

Abstract
Background:
Extracellular vesicles (EVs) from heart stromal/progenitor cells modulate innate immunity, with salutary effects in a variety of cardiac disease models. Little is known, however, about the effects of these EVs on adaptive immunity.
Methods:
Ex vivo differentiation of naïve CD4+ T cells was conducted to assess the effect of EVs on cytokine production and proliferation of Th1, Th2, Th17, and regulatory T (Treg) cells. These effects were further tested in vivo using the experimental autoimmune myocarditis (EAM) model.
Results:
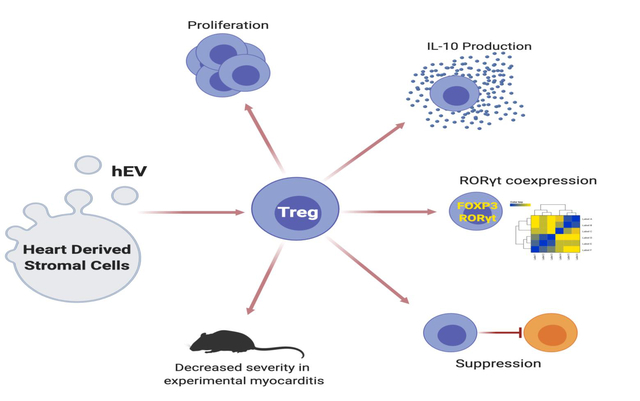
Using differentiated CD4+ T cells, we show that EVs secreted by human-derived heart stromal/progenitor cells selectively influence the phenotype, activity, and proliferation of regulatory T (Treg) cells. Exposure of Treg cells to EVs results in faster proliferation, augmented production of IL-10, and polarization toward an intermediate FOXP3+RORγt+ phenotype. In experimental autoimmune myocarditis, EVs attenuate cardiac inflammation and functional decline, in association with increased numbers of splenic IL10+-Treg cells.
Conclusions:
T cell modulation by EVs represents a novel therapeutic approach to inflammation, harnessing endogenous immunosuppressive mechanisms that may be applied in solid organ transplantation, graft-versus-host disease, and autoimmune disorders.
Keywords: extracellular vesicles, inflammation, regulatory T cells, CD4+ T cells
Graphical Abstract
Introduction
CD4+ T cells are essential mediators of adaptive immunity, capable of mounting inflammatory responses against microbial pathogens, maintaining surveillance against tumors, and establishing long-term immunologic memory. During initial activation, naïve CD4+ T cells can differentiate into various effector T cells (e.g., Th1, Th2, Th17), depending on the cytokine milieu and expression of lineage-specific transcription factors.1 In addition to these conventional effector subtypes, CD4+ T cells can differentiate into regulatory T (Treg) cells, which suppress various immune responses and represent potential therapeutic agents in solid organ transplantation, graft-versus-host disease, and autoimmune disorders.2 Conventional effector T cells and Treg cells are defined by their cytokine profiles and function, but there is substantial plasticity, such that a particular cell type can switch its cytokine profile and phenotype.3 Equally important is the ability of Treg cells to adopt features that overlap with effector T cells (e.g., Th17-like Treg cell), which enables tailored, cell-specific suppression.4
Environmental cues play an important role in differentiation, function, and plasticity of CD4+ T cells. Extracellular vesicles (EVs) can influence cells of adaptive immunity,5, 6 but mechanistic details remain incomplete. Given their disease-modifying bioactivity in multiple inflammatory disease models,7, 8 EVs secreted by heart stromal/progenitor cells (hEVs) merit particular attention in terms of innate and adaptive immunity. Within innate immunity, hEVs have already demonstrated immunomodulatory properties by polarizing macrophages toward an anti-inflammatory, pro-reparative phenotype.9–12 These phenotypic changes led to cardioprotective effects in animal models of myocardial ischemia. Little is known, however, about the effects of hEVs in adaptive immunity. Here, we demonstrate that hEVs increase proliferation of Treg cells, augment their production of IL-10, and reprogram Treg cells into an intermediate FOXP3+RORγt+ phenotype with suppressive capacity.
Materials and Methods
Detailed methods are provided in the Supplementary Material. All datasets used for this study are available upon reasonable request.
