,5-F2C6H3, 4f | 0.68 | 19.2 | 18.9 |
Hammett σp parameters for substituents.
Values are obtained using UM06/6–311+G(d,p), SMD: DMF//UB3LYP/6–31G(d).
Figure 12.

Activation free energy of a radical attack for a set of ‘pyridinium cations vs Hammett parameters. Data from Table 7. Black and white markers represent N-Cy and N-i-Pr, respectively.
Examination of the frontier molecular orbitals interacting during the process provide insight into these electronic effects. Positioning of the alkyl group almost perpendicular to the plane of the pyridine ring in the transition state suggests that a key interaction is between the SOMO of a radical and the LUMO of a pyridine (Figure 13). The energetic effect of the overlap is inversely proportional to the initial energy separation d.18 Electron-withdrawing groups in a pyridine system lower the LUMO energy, decreasing the LUMO-SOMO gap (d on the diagram), thus enhancing the interaction and stabilizing the corresponding association transition state. This relationship also suggests that electron-rich radicals should be more reactive in the association process.
Figure 13.

Example of the radical dissociation transition state and corresponding MO interaction diagram. Energetic effect of the overlap is ε, initial energy SOMO-LUMO gap is d.
Overall Effects on Reduction and C–N Bond Cleavage.
A specific approach to the design of the pyridinium moiety for the C–N bond activation depends on the type of reduction of pyridinium salts. If reduction of the salt is performed in a fashion orthogonal to the further thermal chemical reaction (i.e., electrochemical or photochemical generation of pyridyl radicals), then the reactivity of the compound in C–N homolytic bond cleavage is determined only by the corresponding radical dissociation activation energy (on Figure 1). In this case, bulky and electron-rich pyridine groups will facilitate the activation. Based on our study, it appears that the electronic character of the pyridine moiety has only a minor effect in this case.
However, if reduction contributes to the overall thermal chemical transformation, then the rate of activation is controlled by the dissociation transition state energy relative to the pyridinium (ΔG‡RD rel in Figure 1). Thus, one should consider electronic and steric effects over the sequence of reduction and dissociation steps. Because the activation energy of the dissociation step is weakly favored by electron-donating substituents and reduction is heavily favored by electron-withdrawing substituents, it should be possible to accelerate the overall process using electron-withdrawing substituents. Indeed, a Hammett correlation using ΔG‡RD rel reveals a large negative ρ value (Figure 14). The electronic effects are very pronounced and must be accounted for accordingly in design of any process. Notably, this correlation is the strongest we have observed, likely because the relative energy of the pyridyl radical 2 can either be higher or lower than that of the pyridinium cation, obfuscating the dependence of individual steps on the electronics and sterics (Figure 1). It is the overall transition state barrier from the pyridinium cation (1) to the radical dissociation transition state (3) that is key to understanding these systems.
Figure 14.

Relative activation free energy of radical dissociation for pyridinium systems vs Hammett parameters. Note that the values of relative activation free energies obtained using relative Δ𝐺0RED (see Table 1) via Δ𝐺≠𝑅𝐷 𝑟𝑒𝑙=Δ𝐺0RED+Δ𝐺≠𝑅𝐷. Absolute values of Δ𝐺≠𝑅𝐷 𝑟𝑒𝑙 will depend on the exact nature of the reductant used. Black and white markers represent N–i-Pr and N-Cy respectively.
