

Abstract
SCN8A epileptic encephalopathy is a devastating epilepsy syndrome caused by mutant SCN8A, which encodes the voltage-gated sodium channel NaV1.6. To date, it is unclear if and how inhibitory interneurons, which express NaV1.6, influence disease pathology. Using both sexes of a transgenic mouse model of SCN8A epileptic encephalopathy, we found that selective expression of the R1872W SCN8A mutation in somatostatin (SST) interneurons was sufficient to convey susceptibility to audiogenic seizures. Patch-clamp electrophysiology experiments revealed that SST interneurons from mutant mice were hyperexcitable but hypersensitive to action potential failure via depolarization block under normal and seizure-like conditions. Remarkably, GqDREADD-mediated activation of WT SST interneurons resulted in prolonged electrographic seizures and was accompanied by SST hyperexcitability and depolarization block. Aberrantly large persistent sodium currents, a hallmark of SCN8A mutations, were observed and were found to contribute directly to aberrant SST physiology in computational modeling and pharmacological experiments. These novel findings demonstrate a critical and previously unidentified contribution of SST interneurons to seizure generation not only in SCN8A epileptic encephalopathy, but epilepsy in general.
SIGNIFICANCE STATEMENT SCN8A epileptic encephalopathy is a devastating neurological disorder that results from de novo mutations in the sodium channel isoform Nav1.6. Inhibitory neurons express NaV1.6, yet their contribution to seizure generation in SCN8A epileptic encephalopathy has not been determined. We show that mice expressing a human-derived SCN8A variant (R1872W) selectively in somatostatin (SST) interneurons have audiogenic seizures. Physiological recordings from SST interneurons show that SCN8A mutations lead to an elevated persistent sodium current which drives initial hyperexcitability, followed by premature action potential failure because of depolarization block. Furthermore, chemogenetic activation of WT SST interneurons leads to audiogenic seizure activity. These findings provide new insight into the importance of SST inhibitory interneurons in seizure initiation, not only in SCN8A epileptic encephalopathy, but for epilepsy broadly.
Keywords: depolarization block, epilepsy, interneuron, ion channel, seizure, voltage-gated sodium channel
Introduction
SCN8A epileptic encephalopathy is a severe genetic epilepsy syndrome caused by de novo mutations in the SCN8A gene, which codes for the voltage-gated sodium channel isoform NaV1.6 (Veeramah et al., 2012; Ohba et al., 2014; Larsen et al., 2015; Gardella et al., 2018). Patients exhibit a panoply of devastating symptoms, including refractory epilepsy, cognitive impairment, and motor dysfunction, and have a substantial risk of sudden unexpected death in epilepsy (Veeramah et al., 2012; Ohba et al., 2014; Larsen et al., 2015; Gardella et al., 2018).
Previous studies using patient-derived SCN8A mutations have sought to understand how these de novo mutations alter voltage-gated sodium channel function, intrinsic excitability of single neurons, and network dynamics underlying behavioral seizures (de Kovel et al., 2014; Estacion et al., 2014; Patel et al., 2016; Wagnon et al., 2016; Lopez-Santiago et al., 2017; Ottolini et al., 2017; Baker et al., 2018; Bunton-Stasyshyn et al., 2019; Wengert et al., 2019). Understanding the circuit-basis for SCN8A epileptic encephalopathy requires evaluation of the various cellular subpopulations which comprise neural circuits. Although NaV1.6 is expressed in inhibitory interneurons (Lorincz and Nusser, 2008; Li et al., 2014; Makinson et al., 2017), ∧insight into the mechanisms of network dysfunction in SCN8A epileptic encephalopathy is greatly limited by the current lack of understanding how inhibitory interneuron function is affected by mutant NaV1.6 expression. Interneurons typically constrain network excitability and prevent seizures, and interneuron hypoactivity is a general mechanism of epilepsy (Bernard et al., 2000; Markram et al., 2004; Kumar and Buckmaster, 2006; Stafstrom, 2013). With respect to sodium channel epileptic encephalopathies, hypofunction of inhibitory interneurons has typically been associated with loss-of-function SCN1A mutations, which result in Dravet syndrome (Rhodes et al., 2004; Escayg and Goldin, 2010; Cheah et al., 2012; Tai et al., 2014). Previous studies using mouse models of Dravet syndrome have confirmed that expression of the mutant SCN1A allele in GABAergic interneurons is sufficient to recapitulate the key phenotypes of Dravet syndrome (Cheah et al., 2012). Measurements of intrinsic excitability revealed impaired excitability of parvalbumin-positive and somatostatin-positive (SST) inhibitory interneurons at certain key points in development (Tai et al., 2014; Favero et al., 2018). The phenotypic severity of Dravet syndrome suggests that seemingly small changes to inhibitory interneuron excitability (reduction of maximum firing frequency by ∼20%) may have profound effects for overall network excitability and seizure behavior (Tai et al., 2014).
In this study, we used two mouse models of patient-derived SCN8A mutations: N1768D (Scn8aD/+), which allows global heterozygous expression of the N1768D pathogenic Scn8a variant; and a conditional mouse in which the expression of R1872W (Scn8aW/+) mutation is dependent on Cre recombinase. Both of these mice were generated using patient-derived SCN8A variants and well recapitulate the primary manifestations of SCN8A epileptic encephalopathy: Mice globally expressing either mutation exhibit spontaneous seizures which occasionally lead to sudden unexpected death (Wagnon et al., 2015; Bunton-Stasyshyn et al., 2019; Wenker et al., 2021). Recently, we demonstrated that both models are susceptible to audiogenic seizures, providing a powerful experimental approach to assess seizure susceptibility (Wengert et al., 2021; Wenker et al., 2021). Using the conditionally expressed R1872W pathogenic variant, our previous studies identified that forebrain excitatory neuron expression of R1872W was sufficient for spontaneous seizure development and premature lethality (Bunton-Stasyshyn et al., 2019). Although that study suggested that cortical excitatory neurons are primary drivers of SCN8A epileptic encephalopathy pathology, the potential contribution of specific inhibitory interneurons remains to be investigated.
Using these two mouse models of epileptic encephalopathy, we report that specific expression of R1872W in SST interneurons (Scn8a-SSTW/+), but not expression in forebrain excitatory neurons (Scn8a-EMX1W/+), results in audiogenic seizures, indicating that abnormal activity of SST interneurons uniquely contributes to seizures in SCN8A epileptic encephalopathy. SST interneurons from both mice harboring SCN8A variants showed an initial steady-state hyperexcitability, followed by action potential (AP) failure via depolarization block. Dual-cell recordings revealed that mutant SST interneurons are particularly sensitive to depolarization block under seizure-like conditions, and that depolarization block occurs coincidently with pyramidal neuron ictal discharges. Counterintuitively, GqDREADD-mediated chemogenetic activation of WT control SST interneurons was sufficient to facilitate initial hyperexcitability followed by depolarization block and status epilepticus in the mice, indicating the proconvulsant effect of aberrant SST physiology. Recordings of voltage-gated sodium channel activity identified elevated persistent sodium currents (INaP) in mutant SST interneurons. Computational modeling revealed that elevating INaP was singly sufficient to induce initial hyperexcitability and premature entry into depolarization block. Last, we validated the model predictions by demonstrating that application of veratridine, a pharmacological activator of INaP, to WT SST interneurons induces premature depolarization block. Overall, our results reveal a mechanism by which SST interneurons contribute to seizures in SCN8A epileptic encephalopathy.
Materials and Methods
Ethics approval
All procedures were conducted in accordance with University of Virginia's Animal Care and Use Committee guidelines.
Mouse husbandry and genotyping
Scn8aD/+ and Scn8aW/+ were generated as previously described and maintained through crosses with C57BL/6J mice (Jax, #000664) to keep all experimental mice on a C57BL/6J genetic background (Wagnon et al., 2015; Bunton-Stasyshyn et al., 2019). Cell type-specific expression of R1872W was achieved using males homozygous or heterozygous for the R1872W allele and females homozygous for EIIa-Cre (Jax, #003724), EMX1-Cre (Jax, #005628), or SST-Cre (Jax, #013044) to generate mutant mice (Scn8a-EIIaW/+, Scn8a-EMX1W/+, and Scn8a-SSTW/+, respectively) (Bunton-Stasyshyn et al., 2019). Because certain transgenic mice entail the insertion of Cre directly into the coding sequence, for all experiments we used WT controls that contained the same Cre allele, but lacked the allele encoding the Scn8a variant. In particular, control mice for audiogenic experiments in Scn8a-EIIaW/+ and Scn8a-EMX1W/+ were separate litters lacking the R1872W allele; and for Scn8a-SSTW/+, they were littermate controls lacking the R1872W allele. Fluorescent labeling of SST interneurons was achieved by first crossing male Scn8aD/+ or Scn8aW/+ mice with a Cre-dependent tdTomato reporter (Jax, #007909) and then with female mice homozygous for SST-Cre. Experimental groups used roughly equal numbers of male and female mice to control for any potential sex differences, and no differences because of sex were observed. For chemogenetic experiments, male mice heterozygous for GqDREADD allele (Jax, #026220) were crossed with female mice homozygous for SST-Cre. All genotyping was conducted through using Transnetyx automated genotyping PCR services.
Audiogenic seizure test
To test for audiogenic seizures, mice were taken from their home cage and transferred to a clean test cage where they were allowed to acclimate for ∼20 s before the onset of the acoustic stimulus. Similar to a method described previously (Martin et al., 2020), a sonicator (Branson 200 ultrasonic cleaner) was used to produce the audiogenic stimulus directly adjacent to the test cage. The stimulus duration lasted for 50 s or until the animal had a behavioral seizure. Scn8a-EIIaW/+ mice, which typically exhibit sudden death by P17 (Bunton-Stasyshyn et al., 2019), were tested between P13 and P16. Similarly, Scn8a-Emx1W/+, which often exhibit sudden death by P28 (Bunton-Stasyshyn et al., 2019), were tested between P21 and P24, a time point in which Scn8aD/+ mice exhibit audiogenic seizures (Wengert et al., 2021). Scn8a-SSTW/+ mice were tested at 7-10 weeks. Videos of audiogenic seizure tests were recorded with a laptop webcam, and the duration of seizure phases was analyzed by taking the time in seconds that the mouse spent in each of the phases: a wild-running phase characterized by fast circular running throughout the cage, a tonic phase characterized by hindlimb extension and muscle rigidity, a clonic phase typified by myoclonic jerking of the hindlimbs, and recovery exemplified when the mouse ceased myoclonic jerking and righted itself.
Immunohistochemistry
Brain tissue for immunohistochemistry was processed as follows: Mice were anesthetized and transcardially perfused with 10 ml ice-cold Dulbecco's PBS (DPBS) followed by 10 ml ice-cold 4% PFA. Sagittal brain sections were immersed in 4% PFA for 2 h at 4°C and then stored in DPBS with 0.1% sodium azide. Brains were embedded in 2% agarose, and 40 µm sections were obtained using a vibratome (Leica Microsystems, VT1200). Sections were incubated with rat anti-SST (EMD Millipore, MAB#354) diluted in 2% donkey serum (Jackson ImmunoResearch Laboratories) with 0.1% Triton X (Sigma-Aldrich) at a concentration 1:1000 in DPBS. The secondary antibody, donkey anti-rat AlexaFluor-488, was diluted 1:1000 in donkey serum (2%) and Triton-X (0.1%) in DPBS. Sections were stained free-floating in primary antibody on a shaker at 4°C overnight and with secondary antibody for 1 h at room temperature the following day. Tissues were counterstained with NucBlue Fixed Cell ReadyProbes Reagent (DAPI) (Thermo Fisher Scientific, catalog #R37606) included in the secondary antibody solution. Tissues were mounted on slides using AquaMount (Polysciences).
Brain slice preparation
Preparation of acute brain slices for patch-clamp electrophysiology experiments was modified from standard protocols previously described (Baker et al., 2018; Bunton-Stasyshyn et al., 2019; Wengert et al., 2019). Mice were anesthetized with isoflurane and decapitated. The brains were rapidly removed and kept in chilled ACSF (0°C) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 0.5 L-ascorbic acid, 10 glucose, 25 NaHCO3, and 2 Na-pyruvate (Sigma). The slices were continuously oxygenated with 95% O2 and 5% CO2 throughout the preparation; 300 µm coronal or horizontal brain sections were prepared using a Leica Microsystems VT1200 vibratome. Slices were collected and placed in ACSF warmed to 37°C for ∼30 min and then kept at room temperature for up to 6 h.
Electrophysiology recordings
Brain slices were placed in a chamber continuously superfused (∼2 ml/min) with continuously oxygenated recording solution warmed to 32 ± 1°C. Cortical layer V SST interneurons were identified as red fluorescent cells, and pyramidal neurons were identified based on absence of fluorescence and pyramidal morphology via video microscopy using a Carl Zeiss Axioscope microscope. Whole-cell recordings were performed using a Multiclamp 700B amplifier with signals digitized by a Digidata 1322A digitizer. Currents were amplified, low-pass filtered at 2 kHz, and sampled at 100 kHz. Borosilicate electrodes were fabricated using a Brown-Flaming puller (model P1000, Sutter Instruments) to have pipette resistances between 1.5 and 3.5 mΩ.