Ex vivo T cell experiments
Ex vivo differentiation of naïve CD4+ T cells was conducted to assess the effect of hEVs on phenotype and behavior of conventional effector CD4+ T cells (Th1, Th2, Th17) and Treg cells. Naïve CD4+ T cells were isolated from FOXP3-IRES-mRFP mice (The Jackson Laboratory), polarized, and co-cultured with either hEVs or vehicle. Rates of proliferation, cytokine production, and the global transcriptome were analyzed.
Experimental autoimmune myocarditis
Experimental autoimmune myocarditis (EAM) was induced in female Lewis rats (Charles River) and female BALB/c mice (Taconic) using purified cardiac myosin and αMHC peptide, respectively, emulsified in complete Freund’s adjuvant. Animals were injected with either hEVs or vehicle on day 10 and were euthanized on days 21–28 for functional and immunologic analyses. All animal protocols were approved by the local Institutional Animal Care and Use Committee (IACUC: 008416, 005460, 007407).
Extracellular vesicles
hEVs used in the ex vivo T cell experiments and mouse EAM experiments were isolated from heart stromal/progenitor cells grown from human endomyocardial biopsies, following approval by the local Institutional Review Board (IRB: 00014713). hEVs used in rat EAM experiments were isolated from rat cardiac cells, to facilitate direct comparison with published work using these cells in rat EAM.13 Regardless, hEVs from heart stromal/progenitor cells of different species appear to be roughly bioequivalent14–17 and work well in xenogeneic applications.9, 10, 18
Results
Heart-derived EVs (hEVs) influence T cell proliferation in a cell-specific and dose-dependent manner
Naïve CD4+ T cells were isolated from mice (Figure 1A,B) and differentiated into conventional effector T cells (Th1, Th2, Th17) or induced regulatory T (iTreg) cells (Figure S1A,B). After 5 days of culture, we determined the effects of varying doses of human hEVs (or vehicle) on proliferation rates of each T cell population. hEVs were isolated from media conditioned with heart-derived stromal/progenitor cells for 15 days and had a mean and mode size of ~130 nm and ~120 nm, respectively (Figure 1C,D). Characterization of hEVs demonstrated expression of canonical EV markers: CD9, CD63, and CD81 (Figure 1E). Compared to controls, Th1 and Th2 cells exposed to hEVs had similar rates of proliferation, while Th17 cells had a 7–11% reduction in proliferation (Figure 1F,G). In contrast, iTreg cells exposed to hEVs (iTreg-hEV) proliferated at higher rates compared to iTreg cells exposed only to vehicle (iTreg-Ctrl) (Figure 1F,G). At a cell-to-EV ratio of 1:1000, hEVs elicited a strong proliferative response (iTreg-hEV 75.3 ± 0.6% versus iTreg-CTL 62.5 ± 1.5%, p < 0.0001) (Figure 1H, Figure S1C-F). Notably, exposure to hEVs resulted in distinct, dose-dependent responses among the different types of CD4+ T cells: no change in proliferation of Th1 and Th2 cells, decrease in proliferation of Th17 cells, and increase in proliferation of iTreg cells. Thus, the effects of hEVs on iTreg cells appear to be specific.
Figure 1. Dose-dependent and cell-specific response in proliferation after hEV exposure.

A. Experimental outline for in vitro T cell analysis: isolation of the total CD4+ T cell population from FOXP3 reporter mice, enrichment of naïve CD4+ T cells, and subsequent differentiation into conventional effector T cells (Th1, Th2, Th17) and induced regulatory T (iTreg) cells. Following differentiation, cells were cultured with either vehicle or human hEVs. B. Sorting strategy for naïve CD4+ T cells (red gate). C. Particle concentration and mean and mode sizes of hEVs by nanoparticle tracking analysis. D. Transmission electron microscopy images of hEVs. Bars, 200 nm. E. Analysis of EV markers, including CD9, CD63, and CD81, by western blot (n=1/group). F. Proliferation of effector T cells and iTreg cells after exposure to vehicle or varying doses of hEVs (ratio expressed as cell-to-EV). Top two rows are light microscopy images of the cells. Bars, 100 μm. Bottom rows represent quantification by flow cytometry using dilution of the proliferation dye, CellTrace Violet (CTV). G. Quantification of the change in the rate of proliferation with increasing doses of hEVs relative to vehicle control. %Δ in proliferation = [(proliferation with hEV – proliferation with vehicle)/proliferation with vehicle] x 100. H. Proliferation of iTreg cells following exposure to hEVs (cell-to-EV ratio of 1:1000) and vehicle control (****P < 0.0001, n=6–7/group, two-tailed Student’s t-test). Data are mean ± SEM.
