

Abstract
In the anaerobic ergothioneine biosynthetic pathway, a rhodanese domain containing enzyme (EanB) activates tne hercynine’s sp2 ε-C-H Dona ana replaces it with a C-S bond to produce ergothioneine. The key intermediate for this trans-sulfuration reaction is the Cys412 persulfide. Substitution of the EanB-Cys412 persulfide with a Cys412 perselenide does not yield the selenium analog of ergothioneine, selenoneine. However, in deuterated buffer, the perselenide-modified EanB catalyzes the deuterium exchange between hercynine’s sp2 ε-C-H bond and D2O. Results from QM/MM calculations suggest that the reaction involves a carbene intermediate and that Tyr353 plays a key role. We hypothesize that modulating the pKa of Tyr353 will affect the deuterium-exchange rate. Indeed, the 3,5-difluoro tyrosine containing EanB catalyzes the deuterium exchange reaction with kex of ~10-fold greater than the wild-type EanB (EanBWT). With regards to potential mechanisms, these results support the involvement of a carbene intermediate in EanB-catalysis, rendering EanB as one of the few carbene-intermediate involving enzymatic systems.
Keywords: ergothioneine, selenonine, anaerobic biosynthesis, carbene intermediate, unnatural amino acid (uAA), QM/MM calculation
Graphical Abstract
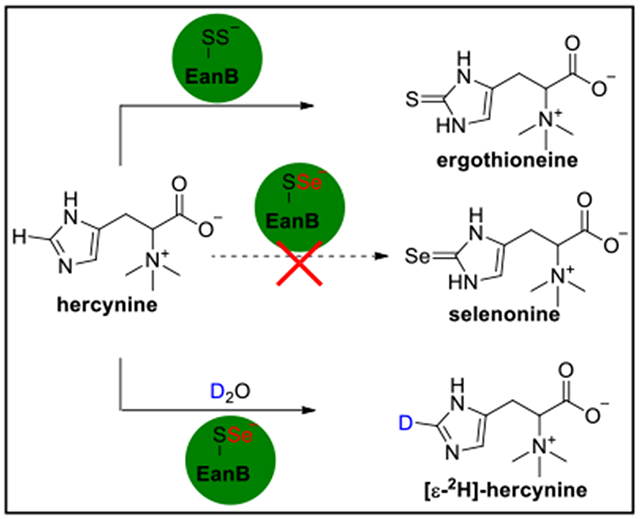
INTRODUCTION
Ergothioneine (5) is a thiol-histidine derivative produced in fungi and bacteria. Animals do not synthesize ergothioneine, however, human and animal tissues enrich ergothioneine to milimolar levels through an ergothioneine-specific transporter (OCTN1).1 As a result, ergothioneine and glutathione (GSH) are among the most abundant cellular thiols in animals2 and together, they maintain a proper redox environment under a wide range of conditions.2,3 Ergothioneine has been suggested to have health benefits for many human disorders, including rheumatoid arthritis, Crohn’s disease, diabetes, cardiovascular diseases, and neurodegeneration and touted as a longevity vitamin.3–13 Ergothioneine’s biosynthetic pathways are also of significant interest given that sulfur-containing natural products are widely distributed in nature, yet the biosynthetic details for the vast majority of them are poorly understood.14–18
Both aerobic and anaerobic ergothioneine biosynthetic pathways are reported.18–20 In the aerobic pathways,21–24 non-heme iron sulfoxide synthases catalyze the oxidative C-S bond formation reactions and there are four types of sulfoxide synthases. Type I enzymes use γ-glutamylcysteine (γ-Glu-Cys) and trimethylhistidine (hercynine) as the substrate (EgtB-catalysis, Scheme 1A).21 Type II Chloracidobacterium thermophilum EgtB accept either γ-Glu-Cys or Cys as the substrate (EgtBcth in Scheme 1B).25,26 Type III fungal Egt1s accept cysteine and hercynine as the substrates (Egt1-catalysis, Scheme 1A).22 Most type IV sulfoxide synthases, such as OvoA from Erwinia tasmaniensis found in the biosynthesis of ovothiol,27 accept cysteine as the substrate (Scheme 1C).28 Interestingly, OvoA accepts both histidine and hercynine as the substrate, but the product C-S bond regio-selectivity is different for histidine and hercynine (Scheme 1C).29 In the anaerobic ergothioneine biosynthetic pathway, a rhodanese (EanB)19 catalyzes the trans-sulfuration reaction using polysulfide as the direct substrate (Scheme 1D).20
Scheme 1. Different ergothioneine biosynthetic pathways discovered in recent years.
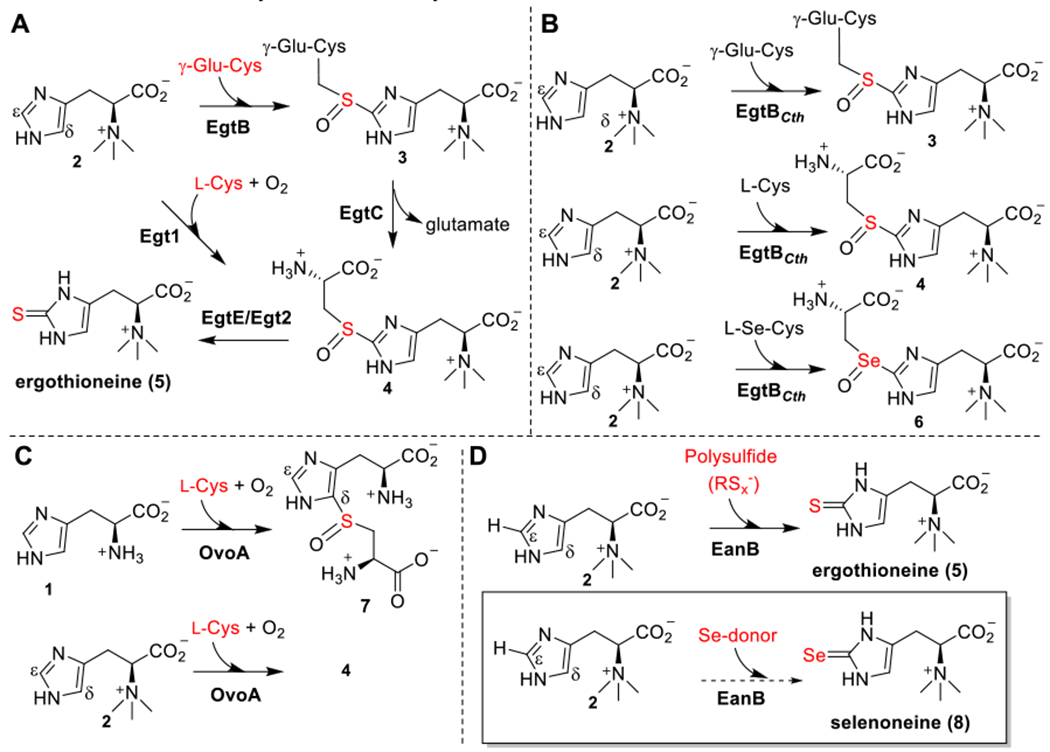
(A) Type I and type III sulfoxide synthases from ergothioneine biosynthesis (EgtB and Egt1 from aerobic pathways). (B) Type II sulfoxide synthases from ergothioneine biosynthesis, in which EgtBcth could accept Cys, γ-Glu-Cys or selenocysteine as the substrate. (C)Type IV sulfoxide synthase from ovothiol biosynthesis, OvoA, which could also catalyze the oxidative coupling between hercynine and Cys. (D) The C-S bond formation reaction from the anaerobic ergothioneine biosynthesis, in which a rhodanese (EanB) catalyzes the direct replacement of the C-H bond by a C-S bond using polysulfide as the substrate.
Selenoneine (8, Scheme 1D) is an analog of ergothioneine, in which the sulfur atom is replaced by selenium.30–32 Given the beneficial roles of ergothioneine in human health and the function of selenium as an essential micronutrient, there is a growing interest in synthesizing selenoneine and characterizing its biological functions. Selenoneine is proposed to play a role in methyl mercury detoxification.33–35 When supplemented with sodium selenate in the growth medium, fission yeast, Schizosaccharomyces pombe, produce selenoneine.36,37 Consistent with the fact that many enzymes in the biosynthesis of sulfur-related natural products could also produce their selenium analogs,38,39 recently, Seebeck and coworkers demonstrated that some sulfoxide synthases from the ergothioneine aerobic biosynthetic pathway use selenocysteine as the substrate (e.g., EgtBcth, Scheme 1B), while the activity is low.40 Herein, we investigate the anaerobic ergothioneine biosynthetic pathway (Scheme 1D) for selenoneine biosynthesis. Surprisingly, the Cys412 perselenide containing EanB does not produce selenoneine, while deuterium exchange occurs between hercynine’s sp2 ε-C-H bond and D2O when D2O buffer is used. QM/MM calculations predict the involvements of a carbene intermediate in EanB-catalysis and that Tyr353 plays a key role. Substitution of the Tyr353 with 3,5-difluoro tyrosine, via the amber suppressor mediated unnatural amino acid incorporation method, increases in reactivity supports the importance of Tyr353 in EanB-catalysis. When all of these results are considered in the contexts of a few proposed mechanistic models, they highly suggest the involvement of a carbene intermediate in EanB-catalysis.
RESULTS
Cysteine polysulfide produced by cystathionine-β-lyase as the sulfur source in EanB-catalysis.
In the anaerobic ergothioneine biosynthetic pathway, EanB catalyzes the hercynine to ergothioneine transformation, in which the unreactive ε-C-H bond of hercynine is replaced by a C-S bond in one-step (Scheme 1D). Since many enzymes in the biosynthesis of sulfur-related natural products can also produce their selenium analogs,38,39 we tested the ability of EanB to produce selenoneine (8, Scheme 1D). We first focused on identifying a proper selenium donor. Our recent study on EanB-catalysis indicated that inorganic polysulfides serve as the direct sulfur source, suggesting that polyselenide might share a similar function.20 However, the polyselenide synthetic conditions are usually incompatible with the enzymatic reaction conditions.41,42 Therefore, we explored enzymatic systems. Cystathionine-β-lyase is a PLP-dependent enzyme that catalyzes the l-cysthathionine to l-homocysteine transformation.43 Cystathionine-β-lyase (e.g., E.coli MetC) also uses cystine as the substrate to produce cysteine persulfide (9 → 10, Figure 1A).44,45 The cysteine persulfide undergoes disproportionation to produce cysteine polysulfides (10 → 11, Figure 1A).46 To examine whether cysteine per(poly)sulfide serves as the direct sulfur source in EanB-catalysis, we overexpressed the E. coli cystathionine-β-lyase encoded by the MetC gene in E. coli (Figure S1). Cysteine polysulfide (11, Figure 1) was then produced in situ using cystathionine-β-lyase catalysis with cystine (9) as the substrate.
Figure 1. Cysteine polysulfide as the sulfur source in EanB-catalysis.
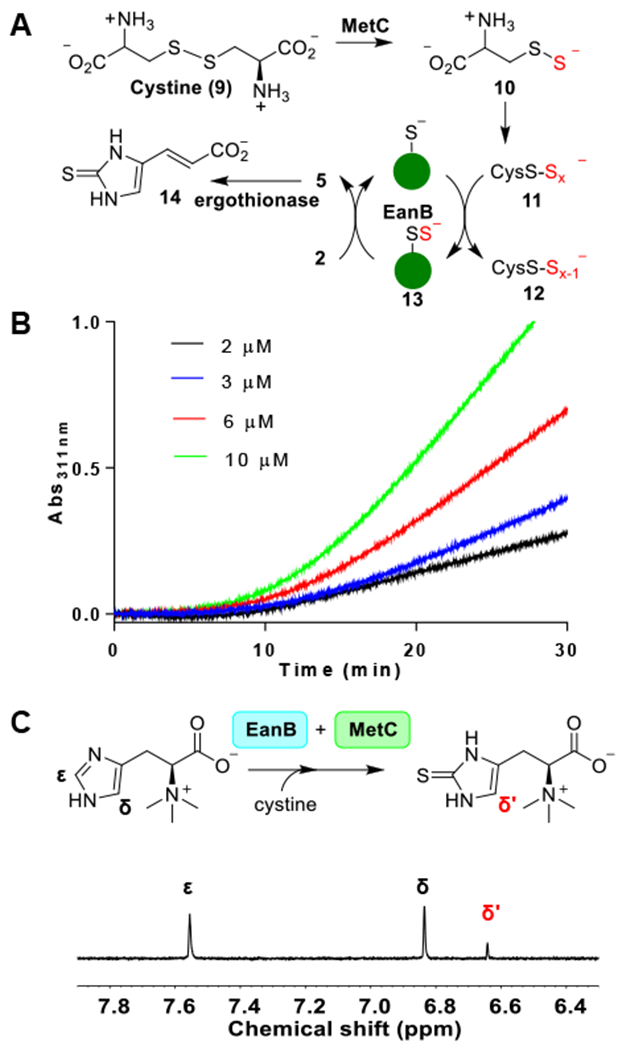
(A) EanB-MetC-ergothionase coupled reaction scheme; (B) Kinetic trace of MetC-EanB-ergothionase coupled assay. A typical 1-mL assay contained 4 nM MetC (3 equivalents of EanB activity), 1 mM cystine, 1 mM hercynine, 400 nM ergothionase (1000-fold of EanB activity) and varying concentration of EanB (2-10 μM) in 10 mM Tris-HCl, pH 8.0. The reaction was monitored at 311 nm. (C) 1H-NMR assay of EanB-MetC coupled reaction.
