

Abstract
Senescent cells (SNCs) degenerate the fibrous cap that normally prevents atherogenic plaque rupture, a leading cause of myocardial infarction and stroke. Here we explored the underlying mechanism using pharmacological or transgenic approaches to clear SNCs in the Ldlr−/− mouse model of atherosclerosis. SNC clearance reinforced fully deteriorated fibrous caps in highly advanced lesions, as evidenced by restored vascular smooth muscle cell (VSMC) numbers, elastin content, and overall cap thickness. We found that SNCs inhibit VSMC promigratory phenotype switching in the first interfiber space of the arterial wall directly beneath atherosclerotic plaque, thereby limiting lesion entry of medial VSMCs for fibrous cap assembly or reinforcement. SNCs do so by antagonizing IGF-1 through the secretion of insulin-like growth factor-binding protein 3 (Igfbp3). These data indicate that the intermittent use of senolytic agents or IGFBP-3 inhibition in combination with lipid lowering drugs may provide therapeutic benefit in atherosclerosis.
Cellular senescence protects against cancer and promotes tissue repair1 and regeneration2,3, two properties that contribute to reproductive fitness and may explain why the senescence program was selected for during evolution. However, senescence is not strictly beneficial, as SNCs accumulating in tissues and organs with aging and at sites of age-related pathologies have been linked to tissue degeneration and dysfunction4–6. Growing evidence indicates that SNCs exert their diverse beneficial and detrimental biological effects on tissue architecture and function through their secretome, commonly referred as the senescence associated secretory phenotype (SASP). However, the SASP is highly complex, dynamic, and senescence inducer- and cell type-dependent, and the SASP components through which SNCs establish specific biological effects in tissues remain largely unknown.
To explore such mechanisms, we focused on atherogenic lesions, which have been shown to contain multiple types of SNCs, including endothelial-like, foamy VSMC-like, and foamy macrophage-like SNCs7. SNCs accumulate throughout atherogenesis and are found in initiation-, growth- and destabilization-stage atheromas of hypercholesteremic Ldlr−/− mice. SNCs in atherosclerotic plaques can be eliminated pharmacologically with the senolytic drug ABT2635,8,9, which targets the B cell lymphoma 2 (BCL-2) protein family members BCL-2, BCL-xL, and BCL-w, as well as with various transgenic senolysis approaches, including INK-ATTAC- and 3MR-based systems7. By eliminating SNCs as they accumulate during atherogenesis, hypercholesteremic Ldlr−/− mice develop fewer lesions and lesions that do form are smaller and more stable.
Key to plaque stability is the fibrous cap, a specialized plaque-covering structure established as part of the vessel injury response to atherogenesis. SNCs in atherogenic lesions have been associated with fibrous cap thinning, a degenerative process that can rupture plaques and cause myocardial infarction or stroke7. Reparative VSMCs are a major cell type within the fibrous cap, raising the possibility that SNCs prompt cap thinning by targeting this population of cells. We sought to investigate this hypothesis not only because it might reveal that in addition to stimulating tissue repair, such as in wound healing, SNCs may also inhibit innate repair mechanisms, but also because it may identify new, therapeutically-targetable biology in atherosclerosis, a disease with considerable residual mortality despite medical management of proatherogenic dyslipidemia10,11. In these studies, we used the Ldlr−/− preclinical mouse model, which develops numerous atherogenic lesions, including highly advanced lesions in the aortic arch. Since this model requires continuous high-fat-diet (HFD) feeding for plaque development, switching to an LFD after the establishment of atherosclerotic lesions can be conveniently used to mimic the clinically-relevant context of atheromas subject to medical management of circulating pro-atherogenic lipids.
Results
Senolysis reverses fibrous cap deterioration
First, we sought to determine how a combination of dyslipidemia management and SNC clearance impacts the fibrous cap structure in established, but not yet end-stage, atherosclerotic plaques. To this end, we placed Ldlr−/− mice on HFD for 12 weeks, followed by a switch to LFD for 9 weeks with concurrent ABT263 or vehicle (Veh) administration daily during the first, fourth and seventh week of LFD feeding (Fig. 1a). This ABT263 treatment protocol did not impact circulating immune cell profiles (Supplementary Fig. 1a–g). Aortic plaques of 12-week HFD fed Ldlr−/− mice exhibited senescence-associated β-galactosidase activity (SA β-Gal), an established marker for SNCs in atherosclerotic lesions7, particularly at plaque boundaries (Fig. 1b and Extended Data Fig. 1a,b), which was retained during 9 weeks of LFD feeding and Veh treatment. Sectioning and H-E staining of aortic plaques showed that fibrous cap thickness decreased by ~25% over this period, indicative of disease progression despite dyslipidemia management (Fig. 1c,d). Plaque size, as measured by en face measurement, did not increase with LFD feeding (Fig. 1e). Aortic arch plaques from corresponding ABT263-treated mice had markedly reduced SA β-Gal staining, fewer foam cells, and did not undergo cap thinning (Extended Data Fig. 1a–d). Importantly, prevention of cap thinning only required a single 7-day ABT263 treatment cycle (Fig. 1a,c,d) and was sustained for at least 26 weeks with senolysis in LFD weeks 1 and 4 (Fig.1f–j). Furthermore, the ability of ABT263 to preserve cap thickness upon LFD switching was confirmed in brachiocephalic artery lesions (Extended Data Fig. 2). Fibrous caps of aortic arch lesions contained numerous smooth muscle actin+ (Sma+) cells after 12 weeks of HFD, which were substantially reduced after the 9 weeks of LFD feeding with Veh treatment, but preserved with ABT263 treatment (Extended Data Fig. 3a,b). Moreover, fibrous cap elastin content was preserved with ABT263 but not with Veh, as assessed by Verhoeff-Van Gieson staining (Extended Data Fig. 3c,d). Consistent with this, caps of ABT263 treated lesions contained a larger proportion of vimentin (Vim)-positive Sma+ cells of a matrix-producing phenotype (Extended Data Fig. 3e,f). Thus, fibrous cap thinning following lipid normalization is driven by SNCs and characterized by a loss of VSMCs and their extracellular matrix production.
Figure 1: ABT263 blocks fibrous cap thinning through depletion of SNCs.
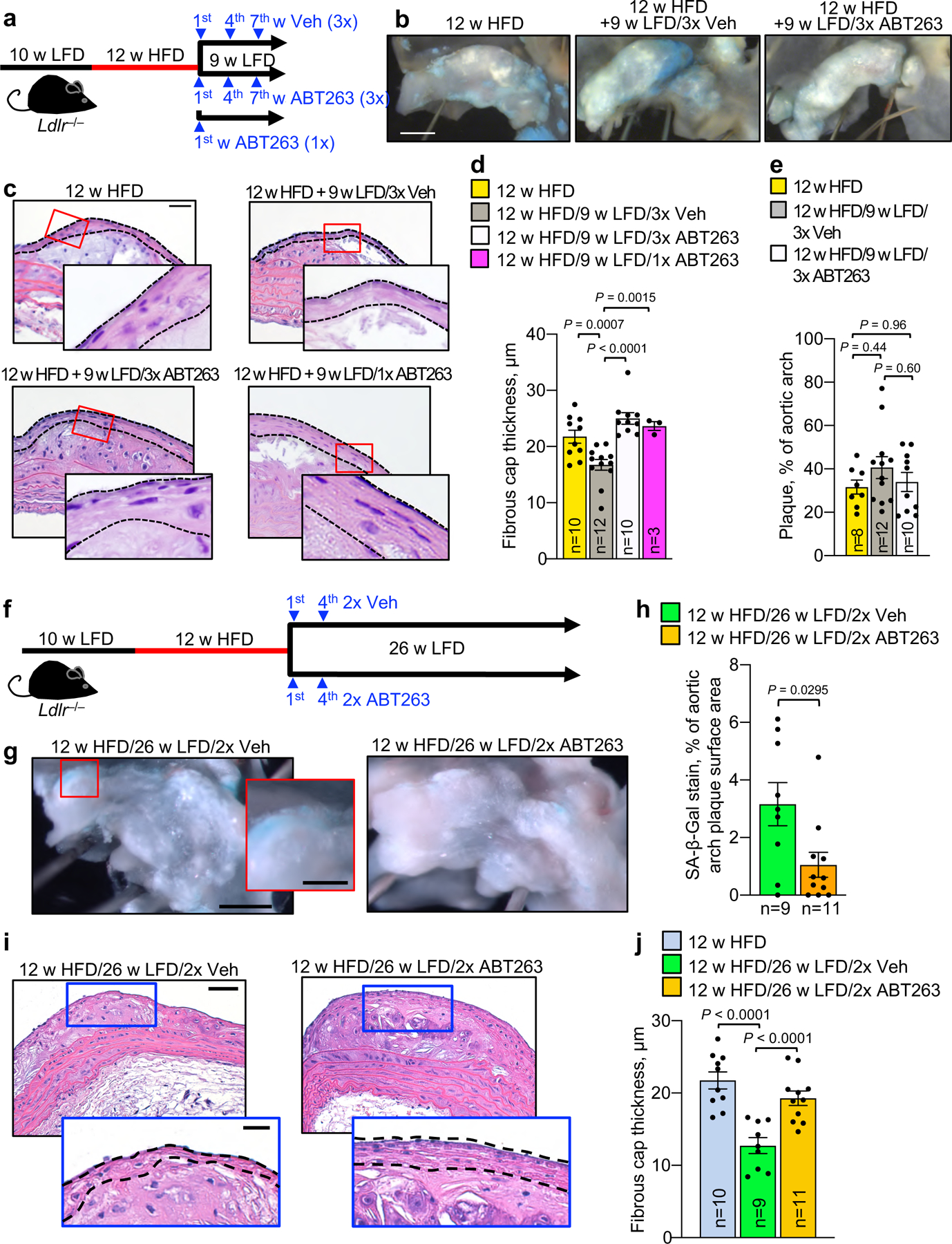
a, Schematic of the experimental design for data in b-e. b, Representative SA β-Gal staining of the indicated aortic arch lesions. SA β-Gal staining is quantified in Extended Data Fig. 1b. c, Representative H-E staining of aortic arch plaque from the indicated mice. Dashed black lines indicate the fibrous cap. d, Quantification of the average fibrous cap thickness in mice of indicated groups. e, Quantification of plaque burden in the aortic arch by en face area measurement in mice of indicated groups. “n” refers in all panels to number of mice. f, Schematic of the experimental design for data in g-j. g, Representative SA β-Gal staining of 12-week HFD aortic arch plaques remodeling for 26 weeks on LFD with either ABT263 or Veh administration during LFD weeks 1 and 4. h, Quantification of the SA β-gal-positive surface area of aortic arch plaques in mice of indicated groups. i, Representative H-E staining of the aortic arch plaque (top) and fibrous cap (bottom inset, dashed lines indicate fibrous cap) in ABT263- or Veh-treated 26-week LFD remodeling mice. j, Quantification of fibrous cap thickness in mice of indicated groups, of which panel i is an example. 12-week HFD group data is also shown in Fig. 1d and reproduced for context. “n” refers in all panels to number of mice. Statistics in panel h were performed by unpaired, two-tailed Student’s t-test with Welch’s correction. Statistics in panel d, e, and j were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for all indicated comparisons, including use of 12 w HFD baseline data for fibrous cap thickness in Fig. 1d, Fig. 1j, and Fig. 2c; and, 12 w HFD baseline en face plaque burden measurements in Fig. 1e and Fig. 2b. Error bars represent s.e.m. Scale bars are 1 mm (b and g, main), 50 μm (c), 0.33 mm (g, inset), 50 μm (i, top), and 20 μm (i, bottom).
Next, we asked whether SNC clearance might restore fibrous cap thickness in highly advanced lesions with terminally-thinned caps. To this end, we focused on inner aortic arch lesions, as these are among the first to form in hypercholesteremic Ldlr−/− mice and the largest in size. Upon prolongation of HFD feeding from 12 to 24 weeks, these lesions markedly increased in size and showed profound cap thinning (Fig. 2a–c). No further thinning occurred during the 9-week LFD interval, suggesting that fibrous caps were effectively maximally-thinned. ABT263 administration during the 9 weeks of LFD feeding significantly increased fibrous cap thickness in mature plaques, a result that was reproducible with INK-ATTAC or p16-3MR transgene-mediated elimination of p16+ SNCs (Fig. 2d–e). Both ABT263 and INK-ATTAC reduced SA β-gal content in plaques (Fig. 2b and Supplementary Fig. 5a–c). Caps thickened by either ABT263 or AP20187 (AP) contained more VSMCs and elastic fibers, as evidenced by Sma immunolabeling and Verhoeff-Van Gieson staining, respectively (Fig. 2f–i). ABT263 treatment did not impact immune-cell or atherogenic lipid profiles in the circulation (Supplementary Fig. 1a–h). Collectively, these results indicate that senolysis is not only capable of blocking fibrous cap thinning, but can, in extremely thin-capped fibroatheromas, thicken a thin fibrous cap by enhancing VSMC content and ECM deposition.
Figure 2: Fibrous caps of advanced atheromas increase VSMC content and thicken with senolysis.
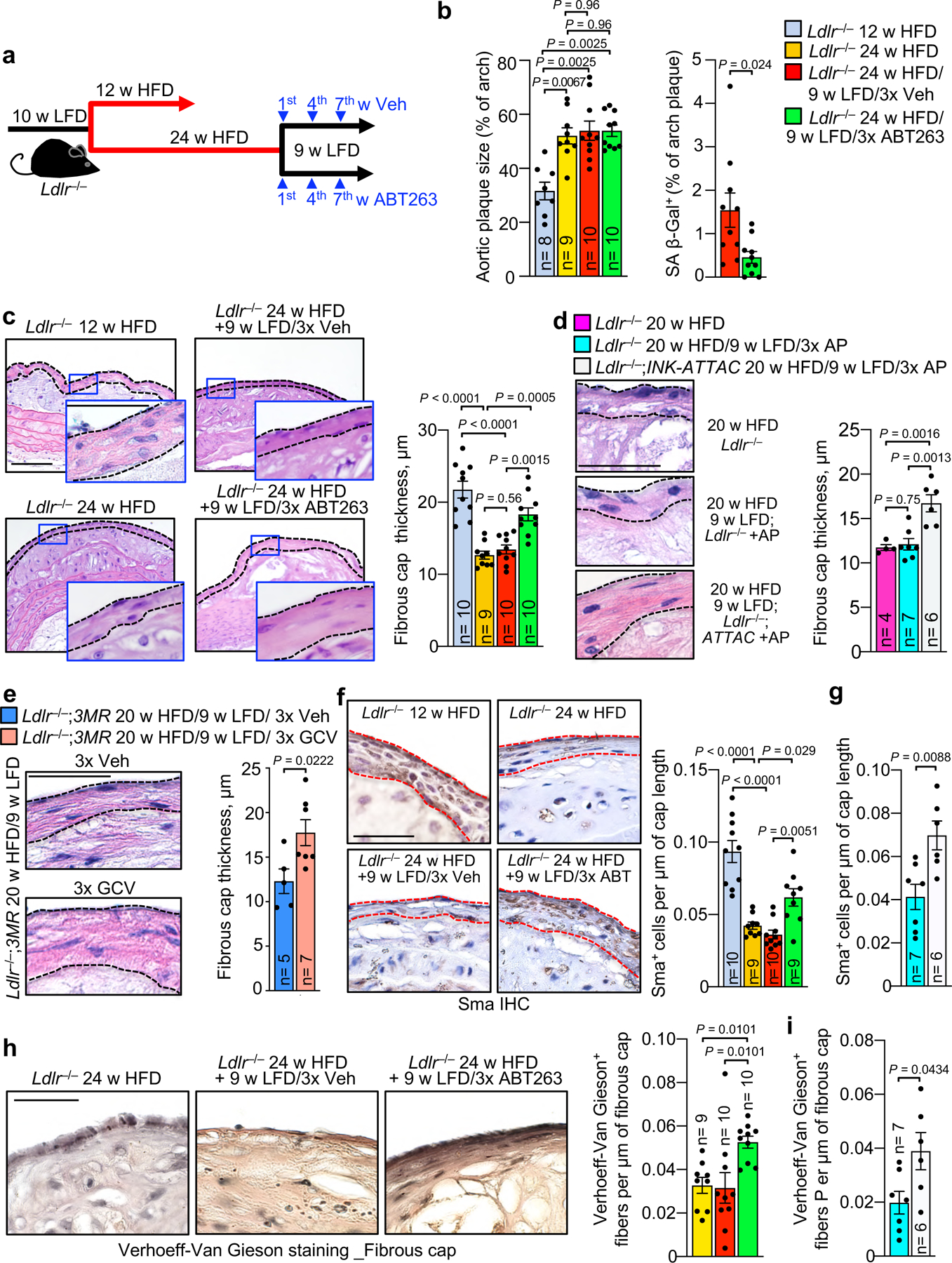
a, Schematic of the experimental design for experiments presented in b and c. b, Total lesion area and % lesional SA β-Gal+ area in aortic arch for the indicated mouse cohorts. c, (Left) Images of representative H-E stained inner aortic arch lesions of the indicated mice. (Right) Quantification of cap thickness in inner aortic arch lesions of the indicated mice (legend as in b). d and e, (Left) Representative H-E stained images of fibrous caps from indicated mice. (Right) Quantification of cap thickness in inner aortic arch lesions of the indicated mice. f, Immune staining (left) and quantification (right) for Sma+ cell number per length of fibrous cap in fibrous caps from groups indicated in c. g, Quantification of Sma+ cells in fibrous caps of mice in indicated groups (legend as in d). h, Verhoeff-Van Gieson staining (left) and elastin fine-fiber quantification for mice of indicated groups (legend as in b). i, Verhoeff-Van Gieson staining quantification for mice of groups as indicated (legend as in d). “n” refers in all panels to number of mice. Panels b (left), c, d, f and h were analyzed by one-way ANOVA with Holm-Sidak multiple comparison correction. Panel b includes data presented in Fig. 1e for context (12 w HFD baseline). Panel c includes data presented in Fig. 1d and 1j for context (12 w HFD baseline). Panel f includes data presented in Extended Data Fig. 3b for context (12 w HFD baseline). For both panels b and c, ANOVA analysis includes all indicated comparisons across Fig.1 and 2. For panel f, ANOVA analysis includes all indicated comparisons across Fig. 2f and Extended Data Fig. 3b. Panels b (right), e, g, and i were analyzed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. Scale bars: for c are 100 μm (main) and 50 μm (inset); for d, 50 μm; for e, 50 μm; for f, 50 μm; for h, 40 μm.
SNCs inhibit VSMC promigratory switching and promote calcification
To explore the underlying mechanisms, we sequenced RNA from aortic arch plaques of 12-week HFD-fed Ldlr−/− mice treated for one week daily with ABT263 or Veh, and then permitted to remodel for two further weeks on LFD (Fig. 3a). ABT263 treatment yielded 132 differentially expressed genes (DEGs; Extended Data Fig. 4a, Supplementary Table 1). A high proportion of the upregulated DEGs were VSMC markers, including markers for dedifferentiated VSMCs, suggesting that fibrous cap maintenance or restoration with ABT263 treatment might involve VSMC phenotypic switching from the contractile to a migratory, proliferative, and ECM-producing phenotype (Extended Data Fig. 4b). Among these factors were an inhibitor of procontractile Activin signaling (Vwc212–14), two receptors for prosynthetic ligands (EdnRA15 and LepR16), the prosynthetic ligand Bmp317,18, and two epigenetic initiators of synthetic switching (Prdm619 and the myocardin-inhibitor Tshz320). Furthermore, multiple procalcification genes (Sp721, Col2a122, Col10a123, Ibsp24, Clec3a25, Tgm126 and Chad27) were among the DEGs downregulated with ABT263 treatment, while inhibitors of ectopic bone formation (Bmp328 and Igfbp629,30) were upregulated, raising the possibility that SNC clearance might boost lesional VSMC numbers by preventing their transdifferentiation into calcification-prone, chondrocyte-like cells (Extended Data Fig. 4c). To test this proposition, we assessed whether ABT263 treatment inhibited plaque calcification using alizarin-red staining in conjunction with routine H-E. In keeping with prior reports of lipid normalization-induced calcification31,32, the switch of Ldlr−/− mice fed HFD for 12, but not 24, weeks onto LFD for 9 additional weeks increased lesion calcification (Fig. 3b,c). As predicted by our RNA-seq data, ABT263 treatment indeed attenuated LFD-induced calcification at the 12-week HFD timepoint.
Figure 3: SNCs inhibit promigratory switching and neointimal recruitment of medial VSMCs.
