

Abstract
Background
Maturity-onset diabetes of the young (MODY) is commonly misdiagnosed as type 1 or type 2 diabetes. Common reasons for misdiagnosis are related to limitations in genetic testing. A precise molecular diagnosis is essential for the optimal treatment of patients and allows for early diagnosis of their asymptomatic family members.
Objective
The aim of this study was to identify rare monogenic variants of common MODY genes in Japanese pediatric patients.
Methods
We investigated 45 Japanese pediatric patients based on the following clinical criteria: development of diabetes before 17 years of age, a family history of diabetes, testing negative for glutamate decarboxylase-65 (GAD 65) antibodies and insulinoma-2-associated autoantibodies (IA-2A), no significant obesity, and evidence of endogenous insulin production. Genetic screening for MODY1 (HNF4α), MODY2 (GCK), MODY3 (HNF1α), and MODY5 (HNF1β) was performed by direct sequencing followed by multiplex ligation amplification assays.
Results
We identified 22 missense variants (3 novel variants) in 27 patients (60.0%) in the GCK, HNF4α, and HNF1α genes. We also detected a whole exon deletion in the HNF1β gene and an exon 5–6 aberration in the GCK gene, each in one proband (4.4%). There were a total of 29 variations (64.4%), giving a relative frequency of 53.3% (24/45) for GCK, 2.2% (1/45) for HNF4α, 6.7% (3/45) for HNF1α, and 2.2% (1/45) for HNF1β genes.
Conclusions
Clinicians should consider collecting and assessing detailed clinical information, especially regarding GCK gene variants, in young antibody-negative patients with diabetes. Correct molecular diagnosis of MODY better predicts the clinical course of diabetes and facilitates individualized management.
1. Introduction
Maturity-onset diabetes of the young (MODY) (MIM 606391) is traditionally characterized by early onset (before 25 years of age) and a heterogeneous autosomal dominant form of inheritance. It accounts for 1% to 6.5% of pediatric diabetes cases in Europe in nationwide population-based studies [1–4]. However, the prevalence in Japanese pediatric patients is not clear, because the diagnostic rates have primarily been low in studies of adult patients [5].
MODY is often misdiagnosed as type 1 (T1D) or type 2 diabetes (T2D) because of the overlap of clinical characteristics [6, 7]. Patients are, therefore, often not offered an optimal treatment regimen [8]. Misdiagnosis of MODY arises because the phenotype of monogenic diabetes is not sufficiently distinctive to allow for easy clinical differentiation of patients with MODY from those with the more common forms of diabetes. Thus, the molecular genetic diagnosis of MODY is important for clinical decisions, treatment, and genetic counseling [9–11]. Currently, however, the rate of genetic diagnosis of monogenic diabetes is low because of the limited phenotype information available and the high costs of genetic testing. These are the typical challenges involved in differentiating patients, and a molecular genetic diagnosis can help identify patients with MODY.
Based on linkage analysis, MODY is defined by variants in different genes, of which at least 13 genes are known on OMIM. Almost all MODY cases are attributable to highly penetrant variants in the four most common MODY genes, namely, MODY1, 2, 3, and 5 (specifically hepatocyte nuclear factor 4-alpha (HNF4α; MODY1), glucokinase (GCK; MODY2), hepatocyte nuclear factor 1-alpha (HNF1α; MODY3), and hepatocyte nuclear factor 1-beta (HNF1β; MODY5)) in Caucasians [12]. Studies on Japanese pediatric patients with MODY have shown a variable prevalence of the different MODY subtypes. In Japan, MODY1, 2, 3, and 5 are the most common forms, while MODY4 and 6 are rare. However, there may be patients with MODY that have not been identified, especially with MODY2.
In this study, we focused on the four most common MODY genes (HNF4α, GCK, HNF1α, and HNF1β) and performed screening analysis of 45 Japanese pediatric patients who were between 2.8 and 17 years of age at diagnosis. Each genetic subtype of MODY demands a different management and treatment strategy. Thus, genetic diagnosis is the key to adequate treatment and requires proper interpretation and assessment of both known and novel variants.
2. Materials and Methods
2.1. Subjects and Clinical Studies
We recruited 45 (17 males and 28 females) unrelated Japanese probands with diabetes clinically characterized as MODY. Members of each family participated. The probands with diabetes were enrolled across 14 unrelated hospitals between 2009 and 2017 in Japan.
Probands were between 2.8 and 17 years of age at the time of diagnosis. In this study, we selected the MODY subjects for genetic testing, based on the following clinical criteria: (1) onset of diabetes before 25 years of age, (2) a family history of diabetes, (3) testing negative for glutamate decarboxylase-65 (GAD 65) antibodies and insulinoma-2-associated autoantibodies (IA-2A), (4) no significant obesity, and (5) evidence of endogenous insulin production.
The clinical diagnosis of MODY was based on the clinical features, laboratory records, and the guidelines of the Expert Committee Report of the American Diabetes Association. Hemoglobin (Hb) A1c levels were measured according to the National Glycohemoglobin Standardization Program (NGSP) [13] using the International System of Units (SI) [14]. The clinical characteristics of the enrolled patients are provided in Table 1.
Table 1.