To monitor ergothioneine production, we used the ergothionase coupling assay reported by us previously.20 Indeed, cysteine polysulfide generated from MetC supported EanB catalysis as revealed by the formation of thiolurocanic acid (14) (Figure 1B). In the EanB-MetC-ergothionase coupled reaction kinetic trace, there is a ten minute lag-phase, which correlates with the need of producing cysteine polysulfide from cysteine persulfide through the disproportionation reaction 10 → 11, Figure 1A). To provide further evidence to support the production of cysteine polysulfide, aliquots of the reaction mixture at various time points were withdrawn, derivatized by iodoacetamide and then analyzed by LC/MS analysis (Figure S2). Indeed, cysteine persulfide and trisulfide derivatives were detected (Figure S2A–B). As an additional line of evidence to support the production of ergothioneine using cysteine polysulfide from MetC-catalysis as the direct sulfur source, we have also analyzed the MetC-EanB coupled reaction using the 1H-NMR assay. In the 1H-NMR spectrum, the peak with a chemical shift of ~6.6 pm is the signal for the δ-H of ergothioneine’s imidazole side-chain (Figure 1C). Therefore, the cysteine polysulfide generated in situ from cystine by MetC-catalysis indeed could serve as the direct sulfur source in EanB catalysis.
Producing Cys412 perselenide-containing EanB.
After demonstrating that the MetC-cystine reaction serves as the sulfur source in EanB-catalysis (Figure 1A), we tested whether EanB catalyzes the production of selenoneine (8, Scheme 1D) according to the experiment outlined in Figure 2A, since MetC also accepts selenocystine as a substrate.47,48 In this reaction, selenocystine (15) is the selenium source. After the production of perselenide (16) from selenocystine (15), it may disproportionate to polyselenide (17) and then transfer the terminal selenium to the EanB’s active cysteine (Cys412). The resulting Cys412-selenide (19, Figure 2A) then serves as the selenium source for EanB-catalysis.
Figure 2. Modification of EanB’s active site cysteine by selenium.
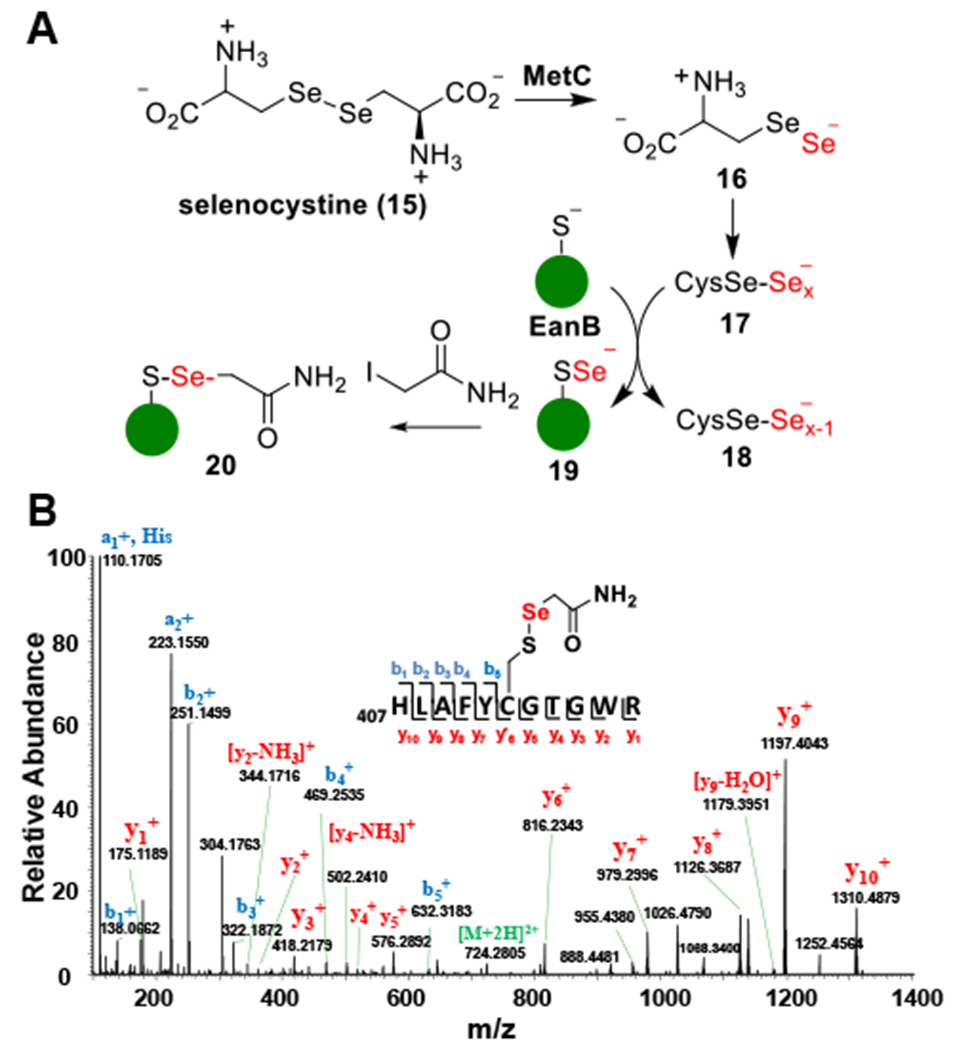
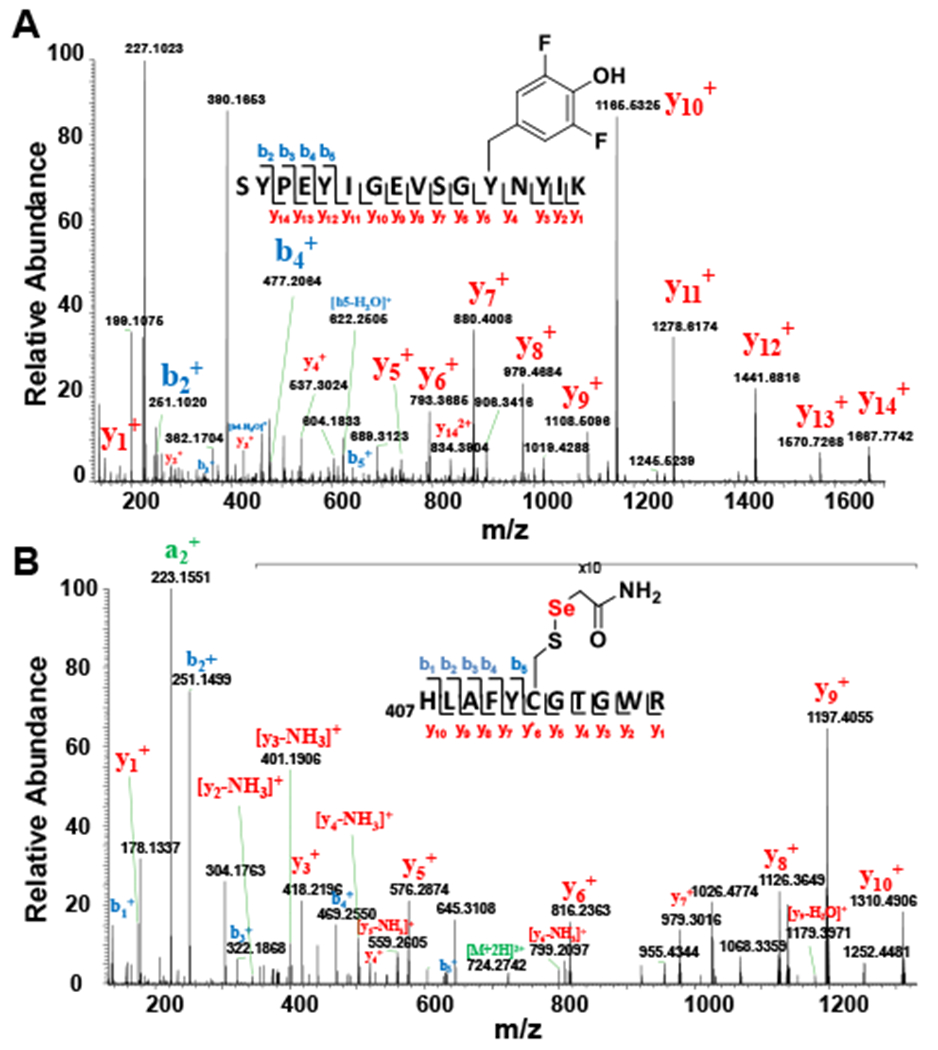
(A) Experimental outline for detecting Cy412 perselenide modification. (B) MS/MS spectrum of tryptic peptides from EanB-MetC coupled reaction using selenocystine as substrate. The Peptide fragment ion (residue 407 to residue 417) with [M+H]2+ at 724.2805 (predicted mass-charge ratio: 724.2776) identified Cys412 with a Cys412-S-Se-acetamide modification, which suggested the formation of the Cys412 perselenide modification during the EanB-MetC coupled reaction.
To create the Cys412 perselenide intermediate, we incubated EanB with MetC in the presence of excess selenocystine for 15 minutes. Small molecules and proteins were then separated by size exclusion chromatography. The protein portion was derivatized using iodoacetamide and subjected to tryptic digestion. The resulting peptides were then characterized by tandem mass spectrometry. MS/MS analysis of the Cys412 containing peptide clearly indicates that the EanB active site cysteine (Cys412) was modified by selenium when MetC and selenocystine were used as the selenium source (Figure 2B).
Computational analysis of EanB-catalysis.
After demonstrating the successful production of the Cys412 perselenide containing EanB, we examined the production of selenoneine (8) in the presence of hercynine under both multiple and single turnover conditions. The reactions were monitored by both 1H-NMR and mass spectrometry. No selenoneine (8) was detected (Figure S3). To understand the reasons behind this lack of reactivity, we analyzed the reaction using computational methods. In our previous work, using efficient hybrid quantum mechanics/molecular mechanic methods (QM/MM) metadynamics simulations, we examined three possible reaction pathways for EanB-catalysis (Scheme 2).20 In all three pathways, the S-S bond cleavage is the rate-determining step, while Tyr353 participates in the key activation step that is a prerequisite for the subsequent S-S bond cleavage. After the imidazole’s side-chain is activated by protonation using Tyr353, the reaction may proceed by persulfide attack on the protonated imidazole, followed by deprotonation at the ε-position, and S-S bond cleavage to produce ergothioneine (Path I, Scheme 2). Path I is a step-wise process, and we have also evaluated the concerted mechanism (Path II, Scheme 2), where, IM-1s can be directly converted to the product in which the S-S bond is partially cleaved and the ε-H is shared by OTyr353 and the imidazole ε-carbon. Our QM/MM analysis indicate that Path II has an activation energy that is much higher than that of the step-wise pathway (Path I, Scheme 2).20 Alternatively, the deprotonated Tyr353 may extract the proton of imidazole’s ε-carbon to produce a carbene intermediate (Path III, Scheme 2). From our QM/MM analysis, the ΔGa for both the sequential pathway (Path I Scheme 2) and the carbene pathways (Path III, Scheme 2) are at a comparable level.
Scheme 2.