Intrinsic excitability recordings
Current-clamp recordings of neuronal excitability were collected in ACSF solution identical to that used for preparation of brain slices. The internal solution contained the following (in mm): 120 K-gluconate, 10 NaCl, 2 MgCl2, 0.5 K2EGTA, 10 HEPES, 4 Na2ATP, 0.3 NaGTP, pH 7.2 (osmolarity 290 mOsm). Intrinsic excitability was assessed using methods adapted from those previously described (Ottolini et al., 2017; Wengert et al., 2019). Briefly, resting membrane potential was manually recorded immediately going whole-cell and confirmed using a 1 min gap-free recording of the neuron at rest. Current ramps from 0 to 400 pA over 4 s were used to calculate passive membrane and AP properties, including threshold as the point at which membrane potential slope reaches 5% of the maximum slope, upstroke and downstroke velocity, which are the maximum and minimum slopes on the AP, respectively; the amplitude, which was defined as the voltage range between AP peak and threshold; the APD50, which is the duration of the AP at the midpoint between threshold and peak; input resistance, which was calculated using a −20 pA pulse in current-clamp recordings; and the rheobase, which was operationally defined as the maximum amount of depolarizing current that could be injected into the neurons before eliciting APs. AP frequency–current relationships were determined using 500 ms current injections ranging from −140 to 600 pA. APs were counted only if the peak of the AP was >0 mV. The threshold for depolarization block was operationally defined as the current injection step that elicited the maximum number of APs (i.e., subsequent current injection steps of greater magnitude resulted in fewer APs because of entry into the depolarization block). For neurons that did not undergo depolarization block within our current injection protocol, the depolarization block threshold was therefore set to the maximum current injection magnitude of 600 pA.
Outside-out voltage-gated sodium channel recordings
Patch-clamp recordings in the outside-out configuration were collected using a protocol modified from an approach previously described (Ottolini et al., 2017). The recording solution was identical to that used for brain-slice preparation. The internal solution for all voltage-gated sodium channel recordings contained the following (in mm): 140 CsF, 2 MgCl2, 1 EGTA, 10 HEPES, 4 Na2ATP, and 0.3 NaGTP with the pH adjusted to 7.3 and osmolality to 300 mOsm. Voltage-dependent activation and steady-state inactivation parameters were recorded using voltage protocols previously described (Wengert et al., 2019).
Voltage-gated sodium channel recordings in acutely dissociated neurons
Acute brain slices were transferred to a solution containing oxygenated HBSS (Invitrogen) solution with calcium and magnesium supplemented with 0.2 mm ascorbic acid and 1 mg/ml of protease type XIV (Sigma). The surface of the solution was continuously blown over with 100% O2. After ∼23 min of incubation, slices were transferred to enzyme-free HBSS solution and washed 3 times before the deeper layers of the somatosensory cortex were carefully dissected free and triturated using fire-polished glass pipettes of decreasing aperture. Neurons were plated onto fibronectin-coated coverslips for electrophysiological recordings. SST interneurons were again identified for patching by TdTomato fluorescence. Recordings of sodium channel currents were collected as previously described (Lopez-Santiago et al., 2017) using a recording solution containing the following (in mm): 30 NaCl, 1 BaCl2, 2 MgCl2, 45 CsCl, 0.2 CdCl2, 1 CaCl2, 10 HEPES, 20 TEA-Cl, and 100 glucose (pH 7.35; and osmolarity 305-310 mOsm).
Persistent sodium current recordings
The recording solution was modified with a reduced sodium concentration to allow for proper voltage control through the entire range of command voltages (Royeck et al., 2008; Wengert et al., 2019). It contained the following (in mm): 50 NaCl, 90 TEACl, 10 HEPES, 3.5 KCl, 2 CaCl2, 2 MgCl2, 0.2 CdCl2, 4 4-aminopyridine (4-AP), 25 D-glucose. Steady-state persistent sodium currents were elicited using a voltage ramp (20 mV/s) from −80 to −20 mV. After collecting recordings at baseline, the procedure was repeated in the presence of 1 µm TTX (Alomone Labs) to completely isolate the sodium current. TTX-subtracted traces were analyzed by extracting the current at each mV from −80 mV to −20 mV. The half-maximal voltage for activation of the current was calculated as previously described (Wengert et al., 2019). Any recordings in which the neuron escaped steady-state voltage control were discarded before TTX application.
Resurgent sodium current recordings
Resurgent sodium currents were recorded as previously described (Barker et al., 2017; Wengert et al., 2019) using a solution containing the following (in mm): 100 NaCl, 26 NaHCO3, 19.5 TEA-Cl, 3 KCl, 2 MgCl2, 2 CaCl2, 2 BaCl2, 0.1 CdCl2, 4 4-AP, and 10 glucose (pH of 7.4; ∼305 mOsm). SST interneurons were held at −100 mV, depolarized to 30 mV for 20 ms, then stepped to voltages between −100 and 0 mV for 40 ms to observe the TTX-sensitive resurgent current.
Dual-cell recordings
Gap-free recordings of pairs of one pyramidal and one SST interneuron <200 µm away were used to examine cell interplay during seizure-like events. Seizure-like events were evoked using Mg2+-free ACSF containing either 50 or 100 µm 4-AP. Seizure-like depolarization block events were defined in this context as instances in which the membrane potential reached a stable value above AP threshold, which occurred simultaneously with burst of APs in the nearby pyramidal neuron.
Computational modeling
A single-compartment conductance-based neuronal model was modified based on one used previously (Nowacki et al., 2012). The dynamics of the neuronal voltage are described by the following:
where V is the transmembrane voltage, C is the membrane capacitance, Iapp is the applied current density, INaT/NaP are the transient and persistent sodium current densities, respectively, and IKT/KP are the transient and persistent potassium currents densities, respectively. These currents are specified by the following functional forms:
where gX represents the maximal conductance of channel type , mX. represents the fraction of open activation gates of channel type X, with hX corresponding to the fraction of open inactivation gates, where appropriate, and VY is the reversal (Nernst) potential of ionic species The dynamics of the gating variables obey the following:
where are the timescales of the activation and inactivation of channel type X, respectively; and and are its steady-state activation and inactivation curves, which are themselves described by Boltzmann functions of the following form:
where are the thresholds for activation and inactivation and are the associated sensitivities around this threshold. The activation of both types of sodium channel is assumed to be instantaneous, meaning that always. The parameter units and values are given in Table 1. All model simulations were performed in MATLAB R2020a. Code to replicate these simulations is available to download via Git from https://git.exeter.ac.uk/kcaw201/depolarizationblock.
Table 1.
Parameter units and values
| Parameter | Unit | Value | Parameter | Unit | Value |
|---|---|---|---|---|---|
| mmho/cm2 | 130 | mV | 60 | ||
| mmho/cm2 | [0-0.4] | mV | −85 | ||
| mmho/cm2 | 10 | mV | −65 | ||
| mmho/cm2 | 1.65 | ms | 1 | ||
| mmho/cm2 | 0.02 | ms | 75 | ||
| mV | −37 | ms | 1 | ||
| mV | −47 | ms | 1400 | ||
| mV | −5.8 | mV | 5 | ||
| mV | −30 | mV | 3 | ||
| mV | −70 | mV | 11.4 | ||
| mV | −68 | mV | 10 | ||
| mA/cm2 | [0-0.4] | mV | −7 | ||
| µF/cm2 | 1 | mV | −9.7 |
Bifurcation analysis
Bifurcation analysis was performed in DDE-BIFTOOL version 3.1.1, which is a MATLAB-based numerical software package for numerical continuation; and in AUTO-07P, which is a Fortran-based numerical continuation program.
In vivo seizure monitoring
Custom electrocorticogram (ECoG) headsets (PlasticsOne) were implanted in 7- to 10-week-old SST-Cre+/−; GqDREADD+/− or SST-Cre; GqDREADD−/− mice using standard aseptic surgical techniques. Anesthesia was induced with 5% and maintained with 0.5%-3% isoflurane. Adequacy of anesthesia was assessed by lack of toe-pinch reflex. A midline skin incision was made over the skull, and burr holes were made at the lateral/rostral end of both the left and right parietal bones to place EEG leads, and at the interparietal bone for a reference and ground electrodes. A headset was attached to the skull with dental acrylic (Jet Acrylic; Lang Dental). Mice received postoperative analgesia with ketoprofen (5 mg/kg, i.p.) and 0.9% saline (0.5 ml i.p.) and were allowed to recover a minimum of 2-5 d before seizure-monitoring experiments.
Mice were then individually housed in custom-fabricated chambers and monitored for the duration of the experiment. The headsets were attached to a custom low-torque swivel cable, allowing mice to move freely in the chamber. ECoG signals were amplified at 2000× and bandpass filtered between 0.3 and 100 Hz, with an analog amplifier (Neurodata model 12, Grass Instruments). Biosignals were digitized with a Powerlab 16/35 and recorded using LabChart 7 software (AD Instruments) at 1 kS/s. Video acquisition was performed by multiplexing four miniature night vision-enabled cameras and then digitizing the video feed with a Dazzle Video Capture Device (Corel) and recording at 30 fps with LabChart 7 software in tandem with biosignals.
In seizure monitoring experiments, ∼30 min of baseline ECoG signal were recorded before the mice were injected with vehicle or CNO (0.2, 1, and 5 mg/kg). Video ECoG was continuously recorded until the ECoG and behavioral manifestations of status epilepticus ended (typically ∼8 h). Mice had access to food ad libitum but were restricted from water to protect against headset damage and animal injury during status epilepticus. Power analysis was performed using Spike2 version 7.17 (Cambridge Electronic Design), using FFT size of 1024 points (Hanning method), which provides a 0.9766 Hz resolution across intervals of 1.024 s. To obtain values for the CNO dose–response (see Fig. 4D), the peak power in band 2-10 Hz after injection was normalized to pre-injection levels.
Figure 4.
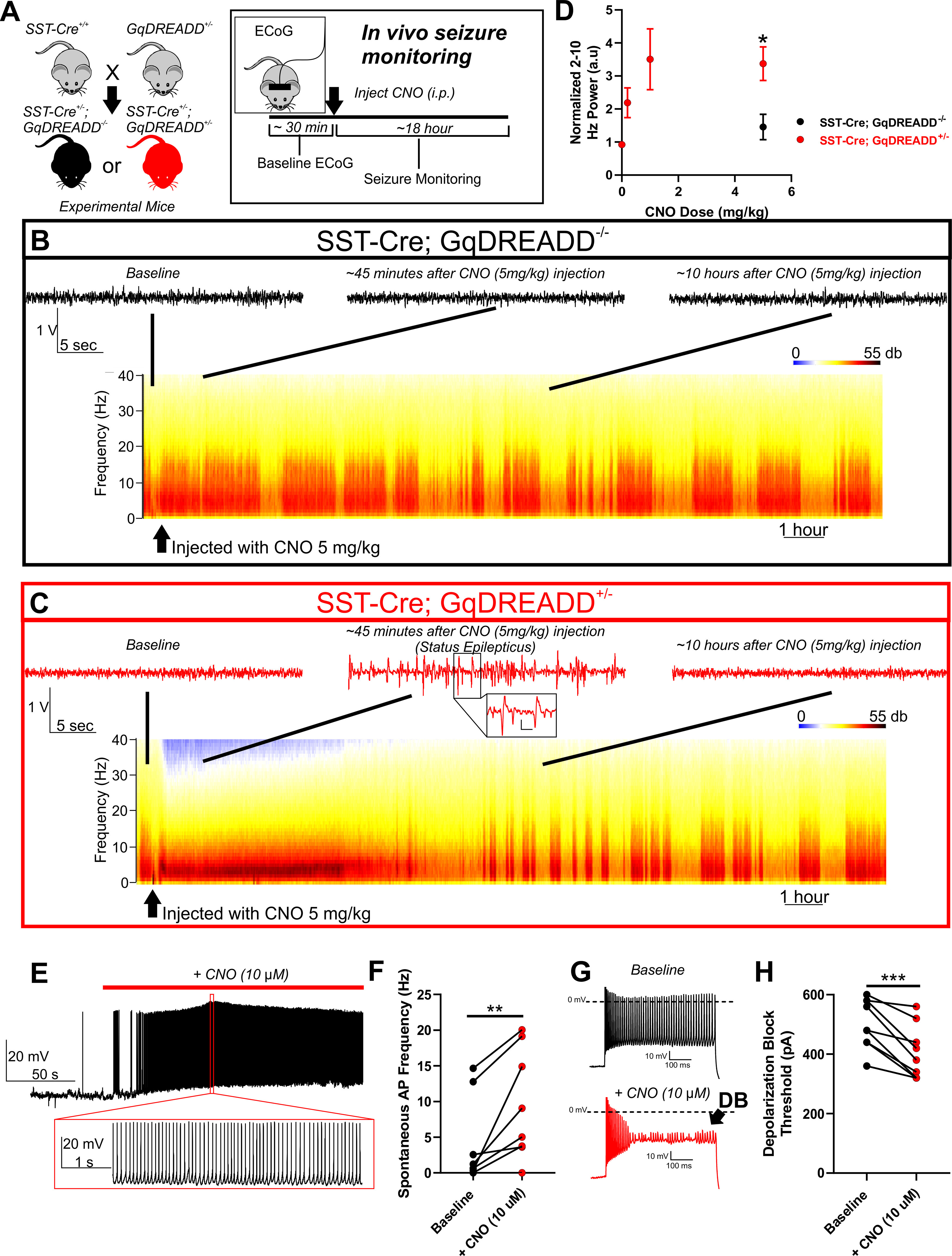
Chemogenetic activation of SST interneurons in WT mice is sufficient to induce seizures. A, Breeding strategy: Female mice homozygous for SST-cre (SST-cre+/+) were bred with male mice heterozygous for a floxed GqDREADD allele (GqDREADD+/−) to produce offspring that were either SST-Cre+/−; GqDREADD−/− control mice (black) or SST-Cre+/−; GqDREADD+/− experimental mice (red). For in vivo seizure monitoring, ECoG was recorded from each mouse for ∼30 min of baseline activity, then treated with vehicle or CNO (i.p. at 0.2, 1, and 5 mg/kg), and monitored for seizure activity for ∼18 h. B, C, Example relative power spectra of ECoG activity for ∼18 h with representative ECoG traces before CNO injection (baseline), ∼45 min after CNO injection, and ∼10 h after CNO injection. B, Example traces indicating that, in mice lacking the GqDREADD receptor (black), CNO treatment (5 mg/kg) did not lead to seizure behavior or changes in ECoG activity. C, Example traces reveal that, on CNO administration, GqDREADD+/− mice (red) exhibited highly synchronized ECoG activity and spike-wave discharges indicative of status epilepticus, which recovers within ∼8 h. Inset, Expanded view of example spike-wave discharges. Calibration: 0.25 V, 0.2 s. D, Normalized 2-10 Hz power relative to pre-injection baseline observed in response to vehicle and CNO (0.2, 1, and 5 mg/kg). SST-Cre; GqDREADD+/− mice (n = 5 for vehicle, 0.2, and 1 mg/kg, n = 8 for 5 mg/kg; red) experienced a robust and dose-dependent increase in 2-10 Hz power. SST-Cre; GqDREADD−/− control mice (black) did not show a significant increase in 2-10 Hz power on administration of 5 mg/kg CNO (n = 4) and was significantly less than SST-Cre; GqDREADD+/− mice. *p < 0.05 (unpaired t test). E, Representative example trace of an SST-Cre; GqDREADD+/− SST interneuron showing spontaneous increase in excitability in response to bath application of CNO (10 μm; red bar). Expanded view illustrates the high firing frequency during CNO exposure. F, Spontaneous firing frequencies of SST interneurons from SST-Cre; GqDREADD+/− mice before (black) and after (red) treatment with CNO (10 μm; n = 8, 4 mice). **p < 0.01 (paired t test). G, Example traces of SST interneuron excitability from an SST-Cre; GqDREADD+/− mouse in response to a 600 pA current injection before (black), and after (red) CNO (10 μm) bath application. Premature depolarization block (DB, arrow) is observed after CNO treatment. H, Depolarization block threshold for each SST-Cre; GqDREADD+/− SST interneuron before (black) and after (red) treatment with CNO (10 μm; n = 8, 4 mice). ***p < 0.001 (paired t test).