CONCLUSIONS
In summary, this analysis reveals the fundamental trends affecting the individual steps as well as the overall process (Figure 15). The predominant control factor in the reduction of the pyridinium cation is electronic in nature, which could arise from substituents at any position of the pyridinium ring. A somewhat lesser dependence on steric factors is seen with larger groups at the 2,6-positions favoring reduction. On the other hand, the dissociation step exhibits a weak electronic dependence with electron-donating groups at any position of the pyridinium ring favoring dissociation. The influence of sterics on this step is very strong, and easily outweighs the electronic influence, with large groups at the 2,6-positions favoring and accelerating dissociation. With respect to the overall process from the pyridinium cation to the alkyl radical, the electronic effects cancel, leading to more facile overall activation with electron-withdrawing groups. On the other hand, the steric effects are synergistic with larger groups promoting both steps and the overall process. Because the reduction is fast, emphasis should be placed on derivatives that promote dissociation to generate the alkyl radical. This knowledge creates a framework to enable the design of new pyridinium salts to meet the limitations that each mode of activation – transition metal catalysis, photoredox catalysis, or photoactivation via EDA complexes – currently encompasses.
Figure 15.

Computationally identified reactivity trends for substituted N-alkylpyridinium salts.
Supplementary Material
ACKNOWLEDGMENT
M.C.K. thanks the NIH (R35 GM131902) for financial support and XSEDE (TG-CHE120052) for computational support. M.P.W. thanks the NIH (R35 GM131816) for financial support. J.C.T. thanks the Chemistry-Biology Interface program (NIH T32-GM133395). J.R. thanks the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences EPSCoR and Catalysis programs under Award Number DESC-0001234. Data were acquired at UD on instruments obtained with assistance of NSF and NIH funding (NSF CHE0421224, CHE1229234, CHE0840401, and CHE1048367; NIH P20 GM104316, P20 GM103541, and S10 OD016267).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. FAIR Data is available as Supporting Information for Publication and includes the primary NMR FID files for compounds: 1a-Cy–1e-Cy and 1g-Cy. See FID for Publication for additional information.
Experimental procedures and spectral data (PDF).
Accession Codes
CCDC 2044313 and 2044314 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing da-ta_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: +44 1223 336033.
REFERENCES
- 1.A search of Pfizer’s internal chemical store revealed over 47,000 alkyl primary amines vs. about 28,000 primary and secondary alkyl halides. ·Garnsey MR, Pfizer Worldwide Research and Development, 2018. [Google Scholar]
- 2.For alkyl-NH2 (1° and 2°), 1237920; Data from eMolecules Database accessed via REAXYS (April 8, 2019).
- 3. (a).Lawrence SA, Amines: Synthesis, Properties and Applications. Cambridge University Press: New York, NY, 2004; [Google Scholar]; (b) Nugent TC, Chiral Amine Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2010; [Google Scholar]; (c) Ruiz-Castillo P; Buchwald SL, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116 (19), 12564–12649. 10.1021/acs.chemrev.6b00512; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McGrath NA; Brichacek M; Njardarson JT, A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87 (12), 1348–1349. 10.1021/ed1003806; [DOI] [Google Scholar]; (e) Liu Y; Ge H, Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 2017, 9 (1), 26–32. 10.1038/nchem.2606. [DOI] [Google Scholar]