Subject characteristics (n = 45).
| Phenotype | Subjects |
|---|---|
| Male/female | 17/28a |
| Age (years) at the time of diagnosis | 10.5 ± 3.66 |
| Age at diabetes onset | |
| ≤5 years | 5/1a |
| >5 years to ≤10 years | 4/10 |
| >10 years to ≤15 years | 8/15 |
| >15 years to ≤25 years | 0/2 |
| Blood glucose concentrations (mg/dL) | |
| OGTT 0 min | 131.8 ± 31.31 |
| OGTT 60 min | 227.5 ± 62.04 |
| OGTT 120 min | 205.7 ± 56.32 |
| IRI (μU/mL) | |
| OGTT 0 min | 5.5 ± 3.21 |
| OGTT 60 min | 37.7 ± 26.95 |
| OGTT 120 min | 50.4 ± 33.95 |
| HbA1c (%) at diagnosis (NGSP) | 7.53 ± 1.69 |
| mmol/mol (SI) | 59 |
| bBody mass index (kg/m2)/obesity index (%) | 18.1 ± 2.73/−0.55 ± 0.19 |
| C-peptide (ng/mL) (pre) | 1.8 ± 1.20 |
| β-Cell antibodies (GAD/IA-2A) | No |
| Diabetic ketoacidosis | 1/45 |
| Urine glucose | 39/46 (86.7%) |
| cFamily history of diabetes (%) | |
| In one parent | 40/45 (88.9%) |
| In a second-degree relative | 33/45 (73.3%) |
| In a third-degree relative | 14/45 (31.1%) |
| In a sibling | 4/456 (8.9%) |
Age at the time of diagnosis, blood glucose concentrations, HbA1c, and insulin dose are represented as means ± standard deviation. aLeft/right denotes male/female. bAll subjects were non-obese. cNumber of subjects with a sibling, or first–third-degree relative with type 1 or type 2 diabetes. NGSP: National Glycohemoglobin Standardization Program; SI: International System of Units; ND: no data.
Control samples (n = 100) were mainly recruited from the Health Science Research Resources Bank of the Japanese Collection of Research. These were members of the general population who underwent medical checkups to confirm that they had no family history of diabetes. Genomic DNA was extracted from the subjects with diabetes using a standard protocol.
The study was approved by the Ethics Committee of the Shikoku Medical Center for Children and Adults (Kagawa, Japan) (approval No. H20-31). This study was conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from the parents/guardians of the children and/or the subjects themselves.
2.2. Identification of MODY Genes
The most common forms of MODY are MODY1–3 and MODY5, while MODY4 and MODY6 are rare in the Japanese population (6). Thus, we performed mutational screening for the four relevant genes in MODY1, MODY2, MODY3, and MODY5 (HNF4α (gene ID: 3172), GCK (gene ID: 2645), HNF1α (gene ID: 6927), and HNF1β (gene ID: 6928), respectively) by a direct sequencing method, as described in our previous report [15]. The primers used are listed in Supplemental Table 1. The three novel variants were identified by comparison with the results of 100 control subjects.
All sequences were compared with the following GenBank reference sequences using GENETYX®Win v8.0 (Genetyx, Tokyo, Japan): NM_175914.3, NM_000457.3, and NP_787110.2 for HNF4α; NM_000162.3 and NP_000153.1 for GCK; NM_000545.5 and NP_000536.5 for HNF1α; and NM_000458.2 and NP_000449.1 for HNF1β.
2.3. In Silico Analysis for Novel Variants
Possible functional effects of the identified variants, especially the unknown variants, were identified with two web-based programs, PolyPhen 2 and SIFT. Potential effects on splicing were evaluated with HSF (Human Splicing Finder). Clinical information was evaluated with HGMD Professional, ClinVar, and ExAC. Sequence conservation was evaluated with HGMD Professional.
2.4. Multiplex Ligation Amplification (MLPA) Assay
We examined DNA copy number variations in HNF4α, GCK, HNF1α, and HNF1β genes in the probands in whom definite mutations were not identified by sequencing. The MLPA reactions were performed according to the manufacturer's general instructions using SALSA MLPA Probemix P241-B1 MODY Mix1 (MRC-Holland, Amsterdam, Netherlands) and were analyzed on a 3130 Genetic Analyzer (Applied Biosystems). DNA from three human genome control samples, namely, male, female, and mixed (male + female) (Promega, WI, USA), was used as reference DNA.
2.5. Custom Array Comparative Genomic Hybridization (CGH) Assay
To confirm copy number aberrations, we also performed a custom CGH microarray specialized for the HNF1β gene using the Agilent array database ver. 6.5 (http://earray.chem.agilent.com/array/; Agilent Technologies, CA, USA). The high-resolution array CGH was used to design a probe set covering the Chr17 genomic region (31,800,001–38,100,000) mapping around the HNF1β gene. This probe set included 31,500 probes on 17q12 (average spacing = 200 bp) and was duplicated to confirm the results and attain the 1 × 244K array format. The protocols used for probe labeling, hybridization, and data analysis were described by Cell Innovator Corp.
3. Results
3.1. Patient Characteristics
The clinical characteristics of the 45 probands are summarized in Table 1. All probands were diagnosed before 20 years of age; the average age was 10.5 ± 3.66 years at the time of diagnosis. The incidence of a family history of diabetes was 100%, and 73.3% (33/45) spanned three generations. HbA1c analysis and oral glucose tolerance tests (OGTT) in probands revealed hyperglycemia during fasting (at OGTT 0 min) without ketoacidosis (except in one proband). The diagnosis of hyperglycemia in most probands was either fortuitous or made using the screening system based on glycosuria in schools.