Analysis
All patch-clamp electrophysiology data were analyzed using custom MATLAB scripts and/or ClampFit 10.7. All statistical comparisons were made using the appropriate test using GraphPad Prism 8. Categorical data, including audiogenic seizure susceptibility and spontaneous AP firing, were analyzed using a Fisher's exact test. For membrane and AP properties, spontaneous firing frequency, peak sodium currents, and half-maximal voltages, mouse genotypes were compared either by one-way ANOVA followed by Dunnett's multiple comparisons test when the data were normally distributed and by the nonparametric Kruskal–Wallis test followed by Dunn's multiple comparisons test when the data were not normally distributed. A two-way ANOVA followed by Tukey's test for multiple comparisons was used to compare groups in experiments in which repetitive measures were made from a single cell over various voltage commands or current injections. Cumulative distribution (survival) plots were analyzed by the Log-rank Mantel-Cox test. One-sided t tests were only used in the CNO physiology experiments where a clear directional hypothesis was articulated, and paired t tests were used in cases where recordings could be collected before and after bath application of a drug (i.e., veratridine or CNO). Data are presented as individual data points and/or mean ± SEM.
Results
Using a mouse model of SCN8A epileptic encephalopathy in which the patient-derived mutation R1872W is expressed in a Cre-dependent manner, we sought to determine the cell type-specific contribution to audiogenic seizure susceptibility extending our previous findings that mouse models of SCN8A epileptic encephalopathy exhibit audiogenic seizures (Wengert et al., 2021; Wenker et al., 2021). Acoustic stimulation of mice globally expressing the R1872W Scn8a mutation (Scn8a-EIIaW/+) caused audiogenic seizures (7 of 8 mice; 87.5%; Fig. 1A–C). Seizures were characterized by a period of wild-running, a tonic phase associated with hindlimb extension, and sudden death (Fig. 1A). To our surprise, expression of the R1872W Scn8a mutation in forebrain excitatory neurons (Scn8a-EMX1W/+) did not reliably convey susceptibility to audiogenic seizures, with only 1 of 9 mice exhibiting a single brief bout of wild-running (p < 0.001 Fisher's exact test; Fig. 1B,C,E). In contrast, expression of the R1872W Scn8a mutation selectively in SST-positive inhibitory interneurons (Scn8a-SSTW/+) was sufficient to induce an audiogenic seizure in 18 of 22 (∼82%), typified by a wild-running phase, a clonic phase characterized by convulsions and jerking of limbs, followed by recovery (Fig. 1A–C). Audiogenic seizures were never observed in WT littermate control mice (N = 26). These results indicate that expression of R1872W specifically in SST interneurons is sufficient to induce audiogenic seizures.
Figure 1.
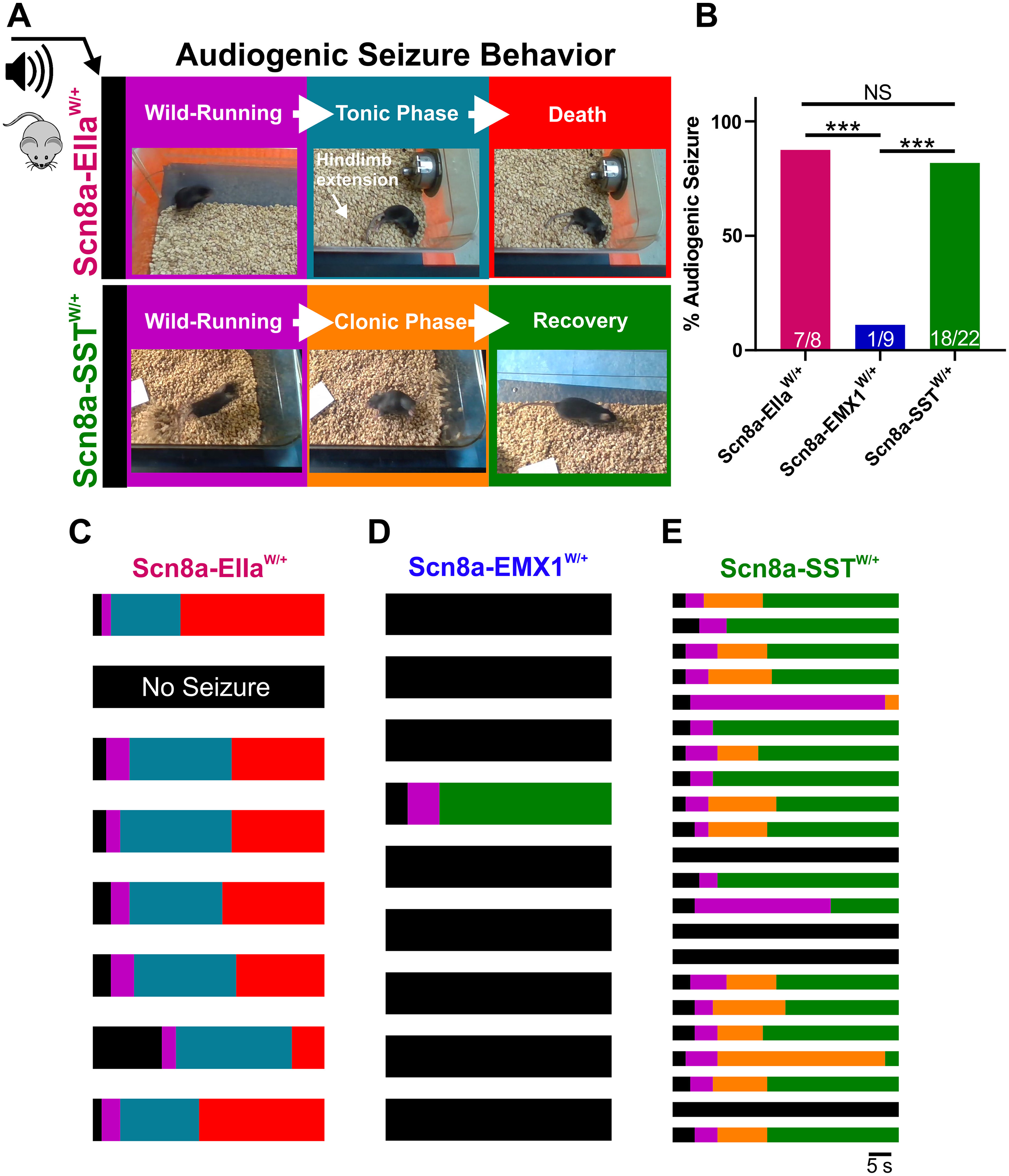
SST interneuron-specific expression of mutant Scn8a is sufficient for susceptibility to audiogenic seizures. A, Audiogenic seizure behavior in mice with cell type-specific expression of R1872W Scn8a mutation. Upon high-intensity acoustic stimulation, Scn8a-EIIaW/+ mice exhibit wild-running (purple) followed by a tonic phase characterized by hindlimb extension (blue), which is followed by collapse of breathing and death (red). Scn8a-SSTW/+ mice exhibit wild-running (purple) but progress to a convulsive clonic phase characterized by repetitive shaking and limb-jerking (orange) followed by recovery (green). B, Propensity of audiogenic seizures in Scn8a-EIIaW/+ (magenta: ∼87%), Scn8a-EMX1W/+ (∼11%), and Scn8a-SSTW/+ (green: ∼82%) mice. *p < 0.01, ***p < 0.001, Fisher's exact test. C-E, Color-coded raster plots for seizure behavior of individual Scn8a-EIIaW/+ (C), Scn8a-EMX1W/+ (D), and Scn8a-SSTW/+ (E) mice.
We next sought to determine the impact of N1768D and R1872W patient-derived SCN8A mutations on SST interneuron function with a goal to better understand how alterations in SST interneuron excitability could lead to seizure susceptibility. We conducted whole-cell patch-clamp electrophysiology recordings of SST interneurons from layer V somatosensory cortex from adult Scn8aD/+, Scn8a-SSTW/+, and WT control mice (littermates from both groups pooled), identified via TdTomato fluorescence (Fig. 2A). Relative to WT controls, a greater proportion of SST interneurons from Scn8aD/+ and Scn8a-SSTW/+ were spontaneously active (p < 0.001; Fisher's exact test; Fig. 2B–E) each with significantly greater frequency of spontaneous APs (p < 0.05 and p < 0.01 relative to WT; Kruskal–Wallis test followed by Dunn's test; Fig. 2F). Analysis of membrane and AP properties revealed that, relative to WT, both Scn8aD/+ and Scn8a-SSTW/+ SST interneurons had depolarized resting membrane potentials and decreased rheobases likely contributing to their steady-state hyperexcitability (Table 2). Additionally, Scn8a-SSTW/+ interneurons had altered upstroke and downstroke velocities and increased AP duration relative to WT interneurons (APD50; Fig. 2G–I; Table 2).
Figure 2.