- 4. (a).Ouyang K; Hao W; Zhang WX; Xi Z, Transition-Metal-Catalyzed Cleavage of C-N Single Bonds. Chem. Rev. 2015, 115 (21), 12045–12090. 10.1021/acs.chemrev.5b00386; [DOI] [PubMed] [Google Scholar]; (b) Pound SM; Watson MP, Asymmetric Synthesis via Stereospecific C–N and C–O Bond Activation of Alkyl Amine and Alcohol Derivatives. Chem. Commun. 2018, 54 (87), 12286–12301. 10.1039/C8CC07093H; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu J; Bercher OP; Talley MR; Watson MP, Nickel-Catalyzed, Stereospecific C–C and C–B Cross-Couplings via C–N and C–O Bond Activation. ACS Catal. 2021, 11 (3), 1604–1612. 10.1021/acscatal.0c05484; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li M-B; Wang Y; Tian S-K, Regioselective and Stereospecific Cross-Coupling of Primary Allylic Amines with Boronic Acids and Boronates through Palladium-Catalyzed C–N Bond Cleavage. Angew. Chem., Int. Ed. 2012, 51, 2968–2971. 10.1002/anie.201109171; [DOI] [PubMed] [Google Scholar]; (e) Moragas T; Gaydou M; Martin R, Nickel-Catalyzed Carboxylation of Benzylic C−N Bonds with CO2. Angew. Chem., Int. Ed. 2016, 55 (16), 5053–5057. 10.1002/anie.201600697; [DOI] [PubMed] [Google Scholar]; (f) Liao LL; Cao GM; Ye JH; Sun GQ; Zhou WJ; Gui YY; Yan SS; Shen G; Yu DG, Visible-Light-Driven External-Reductant-Free Cross-Electrophile Couplings of Tetraalkyl Ammonium Salts. J. Am. Chem. Soc. 2018, 140 (50), 17338–17342. 10.1021/jacs.8b08792; [DOI] [PubMed] [Google Scholar]; (g) Jiang W; Li N; Zhou L; Zeng Q, Copper-Catalyzed Stereospecific C–S Coupling Reaction of Enantioenriched Tertiary Benzylic Amines via in Situ Activation with Methyl Triflate. ACS Catal. 2018, 8 (11), 9899–9906. 10.1021/acscatal.8b03032; [DOI] [Google Scholar]; (h) Scharfbier J; Gross BM; Oestreich M, Stereospecific and Chemoselective Copper-Catalyzed Deaminative Silylation of Benzylic Ammonium Triflates. Angew. Chem., Int. Ed. 2020, 59 (4), 1577–1580. 10.1002/anie.201912490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katrizky AR; Marson CM, Pyrylium Mediated Transformations of Primary Amino Groups into Other Functional Groups. Angew. Chem., Int. Ed. 1984, 23, 420–429. [Google Scholar]
- 6.Basch CH; Liao J; Xu J; Piane JJ; Watson MP, Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C–N Bond Activation. J. Am. Chem. Soc. 2017, 139 (15), 5313–5316. 10.1021/jacs.7b02389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. (a).Li Y-N; Xiao F; Guo Y; Zeng Y-F, Recent Developments in Deaminative Functionalization of Alkyl Amines. Eur. J. Org. Chem. 2021, 2021, 1215–1228. 10.1002/ejoc.202001193; [DOI] [Google Scholar]; (b) Kong D; Moon PJ; Lundgren RJ, Radical Coupling from Alkyl Amines. Nat. Catal. 2019, 2, 473–476. [Google Scholar]