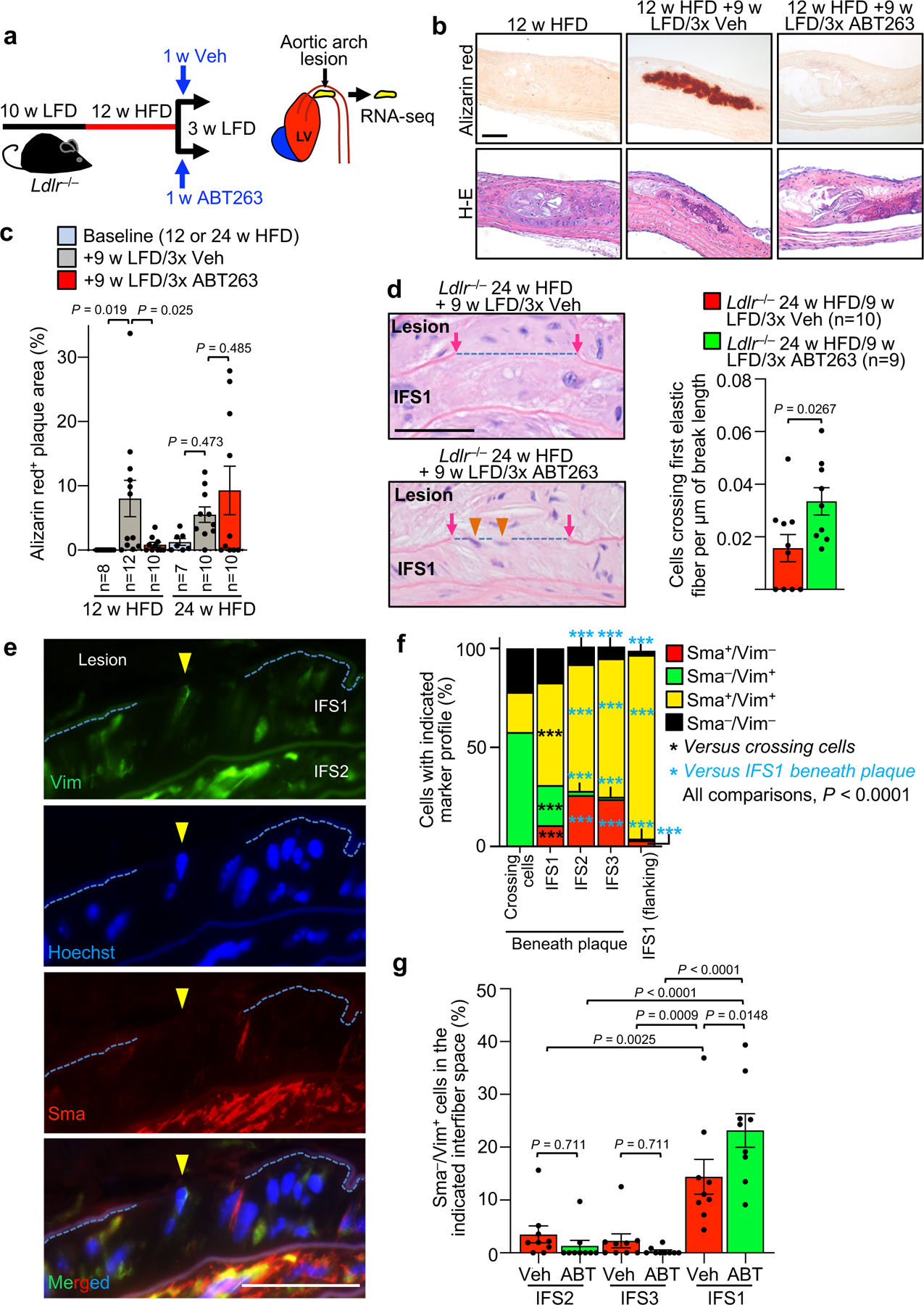
a, Schematic of short-term ABT263 treatment model to produce aortic arch lesions with and without SNCs for RNA-seq analysis. b, Alizarin red stained examples for the indicated groups quantified in panel c. c, Quantification of alizarin-red staining for calcification on Ldlr−/− mice with 12 and 24-weeks HFD feeding, as well as mice fed in this fashion followed by 9-weeks LFD and 3x Veh or 3x ABT treatment. d, (Left) Images of H-E-stained lesions of the indicated mice highlighting cells (orange arrowheads) crossing from IFS1 into the neointima through breaks (blue dashed line) in the first elastic fiber (see Fig. 1a for schematic representation of the experimental design). (Right) Quantification of number of cells crossing the first elastic fiber normalized to length of fiber breaks in plaques of the indicated mice. e, Representative example of Vim+/Sma− cell crossing between plaque and aortic wall through a gap in the first elastic fiber (quantified in panel f). f, Vim and Sma expression profile of cells crossing the first elastic fiber into the intima, cells in IFS1–3 beneath plaque, and cells residing in IFS1 but not underneath plaque (flanking). ***, p <0.001. Beneath plaque: n=74 crossing cells; n=5625 IFS1 cells; n=1398 IFS2 cells; n=1253 IFS3 cells. Flanking plaque, n=337 IFS1 cells. g, Quantification of Sma−/Vim+ cells in IFS1–3 of 24-week HFD remodeling for 9 weeks on LFD with 3 cycles Veh or ABT263 (legend as in d; Veh n=9, ABT n=9). “n” in all panels represents individual mice. Error bars represent s.e.m. Panel d was analyzed by unpaired, two-tailed t-test with Welch’s correction. Panel f was analyzed by global χ2, followed up by individual two-tailed Fischer’s exact tests. Panels c and g were analyzed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. *, p <0.05; **, p <0.01; ***, p <0.001. Scale bars for d and e are 40 μm. Scale bar in b is 100 μm. Error bars represent s.e.m.
RNAseq also revealed that ABT263 downregulated the expression of several secreted inflammatory factors (Ccl533, Prf134, Ltb35, Hfe36, and SerpinB1037) while increasing expression of anti-atherosclerotic inhibitors of inflammatory macrophage polarization (Il3338–40, Aldh1a241, and Lyve142), suggestive of reduced plaque inflammation (Extended Data Fig. 4d). Because inflammation in atherosclerosis has been associated with VSMC apoptosis, we tested the hypothesis that senolysis might act to thicken maximally-thinned caps by increasing VSMC content of lesions through decreased VSMC apoptosis, but Sma/TUNEL staining indicated that this possibility was unlikely (Extended Data Fig. 4e).
To understand how SNCs might promote cap thinning beyond stimulating VSMC transdifferentiation into chondrocyte-like cells, we further examined the ABT263-induced contractile-to-promigratory VSMC switching phenotype predicted by our RNA-seq data. Histological evaluation of aortic lesions revealed that more cells appeared to be crossing from the first interfiber space (IFS1) beneath the plaque into the lesion across breaks in the first elastic fiber when 24-week HFD fed Ldlr−/− mice were ABT263-treated during the ensuing 9-week LFD feeding period (Fig. 3d). Immunofluorescence costaining of lesions for the contractile marker Sma and the migratory marker Vim showed that, regardless of the treatment regimen, most crossing cells were Sma−/Vim+, characteristic of promigratory VSMCs (Fig. 3e,f and Extended Data Fig. 5a–d). Sma−/Vim+ cells were rare in other interfiber spaces underneath plaque as well as scarce in IFS1 immediately adjacent to plaque-bearing areas (Fig. 3f and Extended Data Fig. 5a–c), indicating that contractile-to-promigratory VSMC switching is restricted to the IFS1 underneath a lesion. Importantly, ABT263 treatment stimulated such switching (Fig. 3g), providing a plausible explanation for the observed increase in cells crossing the first elastic fiber beneath plaques. Similar results were obtained in complementary analyses on 12-week HFD fed Ldlr−/− mice treated with ABT263 or Veh during 9 weeks of LFD feeding (Extended Data Fig. 6a–c) and 20-week HFD fed Ldlr−/−;INK-ATTAC mice treated with AP or Veh during 9 weeks of LFD feeding (Extended Data Fig. 6d–f). Furthermore, RNA-seq on non-plaque bearing aortic arch inner curvature tissue from LFD-fed Ldlr−/− mice treated for one week with either ABT263 or Veh yielded 25 DEGs (Supplementary Fig. 3; Supplementary Table 2), none of which were linked to VSMC phenotypic switching. GSEA on these RNA-seq data revealed that of the 57 pathways predicting that ABT263-treatment of atherogenic lesions is anti-calcific and anti-apoptotic and stimulates VSMC phenotype switching, only two were altered with ABT treatment of non-atherogenic aortic tissue (Supplementary Table 3), further indicating that ABT263 does not promote dedifferentiation of contractile medial VSMCs without an overlying lesion.
SNCs inhibit medial VSMC migration throughout atherogenesis
End-stage lesions are highly complex in that they contain VSMCs, endothelial cells, macrophages, and other immune cells, as well as phenotypically altered versions of several of these cells types resulting from transdifferentiation7,43,44. To further test the concept that SNCs contribute to lesion pathogenesis by disrupting reparative functions of VSMCs, we analyzed less complex early-stage inner aortic arch lesions of Ldlr−/− mice present after 4 weeks HFD feeding (Fig. 4a). In the absence of senolysis, these lesions showed strong SA β-Gal activity, whereas ABT263 treatment yielded lesions that were smaller and had low SA β-Gal staining (Supplementary Fig. 4a,b). Aortic arch lesions of Veh-administered Ldlr−/− mice were indeed simple in that we predominately observed SA β-Gal-positive and -negative macrophage-like foam cells in the subendothelial space and no discernable fibrous cap (Fig. 4b and Supplementary Fig. 4c,d). In contrast, lesions of ABT263-treated Ldlr−/− mice possessed markedly fewer subendothelial foam cells and, consistent with efficient senolysis, a lower percentage of foam cells with SA β-Gal-derived X-gal crystals. Instead, these aortic lesions had greater numbers of cells traversing the first elastic lamina, accumulating in the subendothelium, and producing elastin (Fig. 4b–d). To validate that these matrix-secreting subendothelial cells originate from medial VSMCs undergoing phenotype switching, we used Myh11-CreERT2;CAG-LSL-tdTomato-WPRE (Ai14) Ldlr−/− mice to perform SMC lineage tracing (Fig. 5a). Following tamoxifen (Tam) treatment to induce tdTomato expression in SMCs by deletion of the loxP-STOP-loxP (LSL) cassette, mice were HFD-fed for 33 days accompanied by ABT263 or Veh treatment. We found that most lesional Sma+ cells were tdTomato+ regardless of treatment, with ~75% and ~90% of Sma+ lesional cells expressing tdTomato in Veh- and ABT263-treated lesions, respectively (Fig. 5b,c). Consistent with increased medial VSMC migration, tdTomato+ cell numbers were markedly increased in lesions of ABT263-treated mice normalized to either total lesion cellularity or lesion length (Fig. 5d,e). Very few tdTomato+/Sma+ cells within lesions were EdU+ or TUNEL+ in both Veh and ABT263 treated mice, indicating that the increase in media-derived VSMC content upon senolysis is unlikely due increased cell proliferation and/or decreased apoptosis (Fig. 5f,g and Extended Data Fig. 7a).
Figure 4: SNCs suppress innate repair functions of medial VSMCs beginning with the earliest stages of atherogenesis.
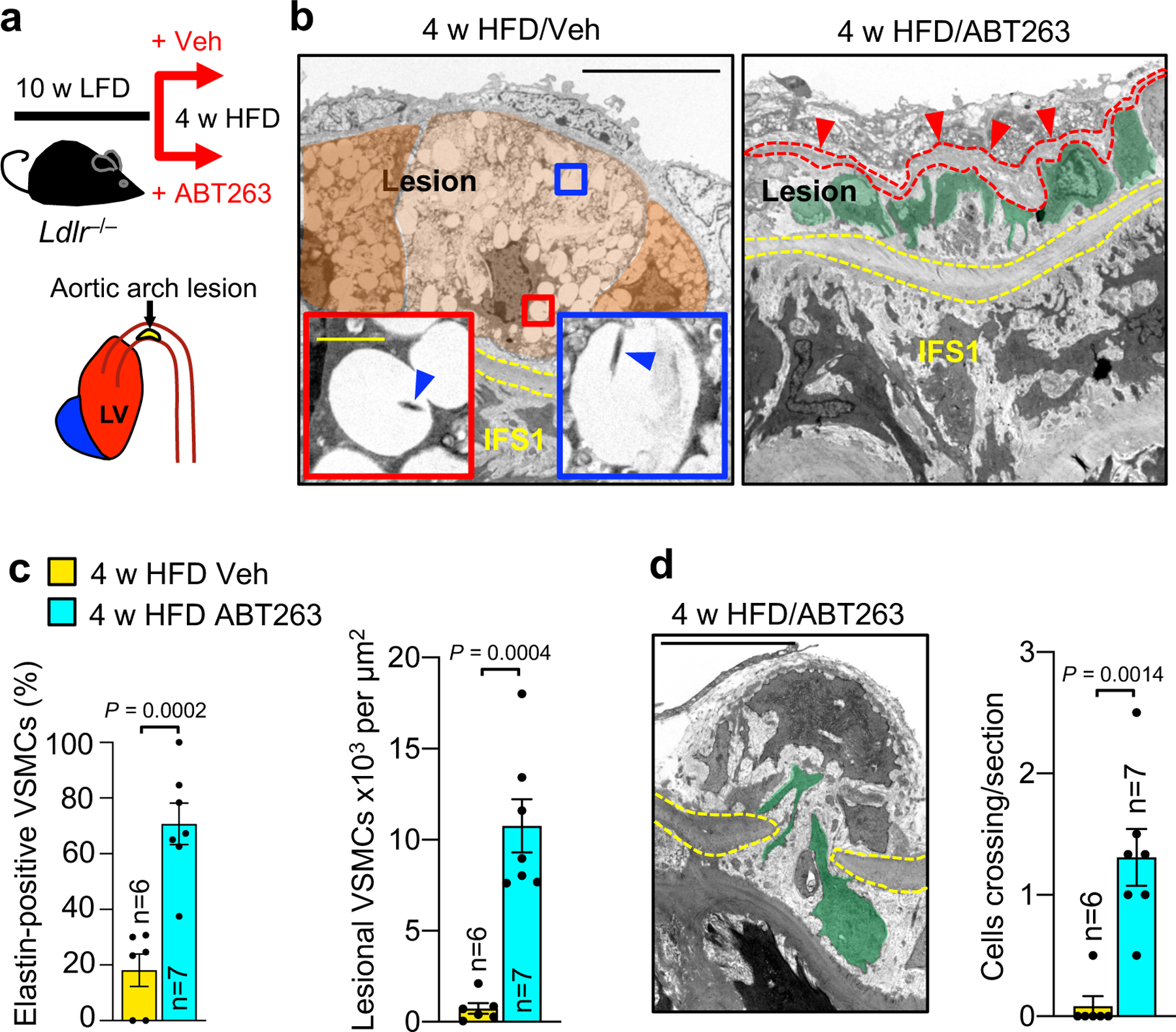
a, Schematic of the experimental design to study the impact of ABT263-mediated senolysis on lesions developing in the inner aortic arch of 4-week HFD fed Ldlr−/− mice. b, Representative TEM micrographs of SA β-Gal-stained aortic arch lesions of the indicated Ldlr−/− mice. Yellow dashed lines denote the first elastic fiber. Orange masks in the Veh-treated panel highlight borders of foamy macrophage-like cells and blue arrowheads in insets indicate X-gal crystals. In the ABT263-treated panel, green masks indicate VSMC-like cells, and red arrowheads indicated elastin deposition (marked with interrupted line). These parameters are quantified in panel c. c, (Left) Percentage of elastin+ VSMC-like cells and (Right) lesional VSMC-like cell content from the indicated groups. An elastin+ VSMC-like cell is one making close contact with extracellular, particulate (non-fiber) elastin. d, (Left) Representative image of a VSMC (green mask) traversing a break in the first elastic fiber (yellow dashed line) in an ABT-treated 4-week HFD plaque. (Right) Quantification of first elastic fiber-crossing cells (legend as in c). “n” in all panels represents individual mice. All panels were analyzed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. Scale bars for b are 10 μm (main panels) and 1 μm (insets). Scale bar in d is 5 μm.
Figure 5: VSMC lineage tracing approaches reveal senolysis enhances medial VSMC phenotypic switching and migration into lesions.
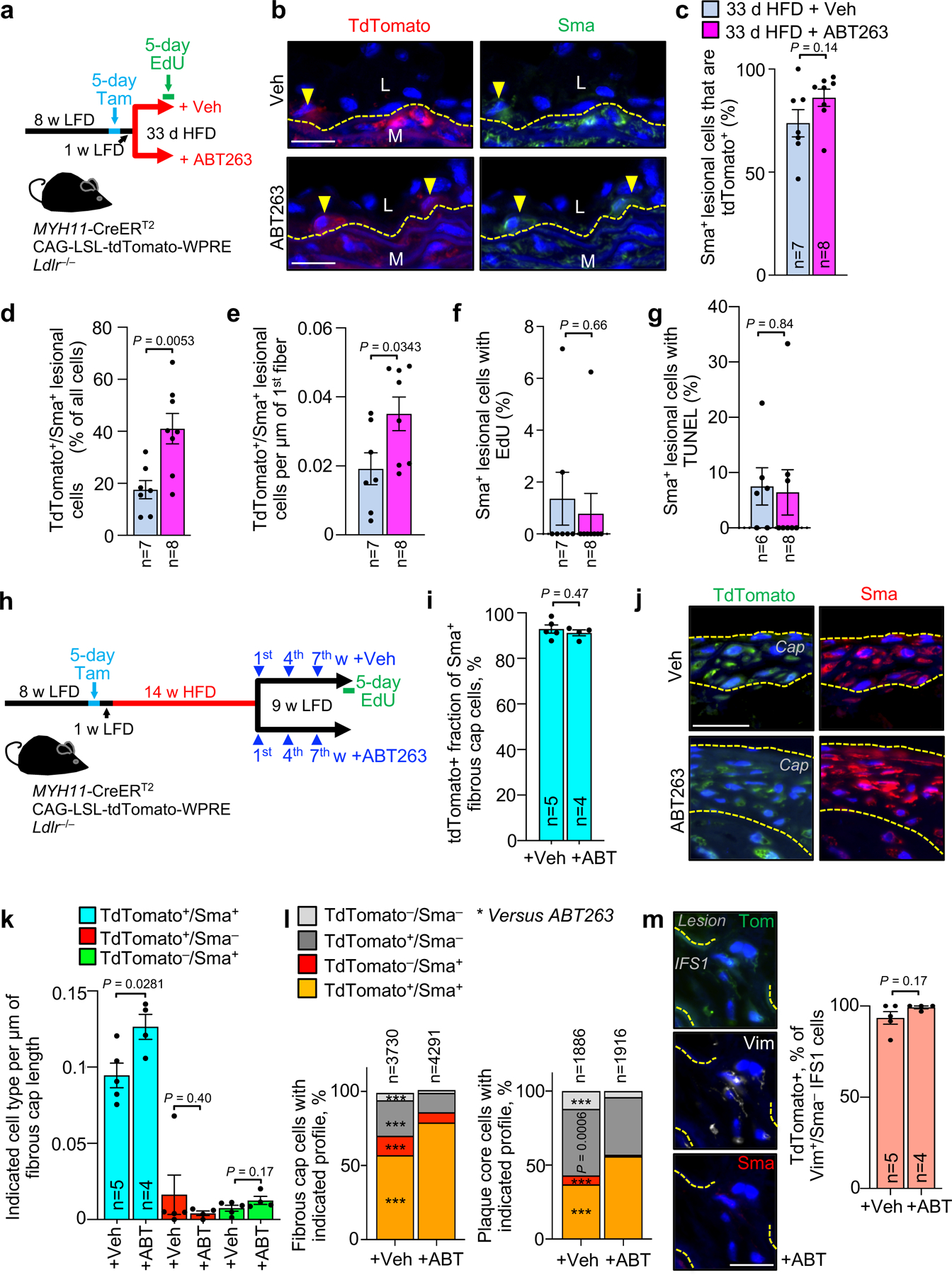
a, Schematic of the experimental design for data in b-g. b, Representative images of inner aortic arch lesions of the indicated mice (quantified in c) immunolabeled for tdTomato and Sma. L, lesion; M, media. Arrowheads mark tdTomato+/Sma+ cells; interrupted line mark first elastic fiber. c, Quantification of the percentage of Sma+ neointimal cells in inner aortic arch lesions of mice indicated in a that are also tdTomato+. d, Quantification of the percentage of neointimal cells that are tdTomato+/Sma+ in inner aortic arch lesions of mice indicated in a. e, Quantification of tdTomato+/Sma+ neointimal cells in lesions of mice indicated in a normalized to plaque burdened length of 1st elastic fiber. f, Percentage of neointimal Sma+ cells in mice from a incorporating EdU. g, Percentage of neointimal Sma+ cells in mice from a showing TUNEL+ nuclei. h, Schematic of the experimental design for data in i-m. i, Quantification of tdTomato+ percentage of Sma+ fibrous cap cells of mice from indicated groups. j, Representative images of tdTomato+/Sma+ cells in fibrous caps (bracketed in yellow dashed lines; quantified in k) from indicated groups. k, Quantification of frequency of cells of indicated tdTomato and Sma status per length of fibrous cap for indicated groups. l, Proportion of cells of indicated Sma and tdTomato status in fibrous cap (left) and plaque core (right) from inner aortic arch plaques of mice indicated in h. ***, p <0.001. m, (Left) Representative images of an IFS1 Vim+/Sma−/tdTomato+ cell and (Right) Quantification of the percentage of IFS1 Vim+/Sma− cells that are tdTomato+ for the indicated groups. All panels were analyzed by unpaired, two-tailed t-test with Welch’s correction, but panel l, which was analyzed by global x2, followed up by individual two-tailed Fischer’s exact tests for each combination of Sma and Tom status cells. Error bars represent s.e.m. *, p <0.05; **, p <0.01; ***, p <0.001. “n” always represents number of mice, excepting panel l, where “n” is total cell count. Scale bars in b, j, and m are all 20 μm.
