

Abstract
p53 is the most frequently mutated gene in squamous cell carcinomas (SCCs) of the skin and head and neck. Certain p53 mutations are oncogenic and promote invasion and metastasis in SCCs. However, it is unclear how the oncogenic function of mutant p53 is modulated by other molecular alterations that co-exist in SCCs. Here, we show that deletion of the p53 gene and activation of an endogenous p53R172H gain-of-function mutation in the skin induce carcinomas with similar kinetics and penetrance. Deletion of p53 induced primarily well differentiated SCCs. However, most of the tumors induced by p53R172H were poorly differentiated SCCs, the only metastatic tumors in this model. These tumors expressed higher levels of Cyclin D1 than the well differentiated SCCs and spindle carcinomas that developed in these mice. Unexpectedly, metastasis was not observed in mice that developed spindle carcinomas, which expressed high levels of the tumor suppressors p16Ink4a and p19Arf, encoded by Cdkn2a, a gene frequently deleted in human SCCs. Remarkably, deletion of the Cdkn2a gene in p53R172H-induced SCCs promoted a dramatic increase in metastasis rates and a shorter survival in mice that developed these tumors, compared with those observed in mice with tumors in which Cdkn2a was deleted in the presence of a p53 loss-of-function mutation or wild-type p53. Accordingly, the survival of patients with head and neck SCCs bearing co-occurring high-risk p53 mutations and CDKN2A homozygous deletions was much shorter than that of patients with tumors in which high-risk p53 mutations did not contain CDKN2A homozygous deletions, or that of patients with tumors in which homozygous CDKN2A deletions co-existed with either low-risk p53 mutations or potential loss-of-function mutations in p53. These findings genetically identify a population of SCC patients with worst outcomes and will help predict outcomes according to the p53 status and alterations in CDKN2A.
Keywords: p53, Cdkn2a, metastasis, squamous cell carcinoma
INTRODUCTION
The TP53 gene (also known as p53) is the most frequently mutated gene in squamous cell carcinomas (SCCs). Recent whole-exome sequencing of cutaneous SCCs (cSCCs) and head and neck SCCs (HNSCCs) identified p53 mutations in over 70% of the cases [1–5]. In addition, these studies uncovered broadly similar mutational profiles for cSCC and HNSCC, with potential tumor suppressor genes such as p53, CDKN2A and NOTCH1 being the most frequently mutated genes.
Most p53 mutations found in human cancers are missense mutations that encode mutant forms of p53. Some of the p53 mutations most commonly found in human cancers can transform or promote invasion in p53-null cells, including SCCs, as a result of acquired gain-of-function (GOF) properties [6,7]. However, human cancers display a broad p53 mutational spectrum, as almost every amino acid within the DNA binding domain has been found mutated in tumors [8]. These mutations encode altered forms of p53 that may differ structurally and functionally [9–11]. Different classification systems have been developed to identify p53 mutations that confer poor prognosis in HNSCC patients [12,13]. These studies demonstrated that a subset of p53 mutations are associated with more aggressive phenotypes, whereas other p53 mutations are linked with a more indolent pattern of tumor progression. Our recently developed system (EAp53) identified p53 mutations with GOF phenotypes, termed high-risk, that are highly prognostic of poor overall survival [13]. Similarly, outcomes from mouse models in which an endogenous p53R172H mutation (equivalent to human p53R175H, a hotspot GOF mutation in human cancer) or deletion of p53 were induced in Ras-initiated tumors support an oncogenic role for p53R172H during skin and oral SCC development [14,15]. Thus, mutant p53 may emerge as a primary oncogene in SCC. In those mice the oncogenic function of mutant p53 was found to be partially dependent on oncogenic Ras, but it is unclear how p53 mutations affect SCC development when activated in the absence of additional genetic alterations, and what effect their interaction with other specific genetic alterations has on the biology of SCCs.
Recently, global genomic analyses identified the tumor suppressor CDKN2A as the second most commonly altered gene in cSCCs and HNSCCs [1–5]. CDKN2A encodes two negative regulators of proliferation, p16INK4A and p14ARF (p19Arf in mice), which share common exons but are transcribed in alternative reading frames, resulting in the expression of two structurally unrelated proteins [16–18]. Mice deficient in Cdkn2a are predisposed to develop tumors, predominantly lymphomas and sarcomas [19–21]. Although Cdkn2a-null mice rarely developed spontaneous carcinomas, their exposure to chemical carcinogens accelerated carcinoma development, supporting a tumor suppressor role for Cdkn2a in cSCCs [21]. Interestingly, p16INK4A is often found upregulated in cSCCs and frequently associated with reduced differentiation and favorable prognosis [22,23].
In this study, we integrated mouse models of SCC with clinical and genomic analyses of human HNSCCs to investigate the role of p53 and CDKN2A alterations during SCC progression and metastasis.
MATERIALS AND METHODS
Mouse Models
The Neo-p53R172H mice, mice carrying floxed alleles for p53 (p53f) and Cdkn2a (Cdkn2af) and the K5.CrePR1 transgenic mice have been previously described [9,24–26]. Mouse genotyping and activation of conditional alleles was conducted by PCR of genomic DNA purified from mouse tails as described [9,24–26] (Supplementary Figure S1). All animals showing excessive tumor burden per the guidelines of The University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee (IACUC) were euthanized and tumors were harvested for histopathologic and molecular analyses. The thoracic and abdominal cavities were examined for evidence of metastasis. Lungs, liver, spleen and lymph nodes were harvested for histological analysis to assess the presence of metastasis. Experimental mice were generated in a mixed genetic background (C57BL/6/FVB/129Sv) and all comparative studies were conducted using littermates with the appropriate genotypes.
Histology and Immunohistochemistry
Tissues and tumors were fixed in 10% neutral-buffered formalin, transferred to 75% ethanol and embedded in paraffin. Histologic sections (5 µm) were stained with hematoxylin and eosin or processed for immunohistochemical analysis. Tissue sections used for immunohistochemistry were deparaffinized and rehydrated in xylene and alcohol series. Antigen retrieval was performed by incubating the samples in 100mM sodium citrate (pH 6.0). Endogenous peroxidase was blocked with 1% hydrogen peroxide solution. Tissue sections were incubated with primary antibodies for p16Ink4a and Zeb1 (Santa Cruz Biotechnology, Dallas, TX, USA), p19Arf (Abcam, Cambridge, MA, USA), p53 (Leica Microsystems, Buffalo Grove, IL, USA), K14 (BioLegend, San Diego, CA, USA) and E-cadherin (BD Biosciences, Franklin Lakes, NJ). Primary antibodies were detected using the ImmPRESS system (Vector Laboratories, Burlingame, CA, USA). Slides were visualized on a DMLA microscope (Leica Microsystems), and images were captured with an ORCA-ER camera (Hamamatsu, Shizuoka, Japan).
Quantitative Real-time PCR
Total RNA was isolated using TRIzol (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. RNA concentrations were determined with a NanoDrop system (Thermo Scientific, Wilmington, DE, USA). To measure mRNA expression, 1 μg of the total RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative real-time PCR was performed using the RT² SYBR Green qPCR Master Mix (Qiagen, Valencia, CA, USA) according to the manufacturer’s guidelines. Primer sequences (Supplementary Table S1) were designed using the Primer3 software or obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/). The genes expression level was normalized to that of the GAPDH gene in the same sample. Relative mRNA expression was calculated using the 2-ΔΔCt method. miRNAs expression was determined using TaqMan miRNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s protocol, using 10 ng of reverse transcribed total RNA. Quantitative RT-PCR was performed using Taqman Universal PCR Master Mix, No AmpErase UNG (Life Technologies) following the manufacturer’s protocol. miRNA expression levels were normalized to Sno202 mature miRNA. All quantitative real-time PCRs were performed on a CFX96 Real Time PCR Detection System (Bio-Rad, Hercules, CA, USA).