iTreg cells primed with hEVs, but not fibroblast EVs, produce higher levels of IL-10
To see if phenotypic changes accompany the observed changes in proliferation, we stimulated effector T cells and iTreg cells after 5 days of culture and analyzed their signature cytokines. Compared to vehicle, human hEVs had little effect on the expression of characteristic cytokines by conventional effector T cells (Figure 2A, Figure S2A). In contrast, exposure of iTreg cells to hEVs resulted in ~1000-fold increase in expression of Il10, relative to vehicle (Figure 2B, Figure S2B). Expression of other iTreg cell markers was either unchanged (Ctla4, Icos, and Tgfb1) or decreased (Il12a and Ebi3), suggesting that hEVs target specific pathways, rather than indiscriminately activating iTreg cells. At the protein level, analysis of conditioned media revealed much greater secretion of IL-10 by iTreg cells that had been exposed to hEVs (Figure 2C). To determine whether the effects on Il10 expression were specific to hEVs, we compared iTreg cell responses to hEVs with those to human dermal fibroblast EVs, which are similar in size distribution (Figure S2C) but functionally inert in other assay systems.8 Indeed, iTreg cells primed with hEVs outperformed iTreg cells primed with fibroblast EVs in terms of Il10 expression (Figure 2D). Enhanced IL-10 production was evident 5 days after hEV exposure, but not two days earlier (Figure 2E). Thus, hEVs (but not fibroblast EVs) augment production of IL-10 by iTreg cells and increase their proliferation, while little effect is seen across effector T cells.
Figure 2. hEVs augment production of IL-10 by iTreg cells.

A. Expression of signature cytokines by human hEV-primed effector T cells, including Ifng and Tnfa by Th1 cells, Il4 and Il13 by Th2 cells, and Il17a and Il17f by Th17 cells, relative to vehicle-primed cells (n=3–5/group). Data are mean ± SD. B. Quantification of Treg markers and cytokines expressed by iTreg-hEV relative to iTreg-Ctrl (n=5–8/marker). Data are mean ± SD. C. Protein concentration of IL-10 quantified by ELISA in the culture media of iTreg-hEV and iTreg-Ctrl (****P < 0.0001, n = 5/group, two-tailed Student’s t-test). Data are mean ± SEM. D. Expression of Il10 by iTreg cells on day 5 following exposure to hEVs or human dermal fibroblast EVs (fbEVs), relative to iTreg-Ctrl (****P < 0.0001, n = 6–10/group, two-tailed Student’s t-test). Data are mean ± SEM. Gene expression data analyzed using the 2−ΔΔCt method after normalization against Gapdh expression and vehicle controls. E. Representative flow cytometry plots and quantification of intracellular IL-10 staining, as percent of total iTreg cells on days 3 and 5 (ns = 0.1539 between iTreg-Ctrl and iTreg-hEV on day 3; ****P < 0.0001 between iTreg-Ctrl on day 3 and iTreg-Ctrl on day 5; ****P < 0.0001 between iTreg-hEV on day 3 and iTreg-hEV on day 5; ****P < 0.0001 between iTreg-Ctrl on day 5 and iTreg-hEV on day 5; n=5/group, two-way ANOVA with Sidak’s multiple comparisons test). Data are mean ± SEM.