3.2. Genetic Variants in MODY Genes
We identified a total of 22 missense variants (3 novel variants) in 27 patients (60.0%) in the GCK, HNF4α, and HNF1α genes in this study involving a small set of 45 Japanese pediatric MODY patients (Table 2). Additionally, we detected an exon 5–6 deletion in the GCK gene (Figure 1) and a whole coding exon deletion in the HNF1β gene (Figure 2) in one proband each (2.2% each). There were a total of 29 probands (64.4%), giving a relative frequency of 53.3% (24/45) for GCK, 2.2% (1/45) for HNF4α, 6.7% (3/45) for HNF1α, and 2.2% (1/45) for HNF1β genes.
Table 2.
Missense variants and exon deletion of MODY genes in Japanese probands.
| Gene | Protein varianta | Location | Nucleotide changes | Carrier frequency | Clinical information | Possible functional impact | Conservation (protein) | Class | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients | Controls (n = 100) | Refs. | HGMD professional | Protein level | RNA level | |||||||
| Polyphen-2/SIFT | Splice | ESE/ESS | ||||||||||
| GCK | M41T | Exon 2 | c.122 T>C | 1 | 0 | [16] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic |
| T60I | Exon 2 | c.179 C>T | 1 | NA | [17] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic | |
| G72R | Exon 3 | c.214 G>A | 1 | NA | [18] | DM | Possibly damaging/deleterious | Yes | No/possible | Yes | Pathogenic | |
| L77P | Exon 3 | c.230 T>C | 1 | NA | [19] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic | |
| G81C | Exon 3 | c.241 G>T | 1 | 0 | [20] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic | |
| G147D | Exon 4 | c.440 G>A | 1 | 0 | Unknown | — | Possibly damaging/deleterious | No | No/no | — | Unknown significant | |
| T206M | Exon 6 | c.617 C>T | 1 | NA | [21] | DM | Possibly damaging/deleterious | No | No/possible | Yes | Pathogenic | |
| G223S | Exon 6 | c.667 G>A | 2 | 0 | [21–22] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic | |
| T228M | Exon 7 | c.683 C>T | 4 | 0 | [23] | DM | Possibly damaging/deleterious | No | Possible/possible | Yes | Pathogenic | |
| Q239R | Exon 7 | c.716 A>G | 1 | 0 | [24] | DM | Benign/tolerated | No | Possible/no | No | Likely pathogenic | |
| N254D | Exon 7 | c.760 A>G | 1 | 0 | Unknown | — | Possibly damaging/deleterious | No | Possible/no | — | Unknown significant | |
| L324P | Exon 8 | c.971 T>C | 1 | NA | [25] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Likely pathogenic | |
| S336X | Exon 8 | c.1,007 C>A | 1 | 0 | [16–20] | DM | Possibly damaging/deleterious | No | Possible/possible | — | Pathogenic | |
| R377H | Exon 9 | c.1,130 G>A | 1 | 0 | [19] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Likely pathogenic | |
| R377S | Exon 9 | c.1,129 C>A | 2 | NA | [16] | DM | Possibly damaging/deleterious | Yes | No/possible | Yes | Pathogenic | |
| S383L | Exon 9 | c.1,148 C>T | 1 | NA | [26] | DM | Possibly damaging/deleterious | No | No/possible | No | Pathogenic | |
| R392C | Exon 9 | c.1,174 C>T | 1 | 0 | [27] | DM | Possibly damaging/deleterious | No | Possible/possible | No | Likely pathogenic | |
| A454V | Exon 10 | c.1,361 C>T | 1 | NA | [27] | DM | Possibly damaging/deleterious | Yes | No/possible | Yes | Pathogenic | |
| E5-6 deletion | Exon 5-6 | — | 1 | NA | [16] | DM | — | — | — | — | Pathogenic | |
|
| ||||||||||||
| HNF4α | V251G | Exon 7 | c.752 T>G | 1 | 0 | Unknown | — | Possibly damaging/deleterious | Yes | Possible/no | — | Unknown significant |
|
| ||||||||||||
| HNF1α | A116V | Exon 2 | c.347 C>T | 1 | NA | [24] | DM | Possibly damaging/deleterious | Yes | Possible/possible | Yes | Pathogenic |
| R131W | Exon 2 | c.391 C>T | 1 | 0 | [28] | DM | Possibly damaging/deleterious | Yes | No/possible | Yes | Pathogenic | |
| R271W | Exon 4 | c.811 C>T | 1 | NA | [29] | DM | Possibly damaging/deleterious | No | Possible/no | Yes | Pathogenic | |
|
| ||||||||||||
| HNF1β | Whole gene deletion | — | 1 | NA | [30] | DM | — | — | — | — | Pathogenic | |
Amino acid numbers and nucleotide changes are based on NCBI RefSeq NM_175914.3 and NP_787110.2 for HNF4α, NM_000162.3 and NP_000153.1 for GCK, and NM_000545.5 and NP_000536.5 for HNF1α. aBold font denotes novel missense mutations in diabetes-specific missense mutations. All changes were heterozygous. NA: not analyzed. Possible functional effects of identified variants, especially unknown (novel) variants, were identified with two web-based programs, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html). Potential effects on splicing were evaluated with HSF (Human Splicing Finder; http://www.umd.be/HSF3/technicaltips.html). Clinical information was evaluated with HGMD professional (http://www.hgmd.cf.ac.uk/) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). Sequence conservation was evaluated with HGMD professional (http://www.hgmd.cf.ac.uk/).