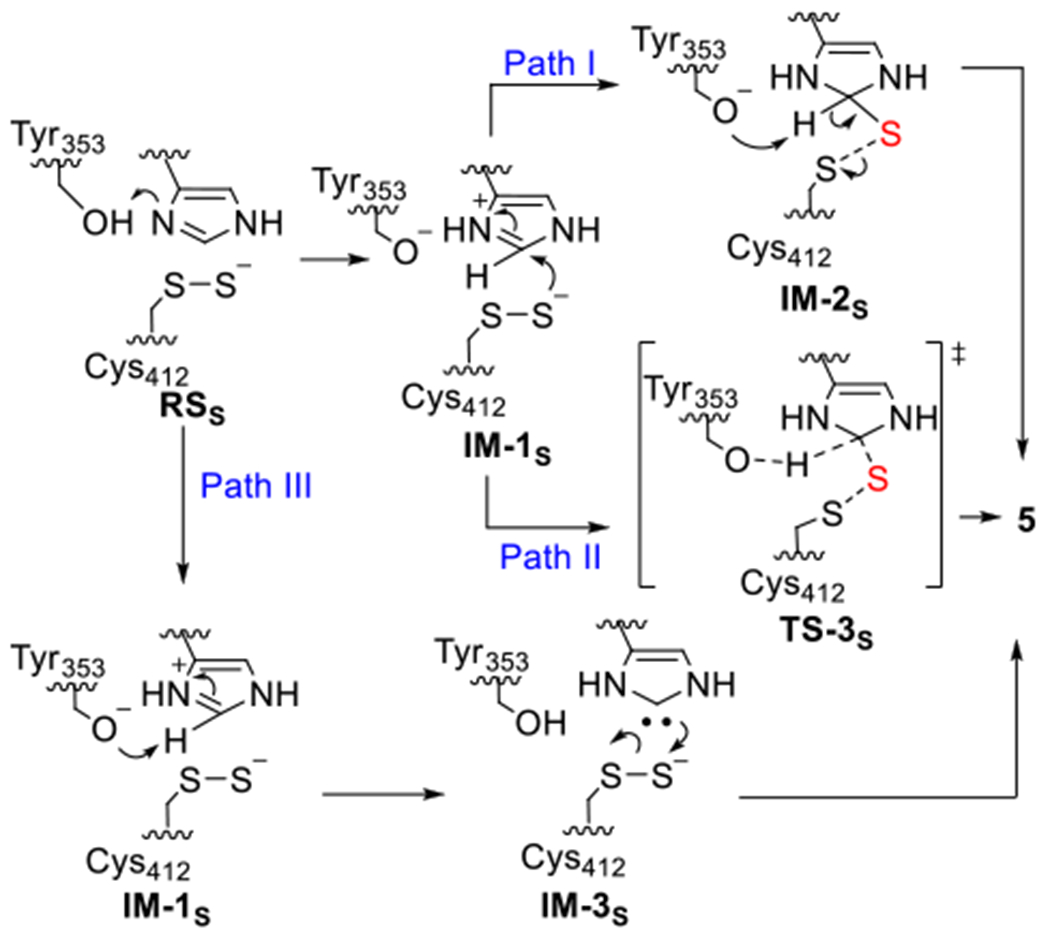
The possible pathways for EanB catalysis proposed by QM/MM metadynamics simulation in our previous study.20 Subscript “S” represents intermediates of the C-S bond formation step in EanB catalysis. Adapted with permission from reference 20. Copyright 2020, American Chemical Society.
Carbenes are key intermediates in many synthetic organic transformations.49–52 However, only limited cases of enzymatic reactions involving carbene intermediates are reported, including thiamine diphosphate dependent enzymes,53,54 and orotidine 5′-phosphate decarboxylase.51,52,55–62 To further confirm our previous QM/MM study results and to understand the difference between sulfur-transfer and selenium-transfer in EanB-catalysis, we conducted QM cluster model calculations at the CPCM/B3LYP-D3/6-31+G(d,p) level of theory,63–68 as further described in Computational Methods. As shown in Figure 3A, IM-1S (with a deprotonated Tyr353) and IM-3S (the carbene intermediate) are stable local minima on the DFT potential energy surface, while the tetrahedral intermediate (IM-2S), is not stable and readily falls back to IM-1S, suggesting that Path I is not be a favorable pathway under cluster model calculations. The existence of the tetrahedral intermediate (IM-2S) in the QM/MM simulation may be a consequence of the constrained C-H bond.20 The observation of the Cys412 hercynine trisulfide adduct in the crystal structure of the EanBY353A mutant (PDB: 6KU2) is likely a consequence of removing Tyr353, which is the key residue that initiates the S-S bond cleavage. For EanBY353A mutant, its activity is several orders of magnitude less than that of EanBWT. In addition, the crystallization process takes days to a week, the observation of the covalent intermediate in is EanBY353A mutant crystal structure certainly consistent with pathway II, while other options may not be ruled out yet.
Figure 3. The potential energy profile for EanB-catalysis using a cluster model at the B3LYP-D3/6-31+G(d,p) level of theory.
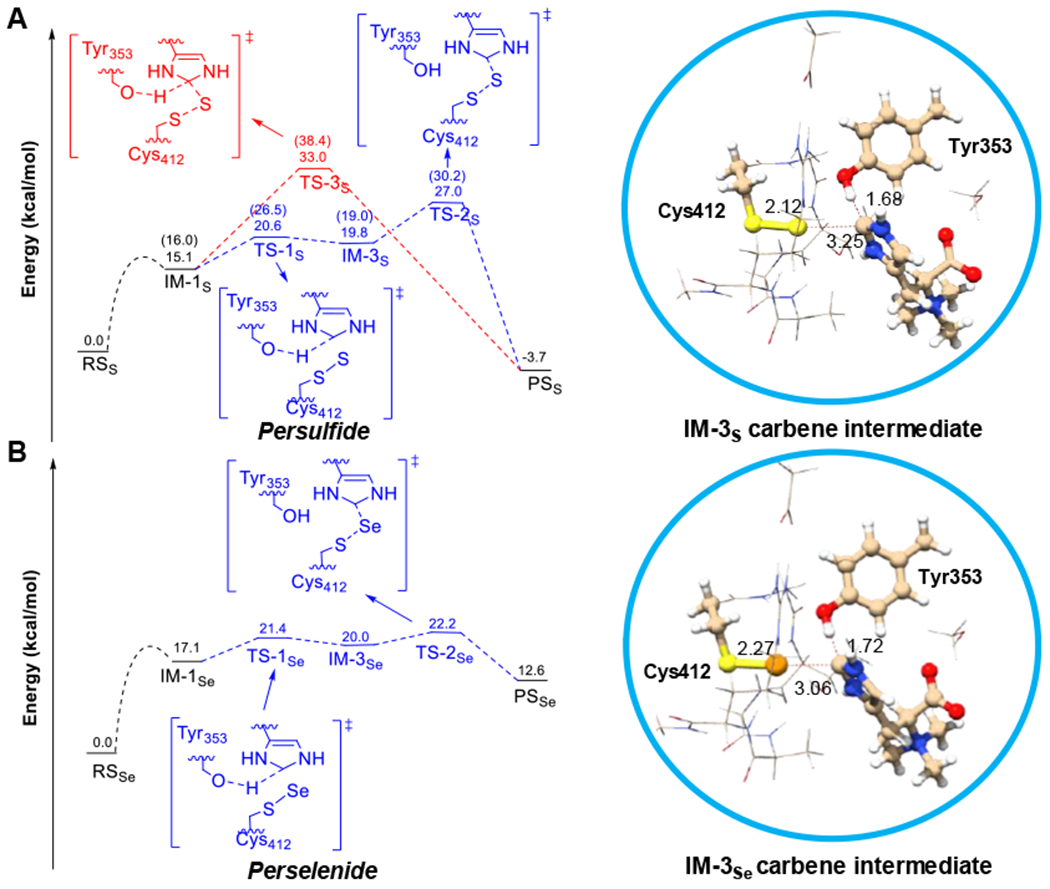
The protein environment was described by CPCM with a dielectric constant of 4.0. (A) EanB with Cys412 persulfide; Note: the numbers in the bracket are free energies (ΔG) from previous DFTB3/MM metadynamics simulation. (B) EanB with the Cys412 perselenide. Subscript “S” represents species in C-S bond formation of EanB catalysis and subscript “Se” represents species in C-Se bond formation of EanB catalysis.
Based on the cluster model calculations (Figure 3A), IM-1S is directly converted to the product through a “concerted” transition state, where the S-S bond is partially cleaved and the ε-H is shared by OTur353 and the ε-C. The energy barrier for this step is fairly high, 33.0 kcal/mol (TS-3S in Figure 3A). Alternatively, a carbene intermediate (IM-3S in Figure 3A) is formed during the conversion of IM-1S to IM-3S with an energy barrier of 20.6 kcal/mol (TS-1S in Figure 3A), suggesting that the deprotonation of ε-C by the deprotonated Tyr353 is energetically feasible. In the subsequent step, i.e., the conversion of the carbene intermediate (IM-3S in Figure 3A) to the product state (PSS in Figure 3A), the energy barrier is 27.0 kcal/mol (TS-2S in Figure 3A), which is lower than that of the concerted pathway (TS-3S in Figure 3A, 33.0 kcal/mol), suggesting that the concerted path is not the favorable pathway based on the cluster model calculations; this is consistent with our previous QM/MM metadynamics simulations. Therefore, calculations from two different methods (both QM/MM and QM cluster models) suggest that a carbene involving mechanism is feasible and that the rate-limiting step is the S-S bond cleavage and C-S bond formation starting from the carbene intermediate (IM-3S in Figure 3A).
In our reaction using the Cys412-perselenide EanB as the catalyst, there is no selenoneine production. To understand the differences between the sulfur and selenium transfer reactions, we examined the selenium transfer reaction using cluster models as we did in the sulfur transfer reaction (Figure 3A). The relative electronic energies (ΔE) for each species of EanB-perselenide (IM-1Se and IM-3Se, Figure 3B) are comparable to those of EanB-persulfide (IM-1S and IM-3S, Figure 3A), except for the product state (PSS and PSSe), as further discussed below. Specifically, the energy barrier (ΔE‡) for the carbene intermediate formation step for the perselenide intermediate (IM-1Se to IM-3Se) is 21.4 kcal/mol (Ts-1Se in Figure 3B), which is comparable to 20.6 kcal/mol (Ts-1S in Figure 3A) in the corresponding persulfide transformation (IM-1S to IM-3S, Figure 3A). However, the energetics of ergothioneine and selenoneine productions are quite different. The energy of the PSs, EanB with ergothioneine (5) relative to the reactant state (RSS), EanB persulfide with hercynine (2), is −3.7 kcal/mol. By contrast, the energy of the PSSe, EanB catalyzed selenoneine (8) formation relative to the RSSe, EanB perselenide with hercynine (2), is 12.6 kcal/mol, suggesting that the reaction intermediates fall back to the substrate side; this provides an explanation for the lack of selenoneine production.
EanB-catalyzed deuterium exchange at the ε-carbon of hercynine’s imidazole side-chain.
Our selenium transfer computational results (Figure 3B) imply that the reverse reaction is preferred in the EanB-catalyzed selenium transfer reaction. These results led to the hypothesis that if EanB-catalysis does involve a carbene intermediate, we will observe a deuterium exchange at hercynine’s imidazole ε-position when the selenium transfer reaction is conducted in D2O buffer. Imidazol-2-yl carbene is difficult to produce in water because the pKa of the corresponding C-H bond of imidazole is 23.8.69 In the absence of a catalyst, at 25 °C, the deuterium exchange is a very slow process in D2O and there is no noticeable deuterium exchange at room temperature after 16 hours (Figure S4A). Even when the mixture was heated up to 80 °C, it took ~8 hours for 3 mM hercynine to achieve >95% deuterium exchange at the ε-C-H bond (Figure S4B).
To test for deuterium exchange in EanB-catalysis, we conducted three sets of experiments. In the first experiment, we incubated the EanB-hercynine mixture in D2O buffer (50 mM potassium phosphate (KPi) buffer in D2O with a pD of 8.22) and the process was monitored by 1H-NMR spectroscopy. In the second set of experiments, the mixture contained hercynine along with MetC and selenocystine in 50 mM KPi buffer in D2O with pD of 8.22. In the third set of experiments, the mixture contained hercynine along with EanB, MetC and selenocystine in 50 mM KPi buffer in D2O with pD of 8.22 and again, the reaction was monitored by 1H-NMR spectroscopy. The results of experiment I are shown in Figure S5 and after 16 hours at 25 °C, the relative intensity of the two signals at ~7.6 ppm and 6.8 ppm remain largely unchanged. The signal at ~7.6 ppm is from the hercynine’s ε-C-H bond and the signal at ~6.8 ppm is from the δ C-H bond. The ratio between these two signals is roughly 0.85:1, which might be due to the relaxation property differences between these two C-H bonds. To provide evidence to further confirm this result, we analyzed the sample at 0 hour and 16 hours by mass spectrometry (Figure S6 and Figure S7) and indeed, the level of deuterium exchange is minimal (< 0.1% at 0 hour and 16 hours). In the second set of experiments (Figure 4A), compared to the first set of experiment, EanB was missing while MetC and its substrate selenocystine were included. As shown in Figure 4A and Figure S6, there was no obvious deuterium exchange either and after 16 hours, the level of deuterium exchange was not detectable (< 0.1%). In the third set of experiments, we included EanB, MetC, hercynine, and selenocystine. As reported in the previous section, this reaction mixture did not produce selenoneine. However, the 1H-NMR signal at ~7.6 ppm disappeared over time (Figure 4B). Further characterization using high resolution mass spectrometry revealed that at hour 16, deuterium exchange reaches 87.9 %. Notably, the reaction mixture contains ~6% H2O because the EanB and MetC samples in H2O buffer were introduced into the reaction mixture.