Infusion of hEVs attenuates disease severity and expands the IL10+-Treg cell population in a model of myocarditis
To test whether hEVs affect the Treg cell population and IL-10 production in vivo, we investigated experimental autoimmune myocarditis (EAM), a myosin-induced and T-cell driven model of myocarditis.19 Heart-derived stromal/progenitor cells have previously been shown to attenuate inflammation and preserve cardiac function in a rat model of EAM;13 we found similar effects with hEVs derived from rat hearts. Following induction of disease (Figure 3A,B), rats receiving hEVs exhibited improved left ventricular ejection fraction (Figure 3C) and less cardiac inflammation (Figure 3D). Although we did not characterize the inflammatory infiltrate in our study, rats with EAM receiving the parent stromal/progenitor cells have been shown to exhibit decreased CD3+ lymphocyte infiltrates.13
Figure 3. hEVs from rats attenuate left ventricular functional decline and inflammation in experimental autoimmune myocarditis.

A. Timeline of the experimental autoimmune myocarditis (EAM) model in rats. On day 10, rats received either a vehicle infusion (EAM+vehicle) or hEVs (EAM+hEV). B. Representative immunofluorescence images of cardiac tissue in healthy controls (Ctrl) and diseased animals (EAM). Bar, 25 μm for Ctrl; 75 μm for EAM. C. Representative echocardiographic M-mode images in short axis on day 28 with quantification of left ventricular function (LVEF), left ventricular end systolic diameter (LVESD), and left ventricular end diastolic diameter (LVEDD). (LVEF: ****P < 0.0001 between Ctrl and EAM+vehicle on day 28; **P = 0.0032 between EAM+vehicle and EAM+hEV on day 28. LVESD: ****P<0.0001 between Ctrl and EAM+vehicle; ns = 0.0735 between EAM+vehicle and EAM+hEV on day 28. LVEDD: *P = 0.0237 between Ctrl and EAM+vehicle; ns = 0.9994 between EAM+vehicle and EAM+hEV on day 28; n = 10–12/group, two-way ANOVA with Tukey’s multiple comparisons test). Data are mean ± SEM. D. Representative cardiac cross sections using hematoxylin and eosin staining and a semiquantitative assessment of inflammation. Bars, 200 μm. Data are individual scores with the median denoted by a bar.
In EAM mice, where T cell populations can be better-sorted than in rats,20, 21 we tested whether human hEVs can expand the overall Treg cell population and IL-10 production, as our ex vivo experiments have shown. Human-derived, rather than murine-derived, cells were used as the EV source to explore the translational value of hEVs. Mice received hEVs on day 10 after induction of disease and were euthanized on day 21 (Figure 4A), a time point that corresponds with peak inflammation.22 Animals receiving hEVs weighed slightly more overall, but splenic sizes and overall splenocyte counts were similar in both groups (Figure 4B,C). Among splenocytes, there were no significant differences in CD4+ cells or CD4+FOXP3+ cells (Figure 4D-F), but the number of CD4+FOXP3+IL10+ cells was significantly higher in animals that received hEVs, relative to vehicle controls (Figure 4G). Within the hearts of EAM mice, transcription levels of Foxp3 were increased by ~2 to 3-fold in animals that received hEVs, while transcription levels of Il10 were similar (Figure S3). Thus, hEV augment IL-10-producing Treg cells in vitro and within mouse spleens in vivo.
Figure 4. Increased number of IL-10+ Treg cells within spleens of mice with experimental autoimmune myocarditis following hEV infusion.

A. Experimental design for autoimmune myocarditis, which involves induction of disease on days 0 and 7, and intravenous injection of human hEVs or vehicle on day 10. B. Total mass on day 0 (baseline) and day 21 (end point) and spleen mass on day 21 of animals in the vehicle control and hEV groups (ns = 0.2019 between control and hEV groups for animal mass on day 0; **P = 0.0097 between control and hEV groups for animal mass on day 21; ns = 0.2019 between control and hEV groups for spleen mass on day 21; n=8/group, two-tailed Student’s t-test). Data are mean ± SEM. C. Total number of splenocytes per mg of spleen tissue (ns = 0.9201 between control and hEV groups; n=8/group, two-tailed Student’s t-test). Data are mean ± SEM. D. Representative flow cytometry plots from spleens of vehicle- and hEV-treated animals on day 21. E. Quantification of the total number of CD4+ cells per mg of spleen tissue after gating on live cells (ns = 0.4175, n=8/group, two-tailed Student’s t-test). Data are mean ± SEM. F. Quantification of the number of CD4+FOXP3+ cells per mg of spleen tissue (ns = 0.6178, n=8/group, two-tailed Student’s t-test). Data are mean ± SEM. G. Quantification of the number of CD4+FOXP3+IL10+ cells per mg of spleen tissue (*P = 0.0487, n=8/group, two-tailed Student’s t-test). Data are mean ± SEM.