Western Blot Analysis
Total protein lysates were prepared by homogenizing the samples in Laemmli protein loading buffer and heating for 10min at 100°C. Equal amounts of protein from each sample were separated by SDS-PAGE and transferred onto Hybond-P membranes (GE Healthcare Life Sciences, Piscataway, NJ). The membranes were incubated with specific antibodies for p16Ink4a and actin (Santa Cruz Biotechnology), p19Arf (Abcam), and vimentin, Erk, p-Erk and Cyclin D1 (Cell Signaling Technology, Danvers, MA, USA). After the primary antibodies were washed, the signal was detected by incubating the membranes with secondary antibodies labeled with horseradish peroxidase and visualized with the ECL Plus chemiluminescent detection system (GE Healthcare Life Sciences).
Analysis of Genomic and Clinical Data from Human HNSCCs
The genomic status for the p53 and CDKN2A genes, and clinical data for 432 HPV[-] HNSCCs for which complete mutation and clinical information was available from The Cancer Genome Atlas (TCGA) was acquired from the 28 January 2016 update of the Firebrowse database (http://firebrowse.org). The HPV status of the tumors was obtained from Tang et al. [27] and from Firebrowse. The p53 mutations were classified as high- and low-risk according to the EAp53 classification system [13] (Supplementary Table S2). Nonsense, frameshift, indels and splice site mutations were grouped under the non-missense category.
Statistics
Tumor development in mice and patient survival was represented using Kaplan–Meier plots, and the differences between groups were assessed by the log-rank test. Gene expression in the different groups and tumor phenotypes were compared using the 2-tailed Student t-test. Error bars represent the mean ± standard error of the mean. Difference in metastasis between the groups was assessed by the Chi-square method. P-values <0.05 were considered statistically significant.
RESULTS
The Oncogenic GOF of p53R172H Promotes SCC Progression and Metastasis, but not SCC Initiation
p53 mutations have been found in epidermal cells of sun-exposed skin and in premalignant lesions that may be SCC precursors, suggesting that they are early events in SCC development [28–32]. However, it is unclear how p53 GOF and loss-of-function (LOF) mutations affect SCC development when the p53 mutations arise in the earliest stages, before other genetic alterations accumulate.
To address this question, we generated mice in which the p53R172H allele or deletion of p53 were activated in mouse stratified epithelia. The p53R172H mutation was induced after crossing mice that carry a conditional p53R172H allele (Neo-p53R172H mice) with K5.CrePR1 mice [9,26]. The remaining wild-type (wt) p53 allele was deleted after additional crossings with floxed-p53 (p53f) mice [24] to generate p53R172H/f mice. To induce homozygous deletion of p53, we generated mice with both copies of the p53f allele, in the presence of K5.CrePR1 (p53f/f mice). In K5.CrePR1 mice, CrePR1 is expressed in stratified epithelia with focal leaky activity. We further induced Cre in the skin by topical application of RU486 (1mg/ml) daily for 5 days.
We found that p53R172H/f and p53f/f mice developed carcinomas with similar kinetics and tumor multiplicity, with an average latency of 15–16 months (Figures 1A and 1B). Specifically, 79% of the p53f/f mice and 72% of the p53R172H/f mice developed skin or oral SCCs. Over 60% of the mice of both genotypes developed skin tumors, and 25% of the p53f/f mice and 16% of the p53R172H/f mice developed oral tumors, in most cases with concurrent skin tumors (Supplementary Table S3). The majority (12/18, 67%) of the skin tumors that developed in p53f/f mice (p53−/− tumors) were well to moderately differentiated SCCs (wSCCs), 11% (2/18) of the p53−/− tumors were poorly differentiated SCCs (pSCCs) and 22% of them (4/18) were spindle cell carcinomas (Figures 1C and 1D). In contrast, most of the skin tumors (11/20, 55%) that developed in p53R172H/f mice (p53R172H/− tumors) were pSCCs, only 20% of them (4/20) were wSCCs and 25% (5/20) were spindle cell carcinomas (Figure 1D).
Figure 1.
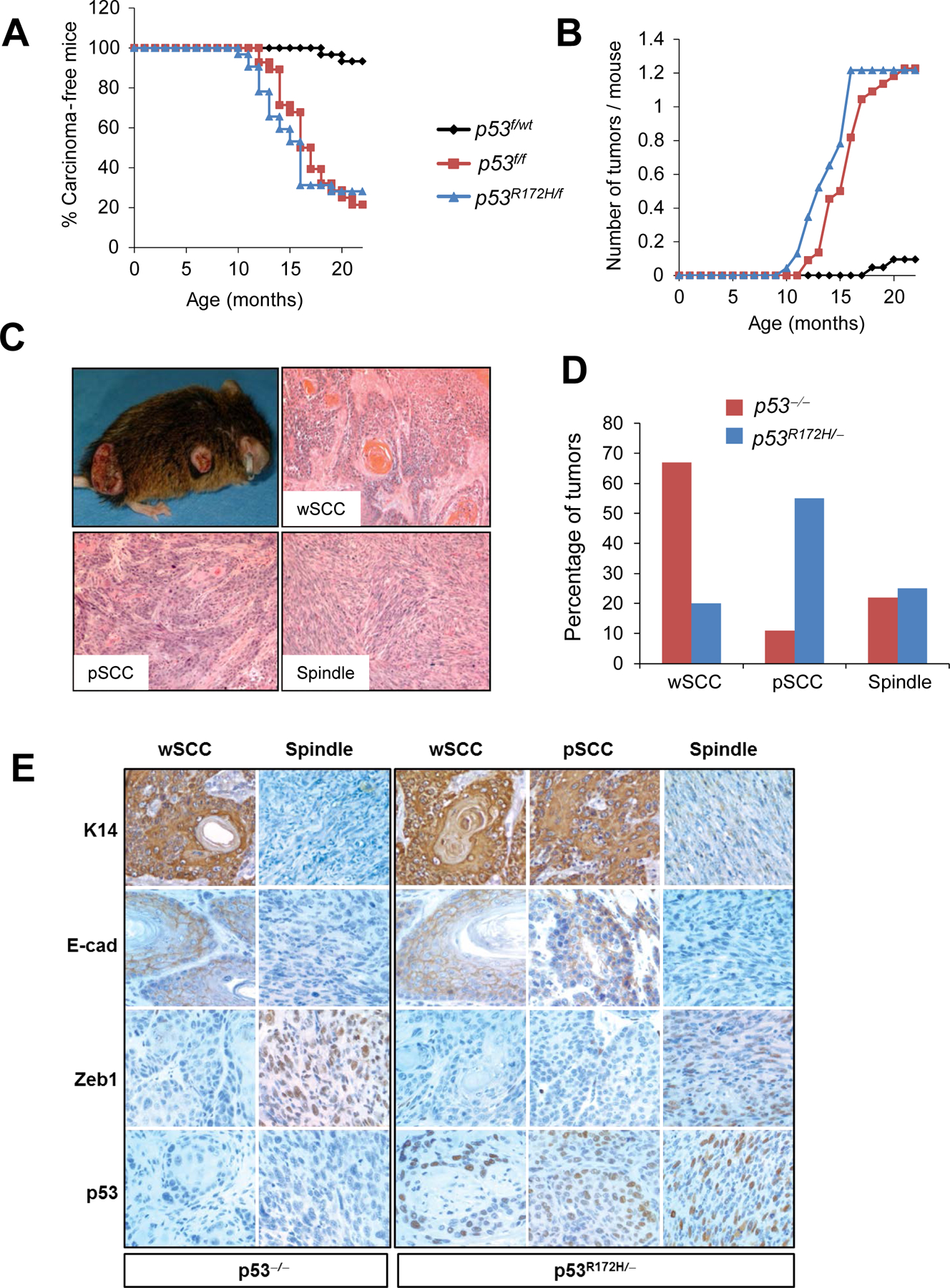
Activation of p53R172H in the skin induces SCC progression. (A) Kinetics of carcinoma development in mice with the indicated genotypes. Note that p53R172H/f mice (n=32) and p53f/f mice (n=28) developed SCCs with similar kinetics. Mice with wild-type p53 (p53f/wt) (n=30) were used as controls. P< 0.05 for the following comparisons: p53R172H/f vs. p53f/wt and p53f/f vs. p53f/wt. (B) Kinetics of tumor multiplicity showing number of tumors per mouse over time. (C) Gross appearance and histology of skin carcinomas induced by p53 mutations. The three main tumor phenotypes observed are represented. (D) Quantification of tumor phenotypes observed in p53R172H/- and p53−/− carcinomas. P< 0.005 for p53R172H/- tumors with pSCC phenotype compared to p53−/− tumors with pSCC phenotype. (E) Immunohistochemical analysis of Keratin 14 (K14), E-cadherin (E-cad), Zeb1 and p53 in the indicated tumors.