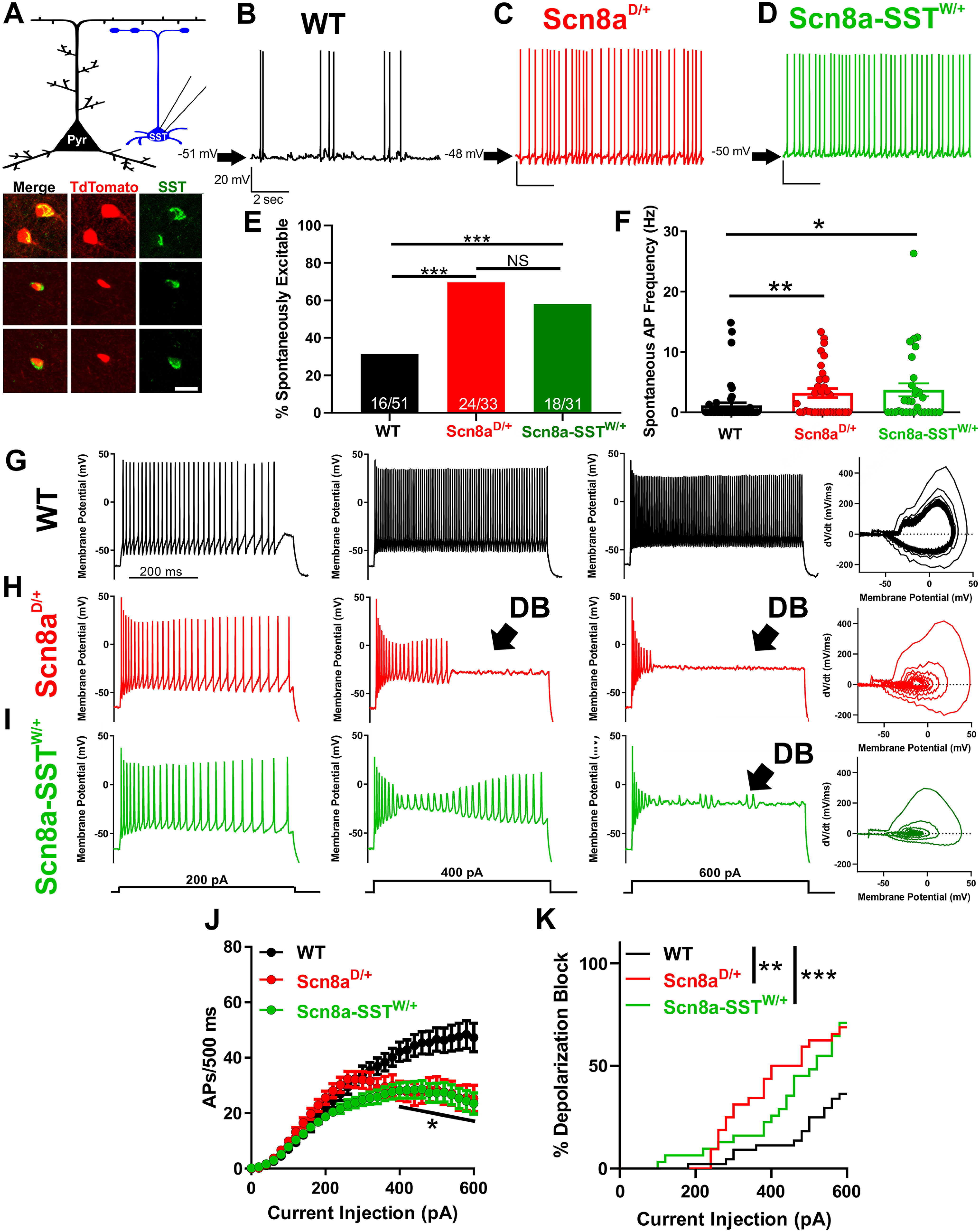
Scn8aD/+ and Scn8a-SSTW/+ SST interneurons are hyperexcitable and readily enter depolarization block. A, Whole-cell recordings collected from WT, Scn8aD/+, and Scn8a-SSTW/+ somatosensory layer V SST interneurons (blue). Example immunohistochemistry images showing colocalization of TdTomato (red) and SST (green) immunofluorescence. Scale bar, 20 μm. B-D, Representative example traces of spontaneous excitability of WT (B; black), Scn8aD/+ (C; red) and Scn8a-SSTW/+ (D; green) SST interneurons. Arrows indicate membrane potential between spontaneous APs. E, Only 16 of 51 (∼31%) WT SST interneurons were spontaneously excitable, whereas 24 of 33 (∼73%) Scn8aD/+ and 18 of 31 (∼58%) Scn8a-SSTW/+ spontaneously fired APs (***p < 0.01 by Fisher's exact test). F, Average spontaneous firing frequencies for WT (black), Scn8aD/+ (red), and Scn8a-SSTW/+ (green) SST interneurons. *p < 0.05, **p < 0.01, Kruskal–Wallis test followed by Dunn's multiple comparisons. G-I, Representative traces for WT (G; black), Scn8aD/+ (H; red), and Scn8a-SSTW/+ (I; green) SST interneurons eliciting APs in response to 500 ms current injections of 200, 400, and 600 pA. Depolarization block is indicated (arrow; DB) in the Scn8aD/+ (H; red) and Scn8a-SSTW/+ (I; green) interneurons. Right, Phase plot corresponding to the 600 pA current traces for WT (G; black), Scn8aD/+ red (H; red), and Scn8a-SSTW/+ (I; green). J, Average number of APs elicited relative to current injection magnitude for WT (black; n = 51, 10 mice), Scn8aD/+ (red; n = 33, 6 mice), and Scn8a-SSTW/+ (green; n = 31, 4 mice). At current injections >400 pA, both Scn8aD/+ and Scn8a-SSTW/+ AP frequencies were reduced relative to WT. *p < 0.05, two-way ANOVA followed by Tukey's correction for multiple comparisons. K, Cumulative distribution of SST interneuron entry into depolarization block relative to current injection magnitude for WT (black), Scn8aD/+ (red), and Scn8a-SSTW/+ (green) mice. Curve comparison by log-rank Mantel-Cox test (**p < 0.01).
Table 2.
Membrane and AP properties of WT, Scn8aD/+, and Scn8a-SSTW/+ SST inhibitory interneurons
| Vm (mV) | AP threshold (mV) | Upstroke velocity (mV/ms) | Downstroke velocity (mV/ms) | AP amplitude (mV) | APD50 (ms) | Input resistance (mΩ) | Rheobase (pA) | |
|---|---|---|---|---|---|---|---|---|
| WT (n = 52) | −60.2 ± 1.1 | −38.5 ± 0.7 | 286 ± 13 | −137 ± 9 | 72.8 ± 1.6 | 0.73 ± 0.03 | 250 ± 16 | 63 ± 9 |
| Scn8aD/+ (n = 33) | −52.9 ± 1.5*** | −38.4 ± 0.8 | 290 ± 18 | −131 ± 7 | 76.7 ± 1.4 | 0.69 ± 0.03 | 298 ± 20 | 37 ± 7* |
| Scn8a-SSTW/+ (n = 31) | −54.4 ± 1.6** | −40.8 ± 1.2 | 231 ± 2.8* | −87 ± 5.7*** | 73.9 ± 2.0 | 1.0 ± 0.06*** | 250 ± 15 | 37 ± 5* |
Data are mean ± SEM.
*p < 0.05,
**p < 0.01,
***p < 0.001, compared with WT using one-way ANOVA followed by Dunnett's multiple comparisons test.
We subjected each neuron to a range of depolarizing current injections to characterize intrinsic excitability. Relative to WT controls (n = 51, 10 mice), SST interneurons from both Scn8aD/+ (n = 33, 6 mice) and Scn8a-SSTW/+ mice (n = 31, 4 mice) exhibited progressive AP failure at current injections >400 pA as the interneurons entered depolarization block: At 600 pA, WT SST interneurons fired 47 ± 5 APs/500 ms (94 Hz), Scn8aD/+ fired 25 ± 5 APs/500 ms (50 Hz), and Scn8a-SSTW/+ fired 23 ± 4 APs/500 ms (46 Hz; p < 0.001, two-way ANOVA with Tukey's correction for multiple comparisons; Fig. 2G–J). Over the range of current injection magnitudes, both Scn8aD/+ and Scn8a-SSTW/+ interneurons were significantly more prone to depolarization block than WT counterparts (p < 0.01; Log-rank Mantel-Cox test; Fig. 2K).
Prior studies have described cellular interplay between excitatory pyramidal neurons and fast-spiking interneurons during brain-slice seizure-like activity and in vivo seizures in which interneuron depolarization block occurs coincidentally with pyramidal neuron ictal discharges (Swadlow, 2003; Ziburkus et al., 2006; Cammarota et al., 2013; Jayant et al., 2019; Parrish et al., 2019). To test whether SST interneurons expressing Scn8a mutations also exhibit depolarization block seizure-like events coincident with pyramidal ictal discharges, we simultaneously recorded from an SST interneuron and a nearby pyramidal neuron and induced seizure-like activity by applying a bath solution with 0 Mg2+ and 4-AP (50 µm) (Ziburkus et al., 2006). Under these conditions, although neurons in all groups increased their excitability, we observed a significantly greater propensity for Scn8aD/+ (n = 12, 3 mice; Fig. 3B) and Scn8a-SSTW/+ (n = 10, 3 mice; Fig. 3C) SST interneurons to spontaneously exhibit depolarization block events compared with WT SST interneurons (n = 9, 3 mice; Fig. 3A), supporting our findings that expression of mutant SCN8A renders SST interneurons prone to depolarization block (p < 0.01, log-rank Mantel-Cox test; Fig. 3A–C,E). Consistent with previous reports in fast-spiking interneurons (Swadlow, 2003; Ziburkus et al., 2006; Cammarota et al., 2013; Jayant et al., 2019; Parrish et al., 2019), instances of SST interneuron depolarization block were entirely concomitant with seizure-like ictal discharges of nearby layer V pyramidal neurons (Fig. 3B–D). Only when the bath concentration of the proconvulsant 4-AP was increased to 100 µm did we observe similar ictal-discharges of pyramidal neurons and depolarization block events in WT SST interneurons (Fig. 3D,E). These results demonstrate not only that mutant SST interneurons are hypersensitive to depolarization block in the seizure-like context, but also that simultaneous SST depolarization block is coincident with pyramidal neuron bursting activity, a hallmark of seizure-like activity.
Figure 3.
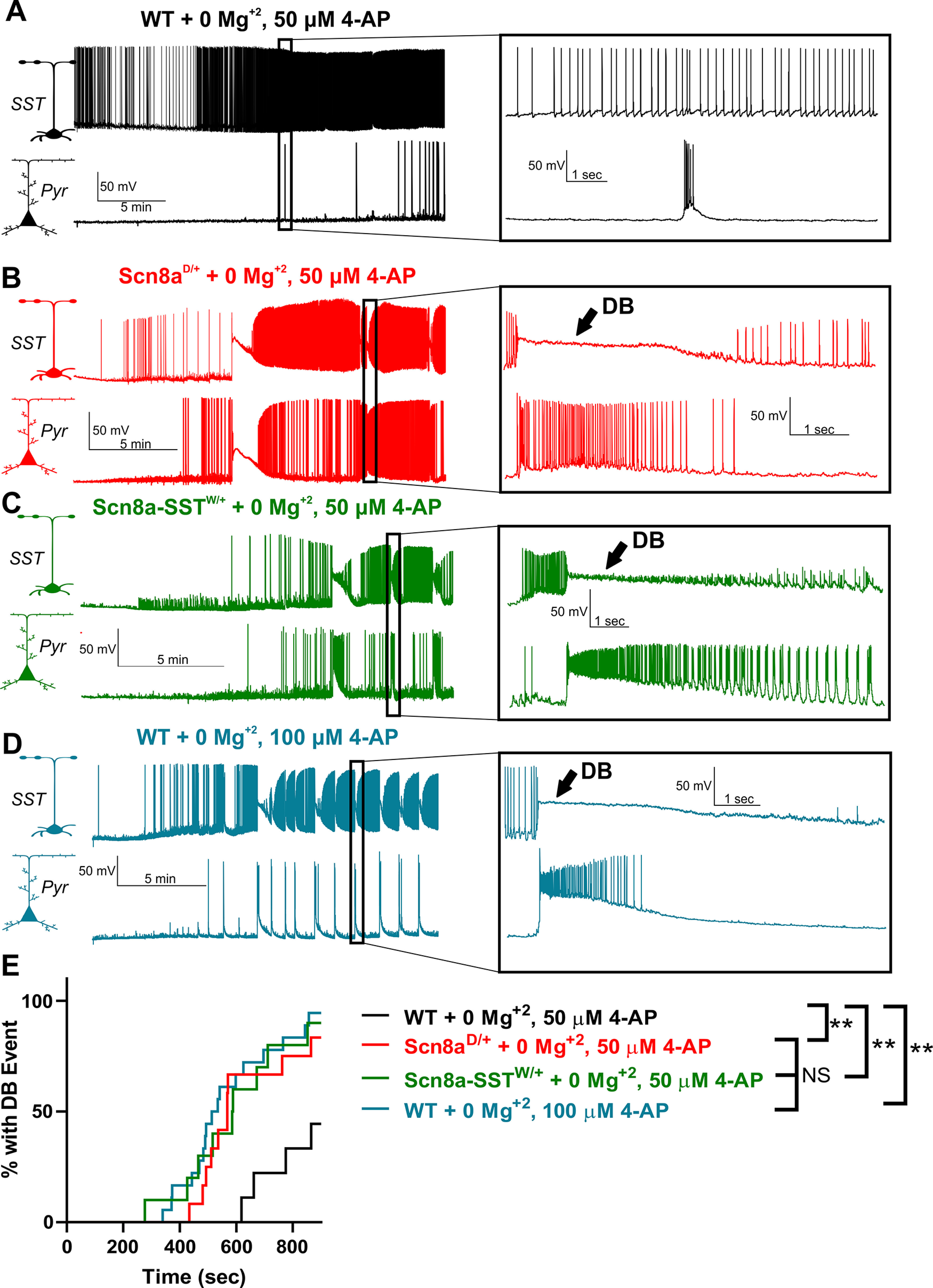
Hypersensitivity of mutant SST interneurons to depolarization block during seizure-like activity. A-D, Representative example traces of WT (A; black), Scn8aD/+ (B; red), and Scn8a-SSTW/+ (C; green) SST-pyramidal neuron pairs exposed to bath solution containing 0 Mg2+, 50 μm 4-AP (A-C) and WT SST-pyramidal neuron pairs exposed to 0 Mg2+, 100 μm 4-AP (D; blue). Expanded views represent example synchronous SST interneuron depolarization block and pyramidal neuron ictal discharge events (B-D), which were not present in most neuronal pairs in the control group (A). E, Cumulative distribution plot of seizure-like events over time reveals that Scn8aD/+ + 0 Mg2+; 50 μm 4-AP (red), Scn8a-SSTW/+ + 0 Mg2+; 50 μm 4-AP (green), and WT + 0 Mg2+; 100 μm 4-AP (blue) were hypersensitive to depolarization block-mediated seizure-like events relative to WT + 0 Mg2+; 50 μm 4-AP neuron pairs (black; **p < 0.01; Log-rank Mantel-Cox test).