Transcriptome analysis of iTreg-hEV reveals an intermediate phenotype with features of Treg and Th17 cells
Given the changes observed among iTreg cells exposed to hEVs, we sought to further characterize this cell population. In a global transcriptome analysis (Figure 5A), iTreg cells upregulated expression of Rorc (RORγt), Il17a, and Il17f in response to human hEVs (Figure 5B), a profile characteristic of Th17 cells.23, 24 Meanwhile, iTreg-hEV preserved their expression of Il2ra (CD25), Foxp3, Ctla4, and Tgfb1 (Figure 5B,C), which are defining markers for Treg cells. Consistent with Figure 2, iTreg-hEV expressed much higher levels of Il10 (Figure 5B), which acts not only to suppress inflammation but also to maintain the Treg phenotype.25 To exclude the possibility that hEVs are polarizing Treg cells toward T regulatory 1 (Tr1) cells, which also secrete high levels of IL-10, we assessed the expression of LAG-3 and CD49b (characteristic markers of Tr1 cells).26 Expression levels of LAG-3 and CD49b were similar between iTreg-Ctrl and iTreg-hEV (Figure S4), suggesting that iTreg-hEV are distinct from Tr1 cells. Taken together, exposure to hEVs induces an intermediate phenotype, sharing characteristics of FOXP3+ Treg cells and RORγt+ Th17 cells (Figure 5D). Treg cells are known to express both FOXP3 and RORγt,27–29 transcription factors that together allow for selective suppression of Th17 cells but, in order to do so, additionally require transcription factor c-Maf (encoded by Maf).30 Indeed, expression of Maf was much higher among iTreg-hEV, compared to iTreg-Ctrl (Figure 5B). Finally, we noted upregulation of Il21, a cytokine that stimulates proliferation of activated T cells.31 Thus, the transcriptome profile of iTreg-hEV points to an intermediate FOXP3+RORγt+ cell type, capable of enhanced proliferation upon activation.
Figure 5. iTreg cells exposed to human hEVs exhibit an intermediate FOXP3+RORγt+ phenotype with Treg cell and Th17 features.

A. Transcriptome analysis of iTreg-Ctrl and iTreg-hEV (n=3/group). B. RNA-sequence volcano plot with –log10[adjusted P value] (y axis) and log2[fold change] (x axis), highlighting significantly upregulated (red) and downregulated (blue) genes in iTreg-hEV, relative to iTreg-Ctrl. C. Log10 expression of Foxp3 in naïve T cells (Th0), iTreg-Ctrl, and iTreg-hEV, relative to average Foxp3 expression among Th0 cells (n=4–6/group). Data are mean ± SEM. D. Merged contour plots for FOXP3 and RORγt expression in iTreg-Ctrl and iTreg-hEV. Plots are representative of two independent experiments.
iTreg-hEV cells exhibit suppressive capacity in vitro
Given the changes in iTreg cells toward an intermediate FOXP3+RORγt+ phenotype, we wanted to assess whether human hEVs alter iTreg cells’ suppressive ability using an in vitro assay with responder CD8+ T cells, antigen presenting cells, and iTreg cells.29 Both iTreg-Ctrl (Figure 6A,B) and iTreg-hEV (Figure 6C,D) suppressed proliferation of CD8+ T cells, verifying that the central immunosuppressive phenotype is not impeded by hEVs. Therefore, iTreg-hEV represent an intermediate FOXP3+RORγt+ cell type with enhanced proliferation and IL-10 production and preserved suppressive capacity.
Figure 6. iTreg-hEV maintain suppressive capacity in vitro.