E-cadherin and keratin 14 (K14) were highly expressed in wSCC and pSCCs, but they were absent in spindle tumors (Figure 1E and Supplementary Figure S2). Unlike wSCC and pSCCs, spindle tumors expressed the mesenchymal marker Zeb1 (Figure 1E and Supplementary Figure S2).
These results indicate that mutant p53R172H and deletion of p53 predisposed to SCC with similar potential when these alterations were activated in the absence of additional genetic alterations. However, p53R172H tumors progressed to a poorly differentiated phenotype that proved to be more aggressive. Indeed, we observed that 28% (5/18) of the p53R172H/f mice analyzed developed metastasis, compared to only 6% (1/18) of the p53f/f mice. Remarkably, metastases developed only in mice carrying pSCCs, with 50% of the mice with pSCC developing metastasis (5 of 10 of the p53R172H/f mice and 1 of 2 of the p53f/f mice). In contrast, metastases were not observed in mice that developed wSCC or spindle carcinomas.
Overexpression of Cyclin D1 in pSCCs Induced by p53 Mutations
Since mutations in Ras genes were previously found to cooperate with p53R172H during SCC progression and metastasis [14], we sequenced H-ras and K-ras in p53R172H/− and p53−/− SCCs. While mutations in K-ras were not observed, H-ras mutations were found in 10% of the tumors analyzed, all of them p53R172H/− (3/16; one wSCC and two pSCCs) (Figure 2A). Subsequently, we analyzed whether Erk might be activated in the tumors independently of Ras mutations. Surprisingly, we observed that Erk activity was blocked in p53R172H/− and p53−/− SCCs, irrespective of the Ras mutational status (Figure 2B). Moreover, we observed that the tumor suppressor p16Ink4a was induced in the majority of the SCCs analyzed (Figure 2B).
Figure 2.
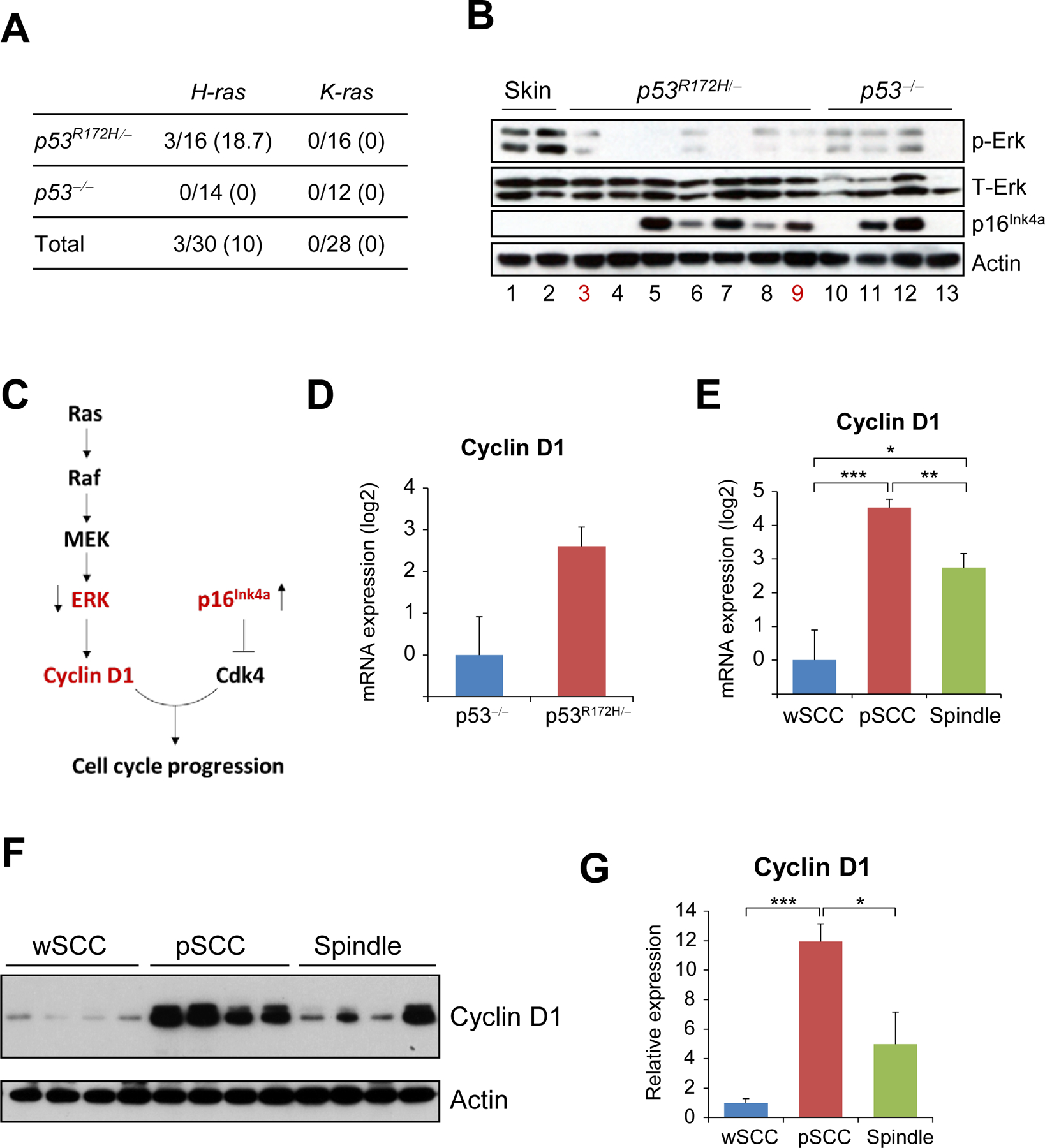
Overexpression of Cyclin D1 in pSCCs induced by p53 mutations. (A) Mutation rates for H-ras and K-ras in p53R172H/- and p53−/− tumors. The data represents number of tumors with mutations in each gene, with percentages shown in parentheses. (B) Western blot analysis of the indicated proteins and tissue samples. Lane numbers in red indicate tumors with Ras mutation; all other samples had wt Ras. (C) Schematic representation of the interaction between ERK and p16Ink4a. The arrows next to ERK and p16Ink4a show the relative expression found in tumors induced by p53 mutations. (D) mRNA expression of Cyclin D1 in p53R172H/- tumors (n=13) and p53−/− tumors (n=11). Note that the difference is not significant. (E) Cyclin D1 mRNA expression was compared in tumors with the indicated tumor phenotypes: wSCC (n=8), pSCC (n=8), spindle (n=8). (F) Western blot analysis of Cyclin D1 in tumors with the indicated phenotypes. Actin was used as a loading control. (G) Quantification of the Cyclin D1 expression in the western blots shown on (F), normalized to actin. *P< 0.05, **p< 0.005, ***p< 0.0005.