Next, we assessed the extent to which ABT263 treatment impacts medial VSMC content in fibrous cap structures. To this end, we switched tamoxifen-treated Myh11-CreERT2;CAG-LSL-tdTomato-WPRE Ldlr−/− mice that had been on HFD for 14 weeks to a 9-week LFD and treated them with ABT263 or Veh during LFD weeks 1, 4, and 7 (Fig. 5h). Before sacrificing the animals, we administered EdU for five consecutive days (Fig. 5h). We found that nearly all Sma+ cells in fibrous cap structures were tdTomato+ regardless of SNC status (Fig. 5i), although caps of ABT263-treated animals had more tdTomato+/Sma+ cells than those of Veh-treated mice (Fig. 5j,k). EdU colocalization with Sma and tdTomato revealed that ABT263 treatment did not increase EdU incorporation among tdTomato+ cells in the cap or core, irrespective of their Sma status (Extended Data Fig. 7b–d), suggesting that a direct effect of senolysis on proliferation is unlikely to be responsible for the increased number of Sma+/tdTomato+ fibrous cap cells seen with SNC clearance. Likewise, TUNEL costaining showed no effect of ABT263 on cell death amongst tdTomato+ cells, regardless of Sma status (Extended Data Fig. 7e–g). However, the proportion of tdTomato+ cells expressing Sma within both the cap and core was increased with ABT263 treatment (Fig. 5l), which together with decreased lesional calcification (Extended Data Fig. 4b–d), is in keeping with the idea that SNCs promote differentiation of lesional VSMCs towards alternative cell fates.
Immunostaining experiments for Vim and Sma presented in Fig. 3e indicate that Vim+/Sma− cells are enriched in the IFS1 directly beneath atherogenic lesions and the most prominent cell population residing in elastic lamina breaks between IFS1 and the lesion. To conclusively examine whether these cells are of medial VSMC origin, we performed immunostainings for Sma and Vim on lesions from the abovementioned tamoxifen-treated Myh11-CreERT2;CAG-LSL-tdTomato-WPRE Ldlr−/− mice placed on HFD for 14 weeks and then on LFD for 9 weeks with or without senolysis. We found the overwhelming majority of Vim+/Sma− cells in the IFS1 beneath lesions or crossing through breaks in the lamina between IFS1 and the lesion to be Tomato+ (Fig. 5m), indicating that they are dedifferentiated promigratory VSMCs. We complemented this analysis with a costaining for Sma and an additional contractile marker Myh11 (smooth muscle myosin heavy chain). We found that, as with Vim+/Sma− cells (Fig. 3e–g and Fig. 5m), IFS1 beneath lesions accumulates more Sma−/Myh11− tTdTomato+ VSMCs compared to IFS2, and that ABT263 treatment further increases the amount of Sma−/Myh11− tdTomato+ VSMCs in IFS1 (Extended Data Fig. 7h,i). In a final set of immunostainings on lesions of tamoxifen-treated Myh11-CreERT2;CAG-LSL-TdTomato-WPRE Ldlr−/− mice with or without senolysis, we sought to exploit the ability to trace medial VSMCs after their relocation to the core to determine the impact of ABT263 on VSMC transdifferentiation to the osteochondrogenic lineage. We found that the proportion of tdTomato+ cells in the core expressing the osteochondrogenic lineage marker Runx145,46 to be markedly reduced with ABT263 treatment, a finding the is consistent with the observed reduction in calcification (Extended Data Fig. 7j).
Medial VSMC migration is a rapid response to lesion formation
To verify that the actions of ABT263 in early lesions were due to senolysis, we employed the transgene-mediated SNC elimination system p16-3MR, which kills p16+ SNCs in atherosclerotic plaques when exposed to ganciclovir (GCV)7. In these experiments, we shortened the HFD feeding time from 4 weeks to 9 days, to also assess whether switching is an immediate early response to HFD-induced vascular injury (Fig. 6a). Indeed, GCV-treated Ldlr−/−;3MR mice showed significantly increased VSMCs residing in fatty streak remnants due to crossing of the first elastic lamina compared to control animals (Fig. 6b–e). This is a rapid response of VSMCs to SNC killing, as shown by a similar enhancement of subendothelial VSMC numbers and IFS1 VSMCs crossing the innermost elastic lamina (Fig. 6f–j) when SNC clearance with p16-3MR was performed for 3 days after formation of 9-day HFD lesions. Collectively, these observations indicate that VSMCs in IFS1 under emerging atherosclerotic lesions are primed for recruitment into the subendothelial space to normalize tissue architecture, and that senescent foam cells counteract this latent, early repair process.
Figure 6: 3MR-mediated senolysis prompts rapid VSMC migration during early atherogenesis.
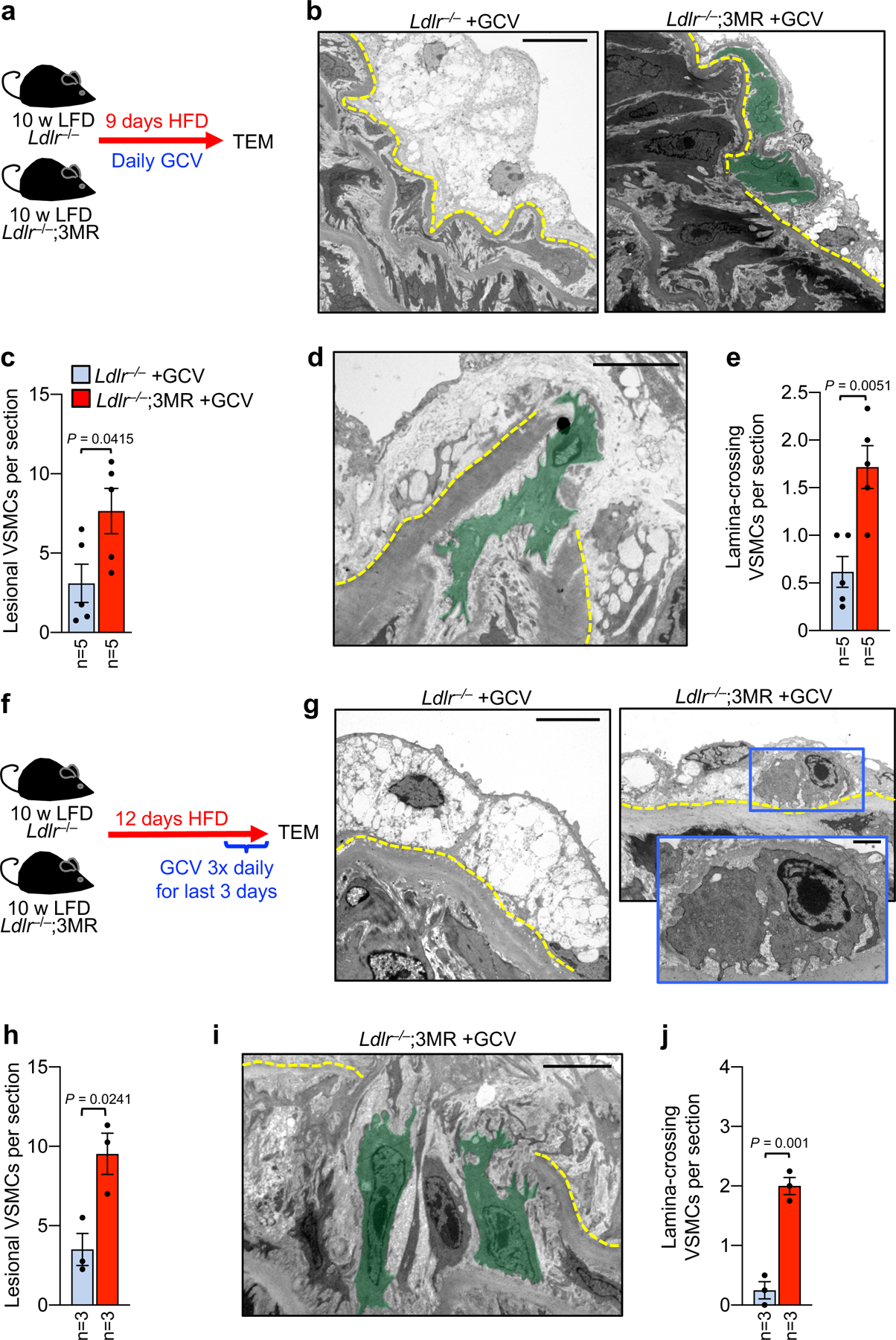
a, Schematic of 9-day HFD plaque formation assay using the p16–3MR transgene and ganciclovir (GCV) to constitutively clear SNCs. b, Representative transmission electron micrographs of indicated groups showing foam cell macrophage-rich fatty streak (left) and VSMCs (green masks, right) infiltrating the subendothelium (quantified in c). c, Quantification of VSMC infiltration in mice of indicated groups. d, Representative electron micrograph of a medial VSMC traversing the first elastic lamina in a 9-day HFD Ldlr−/−;3MR mouse treated with GCV (quantified in e). e, Number of VSMCs crossing the first elastic lamina in mice of indicated groups (legend as in c). f, Schematic of the experimental approach designed to test the acute impact of high-intensity SNC clearance on pre-existing early inner aortic arch lesions. g, Representative transmission electron micrographs of indicated groups showing a foam cell macrophage-rich fatty streak in the absence of senolysis (left) and VSMC presence in the subendothelium with SNC clearance (right, magnified in inset; quantified in h). h, Quantification of VSMCs in the subendothelial space of the indicated mice. i, Representative TEM micrograph of IFS1 VSMCs (green masks; quantified in j) crossing the first elastic lamina (yellow dashed line) in a 9+3-day HFD Ldlr−/−;3MR mouse treated with GCV. j, Number of VSMCs crossing the first elastic lamina in mice from indicated groups. Error bars represent s.e.m.. Statistics in all panels were performed by unpaired, two-tailed t-test with Welch’s correction. “n” in all cases is the number of mice. Scale bar in b is 10 μm; in d, 2 μm; in g, 5 μm; and, in i, 5 μm. Scale bar in g (inset) is 1 μm.
To probe whether early lesions are reversible with dyslipidemia management and the potential impact of SNCs on fibrous cap assembly, we placed Ldlr−/− mice on HFD for 4 weeks followed by 6 weeks of LFD-feeding to normalize dyslipidemia in conjunction with either Veh or ABT263 administration during LFD weeks 1 and 4 (Extended Data Fig. 8a). Instead of reversing, 4-week aortic lesions continued to grow considerably during the LFD-feeding interval, indicating that diet-induced dyslipidemia is not required to drive early-stage plaque progression (Extended Data Fig. 8b,c). TEM revealed that, during the 6-week LFD-remodeling interval, Veh-treated lesions accumulate foam cells and begin to develop a fibrous cap (Extended Data Fig. 8d–i). In contrast, fatty streaks treated with ABT263 possess fewer foam cells than Veh-treated Ldlr−/− mice. As with preventative ABT263-administration, lesions remodeling under LFD conditions with ABT263 showed more VSMCs traversing the innermost elastic lamina, accumulating in the lesion, and secreting elastin than with lipid normalization alone (Extended Data Fig. 8e–h), resulting in a thicker fibrous cap (Extended Data Fig. 8i). These data further provide further evidence that early fibrous cap formation depends on the recruitment of medial VSMCs into the subendothelial space and is inhibited by SNCs regardless of lipid status.
Igfbp3 inhibits VSMC promigratory switching
To understand how lesional SNCs inhibit recruitment of cap-forming medial VSMCs, we speculated on a potential role for insulin-like growth factor binding protein 3 (Igfbp3) for several reasons. First, Igfbp3 is known to sequester and neutralize IGF-1, a potent mediator of VSMC dedifferentiation and migration47–49. Second, Igfbp3 is expressed at elevated levels in SNCs and an integral part of the SASP50–52. Third, Igfbp3 has been shown to induce a senescence-like phenotype in neighboring cells53. Fourth, increasing IGF-1 levels in ApoE−/− mice by systemic infusion or recombinant protein or transgenic overexpression in VSMCs increases plaque stability in addition to reducing plaque burden54,55. Consistent with the hypothesis that Igfbp3 is a critical SASP factor in the suppression of VSMC migration, treatment with ABT263 significantly reduced Igfbp3 levels in 24-week atherosclerotic lesions (Fig. 7a). Importantly, IHC for Igfbp3 uncovered that a very high proportion of SA β-Gal+ lesional cells stained positive for the IGF inhibitor (Fig. 7b). A subset of SA β-Gal− lesional cells was also Igfbp3+ (Fig. 7b). Clearance of SNCs with ABT263 caused a marked reduction in SA β-Gal− cells expressing Igfbp3, suggesting that SNCs induce Igbp3 in neighboring cells through a paracrine mechanism. Quantitative PCR-based analysis of RNA extracted from brachiocephalic arteries of 12-week HFD fed Ldlr−/− mice treated with ABT263 or Veh during 3-weeks of subsequent LFD feeding revealed that senolysis reduced Igfbp3 transcripts, but not transcripts of Igfbp2, 5, or 6, suggesting that SNCs selectively increase lesional Igfbp3 expression (Fig. 7c).
Figure 7: Senolysis depletes lesional Igfbp3 to promote promigratory VSMC phenotype switching of medial VSMCs.
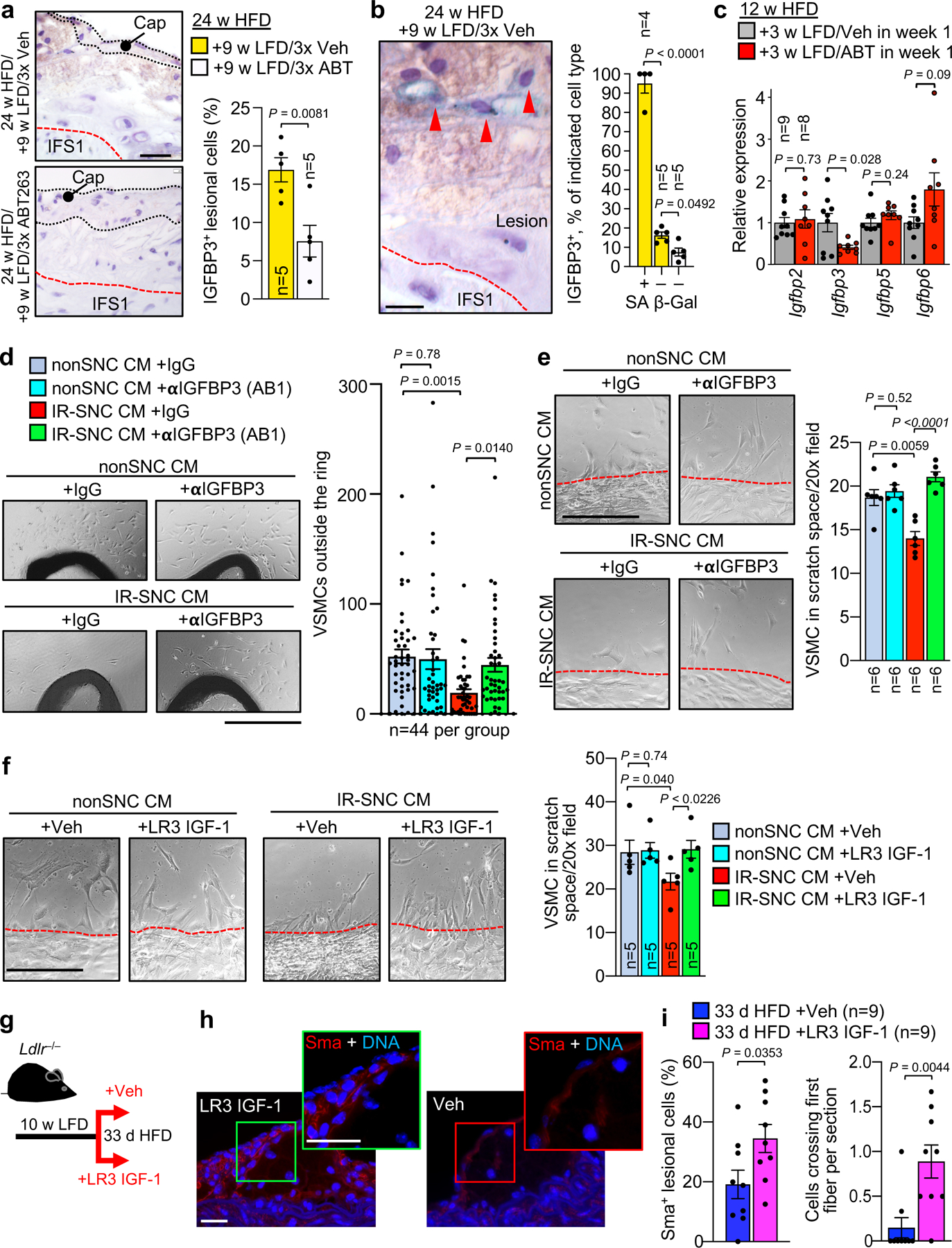
a, (Left) Representative images of lesions of the indicated mice immunostained for Igfbp3. (Right) Quantification of percentage Igfbp3+ cells in lesions of the indicated mice. b, (Left) Representative images of Igfbp3+/SA β-Gal+ lesional cells (arrowheads). (Right) Quantification of Igfbp3+ cell frequency among SA β-Gal+ and SA β-Gal− cells from indicated groups (legend as in a). c, Relative expression of Igfbp genes in brachiocephalic arteries of the indicated Ldlr−/− mice. d, (Left) Representative images of VSMCs outgrowing from aortic rings (AR) of wildtype C57BL/6 mice treated with the indicated conditioned media (CM). αIGFBP3 (AB1), ab193910. (Right) Quantification of outgrowing VSMCs in the indicated treatment groups. e, (Left) Representative images of human aortic VSMCs emigrating into scratch wound space with indicated CM. (Right) Quantification of emigrating VSMCs in the indicated experimental groups (legend as in d). f, (Left) Representative images of human aortic VSMCs emigrating into scratch wound space with indicated CM. (Right) Quantification of emigrating VSMCs in the indicated experimental groups. LR3 IGF-1. g, Schematic of experiments presented in h and i. h, Representative Sma immunostaining in the indicated early lesions. i, (Left) Quantification of neointimal Sma+ cells in lesions of the indicated mice. (Right) Quantification of cells crossing the first elastic fiber in indicated groups. Statistics in panels b and d were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Statistics in panels e and f were performed by RM one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. All other panels were analyzed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. “n” refers to individual mice in a, b, c, and i; individual aortic rings in d; and, CM from independent MEF lines in e and f. Scale bar in a is 20 μm; b, 10 μm; d, 500 μm; e and f, 200 μm; and, g (main and inset), 20 μm.
To probe the role of the SASP and its constituent Igfbp3 in inhibiting VSMC migration, we utilized two established in vitro migration assays: outgrowth of VSMCs from ex vivo cultured aortic rings and scratch wounding of confluent VSMC cultures56–58. In the first assay, conditioned medium from irradiated senescent-MEFs (IR-SNC CM), reduced the number of outgrowing VSMCs surrounding murine aortic rings versus rings treated with CM from proliferating MEFs (nonSNC CM) (Fig. 7d and Extended Data Fig. 9a,b). Importantly, normalization of VSMC outgrowth in IR-SNC CM was observed with the addition of Igfbp3-neutralizing antibodies. In contrast, antibody-mediated Igfbp3 neutralization in nonSNC CM had no discernable impact on VSMC outgrowth (Fig. 7d). Likewise, in scratch wound assays, IR-SNC CM repressed emigration of human VSMCs into the bare wound space, yet not in the presence of Igfbp3-neutralizing antibody (Fig. 7e and Extended Data Fig. 9c), providing further evidence for the idea that SNCs act to inhibit VSMC migration through the SASP, with Igfbp3 as a critical SASP factor. Two additional observations supported this mechanism. First, supplementation of IR-SNC CM with recombinant LR3 IGF-1, a potent IGF-1 variant with reduced Igfbp binding affinity47, normalized VSMC migration in VSMC treated with IR-SNC CM, yet had no impact on VSMC migration in the context of nonSNC CM (Fig. 7f and Extended Data Fig. 9c). Second, IR-CM increased Sma levels and suppressed VIM expression of cultured human VSMCs, indicative of maintenance of the contractile state, a phenomenon prevented by the presence of LR3 IGF-1 or Igfbp3-neutralizing antibody (Extended Data Fig. 9d,e).
We further probed this mechanism in vivo by administering LR3 IGF-1 or Veh to Ldlr−/− mice during a 33-day HFD feeding period (Fig. 7g). Examining lesions in the aortic arch inner curvature, we found that the number of Sma+ VSMCs in the subendothelium were two-fold higher in LR3 IGF-1-treated mice than in Veh-treated counterparts, which correlated with an increase in cells crossing the first fiber (Fig. 7h,i). LR3 IGF-1 treatment did not affect lesion size or overt SA β-Gal activity (Extended Data Fig. 9f).