Figure 4. 1H-NMR analysis of hercynine’s imidazole side-chain ε-C-H bond deuterium exchange.
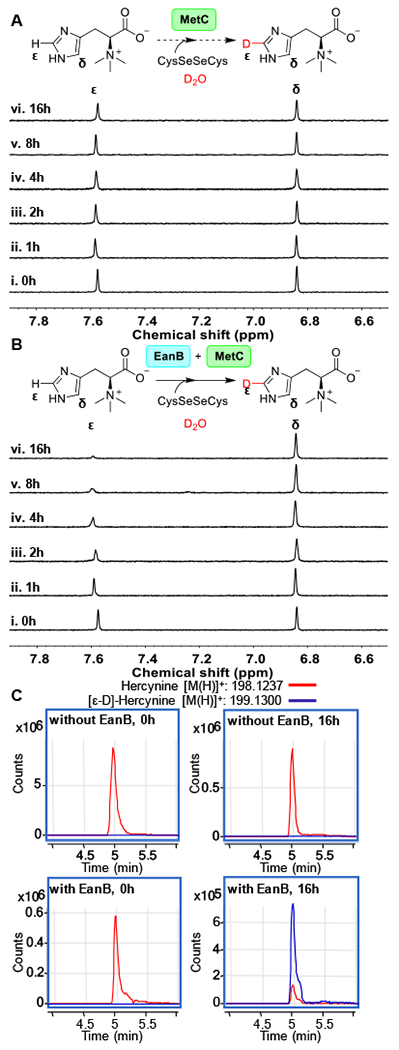
(A) 1H-NMR spectra over time for the reaction mixture containing 3 mM hercynine, selenocystine saturated solution, 0.5 μM MetC in 50 mM KPi buffer in D2O (pD = 8.22). (B) 1H-NMR spectra overtime for the reaction mixture containing 3 mM hercynine, selenocystine saturated solution, 0.5 μM MetC, and 50 μM of EanB in 50 mM KPi buffer in D2O (pD = 8.22); (C) The extracted LC-MS ion chromatograph of [ε-D]-hercynine and [ε-H] hercynine in the EanB-MetC coupled reaction mixture using selenocystine as the substrate in D2O buffer. The percentage of [ε-D]-hercynine is quantified based on the peak area between [ε-D]-hercynine (m/z: 199.1300) and hercynine (m/z: 198.1237) from the extracted ion chromatograph.
Cys412 is essential for carbene formation.
Results in the previous section clearly indicated that the hercynine’s ε C-H bond deuterium exchange in D2O buffer is EanB-catalysis dependent. For the reaction conditions used in Figure 4B studies, it leads to a Cys412 perselenide intermediate formation (Figure 2). Neither EanB nor MetC with selenocystine alone led to a noticeable amount of hercynine’s imidazole side-chain ε C-H bond deuterium exchange with D2O. On the other hand, MS/MS analysis showed that the perselenide modification on EanB by MetC occurs on other EanB cysteine residues (Cys116, Cys184, Cys339 and Cys370. Figure S8–S11) possibly because they are solvent exposed. To provide an additional line of evidence to support the importance of Cys412-perselenide in this deuterium exchange reaction, we repeated the experiment in Figure 4B by replacing EanBWT with EanBC412-only mutant where all of the other four cysteine resides (Cys116, Cys184, Cys339, and Cys370) were replaced with alanine. Similar to the deuterium exchange experiment reported in Figure 4B, in the reaction mixture containing hercynine, EanBC412-only mutant, MetC and selenocystine in 50 mM KPi buffer in D2O with pD of 8.22, the level of deuterium incorporation reaches 83.9 % after 16 hours (Figure S12–S14).
To provide another line of evidence to support the EanB-activity dependence for the observed hercynine deuterium exchange with D2O, we also repeated the experiment using EanBC412S mutant. In the reaction mixture containing hercynine, EanBC412S mutant, MetC and selenocystine in 50 mM KPi buffer in D2O with pD of 8.22, we did not detect deuterium exchange after 16 hours (Figure S15)
Modulate the deuterium exchange rate using unnatural tyrosine-containing EanB.
Our computational studies (Scheme 2 & Figure 3) suggest that the protonation of hercynine’s imidazole ring π-N is a key activation step for EanB-catalysis. This is consistent with our report on the crystal structure of the EanB«hercynine«Cys412 persulfide tertiary complex.20 From this structure, Tyr353 is most likely the active site Lewis acid responsible for hercynine’s imidazole protonation. Indeed, in our previous report, when Tyr353 is mutated to phenylalanine or alanine, the EanB ergothioneine production activity is reduced by orders of magnitude with negligible ergothioneine production.20 To provide additional evidence to support the importance of Y353 in EanB-catalysis, we replaced Tyr353 by tyrosine analogs using the amber suppressor mediated unnatural amino acid incorporation method. Histidine’s imidazole side-chain pKa is ~6.0, while tyrosine’s side-chain pKa is ~10. If Tyr353 is the Lewis acid as suggested by our crystal structure and our computational prediction, there is a mismatch of 4 units unless the pKas of hercynine and tyrosine are significantly perturbed in the EanB active site. Based on this analysis, we replaced Tyr353 by 3,5-difluoro tyrosine (F2Y) because the pKa of F2Y is ~2.8 lower than that of tyrosine.70–72
F2Y was enzymatically synthesized using tyrosine phenol lyase (TPL) as previously reported.73–75 The identity of F2Y was confirmed by 1H-NMR, 13C-NMR and mass spectrometry analysis (Figure S16). To incorporate F2Y in EanB and replace the Tyr353, an engineered aaRS/tRNA pair from Methanococcus jannaschii was used to specifically recognize F2Y.75 The EanBY353F2Tyr mutant was overexpressed and the successful incorporation of F2Y was confirmed by peptide tandem MS/MS and SDS-PAGE analysis (Figure 5A and Figure S17). The kinetics parameters of EanBY353F2Tyr catalyzed ergothioneine formation (using polysulfide and hercynine as the substrate, kcat = 0.42 ± 0.01 min−1, and Km,her = 82.8 ± 9.1 μM) are comparable to that of EanB (Figure S18). This is not unexpected because the EanB rate-limiting step is the S-S cleavage and C-S bond formation from the carbene intermediate. However, lowering the pKaof Tyr353 might affect the deuterium exchange rate if indeed a carbene intermediate is involved in EanB-catalysis. Therefore, we measured the hercynine deuterium exchange rate using EanBY353F2Tyr as the catalyst. To produce Cys412-perselenide containing EanBY353F2Tyr, we incubated the EanBY353F2Tyr variant with MetC in the presence of excess selenocystine for 15 minutes. The presence of the Cys412-perselenide was analyzed by mass spectrometry after the the EanBY353F2Tyr variant was derivatized by iodoacetamide as described in Figure 2A. After tryptic digestion, the resulting peptides were then characterized by tandem mass spectrometry. MS/MS analysis of the Cys412 containing peptide clearly indicated that the active site cysteine (Cys412) in EanBY353F2Tyr was modified by selenium when MetC and selenocystine were present as the selenium source (Figure 5B). After demonstrating the formation of the Cys412 perselenide in the EanBY353F2Tyr variant, we conducted the hercynine deuterium exchange experiment with a mixture containing hercynine, EanBY353F2Tyr, MetC, and selenocystine in 50 mM KPi D2O buffer with pD of 8.22. We firstly monitored the deuterium exchange using 1H-NMR as we did previously (Figure 4). Interestingly, the hercynine’s deuterium exchange is much more efficient in the EanBY353F2Tyr variant relative to EanBWT (Figure 6). In order to reliably capture the deuterium exchange process, we lowered the concentration of the EanBY353F2Tyr variant to a level of ~ ¼ of the concentration used for EanBWT and EanBC412-only in the previous experiments (Figure 4 and Figure S12). Even under this condition, the hercynine deuterium exchange still occurs much faster in the EanBY353F2Tyr variant relative to that of the EanBWT (Figure S19 and Figure S20). Even with a reduced amount of enzyme, the EanBY353F2Tyr reaction reached the deuterium exchange equilibrium in 8 hours while it took 16 hours for the EanB reaction to reach this level. We then quantitatively measured the hercynine-D2O deuterium exchange rate using EanB or EanBY353F2Tyr as the catalyst by the method described in orotidine 5′ - monophosphate decarboxylase studies.51,52,55–62
Figure 5. MS/MS analysis of EanBY353F2Tyr mutant.
(A) Peptide fragment (residue 342 – residue 357) with [M+H]2+ as 959.4395 (predicted m/z: 959.4395) identified 3,5-difluro tyrosine residue at position 353, which suggests the successful incorporation of 3,5-difluro tyrosine at position 353 in EanB (B) MS/MS spectrum of tryptic peptides from EanBY353F2Tyr MetC coupled reaction using selenocystine as substrate. The Peptide fragment (residue 407 to residue 417) with [M+H]2+ as 724.2773 (predicted m/z: 724.2776) identified Cys412 as Cys412-S-Se-acetamide modification, which suggests the Cys412 perselenide modification during the EanBY353F2Tyr-MetC coupled reaction.
Figure 6. Hercynine’s deuterium exchange with D2O when EanBY353F2Tyr variant is used as the catalyst and MetC using selenocystine as the selenium source.
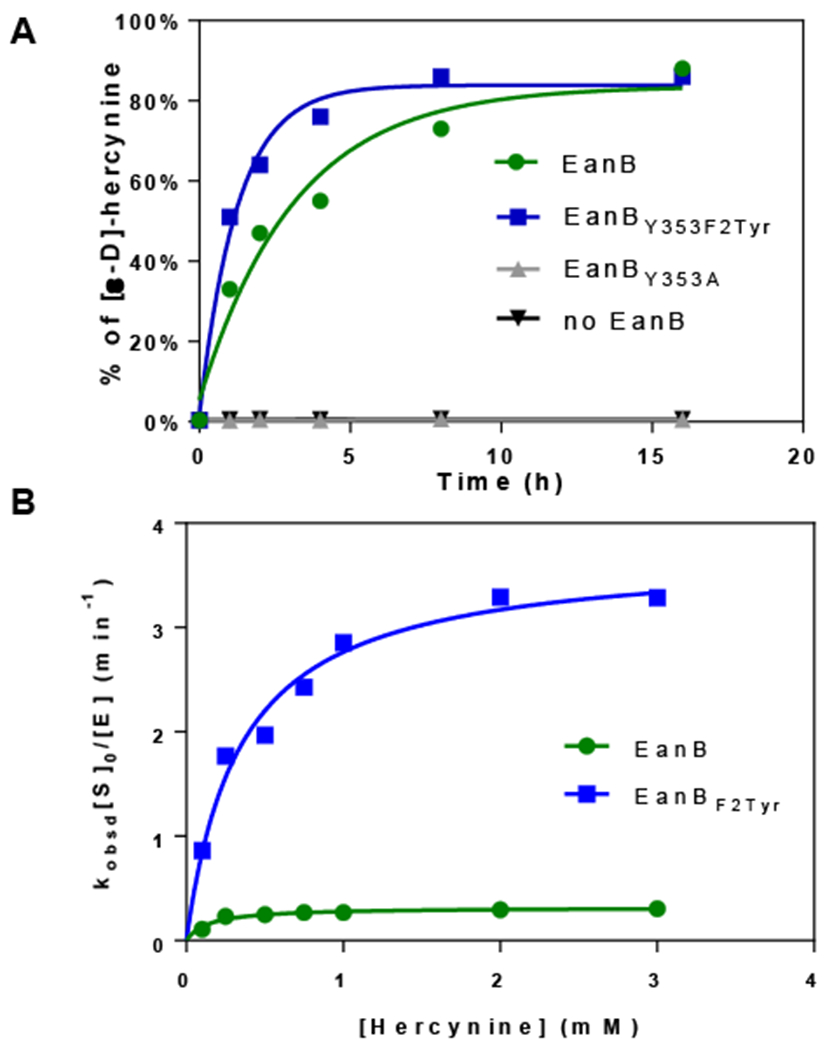
(A) Time course of the [ε-D]-hercynine formation under multiple turn-over conditions. The reaction mixture contained 50 μM of EanBY353A, EanBWT, or 12.5 μM EanBY353F2Tyr variant. (B) Hercynine’s deuterium exchange with D2O rate at various hercynine concentrations using EanB or EanBY353F2Tyr. The 1-ml reaction mixture contains 8 ~ 50 μM EanBWT (or 0.65 ~ 6.5 μM EanBY353F2Tyr), 100 ~ 3000 μM hercynine, selenocystine saturated solution (1 mg powder added), 0.5 μM MetC in 50 mM KPi buffer, with pD of 8.22. The Kobsd at different hercynine concentration is calculated based on the slope from the semilogarithmic plot of the reaction conversion vs time.