Figure 1.
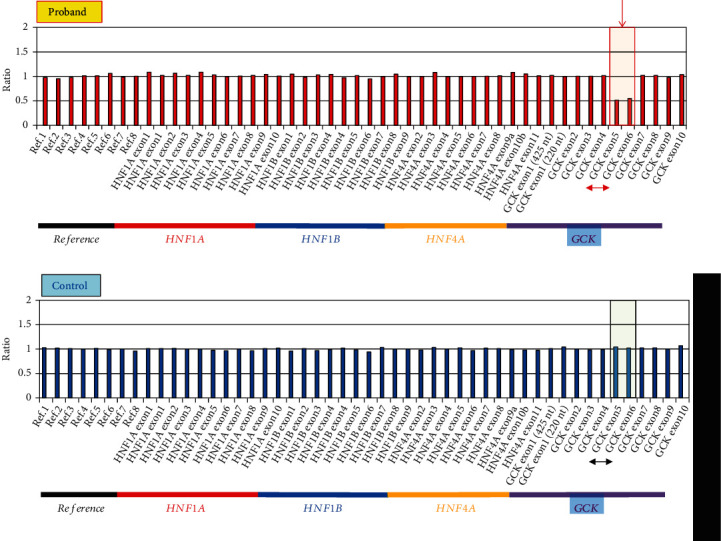
Detection of the exon 5–6 hemizygous deletion in the GCK gene in leukocyte genomic DNA by MLPA assay. Graphical representation of the GCK gene. Chromosome X and Y probes were normalized to the three control samples in the proband (a) and control (b). GCK probes in exons 5 and 6 are expressed in half dosages in the proband, indicating the deletion of exon 5–6. The exon 5–6 deletion is marked with a red arrow and square.
Figure 2.
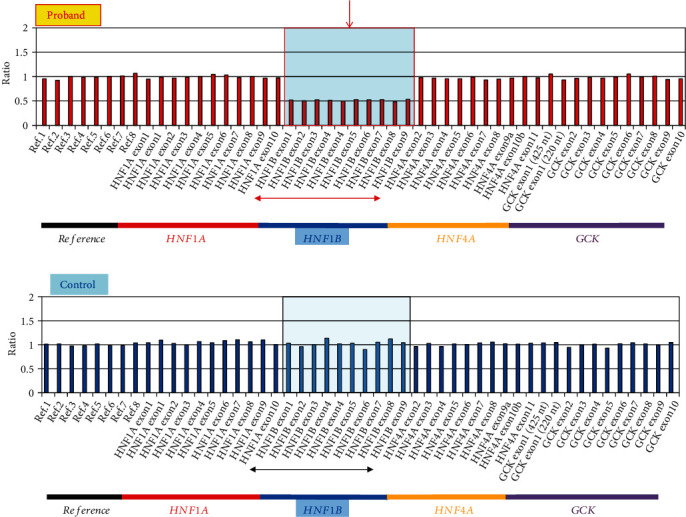
Detection of a whole hemizygous deletion in the HNF1β gene in leukocyte genomic DNA by MLPA assay. Graphical representation of the HNF1β gene. Chromosome X and Y probes were normalized to the control in the proband (a) and control (b). MLPA results show that all HNF1β probes are expressed in half dosages in the proband relative to the expected heights indicating a whole-gene hemizygous deletion change in HNF1β. The whole-gene deletion of HNF1β is marked with a red arrow and square.
3.2.1. Screening for the GCK Gene
We identified 18 missense variants (2 novel and 16 previously described variants [16–27] in 23 probands (Table 2). Most of these variants were found in one proband, while G223S and R377S were found in two probands and T228M was identified in four probands. Of these missense changes, two (G147D and N254D) were novel (Supplemental Figure 1_A, B), and all changes were heterozygous. Neither of the novel missense changes (G147D and N254D) were identified in the 100 healthy controls, suggesting that these are diabetes-specific variants. The 18 variant sites bearing the missense variants were conserved across species (data not shown).
We identified an exon 5–6 hemizygous deletion in the GCK gene in leukocyte genomic DNA from one proband (Figure 1). The GCK gene variants had the highest frequency among the MODY gene variants.
3.2.2. Screening for the HNF4α Gene
We identified one missense heterozygous variant in the HNF4α gene. This missense variant, V251G (exon 7), was novel (Supplemental Figure 1_C) and found in one proband. This novel missense change was not identified in the healthy controls, suggesting that it is a diabetes-specific variant.
3.2.3. Screening for the HNF1α Gene
We identified three known missense variants in the HNF1α gene [24, 28, 29]. These changes, A116V, R131W, and R271W, which occurred in one proband each, were all heterozygous. The other missense changes identified (I27L (exon 1) and S487N (exon 7); data not shown) were also detected in healthy controls (I27L: 52% (heterozygous) and 23% (homozygous); S487N: 59% (heterozygous) and 22% (homozygous) in controls), suggesting that they are polymorphism changes.