To test whether depolarization block of SST interneurons directly contributes to behavioral seizures, we generated mice in which the GqDREADD excitatory chemogenetic receptor was specifically expressed in SST interneurons (SST-Cre; GqDREADD+/−; Fig. 4A). GqDREADD receptors were activated using CNO (i.p.) at 0.2, 1, and 5 mg/kg and changes in ECoG activity monitored (Fig. 4A). In control mice (SST-Cre; GqDREADD−/−), CNO administration at 5 mg/kg did not alter the ECoG or induce seizure behavior (Fig. 4B). In contrast, SST-Cre; GqDREADD+/− mice exhibited a robust hypersynchronization of ECoG activity characterized by an increase in low-frequency (2-10 Hz) power and displayed behavioral manifestations associated with status epilepticus, including loss of balance, and clonic jerks (Fig. 4C; Movie 1). The fact that the effect of CNO administration in SST-Cre; GqDREADD+/− mice was dose-dependent (p < 0.05 comparing SST-Cre;GqDREADD−/−; black; n = 4 and SST-Cre;GqDREADD+/−; red; n = 8; unpaired t test; Fig. 4D) taken alongside the absence of effect of CNO in SST-Cre;GqDREADD−/− mice supports our notion that GqDREADD-mediated activation of SST interneurons is sufficient to induce prolonged seizures (i.e., status epilepticus) in mice expressing WT sodium channels. In support of our hypothesis, recordings from SST interneurons in SST-Cre;GqDREADD+/− mice demonstrated an increase in spontaneous excitability (p < 0.01; n = 8, 4 mice; paired t test; Fig. 4E,F) and premature depolarization block in response to depolarizing current injections (p < 0.001; n = 8, 4 mice; paired t test; Fig. 4G,H) after bath application of CNO (10 µm). These findings thus resembled the physiological phenotype observed in mutant SCN8A-expressing SST interneurons (Fig. 2).
Behavioral response to CNO (5 mg/kg) in SST-Cre;GqDREADD-/- control and SST-Cre; GqDREADD+/- experimental mice. Representative example video clips of an SST-Cre; GqDREADD-/- control mouse (top) and an SST-Cre; GqDREADD+/- experimental mouse (bottom) ∼20 minutes after injection with CNO (5 mg/kg). The SST-Cre; GqDREADD+/− mouse exhibits jerking movement and loss of balance indicative of status epilepticus.
Previous studies have found that pathogenic SCN8A variants lead to an augmented steady-state INaP (Baker et al., 2018; Bunton-Stasyshyn et al., 2019; Wengert et al., 2019; Wengert and Patel, 2021). We therefore recorded INaP directly in brain slice neurons using slow voltage ramps to maintain voltage control under steady-state conditions (Fig. 5A). We found that the ramp-elicited INaP from Scn8aD/+ (63 ± 8 pA; n = 13, 4 mice) and Scn8a-SSTW/+ (68 ± 8 pA; n = 15, 3 mice) SST interneurons were augmented relative to WT interneurons (38 ± 4 pA; n = 12, 4 mice; p < 0.05, one-way ANOVA followed by Dunnett's multiple comparisons test; Fig. 5B–E). Half-maximal voltages (V1/2) were not different between groups (Fig. 5F). We also recorded the resurgent sodium current (INaR), another component of the overall sodium current known to contribute to fast-spiking physiology in various neuronal populations (Raman and Bean, 1997; Khaliq et al., 2003; Patel et al., 2016; Ottolini et al., 2017; Wengert et al., 2019) (Fig. 5G–K). Although we observed an INaR in SST interneurons, we did not detect differences in voltage-current relationship (Fig. 5J) or peak INaR magnitude (Fig. 5K) between WT (n = 12, 4 mice), Scn8aD/+ (n = 9, 3 mice), and Scn8a-SSTW/+ (n = 10, 3 mice) SST interneurons. Together, these results suggest that increases in INaP current magnitude contribute to the altered excitability observed in the mutant SST interneurons and subsequent entry into depolarization block.
Figure 5.
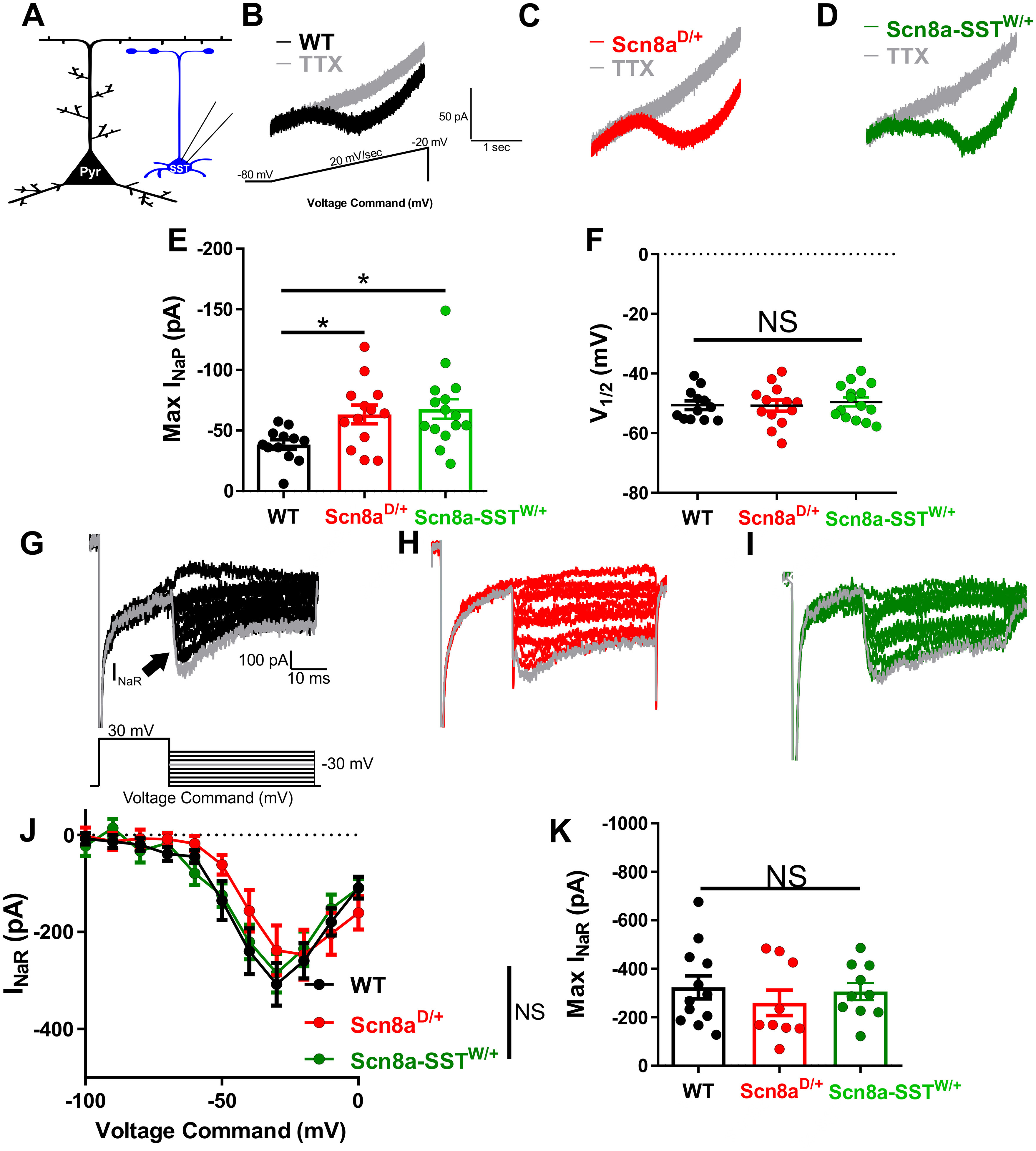
Elevated persistent sodium currents in Scn8aD/+ and Scn8a-SSTW/+ SST interneurons. A, Whole-cell recordings were collected from SST interneurons (blue) to measure whole-cell persistent sodium currents. B-D, Example traces of steady-state INaP evoked by slow voltage ramps (−80 mV to −20 mV at 20 mV/s) before (black, red, or green) and after addition of TTX (500 nm; gray) for WT (B; black), Scn8aD/+ (C; red), and Scn8a-SSTW/+ (D; green) SST interneurons. E, Elevated maximum INaP in Scn8aD/+ SST interneurons (red; n = 13, 4 mice; *p < 0.05) and Scn8a-SSTW/+ (green; n = 14, 3 mice; *p < 0.05) compared with WT SST interneurons (black; n = 12, 4 mice; one-way ANOVA followed by Dunnett's multiple comparisons test). F, Half-maximal voltage of activation between WT, Scn8aD/+, and Scn8a-SSTW/+ SST interneurons was not significantly different between groups (NS; p > 0.05; one-way ANOVA followed by Dunnett's multiple comparisons test). G-I, Example traces of TTX-subtracted INaR for WT (G, black), Scn8aD/+ (H, red), and Scn8a-SSTW/+ (I, green), evoked by voltage commands in which the cell was stepped to membrane potentials of −100 to 0 mV, increments of 10 mV for 40 ms after first being stepped to 30 mV for 20 ms. The INaR (arrow) is observed for steps to −50 mV through 0 mV. J, No differences were observed in the INaR current–voltage relationship between WT (black; n = 12, 4 mice), Scn8aD/+ (red; n = 9, 3 mice), and Scn8a-SSTW/+ (green; n = 10, 3 mice). K, Maximum INaR magnitudes were not different between groups.
We also sought to characterize the macroscopic sodium current in SST interneurons from WT, Scn8aD/+, and Scn8a-SSTW/+ mice, which because of its magnitude and activation speed cannot be properly voltage-clamped in brain slice preparations. First, we examined the transient sodium current by recording excised somatic patches from SST interneurons which allowed for proper voltage control of the macroscopic sodium current (Fig. 6A). No differences in current density, voltage-dependent activation, or steady-state inactivation between WT (n = 9, 4 mice), Scn8aD/+ (n = 13, 5 mice), and Scn8a-SSTW/+ (n = 8, 3 mice) SST interneurons were detected (Fig. 6A–G; Table 3). Excised somatic patches may not fully represent the complete somatic expression of voltage-gated sodium channels. As such, we additionally recorded voltage-gated sodium channel currents from acutely dissociated SST interneurons from WT (n = 7, 3 mice), Scn8aD/+ (n = 7, 3 mice), and Scn8a-SSTW/+ (n = 11, 3 mice; Fig. 6H). Similar to our excised somatic patch recordings, current density, voltage-dependent activation, or steady-state activation were not different between the groups (Fig. 6I–N; Table 4). Interestingly, the magnitude of INaP was not different in acutely dissociated SST interneurons (Fig. 6O). Considering that within GABAergic interneurons NaV1.6 is exclusively expressed within distal subcellular regions including the axon initial segment and axon (Lorincz and Nusser, 2008), it is possible that these measurements of the transient current, which primarily record activity from somatic voltage-gated sodium channels, do not fully represent the contribution of axonal NaV1.6 channels as observed when using slow ramp voltages in intact neurons (Fig. 5A–D).
Figure 6.