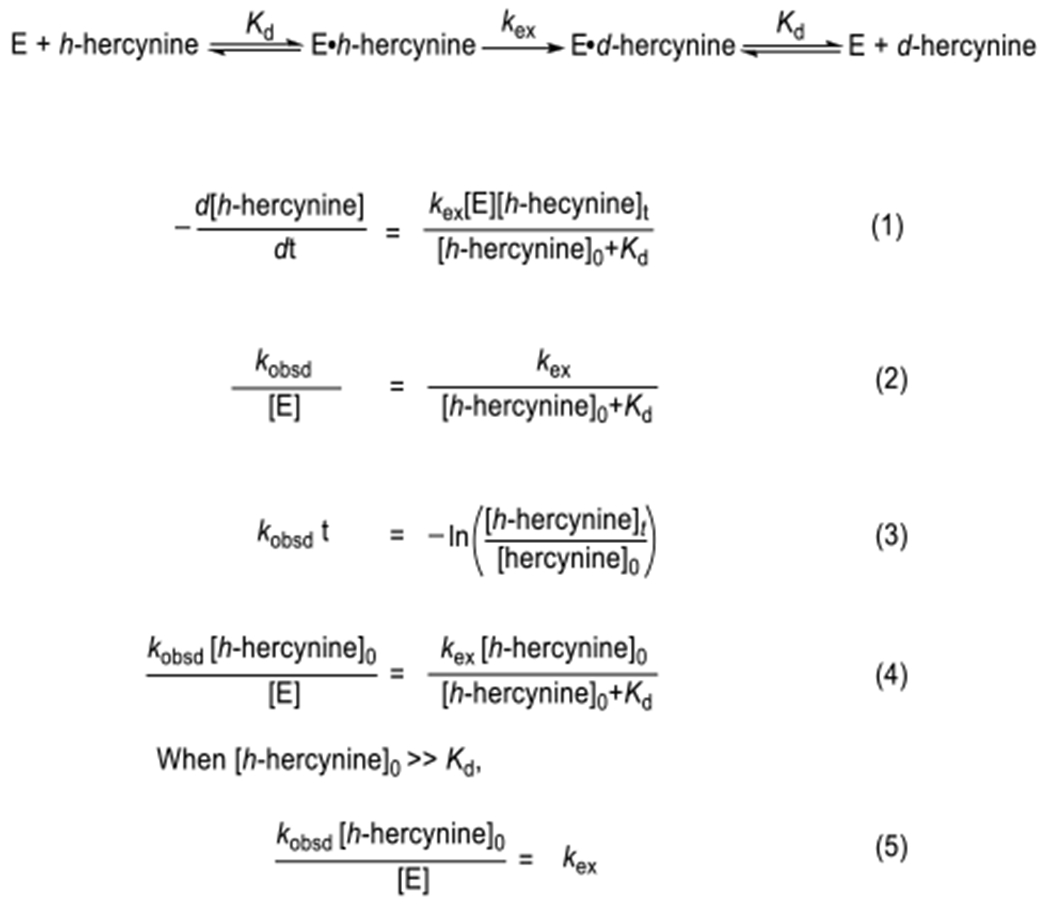
We propose the EanB-catalyzed hercynine deuterium exchange reaction in Scheme 3. The reaction rate of EanB-catalyzed deuterium exchange is given by equation (1), where the kex (S−1) is the deuterium exchange step rate constant and Kd is the dissociation constant for the EanB•h-Her complex. The kinetic parameters correlate to the observed 2nd order reaction rate shown in equation (2), where the [hercynine]0 is the total hercynine concentration regardless of its isotope form. Specifically, the observed first order reaction rates (kobsd) are calculated based on hercynine’s deuterium exchange ratio over different time points, which is shown in equation (3). Furthermore, the equation (2) can be converted to equation (4), which represents more a familiar dependence of initial enzymatic rate versus total substrate concentration.
Scheme 3.
Equations for the EanB-catalyzed hercynine deuterium exchange rate quantification
We re-analyzed the reaction based on the Scheme 3. Specifically, we withdrew aliquots of the reaction mixture at various time points (25 minutes, 45 minutes, 65 minutes and 85 minutes, the time intervals were chosen to cover 4 data points before the deuterium exchange reaction reached ~50 % conversion) and quantitatively analyzed the samples by high resolution mass spectrometry to measure the ratio between hercynine and deuterium-labeled hercynine. We calculated the kobsd from the slope of a semi-logarithmic plot of reaction conversion versus time and fit the results to equation (4) to obtain the kinetics parameters. The kinetics of hercynine deuterium exchange by EanBWT and EanBY353F2Tyr are shown in Figure 6B. The kinetics parameters are kex = 0.34 ± 0.01 min−1 and Kd = 160.7 ± 18.6 μM for EanBWT and kex = 3.72 ± 0.24 min−1 and Kd = 337.4 ± 69.1 μM for EanBY353F2Tyr. Therefore, the exchange rate constant (kex) of the EanBY353F2Tyr variant is ~10 fold greater than that of the EanB. The comparative studies between EanBWT and EanBY353F2Tyr variant clearly indicated that Tyr353 is playing a key role in EanB activation and related to the observed deuterium exchanged reaction in D2O buffer.
DISCUSSION
Despite the significance of sulfur-containing molecules in nature, many of their biosynthetic pathways are poorly understood.14,16–18,20,76,77 Therefore, the biosynthesis of sulfur-containing natural products and the mechanistic studies of newly discovered C-S bond formation reactions are at the frontier of enzymology.14,18,78 Our recent biochemical and structural analyses indicate that the small molecular polysulfide is the direct sulfur source for EanB-catalysis in the ergothioneine biosynthesis. Pentasulfide has been observed in the crystal structure of a Radical SAM enzyme (MiaB).79 However, it is generally believed that the iron-sulfur cluster attached sulfide/polysulfide is the substrate in MiaB catalysis.79,80 Therefore, EanB is the first reported case of an enzyme using polysulfide as the direct sulfur source in a natural product biosynthesis. Our previous QM/MM metadynamics simulations and the QM cluster model calculations in this report examined three potential mechanistic pathways for EanB-catalysis. The imidazole’s ε-position C-H bond is an unactivated position with a pKa of 23.8 based on studies from model systems.69 In EanB-catalysis, hercynine must activate first and our QM/MM metadynamics simulation indicate that protonation of hercynine’s Nπ atom by Tyr353 is a key activation step. After the imidazole’s side-chain is activated by protonation, the reaction proceeds to produce a C-S bond via either a sequential (Path I) or concerted (Path II) mechanism (Scheme 2). Alternatively, the C-S bond forms via a carbene intermediate (Path III, Scheme 2). Seebeck et al. also considered the possibility of a carbene intermediate in EanB-catalysis, but suggested that this carbene pathway is unlikely.19,81 However, from our QM/MM simulation, the activation barrier for the sequential pathway (Path I) is at a level comparable to that of the carbene-involved reaction, suggesting that the carbene pathway should be considered.
In enzymatic systems, carbene intermediates are proposed in only a few limited cases, including thiamine diphosphate dependent enzymes,53,54 orotidine 5′-phosphate decarboxylase,51,52,55–62,82–92 and some engineered p450 variant-catalyzed unnatural reactions.93 Recently, Meyer et al. report crystallographic evidence for a carbene intermediate in thiamine diphosphate dependent enzymes.53 Biochemically, in both thiamine diphosphate dependent enzymes and orotidine 5′-phosphate decarboxylase, the presence of a carbene intermediate is inferred from a deuterium exchange reaction in D2O buffer.51,52,55–62 In this work, using Cys412 perselenide-containing EanB, we have successfully detected hercynine’s ε-position C-H bond deuterium exchange. The pKa of this C-H bond is estimated to be 23.8, which is higher than 19.5 of thiazole in the thiamine diphosphate cofactor.69 Therefore, formation of an imidazol-2-yl carbene maybe even more challenging. Imidazole carbenes have been produced using synthetic organic approaches and are key reagents for a wide range of chemical transformations.49,50 To the best of our knowledge, imidazole carbenes have not yet been reported in enzymatic systems.
Based on our previous QM/MM metadynamics simulation, Tyr353 plays a key role in activating hercynine’s imidazole for the EanB-catalyzed trans-sulfurration reaction. It is worth noting that, for Tyr353, with its phenol group pKa higher than the ∏-N of imidazole, the activation free energy is high, in the magnitude of ~20.0 kcal/mol, as discussed in our previous paper.20 If Tyr353 plays a key role in this activation step, replacement of Tyr353 with an unnatural amino tyrosine with a lower pKa may affect the deuterium exchange rate. Indeed, when we replaced Tyr353 with its 3,5-difluoro tyrosine analog, the deuterium exchange rate constant kex increases by ~10-fold. Once the hercynine’s imidazole activates, subsequent ε-position C-H bond deprotonation requires a base. We propose that the base is the deprotonated Tyr353. Tyr353 functions as both the Lewis acid in the hercynine activation step, and as a Lewis base in the ε-position C-H deprotonation step.
Beside Tyr353, the Cys412 perselenide or persulfide may serve as the base to deprotonate the hercynine’s ε-position C-H bond to produce the carbene intermediate. However, we note that the reported pKa for cysteine persulfide is about 4.3, suggesting that the neutral persulfide is unlikely to be highly populated in the active site.94 Therefore, Tyr353 is more likely to be the protonation source for hercynine imidazole activation. We are aware that the pKa might be perturbed in the enzyme environment, therefore neutral persulfide may exist. To test this option, we have examined the intrinsic reactivity of neutral persulfide with imidazole using small molecule models and results from these calculations suggest that the reaction between neutral persulfide and imidazole is indeed energetically feasible. Relevant discussions and Figures have been added to the Supporting Information. To understand the possible mechanism, we examined the stability of intermediates and explored the potential energy surface of neutral persulfide with imidazole, as shown in Figure S24 to S30. The results suggest that the reaction likely still involves a carbene intermediate and the reaction pathways are similar to those of negatively charged persulfide (Cys-S-S−). In addition, our cluster model calculations suggest that neutral persulfide or perselenide (i.e. S-S-H or S-Se-H) is stable with the carbene intermediate if Tyr353 is in its protonated state, similar to the findings obtained from our small model calculations. The transition state structures for persulfide and neutral persulfide in the rate-limiting step are similar in C-S and S-S distances, though for neutral persulfide, the TS is “synchronous”: C-S bond formation and proton transfer from one sulfur to the other occur at the same time. As a result, the intrinsic energy barrier for neutral persulfide is higher than that of persulfide. Therefore, negatively charged persulfide is likely the optimal choice in EanB enzyme reaction. Nevertheless, for a thorough understanding, accurate QM/MM free energy calculations are necessary and will be reported in the future.
The observation of deuterium exchange in EanB-catalysis is consistent with the proposed carbene mechanism (Scheme 2). However, if the reaction is fully reversible, in D2O buffer, ergothioneine or selenoneine to hercynine transformation will also lead to deuterium incorporation into hercynine [PSs (or PSSe) → hercynine transformation]. To test whether the observed hercynine deuterium exchange observed in EanB-catalysis in D2O buffer is due to this reverse reaction, we conduct another set of experiments and two reactions were setup in parallel. In the first reaction, 3 mM hercynine and ergothioneine were mixed in 50 mM KPi D2O buffer. In the second reaction, 3 mM hercynine and ergothioneine were mixed with 50 μM EanB in 50 mM KPi D2O buffer. Both of the reactions were monitored by NMR assay and MS quantification for up to four days. For the first reaction, in the absence of EanB-catalysis, hercynine deuterium exchange is extremely slow Figure 4A [after 4 days, there is less a very small amount of deuterium incorporation (<10%)]. Interestingly, for the second reaction (hercynine/ergothioneine mixture in the presence of EanB enzyme), after 4 days, there was ~ 40% conversion of [ε-2H]-hercynine (Figure S21) and kex of hercynine deuterium exchange for this EanB reverse sulfur transfer reaction was ~0.005 min−1, which is two orders of magnitude lower than the kex of EanBWT and three orders of magnitude lower than the kex of EanBY353F2Tyr using MetC selenocystine as intermediate. More importantly, based on our calculation, the S-S or S-Se bond cleavage in EanB-catalysis is the rate-limiting step. Therefore, if the the hercynine deuterium exchange observed in EanB-catalysis is due to the reverse reaction, kex will be either at a level comparable to that of kcat or smaller than kcat. However, kex for hercynine deuterium exchange in EanBY353F2Tyr is ~ 10 x of kcat. Therefore, even if there will be hercynine deuterium exchange from the reverse reaction (ergothioneine → hercynine), it accounts for ~1% or less of the observed deuterium incorporation into hercynine (Figure S21). Therefore, the involvement of carbene intermediate maybe the key factor leading to the observed hercynine deuterium exchange in D2O buffer.