3.2.4. Screening for the HNF1β Gene
No missense changes were identified in the HNF1β gene. However, we identified a known whole-gene hemizygous deletion in HNF1β in leukocyte genomic DNA from one proband by MLPA, as peaks for whole exons in HNF1β were half the expected height [30] (Figures 2(a) and 2(b)). Unfortunately, we could not obtain DNA from parents or other family members of this proband at the time of this study. Therefore, we were unable to screen for this change in relatives of this proband.
To confirm this observation and assess the breakpoints of hemizygous deletion in HNF1β, we performed whole-genome array CGH. We confirmed a 1.7-Mb hemizygous deletion on chromosome 17q12 (Supplemental Figure 2). A magnified view of the array CGH indicated that the genomic deletion included the HNF1β gene and 15 other genes (LHX1, ZNHIT3, MYO19, PIGW, GGNBP2, DHRS11, MRM1, AATF, ACACA, C17orf78, TADA2A, DUSP14, SYNRG, DDX52, and LOC284100).
3.3. Family History Analysis
In this study, although DNA of all of the probands' parents was not available for genetic testing, the family histories of all probands were based on genetic counseling information. Among the 29 probands with variants (GCK = 24, HNF1α = 3, HNF4α = 1, and HNF1β = 1), parental DNA was available for only 17 probands (Supplemental Figure 3).
Among these 17 probands, DNA from at least three generations was available for only one family. All families had a heterozygous variant consistent with that of their child in 2 consecutive generations. In one family with the L324P variant in the GCK gene, the proband's mother had normal alleles, while DNA from one parent (F-42_I.1) was not available. Therefore, care should be taken, as that parent may also have MODY variants.
3.4. Clinical Characteristics of Patients with Variants in Four Genes and MODY X (with Unknown Genetic Etiology)
Based on the screening results, the clinical characteristics of the affected individuals with GCK, HNF1α, and HNF4α variants are presented in Supplemental Table 2. The age at diagnosis of diabetes was higher in HNF4α- and HNF1β-MODY probands than in the other MODY types, including MODY X. MODY X probands exhibited higher fasting glucose levels and glucose levels at 60 min during an OGTT compared with those of GCK-, HNF1α-, and HNF4α-MODY probands. Insulin secretion, as assessed by the 60 min insulin-to-glucose ratio during an OGTT, was lower in HNF4α-MODY patients than in other MODY probands, indicating the presence of a more severe glucose-stimulated insulin secretory defect, although the comparison was made against only one subject. Further studies that include other HNF4α-MODY probands are required to completely rule this result out. In contrast, GCK-MODY patients displayed increased insulin secretion at 60 min during the OGTT relative to the levels in the other MODY groups. The prevalence of obesity and HbA1c was not different between groups.
The sample size for this study was small. Our mutational analysis of only GCK, HNF4α, HNF1α, and HNF1β genes is not sufficient to strictly confirm the susceptibility mutation in patients with an onset of diabetes at an early age. Due to the small number of MODY cases, which represent only a small percentage of pediatric diabetes cases, and the high costs of genetic analysis, it is not easy to increase the sample size and include other candidate genes for analysis.
4. Discussion
Correct genetic diagnosis of MODY is important because it better predicts the clinical course of diabetes and facilitates individualized management. In this study, 22 missense variants in the GCK, HNF4α, and HNF1α genes and 2 deletions in the GCK and HNF1β genes were identified in 29 probands (64.5%). These included three novel variants affecting the amino acid residues G147D and N254D in GCK and V251G in HNF4α. Although these variants occurred rarely and were each found in only one individual, none of these variants were found in control patients. Based on these observations and predicted functional effects from in silico analysis, we concluded that these variants are putatively associated with MODY phenotypes in Japanese pediatric patients.
GCK-MODY causes a mild, asymptomatic diabetes and stable fasting hyperglycemia usually requiring no specific treatment. Generally, the incidence of GCK variants has been reported less frequently than other MODY types in Japanese patients [20]. However, the incidence is mainly based on studies on adult subjects, and this result is similar to those of reports from other Asian populations [31–33]. In patients with pediatric onset T2D that are young and nonobese and have a family history of MODY or persistent mild fasting hyperglycemia, there may be a genetic variant in the GCK gene [6, 34]. Thus, we hypothesized that GCK gene variants would be common in nonobese patients without autoantibodies with asymptomatic diabetes in school medical examinations.
Indeed, according to a systematic approach using detailed laboratory data and family history, we identified 19 GCK variants in 24 probands. This frequency was higher than that of other MODY genes. A GCK variant should be considered if there is significant family history. Most GCK gene variants are generally single nucleotide variants, and GCK deletions are rare [35]. In this study, we identified one small deletion encompassing exon 5–6 of the GCK gene in one proband (F-17 in Supplemental Figure 3). This deletion was also identified in this proband's father, indicating cosegregation with the onset of diabetes.
In Japan, variants in the HNF1α gene are the most commonly identified MODY genes, which is consistent with studies on the European population [5]. Variants in the HNF1α/HNF4α genes display very similar clinical characteristics. The most striking features of HNF1α-/HNF4α-MODY are that they result in a reduced insulin secretory response to glucose but marked sensitivity to low-dose sulfonylureas [36] and cause progressive pancreatic β-cell dysfunction and increasing hyperglycemia that leads to diabetes in early adult life. These features are similar our patients with HNF1α-/HNF4α-MODY. The patient with the HNF4α variant revealed a novel heterozygous variation at V251G (exon 7). Unfortunately, we were unable to obtain DNA from the parents or sibling of this proband at the time of this study.