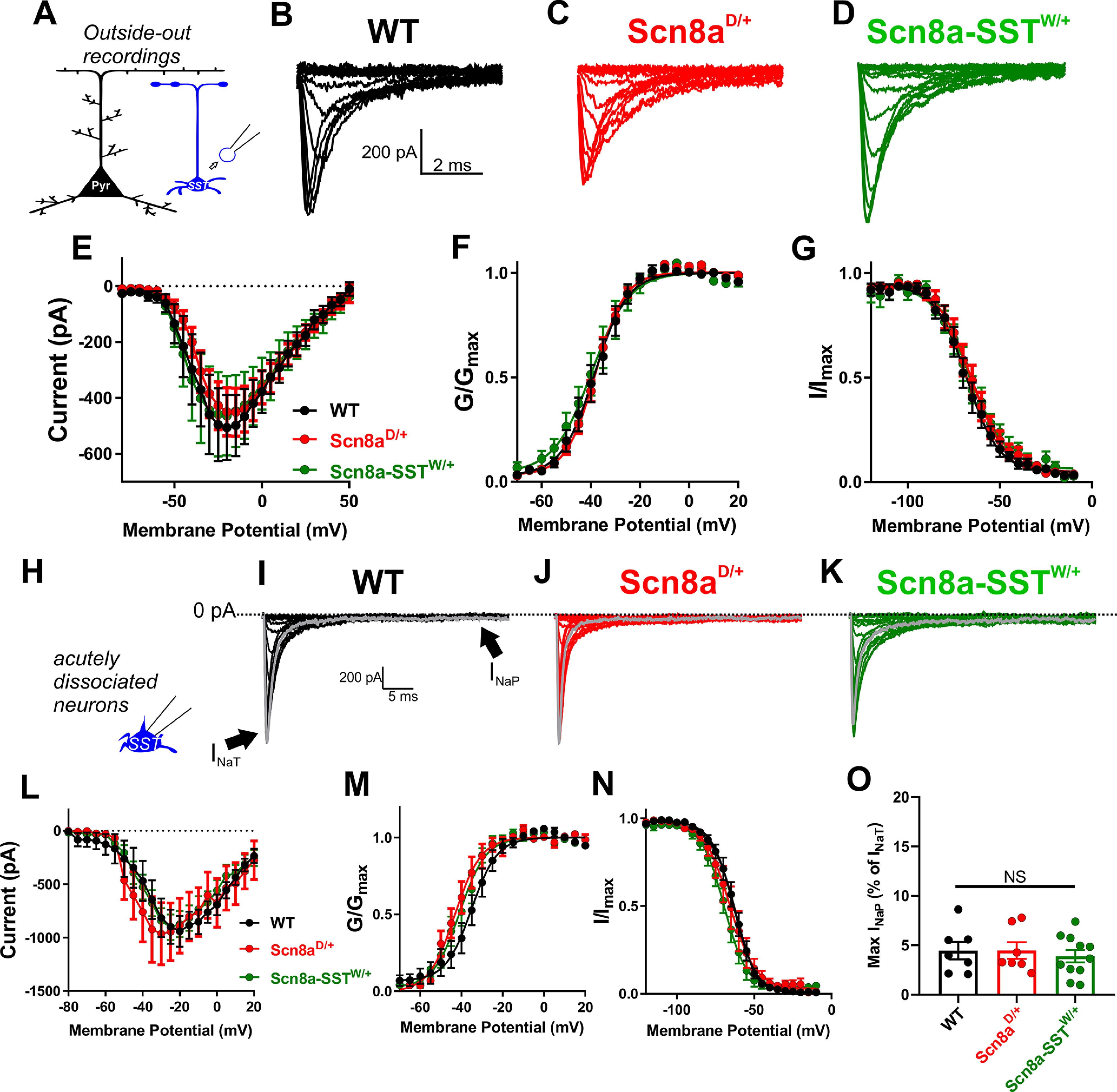
Somatic voltage-gated sodium currents in WT, Scn8aD/+ and Scn8a-SSTW/+ SST interneurons. A, Somatic transient sodium current was assessed in SST interneurons (blue) using patch-clamp recordings in the outside-out configuration. B-D, Example traces for family of voltage-dependent sodium currents recorded from outside-out excised patches from WT (B; black), Scn8aD/+ (C; red), and Scn8a-SSTW/+ (D; green) SST interneurons. E, Current–voltage relationship for WT (black; n = 9, 4 mice), Scn8aD/+ (red; n = 13, 5 mice), and Scn8a-SSTW/+ (green; n = 8, 3 mice) SST interneurons. F, G, Voltage-dependent conductance and steady-state inactivation curves for WT (black), Scn8aD/+ (red), and Scn8a-SSTW/+ (green) SST interneurons. H, Voltage-gated sodium channel currents were also examined in acutely dissociated neurons, which remove much of the distal neuronal processes. I-K, Example traces for family of voltage-dependent sodium currents recorded from acutely dissociated SST interneurons from WT (I; black), Scn8aD/+ (J; red), and Scn8a-SSTW/+ (K; green) mice. Arrows indicate the fast transient (INaT) and persistent (INaP) sodium currents. L, Current–voltage relationship for WT (black; n= 7, 3 mice), Scn8aD/+ (red; n = 7, 3 mice), and Scn8a-SSTW/+ (green; n = 11, 3). M, N, Boltzmann curves for voltage-dependent activation (M) and steady-state inactivation (N). O, Maximum magnitude of the INaP elicited by voltage steps in acutely dissociated SST interneurons.
Table 3.
Voltage-gated sodium channel parameters from somatic outside-out recordings WT, Scn8aD/+, and Scn8a-SSTW/+ SST inhibitory interneurons
| Peak current (pA) | Activation V1/2 (mV) K (mV) | Inactivation V1/2 (mV) K (mV) | Decay tau (ms) | |||
|---|---|---|---|---|---|---|
| WT (n = 13) | −505 ± 117 | −38.7 ± 2.2 | 5.1 ± 0.4 | −67.9 ± 2.7 | 9.6 ± 0.9 | 0.54 ± 0.04 |
| Scn8aD/+ (n = 9) | −451 ± 87 | −37.7 ± 1.6 | 5.9 ± 0.6 | −65.0 ± 3.1 | 10.1 ± 0.7 | 0.66 ± 0.05 |
| Scn8a-SSTW/+ (n = 8) | −498 ± 153 | −40.4 ± 2.9 | 5.9 ± 0.8 | −65.8 ± 2.8 | 11.2 ± 1.0 | 0.68 ± 0.05 |
Data are mean ± SEM. All comparisons with WT were nonsignificant using a one-way ANOVA followed by Dunnett's multiple comparisons test.
Table 4.
Voltage-gated sodium channel parameters recordings from acutely dissociated WT, Scn8aD/+, and Scn8a-SSTW/+ SST inhibitory interneurons
| Peak current (pA/pF) | Activation V1/2 (mV) K (mV) | Inactivation V1/2 (mV) K (mV) | Decay tau (ms) | |||
|---|---|---|---|---|---|---|
| WT (n = 10) | −941 ± 146 | −36.4 ± 3.5 | 4.7 ± 0.5 | −63.1 ± 1.9 | 6.5 ± 0.7 | 0.83 ± 0.10 |
| Scn8aD/+ (n = 10) | −907 ± 265 | −42.3 ± 2.7 | 3.5 ± 0.3 | −65.2 ± 3.8 | 6.4 ± 0.4 | 0.70 ± 0.08 |
| Scn8a-SSTW/+ (n = 13) | −902 ± 119 | −41.1 ± 2.5 | 3.8 ± 0.3 | −69.2 ± 2.4 | 6.2 ± 0.5 | 0.75 ± 0.06 |
Data are mean ± SEM. All comparisons with WT were nonsignificant using a one-way ANOVA followed by Dunnett's multiple comparisons test.
To test our hypothesis for the importance of INaP in controlling SST interneuron excitability, we used a single-compartment conductance-based computational neuron model to examine neuronal frequency-current relationships as we varied the magnitude of the persistent sodium conductance (gNaP). At low levels of gNaP, neuronal firing frequency increased with the injected current magnitude over a large range of depolarizing current injections, reaching a maximum of 96 APs/500 ms (Fig. 7A–C). In support of our hypothesis, we found that increasing the magnitude of the gNaP led to a decrease in rheobase current and a corresponding facilitation of AP firing at low-magnitude current injections, followed by premature AP failure via entry into depolarization block as the current injection magnitude was increased (Fig. 7A–C). Although the neuronal model used is overly simple in both geometric and ionic terms relative to SST interneurons in vivo, the sufficiency of the model to reproduce both features of mutant SST interneuron excitability is strong evidence that an elevated INaP is directly sufficient for initial hyperexcitability followed by premature entry into depolarization block.
Figure 7.
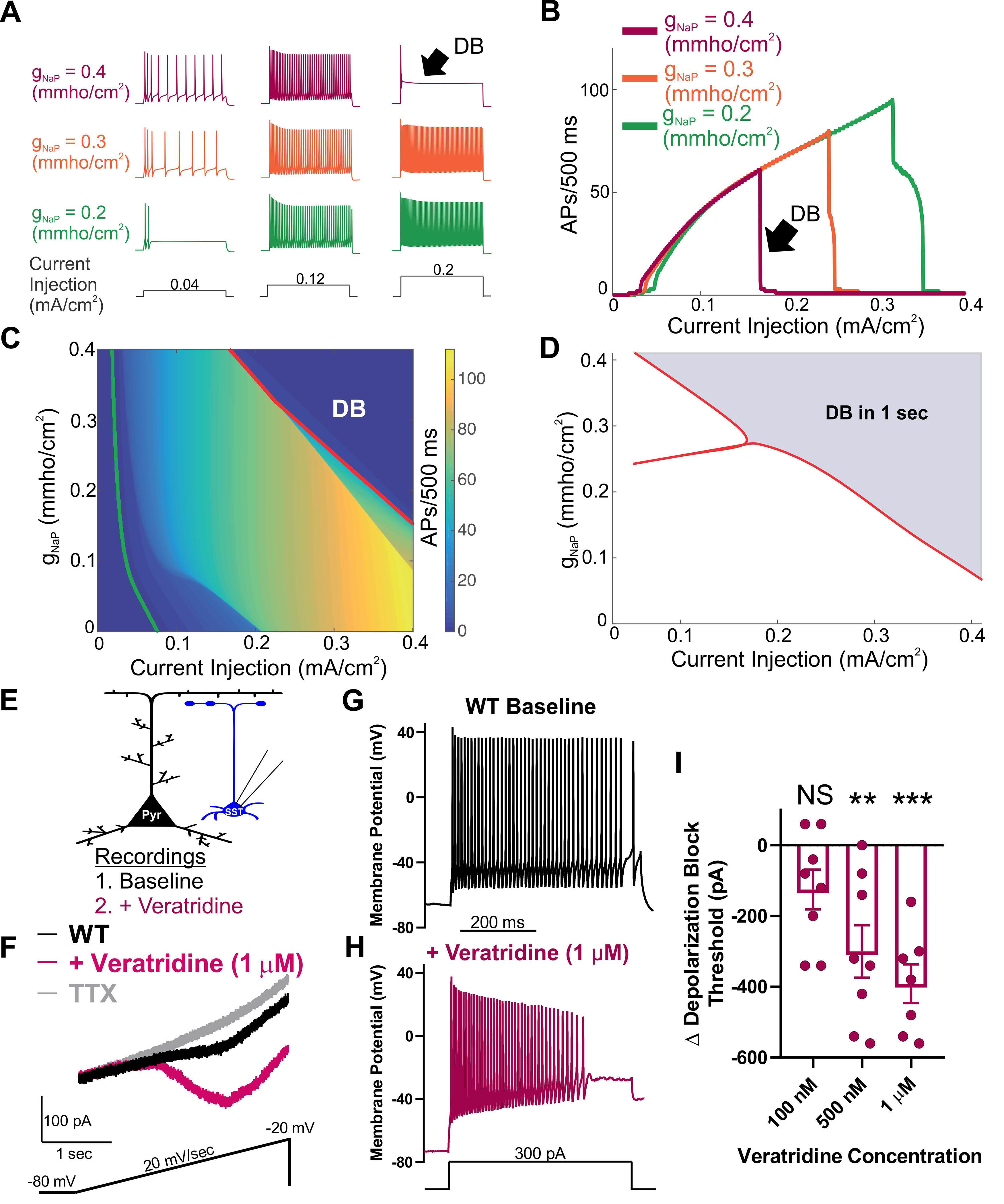
Augmentation of the persistent sodium current induces depolarization block. A, Example traces of APs elicited in response to 500 ms current injections in an in silico neuronal model. The maximal persistent sodium conductance was varied from gNaP = 0.2 mmho/cm2 (green), through gNaP = 0.3 mmho/cm2 (orange) to gNaP = 0.4 mmho/cm2 (magenta), while the magnitude of the current injection was increased from 0.04 mA/cm2 (left), through 0.12 mA/cm2 (middle) to 0.2 mA/cm2 (right). Increasing gNaP increased the number of APs observed over the stimulation period at low current magnitudes, and led to failure of AP initiation at higher current injection magnitudes (black arrow, DB). B, Number of APs observed over the 500 ms stimulus relative to the current magnitude. As gNaP increases, the current magnitude required to induce depolarization block (black arrow, DB) decreases. C, Heat map showing the number of APs as both gNaP and the current injection magnitude are varied. Also shown is the rheobase current required to induce at least one AP (green line) and the curve at which the neuron ceases to generate repetitive APs as it enters depolarization block (red line, DB). D, Two parameter bifurcation showing the location of dynamic transitions leading to depolarization block as the gNaP and the current magnitude are varied. Red line indicates the shaded region of parameter space in which depolarization block occurs (over a 1s current pulse) from the unshaded region in which it does not. E, Experimental design: Whole-cell recordings were made from an SST interneuron (blue) before (baseline) and after bath application of the INaP current activator, veratridine (magenta). F, Example trace of INaP current before (black), after application of veratridine (1 μm; magenta) followed by application of TTX (500nM: gray). G, H, Example traces of a WT SST interneuron before (G; black) and after (H; magenta) bath application of INaP current-activator veratridine (1 μm). I, Shift in depolarization block threshold in response to bath-applied veratridine at 100 nm (n = 8, 3 mice), 500 nm (n = 8, 3 mice), and 1 μm (n = 7, 3 mice). **p < 0.01, ***p < 0.001, comparing before and after treatment with veratridine (paired t test).