- 8. (a).Guan W; Liao J; Watson MP, Vinylation of Benzylic Amines via C–N Bond Functionalization of Benzylic Pyridinium Salts. Synthesis 2018, 50 (16), 3231–3237. 10.1055/s-0037-1610084; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoerrner ME; Baker KM; Basch CH; Bampo EM; Watson MP, Deaminative Arylation of Amino Acid-derived Pyridinium Salts. Org. Lett. 2019, 21 (18), 7356–7360. 10.1021/acs.orglett.9b02643; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liao J; Basch CH; Hoerrner ME; Talley MR; Boscoe BP; Tucker JW; Garnsey MR; Watson MP, Deaminative Reductive Cross-Electrophile Couplings of Alkylpyridinium Salts and Aryl Bromides. Org. Lett. 2019, 21 (8), 2941–2946. 10.1021/acs.orglett.9b01014; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liao J; Guan W; Boscoe BP; Tucker JW; Tomlin JW; Garnsey MR; Watson MP, Transforming Benzylic Amines into Diarylmethanes: Cross-Couplings of Benzylic Pyridinium Salts via C–N Bond Activation. Org. Lett. 2018, 20 (10), 3030–3033. 10.1021/acs.orglett.8b01062; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Plunkett S; Basch CH; Santana SO; Watson MP, Harnessing Alkyl Pyridinium Salts as Electrophiles in De-aminative Alkyl-Alkyl Cross-Couplings. J. Am. Chem. Soc. 2019, 141 (6), 2257–2262.; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang J; Hoerrner ME; Watson MP; Weix DJ, Nickel-Catalyzed Synthesis of Dialkyl Ketones from the Coupling of N-Alkyl Pyridinium Salts with Activated Carboxylic Acids. Angew. Chem., Int. Ed. 2020, 59 (32), 13484–13489. 10.1002/anie.202002271; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Martin-Montero R; Yatham VR; Yin H; Davies J; Martin R, Ni-catalyzed Reductive Deaminative Arylation at sp(3) Carbon Centers. Org. Lett. 2019, 21 (8), 2947–2951. 10.1021/acs.orglett.9b01016; [DOI] [PubMed] [Google Scholar]; (h) Yi J; Badir SO; Kammer LM; Ribagorda M; Molander GA, Deaminative Reductive Arylation Enabled by Nickel/Photoredox Dual Catalysis. Org. Lett. 2019, 21 (9), 3346–3351. 10.1021/acs.orglett.9b01097; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yue H; Zhu C; Shen L; Geng Q; Hock KJ; Yuan T; Cavallo L; Rueping M, Nickel-catalyzed C–N bond activation: activated primary amines as alkylating reagents in reductive cross-coupling. Chem. Sci. 2019, 10, 4430–4435. 10.1039/c9sc00783k; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ni S; Li C-X; Mao Y; Han J; Wang Y; Yan H; Pan Y, Ni-catalyzed Deaminative Cross-electrophile Coupling of Katritzky Salts with Halides via C–N Bond Activation. Sci. Adv. 2019, 5, eaaw9516.; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Sun SZ; Romano C; Martin R, Site-Selective Catalytic Deaminative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2019, 141 (41), 16197–16201. 10.1021/jacs.9b07489. [DOI] [PubMed] [Google Scholar]
- 9. (a).Ociepa M; Turkowska J; Gryko D, Redox-Activated Amines in C(sp3)–C(sp) and C(sp3)–C(sp2) Bond Formation Enabled by Metal-Free Photoredox Catalysis. ACS Catal. 2018, 8 (12), 11362–11367. 10.1021/acscatal.8b03437; [DOI] [Google Scholar]; (b) Klauck FJR; James MJ; Glorius F, Deaminative Strategy for the Visible‐Light‐Mediated Generation of Alkyl Radicals. Angew. Chem., Int. Ed. 2017, 56 (40), 12336–12339. 10.1002/anie.201706896; [DOI] [PubMed] [Google Scholar]; (c) Klauck FJR; Yoon H; James MJ; Lautens M; Glorius F, Visible-Light-Mediated Deaminative Three-Component Dicarbofunctionalization of Styrenes with Benzylic Radicals. ACS Catal. 2019, 9 (1), 236–241. 10.1021/acscatal.8b04191; [DOI] [Google Scholar]; (d) Jiang X; Zhang MM; Xiong W; Lu LQ; Xiao WJ, Deaminative (Carbonylative) Alkyl-Heck-type Reactions Enabled by Photocatalytic C-N Bond Activation. Angew. Chem., Int. Ed. 2019, 58 (8), 2402–2406. 10.1002/anie.201813689; [DOI] [PubMed] [Google Scholar]; (e) Zhang M-M; Liu F, Visible-light-mediated allylation of alkyl radicals with allylic sulfones via a deaminative strategy. Org. Chem. Front. 2018, 5 (23), 3443–3446. 10.1039/C8QO01046C; [DOI] [Google Scholar]; (f) Zhu Z-F; Zhang M-M; Liu F, Radical alkylation of isocyanides with amino acid-/peptide-derived Katritzky salts via photoredox catalysis. Org. Biomol. Chem. 2019, 17 (6), 1531–1534. 10.1039/C8OB02786B. [DOI] [PubMed] [Google Scholar]