Igfbp3 inhibits medial VSMC dedifferentiation in explanted plaque
Next, we investigated the role of Igfbp3 in suppressing VSMC promigratory phenotype switching using ex vivo culture of atherosclerotic plaques59. We collected plaque-rich aortic arches from Ldlr−/− mice that were fed HFD for 12 weeks and then placed on LFD for 2 weeks. Rings prepared from these arches were cultured for 18 h in the presence of anti-Igfbp3 or IgG control antibody (Fig. 8a). Immunolabeling of sections prepared from these explants revealed that Igfbp3-neutralizing antibody markedly increased the number of Vim+/Sma− cells in IFS1 beneath plaques while simultaneously reducing the number of Vim−/Sma+ cells (Fig. 8b), further supporting the conclusion that Igfbp3 plays a central role in suppressing lesional recruitment of medial VSMCs. To assess potential applicability of these findings to human atherosclerosis management, we collected aortic and femoral endarterectomy specimens prepared as paired serial slices from these lesions, and cultured them in the presence of IgG or one of two distinct IGFBP3-neutralizing antibodies for 2 days (Fig. 8c–e). IGFBP3 neutralization increased the number of SMA+ cells expressing VIM in human atheroma explants, regardless of their site of origin (Fig. 8d,e), implying that a key mechanism by which SNCs suppress VSMC promigratory switching in murine plaques may be conserved in human disease tissue. Of the 12 human endarterectomy specimens analyzed in this fashion, 11 were from patients being treated with a statin, indicating that promigratory switching of SMA+ cells by IGFBP3 neutralization is a phenomenon that occurs on top of the therapeutic effects of lipid-lowering drugs.
Figure 8: Igfbp3 neutralization promotes promigratory switching of VSMC in mouse and human explant atheromas.
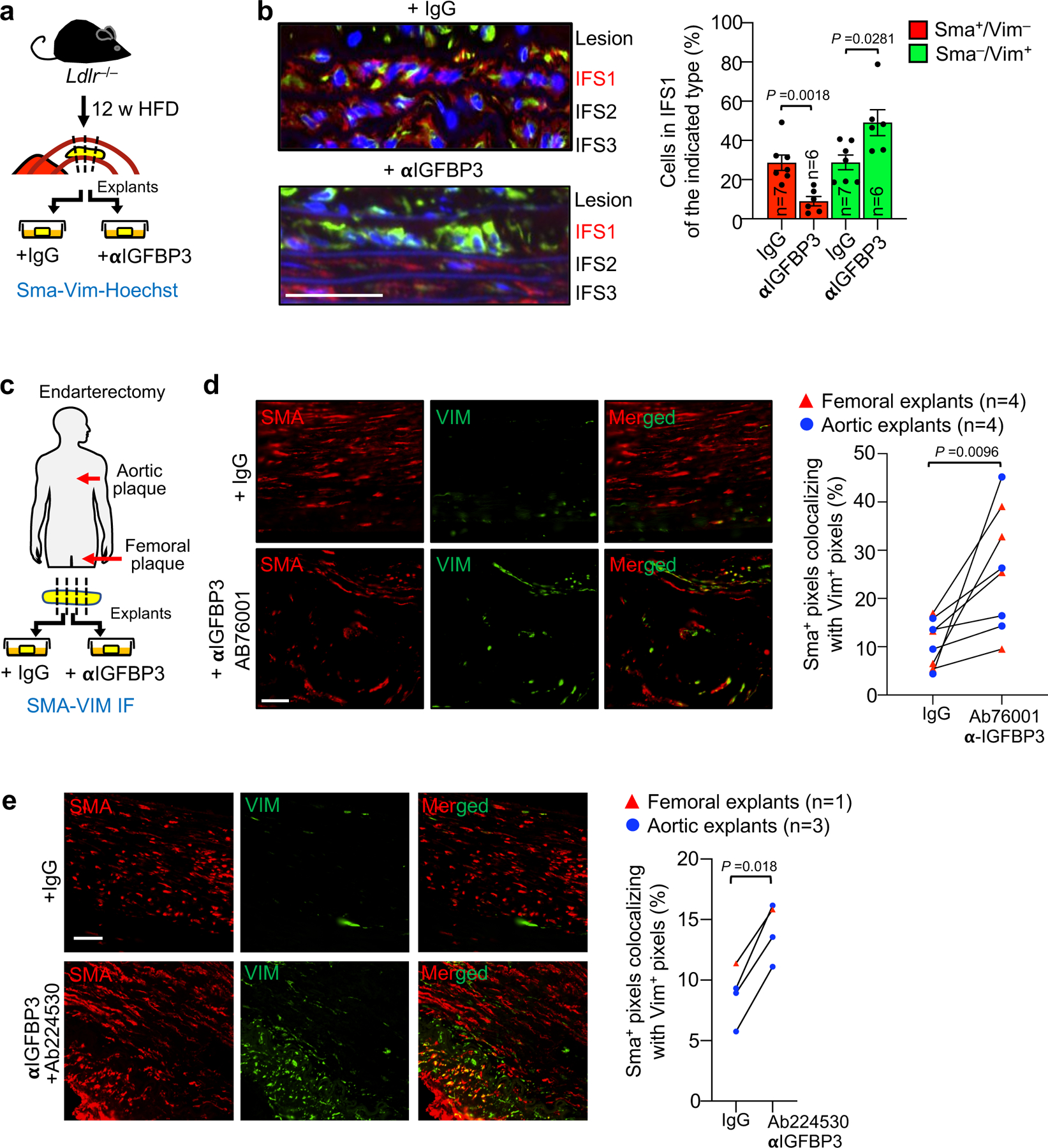
a, Schematic of in vitro Igfbp3 neutralization experiment in aortic arch plaque from Ldlr−/− mice. b, (Left) Representative images of explanted lesions from Ldlr−/− mice cultured in the presence of the indicated antibodies and costained for Vim and Sma. Neointima flanking the media (lesion) and IFS1–3 are shown. (Right) Quantification of the percentage Sma+/Vim− and Sma−/Vim+ cells in IFS1 of the indicated explants. Vim+/Sma+ frequency is not shown as it was unchanged with treatment. c, Schematic of in vitro Igfbp3 neutralization experiment in aortoiliac and femoral plaque from human endarterectomy patients. d, (Left) Representative images of the indicated explanted human plaque cultured in the presence of the indicated antibodies and costained for Vim and Sma. (right) Quantification of colocalization of Vim and Sma signal in human plaque treated in this fashion. e, (Left) Representative images of the indicated explanted human plaque cultured in the presence of the indicated antibodies and costained for Vim and Sma. (right) Quantification of colocalization of Vim and Sma signal in human plaque treated in this fashion. Statistics in panels b, d, and e were performed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. “n” represents explanted aortic plaque-bearing rings in b, and endarterectomy patients in d and e. Scale bars in b, d, and e are 50 μm.
Discussion
Despite effective medical management of dyslipidemia, patients with advanced atherosclerosis face significant residual risk of death or disability from fibrous cap disruption10,11. Our results suggest that SNCs in atheromas may explain this observation by acting through a novel lipid-independent mechanism: antagonism of IGF-1 mediated VSMC switching from a contractile to a promigratory state through the paracrine actions of a core SASP component, Igfbp3, thereby impairing fibrous cap formation, maintenance, and restoration (Extended Data Fig. 10). Our findings support the concept that, despite emerging evidence for SASP complexity and heterogeneity, individual SASP factors may disproportionately contribute to key pathological features of major age-related diseases. The observation that the beneficial effects of senolysis on the fibrous cap last at least six months in mice suggests that intervals between treatment sessions can be long, thereby possibly minimizing risk for negative side effects of senolytic drugs such as those targeting Bcl2 family members. Additionally, the observation that lesions of all stages respond to senolysis by VSMC promigratory phenotype switching may be favorable for human therapy, in which lesions at a spectrum of development are likely present. Collectively, these results suggest that intermittent pharmacological senolysis or neutralization of key SASP components, such as IGFBP3, could complement lipid-lowering interventions in patients with atherosclerosis to simultaneously block plaque growth and stimulate VSMC-dependent fibrous cap repair and maintenance. Extrapolating atherosclerosis studies from mice to humans can be challenging due to fundamental differences between atherogenesis in mice and humans60,61. However, the observation that inhibition of IGFBP3 in human atherogenic lesions induces expression of a key promigratory VSMC marker, VIM, holds the promise that other mechanistic aspects of senolysis observed in murine lesions, such as increased ECM deposition by lesional VSMCs, may be conserved across species.
SNCs that accumulate during early atherogenesis not only enhance foam cell formation7, but also inhibit promigratory phenotype switching of medial VSMCs. Although early stage lesions lack a fibrous cap even with senolysis, once these structures start to develop, they do so more robustly when SNCs are subject to elimination. We find that lesional recruitment of medial VSMC continues into the most advanced stages of disease in mice, with senolysis continuing to stimulate this process. The question can be raised as to what extent this mechanism is relevant to preventing cap rupture, particularly with recently developed transgenic approaches providing evidence that lesional VSMCs undergo clonal expansion from relatively few migration-competent (dedifferentiated) precursors62–64. Single-cell RNA sequencing indicates that these “pioneer” VSMCs subsequently redifferentiate into contractile VSMCs as well as transdifferentiate into macrophage-like and osteochrondrogenic descendants65,66. These approaches were not integrated in our study, so whether there is an impact of senolysis on clonal expansion of VSMCs or their absolute redifferentiation trajectories remain to be determined. Our short-term EdU incorporation experiments indicate that media-derived lesional VSMCs do proliferate in both the core and the cap, supporting a cap repair model in which migration is followed by proliferation. However, such proliferation is not likely to explain why removing SNCs promotes cap maintenance and repair because senolysis did not increase the percentage of cycling cells among lesional VSMCs. Likewise, apoptotic death of media-derived VSMC is observed, albeit at low rates, but, like proliferation, is unaffected by senolysis. Apart from stimulating recruitment of medial VSMCs into lesions, senolysis may act to preserve the VSMC status of these cells by inhibiting their transdifferentiation to other cell types60,61. Although the current study by no means comprehensively addresses the impact of senolysis on transdifferentiation, three lines of evidence suggest that inhibition of VSMC transdifferentiation might be an important contributing mechanism to fibrous cap thickening with senolysis (Extended Data Fig. 10). First, ABT treatment reduced the proportion of lesional VSMCs expressing the osteochondrogenic marker Runx1, an observation that correlated with reduced calcification in atherogenic lesions present after 12 weeks of HFD feeding. Second, ABT-treated mice show a higher proportion of Sma expression among wall-derived VSMC within lesions, suggesting their VSMC phenotype is preserved. Third, senolysis promotes expression of the signature contractile VSMC protein Sma in media-derived VSMCs residing in the fibrous cap. In future studies, it will be of interest to determine whether senolysis impacts VSMC phenotype switching to a lineage characterized by macrophage markers, as preventing such transdifferentiation by inactivating Klf4 has been shown to increase in fibrous cap thickness and VSMC content much like SNC clearance67.
In closing, while previous studies have implicated SNCs in tissue regeneration, here we provide evidence that they exert the opposite biological effect in atherogenesis in that they suppress the innate cap-reparative properties of medial VSMCs. By exploring how SNCs exert this effect, we identified new, therapeutically-targetable biology in a major human disease with considerable residual mortality despite medical management of dyslipidemia. It will therefore be important to develop an in-depth understanding of how other new concepts relevant to atherogenesis such as VSMC clonality and the various types of VSMC transdifferentiation interface with senescence.
METHODS
Animal Models
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic and conducted according to ethical guidelines per AALAC. C57BL/6 Ldlr−/− mice were purchased from the Jackson Labs (stock number 002207) and interbred to generate homozygous Ldlr−/− female mice for experiments. C57BL/6 p16-3MR transgenic mice1 were bred to C57BL/6 Ldlr−/− to generate Ldlr+/–;3MR mice, which were then bred to C57BL/6 Ldlr−/− mice to produce Ldlr−/−;3MR males. Female Ldlr−/−;3MR mice used in experiments were generated by breeding Ldlr−/−;3MR males to C57BL/6 Ldlr−/− females. C57BL/6 INK-ATTAC transgenic mice68 were bred to C57BL/6 Ldlr−/− to generate Ldlr+/–;INK-ATTAC mice, which were then bred to C57BL/6 Ldlr−/− mice to produce Ldlr−/−;INK-ATTAC males. Female Ldlr−/−;INK-ATTAC mice used in experiments were generated by breeding Ldlr−/−;INK-ATTAC males to C57BL/6 Ldlr−/− females. C57BL/6.FVB Myh11-CreERT2 (stock number 019079) and C57BL/6 CAG-LSL-tdTomato-WPRE (Ai14; stock number 007908) mice were acquired from Jackson Labs, crossed onto a C57BL/6 Ldlr+/– background and then interbred to generate Myh11-CreERT2 CAG-LSL-tdTomato-WPRE Ldlr−/− male mice for experiments69. All mice were housed in a specific-pathogen free (SPF) facility, with a 12 h light/dark cycle and ad libitum access to food and water for the duration of experiments.
Atherosclerosis Induction and Senolysis Methods
C57BL/6 Ldlr−/− female mice were raised to ~10 weeks of age on standard irradiated non-atherogenic chow diet (LabDiet #5053; 13.205% calories from fat), at which point they were enrolled to one of several experimental designs. HFD (TD. 88137; 42% calories from fat) was obtained irradiated from Harlan-Teklad, stored at 4 °C until use, and exchanged weekly. In all cases, low-fat diet (LFD) refers to the above standard non-atherogenic chow diet.
ABT263 was obtained from Synnovator or Selleck, formulated in Veh (PBS with 15% DMSO; 7% Tween-20), and delivered by intraperitoneal (IP) injection at a dose of 100 mg/kg in all experiments. For intermediate-disease remodeling experiments, female Ldlr−/− mice were placed on HFD for 12 weeks, followed by 9 weeks of LFD with concurrent treatment with ABT263 or Veh either during LFD-feeding week 1 (1x ABT263) or weeks 1, 4, and 7 (3x ABT263). For durability testing of intermediate-disease remodeling, female Ldlr−/− mice were placed on HFD for 12 weeks, followed by 26 weeks of switch to LFD with concurrent treatment with ABT263 or Veh during LFD-feeding weeks 1 and 4 (2x ABT263). For late-disease remodeling experiments, female Ldlr−/− mice were placed on HFD for 24 weeks, followed by 9 weeks of LFD with concurrent treatment with ABT263 or Veh during LFD-feeding weeks 1, 4, and 7 (3x ABT263). For the aforementioned studies, 100 mg/kg ABT263 or volume-matched Veh were delivered by IP once-daily during injection weeks.
For late-disease remodeling experiments with INK-ATTAC, female Ldlr−/− and Ldlr−/−;INK-ATTAC mice68 were placed on HFD for 20 weeks, followed by 9 weeks of LFD feeding with concurrent treatment with AP20187 (2 mg/kg) in Veh (4% EtOH; 10% PEG; 1.72% Tween-20 in PBS) delivered by IP injection once-daily during LFD-feeding weeks 1, 4, and 7. For late-disease remodeling experiments with p16-3MR, female Ldlr−/−;3MR mice placed on HFD for 20 weeks, followed by 9 weeks of LFD feeding with concurrent IP injection daily with vehicle or ganciclovir (5 mg/kg) in PBS vehicle during LFD-feeding weeks 1, 4, and 7.
For analysis of inner aortic curvature atherosclerotic lesions in prevention experiments with ABT263, female Ldlr−/− mice were placed on HFD for 4 weeks. During HFD administration, mice were treated with 100 mg/kg ABT263 or equivalent volume Veh constitutively, once-per-day, on a 5-days-on/2-days-off schedule. To study cap-formation in early inner aortic curvature atherogenic lesions, female Ldlr−/− mice were placed on HFD for 4 weeks to induce lesions, followed by LFD feeding for a further 6 weeks. During this LFD-feeding interval, mice were treated with 100 mg/kg ABT263 or Veh once-daily during LFD-feeding weeks 1 and 4.
To analyze the impact of IGF-1 on inner aortic curvature atherogenic lesions, female Ldlr−/− mice were placed on HFD for 33 days and administered recombinant LR3 IGF-1 (ProspecBio, cat. no. CYT-022) by IP injection (0.3 mg/kg) in 10 mmol/l HCl/PBS Veh on a 5 d on/2 d off schedule for 3 weeks, and then daily for the final 10 days. During the last 5 days, EdU (50 mg/kg) was co-administered with IP LR3 IGF-1 or Veh (10 mmol/l HCl/PBS) injection.
For VSMC tracing studies, 8-week-old male Myh11-CreERT2 CAG-LSL-tdTomato-WPRE Ldlr−/− mice were treated once-daily by gavage with 50 mg/kg Tamoxifen (Sigma-Aldrich) in corn oil/5% EtOH for 5 days. Following one week of Tamoxifen withdrawal, mice were placed on HFD for 33 days on a 5-days-on/2-days-off treatment with ABT263 or Veh. Alternatively, following one week of Tamoxifen withdrawal, mice were placed on HFD for 14 weeks, followed by 9 weeks LFD feeding with ABT administration once-daily with with ABT263 or Veh during LFD feeding weeks 1, 4, and 7. During the final 5 days, EdU (50 mg/kg in PBS once-daily) was administered by tail vein injection to assess VSMC proliferation.
Female Ldlr−/−;3MR and Ldlr−/− mice for analysis of the vessel injury response to atherogenesis in emerging inner aortic curvature lesions were placed on HFD for 9 day and treated daily with ganciclovir (GCV; 25 mg/kg in PBS). To study timing of VSMC migration in response to senolysis, female Ldlr−/−;3MR and Ldlr−/− mice were placed on HFD for 12 days and treated with GCV (25 mg/kg in PBS, thrice daily) during the last three days.
Blood Analysis
No more than 24 h prior to sacrifice, blood was collected using retro-orbital sinus puncture with a heparinized glass capillary into an EDTA-treated microcentrifuge tube. Whole blood was immediately subjected to gross circulating cell analysis using the Hemavet 950 instrument (Drew Scientific Inc., Miami Lakes, FL, USA). Plasma was isolated by centrifugation in EDTA-treated microcentrifuge tubes for 15 min at 4 °C and 3500 g. Lipid analysis was performed by the Mayo Clinic Immunochemical Core Laboratory (ICL) using high-performance liquid chromatography.
SA β-Gal Staining and GAL-EM
SA β-Gal staining was performed using a kit (Cell Signaling; cat. no. 9860S) per the manufacturer’s instructions with slight modification. Briefly, whole aortas were excised, opened lengthwise, and stored in PBS on wet ice until fixation, then fixed for 15 min at room temperature (RT) in kit fixative. Aortas were then washed 2 × 5 min in ice-cold PBS to remove excess fixative and transferred to pH 6.0 SA β-Gal staining solution at 37 °C for 12 h.
SA β-Gal stained aortas intended for routine histological analysis were post-fixed in 10% neutral buffered formalin for 12 h at RT before inner-curvature aortic arch plaque microdissection, standard dehydration, paraffin infiltration, embedding, and sectioning. SA β-Gal stained aortas intended for GAL-EM analysis were post-fixed in Trump’s fixative for 4 h at RT. The inner-curvature aortic arch plaque was then microdissected and processed for EM (dehydration through xylene-alcohol series, followed by osmium-tetroxide counterstaining, infiltration with Epon resin, and sectioning). Between 1 and 4 plaque-bearing thin sections were analyzed per mouse for all plaque parameters. Cells were considered X-Gal+ if they contained one or more needle-shaped or cuboidal electron-dense crystal(s) within a vacuole.
Cell-type identification was as described7 and based on ultrastructural morphology. Briefly, approximately circular cells with numerous vacuolations were considered to be foam cell macrophage-like. Spindle shaped or highly ramified cells with electron-dense cytoplasm rich in endoplasmic reticulum, stress fibers, and with few vacuoles were considered to be VSMC-like.
Immunohistochemical and Immunofluorescent Labeling
Immunohistochemical detection of Sma was performed using standard technique. Briefly, 5 μm thick paraffin sections were rehydrated and antigen retrieval performed using high pH antigen unmasking solution (Vector, H-3301) before blocking with 2% normal goat serum and incubating overnight in primary antibody (mouse-anti-Sma (1:200; Abcam, cat. no. ab7817)) in 2% serum block at 4 °C in a humidified chamber. Sections were then washed, incubated with biotin-conjugated goat-anti-rabbit secondary, washed, incubated with avidin-conjugated horseradish peroxidase enzyme, and developed with DAB kit (Vector, SK-4100). Sections were counterstained with dilute haemotoxylin. IHC staining for Igfbp3 was performed as above, with the exception of retrieval in low pH antigen unmasking solution (Vector, H-3300) and incubating overnight with rabbit-anti-Igfbp3 (1:200; Abcam, cat. no. ab76001).