A. Representative histograms show proliferation of CD8+ T cells after co-culture with antigen presenting cells and iTreg-Ctrl at various ratios. B. Quantification of proliferation and suppression of CD8+ T cells co-cultured with iTreg-Ctrl; n=4/group. Data are mean ± SEM. % Suppression = [(% proliferating with no iTreg - % proliferating with iTreg)/% proliferating with no iTreg) x 100]. C. Representative histograms show proliferation of CD8+ T cells after co-culture with antigen presenting cells and iTreg-hEV at various ratios. D. Quantification of proliferation and suppression of CD8+ T cells co-cultured with iTreg-hEV; n=3–4/group. Data are mean ± SEM. % Suppression = [(% proliferating with no iTreg - % proliferating with iTreg)/% proliferating with no iTreg) x 100].
Discussion
EVs are nanometer-sized, lipid-bilayer particles laden with noncoding RNAs and a diverse signaling cargo.32 In the context of immunity, EVs derived from both immune and non-immune parent cells have demonstrated bioactivity in dendritic cells, monocytes, macrophages, natural killer cells and T lymphocytes.6, 33 Exposure to EVs can induce either activating or inhibitory effects, depending on the origin and state of the parent cell that produced the EVs. Thus, the underlying mechanisms of EV immunomodulation are diverse and warrant further investigation to better define their therapeutic and diagnostic potential.
Here, we have demonstrated that hEVs elicit cell-specific and dose-dependent responses among effector and regulatory CD4+ T cells. The biological effect of hEVs was most pronounced in Treg cells, with increased rates of proliferation, augmented production of IL-10, and modified expression of lineage-specific transcription factors. Effects of hEVs on cells of innate immunity are well-described; for example, hEVs polarize macrophages toward a distinctive phenotype, with decreased expression of proinflammatory genes (Nos2 and Tnf), increased expression of anti-inflammatory genes (Arg1, Il4ra, Tgfb1), and enhanced efferocytosis.10, 11 These changes are mediated through transfer of EV cargo, including noncoding RNAs and micro-RNAs (e.g., Y RNA fragments and miR-181b). Fragments of Y RNA are of particular interest, as they constitute the largest fraction of small RNAs within hEVs. The most abundantly expressed fragment, EV-YF1, induces production and secretion of IL-10 by macrophages, with downstream cardioprotective effects.9 Therefore, this RNA fragment could potentially account for the increase in IL-10 we have observed, but this possibility remains untested. In terms of proliferation, miR-155 is known to promote Treg cell expansion and therefore could be the responsible factor within hEV cargo, but this too is conjectural.34
The increase in IL-10 production by Treg cells, coupled with their enhanced proliferation, can theoretically dampen systemic and local inflammation. Although Treg cells employ other mechanisms of suppression (e.g., TGFβ, IL-35, CTLA4, CD25), only expression of IL-10 was increased following hEV administration in our study (Figure 2B, Figure 5B), suggesting that the suppressive effects we observed in vitro were related to changes in IL-10. In EAM, we showed that hEVs have anti-inflammatory and cardioprotective effects, which correlated with increased numbers of IL10+-Treg cells. Further characterization of iTreg-hEV revealed a transcriptional profile with intermediate features of Treg and Th17 cells. Dual expression of FOXP3 and RORγt by Treg cells has been described in rodents and humans.27–29, 35 In humans, FOXP3+RORγt+ Treg cells are capable of producing IL-17, yet retain their suppressive function.35, 36 Other studies have demonstrated enhanced suppressive function by FOXP3+RORγt+ Treg cells.29 Because Treg cells can acquire features specific to the type of cell they aim to suppress,4 FOXP3+RORγt+ Treg cells are well-suited to suppress Th17 cells. This is particularly important in EAM, where higher levels of Th17 cells account for increased EAM susceptibility.37 Furthermore, Th17-mediated inflammation favors adverse cardiac remodeling and progression to dilated cardiomyopathy in mice and humans with myocarditis.38, 39 Thus, the intermediate FOXP3+RORγt+ phenotype induced by hEV represents a potential mechanism for the functional benefits observed in EAM, but further investigation (e.g., depletion and adoptive transfer studies) is needed to test this possibility.