HNF1β-MODY is characterized by renal dysfunction, including renal cysts, renal dysplasia, pancreatic atrophy, and genital abnormalities [37]. We identified a hemizygous whole-gene deletion in the HNF1β gene in a 17-year-old girl with nephropathy, renal cysts in the left kidney, and absence of a right kidney as identified by computed tomography scans, along with loss of the vagina after following the clinical characteristics of high glucose levels and impaired insulin secretion. The results of genetic testing were consistent with our speculation about the abnormality in the HNF1β gene. Our data suggests that according for the HNF1β variant, the typical phenotype is more important than the family history, because this case was detected without family analysis.
Generally, approximately 60% or more of clinically diagnosed MODY cases can be attributed to a single gene. However, the remaining 20% to 30% of MODY cases are likely due to an unidentified gene, as in MODY-X cases in Caucasians [38], and this percentage is even higher in Asian (including Japanese) populations [39]. In Japanese populations, MODY-X may account for 20–50% of patients with MODY. Recently, a clinical prediction model has been developed to predict the probability of MODY in young patients [40]. Tools such as the MODY calculator could be of significant assistance in considering a MODY diagnosis in the first steps. This tool is available at http://www.diabetesgenes.org/content/mody-probability-calculator. Rapid genetic MODY diagnosis is important, but diagnosing MODY subtypes is difficult for physicians, and most patients with MODY remain undiagnosed. In current diagnostic testing, next-generation sequencing (NGS) technology provides the potential for simultaneous analysis of the known disease genes in a single assay [31, 41]. We are now planning to develop a targeted NGS assay to identify variants causing monogenic diabetes for patients with MODY without known variants.
The results of this study suggested that improved clinical specificity can allow for better detection of MODY variants. Interestingly, we identified 20 patients (83.3%) with variants in the GCK gene (all but 4 cases) or HNF1β deletion (100%) by single target gene analysis. These patients with variants can be associated with diabetes phenotypes or clinical features involving the GCK or HNF1β gene, such as age of onset of diagnosis, level of secretion of insulin during an OGTT, or renal dysplasia or abnormality. In contrast, it was difficult to identify the variant for probands who could not be diagnosed with a specific MODY subtype due to limited phenotype information. We were only able to identify the variant against a single gene in one case (5.0%, 1/20). Thus, there are close genotype-phenotype correlations between the genetic variant and the clinical features. We strongly suggest that “genotype-phenotype-specific candidate gene selection” is important in order to reduce the unidentified variants, and screening will become more cost-effective with advances in research.
5. Conclusion
A molecular genetic diagnosis of disease-causing genes allows for more appropriate management and can help identify affected and at-risk family members. Diagnosing MODY is a challenge for physicians, and the majority of cases remain unidentified. A nationwide and systematic approach is required for the rapid diagnosis and appropriate management of MODY.
Acknowledgments
We sincerely thank the many pediatricians listed in the following for providing the resources to conduct this study: Dr. Ogawa Y., Niigata University School of Medicine; Dr. Miyagawa S., NHO Kure Medical Center; Dr. Iwase T., Kagawa University Hospital; Dr. Jinno K., JR Hiroshima Hospital; Dr. Takemoto K. & Hamada J., Ehime University School of Medicine; Dr. Amemiya S., Saitama Medical University Faculty of Medicine; Dr. Sugihara S., Tokyo Women's Medical University Medical Center East; Dr. Kikuchi N., Yokohama City Minato Red Cross Hospital; Dr. Izumida I., Niigata Prefectural Shibata Hospital; Dr. Araki M., Kochi Medical School Hospital (all of the Department of Pediatrics); and Dr. Ibata T., Department of Endocrinology & Metabolism, Kagawa University Hospital, as well as the participating patients and their families. We also thank the physicians and pediatricians who collected the blood samples and clinical/biological data.
Abbreviations
- MODY:
Maturity-onset diabetes of the young
- MODY1:
Hepatocyte nuclear factor 4-alpha (HNF4α)
- MODY2:
Glucokinase (GCK)
- MODY3:
Hepatocyte nuclear factor 1-alpha (HNF1α)
- MODY5:
Hepatocyte nuclear factor 1-beta (HNF1β)
- T1D:
Type 1 diabetes
- T2D:
Type 2 diabetes
- MLPA:
Multiplex ligation amplification
- CGH:
Comparative genomic hybridization
- OGTT:
Oral glucose tolerance tests
- NGS:
Next-generation sequencing.
Data Availability
The confidential data used to support the findings of this study are restricted by the ethics committee in order to protect patient privacy.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Supplementary Materials
Supplemental Table 1: primers used for HNF4α, GCK, HNF1α, and HNF1β genes for sequencing analysis. Supplemental Table 2: clinical characteristics of MODY cases with or without variants in each gene. Supplemental Figure 1: sequences of two novel GCK gene mutations and one novel HNF4α gene mutation. Supplemental Figure 2: results of array CGH (human genome CGH array 244K). Supplemental Figure 3: pedigrees of the families of the probands with mutations in GCK, HNF1α, HNF4α, and HNF1β genes, respectively.