For immunofluorescent co-labeling of Vim and Sma, 5 μm thick paraffin sections from SA β-Gal stained intermediate or late-disease remodeling mice were deparaffinized by two 25-min incubations in 50°C xylene before standard rehydration to 70% EtOH, 5 min incubation in 0.85% NaCl/dH2O, and transfer to PBS. Sections were then pre-fixed in 4% PFA for 7.5 min; antigen retrieved via Proteinase K (10 μg/ml) for 10 min; post-fixed in 4% PFA for 5 min; blocked with mouse-on-mouse blocking reagent (Vector Labs; cat. no. MKB-2213) for 30 min; blocked with 2% NGS for 30 min; and, incubated in 1:150 Sma (Abcam, cat. no. ab7817) and Vim (Abcam, cat. no. ab92547) primary antibodies overnight at 4C before secondary staining. Samples were Hoechst stained, washed, and mounted in Vectashield with 1:100 Hoechst to highlight both nuclei and retain Hoechst staining of elastic fibers. For all experiments, both Veh and ABT-treated mice were stained and imaged in parallel on identical exposure settings, and at least 2 sections separated by at least 200 μm were assessed. IFS1 is the space between the first pair of elastic fibers as identified by Hoechst; IFS2, the second pair; and, IFS3, the third pair. For IFS1 scoring, all cells beneath plaque on all sections were scored. For IFS2 and IFS3, at least one section from all mice was scored for all such cells. For flanking region scoring, no more than 20 IFS1 nuclei beyond the plaque edge were assessed from 1–3 sections per mouse.
For Sma+ scoring of fibrous cap, all fibrous cap cells throughout 2–3 sections separated by 200 μm or more were scored and normalized to the length of the total plaque luminal surface. For 12 w HFD baseline, 4 mice were scored by IHC and 6 by IF for Sma; for 12 w HFD +9w Veh, 4 mice were scored by IHC and 5 by IF for Sma; for 12 w HFD +9 w ABT 4 mice were scored by IHC and 6 by IF for Sma; for 24 w HFD baseline, 4 mice were scored by IHC and 5 by IF for Sma; for 24 w HFD +9 w Veh, 5 mice were scored by IHC and 5 by IF for Sma; for 24 w HFD +9 w ABT 5 mice were scored by IHC and 5 by IF for Sma; for 12 w HFD/9 w LFD/1xABT, 3 mice were scored by IHC. IF quantifications of Sma content were performed on Sma/TUNEL costained material (see Histological Analyses subsection for protocol), excepting the 24 w HFD baseline, which was performed on Vim/Sma costained material (see above paragraph for method).
Immunofluorescent staining of aortic inner curvature lesions of Myh11-CreERT2 CAG-LSL-tdTomato-WPRE Ldlr−/− males for Sma and tdTomato was as follows. After euthanasia, 33d HFD-fed aortas were flushed twice with PBS, fixed in ice-cold 4% PFA/PBS for 2 h, and infiltrated for 12 h in 30% sucrose. Next, aortas were pinned en face in 4% PFA at RT for 6 h to flatten, trimmed, and processed for paraffin embedding. 5 μm sections of paraffin-embedded aortic arch were prepared at 200 μm intervals throughout the length of the arch and screened by routine H-E to identify plaque-bearing positions. Sections were deparaffinized by two 25-min incubations in 50 °C xylene before standard rehydration to 70% EtOH, 5 min incubation in 0.85% NaCl/dH2O, and transfer PBS. Sections were then pre-fixed in 4% PFA for 7.5 min; antigen retrieved via Proteinase K (10 μg/ml) for 10 min; post-fixed in 4% PFA for 5 min; and, blocked with 2% NGS for 30 min. Sma and tdTomato in these sections were then labelled with Sma (Abcam, mouse, 1:150; cat. no. ab7817) and RFP (Rockland, rabbit, cat. no. 600-401-379; 1:200) overnight at 4C before secondary staining. Samples were Hoechst stained, washed, and mounted in Vectashield with 1:100 Hoechst to highlight both nuclei and retain Hoechst staining of elastic fibers.
For triple-staining of Sma, tdTomato, and Vim, 14-week HFD fed/9-week LFD-fed aortas aortas were flushed twice with PBS, fixed in ice-cold 4% PFA/PBS for 2 h, and infiltrated for 12 h in 30% sucrose. Next, aortas were pinned en face in 4% PFA at RT for 6 h to flatten, trimmed, and processed for paraffin embedding. 5 μm sections of paraffin-embedded aortic arch were prepared at 200 μm intervals throughout the length of the arch. Sections were then pre-fixed in 4% PFA for 7.5 min; antigen retrieved via Proteinase K (10 μg/ml) for 10 min; post-fixed in 4% PFA for 5 min; blocked with mouse-on-mouse blocking reagent (Vector Labs; cat. no. MKB-2213) for 30 min; blocked with 2% NGS for 30 min. Sma and Vim in these sections were then labelled with Sma (Abcam, mouse, 1:150; cat. no. ab7817) and Vim (Abcam, rabbit, 1:150; cat. no. ab92547) overnight at 4C, followed by labeling with secondaries. Following 2-minute fix in 4% PFA, samples were incubated for 1.5 h at RT in fluorescein-conjugated anti-RFP antibody (Rockland, rabbit, cat. no. 600-402-379; 1:200) before Hoechst staining, washing, and mounting in Vectashield with 1:100 Hoechst to highlight both nuclei and retain Hoechst staining of elastic fibers. For double staining of RFP and Runx1, tissues were prepared as above and incubated overnight at 4 °C in anti-Runx1 (Abcam, rabbit, 1:150; cat. no. ab35962) followed the next day by anti-RFP detection as above. For triple staining of Sma, RFP, and Myh11, tissues were prepared as above and incubated overnight at 4 °C in anti-Sma (Abcam, mouse, 1:150; cat. no. ab7817) and anti-Myh11 (Proteintech, rabbit, 1:100; cat. no. 21404-1-AP), followed the next day by anti-RFP detection as above with fluorescein-conjugated anti-RFP (1:100).
Histological Analyses
For all comparisons of the effects of senolysis, measurements were performed by an observer blinded to treatment group.
Fibrous cap thickness measurements:
Cap thickness was measured on routine H-E-stained 5 μm-thick paraffin sections using 20x images and ImageJ software. The fibrous cap was operationally defined as largely uninterrupted strip of eosinophilic material with a higher density of nuclei than the plaque core. A single foam cell macrophage or small cholesterol crystal cleft was not considered to be a disruption and were included in measurements. Per mouse, between 1 and 4 sections were measured. Each plaque-bearing section thus measured was separated from others from the same mouse (if applicable) by 200 μm. Multiple sections were averaged to produce per-mouse fibrous cap thickness measurements where applicable. For Extended Data Fig 8i, images completely covering lesion area were obtained on up to 5kX magnification by transmission electron microscopy for 1–3 positions separated by 200 μm and scored for cap thickness. By TEM, fibrous cap is defined as a region of cells with VSMC morphology interrupted by no more than a single cell with foam cell macrophage morphology. At least 10 length measurements were taken per section for both H-E and TEM quantifications.
Quantification of cells traversing the innermost elastic lamina:
Briefly, images of all elastic fiber breaks per section for at least two H-E-stained plaque sections per mouse were measured for the length between elastic fiber free ends and used to calculate the average length of disrupted innermost elastic fiber per break. Total nuclei intersecting a straight line joining each pair of free fiber ends were then counted. The number of medial cell nuclei crossing the first fiber was then normalized to total gap length. Sections with no breaks and no cells crossing the first elastic lamina are considered as “0 cells/μm of break length”, but are not considered in the average break length calculation. For TEM-based measurements, total fiber crossing cells per plaque-bearing section were assessed by inspecting the first elastic fiber in 1 and 4 sections per mouse.
Verhoeff-Van Gieson-positive fine elastin fibers:
Density of elastin fine fibers was measured on 5 μm-thick paraffin sections stained with Verhoeff-Van Gieson per the manufacturer’s protocol (Polyscientific R&D, cat. No. k059). Using a 100x oil immersion lens, all Verhoeff-Van Gieson fine fibers of any length were manually counted across the entire fibrous cap and then normalized to total length of the lumen-facing surface of the lesion. 1–3 sections separated by 200 μm (if applicable) were used to produce per-mouse elastin fine fiber density measurements.
Alizarin red staining:
Staining was performed on 5 μm thick paraffin sections. Briefly, slides were rehydrated through xylene-ethanol-water gradient, stained for 2 min in 2% alizarin red S/dH2O solution, cleared with acetone, and dehydrated in 1:1 mixture of acetone and xylene, before mounting in Cytoseal. Bright red-orange material was considered alizarin red-positive, and normalized to total neointimal area. Between 1 and 4 sections separated by 200 μm or more, when applicable, were scored and averaged to derive per-mouse alizarin-red positive area measurements.
TUNEL staining:
Paraffin sections of plaque were stained for TUNEL positivity according to the manufacturer’s instructions (DeadEnd fluorometric TUNEL kit, Promega, cat. no. G3250 for Extended Data Fig. 6e; In situ cell death detection kit, fluorescein, Roche, cat. no. 11684795910 for Extended Data Fig. 7e–g), followed by overnight staining for Sma at 4C (Abcam, mouse, 1:250; cat. no. ab7817) or Sma and RFP (Rockland, rabbit, 1:200; cat. no. 600-401-379). For Extended Data Fig. 4e, at least two sections per mouse separated by at least 200 μm or more were analyzed for frequency of Sma+/TUNEL+ cells as the percentage of total Sma+ cells throughout the lesion. For Extended Data Fig. 7e–g, at least 1 section per mouse was so analyzed. A cell was considered TUNEL+ if bright TUNEL signal overlapped completely with Hoechst nuclear counterstain.
EdU staining:
Paraffin sections of plaque were stained for EdU positivity according to the manufacturer’s instructions (Click-iT EdU cell proliferation kit, Thermo-Fischer, cat. no. C10337 Alexa-488 conjugated) followed by overnight staining with anti-Sma at 4C (Abcam, mouse, 1:150; cat. no. ab7817; Fig. 5b–g) or anti-Sma/anti-RFP (1:150 and 1:200, respectively; Extended Data Fig. 7b–d). One to two sections per mouse separated by at least 200 μm or more were analyzed for frequency of Sma+/EdU+ cells as the percentage of total Sma+ or Sma+/tdTom+ cells throughout the lesion.
Generation of Conditioned Media
MEFs were generated at embryonic day 13.5. Cells were expanded in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, L-glutamine, non-essential amino-acids, sodium pyruvate, gentamicin and β-mercaptoethanol, at 3% O2 and used for experiments at passage (P)5. To establish IR-senescent MEFs, subconfluent cells were exposed to 10 Gy γ-radiation (137Caesium source) and cultured for 10 days. Confluent cell layers of IR-SNCs and replication-competent (nonSNC) controls were washed twice with PBS before addition of 5 ml of DMEM/F12 (0.5% FBS) per T75. After 48 h of conditioning, medium was harvested, filtered through a 0.2 μm syringe filter, aliquoted and stored at −80 °C until use.
Neutralizing Antibody Preparation
For experiments in Fig. 8b and d and Extended Data Fig. 9b–d, polyclonal rabbit IgG control (Abcam, cat. no. AB171870) and polyclonal rabbit α-IGFBP3 Ab (Abcam, cat. no. AB76001) were stored at 4 °C until use. To avoid freeze/thaw cycling and remove stabilizing excipients such as azides, prior to application to cultured tissue or cells, an appropriate quantity of antibody was washed 5 times with a 5x volume of sterile PBS using 5 kD size-exclusion columns (Vivaspin 500, GE Healthcare, cat. no. 28-9322-23) in a 4 °C microcentrifuge (12,000 RCF, 15 min), resuspended in PBS to a final concentration of 1 μg/μl, and stored at 4 °C until use for no more than 24 h. For experiments in Fig. 8e, recombinant α-Igfbp3 Ab (Abcam, cat. no. AB224530; clone: EPR18680-153 without carrier) or polyclonal rabbit IgG control (Abcam, cat. no. AB171870) were purified as above on receipt, and stored in individual aliquots at −20 °C to avoid freeze-thaw effects. For experiments in Fig. 7d–f, polyclonal rabbit IgG control (Abcam, cat. no. AB171870) and recombinant α-Igfbp3 Ab (Abcam, cat. no. ab193910; clone: EPR18680–153 with carrier) were purified as above on receipt, and stored in individual aliquots at −20 °C to avoid freeze-thaw effects.
Human VSMC Migration Assays
Primary, normal human aortic VSMCs were obtained from the American Type Culture Collection (cat. no. ATCC PCS-100-012; female donor) and expanded for 4 passages in vascular cell basal media (cat. no. ATCC PCS-100-030) supplemented with growth factors (vascular smooth muscle cell growth kit; cat. no. ATCC PCS-100-042). All experiments in Extended Data. Fig. 9 were done on VSMCs between P4 and P10.
For IF co-staining of Sma and Vim, 1.8 × 103 VSMC were seeded per well on glass chamber slides in VSMC basal media and allowed to adhere for 12 h. Culture medium was then exchanged for 20 h with 200 μL (50% fresh VSMC basal media and 50% one of the following: IR-SNC CM with polyclonal rabbit IgG control (Abcam, cat. no. AB171870), nonSNC CM with polyclonal rabbit IgG control, IR-SNC CM with rabbit α-Igfbp3 Ab (Abcam, cat. no. AB76001) or IR-SNC CM with recombinant LR3 IGF-1). Wells without LR3 IGF1 included volume-matched LR3 vehicle. End concentration of all antibodies was 4 μg/ml; end concentration of LR3 IGF-1 was 1.2 μg/ml. Cells were fixed for 12 minutes in 4% PFA/PBS, permeabilized in 0.5% Triton X-100/PBS for 5 minutes at room temperature, and stained for 30 minutes at room temperature for Sma and Vim (1:300). Using ImageJ, 25–50 cells per line of CM were scored for integrated signal density of Sma and Vim to generate a per-line average signal intensity. These intensity measurements were normalized to Div+IgG/LR3 Veh signal from 1–3 concurrently imaged lines
For scratch wound assays of VSMC migration, 40 × 103 VSMC (ED Fig. 9c; 24 well tissue culture plate) were seeded per well in fresh VSMC basal media and grown to confluence. A P20 pipette tip was used to make a single linear scratch across the well. Culture medium was immediately changed to fresh VSMC basal media, to which was added an equal volume of either IR-SNC CM with polyclonal rabbit IgG control (Abcam, cat. no. AB171870), nonSNC CM with polyclonal rabbit IgG control, IR-SNC CM with rabbit α-Igfbp3 Ab (Abcam, cat. no. AB76001), or IR-SNC CM with recombinant LR3 IGF-1. Wells without LR3 IGF1 included volume-matched LR3 vehicle. At least three random 20x fields per scratch wound were analyzed at 4 hr post-scratch for VSMC immigrating into the wound space, for a total of 6 scratch wounds per condition analyzed. Each wound was treated with CM derived from a distinct T75 of MEFs cultured per the “Generation of Conditioned Media” methods subsection. End concentration of all antibodies was 4 μg/ml; end concentration of LR3 IGF-1 was 1.2 μg/ml. Experiments in Fig.7e–f we performed as above, except 17 × 103 VSMC were seeded in 48 well tissue culture plate and grown to confluence; ab193910 (Abcam; rabbit recombinant monoclonal) was used as the IGFBP3 neutralizing antibody; nonSNC CM containing ab193910 was used as an additional control; VSMC from a distinct donor (American Type Culture Collection; cat. no. ATCC PCS-100-012; female donor) were used between passage 3–4; and 6 and 5 scratch wounds per treatment group, respectively, were analyzed for Fig. 7e and f.
Murine Aortic Ring Cultures
For aortic ring explant experiments in Extended Data Fig. 9b, thoracic aortas from 6-week-old C57BL/6 female mice were briefly perfused with ice-cold PBS and then excised. ~1 mm thick rings were collected and transferred to PBS on wet ice. A 48-well plate was then prepared with wells containing a total volume of 300 μl media (50% fresh DMEM/F12 (20% FBS) and 50% conditioned media (0.5% FBS), supplemented to obtain 20% FBS concentration during the assay, and either 5 μg/mL rabbit IgG (Abcam, cat. no. AB171870) or 5 μg/ml α-Igfbp3 (Abcam, cat. no. AB76001). Conditioned media in this experiment was an equal-volume mixture of CM from three T75 flasks (distinct MEF lines) cultured per the “Generation of Conditioned Media” methods subsection. Aortic rings were transferred to this plate, and left to incubate undisturbed at 37 °C in 5% CO2 incubator. At 72 h, the number of outgrowing VSMCs beyond each aortic ring’s circumference was manually counted at 20x magnification. For aortic ring explant experiments in Fig. 7d, the method was as above, except ab193910 (Abcam; rabbit recombinant monoclonal) was used as the IGFBP3 neutralizing antibody; nonSNC CM containing ab193910 was included as an additional control; and, thoracic aortas from 6–10-week-old C57BL/6 female mice were used.
Murine Plaque Explant Igfbp3-neutralization and Vim/Sma Colabeling
Female Ldlr−/− mice were fed HFD for 12 weeks, then switched to LFD for 2 weeks to normalize circulating lipid profile. After CO2 euthanasia, aortas were flushed 2x with ice-cold PBS, the aortic arch removed, and ~1 mm thick cross-sectional rings of plaque-bearing aortic arch explanted to 125 μl DMEM/F12 with 0.5% FBS. Either 8 μg/ml of rabbit α-Igfbp3 (Abcam, cat. no. AB76001) or polyclonal rabbit IgG control (Abcam, cat. no. AB171870) antibody was added per well and incubated in a humidified chamber at 37 °C/5% CO2 for 18 h. Rings were then fixed at RT in 10% neutral buffered formalin for 12 h and processed for paraffin embedding. One 5 μm thick paraffin section per ring was then stained for 12 h at 4 °C with 1:150 rabbit α-Vim and mouse α-Sma, imaged completely at 20x magnification, and the proportion of IFS1 cells with the indicated marker profile determined manually by an operator blinded to sample identity.
Human Plaque Explant IGFBP3-neutralization and Vim/Sma Colabeling
All studies involving human subjects were approved by the Institutional Review Board at Mayo Clinic (protocol #2019-010502). Patients identified as eligible for therapeutic endarterectomy by the Department of Vascular and Endovascular Surgery (9 male, 3 female) provided written informed consent for research use of discarded endarterectomy tissue from either femoral or aortoiliac sites. Tissue was collected into sterile saline and stored at 4 °C for up to 1 h until processing. Serial ~2–3 mm slices were prepared from the explanted material and transferred individually into 450 μl prewarmed DMEM/F12 with 0.5% FBS containing either, for Fig. 8d, 8 μg/ml of rabbit α-Igfbp3 Ab (Abcam, cat. no. AB76001) or 8 μg/ml polyclonal rabbit IgG control (Abcam, cat. no. AB171870); or, for Fig. 8e, 8 μg/ml rabbit recombinant α-Igfbp3 Ab (Abcam, cat. no. AB224530) or 8 μg/ml polyclonal rabbit IgG control (Abcam, cat. no. AB171870). Samples were incubated in a humidified chamber at 37 °C/5% CO2 for 48 h and fixed at 4 °C for 12 h in 4% PFA/PBS (Fig. 8d) or 4 °C for 2 h in 4% PFA/PBS before overnight infiltration in 30% sucrose/PBS at 4 °C (Fig. 8e). Samples were then decalcified by gentle inversion in 15 ml of 10% HCl/10% acetic acid/80% dH2O for 12 h at RT, post-fixed for 2 h at 4 °C in 4%PFA/PBS and processed for paraffin embedding. 5 μm thick paraffin sections were then stained overnight (Fig. 8d) or for 1.5 hrs at room temperature (Fig. 8e) with 1:150 rabbit α-Vim and mouse α-Sma, followed by secondary staining with 1:250 goat-α-rabbit Alexa-488 and goat-α-mouse Alexa-594 for 30 min at RT. Due to high blue-channel autofluorescence, costaining with Hoechst was uninformative. Therefore, colocalization of goat-α-rabbit Alexa-488 and goat-α-mouse Alexa-594 was determined as % co-positive pixels per 20x field using the colocalization function in CellSENS software. An operator blinded to sample identity identified at least three 20x fields containing Sma+ cells per section, which were then imaged for both Sma and Vim without considering Vim status. Both IgG- and α-Igfbp3-treated paired samples were stained and imaged in parallel. Between 1–3 explant slice pairs were analyzed per patient, to generate a per-patient % colocalization with IgG or α-Igfbp3 treatment.