The ability to modulate Treg cells is an attractive therapeutic approach to inflammation that relies on harnessing an endogenous mechanism of immunosuppression. Multiple (>50) clinical trials are exploring this general approach in the setting of solid organ transplantation, graft-versus-host disease, and autoimmune disorders. Most of these trials, however, rely on a challenging adoptive-transfer protocol that involves isolation of autologous Treg cells, expansion of these cells ex vivo, and reinfusion of the expanded population back into the patient.2 This paradigm is limited by the scarcity of Treg cells in the peripheral blood (the most common source for Treg cells), as well as uncertainties about the suppressive and proliferative potential of Treg cells after ex vivo expansion. Treatment with hEVs can potentially harness Treg cells in vivo, 40, 41 circumventing the limitations of adoptive transfer. We already have circumstantial evidence of hEV bioactivity in patients with COVID-19, a syndrome characterized by a maladaptive, hyperinflammatory host response and lymphocytopenia.42 Therapy using the parent cardiac stromal/progenital cells in 4 of 6 such critically-ill patients was associated with clinical improvement, including higher lymphocyte counts and lower levels of inflammatory biomarkers.43 Beyond COVID-19, these parent cells have demonstrated signs of therapeutic efficacy in clinical trials of ischemic44, 45 and dilated46 cardiomyopathy, and in Duchenne muscular dystrophy.47 Our findings that hEVs are able to augment Treg cell proliferation and IL-10 production, along with prior evidence of immunomodulatory effects by the parent cells,12, 13, 48–50 may shed light on the mechanisms underlying disease-modifying bioactivity. Not only in COVID-19 or myocarditis, but more generally for inflammatory disorders involving innate and adaptive immunities, hEVs represent logical next-generation therapeutic candidates.
Supplementary Material
Acknowledgments:
We thank all other members of the Marbán Laboratory for their support; the Cedars-Sinai Medical Center Flow Cytometry Core; the Applied Genomics, Computation, and Translational Core; and Kostas Malliaras for advice on implementing EAM models.
Financial disclosure statement: A. Akhmerov was supported by the National Institutes of Health (NIH) T32 HL116273-07. The work was supported by NIH R01 HL 124074. E. Marbán holds the Mark S. Siegel Family Distinguished Chair of the Cedars-Sinai Medical Center. E. Marbán owns founder’s equity in Capricor Therapeutics. The other authors report no conflicts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010;28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discov 2019;18:749–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010;327:1098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol 2018;19:665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie M, Xiong W, She Z, et al. Immunoregulatory Effects of Stem Cell-Derived Extracellular Vesicles on Immune Cells. Front Immunol 2020;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 2014;14:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marbán E A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat Biomed Eng 2018;2:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibrahim AG, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2014;2:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cambier L, de Couto G, Ibrahim A, et al. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol Med 2017;9:337–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Couto G, Gallet R, Cambier L, et al. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017;136:200–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Couto G, Jaghatspanyan E, DeBerge M, et al. Mechanism of Enhanced MerTK-Dependent Macrophage Efferocytosis by Extracellular Vesicles. Arterioscler Thromb Vasc Biol 2019;39:2082–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Couto G, Liu W, Tseliou E, et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest 2015;125:3147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nana-Leventaki E, Nana M, Poulianitis N, et al. Cardiosphere-Derived Cells Attenuate Inflammation, Preserve Systolic Function, and Prevent Adverse Remodeling in Rat Hearts With Experimental Autoimmune Myocarditis. J Cardiovasc Pharmacol Ther 2019;24:70–7. [DOI] [PubMed] [Google Scholar]
- 14.Aminzadeh MA, Rogers RG, Fournier M, et al. Exosome-Mediated Benefits of Cell Therapy in Mouse and Human Models of Duchenne Muscular Dystrophy. Stem Cell Reports 2018;10:942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrahim AGE, Li C, Rogers R, et al. Augmenting canonical Wnt signalling in therapeutically inert cells converts them into therapeutically potent exosome factories. Nat Biomed Eng 2019;3:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers RG, Fournier M, Sanchez L, et al. Disease-modifying bioactivity of intravenous cardiosphere-derived cells and exosomes in mdx mice. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malliaras K, Li TS, Luthringer D, et al. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 2012;125:100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallet R, Dawkins J, Valle J, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J 2017;38:201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol 1991;147:2141–7. [PubMed] [Google Scholar]