Transitions in dynamical states, such as the entry into depolarization block, can be understood through the application of bifurcation theory. This approach allowed for tracking of the number and properties of steady states (corresponding to the neuron being at rest or in depolarization block) and periodic orbits (corresponding to spiking behavior) as model parameters were varied. Providing further evidence in support of our hypothesis, we observed that, as gNaP was increased, the depolarization block boundary shifted leftward to lower current magnitudes, corroborating our simulation results and providing a more detailed theoretical underpinning for them (Fig. 7D).
To test predictions from the computational model regarding the effect of increasing persistent sodium conductance, we collected recordings of WT SST interneurons before and after application of veratridine. Veratridine is a plant-derived toxin that increases the magnitude of the INaP (Alkadhi and Tian, 1996; Mantegazza et al., 1998; Otoom and Alkadhi, 2000; Bikson et al., 2003; Tazerart et al., 2008; Tsuruyama et al., 2013). In agreement with our hypothesis, veratridine (1 µm) increased the SST interneuron INaP by 68 ± 20 pA (n = 6, 2 mice; *p < 0.05; paired t test; Fig. 7F). In current-clamp recordings, we compared the depolarization block threshold before and after treatment with veratridine at 100 nm, 500 nm, and 1 µm. We observed a dose-dependent reduction in depolarization block threshold in response to veratridine at 500 nm (p < 0.01; paired t test; n = 8, 3 mice) and 1 µm (p < 0.01; paired t test; n = 7, 3 mice), but not at 100 nm (n = 8, 3 mice; paired t test; Fig. 7G–I). Together, our computational and pharmacological evidence demonstrates that the magnitude of the INaP current strongly influences the threshold for depolarization block in SST interneurons.
Discussion
In this study, we have identified a mechanism by which SST interneurons are impaired and contribute to seizures in SCN8A epileptic encephalopathy. We show that (1) expression of the SCN8A mutation R1872W selectively in SST interneurons is sufficient to induce audiogenic seizures, implicating a previously unidentified role for SST interneurons in SCN8A epileptic encephalopathy; (2) gain-of-function SCN8A mutations facilitate SST interneuron intrinsic hyperexcitability and AP failure via depolarization block; (3) rhythmic instances of depolarization block of mutant SST interneurons are coincident with cortical pyramidal neuron ictal discharges under seizure-like conditions; (4) chemogenetic activation and induction of depolarization block of WT SST interneurons induces prolonged seizures, further supporting a critical role for SST interneurons in maintaining balanced neuronal network excitation; and (5) the INaP is elevated in SST interneurons expressing gain-of-function SCN8A mutations, and it directly facilitates depolarization block. These findings provide novel insight into a previously unappreciated role for SST interneurons in seizure initiation, not only in the context of SCN8A epileptic encephalopathy, but to epilepsy more generally.
Previous studies using transgenic mouse models of SCN8A epileptic encephalopathy have focused primarily on the impact of gain-of-function SCN8A mutations on excitatory neurons and how pro-excitatory alterations in sodium channel properties render these neurons hyperexcitable and corresponding networks seizure prone (Lopez-Santiago et al., 2017; Ottolini et al., 2017; Baker et al., 2018; Bunton-Stasyshyn et al., 2019; Wengert et al., 2019). Our finding that cell-type-specific expression of a patient-derived gain-of-function SCN8A mutation (R1872W) in SST interneurons, but not forebrain excitatory neurons, resulted in audiogenic seizures indicates that this neuronal population contributes to seizures in SCN8A epileptic encephalopathy. It is unclear whether patients with SCN8A epileptic encephalopathy experience audiogenic seizures. All patients are typically on sodium channel blockers as a means to controlling their seizures, yet the patients continue to experience seizures and have a high incidence of sudden unexpected death in epilepsy (Veeramah et al., 2012; Gardella et al., 2018; Johannesen et al., 2018). Our results here and previously in the Scn8aD/+ mouse model (Wengert et al., 2021) encourage future investigation into audiological impairment in SCN8A patients. Future studies are warranted to examine how various cortical and subcortical neural circuits are recruited during audiogenic seizures and corresponding behavior.
A proconvulsant impact of inhibitory interneuron hypofunction has been reported previously in the context of Dravet syndrome in which expression of loss-of-function SCN1A results in reduced intrinsic excitability of inhibitory interneurons (Rhodes et al., 2004; Escayg and Goldin, 2010; Cheah et al., 2012; Tai et al., 2014). In contrast, our recordings in SST interneurons expressing gain-of-function SCN8A mutations exhibited an initial hyperexcitability followed by progressive AP failure because of depolarization block. Interestingly, our results were similar to those found in a recent study examining the gain-of-function SCN1A point-mutation T226M, which leads to hypofunction through depolarization block and clinically results in a distinct epileptic encephalopathy more severe than traditional Dravet syndrome (Sadleir et al., 2017; Berecki et al., 2019). Our findings support their interpretation that gain-of-function voltage-gated sodium channel variants can produce functionally dominant-negative effects through depolarization block-mediated AP failure.
Making up 5%-10% of cortical neurons, SST inhibitory interneurons are critical components of cortical microcircuits (Rudy et al., 2011; Nigro et al., 2018). In cortical layer V, SST interneurons have been shown to provide lateral inhibition to local pyramidal neurons on high-frequency AP firing (Silberberg and Markram, 2007). This disynaptic inhibitory mechanism has been suggested to regulate the overall excitability of the cortical column, preventing simultaneous excitability in many pyramidal neurons at one time (Silberberg and Markram, 2007). In our dual-recording experiments, in which an SST interneuron and a nearby pyramidal neuron were recorded simultaneously under seizure-like conditions, we observed that SST interneuron depolarization block events and pyramidal neuron ictal discharges were coincident. Depolarization block of SST interneurons could alter the excitability of nearby pyramidal neurons by transiently silencing synaptic inhibition from SST interneurons, which has been shown to act in concert to strongly constrain the excitability of cortical pyramidal neurons (Safari et al., 2017). An additional, non-mutually exclusive mechanism is that depolarization block in SST interneurons could change the local extracellular ionic environment in a manner that elevates pyramidal neuron excitability. In a previous modeling study of interplay between interneurons and pyramidal neurons during in vitro seizure-like events, elevations in external potassium concentration because of interneuron depolarization block were a critical parameter in achieving good agreement between the model and experimental results (Ullah and Schiff, 2010). Future experiments focused on more completely identifying the mechanisms by which interneurons synchronize with pyramidal neurons under seizure-like conditions are warranted.
An interplay between inhibitory and excitatory neurons via depolarization block of inhibitory interneurons and network seizure-like activity has been previously proposed (Swadlow, 2003; Ziburkus et al., 2006; Cammarota et al., 2013; Parrish et al., 2019). In the study by Parrish et al. (2019), depolarization block of parvalbumin, but not SST, interneurons occurred during seizure-like events in vitro, although recording in the cell-attached configuration might prohibit one to definitively detect the brief instances of depolarization block in SST interneurons observed in our study (Parrish et al., 2019). To our knowledge, our study is the first to show that cortical SST interneurons, similar to other fast-spiking interneurons, have coordinated activity with nearby pyramidal neurons characterized by coincident depolarization block events during pyramidal ictal discharges.
Our chemogenetic experiments further support our hypothesis that hypersensitivity to depolarization block of SST interneurons directly contributes to seizures in vivo. GqDREADD-mediated hyperactivation of WT SST interneurons increased spontaneous excitability and increased susceptibility to depolarization block (similar to the effects of mutant SCN8A expression) and remarkably led to prolonged seizure activity and behavior (i.e., status epilepticus). These surprising results indicate that aberrant SST physiology, namely, increased propensity for depolarization block, is sufficient to induce hypersynchronous ECoG activity and seizures in otherwise normal mice. Additional experiments are warranted to continue to clarify the role that SST interneurons have in various types of epilepsy and whether SST depolarization block is pathogenic in other contexts.
Using numerous methods to explore sodium channel activity, we observed a profound increase in the magnitude of the INaP in both Scn8aD/+ and Scn8a-SSTW/+ SST interneurons measured using slow voltage ramps in brain slice neurons without changes in INaR. Although our experiments using acutely dissociated neurons enabled us to measure transient sodium channel activity, we failed to observe an elevated INaP in the mutant SST interneurons. A likely explanation for this difference is the dissociation procedure itself, which disrupts the integrity of distal subcellular regions, namely, the axon initial segment and axon where NaV1.6 is predominately expressed. The INaP has been intensely investigated since its discovery (Stafstrom et al., 1985; French et al., 1990; Stafstrom, 2007; Wengert and Patel, 2021). It has been ascribed a variety of important physiological and neurocomputational functions, including spike timing (Vervaeke et al., 2006; Osorio et al., 2010), amplification of synaptic inputs (Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Stuart, 1999), and pacemaking (Del Negro et al., 2002; Zhong et al., 2007; Yamada-Hanff and Bean, 2013; Yamanishi et al., 2018). An aberrantly large INaP has also been implicated in various epilepsy syndromes (Lossin et al., 2003; Rhodes et al., 2004; Stafstrom, 2007; Hargus et al., 2011, 2013; Veeramah et al., 2012; Wengert and Patel, 2021). In our recordings of mutant SST interneurons, we found higher rates of spontaneous activity, depolarized resting membrane potential, decreased rheobase, and altered upstroke and downstroke velocity, each of which has been previously associated with an augmented INaP (Stafstrom et al., 1982; Herzog et al., 2001; Yamada-Hanff and Bean, 2013; Ceballos et al., 2017). Moreover, by modifying the magnitude of the INaP alone, our computational model recapitulated both of the primary physiological features of the mutant interneurons, that of initial hyperexcitability at low current injections followed by premature depolarization block at higher current injections. Although we cannot rule out potential off-target effects, our findings that veratridine, a toxin previously used to interrogate the mechanism of the INaP (Alkadhi and Tian, 1996; Mantegazza et al., 1998; Otoom and Alkadhi, 2000; Bikson et al., 2003; Tazerart et al., 2008; Tsuruyama et al., 2013), increased the INaP and induced premature depolarization block in WT SST interneurons, highly supports our computational modeling. Further, our bifurcation analysis revealed that the dynamic transitions from resting to spiking to depolarization block were each dependent on the INaP magnitude, indicating that the INaP magnitude is of general importance for determining neuronal spiking dynamics. Thus, an increased INaP, regardless of particular mechanism, would be predicted to induce hypersensitivity to depolarization block.
Within the class of SST interneurons, there is a great deal of diversity with respect to morphology, gene expression, synaptic input and output, and physiology. In our recordings of cortical layer V SST interneurons, we likely recorded from numerous distinct subpopulations of inhibitory interneurons, including Martinotti cells, basket cells, bitufted, horizontal, and multipolar cells (Yavorska and Wehr, 2016). Ongoing efforts to distinguish subcellular populations within SST-positive interneurons will further refine the interpretation of the results presented here.
In conclusion, previous studies have contributed significant evidence to the notion that SCN8A epileptic encephalopathy is caused by gain-of-function SCN8A mutations, which result in hyperexcitability of excitatory neurons and renders the network hyperexcitable and seizure-prone. In this report, we have demonstrated that dysfunctional SST inhibitory interneurons also contribute to SCN8A epileptic encephalopathy: An elevated INaP in SST inhibitory interneurons results in hyperexcitability and AP failure because of depolarization block. Gain-of-function SCN8A mutations render SST interneurons more sensitive to depolarization block; and under seizure conditions, these instances of SST depolarization block are coincident to pyramidal neuron ictal discharges. Induction of SST interneuron hyperexcitability and depolarization block, even in the absence of an SCN8A mutation, leads to status epilepticus illustrating the critical contribution of SST interneurons to seizures. Restoration of normal excitability in SST inhibitory interneurons may provide a novel therapeutic strategy for patients with SCN8A epileptic encephalopathy.
Footnotes
This was supported by National Institutes of Health Grants R01NS103090 and R01NS120702 to M.K.P. and Grant 1F31NS115451-01 to E.R.W. K.C.A.W. supported by MRC Fellowship MR/P01478X/1. We thank the entire M.K.P. laboratory and others at the University of Virginia for thoughtful feedback on this study and manuscript. K.C.A.W. thanks Jennifer Creaser for assistance in the use of AUTO-07P.
The authors declare no competing financial interests.