We speculated that Erk was inactive because other components of the pathway acting downstream Erk may have been induced through Erk-independent mechanisms, and as a result, Erk was inactivated by negative feedback loops. As Cyclin D1 functions downstream both Erk and p16Ink4a (Figure 2C), and is a known oncogene in human SCC, we analyzed the expression of Cyclin D1 in p53R172H/− and p53−/− tumors. We found that Cyclin D1 mRNA expression was slightly higher in p53R172H/− tumors than in p53−/− tumors, although the difference was not statistically significant (Figure 2D). However, a more detailed analysis of the p53R172H/− tumors revealed that Cyclin D1 mRNA and protein expression was significantly higher in pSCCs than in either wSCCs or spindle tumors (Figure 2E–G). These results suggest that overexpression of Cyclin D1 in p53R172H/− pSCCs may contribute to the aggressive phenotype of these tumors.
Upregulation of p16Ink4a and p19Arf in Spindle Tumors Induced by p53 Mutations
The presence of metastasis in mice with pSCCs, but not in mice with spindle tumors was surprising because spindle cell carcinomas are considered to represent the latest stages in skin carcinogenesis [33]. Although Cyclin D1 expression was highest in pSCCs, spindle tumors expressed higher levels of Cyclin D1 than wSCCs. However, metastasis was not observed neither in wSCCs nor in spindle carcinomas. As p16Ink4a antagonizes the Cyclin D1 function and was found upregulated in a subset of SCCs induced by p53 mutations (Figure 2B), we investigated in more detail the potential role of the Cdkn2a gene products in these tumors.
We observed that the mRNA expression of both p16Ink4a and p19Arf tended to be higher in p53R172H/− tumors than in p53−/− tumors, although the difference was not statistically significant (Supplementary Figure S3A). However, analysis of the expression of p16Ink4a and p19Arf relative to the major tumor phenotypes observed in these mice revealed that spindle tumors expressed the highest levels of mRNA and protein for both p16Ink4a and p19Arf (Figures 3A, 3B, and Supplementary Figure S3B). Of note, although no significant differences were found in direct comparisons between metastatic and non-metastatic pSCCs, non-metastatic pSCCs, but not metastatic pSCCs, expressed significantly higher levels of p16Ink4a compared to wSCC, suggesting a trend toward higher expression of p16Ink4a in non-metastatic pSCCs (Supplementary Figure S4). Immunohistochemical analysis showed that p16Ink4a and p19Arf staining was diffuse or absent in wSCCs and predominantly nuclear in pSCCs and spindle tumors (Figure 3C). Overall, these findings suggest that p16Ink4a and p19Arf were induced during tumor progression, reaching the highest levels in the most undifferentiated tumors. The high expression of p16Ink4a and p19Arf observed in the spindle tumors was surprising, considering the tumor suppressor functions of both p16Ink4a and p19Arf. Based on these data, we hypothesized that upregulation of p16Ink4a and p19Arf in SCCs induced by mutant p53 prevents malignant progression and metastasis, which may explain the lack of metastasis in mice with tumors that expressed high levels of p16Ink4a and p19Arf.
Figure 3.
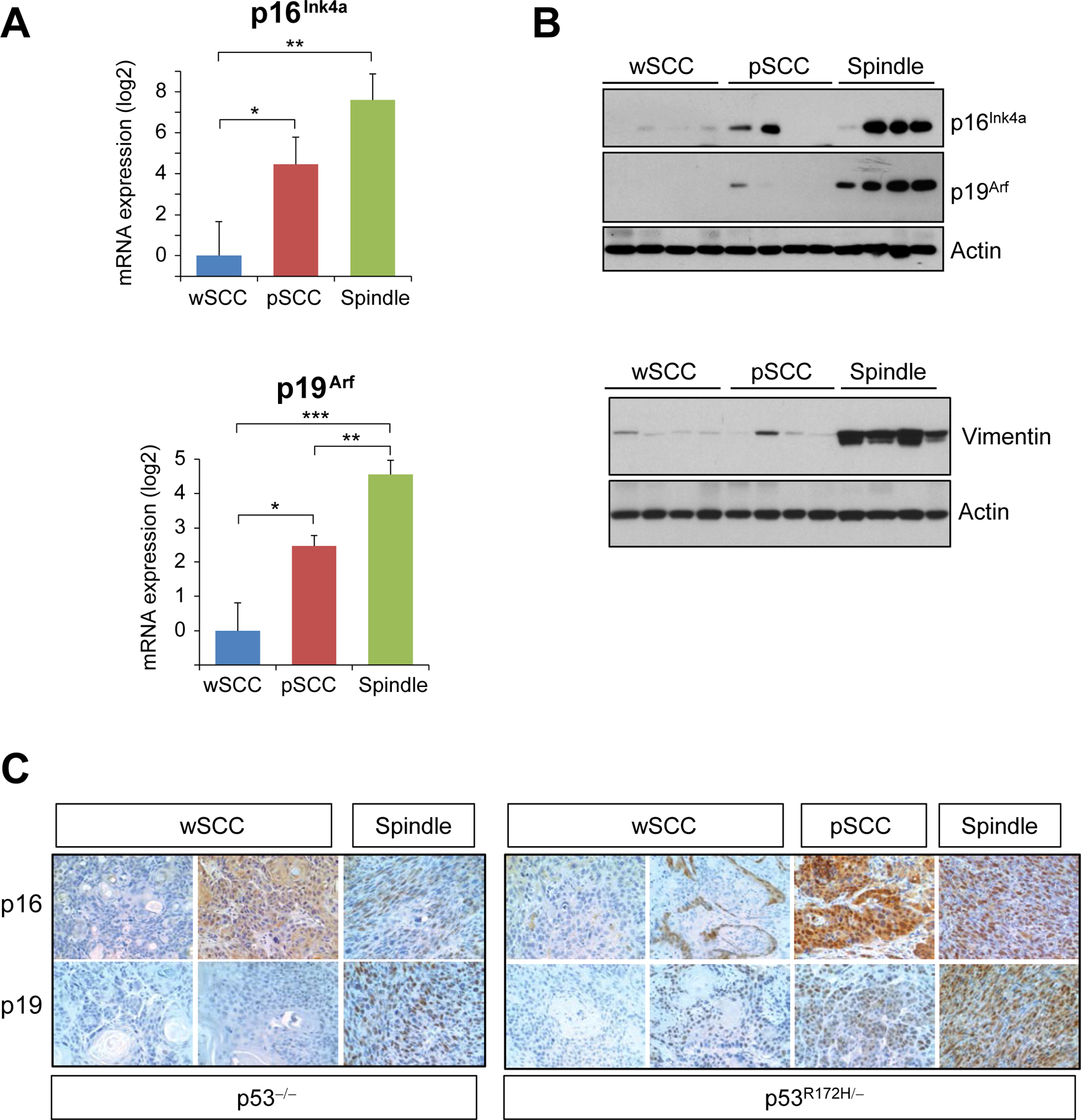
Upregulation of p16Ink4a/p19Arf in spindle carcinomas induced by p53 mutations. (A) mRNA expression levels of p16Ink4a and p19Arf in wSCCs (n=8), pSCCs (n=8), spindle carcinomas (n=8). (B) Western blot analysis of p16Ink4a and p19Arf in p53R172H/- carcinomas (top panel) and vimentin (bottom panel) in tumors with the indicated phenotypes. Note the high expression of the mesenchymal marker vimentin in spindle carcinomas. Actin was used as a loading control. (C) Immunohistochemical analysis for p16Ink4a and p19Arf in tumors with the indicated phenotypes and genotypes. *P< 0.05, **p< 0.005, ***p< 0.0005.
Deletion of Cdkn2a Promotes Metastasis in Skin Carcinomas Induced by p53R172H
To assess the role of p16Ink4a and p19Arf in SCCs induced by p53 alterations we deleted the Cdkn2a gene in stratified epithelia in the presence or absence of p53 GOF or LOF mutations. To delete the Cdkn2a gene in the skin, we crossed mice carrying a floxed-Cdkn2a allele (Cdkn2af) [25] with K5.CrePR1 mice. To induce the additional activation of the p53R172H mutation or p53 deletion, these mice were crossed to p53f and Neo-p53R172H mice to generate the following groups of mice: Cdkn2af/f;p53R172H/f, Cdkn2af/f;p53f/f, Cdkn2af/f;p53wt/wt and Cdkn2awt/wt;p53f/f.