RNA-seq Library Preparation, Sequencing and Bioinformatic Analyses
RNA extraction (RNeasy Micro kit, #74004, Qiagen) was performed according to the manufactures’ instructions. RNA quality and quantity were assessed using Agilent Bioanalyzer RNA 6000 Pico chips (#5067-1513, Agilent Technologies). About 100 ng high-quality RNA from microdissected plaques or 10 ng from microdissected individual inner curvature of aortic arch were subjected to library preparation using the TruSeq RNA Library Prep Kit v2 (#RS-122-2001, Illumina), according to manufacturer’s instructions. The concentration and size distribution of the completed libraries were confirmed using Agilent DNA 1,000 chips (#5067-1504, Agilent Technologies) and Qubit fluorometry (Qubit dsDNA HS, #Q32851, Invitrogen). Libraries were sequenced following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. Flow cells were sequenced as 100 × 2 paired end reads on an Illumina HiSeq 4000 using HiSeq 3000/4000 sequencing kit and HCS 3.3.20 collection software. Base-calling was performed using Illumina’s RTA 2.5.2 software. RNA sequencing was performed at the Mayo Clinic Center for Individualized Medicine Medical Genomics Facility (Mayo Clinic, Rochester, Minnesota). Fastq files of paired-end reads were aligned with Tophat 2.0.14 to the UCSC reference genome mm10 using Bowtie2 2.2.6 with default parameters. Gene level counts were obtained using FeatureCounts 1.4.6 from the SubRead package with gene models from corresponding UCSC annotation packages. Differential expression analysis was performed using R package DESeq2 1.10.1 after removing genes with average raw counts less than 10. Differential expression results are summarized in Supplementary Tables 1 and 2. GSEA analysis results of select pathway categories in aortic arch plaque and LFD-fed inner curvature are summarized in Supplementary Table 3. Genes with false discovery rate (FDR) < 0.05 irrespective of unshrunk log2 fold change as determined by DESeq2 were considered significantly upregulated or downregulated. Heatmaps were generated with Morpheus, Broad Institute (https://software.broadinstitute.org/morpheus) using lfcMLE values and negative Log10 (FDR) values.
RT-qPCR analysis
MEFs:
IR-senescent and non-senescent MEFs were lysed in RLT buffer supplemented with β-mercaptoethanol according to the RNA extraction protocol. RNA extraction (Qiagen, RNeasy Mini kit, cat. no. 74104), cDNA synthesis (Invitrogen, SuperScript III First-Strand Synthesis System, cat. no. 18080051), and reverse transcription quantitative PCR (RT-qPCR) analysis (Applied Biosystem, SYBR Green Real-Time PCR Master Mix, cat. no. 4309155) were performed according to manufacturer’s instructions. The on-column DNase digestion step was avoided during the RNA extraction procedure. Tbp was used as a reference gene. The following primers were used: Igfbp3 fwd 5’ GGAGCAGTACCCGCTGAG 3’; Igfbp3 rev 5’ CACTGATGTTTCCTGGAGCA 3’; Tbp fwd 5’ GGCCTCTCAGAAGCATCACTA 3’; Tbp rev 5’ GCCAAGCCCTGAGCATAA3’.
Brachiocephalic artery:
RNA was extracted from pulverized brachiocephalic arteries (aortic insertion through bifurcation) using a RNeasy Micro kit (Qiagen, cat. no. 74004). cDNA was synthesized from 1 ng RNA per reaction using a Superscript III 1st Strand cDNA synthesis kit (ThermoFisher, cat. no. 18080051). RT-qPCR was performed using SYBR Green PCR master mix (Applied Biosystems, cat. no. 4309155) according to the manufacturer’s protocol and normalized to GAPDH expression. Primers used were: Igfbp3 (fwd: 5’-TAAGAAGAAGCAGTGCCGCC-3’; rev: 5’ TTTCCCCTTGGTGTCGTAGC-3’); Igfbp2 (fwd: 5’-GGGTGCCAAACACCTCAG-3’; rev: 5’-AGGTTGTACCGGCCATGC-3’); Igfbp5 (fwd: 5’- ATACAACCCAGAACGCCAGCT; rev: 5’- ACCTGGGCTATGCACTTGATG-3’); Igfbp6 (fwd: 5’- CAACCCCGAGAGAACGAAGAG-3’; rev: 5’- TCTCCTTTGTAGTCTCCTCCG-3’); and, GAPDH (fwd: 5’-TGCACCACCAACTGCTTA-3’; rev: 5’-TGGATGCAGGGATGATGTTC-3’).
Statistical Analyses
All statistical analyses were performed using Graphpad Prism software, version 8.0. Ordinary one-way ANOVA with Holm-Sidak multiple test correction was used for all comparisons of multiple groups, excepting instances where the exact same sample, such as individual aliquots of MEF conditioned media, were assessed; here, RM one-way ANOVA with Holm-Sidak multiple test correction was used.
Extended Data
Extended Data Fig. 1: ABT263 depletes SNCs and reduces foam cell macrophage content in intermediate-stage lesions.
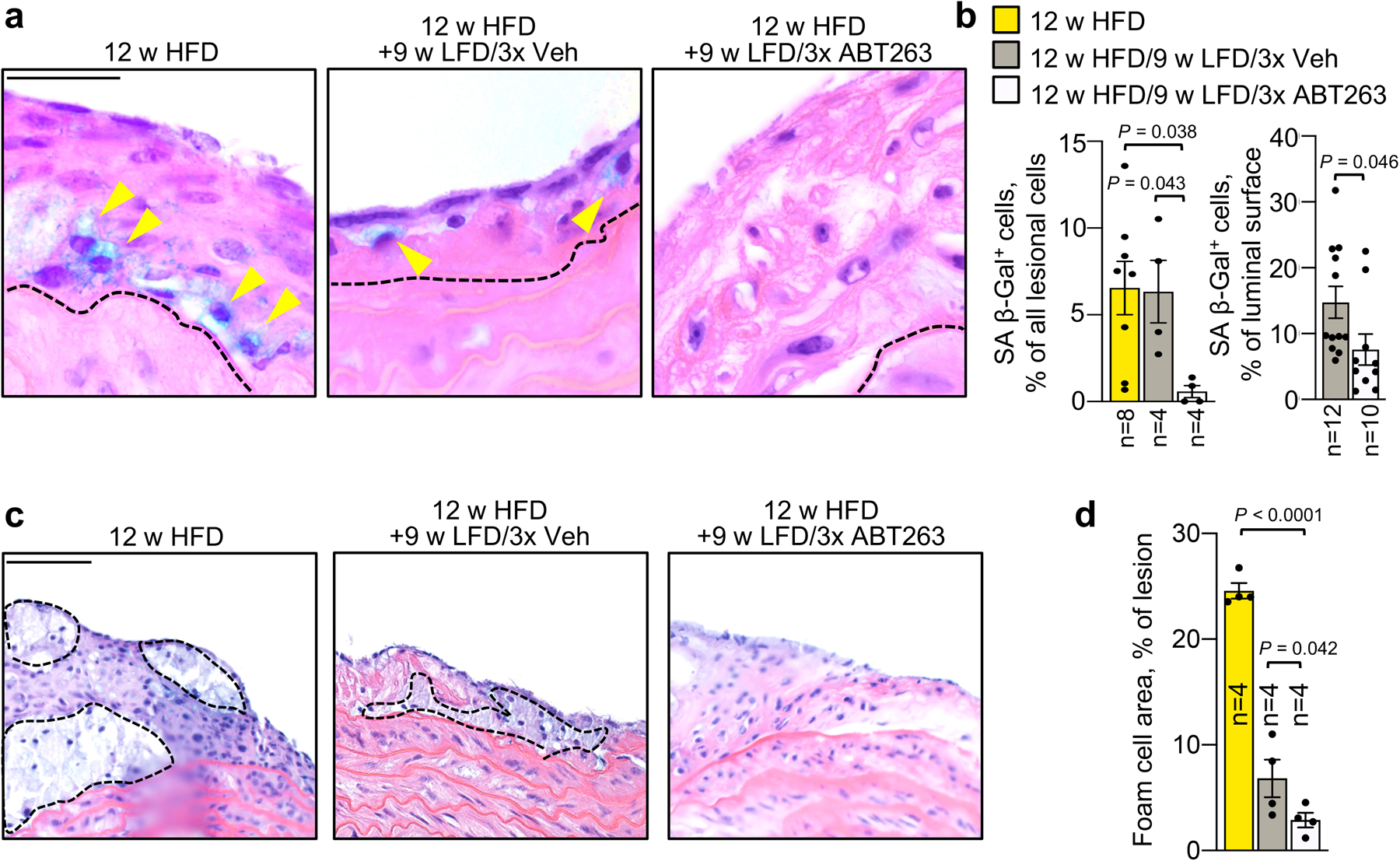
a, Representative SA β-Gal+ cells in H-E stained aortic arch plaques from indicated groups and quantified in b (left). b, (Left) Quantification of SA β-Gal+ cell counts in mice from indicated groups. (Right) En face quantification of SA β-Gal+ lesional surface area in mice from indicated groups. c, Representative foam cell macrophage pockets (black traced areas; quantified in d) in mice from 12-week HFD-fed Ldlr−/− mice and 12-week HFD-fed Ldlr−/− switched to LFD for 9 weeks with either 3x ABT263 or 3x Veh administration. d, Quantification of foam cell macrophage content per section (legend as in b). “n” refers in all panels to number of mice. Statistics in panel b (left) and d were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Panel b (right) was analyzed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. Scale bars are 50 μm (a) and 100 μm (c).
Extended Data Fig. 2: Senolysis blocks fibrous cap thinning in the brachiocephalic artery.
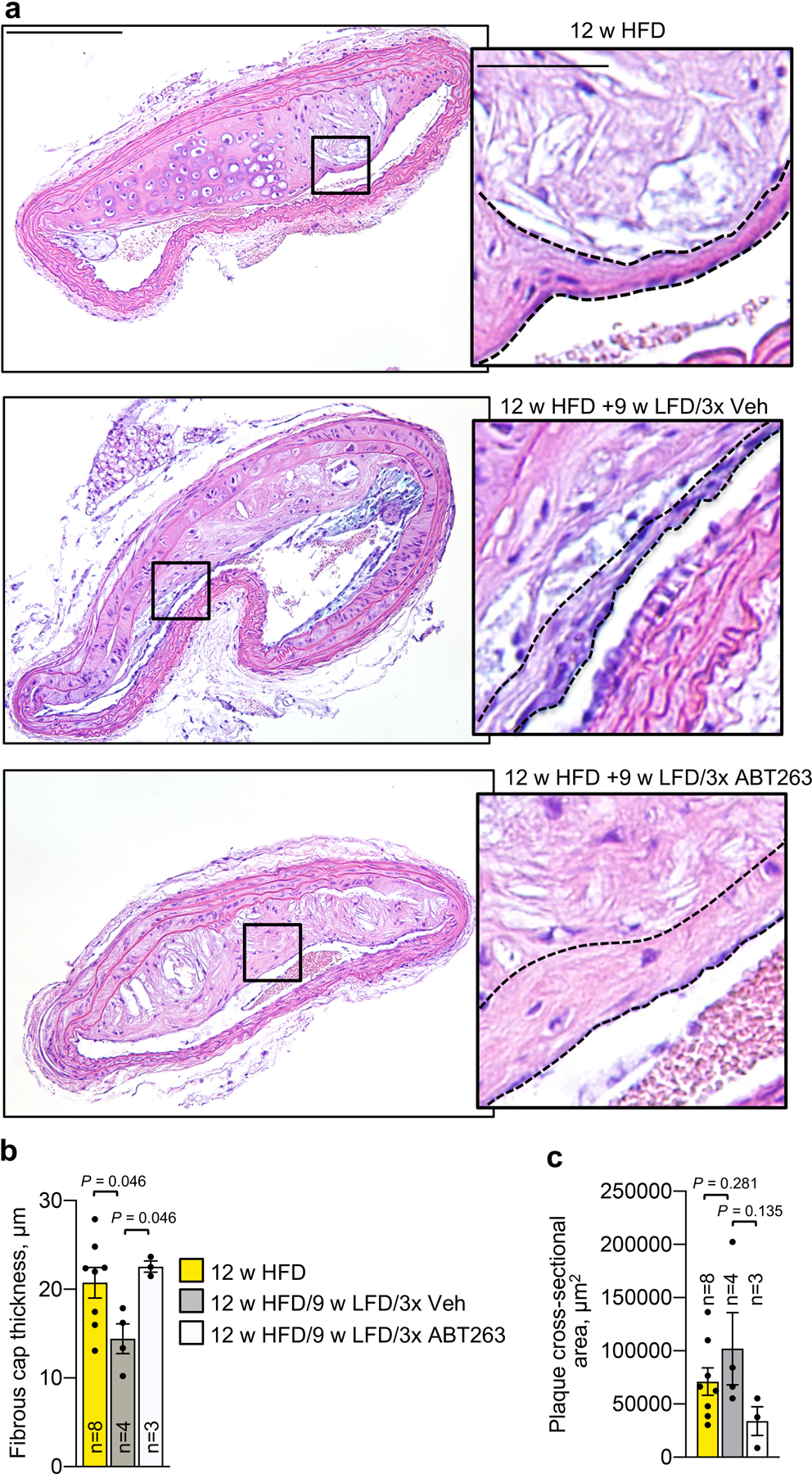
a, Representative H-E staining of brachiocephalic artery sections from 12-week HFD-fed Ldlr−/− mice and 12-week HFD-fed Ldlr−/− switched to LFD for 9 weeks with either administration of 3-cycles ABT263 or 3-cycles Veh (3x Veh); see Fig 1a for experimental design. Insets show a region of fibrous cap, outlined in black dashed lines and quantified in b. b, Quantification of fibrous cap thickness in brachiocephalic arteries from indicated groups. c, Quantification of average brachiocephalic plaque cross-sectional area per section in mice of indicated groups. “n” refers in all panels to number of mice. All statistics were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Error bars represent s.e.m. Scale bars for a are 400 μm (main) and 100 μm (inset).
Extended Data Fig. 3: ABT263 blocks fibrous cap thinning in aortic arch lesions by maintaining elastogenic VSMC numbers.
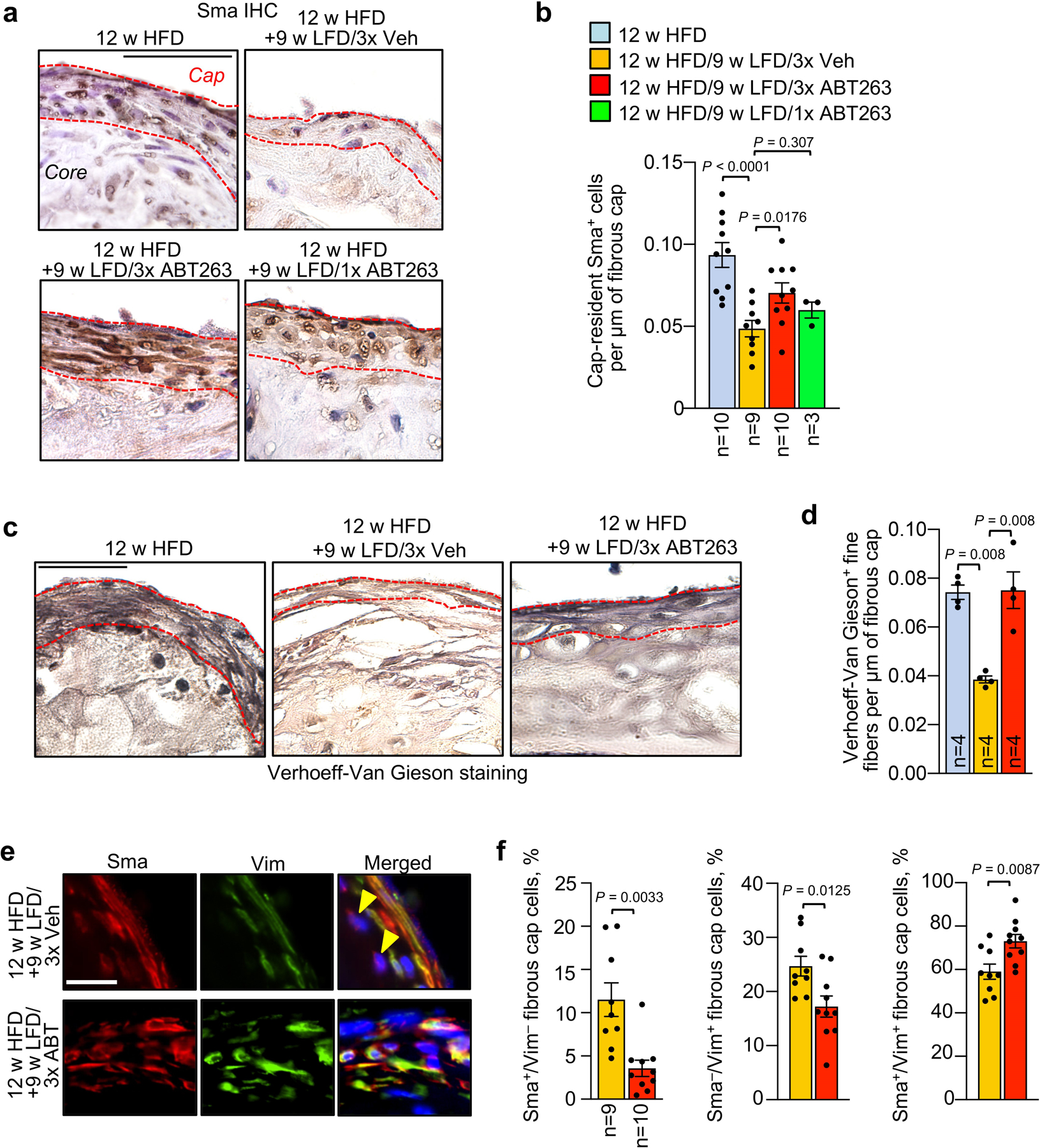
a, Immunohistochemical staining for Sma (quantified in b) in fibrous caps (dashed red lines) from 12-week HFD baseline mice, as well as those remodeling for 9 weeks on LFD with three 7-day cycles of ABT263 or Veh (3x ABT263 and 3x Veh; LFD weeks 1,4, and 7), or one 7-day cycle of ABT263 (1x ABT; LFD week 1). b, Quantification of Sma+ cells per μm of fibrous cap length from the indicated mice. c, Representative Verhoeff-Van Gieson staining of fibrous caps from the indicated groups and quantified in d. d, Quantification of Verhoeff-Van Gieson-positive fine fibers in fibrous caps of the indicated groups (legend as in b). e, Representative Vimentin and Sma costaining in fibrous caps of mice of the indicated groups (quantified in f). Yellow arrowheads indicated Vim−/Sma+ fibrous cap cells. f, Quantification of the percentage of Sma+/Vim− (left), Sma−/Vim+ (middle), and Sma+/Vim+ (right) cells in fibrous caps from indicated groups (legend as in b). Arrowheads: Sma+/Vim− fibrous cap cells. “n” refers in all panels to number of mice. Statistics in panels b and d were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Panel b includes data presented in Fig. 2f for context (12 w HFD baseline); here, ANOVA includes all indicated comparisons across Fig. 2f and Extended Data Fig. 3b. Panel f was analyzed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m.. Scale bar for a is 50 μm; c, 40 μm; and, e, 20 μm.
Extended Data Fig. 4: Transcriptome analysis indicates that ABT263 induces multiple favorable changes in plaque pathogenesis.
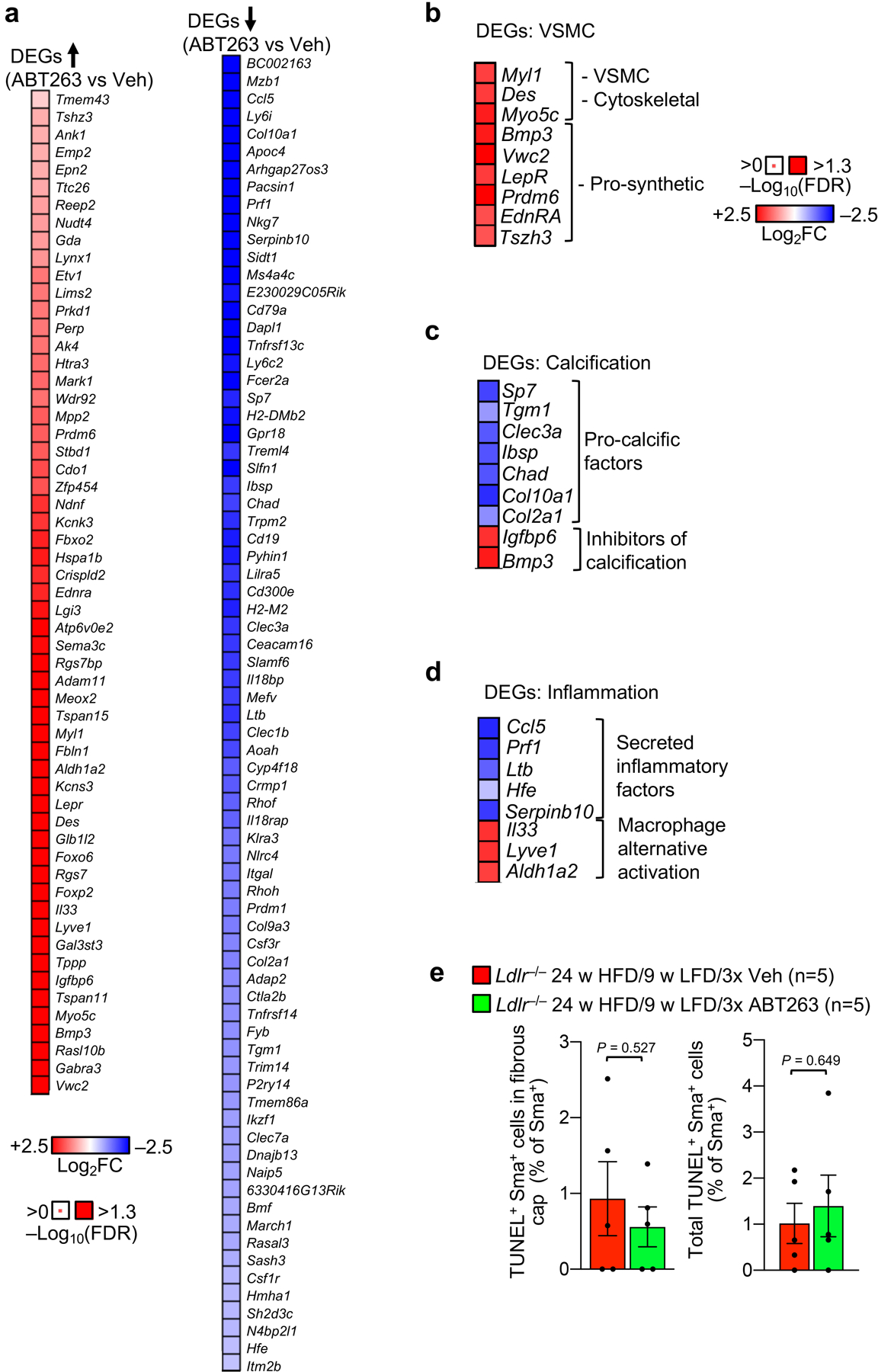
a, Heat map of differentially expressed genes upregulated (left) and downregulated (right) by ABT263 treatment in aortic arch plaque (see Fig. 3a for experimental schematic). b, Heat-map of pro-synthetic VSMC phenotype and muscle function genes upregulated with ABT263 treatment. c, Heat-map of anti-calcification changes in gene expression produced by ABT263 treatment among DEGs. d, Heat-map of pro-inflammatory and macrophage polarization DEGs modulated with ABT263 treatment in aortic arch plaque. e, Percentage of Sma+ cells showing TUNEL+ nuclei in fibrous caps (left) and total plaque (right) of indicated groups. Panel e (left and right) were assessed by unpaired, two-tailed Student’s t-test with Welch’s correction. Error bars represent s.e.m. “n” in all cases represents number of individual mice.