The observation of the Cys412 hercynine trisulfide adduct in the crystal structure of the EanBY353A mutant indicated the possibility of tetrahedral intermediate model (Path I in Scheme 2) .20 Since the crystallization process takes a few days to a eek, it suggests that the formation of Cys412 tetrahedral intermediate is a very slow process. On the other hand, the level of kex in EanB is comparable to or even 10 x higher than that of kcat. Therefore, after all of these factors are taken into consideration, we are inclined to the carbene model of EanB catalysis. Further differentiation between two mechanistic models and potential contribution of deuterium exchange due to the reverse reaction may require additional mechanistic investigations.
In summary, deuterium exchange occurs at the hercynine’s ε-position C-H bond in D2O buffer. This deuterium exchange reaction is EanB-activity dependent based on the following lines of evidence. First, imidazole C(2) deuterium exchange is a very slow process.69 Indeed, even upon increasing the reaction temperature to 80 °C, the reaction requires 8 hours to achieve 95% deuterium exchange for 3 mM hercynine at the ε-position C-H bond in D2O. Second, active EanB is necessary for this reaction. In the EanBY353A mutant, the deuterium exchange does not occur (Figure S22). Third, the Cys412-perselenide intermediate does not lead to selenoneine formation, but instead to catalyze hercynine deuterium exchange with the D2O solvent. The involvement of an imidazole carbene intermediate in this interesting biosynthetic pathway will spur additional EanB studies as well as the investigation of other biotransformation, which may utilize a carbene intermediate as a key step. Finally, selenonine synthesis and its application in both basic95 and translational research3 are ongoing studies in our laboratory.
EXPERIMENTAL SECTION
Materials:
Reagents were purchased from Sigma-Aldrich and Fisher Scientific unless otherwise specified. Hercynine was synthesized following reported procedure.29 NMR spectra were recorded using Agilent 500 (500 MHz VNMRS). The kinetic parameter was determined using Cary Bio-100 spectrophotometer (Agilent).
Overexpression and purification of EanBY353F2Tyr.
BL21(DE3) cells were co-transformed with EanBY353TAG/pET28a and F2YRS/pEVOL (F2YRS/pEVOL was provided by Dr. Jiangyun Wang). One colony was inoculated into 50 mL LB media supplemented with 50 μg/mL kanamycin and 25 μg/mL chloramphenicol at 37 °C overnight. Then, 10-mL of seed culture was then inoculated to 1 L LB media supplemented with 50 μg/mL kanamycin and 25 μg/mL chloramphenicol at 37 °C. When OD600nm reaches 0.6, the media was supplied with arabinose to 0.02% (w/v) final concentration. When OD600nm reached 1.2, protein overexpression was induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) final concentration and the media was supplemented with D-glucose to 5 g/L final concentration, 3,5-difluoro tyrosine to 1 mM final concentration, and 100 mM KPi buffer, pH 7.0. The culture was incubated at 16 °C for 12 hours before harvest by centrifugation.
EanBY353F2Tyr was purified anaerobically. 14 g cells were suspended in 70 ml lysis buffer (100 mM Tris-HCl, 200 mM NaCl, pH 8.0) with 1 mg/mL final concentration of lysozyme. The mixture was incubated on ice with shaking for 30 min. Cells were lysed by sonication. DTT was added to cell lysate to 1 mM final concentration. Cell debris was removed via centrifugation at 20,000 rpm for 30 min, 4 °C. The supernatant was incubated with 5 mL of Ni Sepharose resin (GE Healthcare) for 30 minutes. The column was washed with 25 mL of lysis buffer with 30 mM imidazole and 1 mM DTT followed by 25 mL lysis buffer with 30 mM imidazole. The protein was eluted by lysis buffer containing 250 mM imidazole and concentrated using ultrafiltration (Millipore). Imidazole was removed using a G25 Sephadex size-exclusion column (30×600 mm). The EanBY353F2Tyr containing fractions were combined and concentrated through ultrafiltration.
Tandem MS/MS analysis of EanB.
The EanB Cys412-S-Se modification was achieved from the coupled reaction of EanB with MetC with selenocystine as the selenium source. A 435 μL reaction mixture with 698 μM EanB,7 μM MetC selenocystine saturated solution (1.5 mg powder added) in 150 mM Tris-HCl, pH 8.0 buffer was set up at room temperature for 15 minutes. Then, excess small molecules were removed using G-25 size-exclusion chromatography. EanB with the putative Cys412 perselenide modification was alkylated by iodoacetamide. The 400-μL alkylation reaction contained 106 μM EanB and 2 mM iodoacetamide and was incubated at room temperature for 1 hour in the absence of light. The protein fraction after size exclusion chromatography was digested by Sequencing Grade Modified Trypsin (Promega) at 37 °C in absence of light for overnight. Typically, each 70 μL mixture used 0.8 μg of Trypsin and 80 μg protein in 50 mM Tris-HCl, pH 8.0 buffer. The digested peptide was desalted using a microspin C18 column (The Nest Group).
The peptide was loaded on an in-house packed capillary reverse-phase C18 column connected to an Easy LC 1200 system. The samples were analyzed with a 120min-HPLC gradient from 5% to 35% of buffer B (buffer A: 0.1 % formic acid in water; buffer B: 0.1% formic acid in 80% acetonitrile) at 300 nL•min. The eluted peptides were ionized and directly introduced into a Q-Exactive mass spectrometer using a nano-spray source. Survey full-scan MS spectra (from m/z 100–2000) were acquired in the Orbitrap analyzer with a resolution r = 70,000 at m/z 400. Protein identification and protein post-modification identification were done with Proteome Discoverer 2.3.
EanB-catalyzed hercynine deuterium exchange.
A typical 2-mL reaction mixture contained 50 μM EanB, 3 mM hercynine, selenocystine saturated solution (6.7 mg powder added), and 0.5 μM MetC in 50 mM KPi D2O buffer, pD = 8.22. In order to rule out the non-EanB-catalyzed exchange of the ε-C of hercynine in D2O, a 2-mL reaction mixture with 3 mM hercynine, selenocystine saturated solution (6.7 mg powder added), and 0.5 μM MetC in 50 mM KPi D2O buffer, pD = 8.22 was setup in parallel. The percentage of H2O in the reaction mixtures was 6 % (It came from protein H2O buffer in the protein purification process). The reaction mixture was incubated for 16 hours. 300 uL of sample was taken at various time and analyzed by 1H-NMR assay to monitor the exchange of hercynine’s ε-C-H bond. The ratio between [ε-H]-hercynine and [ε-D]-hercynine at 0 hour and 16 hours were also analyzed by mass spectrometry. The protein from 100 μL was quenched by adding 20 μL 6 M HCl and centrifuged at 15k rpm for 10 minutes. The supernatant was collected and lyophilized. Lyophilized sample was re-dissolved in 100 μL H2O and quantified by LC/MS. The experiment was repeated by two independent trials.
EanB variant-catalyzed hercynine deuterium exchange with D2O.
A typical 2-mL reaction mixture contained 50 μM EanB variants (EanBC412S, EanBC412-only and EanBY353A), 3 mM hercynine, selenocystine saturated solution (6.7 mg powder added), and 0.5 μM MetC, in 50 mM KPi D2O buffer, pD =8.22. In order to rule out the non-EanB-catalyzed exchange of hercynine ε-C-H bond with D2O, a 2-mL reaction mixture with 3 mM Hercynine, selenocystine saturated solution (6.7 mg powder added), and 0.5 μM MetC, in 50 mM KPi D2O buffer, pD =8.22 was setup in parallel. The percentage of H2O in the reaction mixture was 12.5% (It came from protein H2O buffer in the protein purification process). The reaction mixture was incubated for 16 hours. 300 μL of sample was withdrawn at various time and analyzed by 1H-NMR assay to monitor the hercynine ε-C-H deuterium exchange with D2O. The ratio between [ε-H]-hercynine and [ε-D]-hercynine at 0 hour and 16 hours were further analyzed by mass spectrometry. The protein from 100 μL was quenched by adding 20 μL 6 M HCl and centrifuged at 15k rpm for 10 minutes. The supernatant was collected and lyophilized. Lyophilized sample was re-dissolved in 100 μL H2O and quantified by LC-MS.
EanB hercynine deuterium exchange reaction with ergothioneine and D2O.
A typical 2-mL reaction mixture contained 50 μM EanB, 3 mM hercynine, 3 mM ergothioneine, in 50 mM KPi D2O buffer, pD = 8.22. In order to rule out the non-EanB-catalyzed exchange of the ε-C of hercynine in D2O, a 2-mL reaction mixture with 3 mM hercynine, 3 mM ergothioneine, in 50 mM KPi D2O buffer, pD = 8.22 was setup in parallel. The percentage of H2O in the reaction mixtures was 6 % (It came from protein H2O buffer in the protein purification process). The reaction mixture was incubated for 5 days. 300 uL of sample was taken at various time and analyzed by 1H-NMR assay to monitor the exchange of hercynine’s ε-C-H bond. The ratio between [ε-H]-hercynine and [ε-D]-hercynine at different time points were also analyzed by mass spectrometry. The protein from 100 μL was quenched by adding 20 μL 6 M HCl and centrifuged at 15k rpm for 10 minutes. The supernatant was collected and lyophilized. Lyophilized sample was re-dissolved in 100 μL H2O and quantified by LC-MS.
EanBY353F2Tyr-catalyzed hercynine deuterium exchange with D2O.
The conditions are the same as above except that 12.5 μM EanBY353F2Tyr was used in this experiment.
Hercynine deuterium exchange kinetics catalyzed by EanB and EanBY353F2Tyr.
1-ml reaction mixture with variable concentrations of EanB (8 to 50 μM for EanBWT and 0.65 to 6.5 μM Y353F2Tyr variant), 0.5 μM MetC, selenocystine saturated solution (1 mg powder added), and variable concentrations of hercynine (0.1 to 3 mM) in 50 mM KPi D2O buffer, pD 8.22. Four times points (25 minutes, 45 minutes, 65 minutes, and 85 minutes) were chosen to ensure that when the reaction was quenched, there was less than 50% of hercynine deuterium exchange. At multiple time points, a portion of 250 μL reaction mixture was withdrawn and quenched by adding 50 μL 6 M HCl, and centrifuged at 15k rpm for 10 minutes. The supernatant was collected and lyophilized. Lyophilized sample was re-dissolved in 300 μL H2O and quantified by LC/MS.
Computational Methods.
The QM cluster models is a truncated model based on QM/MM calculations (see our previous paper);20 this consists of 136 atoms, including key reacting residues, the hercynine substrate and some residues in α-helix 18 (i.e. Glu345, Tyr353, Tyr375, Tyr411, Cys412, Gly413, Thr414, Gly415, Trp416, Arg417 and Gly418), as shown in Fig. S22. The total charge of the system is −1 e. Six atoms are fixed during geometry optimization to retain the protein structures: γ-C of Glu345, β-C of Tyr353, carbonyl O of Thr414, two terminal C atoms of truncated Trp416 and terminal C atom of truncated Tyr375. All calculations were performed using the Gaussian 16 program96. The Becke, three-parameter, Lee-Yang-Parr exchange-correlation functional (B3LYP)63 with the addition of Grimme’s third version semi-empirical dispersion correction (D3)64 were used with the 6-31+G(d,p) basis set65,66. Geometry optimizations and harmonic vibrational analysis were performed with conductor-like polarizable continuum solvation model67,68 (CPCM, dielectric constant as 4.0 to model the solvation effect of the protein environment).
Mass spectrometry analysis of hercynine deuterium exchange.
The UPLC-MS analysis was performed on an Agilent 1290 UPLC (Agilent, USA) coupled to an Agilent 6530 QTOF mass spectrometer (Agilent, USA) with the electrospray ionization (ESI) source. A Waters ACQUITY UPLC BEH HILIC column (1.7 μm, 2.1 × 100 mm) was used for separation with flow rate at 0.4 mL/min and column temperature at 45 °C. The mobile phases were comprised of (A) 0.2% formic acid and 10 mM ammonium acetate in 50% acetonitrile and (B) 0.2% formic acid and 10 mM ammonium acetate in 95% acetonitrile. The gradient elution was 90% B kept for 1.0 min, followed by a linear gradient to 5% B during 7.0 min and maintained 5% B to 10.0 min, then increased to 90% B in 10.1 min and maintained for 2.9 min. Multiple Reaction Monitoring (MRM) was used to monitor the compounds in the positive ion mode. The injection volume was set to 2μL. The samples were run in auto MS/MS mode. MS acquisition parameters were set as follows: positive ion polarity, capillary voltage was 4000 V, nozzle voltage was 500 V, gas temperature was 250 °C, gas flow was 12 L/min, nebulizer was 35 psi, sheath gas temperature was 300 °C, sheath gas flow was 11 L/min. fragmentor voltage was 200V, skimmer was 65 V, MS and MS/MS mass range of 50-600 m/z, a maximum of 3 precursors per cycle, and a precursor selection threshold of 1000 counts absolute or 0.01% relative. Data acquisition and processing were performed on Agilent Mass hunter workstation software (Agilent, USA). The ratio between [ε-H]-hercynine and [ε-D]-hercynine was quantified by peak area of their extraction ion chromatogram. Additionally, the product from the deuterium experiment was analyzed by LC/MS on a LTQ-FT-ICR (Thermo Fisher Scientific, USA) mass spectrometer. AThermoFisher Hybercarb column (5 μm, 4.6 χ 150 mm) was use for separation with flow rate at 0.5 mL/min. The mobile phase was composed of (A) 0.1 % formic acid in H2O and (B) 0.1 % formic acid in acetonitrile. The gradient elution was 0 % B for 3 min, followed by a linear gradient to 90 % B during 17 min and the gradient was maintained to 22 min, then the grading was decreased to 0 % B and maintained to 30 min. Compounds mass range from 100 to 400 Da was collected and was detected in positive mode.