We observed that mice in which Cdkn2a was deleted in the presence of the p53R172H mutation (Cdkn2af/f;p53R172H/f) developed skin tumors much faster than any other group, including mice in which Cdkn2a and p53 were co-deleted (Cdkn2af/f;p53f/f) (Figure 4A). Remarkably, all tumors that developed upon deletion of Cdkn2a, regardless of the p53 status, were spindle tumors (Figure 4B). In addition to the accelerated tumor rates, we noted that 79% of the Cdkn2af/f;p53R172H/f mice developed metastasis, primarily in liver and/or spleen, compared to 23% of the Cdkn2af/f;p53f/f mice and none of the mice in which Cdkn2a was deleted in the presence of wt p53 (Cdkn2af/f;p53wt/wt) (Figures 4B and 4C and Supplementary Table S4). These findings indicate that Cdkn2a deletion facilitates tumor development when co-occurring with the p53R172H mutation and promotes metastasis in tumors carrying this oncogenic p53 mutation.
Figure 4.
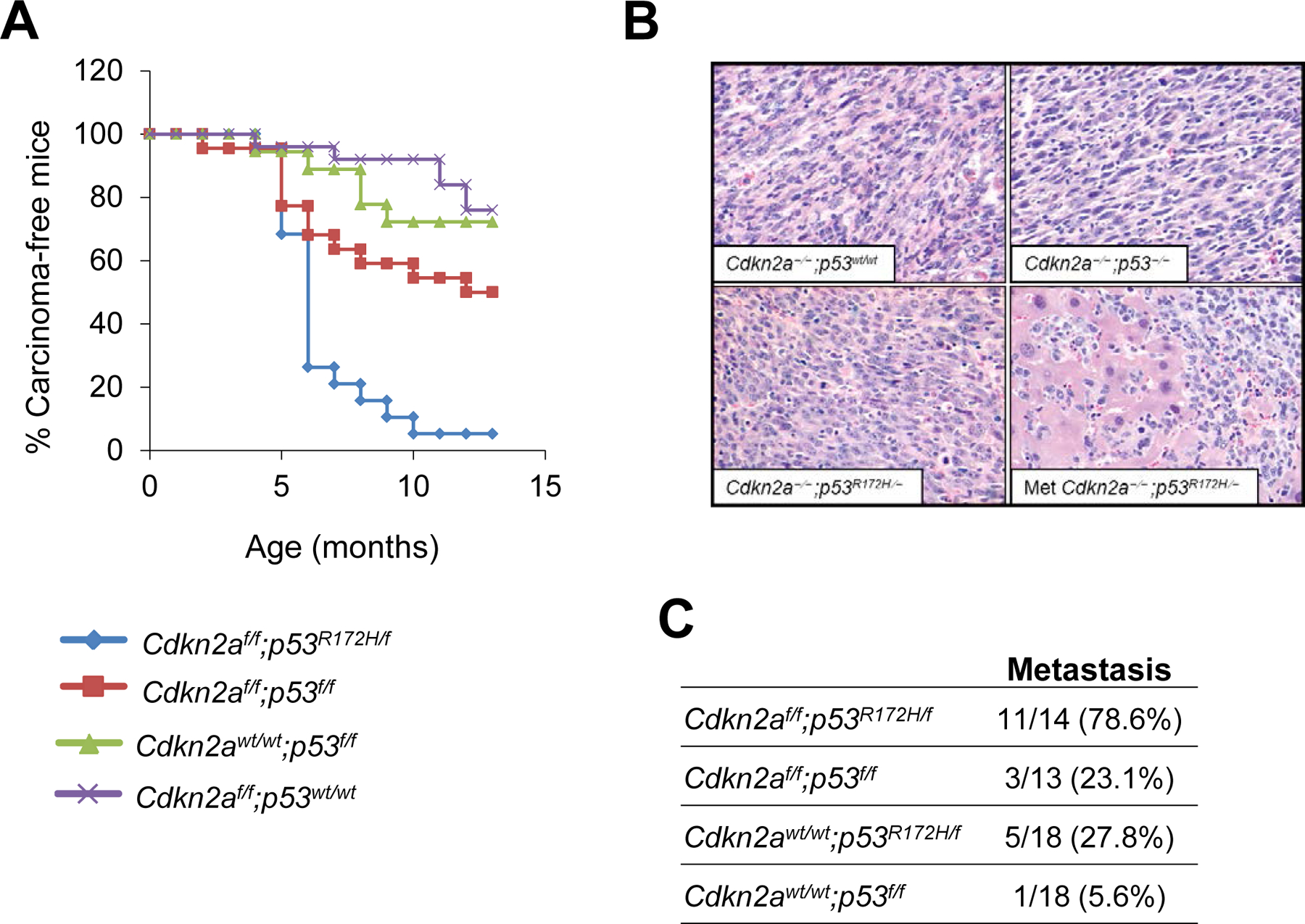
Cdkn2 deletion accelerates skin tumor formation and promotes metastasis in carcinomas induced by mutant p53. (A) Tumor kinetics in mice with the following genotypes: Cdkn2af/f;p53R172H/f (n=22), Cdkn2af/f;p53f/f (n=27), Cdkn2af/f;p53wt/wt (n=30) and Cdkn2awt/wt;p53f/f (n=23). *P< 0.05 for the following comparisons: Cdkn2af/f;p53R172H/f with each of the other groups; Cdkn2af/f;p53f/f with Cdkn2af/f;p53wt/wt. (B) Hematoxylin and eosin staining of primary tumors with the indicated genotypes. Note the spindle appearance of all tumors that lack Cdkn2a, regardless of the p53 status. The lower right panel shows a representative image for a liver metastasis that developed from a tumor induced by deletion of Cdkn2a and activation of p53R172H. (C) Metastasis rates in mice with the indicated genotypes. P<0.05 for comparisons between Cdkn2af/f;p53R172H/f and the rest of the groups.
Molecular analysis revealed a marked downregulation of the mRNA expression of epithelial markers, including Mir200c and E-cadherin, and upregulation of mesenchymal markers such as N-cadherin, vimentin, twist and Zeb1, in all tumors that lacked Cdkn2a, suggesting that these tumors undergo epithelial-to-mesenchymal transition (EMT) (Figure 5A). However, the expression of EMT markers was similar in Cdkn2a-deficient tumors carrying p53R172H, loss of p53 or wt p53, indicating that the spindle phenotype was primarily determined by the Cdkn2a deletion. Immunohistochemical analysis confirmed the loss of epithelial markers and upregulation of mesenchymal markers in Cdkn2a-deficient carcinomas (Figure 5B).
Figure 5.