Extended Data Fig. 5: Contractile to promigratory phenotype switching of medial VSMCs is restricted to IFS1 underneath lesions.
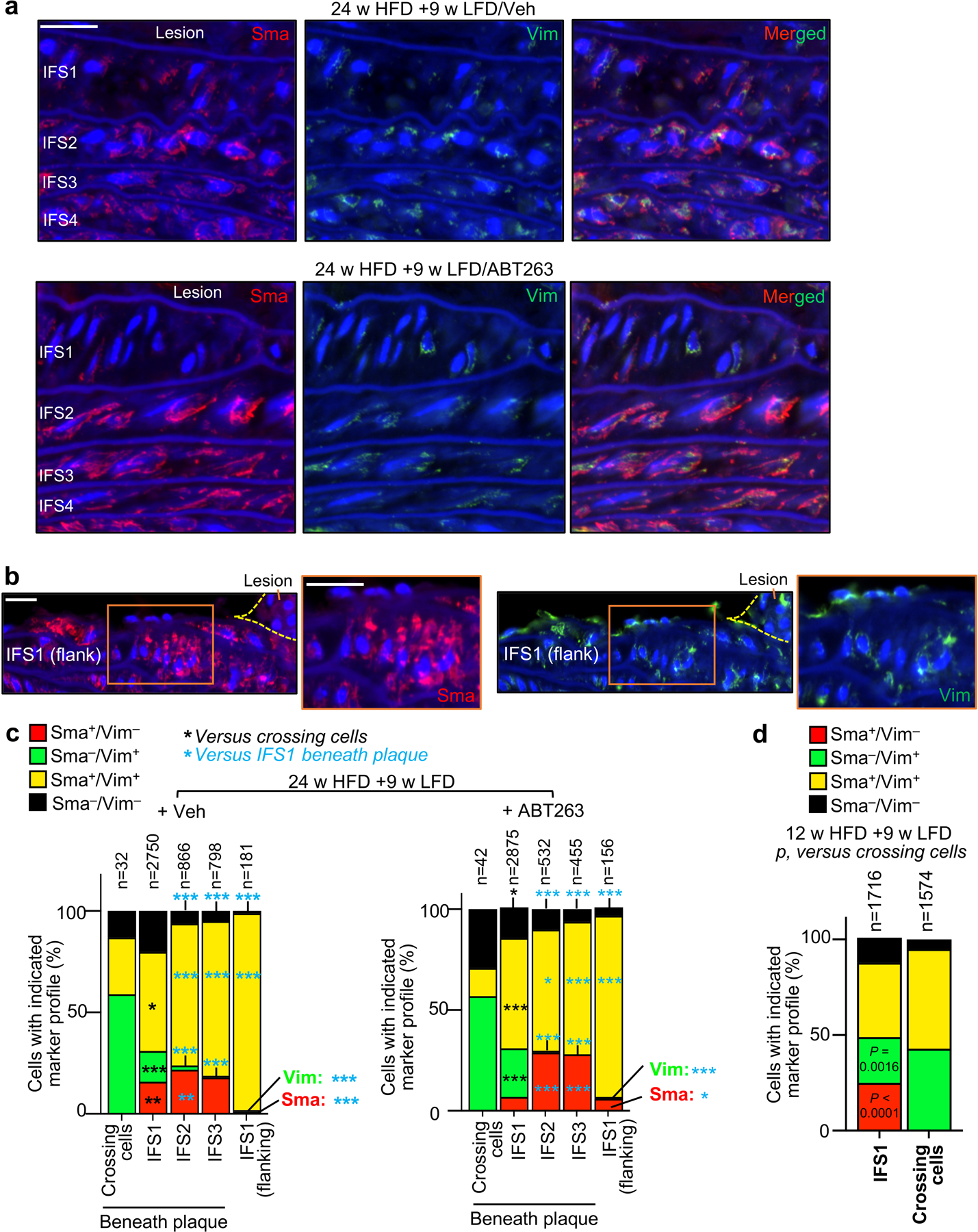
a, Representative images of IFS1–4 beneath plaque of the indicated groups stained for Sma and Vim (quantified in c). b, Representative images of plaque-flanking IFS1 VSMCs stained for Sma and Vim (quantified in c). c, VSMC expression profile of Vim and Sma in cells crossing the first elastic fiber, interfiber spaces (IFS) 1 to 3 beneath plaques, and IFS1 flanking plaques separated by treatment group (ABT or Vehicle administration during LFD feeding weeks 1, 4, and 7, following 24 weeks HFD feeding. *, p <0.05; **, p <0.01; ***, p <0.001. Exact p-values are indicated in the raw data source file. d, VSMC expression profile of Vim and Sma in cells crossing the first elastic fiber versus interfiber space (IFS) 1 aggregated across treatment group (ABT or Vehicle administration during LFD feeding weeks 1, 4, and 7, following 12 weeks HFD feeding). Panels in c and d were analyzed by global x2, followed up by individual two-tailed Fischer’s exact tests. “n” represents individual cells analyzed per aortic compartment. Scale bars in a and b are 20 μm.
Extended Data Fig. 6: Fibrous cap repair is characterized by enhanced migration of medial VSMCs.
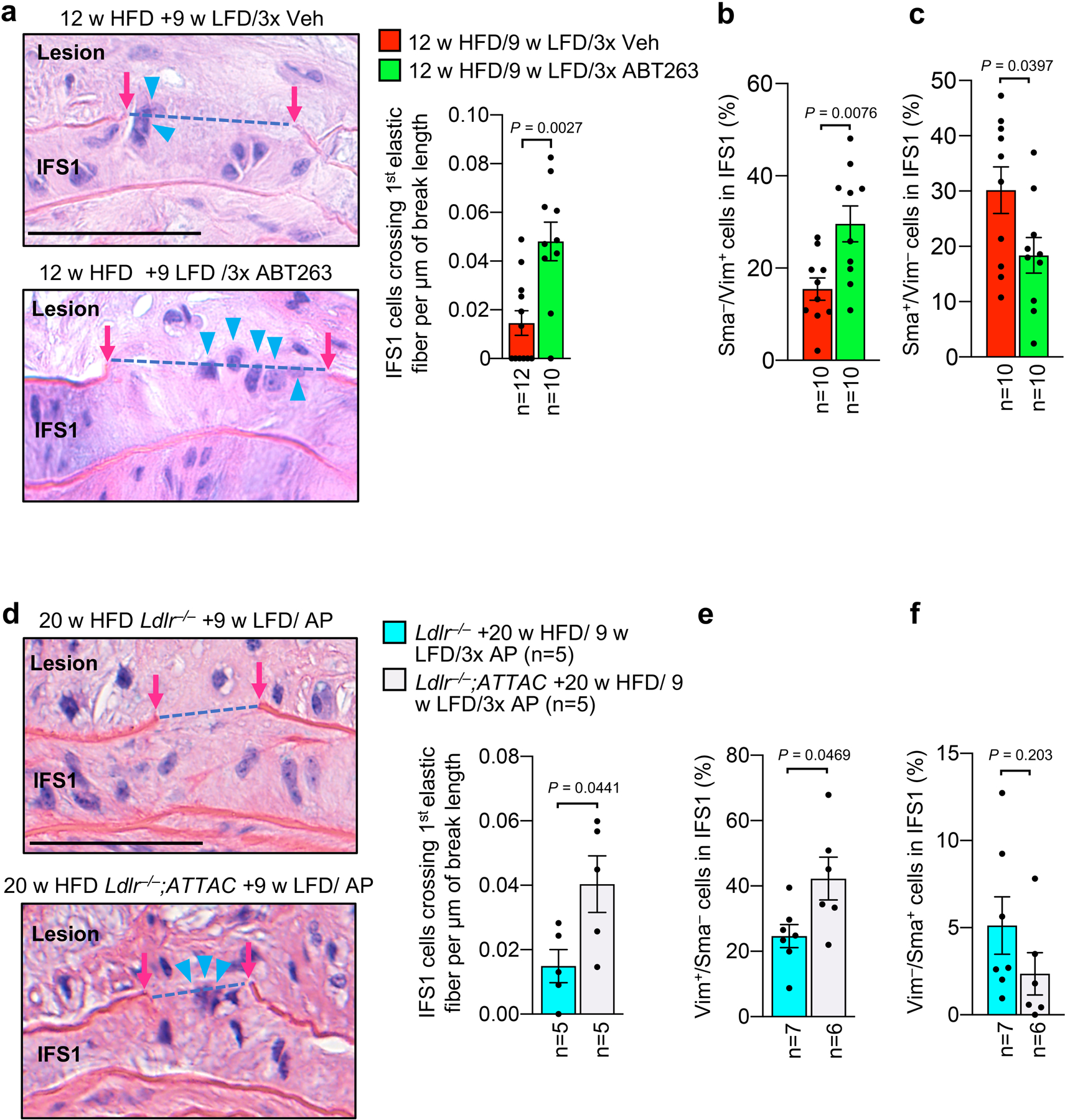
a, (Left) Representative H/E-stained examples of medial cells crossing the first elastic lamina (blue arrowheads) from indicated groups. (Right) Quantification of medial cells crossing the first elastic lamina normalized to elastic fiber break length. b, Quantification of Sma−/Vim+ cells in IFS1 of indicated groups (legend as in a). c, Quantification of Sma+/Vim− cells in IFS1 of indicated groups (legend as in a). d, (Left) Representative H/E-stained examples of medial cells crossing the first elastic lamina (blue arrowheads) from indicated groups. (Right) Quantification of medial cells crossing the first elastic lamina normalized to elastic fiber break length. e, Quantification of Sma−/Vim+ cells in IFS1 of indicated groups (legend as in d). f, Quantification of Sma+/Vim− cells in IFS1 of indicated groups (legend as in a). Statistics in all panels were performed by unpaired, two-tailed t-test with Welch’s correction. Error bars represent s.e.m. “n” in all cases refers to number of individual mice. Scale bars in a and d are 60 μm.
Extended Data Fig. 7: Impact on senolysis on proliferation, viability and osteogenic transdifferentiation of media-derived VSMCs in lesions.
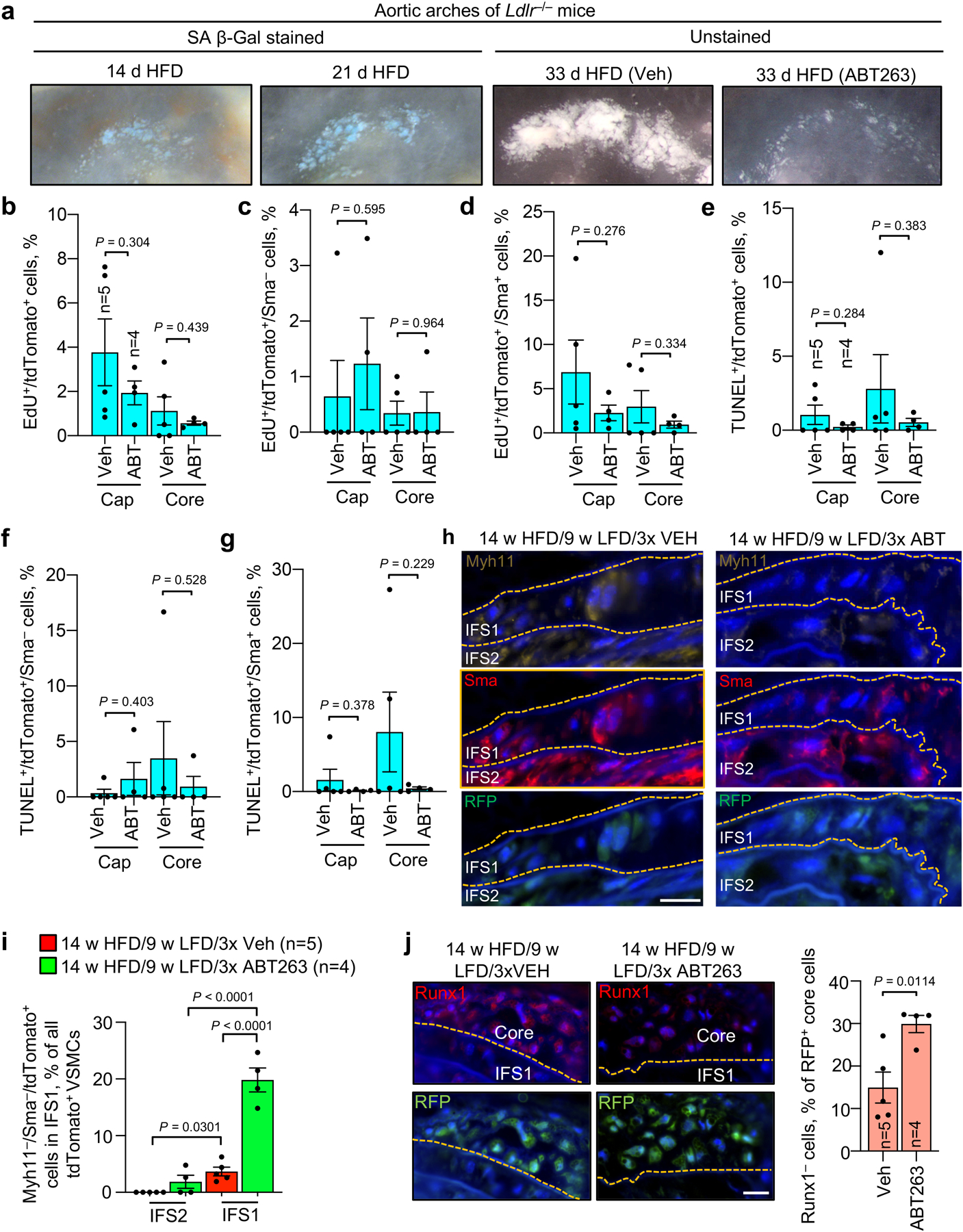
a, Images of early-stage aortic arch lesions after various lengths of HFD feeding (14 and 21 day images were from Ldlr−/− females and 33 day images from Myh11-CreERT2;CAG-LSL-tdTomato-WPRE Ldlr−/− males). Note that 33-day streaks (n=7 mice +Veh/ n=8 mice +ABT) cover a broad spectrum of lesion sizes, including the small-sized foci developing in streaks of 14 (n=4 mice) and 21 day (n=4 mice) HFD-fed mice. b, Quantification of EdU+ percentage of tdTomato+ in fibrous caps and plaque cores for mice of the indicated groups. c, Percentage of Tom+/Sma− cells with EdU among plaque fibrous cap and core cells, with or without ABT263 treatment. d, Percentage of Tom+/Sma+ cells with EdU among plaque fibrous cap and core cells, with or without ABT263 treatment. e, Quantification of TUNEL+ percentage of tdTomato+ cells in fibrous caps and plaque cores for mice of indicated groups. f, Percentage of Tom+/Sma− cells with TUNEL among plaque fibrous cap and core cells, with or without ABT263 treatment. g, Percentage of Tom+/Sma+ cells with TUNEL among plaque fibrous cap and core cells, with or without ABT263 treatment. h, Representative images of Myh11−/Sma+ cells among all tdTomato+ IFS1 and IFS2 cells (quantified in i). i, Quantification of indicated groups from h. j, Representative images (left) and quantification (right) of Runx1 and tdTomato colocalization in mice of indicated groups. Orange dashed line indicates the first elastic fiber. Panels b-g and j (right) were analyzed by unpaired, two-tailed t-tests with Welch’s correction. Panel h was analyzed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. ‘n’ in all cases refers to number of mice. Error bars represent s.e.m. Scale bars are 0.5 mm in a and 20 μm in h and j.
Extended Data Fig. 8: ABT263 accelerates formation of a fibrous cap structure by stimulating migration of medial VSMC.
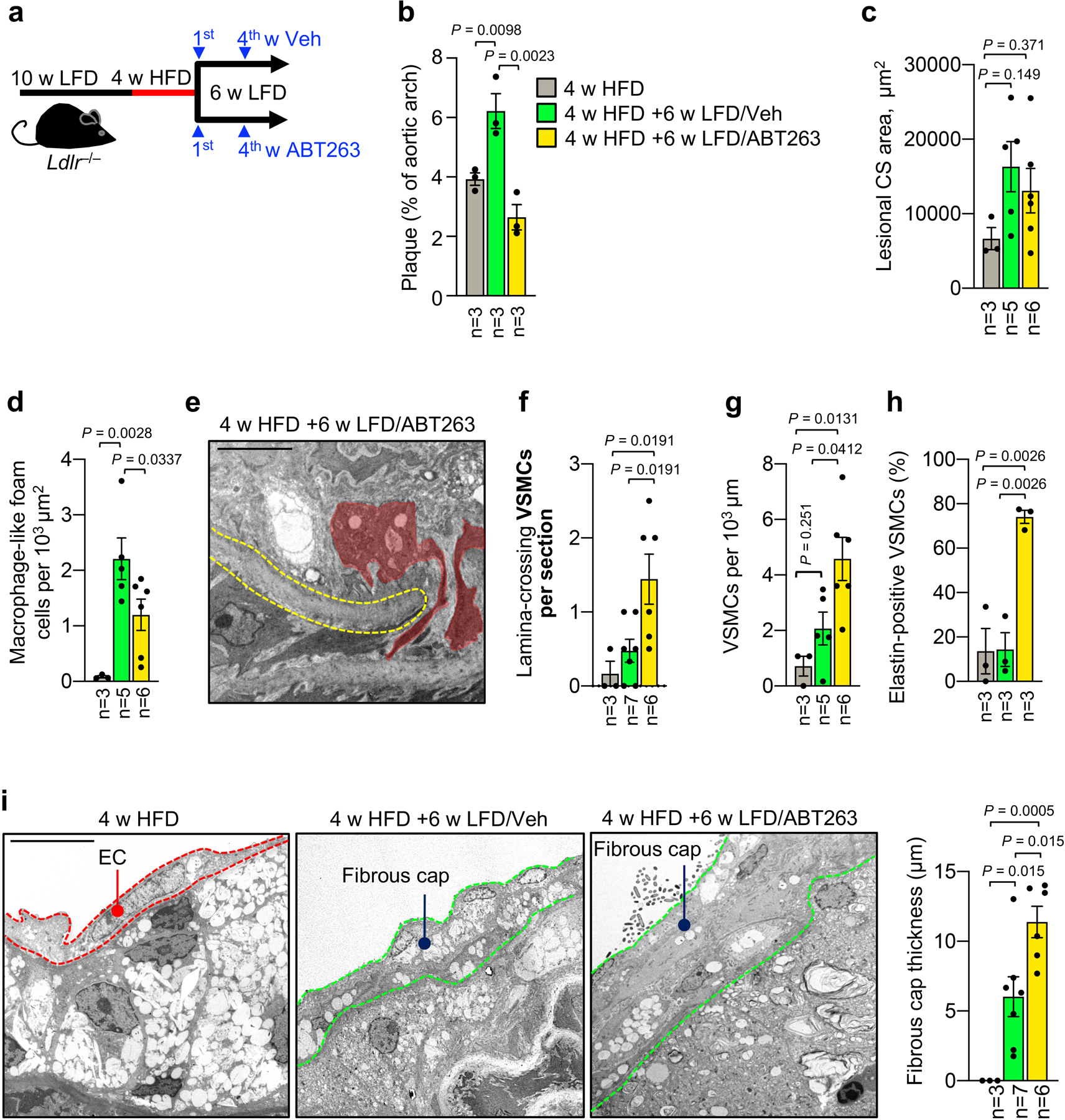
a, Schematic of experiments designed to study the impact of ABT263-mediated senolysis on fibrous cap formation in early inner aortic arch lesions of Ldlr−/− mice after HFD-to-LFD switching. b, Total plaque burden as measured by en face scoring in mice from indicated groups. c, Neointimal cross sectional area as measured by TEM in lesions of indicated mice (legend is as in b). d, Macrophages per neointimal area in mice of indicated groups (legend is as in b). e, Representative image illustrating VSMCs traversing the first elastic lamina (red masks) in a lesion of the indicated Ldlr−/− mouse (quantified in f). f, Quantification of VSMCs crossing the first elastic lamina in mice of indicated groups (legend is as in b). g, VSMCs per μm of neointima in mice of indicated groups (legend is as in b). h, Percentage of elastin-producing VSMCs in mice from indicated groups (legend is as in b). i, (Left) Representative electron micrographs of 4-week HFD simple foam cell lesions and lesions remodeling during 6-week LFD feeding with Veh or ABT263 administration (quantified in right panel). EC, Endothelial cell. (Right) Quantification of fibrous cap thickness (legend is as in b). “n” refers in all panels to number of mice. All analyses were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Error bars represent s.e.m. Scale bars in e and i are 5 μm.