References
- Alkadhi KA, Tian LM (1996) Veratridine-enhanced persistent sodium current induces bursting in CA1 pyramidal neurons. Neuroscience 71:625–632. 10.1016/0306-4522(95)00488-2 [DOI] [PubMed] [Google Scholar]
- Baker EM, Thompson CH, Hawkins NA, Wagnon JL, Wengert ER, Patel MK, George AL, Meisler MH, Kearney JA (2018) The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 59:1166–1176. 10.1111/epi.14196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BS, Nigam A, Ottolini M, Gaykema RP, Hargus NJ, Patel MK (2017) Pro-excitatory alterations in sodium channel activity facilitate subiculum neuron hyperexcitability in temporal lobe epilepsy. Neurobiol Dis 108:183–194. 10.1016/j.nbd.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, Petrou S (2019) SCN1A gain of function in early infantile encephalopathy. Ann Neurol 85:514–525. 10.1002/ana.25438 [DOI] [PubMed] [Google Scholar]
- Bernard C, Cossart R, Hirsch JC, Esclapez M, Ben-Ari Y (2000) What is GABAergic inhibition? How is it modified in epilepsy? Epilepsia 41 Suppl 6:S90–S95. 10.1111/j.1528-1157.2000.tb01564.x [DOI] [PubMed] [Google Scholar]
- Bikson M, Hahn PJ, Fox JE, Jefferys JG (2003) Depolarization block of neurons during maintenance of electrographic seizures. J Neurophysiol 90:2402–2408. 10.1152/jn.00467.2003 [DOI] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RK, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, Bhatia K, Jones JM, Maniaci MR, Parent JM, Goodkin HP, Patel MK, Meisler MH (2019) Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142:362–375. 10.1093/brain/awy324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M, Carmignoto G (2013) Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol 591:807–822. 10.1113/jphysiol.2012.238154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos CC, Roque AC, Leão RM (2017) A negative slope conductance of the persistent sodium current prolongs subthreshold depolarizations. Biophys J 113:2207–2217. 10.1016/j.bpj.2017.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA (2012) Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 109:14646–14651. 10.1073/pnas.1211591109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM, van 't Slot R, van Lieshout S, Nijman IJ, O'Brien JE, Hammer MF, Estacion M, Waxman SG, Dib-Hajj SD, Koeleman BP (2014) Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res 108:1511–1518. 10.1016/j.eplepsyres.2014.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Negro CA, Koshiya N, Butera RJ, Smith JC (2002) Persistent sodium current, membrane properties and bursting behavior of pre-Bötzinger complex inspiratory neurons in vitro. J Neurophysiol 88:2242–2250. 10.1152/jn.00081.2002 [DOI] [PubMed] [Google Scholar]
- Escayg A, Goldin AL (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51:1650–1658. 10.1111/j.1528-1167.2010.02640.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, O'Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, Meisler MH (2014) A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis 69:117–123. 10.1016/j.nbd.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM (2018) A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet syndrome. J Neurosci 38:7912–7927. 10.1523/JNEUROSCI.0193-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW (1990) A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol 95:1139–1157. 10.1085/jgp.95.6.1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardella E, Marini C, Trivisano M, Fitzgerald MP, Alber M, Howell KB, Darra F, Siliquini S, Bölsterli BK, Masnada S, Pichiecchio A, Johannesen KM, Jepsen B, Fontana E, Anibaldi G, Russo S, Cogliati F, Montomoli M, Specchio N, Rubboli G, et al. (2018) The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 91:E1112–E1124. 10.1212/WNL.0000000000006199 [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram EH, Patel MK (2011) Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol Dis 41:361–376. 10.1016/j.nbd.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Nigam A, Bertram EH, Patel MK (2013) Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol 110:1144–1157. 10.1152/jn.00383.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Waxman SG (2001) Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J Neurophysiol 86:1351–1364. 10.1152/jn.2001.86.3.1351 [DOI] [PubMed] [Google Scholar]
- Jayant K, Wenzel M, Bando Y, Hamm JP, Mandriota N, Rabinowitz JH, Plante IL, Owen JS, Sahin O, Shepard KL, Yuste R (2019) Flexible nanopipettes for minimally invasive intracellular electrophysiology in vivo. Cell Rep 26:266–278.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesen KM, Gardella E, Scheffer I, Howell K, Smith DM, Helbig I, Møller RS, Rubboli G (2018) Early mortality in SCN8A-related epilepsies. Epilepsy Res 143:79–81. 10.1016/j.eplepsyres.2018.04.008 [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM (2003) The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 23:4899–4912. 10.1523/JNEUROSCI.23-12-04899.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS (2006) Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci 26:4613–4623. 10.1523/JNEUROSCI.0064-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, Depienne C, Brilstra E, Mang Y, Nielsen JE, Kirkpatrick M, Goudie D, Goldman R, Jähn JA, Jepsen B, Gill D, Döcker M, Biskup S, McMahon JM, Koeleman B, et al. (2015) The phenotypic spectrum of SCN8A encephalopathy. Neurology 84:480–489. 10.1212/WNL.0000000000001211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Tian C, Scalmani P, Frassoni C, Mantegazza M, Wang Y, Yang M, Wu S, Shu Y (2014) Action potential initiation in neocortical inhibitory interneurons. PLoS Biol 12:e1001944. 10.1371/journal.pbio.1001944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Yuan Y, Wagnon JL, Hull JM, Frasier CR, O'Malley HA, Meisler MH, Isom LL (2017) Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci USA 114:2383–2388. 10.1073/pnas.1616821114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z (2008) Cell-type-dependent molecular composition of the axon initial segment. J Neurosci 28:14329–14340. 10.1523/JNEUROSCI.4833-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C, Rhodes TH, Desai RR, Vanoye CG, Wang D, Carniciu S, Devinsky O, George AL (2003) Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J Neurosci 23:11289–11295. 10.1523/JNEUROSCI.23-36-11289.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, Huguenard JR (2017) Regulation of thalamic and cortical network synchrony by Scn8a. Neuron 93:1165–1179.e6. 10.1016/j.neuron.2017.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G (1998) Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurones. J Physiol 507:105–116. 10.1111/j.1469-7793.1998.105bu.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C (2004) Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5:793–807. 10.1038/nrn1519 [DOI] [PubMed] [Google Scholar]
- Martin B, Dieuset G, Pawluski JL, Costet N, Biraben A (2020) Audiogenic seizure as a model of sudden death in epilepsy: a comparative study between four inbred mouse strains from early life to adulthood. Epilepsia 61:342–349. 10.1111/epi.16432 [DOI] [PubMed] [Google Scholar]
- Nigro MJ, Hashikawa-Yamasaki Y, Rudy B (2018) Diversity and connectivity of layer 5 somatostatin-expressing interneurons in the mouse barrel cortex. J Neurosci 38:1622–1633. 10.1523/JNEUROSCI.2415-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacki J, Osinga HM, Tsaneva-Atanasova K (2012) Dynamical systems analysis of spike-adding mechanisms in transient bursts. J Math Neurosci 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Kato M, Takahashi S, Lerman-Sagie T, Lev D, Terashima H, Kubota M, Kawawaki H, Matsufuji M, Kojima Y, Tateno A, Goldberg-Stern H, Straussberg R, Marom D, Leshinsky-Silver E, Nakashima M, Nishiyama K, Tsurusaki Y, Miyake N, Tanaka F, et al. (2014) Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 55:994–1000. 10.1111/epi.12668 [DOI] [PubMed] [Google Scholar]
- Osorio N, Cathala L, Meisler MH, Crest M, Magistretti J, Delmas P (2010) Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol 588:651–670. 10.1113/jphysiol.2010.183798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otoom SA, Alkadhi KA (2000) Epileptiform activity of veratridine model in rat brain slices: effects of antiepileptic drugs. Epilepsy Res 38:161–170. 10.1016/S0920-1211(99)00084-4 [DOI] [PubMed] [Google Scholar]
- Ottolini M, Barker BS, Gaykema RP, Meisler MH, Patel MK (2017) Aberrant sodium channel currents and hyperexcitability of medial entorhinal cortex neurons in a mouse model of SCN8A encephalopathy. J Neurosci 37:7643–7655. 10.1523/JNEUROSCI.2709-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish RR, Codadu NK, Mackenzie-Gray Scott C, Trevelyan AJ (2019) Feedforward inhibition ahead of ictal wavefronts is provided by both parvalbumin- and somatostatin-expressing interneurons. J Physiol 597:2297–2314. 10.1113/JP277749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR (2016) Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 139:2164–2181. 10.1093/brain/aww129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1997) Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17:4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL (2004) Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci USA 101:11147–11152. 10.1073/pnas.0402482101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H (2008) Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol 100:2361–2380. 10.1152/jn.90332.2008 [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee SH, Hjerling-Leffler J (2011) Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71:45–61. 10.1002/dneu.20853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, Devile C, Kurian MA, Mandelstam S, Wirrell E, Nickels KC, Murali HR, Carvill G, Myers CT, Mefford HC, Scheffer IE, DDD Study (2017) Not all SCN1A epileptic encephalopathies are Dravet syndrome. Neurology 89:1035–1042. 10.1212/WNL.0000000000004331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safari MS, Mirnajafi-Zadeh J, Hioki H, Tsumoto T (2017) Parvalbumin-expressing interneurons can act solo while somatostatin-expressing interneurons act in chorus in most cases on cortical pyramidal cells. Sci Rep 7:12764. 10.1038/s41598-017-12958-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE (1995) Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol 74:2220–2224. 10.1152/jn.1995.74.5.2220 [DOI] [PubMed] [Google Scholar]
- Silberberg G, Markram H (2007) Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53:735–746. 10.1016/j.neuron.2007.02.012 [DOI] [PubMed] [Google Scholar]
- Stafstrom CE (2007) Persistent sodium current and its role in epilepsy. Epilepsy Curr 7:15–22. 10.1111/j.1535-7511.2007.00156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE (2013) Jasper's basic mechanisms of the epilepsies, 4th edition. Neurology 81:1883–1884. 10.1212/01.wnl.0000436077.88232.2a [DOI] [Google Scholar]
- Stafstrom CE, Schwindt PC, Crill WE (1982) Negative slope conductance due to a persistent subthreshold sodium current in cat neocortical neurons in vitro. Brain Res 236:221–226. 10.1016/0006-8993(82)90050-6 [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Schwindt PC, Chubb MC, Crill WE (1985) Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J Neurophysiol 53:153–170. 10.1152/jn.1985.53.1.153 [DOI] [PubMed] [Google Scholar]
- Stuart G (1999) Voltage-activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat Neurosci 2:144–150. 10.1038/5698 [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B (1995) Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15:1065–1076. 10.1016/0896-6273(95)90095-0 [DOI] [PubMed] [Google Scholar]
- Swadlow HA (2003) Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb Cortex 13:25–32. 10.1093/cercor/13.1.25 [DOI] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA (2014) Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 111:E3139–E3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazerart S, Vinay L, Brocard F (2008) The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J Neurosci 28:8577–8589. 10.1523/JNEUROSCI.1437-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruyama K, Hsiao CF, Chandler SH (2013) Participation of a persistent sodium current and calcium-activated nonspecific cationic current to burst generation in trigeminal principal sensory neurons. J Neurophysiol 110:1903–1914. 10.1152/jn.00410.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah G, Schiff SJ (2010) Assimilating seizure dynamics. PLoS Comput Biol 6:e1000776. 10.1371/journal.pcbi.1000776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF (2012) De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90:502–510. 10.1016/j.ajhg.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ, Storm JF (2006) Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron 49:257–270. 10.1016/j.neuron.2005.12.022 [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH (2015) Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 24:506–515. 10.1093/hmg/ddu470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK, Meisler MH (2016) Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 3:114–123. 10.1002/acn3.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengert ER, Patel MK (2021) The role of the persistent sodium current in epilepsy. Epilepsy Curr 21:40–47. 10.1177/1535759720973978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengert ER, Saga AU, Panchal PS, Barker BS, Patel MK (2019) Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 158:107699. 10.1016/j.neuropharm.2019.107699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengert ER, Wenker IC, Wagner EL, Wagley PK, Gaykema RP, Shin JB, Patel MK (2021) Adrenergic mechanisms of audiogenic seizure-induced death in a mouse model of SCN8A encephalopathy. Front Neurosci 15:581048. 10.3389/fnins.2021.581048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Teran FA, Wengert ER, Wagley PK, Panchal PS, Blizzard EA, Saraf P, Wagnon JL, Goodkin HP, Meisler MH, Richerson GB, Patel MK (2021) Postictal death is associated with tonic phase apnea in a mouse model of sudden unexpected death in epilepsy. Ann Neurol 89:1023–1035. 10.1002/ana.26053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Hanff J, Bean BP (2013) Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J Neurosci 33:15011–15021. 10.1523/JNEUROSCI.0577-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi T, Koizumi H, Navarro MA, Milescu LS, Smith JC (2018) Kinetic properties of persistent Na+ current orchestrate oscillatory bursting in respiratory neurons. J Gen Physiol 150:1523–1540. 10.1085/jgp.201812100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavorska I, Wehr M (2016) Somatostatin-expressing inhibitory interneurons in cortical circuits. Front Neural Circuits 10:76. 10.3389/fncir.2016.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Masino MA, Harris-Warrick RM (2007) Persistent sodium currents participate in fictive locomotion generation in neonatal mouse spinal cord. J Neurosci 27:4507–4518. 10.1523/JNEUROSCI.0124-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E, Schiff SJ (2006) Interneuron and pyramidal cell interplay during in vitro seizure-like events. J Neurophysiol 95:3948–3954. 10.1152/jn.01378.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Behavioral response to CNO (5 mg/kg) in SST-Cre;GqDREADD-/- control and SST-Cre; GqDREADD+/- experimental mice. Representative example video clips of an SST-Cre; GqDREADD-/- control mouse (top) and an SST-Cre; GqDREADD+/- experimental mouse (bottom) ∼20 minutes after injection with CNO (5 mg/kg). The SST-Cre; GqDREADD+/− mouse exhibits jerking movement and loss of balance indicative of status epilepticus.