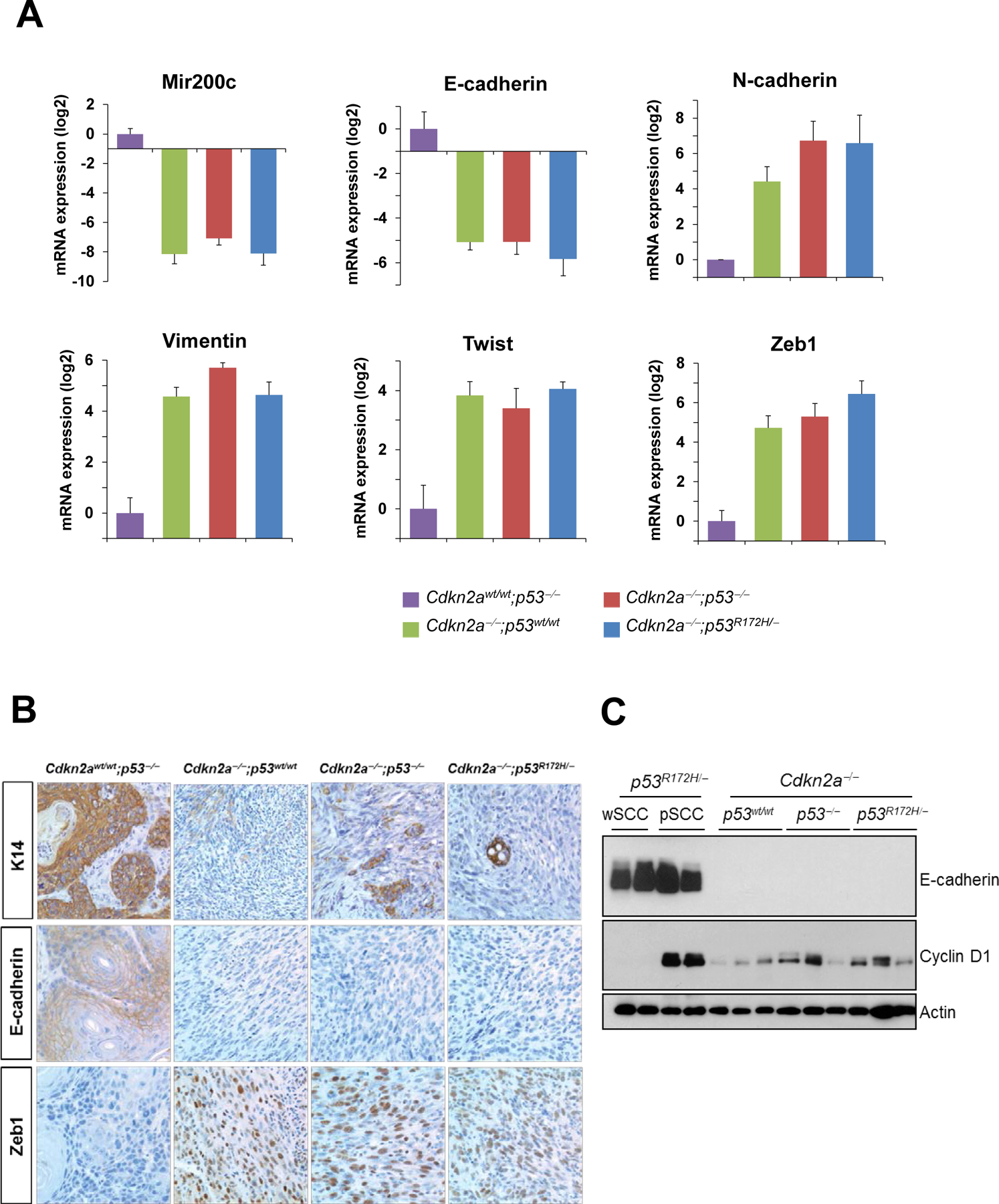
Tumors induced by Cdkn2a deletion exhibit spindle phenotypes. (A) mRNA expression of epithelial and mesenchymal markers in skin tumors with the indicated genotypes. Cdkn2awt/wt;p53−/− wSCCs were used as representative epithelial tumors. Note the downregulation of epithelial markers and induction of mesenchymal markers in tumors lacking Cdkn2a, regardless of the p53 status. The difference between Cdkn2awt/wt;p53−/− tumors and each of the other groups was statistically significant (p<0.05) for all the genes analyzed. The rest of comparisons were not significant. (B) Immunohistochemical analysis for K14, E-cadherin, and Zeb1 in tumors with the indicated genotypes. (C) Western blot analysis of E-cadherin and Cyclin D1 in tumors with the indicated phenotypes and genotypes. Actin was used as a loading control.
Because we previously showed that Cyclin D1 overexpression is associated with malignant progression in SCCs induced by p53 alterations in the context of wt Cdkn2a, we analyzed the expression of Cyclin D1 in tumors with homozygous deletion of Cdkn2a. We observed that Cdkn2a-deficient tumors, regardless of the p53 status, expressed slightly higher levels of Cyclin D1 than wSCCs induced by p53 alterations but lower than p53R172H pSCCs (Figure 5C). The lower levels of Cyclin D1 found in the highly metastatic Cdkn2a−/−;p53R172H/− tumors compared to Cdkn2awt/wt;p53R172H/− pSCCS supports the idea that the metastatic potential of mutant p53 is promoted by either inactivation of Cdkn2a or overexpression of Cyclin D1.
Deletion of CDKN2A Confers Poor Prognosis in Patients with HNSCCs Carrying High-risk p53 Mutations
To determine whether the cooperation between oncogenic p53R172H and deletion of Cdkn2a that we identified in mouse models of SCC is relevant to human disease, we analyzed genomic and clinical data available in TCGA for HNSCCs. Because HPV[+] and HPV[-] HNSCCs are genetically and clinically different entities, and p53 mutations are rarely found in HPV[+] HNSCCs, we focused this analysis on HPV[-] HNSCCs. The p53 mutations were classified as high- and low-risk according to the EAp53 classification system [13]. High-risk p53 mutations were found to promote invasion and metastasis in head and neck cancer cell lines, as they acquired oncogenic gain-of-function properties, while low-risk mutations retained p53 activity and were grouped with wt p53 [13]. Nonsense, frameshift, indels and splice site mutations were grouped in this study under a non-missense category that better resembles p53 loss-of-function.
We observed that p53 mutations were not associated with survival rates in HPV[-] HNSCCs patients (Supplementary Figure 5A). By dividing the p53 mutations into high-risk, low-risk and non-missense, we found that high-risk mutations were marginally associated with lower survival rates, compared with low-risk mutations, but not with non-missense mutations (Supplementary Figure 5B). Interestingly, we found that the overall survival of patients with p53 mutations was not increased significantly in the presence of homozygous deletions of CDKN2A deletion (Figure 6A). However, when the p53 mutations were separated into high-risk, low-risk and non-missense we found that patients with tumors with co-occurring high-risk p53 mutations and homozygous deletion of CDKN2A had a much shorter survival than those in which high-risk p53 mutations were not accompanied by homozygous deletion of CDKN2A (Figure 6B). In contrast, homozygous deletion of CDKN2A did not alter the survival rates of patients with p53 low-risk or non-missense mutations (Supplementary Figure 5C). Moreover, the survival of patients with co-existing high-risk p53 mutations and homozygous deletion of CDKN2A was much shorter than that of patients in which CDKN2A homozygous deletion co-existed with p53 low-risk or non-missense mutations (Figure 6B). These observations indicate that high-risk p53 mutations confer poorer prognosis when the mutations occur in tumors in which the CDKN2A gene is deleted.
Figure 6.