Supplementary Material
ACKNOWLEDGMENT
This work is partially supported by the National Science Foundation (CHE-2004109 to P. Liu) and National Institute of Health (GM-106443 to Q. C.).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
More detailed experimental procedures, protein purification and characterization, and compound synthesis is available in the supporting information file.
Contributor Information
Ronghai Cheng, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Rui Lai, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Chao Peng, National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China.
Juan Lopez, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Zhihong Li, National Facility for Protein Science in Shanghai, Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science, Shanghai 201210, China.
Nathchar Naowarojna, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Kelin Li, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Christina Wong, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Norman Lee, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Stephen A. Whelan, Department of Chemistry, Boston University, Boston, MA 02215, USA
Lu Qiao, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Mark W. Grinstaff, Department of Chemistry, Boston University, Boston, MA 02215, USA; Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA. 0000-0002-5453-3668
Jiangyun Wang, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, People’s Republic of China.
Qiang Cui, Department of Chemistry, Boston University, Boston, MA 02215, USA.
Pinghua Liu, Department of Chemistry, Boston University, Boston, MA 02215, USA.
REFERENCES
- (1).Grundemann D; Harlfinger S; Golz S; Geerts A; Lazar A; Berkels R; Jung N; Rubbert A; Schomig E, Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 5256–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fahey RC, Novel thiols of prokaryotes. Annu. Rev. Microbiol 2001, 55, 333–356. [DOI] [PubMed] [Google Scholar]
- (3).Cheah IK; Halliwell B, Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta, Mol. Basis Dis 2012, 1822, 784–793. [DOI] [PubMed] [Google Scholar]
- (4).Ames BN, Prolonging healthy aging: Longevity vitamins and proteins. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 10836–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Libby P; Ridker PM; Hansson GK, Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [DOI] [PubMed] [Google Scholar]
- (6).Taubert D; Lazar A; Grimberg G; Jung N; Rubbert A; Delank K-S; Perniok A; Erdmann E; Schömig E, Association of rheumatoid arthritis with ergothioneine levels in red blood cells: a case control study. J. Rheumatol 2006, 33, 2139–2145. [PubMed] [Google Scholar]
- (7).Tokuhiro S; Yamada R; Chang X; Suzuki A; Kochi Y; Sawada T; Suzuki M; Nagasaki M; Ohtsuki M; Ono M, An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet 2003, 35, 341–348. [DOI] [PubMed] [Google Scholar]
- (8).Peltekova VD; Wintle RF; Rubin LA; Amos CI; Huang Q; Gu X; Newman B; Van Oene M; Cescon D; Greenberg G, Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet 2004, 36, 471–475. [DOI] [PubMed] [Google Scholar]
- (9).Leung E; Hong J; Fraser AG; Merriman TR; Vishnu P; Krissansen GW, Polymorphisms in the organic cation transporter genes SLC22A4 and SLC22A5 and Crohn’s disease in a New Zealand Caucasian cohort. Immunol. Cell. Biol 2006, 84, 233–236. [DOI] [PubMed] [Google Scholar]
- (10).Kaneko I; Takeuchi Y; Yamaoka Y; Tanaka Y; Fukuda T; Fukumori Y; Mayumi T; Hama T, Quantitative determination of ergothioneine in plasma and tissues by TLC-densitometry. Chem. Pharm. Bull 1980, 28, 3093–3097. [DOI] [PubMed] [Google Scholar]
- (11).Briggs I, Ergothioneine in the central nervous system. J. Neurochem 1972, 19, 27–35. [DOI] [PubMed] [Google Scholar]
- (12).Crossland J; Mitchell J; Woodruff G, The presence of ergothioneine in the central nervous system and its probable identity with the cerebellar factor. J. Physiol 1966, 182, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Moncaster JA; Walsh DT; Gentleman SM; Jen L-S; Aruoma OI, Ergothioneine treatment protects neurons against N-methyl-D-aspartate excitotoxicity in an in vivo rat retinal model. Neurosci. Lett 2002, 328, 55–59. [DOI] [PubMed] [Google Scholar]
- (14).Dunbar KL; Scharf DH; Litomska A; Hertweck C, Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem. Rev 2017, 117, 5521–5577. [DOI] [PubMed] [Google Scholar]
- (15).Fontecave M; Ollagnier-de-Choudens S; Mulliez E, Biological radical sulfur insertion reactions. Chem. Rev 2003, 103, 2149–2166. [DOI] [PubMed] [Google Scholar]
- (16).Kessler D, Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev 2006, 30, 825–840. [DOI] [PubMed] [Google Scholar]
- (17).Mueller EG, Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol 2006, 2, 185–194. [DOI] [PubMed] [Google Scholar]
- (18).Naowarojna N; Cheng R; Chen L; Quill M; Xu M; Zhao C; Liu P, Mini-review: ergothioneine and ovothiol biosyntheses, an unprecedented trans-sulfur strategy in natural product biosynthesis. Biochemistry 2018, 57, 3309–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Burn R; Misson L; Meury M; Seebeck FP, Anaerobic origin of ergothioneine. Angew. Chem. Int. Ed 2017, 56, 12508–12511. [DOI] [PubMed] [Google Scholar]
- (20).Cheng R; Wu L; Lai R; Peng C; Naowarojna N; Hu W; Li X; Whelan SA; Lee N; Lopez J; Zhao C; Yong U; Xue J; Jiang X; Grinstaff MW; Deng Z; Chen J; Cui Q; Zhou J; Liu P, Single-step replacement of an unreactive C–H bond by a C–S bond using polysulfide as the direct sulfur source in the anaerobic ergothioneine biosynthesis. ACS Catal. 2020, 10, 8981–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seebeck FP, In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc 2010, 132, 6632–6633. [DOI] [PubMed] [Google Scholar]
- (22).Hu W; Song H; Her AS; Bak DW; Naowarojna N; Elliott SJ; Qin L; Chen XP; Liu PH, Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus Neurospora crassa ergothioneine biosynthetic pathway. Org. Lett 2014, 16, 5382–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Song H; Hu W; Naowarojna N; Her AS; Wang S; Desai R; Qin L; Chen X; Liu P, Mechanistic studies of a novel C-S lyase in ergothioneine biosynthesis: the involvement of a sulfenic acid intermediate. Sci. Rep 2015, 5, 11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Irani S; Naowarojna N; Tang Y; Kathuria KR; Wang S; Dhembi A; Lee N; Yan W; Lyu H; Costello CE; Liu P; Zhang YJ, Snapshots of C-S cleavage in Egt2 reveals substrate specificity and reaction mechanism. Cell Chem. Biol 2018, 25, 519–529 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Naowarojna N; Irani S; Hu W; Cheng R; Zhang L; Li X; Chen J; Zhang YJ; Liu P, Crystal structure of the ergothioneine sulfoxide synthase from Candidatus Chloracidobacterium thermophilum and structure-guided engineering to modulate its substrate selectivity. ACS Catal. 2019, 9, 6955–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stampfli AR; Goncharenko KV; Meury M; Dubey BN; Schirmer T; Seebeck FP, An alternative active site architecture for O2 activation in the ergothioneine biosynthetic EgtB from Chloracidobacterium thermophilum. J. Am. Chem. Soc 2019, 141, 5275–5285. [DOI] [PubMed] [Google Scholar]
- (27).Naowarojna N; Huang P; Cai Y; Song H; Wu L; Cheng R; Li Y; Wang S; Lyu H; Zhang L; Zhou J; Liu P, In vitro reconstitution of the remaining steps in ovothiol A biosynthesis: C-S lyase and methyltransferase reactions. Org. Lett 2018, 20, 5427–5430. [DOI] [PubMed] [Google Scholar]
- (28).Braunshausen A; Seebeck FP, Identification and characterization of the first ovothiol biosynthetic enzyme. J. Am. Chem. Soc 2011, 133, 1757–9. [DOI] [PubMed] [Google Scholar]
- (29).Song H; Leninger M; Lee N; Liu P, Regioselectivity of the oxidative C–S bond formation in ergothioneine and ovothiol biosyntheses. Org. Lett 2013, 15, 4854–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Klein M; Ouerdane L; Bueno M; Pannier F, Identification in human urine and blood of a novel selenium metabolite, Se-methylselenoneine, a potential biomarker of metabolization in mammals of the naturally occurring selenoneine, by HPLC coupled to electrospray hybrid linear ion trap-orbital ion trap MS. Metallomics 2011, 3, 513–20. [DOI] [PubMed] [Google Scholar]
- (31).Yamashita M; Yamashita Y; Ando T; Wakamiya J; Akiba S, Identification and determination of selenoneine, 2-selenyl-N alpha , N alpha , N alpha -trimethyl-L-histidine, as the major organic selenium in blood cells in a fish-eating population on remote Japanese Islands. Biol. Trace Elem. Res 2013, 156, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yamashita Y; Yabu T; Yamashita M, Discovery of the strong antioxidant selenoneine in tuna and selenium redox metabolism. World J. Biol. Chem 2010, 1, 144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yamashita M; Yamashita Y; Suzuki T; Kani Y; Mizusawa N; Imamura S; Takemoto K; Hara T; Hossain MA; Yabu T; Touhata K, Selenoneine, a novel selenium-containing compound, mediates detoxification mechanisms against methylmercury accumulation and toxicity in zebrafish embryo. Mar. Biotechnol 2013, 15, 559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zayas ZP; Ouerdane L; Mounicou S; Lobinski R; Monperrus M; Amouroux D, Hemoglobin as a major binding protein for methylmercury in white-sided dolphin liver. Anal. Bioanal. Chem 2014, 406, 1121–1129. [DOI] [PubMed] [Google Scholar]
- (35).Reich HJ; Hondal RJ, Why nature chose selenium. ACS Chem. Biol 2016, 11, 821–41. [DOI] [PubMed] [Google Scholar]
- (36).Pluskal T; Ueno M; Yanagida M, Genetic and metabolomic dissection of the ergothioneine and selenoneine biosynthetic pathway in the fission yeast, S. pombe, and construction of an overproduction system. PLoS One 2014, 9, e97774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Turrini NG; Kroepfl N; Jensen KB; Reiter TC; Francesconi KA; Schwerdtle T; Kroutil W; Kuehnelt D, Biosynthesis and isolation of selenoneine from genetically modified fission yeast. Metallomics 2018, 10, 1532–1538. [DOI] [PubMed] [Google Scholar]
- (38).Birringer M; Pilawa S; Flohé L, Trends in selenium biochemistry. Nat. Prod. Rep 2002, 19, 693–718. [DOI] [PubMed] [Google Scholar]
- (39).Stadtman TC, Selenium biochemistry. Annu. Rev. Biochem 1990, 59, 111–27. [DOI] [PubMed] [Google Scholar]
- (40).Goncharenko KV; Fluckiger S; Liao CS; Lim D; Stampfli AR; Seebeck FP, Selenocysteine as a substrate, an inhibitor and a mechanistic probe for bacterial and fungal iron-dependent sulfoxide synthases. Chem-Eur J 2020, 26, 1328–1334. [DOI] [PubMed] [Google Scholar]
- (41).Jing X; Chen C; Deng X; Zhang X; Wei D; Yu L, Design and preparation of poly-selenides: easily fabricated and efficient organoselenium materials for heavy metal removing and recycling. Appl. Organomet. Chem 2018, 32, e4332. [Google Scholar]
- (42).Sharp KW; Koehler WH, Synthesis and characterization of sodium polyselenides in liquid ammonia solution. Inorg. Chem 1977, 16, 2258–2265. [Google Scholar]
- (43).Ejim LJ; Blanchard JE; Koteva KP; Sumerfield R; Elowe NH; Chechetto JD; Brown ED; Junop MS; Wright GD, Inhibitors of bacterial cystathionine β-lyase: leads for new antimicrobial agents and probes of enzyme structure and function. J. Med. Chem 2007, 50, 755–764. [DOI] [PubMed] [Google Scholar]
- (44).Dwivedi CM; Ragin RC; Uren JR, Cloning, purification, and characterization of. beta.-cystathionase from E. coli. Biochemistry 1982, 21, 3064–3069. [DOI] [PubMed] [Google Scholar]