References
- 1.Fendler F., Borowiec M., Baranowska-Jazwiecka A., et al. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia . 2012;55(10):2631–2635. doi: 10.1007/s00125-012-2621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irgens H. U., Molnes J., Johansson B. B., et al. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia . 2013;56(7):1512–1519. doi: 10.1007/s00125-013-2916-y. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd M., Shields B., Hammersley S., et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care . 2016;39(11):1879–1888. doi: 10.2337/dc16-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johansson B. B., Irgens H. U., Molnes J., et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia . 2017;60(4):625–635. doi: 10.1007/s00125-016-4167-1. [DOI] [PubMed] [Google Scholar]
- 5.Yorifuji T., Fujimaru R., Hosokawa Y., et al. Comprehensive molecular analysis of Japanese patients with pediatric–onset MODY-type diabetes mellitus. Pediatric Diabetes . 2012;13(1):26–32. doi: 10.1111/j.1399-5448.2011.00827.x. [DOI] [PubMed] [Google Scholar]
- 6.Ellard S., Bellanné-Chantelot C., Hattersley A. T., European Molecular Genetics Quality Network (EMQN) MODY group Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia . 2008;51(4):546–553. doi: 10.1007/s00125-008-0942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shields B. M., McDonald T. J., Ellard S., Campbell M. J., Hyde C., Hattersley A. T. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia . 2012;55(5):1265–1272. doi: 10.1007/s00125-011-2418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shields B. M., Hicks S., Shepherd M. H., Colclough K., Hattersley A. T., Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia . 2010;53(12):2504–2508. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 9.Nkonge K. M., Nkonge D. K., Nkonge T. N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY) Clinical Diabetes and Endocrinology . 2020;6(1):p. 20. doi: 10.1186/s40842-020-00112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delvecchio M., Pastore C., Giodano P. Treatment options for MODY patients: a systematic review of literature. Diabetes Therapy . 2020;11(8):1667–1685. doi: 10.1007/s13300-020-00864-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broome D. T., Pantalone K. M., Kashyap S. R., Philipson L. H. Approach to the patient with MODY-monogenic diabetes. The Journal of Clinical Endocrinology & Metabolism . 2021;106(1):237–250. doi: 10.1210/clinem/dgaa710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Althari S., Gloyn A. L. When is it MODY? Challenges in the interpretation of sequence variants in MODY genes. The Review of Diabetic Study . 2015;12(3-4):330–348. doi: 10.1900/RDS.2015.12.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.International expert committee. International expert committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care . 2009;32(7):1327–1334. doi: 10.2337/dc09-9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones G. R., Barker G., Goodall I., Schneider H. G., Shephard M. D. S., Twigg S. M. Change of HbA1c reporting to the new SI units. Medical Journal of Australia . 2011;195(1):45–46. doi: 10.5694/j.1326-5377.2011.tb03190.x. [DOI] [PubMed] [Google Scholar]
- 15.Moritani M., Yokota I., Tsubouchi K., et al. Identification of INS and KCNJ11 gene mutations in type 1B diabetes in Japanese children with onset of diabetes before 5 yr of age. Pediatric Diabetes . 2013;14(2):112–120. doi: 10.1111/j.1399-5448.2012.00917.x. [DOI] [PubMed] [Google Scholar]
- 16.Osbak K. K., Colclough K., Saint-Martin C., et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Human Mutation . 2009;30(11):1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- 17.Garin I., Rica I., Estalella I., et al. Haploinsufficiency at GCK gene is not a frequent event in MODY2 patients. Clinical Endocrinology . 2008;68(6):873–878. doi: 10.1111/j.1365-2265.2008.03214.x. [DOI] [PubMed] [Google Scholar]
- 18.Arden C., Trainer A., de la Iglesia N., et al. Cell biology assessment of glucokinase mutations V62M and G72R in Pancreatic β-Cells: Evidence for cellular instability of catalytic activity. Diabetes . 2007;56(7):1773–1782. doi: 10.2337/db06-1151. [DOI] [PubMed] [Google Scholar]
- 19.Estalella I., Rica I., de Nanclares G. P., et al. Mutations in GCK and HNF-1? explain the majority of cases with clinical diagnosis of MODY in Spain. Clinical Endocrinology . 2007;67(4, article 070615230707001) doi: 10.1111/j.1365-2265.2007.02921.x. [DOI] [PubMed] [Google Scholar]
- 20.Yokota I., Moritani M., Nishisho K., Miyoshi T., Kotani Y., Kagami S. Detection of glucokinase gene defects in non-obese Japanese children diagnosed with diabetes by school medical examinations. Endocrine Journal . 2011;58(9):741–746. doi: 10.1507/endocrj.K11E-062. [DOI] [PubMed] [Google Scholar]
- 21.Massa O., Meschi F., Cuesta-Munoz A., et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia . 2001;44(7):898–905. doi: 10.1007/s001250100530. [DOI] [PubMed] [Google Scholar]