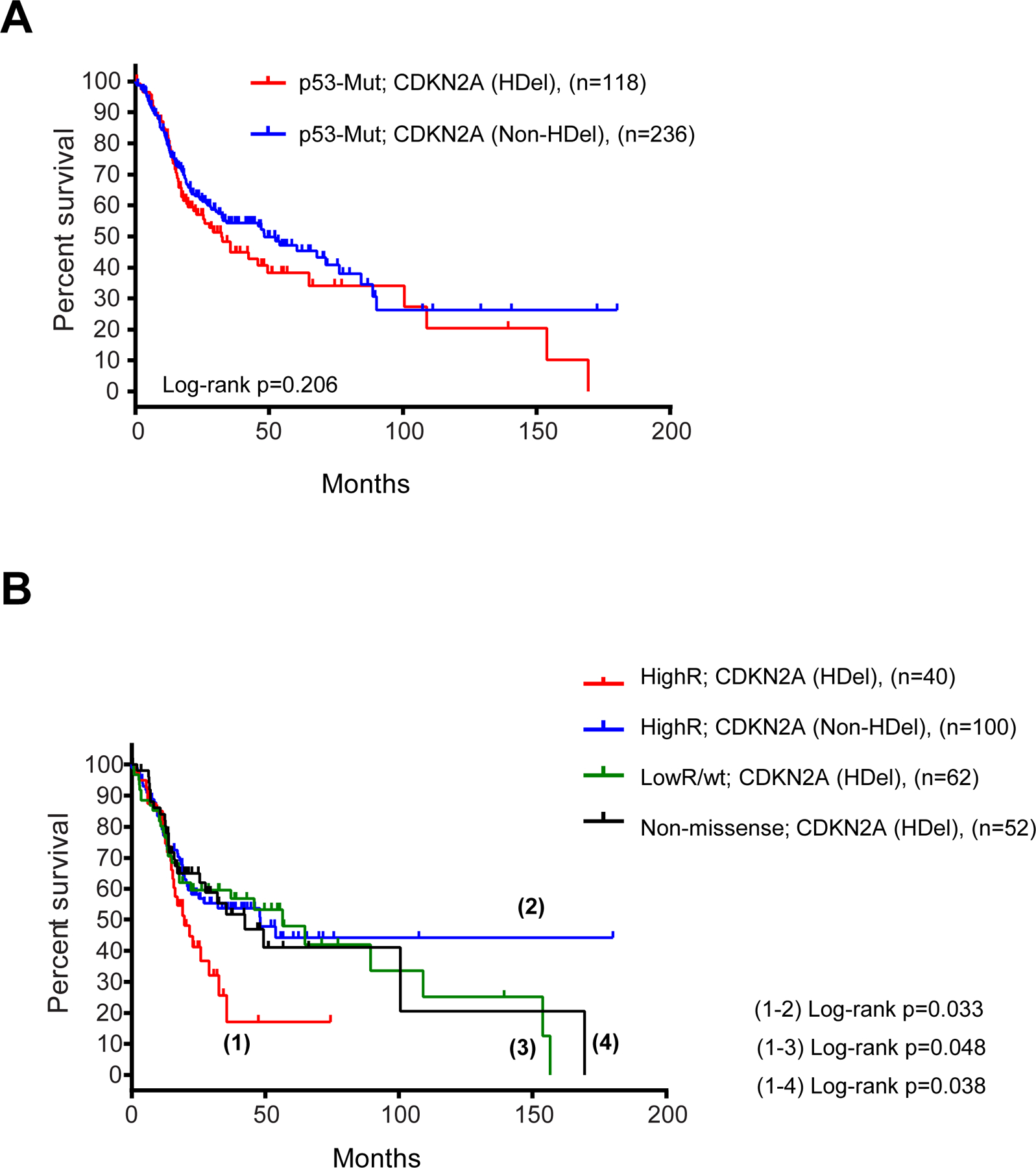
Deletion of CDKN2A confers poor prognosis to HNSCCs that carry high-risk p53 mutations. (A) Kaplan-Meier plot for the survival of HNSCC patients with tumors carrying homozygous deletions (HDel) of CDKN2A or tumors that do not contain homozygous deletions of CDKN2A (Non-HDel). (B) Survival of HNSCC patients with tumors containing the indicated p53 mutations, high-risk (HighR), low-risk (LowR) or non-missense, in the presence or absence of homozygous deletions in CDKN2A, as indicated. Log-rank p values are indicated for the statistically significant comparisons.
DISCUSSION
Mutations in p53 are found in the majority of the HNSCCs and cSCCs. During the past years, clinical and experimental studies have shown that certain p53 mutations are oncogenic. However, the molecular context that determines the oncogenic potential of p53 mutations in SCCs is unclear. This question is particularly relevant in light of recent cancer genome studies showing that p53 mutations co-exist with a wide array of genetic alterations, including homozygous deletions of the CDKN2A gene. This study demonstrates that although both, the mutant p53R172H and deletion of the p53 gene, induced SCCs with similar high penetrance and long latency, p53R172H, but not deletion of the p53, promoted progression to highly metastatic pSCCs. The high levels of Cyclin D1 in the most aggressive tumors suggests that Cyclin D1 overexpression in tumors that carry the oncogenic p53R172H mutation contributes to tumor progression. Notably, the highest expression levels of the tumor suppressors p16Ink4a and p19Arf were preferentially found in non-metastatic tumors, suggesting that p16Ink4a/p19Arf may block metastasis in these tumors. Indeed, deletion of Cdkn2a in tumors induced by p53R172H accelerated tumor development, increased the metastasis rates and resulted in decreased survival. In agreement with these findings, HNSCC patients with tumors with co-occurring high-risk p53 mutations and homozygous deletions in CDKN2A had the worst outcomes.
A lack of gain of function during skin cancer development has been reported for the gain-of-function mutation p53R270H [34]. Since our previous studies demonstrated that p53R172H promoted metastasis in cSCCs initiated by oncogenic K-ras much more frequently than deletion of the p53 gene, it was unclear whether the contrasting effects observed for p53R270H and p53R172H in these reports were mutation-specific or cell-context dependent [14]. Our present studies suggest that the gain-of-function activities of p53R172H in SCCs are silent or minimized in the absence of additional genetic alterations, as tumors induced by p53R172H and deletion of p53 developed with similar kinetics. Since p53R172H SCCs progressed to pSCC and became metastatic much more frequently than those induced by deletion of p53, one may speculate that molecular alterations that accumulate during tumor progression cooperate with mutant p53 during malignant progression. Indeed, we observed that Cyclin D1, a known oncogene in human SCCs [35,36], is overexpressed in pSCCs, the only metastatic tumors in this model. We propose that the high levels of Cyclin D1 in these tumors may overcome the expression of the tumor suppressors p16Ink4a/p19Arf. Conversely, the higher expression of p16Ink4a/p19Arf in spindle tumors may counteract Cyclin D1 and prevent metastasis. Thus, a balance between Cyclin D1 and p16Ink4a may control SCC progression and metastasis. This is also supported by the high metastasis rates observed in tumors induced by co-activating p53R172H and deletion of Cdkn2a.
Traditional chemical carcinogenesis models posit that skin cancers arise through a step-wise process in which benign lesions progress to wSCCs, some of which may convert to pSCCs. A subset of these tumors may undergo EMT and progress to highly aggressive and undifferentiated spindle cell carcinomas [33]. This linear progression model was recently challenged by the notion that a subset of spindle tumors induced by DMBA/TPA may arise from a separate route that does not involve progression from SCCs [37,38]. Remarkably, these spindle tumors showed frequent inactivation of Cdkn2a, unlike wSCCs induced by DMBA/TPA [39,40]. Our results indicate that epidermal deletion of Cdkn2a induces spindle carcinomas that do not seem to evolve from SCCs, suggesting different cells of origin for SCCs and Cdkn2a-deficient spindle tumors. Alternatively, these phenotypes may reflect the potential plasticity of the tumor cells of origin, as differentiating cells are susceptible to being reprogrammed to acquire stem cell features. Since Cdkn2a acts a barrier to prevent stem cell reprograming [41,42], it is also conceivable that SCCs and spindle tumors arise from a common cell of origin, and that loss of Cdkn2a may facilitate reprogramming to cell lineages that, upon transformation, develop into spindle tumors.
It is well-established that EMT induces structural and morphological changes in epithelial tumors that allow for more effective invasion and metastasis, leading to the assumption that acquiring spindle phenotypes is generally linked to malignancy [43]. However, the clinical prognosis of human spindle carcinomas remains uncertain, as they have been associated with favorable outcomes in some studies and with poor prognosis in others [44]. Our results can help reconcile these contrasting views, since spindle carcinomas induced by p53 mutations or by deletion of Cdkn2a did not metastasize, in contrast to the high metastasis rates observed in mice with spindle tumors induced by co-activation of the p53R172H mutation and Cdkn2a deletion.