- 20.Jennings VM, Dillehay DL. The Laboratory Rat (Second Edition): Academic Press; 2006. [Google Scholar]
- 21.Rodriguez-Perea AL, Arcia ED, Rueda CM, Velilla PA. Phenotypical characterization of regulatory T cells in humans and rodents. Clin Exp Immunol 2016;185:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol 2008;99:95–114. [DOI] [PubMed] [Google Scholar]
- 23.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005;6:1123–32. [DOI] [PubMed] [Google Scholar]
- 24.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006;126:1121–33. [DOI] [PubMed] [Google Scholar]
- 25.Murai M, Turovskaya O, Kim G, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 2009;10:1178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gagliani N, Magnani CF, Huber S, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med 2013;19:739–46. [DOI] [PubMed] [Google Scholar]
- 27.Sefik E, Geva-Zatorsky N, Oh S, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 2015;349:993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohnmacht C, Park JH, Cording S, et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 2015;349:989–93. [DOI] [PubMed] [Google Scholar]
- 29.Yang BH, Hagemann S, Mamareli P, et al. Foxp3(+) T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol 2016;9:444–57. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, Pokrovskii M, Ding Y, et al. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018;554:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parrish-Novak J, Dillon SR, Nelson A, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 2000;408:57–63. [DOI] [PubMed] [Google Scholar]
- 32.Wiklander OPB, Brennan MA, Lotvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veerman RE, Gucluler Akpinar G, Eldh M, Gabrielsson S. Immune Cell-Derived Extracellular Vesicles - Functions and Therapeutic Applications. Trends Mol Med 2019;25:382–94. [DOI] [PubMed] [Google Scholar]
- 34.Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 2009;30:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A 2009;106:4793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beriou G, Costantino CM, Ashley CW, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 2009;113:4240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen P, Baldeviano GC, Ligons DL, et al. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol 2012;169:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldeviano GC, Barin JG, Talor MV, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res 2010;106:1646–55. [DOI] [PubMed] [Google Scholar]
- 39.Hua X, Hu G, Hu Q, et al. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation 2020. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, Yeo RWY, Lai RC, Sim EWK, Chin KC, Lim SK. Mesenchymal stromal cell exosome-enhanced regulatory T-cell production through an antigen-presenting cell-mediated pathway. Cytotherapy 2018;20:687–96. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q, Fu L, Liang Y, et al. Exosomes originating from MSCs stimulated with TGF-beta and IFN-gamma promote Treg differentiation. J Cell Physiol 2018;233:6832–40. [DOI] [PubMed] [Google Scholar]
- 42.Akhmerov A, Marbán E. COVID-19 and the Heart. Circ Res 2020;126:1443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh S, Chakravarty T, Chen P, et al. Allogeneic cardiosphere-derived cells (CAP-1002) in critically ill COVID-19 patients: compassionate-use case series. Basic Res Cardiol 2020;115:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makkar RR, Smith RR, Cheng K, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 2012;379:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makkar RR, Kereiakes DJ, Aguirre F, et al. Intracoronary ALLogeneic heart STem cells to Achieve myocardial Regeneration (ALLSTAR): a randomized, placebo-controlled, double-blinded trial. Eur Heart J 2020. [DOI] [PubMed] [Google Scholar]
- 46.Chakravarty T, Henry TD, Kittleson M, et al. Allogeneic cardiosphere-derived cells for the treatment of heart failure with reduced ejection fraction: the Dilated cardiomYopathy iNtervention with Allogeneic MyocardIally-regenerative Cells (DYNAMIC) trial. EuroIntervention 2020;16:e293–e300. [DOI] [PubMed] [Google Scholar]
- 47.Taylor M, Jefferies J, Byrne B, et al. Cardiac and skeletal muscle effects in the randomized HOPE-Duchenne trial. Neurology 2019;92:e866–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grigorian-Shamagian L, Liu W, Fereydooni S, et al. Cardiac and systemic rejuvenation after cardiosphere-derived cell therapy in senescent rats. Eur Heart J 2017;38:2957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aminzadeh MA, Tseliou E, Sun B, et al. Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur Heart J 2015;36:751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallet R, de Couto G, Simsolo E, et al. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 2016;1:14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.