- 10. (a).Wu J; He L; Noble A; Aggarwal VK, Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc. 2018, 140 (34), 10700–10704. 10.1021/jacs.8b07103; [DOI] [PubMed] [Google Scholar]; (b) Hu J; Wang G; Li S; Shi Z, Selective C−N Borylation of Alkyl Amines Promoted by Lewis Base. Angew. Chem., Int. Ed. 2018, 57 (46), 15227–15231. 10.1002/anie.201809608; [DOI] [PubMed] [Google Scholar]; (c) Wu J; Grant PS; Li X; Noble A; Aggarwal VK, Catalyst-Free Deaminative Functionalizations of Primary Amines by Photoinduced Single-Electron Transfer. Angew. Chem., Int. Ed. 2019, 58 (17), 5697–5701. 10.1002/anie.201814452; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) James MJ; Strieth-Kalthoff F; Sandfort F; Klauck FJR; Wagener F; Glorius F, Visible-Light-Mediated Charge Transfer Enables C−C Bond Formation with Traceless Acceptor Groups. Chem. Eur. J. 2019, 25 (35), 8240–8244. 10.1002/chem.201901397. [DOI] [PubMed] [Google Scholar]
- 11.Hu J; Cheng B; Yang X; Loh TP, Transition‐Metal‐Free Deaminative Vinylation of Alkylamines. Adv. Synth. Catal. 2019, 361, 4902–4908. 10.1002/adsc.201900576. [DOI] [Google Scholar]
- 12. (a).Katritzky AR; De Ville G; Patel RC, Carbon-alkylation of simple nitronate anions by N-substituted pyridiniums. Tetrahedron 1981, 37, 25–30. 10.1016/0040-4020(81)85037-5; [DOI] [Google Scholar]; (b) Marquet J; Moreno-Mañas M; Pacheco P; Prat M; Katritzky AR; Brycki B, C-alkylation of β diketones with benzylpyridinium salts. Evidence for chain radical mechanisms. Tetrahedron 1990, 46 (15), 5333–5346. 10.1016/S0040-4020(01)87840-6; [DOI] [Google Scholar]; (c) Grimshaw J; Moore S; Trocha-Grimshaw J, Electrochemical Reactions. Part 26.* Radicals Derived by Reduction of N-Alkylpyridinium Salts and Homologous N,N’-Polymethylenebispyridinium Salts. Cleavage of the Carbon–Nitrogen Bond. Acta Chemica Scandinavica B 1983, 37, 485–489. ; [Google Scholar]; (d) Itoh M; Nagakura S, Preparation ESR spectra and electronic absorption spectra of substituted pyridinyl radicals. Tetrahedron Lett. 1965, 6 (8), 417–422. 10.1016/S0040-4039(00)89970-0. [DOI] [Google Scholar]
- 13.Katritzky AR; Thind SS, The synthesis and reactions of sterically constrained pyrylium and pyridinium salts. J. Chem. Soc., Perkin Trans 1 1980, (0), 1895–1900. 10.1039/P19800001895. [DOI] [Google Scholar]
- 14.Hansch C; Leo A; Taft RW, A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- 15.Charton M, Steric effects. III. Bimolecular nucleophilic substitution. J. Am. Chem. Soc. 1975, 97 (13), 3694–3697. 10.1021/ja00846a023. [DOI] [Google Scholar]
- 16.When σ and ν are each normalized to 1–2.3 ranges, the coefficient is still 40% larger for σ than ν: ΔG0red = –21.52σp – 15.03ν + 76.20.
- 17.When σ and ν are each normalized to 1–2.3 ranges, the coefficient is fourteen times larger for ν than σ: ΔG‡RD = –0.56σp – 8.05ν + 29.05.
- 18.Albright TA; Burdett JK; Whangbo M-H, Concepts of Bonding and Orbital Interaction. In Orbital Interactions in Chemistry, Albright TA; Burdett JK; Whangbo M, Eds. John Wiley & Sons, Inc.: Wiley Online Books, 2013; pp 15–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.