In humans, loss of CDKN2A in SCCs has been found correlated with poor prognosis [45]. Remarkably, our TCGA analysis revealed that CDKN2A deletion is associated with worse outcomes in HNSCCs that also contain high-risk p53 mutations, but not low-risk mutations or potential loss-of-function mutations in p53. These findings support the concept that CDKN2A suppresses the oncogenic function of mutant p53 that promotes malignant progression in SCCs, and the prognostic value of mutant p53 needs to be considered in the context of CDKN2A. Since the survival rates of HNSCCs patients with tumors carrying high risk p53 mutations and homozygous deletion of CDKN2A are extremely low, this is a patient population in urgent need of more effective therapies.
Supplementary Material
ACKNOWLEDGMENTS
The clinical and genomic data for HNSCCs published here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We thank Dennis Roop for the K5.CrePR1 mice, Ronald DePinho for the floxed-Cdkn2a mice, Gigi Lozano for the Neo-p53R172H mice, Anton Berns for the floxed-p53 mice, and Arthur Gelmis for editorial assistance. This study was supported by the National Institutes of Health (NIH) grant DE015344 (C. Caulin), and in part through MD Anderson’s Cancer Center Support Grant CA016672.
Footnotes
Conflict of interest: The authors declare no conflict of interest
SUPPORTING INFORMATION
Additional supporting information consists of five supplementary figures and four supplementary tables. All supplementary figures are included with their corresponding figure legends. All supplementary tables include a header with a short description of the information provided in the tables.
REFERENCE LIST
- [1].Agrawal N, Frederick MJ, Pickering CR et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011; 333: 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stransky N, Egloff AM, Tward AD et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011; 333: 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015; 517: 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pickering CR, Zhou JH, Lee JJ et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res 2014; 20: 6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li YY, Hanna GJ, Laga AC et al. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res 2015; 21: 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dittmer D, Pati S, Zambetti G et al. Gain of function mutations in p53. Nat Genet 1993; 4: 42–46. [DOI] [PubMed] [Google Scholar]
- [7].Zhou G, Wang J, Zhao M et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell 2014; 54: 960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713. [DOI] [PubMed] [Google Scholar]
- [9].Lang GA, Iwakuma T, Suh YA et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872. [DOI] [PubMed] [Google Scholar]
- [10].Hanel W, Marchenko N, Xu S et al. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 2013; 20: 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xu J, Qian J, Hu Y et al. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci Rep 2014; 4: 4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Poeta ML, Manola J, Goldwasser MA et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007; 357: 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Neskey DM, Osman AA, Ow TJ et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res 2015; 75: 1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Caulin C, Nguyen T, Lang GA et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest 2007; 117: 1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Acin S, Li Z, Mejia O et al. Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras. J Pathol 2011; 225: 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993; 366: 704–707. [DOI] [PubMed] [Google Scholar]
- [17].Quelle DE, Zindy F, Ashmun RA et al. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995; 83: 993–1000. [DOI] [PubMed] [Google Scholar]
- [18].Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res 2005; 576: 22–38. [DOI] [PubMed] [Google Scholar]
- [19].Serrano M, Lee H, Chin L. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996; 85: 27–37. [DOI] [PubMed] [Google Scholar]
- [20].Kamijo T, Zindy F, Roussel MF et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997; 91: 649–659. [DOI] [PubMed] [Google Scholar]
- [21].Sharpless NE, Bardeesy N, Lee KH et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001; 413: 86–91. [DOI] [PubMed] [Google Scholar]
- [22].Hodges A, Smoller BR. Immunohistochemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod Pathol 2002; 15: 1121–1125. [DOI] [PubMed] [Google Scholar]
- [23].Nilsson K, Svensson S, Landberg G. Retinoblastoma protein function and p16INK4a expression in actinic keratosis, squamous cell carcinoma in situ and invasive squamous cell carcinoma of the skin and links between p16INK4a expression and infiltrative behavior. Mod Pathol 2004; 17: 1464–1474. [DOI] [PubMed] [Google Scholar]
- [24].Jonkers J, Meuwissen R, van der GH et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29: 418–425. [DOI] [PubMed] [Google Scholar]
- [25].Aguirre AJ, Bardeesy N, Sinha M et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003; 17: 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou Z, Wang D, Wang XJ et al. In utero activation of K5.CrePR1 induces gene deletion. Genesis 2002; 32: 191–192. [DOI] [PubMed] [Google Scholar]
- [27].Tang KW, Alaei-Mahabadi B, Samuelsson T et al. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun 2013; 4: 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Campbell C, Quinn AG, Ro YS et al. p53 mutations are common and early events that precede tumor invasion in squamous cell neoplasia of the skin. J Invest Dermatol 1993; 100: 746–748. [DOI] [PubMed] [Google Scholar]
- [29].Ziegler A, Jonason AS, Leffell DJ et al. Sunburn and p53 in the onset of skin cancer. Nature 1994; 372: 773–776. [DOI] [PubMed] [Google Scholar]
- [30].Nakazawa H, English D, Randell PL et al. UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc Natl Acad Sci U S A 1994; 91: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jonason AS, Kunala S, Price GJ et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A 1996; 93: 14025–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ren ZP, Hedrum A, Ponten F et al. Human epidermal cancer and accompanying precursors have identical p53 mutations different from p53 mutations in adjacent areas of clonally expanded non-neoplastic keratinocytes. Oncogene 1996; 12: 765–773. [PubMed] [Google Scholar]
- [33].Klein-Szanto AJ, Larcher F, Bonfil RD et al. Multistage chemical carcinogenesis protocols produce spindle cell carcinomas of the mouse skin. Carcinogenesis 1989; 10: 2169–2172. [DOI] [PubMed] [Google Scholar]
- [34].Wijnhoven SW, Speksnijder EN, Liu X et al. Dominant-negative but not gain-of-function effects of a p53.R270H mutation in mouse epithelium tissue after DNA damage. Cancer Res 2007; 67: 4648–4656. [DOI] [PubMed] [Google Scholar]
- [35].Jayasurya R, Sathyan KM, Lakshminarayanan K et al. Phenotypic alterations in Rb pathway have more prognostic influence than p53 pathway proteins in oral carcinoma. Mod Pathol 2005; 18: 1056–1066. [DOI] [PubMed] [Google Scholar]
- [36].Nakahara Y, Shintani S, Mihara M et al. Alterations of Rb, p16(INK4A) and cyclin D1 in the tumorigenesis of oral squamous cell carcinomas. Cancer Lett 2000; 160: 3–8. [DOI] [PubMed] [Google Scholar]
- [37].Wong CE, Yu JS, Quigley DA et al. Inflammation and Hras signaling control epithelial-mesenchymal transition during skin tumor progression. Genes Dev 2013; 27: 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Song IY, Balmain A. Cellular reprogramming in skin cancer. Semin Cancer Biol 2014. [DOI] [PMC free article] [PubMed]
- [39].Linardopoulos S, Street AJ, Quelle DE et al. Deletion and altered regulation of p16INK4a and p15INK4b in undifferentiated mouse skin tumors. Cancer Res 1995; 55: 5168–5172. [PubMed] [Google Scholar]
- [40].Pons M, Cigudosa JC, Rodriguez-Perales S et al. Chromosomal instability and phenotypic plasticity during the squamous-spindle carcinoma transition: association of a specific T(14;15) with malignant progression. Oncogene 2005; 24: 7608–7618. [DOI] [PubMed] [Google Scholar]
- [41].Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663–676. [DOI] [PubMed] [Google Scholar]
- [42].Li H, Collado M, Villasante A et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009; 460: 1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–454. [DOI] [PubMed] [Google Scholar]
- [44].Hollmig ST, Sachdev R, Cockerell CJ et al. Spindle cell neoplasms encountered in dermatologic surgery: a review. Dermatol Surg 2012; 38: 825–850. [DOI] [PubMed] [Google Scholar]
- [45].Kusters-Vandevelde HV, Van LA, Verdijk MA et al. CDKN2A but not TP53 mutations nor HPV presence predict poor outcome in metastatic squamous cell carcinoma of the skin. Int J Cancer 2010; 126: 2123–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.