- 22.García-Herrero C. M., Rubio-Cabezas O., Azriel S., et al. Functional characterization of MODY2 mutations highlights the importance of the fine-tuning of glucokinase and its role in glucose sensing. PLoS One . 2012;7(1, article e30518) doi: 10.1371/journal.pone.0030518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoffel M., Froguel P. H., Takeda J., et al. Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proceedings of the National Academy of Science . 1992;89(16):7698–7702. doi: 10.1073/pnas.89.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng M. C., Lee S. C., Ko G. T., et al. Familial early-onset type 2 diabetes in Chinese patients: obesity and genetics have more significant roles than autoimmunity. Diabetes Care . 2011;24(4):663–671. doi: 10.2337/diacare.24.4.663. [DOI] [PubMed] [Google Scholar]
- 25.McKinney J. L., Cao H., Robinson J. F., et al. Spectrum of HNF1A and GCK mutations in Canadian families with maturity-onset diabetes of the young (MODY) Clinical and Investigative Medicine . 2004;27(3):135–141. [PubMed] [Google Scholar]
- 26.Barrio R., Bellanné-Chantelot C., Moreno J. C., et al. Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. Journal of Clinical Endocrinology and Metabolism . 2002;87(6):2532–2539. doi: 10.1210/jcem.87.6.8530. [DOI] [PubMed] [Google Scholar]
- 27.Thomson K. L., Gloyn A. L., Colclough K., et al. Identification of 21 novel glucokinase (GCK) mutations in UK and European Caucasians with maturity-onset diabetes of the young (MODY) Human Mutation . 2003;22(5):417–421. doi: 10.1002/humu.9186. [DOI] [PubMed] [Google Scholar]
- 28.Glucksmann M. A., Lehto M., Tayber O., et al. Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes . 1997;46(6):1081–1086. doi: 10.2337/diab.46.6.1081. [DOI] [PubMed] [Google Scholar]
- 29.Chèvre J. C., Hani E. H., Boutin P., et al. Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia . 1998;41(9):1017–1023. doi: 10.1007/s001250051025. [DOI] [PubMed] [Google Scholar]
- 30.Bellanne-Chantelot C., Clauin S., Chauveau D., et al. Large genomic rearrangements in the hepatocyte nuclear Factor-1β (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes . 2005;54(11):3126–3132. doi: 10.2337/diabetes.54.11.3126. [DOI] [PubMed] [Google Scholar]
- 31.Xu J. Y., Dan Q. H., Chan V., et al. Genetic and clinical characteristics of maturity-onset diabetes of the young in Chinese patients. European Journal of Human Genetics . 2005;13(4):422–427. doi: 10.1038/sj.ejhg.5201347. [DOI] [PubMed] [Google Scholar]
- 32.Liang H., Zhang Y., Li M., et al. Recognition of maturity-onset diabetes of the young in China. Journal of Diabetes Investigation . 2021;12(4):501–509. doi: 10.1111/jdi.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang J. S., Shin C. H., Yang S. W., Jung S. Y., Huh N. Genetic and clinical characteristics of Korean maturity-onset diabetes of the young (MODY) patients. Diabetes Research and Clinical Practice . 2006;74(1):75–81. doi: 10.1016/j.diabres.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Sugihara S., Sasaki N., Amemiya S., Kohno H., Tanaka T., Matsuura N. Analysis of weight at birth and at diagnosis of childhood-onset type 2 diabetes mellitus in Japan. Pediatric Diabetes . 2008;9(4part1):285–290. doi: 10.1111/j.1399-5448.2008.00402.x. [DOI] [PubMed] [Google Scholar]
- 35.Ellard S., Thomas K., Edghill E. L., et al. Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia . 2007;50(11):2313–2317. doi: 10.1007/s00125-007-0798-6. [DOI] [PubMed] [Google Scholar]
- 36.Pearson E. R., Starkey B. J., Powell R. J., Gribble F. M., Clark P. M., Hattersley A. T. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet . 2003;362(9392):1275–1281. doi: 10.1016/S0140-6736(03)14571-0. [DOI] [PubMed] [Google Scholar]
- 37.Edghill E. L., Bingham C., Slingerland A. S., et al. Hepatocyte nuclear factor-1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF-1beta in human pancreatic development. Diabetic Medicine . 2006;23(12):1301–1306. doi: 10.1111/j.1464-5491.2006.01999.x. [DOI] [PubMed] [Google Scholar]
- 38.Johansen A., Ek J., Mortensen H. B., Pedersen O., Hansen T. Half of clinically defined maturity-onset diabetes of the young patients in Denmark do not have mutations in HNF4A, GCK, and TCF1. Journal of Clinical Endocrinology and Metabolism . 2005;90(8):4607–4614. doi: 10.1210/jc.2005-0196. [DOI] [PubMed] [Google Scholar]
- 39.Sujjitjoon J., Jungtrakoon P., Boonyasrisawat W., et al. Molecular genetics of monogenic beta-cell diabetes. Thai Journal of Genetics . 2008;1(2):93–108. [Google Scholar]
- 40.Njølstad P. R., Molven A. To test, or not to test: time for a MODY calculator? Diabetologia . 2012;55(5):1231–1234. doi: 10.1007/s00125-012-2514-4. [DOI] [PubMed] [Google Scholar]
- 41.Alkorta-Aranburu G., Carmody D., Cheng Y. W., et al. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Molecular Genetics and Metabolism . 2014;113(4):315–320. doi: 10.1016/j.ymgme.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: primers used for HNF4α, GCK, HNF1α, and HNF1β genes for sequencing analysis. Supplemental Table 2: clinical characteristics of MODY cases with or without variants in each gene. Supplemental Figure 1: sequences of two novel GCK gene mutations and one novel HNF4α gene mutation. Supplemental Figure 2: results of array CGH (human genome CGH array 244K). Supplemental Figure 3: pedigrees of the families of the probands with mutations in GCK, HNF1α, HNF4α, and HNF1β genes, respectively.
Data Availability Statement
The confidential data used to support the findings of this study are restricted by the ethics committee in order to protect patient privacy.