Extended Data Fig. 9: Senescent cell-derived IGFBP3 inhibits IGF1-mediated promigratory phenotype switching of VSMCs.
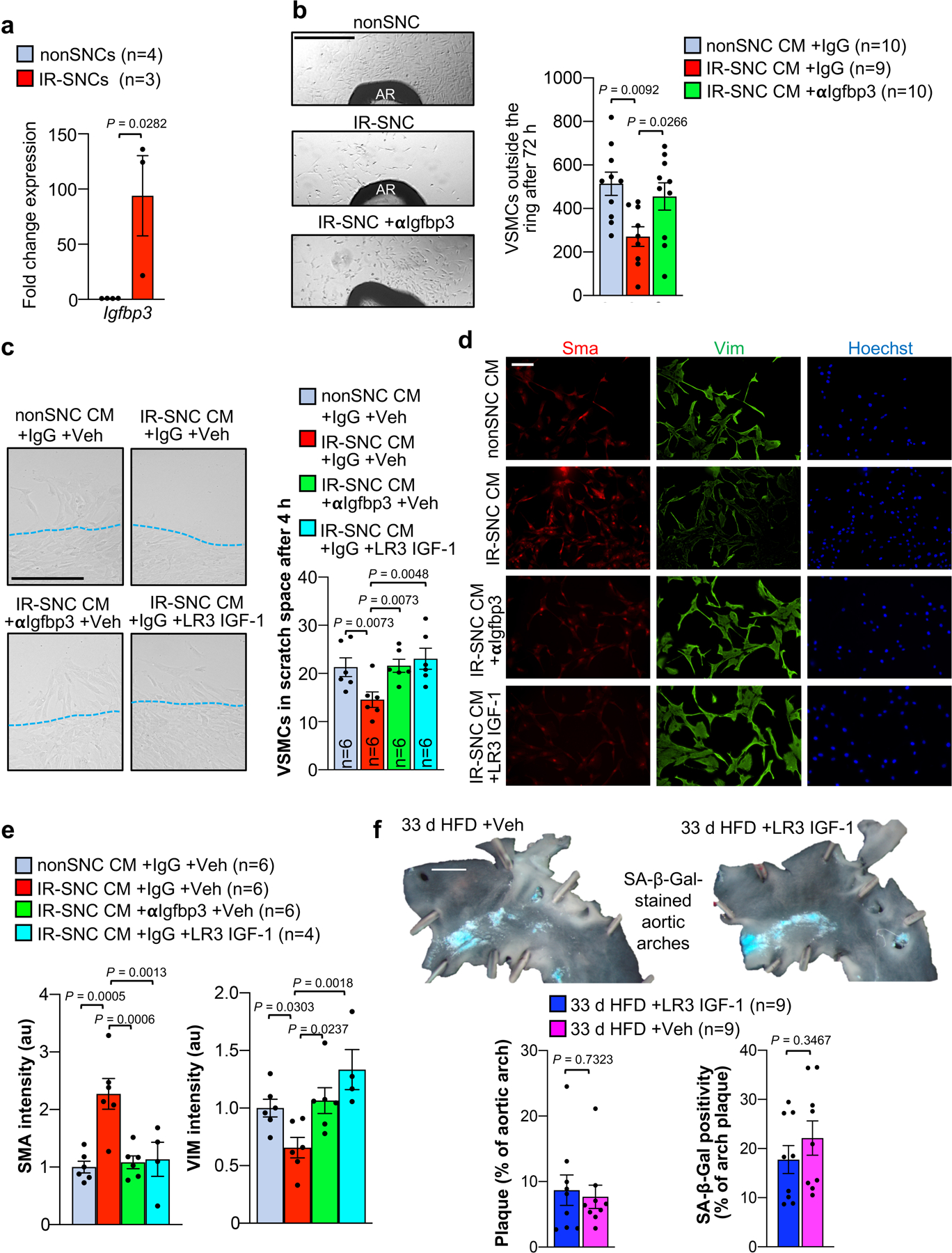
a, Expression of Igfbp3 in the indicated MEFs. b, (Left) Representative images of VSMC outgrowing from aortic rings (AR) of wildtype C57BL/6 mice treated with the indicated conditioned media (CM). (Right) Quantification of outgrowing VSMCs in the indicated treatment groups. c, (Left) Representative images of human aortic VSMCs emigrating into scratch wound space with indicated conditioned media (CM). Red dashed lines indicate cell monolayer/scratch wound boundary. (Right) Quantification of emigrating VSMCs in the indicated experimental groups. LR3 IGF-1, long R3 mutant stabilized recombinant IGF-1. d, Immunofluorescent staining of Vim and Sma in human aortic VSMC with the indicated treatments. CM, conditioned media (quantified in e). e, Quantification of average fluorescent signal intensity of SMA and VIM per cell in human aortic VSMC receiving indicated treatments. f, (Top) Representative images of SA β-Gal stained Ldlr–/– aortic arches from mice fed HFD for 33 d with concurrent administration of either LR3 IGF1 or vehicle control. (Bottom left) Quantification of total plaque burden in the aortic arch and (bottom right) percentage of aortic arch plaque with SA β-Gal positivity in indicated treatment groups. “n” refers individual MEF lines (panel a); individual aortic rings in b; individual mice in panel f; and, individual lines of MEF conditioned media (panels c and e). Panel a was analysed with unpaired, two-tailed t-test. Panel f was analyzed by unpaired, two-tailed t-test with Welch’s correction. Analyses in panels b and e were performed by ordinary one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons, and analysis of panel c was performed by RM one-way ANOVA with Holm-Sidak multiple comparison correction for the indicated comparisons. Error bars represent s.e.m. Scale bar in b is 500 μm; c, 200 μm; d, 100 μm; and, f, 1 mm.
Extended Data Fig. 10: Proposed model for how SNCs suppress the cap repair functions of VSMCs in advanced atherosclerotic lesions.
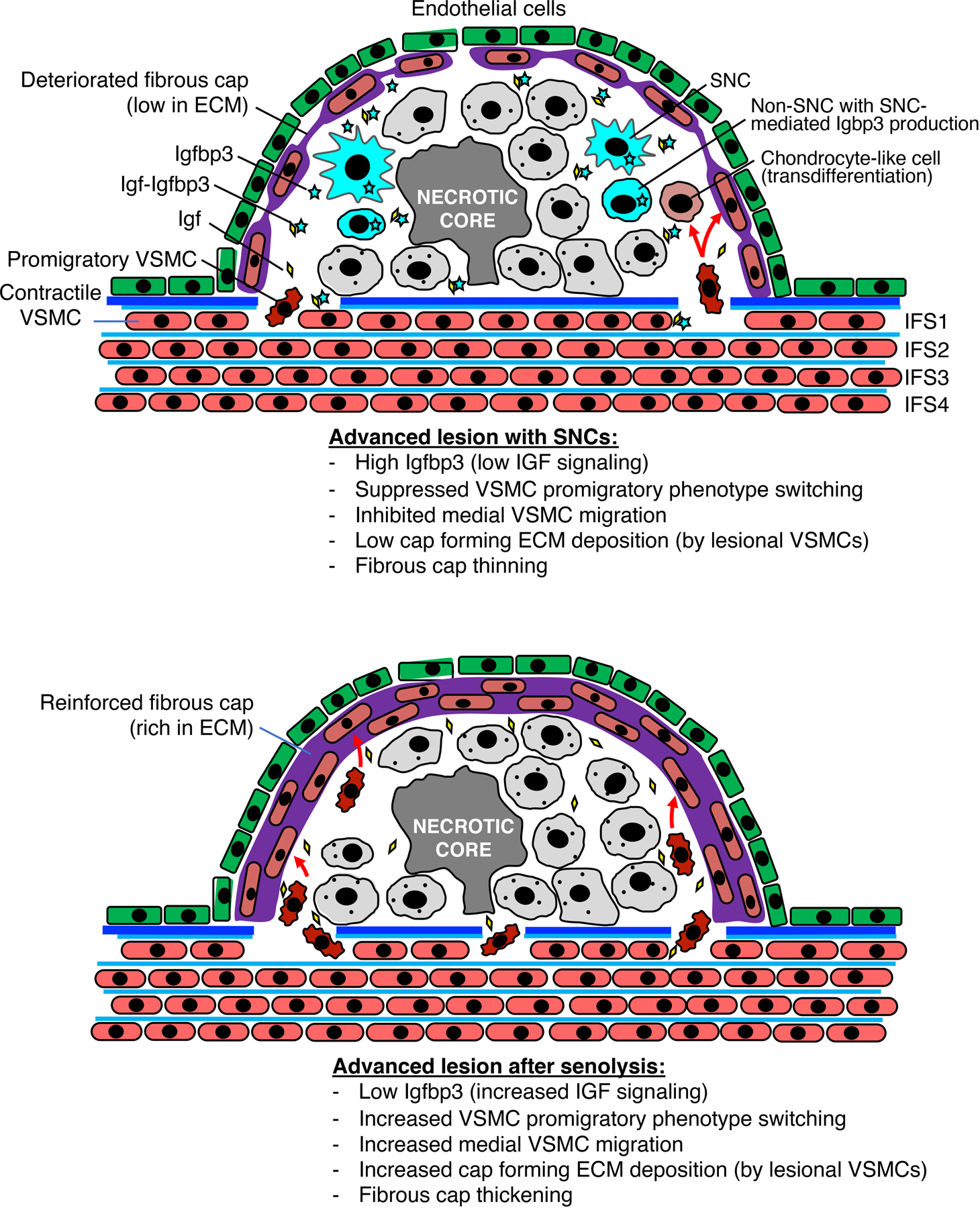
(Top) SNCs inhibit lesional IGF signaling by elevating Igfbp3 levels, thereby suppressing promigratory phenotype switching of contractile VSMCs in the media and their recruitment to the fibrous cap, as well as ECM deposition by VSMCs in the cap, resulting in fibrous cap erosion. (Bottom) SNC clearance reduces lesional Igfbp3 levels, thereby stimulating IGF-mediated promigratory phenotype switching of medial VSMC and their recruitment to the fibrous cap, as well as ECM deposition by VSMCs in the cap, restoring fibrous cap thickness and plaque stability.
Supplementary Material
Supplementary Table 1: Fold changes and adjusted p-values for all 132 differentially expressed genes resulting from 1-week ABT263 treatment of 12-week HFD aortic arch plaques.
Supplementary Table 2: Fold changes and adjusted p-values for all 31 differentially expressed genes resulting from 1-week ABT263 treatment of LFD-fed aortic arch inner curvature samples.
Supplementary Table 3: Normalized enrichment score and FDR q-values for 57 ABT263-modulated pathways related to VSMC function and survival as identified by GSEA as in inner curvature atherosclerotic lesions, with comparison to ABT-treated non-lesioned inner curvature.
Acknowledgments:
The authors would like to thank Dr. Cheryl Conover for helpful discussions.
This work was supported by the Glenn Foundation for Medical Research (J.M.v.D. D.J.B. and H.L.), the Keck Foundation (J.M.v.D.), National Institute of Aging grants AG57493 (J.M.v.D.) and AG049672 (B.G.C.), and the Mayo Clinic Kendall Fellowship (B.G.C).
Footnotes
Competing interests: J.M.v.D. is a cofounder of Unity Biotechnology. J.M.v.D., D.J.B. and B.G.C. are inventors on Mayo Clinic patents licensed to Unity Biotechnology. J.M.v.D., D.J.B. H.L., I.S., and B.G.C. are Unity Biotechnology shareholders. All other authors declare no competing interests.
Data Availability Statement
RNA-seq data have been deposited in the Gene Expression Omnibus under the accession number GSE130382 and GSE167003. All underlying data used for generation of figures are collated in the associated source files.
REFERENCES
- 1.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiche A, et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell stem cell 20, 407–414 e404 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Mosteiro L, Pantoja C, de Martino A & Serrano M Senescence promotes in vivo reprogramming through p16(INK)(4a) and IL-6. Aging Cell 17(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeon OH, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bussian TJ, et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bittencourt MS & Cerci RJ Statin effects on atherosclerotic plaques: regression or healing? BMC medicine 13, 260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reith C & Armitage J Management of residual risk after statin therapy. Atherosclerosis 245, 161–170 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Kloppenburg GT, Grauls GE, Bruggeman CA & Stassen FR Adenoviral activin A expression prevents vein graft intimal hyperplasia in a rat model. Interactive cardiovascular and thoracic surgery 8, 31–34 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Engelse MA, et al. Adenoviral activin a expression prevents intimal hyperplasia in human and murine blood vessels by maintaining the contractile smooth muscle cell phenotype. Circulation research 90, 1128–1134 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Almehmadi A, et al. VWC2 Increases Bone Formation Through Inhibiting Activin Signaling. Calcified tissue international 103, 663–674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepore JJ, Cappola TP, Mericko PA, Morrisey EE & Parmacek MS GATA-6 regulates genes promoting synthetic functions in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology 25, 309–314 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Trovati M, et al. Leptin and vascular smooth muscle cells. Current pharmaceutical design 20, 625–634 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Huang K, et al. MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovascular research 113, 488–497 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Lagna G, et al. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. The Journal of biological chemistry 282, 37244–37255 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, et al. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. American journal of human genetics 98, 1082–1091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin E, et al. TSHZ3 and SOX9 regulate the timing of smooth muscle cell differentiation in the ureter by reducing myocardin activity. PloS one 8, e63721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goettsch C, et al. miR-125b regulates calcification of vascular smooth muscle cells. The American journal of pathology 179, 1594–1600 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Crombrugghe B, et al. Transcriptional mechanisms of chondrocyte differentiation. Matrix biology : journal of the International Society for Matrix Biology 19, 389–394 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Gu J, et al. Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell death & disease 5, e1469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong YC, et al. Silencing of osterix expression by siRNA inhibits aldosteroneinduced calcification of vascular smooth muscle cells in mice. Molecular medicine reports 14, 2111–2118 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Lau D, et al. The cartilage-specific lectin C-type lectin domain family 3 member A (CLEC3A) enhances tissue plasminogen activator-mediated plasminogen activation. The Journal of biological chemistry 293, 203–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beazley KE, et al. Transglutaminase inhibitors attenuate vascular calcification in a preclinical model. Arteriosclerosis, thrombosis, and vascular biology 33, 43–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessle L, et al. The skeletal phenotype of chondroadherin deficient mice. PloS one 8, e63080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu M, Chen G & Li YP TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone research 4, 16009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabbitas B & Canalis E Growth factor regulation of insulin-like growth factor binding protein-6 expression in osteoblasts. Journal of cellular biochemistry 66, 77–86 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Ding W, et al. miR-30e targets IGF2-regulated osteogenesis in bone marrow-derived mesenchymal stem cells, aortic smooth muscle cells, and ApoE−/− mice. Cardiovascular research 106, 131–142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henein M, et al. High dose and long-term statin therapy accelerate coronary artery calcification. International journal of cardiology 184, 581–586 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Nakazato R, et al. Statins use and coronary artery plaque composition: results from the International Multicenter CONFIRM Registry. Atherosclerosis 225, 148–153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braunersreuther V, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arteriosclerosis, thrombosis, and vascular biology 28, 1090–1096 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Hiebert PR, Boivin WA, Zhao H, McManus BM & Granville DJ Perforin and granzyme B have separate and distinct roles during atherosclerotic plaque development in apolipoprotein E knockout mice. PloS one 8, e78939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grandoch M, et al. Deficiency in lymphotoxin beta receptor protects from atherosclerosis in apoE-deficient mice. Circulation research 116, e57–68 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Wessling-Resnick M Iron homeostasis and the inflammatory response. Annual review of nutrition 30, 105–122 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mo Y, et al. Epithelial SERPINB10, a novel marker of airway eosinophilia in asthma, contributes to allergic airway inflammation. American journal of physiology. Lung cellular and molecular physiology 316, L245–L254 (2019). [DOI] [PubMed] [Google Scholar]
- 38.He R, et al. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Molecular immunology 90, 42–49 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Miller AM, et al. IL-33 reduces the development of atherosclerosis. The Journal of experimental medicine 205, 339–346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLaren JE, et al. IL-33 reduces macrophage foam cell formation. Journal of immunology 185, 1222–1229 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Gundra UM, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 123, e110–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho CH, et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circulation research 100, e47–57 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Tabas I & Lichtman AH Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 47, 621–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sage AP, Tsiantoulas D, Binder CJ & Mallat Z The role of B cells in atherosclerosis. Nature reviews. Cardiology 16, 180–196 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Tang CY, et al. Runx1 up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem J 477, 2421–2438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimura A, et al. Runx1 and Runx2 cooperate during sternal morphogenesis. Development 137, 1159–1167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von der Thusen JH, et al. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol 178, 924–934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng J & Du J Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arteriosclerosis, thrombosis, and vascular biology 27, 1744–1751 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Li K, Wang Y, Zhang A, Liu B & Jia L miR-379 Inhibits Cell Proliferation, Invasion, and Migration of Vascular Smooth Muscle Cells by Targeting Insulin-Like Factor-1. Yonsei Med J 58, 234–240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elzi DJ, et al. Plasminogen activator inhibitor 1--insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci U S A 109, 12052–12057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim KS, et al. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell 6, 535–545 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ozcan S, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY) 8, 1316–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vassilieva I, et al. Paracrine senescence of human endometrial mesenchymal stem cells: a role for the insulin-like growth factor binding protein 3. Aging (Albany NY) 12, 1987–2004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sukhanov S, et al. IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arteriosclerosis, thrombosis, and vascular biology 27, 2684–2690 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Shai SY, et al. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe−/− mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arteriosclerosis, thrombosis, and vascular biology 30, 1916–1924 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang C, et al. Regulation of vascular smooth muscle cell proliferation and migration by human sprouty 2. Arteriosclerosis, thrombosis, and vascular biology 25, 533–538 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Yang K & Proweller A Vascular smooth muscle Notch signals regulate endothelial cell sensitivity to angiogenic stimulation. The Journal of biological chemistry 286, 13741–13753 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicosia RF The aortic ring model of angiogenesis: a quarter century of search and discovery. Journal of cellular and molecular medicine 13, 4113–4136 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lebedeva A, et al. Ex vivo culture of human atherosclerotic plaques: A model to study immune cells in atherogenesis. Atherosclerosis 267, 90–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Higashi Y, Gautam S, Delafontaine P & Sukhanov S IGF-1 and cardiovascular disease. Growth Horm IGF Res 45, 6–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR & Mallat Z Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 16, 727–744 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Jacobsen K, et al. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Misra A, et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun 9, 2073 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chappell J, et al. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ Res 119, 1313–1323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pan H, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 142, 2060–2075 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alencar GF, et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 142, 2045–2059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shankman LS, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gomez D, Shankman LS, Nguyen AT & Owens GK Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods 10, 171–177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Fold changes and adjusted p-values for all 132 differentially expressed genes resulting from 1-week ABT263 treatment of 12-week HFD aortic arch plaques.
Supplementary Table 2: Fold changes and adjusted p-values for all 31 differentially expressed genes resulting from 1-week ABT263 treatment of LFD-fed aortic arch inner curvature samples.
Supplementary Table 3: Normalized enrichment score and FDR q-values for 57 ABT263-modulated pathways related to VSMC function and survival as identified by GSEA as in inner curvature atherosclerotic lesions, with comparison to ABT-treated non-lesioned inner curvature.
Data Availability Statement
RNA-seq data have been deposited in the Gene Expression Omnibus under the accession number GSE130382 and GSE167003. All underlying data used for generation of figures are collated in the associated source files.