- (45).Filipovic MR; Zivanovic J; Alvarez B; Banerjee R, Chemical biology of H2S signaling through persulfidation. Chem. Rev 2018, 118, 377–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yadav PK; Martinov M; Vitvitsky V; Seravalli J; Wedmann R; Filipovic MR; Banerjee R, Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc 2016, 138, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Esaki N; Nakamura T; Tanaka H; Suzuki T; Morino Y; Soda K, Enzymic synthesis of selenocysteine in rat liver. Biochemistry 1981, 20, 4492–4496. [DOI] [PubMed] [Google Scholar]
- (48).McCluskey T; Scarf A; Anderson J, Enzyme catalysed α, β-elimination of selenocystathionine and selenocystine and their sulphur isologues by plant extracts. Phytochemistry 1986, 25, 2063–2068. [Google Scholar]
- (49).Bourissou D; Guerret O; Gabbai FP; Bertrand G, Stable carbenes. Chem. Rev 2000, 100, 39–91. [DOI] [PubMed] [Google Scholar]
- (50).Soleilhayoup M; Bertrand G, Stable carbenes, nitrenes, phosphinidenes, and borylenes: past and future. Chem-Us 2020, 6, 1275–1282. [Google Scholar]
- (51).Lee JK; Houk K, A proficient enzyme revisited: the predicted mechanism for orotidine monophosphate decarboxylase. Science 1997, 276, 942–945. [DOI] [PubMed] [Google Scholar]
- (52).Goryanova B; Amyes TL; Gerlt JA; Richard JP, OMP Decarboxylase: phosphodianion binding energy is used to stabilize a vinyl carbanion intermediate. J. Am. Chem. Soc 2011, 133, 6545–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Meyer D; Neumann P; Ficner R; Tittmann K, Observation of a stable carbene at the active site of a thiamin enzyme. Nat. Chem. Biol 2013, 9, 488–U84. [DOI] [PubMed] [Google Scholar]
- (54).Kluger R, Thiamin diphosphate - a mechanistic update on enzymatic and nonenzymic catalysis of decarboxylation. Chem. Rev 1987, 87, 863–876. [Google Scholar]
- (55).Miller BG; Wolfenden R, Catalytic proficiency: The unusual case of OMP decarboxylase. Annu. Rev. Biochem 2002, 71, 847–885. [DOI] [PubMed] [Google Scholar]
- (56).Tsang WY; Wood BM; Wong FM; Wu WM; Gerlt JA; Amyes TL; Richard JP, Proton transfer from C-6 of uridine 5′-monophosphate catalyzed by orotidine 5′-monophosphate decarboxylase: formation and stability of a vinyl carbanion intermediate and the effect of a 5-fluoro substituent. J. Am. Chem. Soc 2012, 134, 14580–14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Toth K; Amyes TL; Wood BM; Chan K; Gerlt JA; Richard JP, Product deuterium isotope effects for orotidine 5 ′-monophosphate decarboxylase: effect of changing substrate and enzyme structure on the partitioning of the vinyl carbanion reaction intermediate. J. Am. Chem. Soc 2010, 132, 7018–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Amyes TL; Wood BM; Chan K; Gerlt JA; Richard JP, Formation and stability of a vinyl carbanion at the active site of orotidine 5‘-monophosphate decarboxylase: pKa of the C-6 proton of enzyme-bound UMP. J. Am. Chem. Soc 2008, 130, 1574–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Goryanova B; Goldman LM; Amyes TL; Gerlt JA; Richard JP, Role of a guanidinium cation–phosphodianion pair in stabilizing the vinyl carbanion intermediate of orotidine 5 ′ -phosphate decarboxylase-catalyzed reactions. Biochemistry 2013, 52, 7500–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Toth K; Amyes TL; Wood BM; Chan K; Gerlt JA; Richard JP, Product deuterium isotope effect for orotidine 5‘-monophosphate decarboxylase: evidence for the existence of a short-lived carbanion intermediate. J. Am. Chem. Soc 2007, 129, 12946–12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sievers A; Wolfenden R, Equilibrium of formation of the 6-carbanion of UMP, a potential intermediate in the action of OMP decarboxylase. J. Am. Chem. Soc 2002, 124, 13986–13987. [DOI] [PubMed] [Google Scholar]
- (62).Appleby TC; Kinsland C; Begley TP; Ealick SE, The crystal structure and mechanism of orotidine 5′ -monophosphate decarboxylase. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Becke A, Density - functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648. [Google Scholar]
- (64).Grimme S; Antony J; Ehrlich S; Krieg H, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- (65).Hehre WJ; Ditchfield R; Pople JA, Self-consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- (66).Francl MM; Pietro WJ; Hehre WJ; Binkley JS; Gordon MS; DeFrees DJ; Pople JA, Self - consistent molecular orbital methods. XXIII. A polarization - type basis set for second row elements. J. Chem. Phys 1982, 77, 3654–3665. [Google Scholar]
- (67).Barone V; Cossi M, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- (68).Cossi M; Rega N; Scalmani G; Barone V, Energies, structures, and electronic properties of molecules in solution with the C - PCM solvation model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (69).Amyes TL; Diver ST; Richard JP; Rivas FM; Toth K, Formation and stability of N-heterocyclic carbenes in water: the carbon acid pKa of imidazolium cations in aqueous solution. J. Am. Chem. Soc 2004, 126, 4366–4374. [DOI] [PubMed] [Google Scholar]
- (70).Oyala PH; Ravichandran KR; Funk MA; Stucky PA; Stich TA; Drennan CL; Britt RD; Stubbe J, Biophysical characterization of fluorotyrosine probes site-specifically incorporated into enzymes: E. coli ribonucleotide reductase as an example. J. Am. Chem. Soc 2016, 138, 7951–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Yu Y; Lv X; Li J; Zhou Q; Cui C; Hosseinzadeh P; Mukherjee A; Nilges MJ; Wang J; Lu Y, Defining the role of tyrosine and rational tuning of oxidase activity by genetic incorporation of unnatural tyrosine analogs. J. Am. Chem. Soc 2015, 137, 4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Seyedsayamdost MR; Reece SY; Nocera DG; Stubbe J, Mono-, di-, tri-, and tetra-substituted fluorotyrosines: new probes for enzymes that use tyrosyl radicals in catalysis. J. Am. Chem. Soc 2006, 128, 1569–1579. [DOI] [PubMed] [Google Scholar]
- (73).Chen L; Naowarojna N; Chen B; Xu ML; Quill M; Wang JY; Deng ZX; Zhao CM; Liu PH, Mechanistic studies of a nonheme iron enzyme OvoA in ovothiol biosynthesis using a tyrosine analogue, 2-amino-3-(4-hydroxy-3-(methoxyl) phenyl) propanoic acid (MeOTyr). ACS Catal. 2019, 9, 253–258. [Google Scholar]
- (74).Chen L; Naowarojna N; Song H; Wang S; Wang JY; Deng ZX; Zhao CM; Liu PH, Use of a tyrosine analogue to modulate the two activities of a nonheme iron enzyme OvoA in ovothiol biosynthesis, cysteine oxidation versus oxidative C-S bond formation. J. Am. Chem. Soc 2018, 140, 4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Minnihan EC; Young DD; Schultz PG; Stubbe J, Incorporation of fluorotyrosines into ribonucleotide reductase using an evolved, polyspecific aminoacyl-tRNA synthetase. J. Am. Chem. Soc 2011, 133, 15942–15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Lin CI; McCarty RM; Liu HW, The biosynthesis of nitrogen-, sulfur-, and high-carbon chain-containing sugars. Chem. Soc. Rev 2013, 42, 4377–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Dong L-B; Rudolf JD; Shen B, Antibacterial sulfur-containing platensimycin and platencin congeners from Streptomyces platensis SB12029. Bioorganic & Medicinal Chemistry 2016, 24, 6348–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Burkhart BJ; Schwalen CJ; Mann G; Naismith JH; Mitchell DA, YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem. Rev 2017, 117, 5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Forouhar F; Arragain S; Atta M; Gambarelli S; Mouesca JM; Hussain M; Xiao R; Kieffer-Jaquinod S; Seetharaman J; Acton TB; Montelione GT; Mulliez E; Hunt JF; Fontecave M, Two Fe-S clusters catalyze sulfur insertion by radical-SAM methylthiotransferases. Nat. Chem. Biol 2013, 9, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Landgraf BJ; Arcinas AJ; Lee KH; Booker SJ, Identification of an intermediate methyl carrier in the radical S-adenosylmethionine methylthiotransferases RimO and MiaB. J. Am. Chem. Soc 2013, 135, 15404–15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Leisinger F; Burn R; Meury M; Lukat P; Seebeck FP, Structural and mechanistic basis for anaerobic ergothioneine biosynthesis. J. Am. Chem. Soc 2019, 141, 6906–6914. [DOI] [PubMed] [Google Scholar]
- (82).Iiams V; Desai BJ; Fedorov AA; Fedorov EV; Almo SC; Gerlt JA, Mechanism of the orotidine 5 ′ -monophosphate decarboxylase-catalyzed reaction: Importance of residues in the orotate binding site. Biochemistry 2011, 50, 8497–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Barnett SA; Amyes TL; McKay Wood B; Gerlt JA; Richard JP, Activation of R235A mutant orotidine 5 ′ -monophosphate decarboxylase by the guanidinium cation: effective molarity of the cationic side chain of Arg-235. Biochemistry 2010, 49, 824–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Wood BM; Amyes TL; Fedorov AA; Fedorov EV; Shabila A; Almo SC; Richard JP; Gerlt JA, Conformational changes in orotidine 5 ′ -monophosphate decarboxylase:“Remote” residues that stabilize the active conformation. Biochemistry 2010, 49, 3514–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Wood BM; Chan KK; Amyes TL; Richard JP; Gerlt JA, Mechanism of the orotidine 5′ -monophosphate decarboxylase-catalyzed reaction: Effect of solvent viscosity on kinetic constants. Biochemistry 2009, 48, 5510–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Chan KK; Wood BM; Fedorov AA; Fedorov EV; Imker HJ; Amyes TL; Richard JP; Almo SC; Gerlt JA, Mechanism of the orotidine 5′ -monophosphate decarboxylase-catalyzed reaction: evidence for substrate destabilization. Biochemistry 2009, 48, 5518–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Toth K; Amyes TL; Wood BM; Chan KK; Gerlt JA; Richard JP, An examination of the relationship between active site loop size and thermodynamic activation parameters for orotidine 5 ′ -monophosphate decarboxylase from mesophilic and thermophilic organisms. Biochemistry 2009, 48, 8006–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Lewis CA Jr; Wolfenden R, Orotic acid decarboxylation in water and nonpolar solvents: a potential role for desolvation in the action of OMP decarboxylase. Biochemistry 2009, 48, 8738–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Lewis CA; Wolfenden R, Indiscriminate Binding by Orotidine 5 ‘-Phosphate Decarboxylase of Uridine 5 ‘-Phosphate Derivatives with Bulky Anionic C6 Substituents. Biochemistry 2007, 46, 13331–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Callahan BP; Wolfenden R, OMP Decarboxylase: An experimental test of electrostatic destabilization of the enzyme– substrate complex. J. Am. Chem. Soc 2004, 126, 14698–14699. [DOI] [PubMed] [Google Scholar]
- (91).Callahan BP; Wolfenden R, Charge development in the transition state for decarboxylations in water: Spontaneous and acetone-catalyzed decarboxylation of aminomalonate. J. Am. Chem. Soc 2004, 126, 4514–4515. [DOI] [PubMed] [Google Scholar]
- (92).Wittmann JG; Heinrich D; Gasow K; Frey A; Diederichsen U; Rudolph MG, Structures of the Human Orotidine-5′ -Monophosphate Decarboxylase Support a Covalent Mechanism and Provide a Framework for Drug Design. Structure 2008, 16, 82–92. [DOI] [PubMed] [Google Scholar]
- (93).Lewis RD; Garcia-Borras M; Chalkley MJ; Buller AR; Houk KN; Kan SBJ; Arnold FH, Catalytic iron-carbene intermediate revealed in a cytochrome C carbene transferase. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Cuevasanta E; Lange M; Bonanata J; Coitiño EL; Ferrer-Sueta G; Filipovic MR; Alvarez B, Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J. Biol. Chem 2015, 290, 26866–26880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Fernandez PL; Murkin AS, Inverse Solvent Isotope Effects in Enzyme-Catalyzed Reactions. Molecules 2020, 25, 1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Gaussian 16 Rev. C.01, Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Wallingford CT, 2016 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.