

Summary:
Transient receptor potential mucolipin 1 (TRPML1) regulates lysosomal calcium signaling, lipid trafficking and autophagy-related processes. This channel is regulated by phosphoinositides and the low pH environment of the lysosome, maintaining calcium levels essential for proper lysosomal function. Recently, several small molecules specifically targeting the TRPML family have been demonstrated to modulate channel activity. One of these, a synthetic antagonist ML-SI3, can prevent lysosomal calcium efflux and has been reported to block downstream TRPML1-mediated induction of autophagy. Here, we report a cryo-EM structure of human TRPML1 with ML-SI3 at 2.9-Å resolution. ML-SI3 binds to the hydrophobic cavity created by S5, S6 and PH1, the same cavity where the synthetic agonist ML-SA1 binds. Electrophysiological characterizations show that ML-SI3 can compete with ML-SA1, blocking channel activation yet does not inhibit PI(3,5)P2 dependent activation of the channel. Consequently, this work provides molecular insight into how ML-SI3 and native lipids regulate TRPML1 activity.
Graphical Abstract
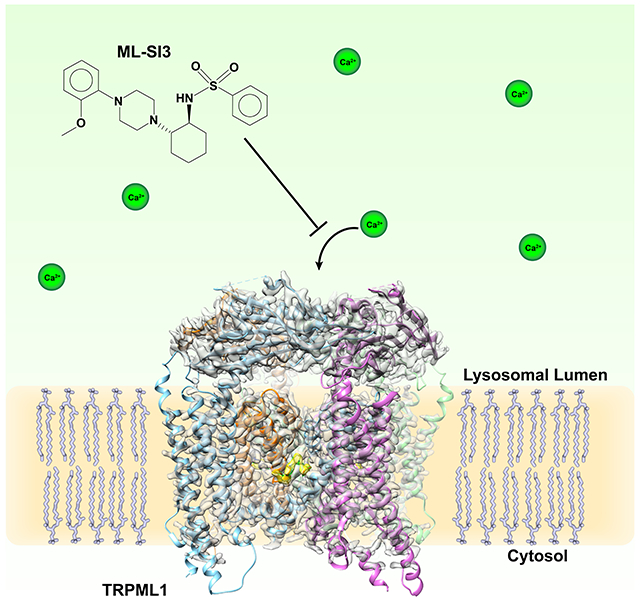
eTOC Blurb
Building on the previously solved TRPML1 structures, Schmiege et al. report the structure of TRPML1 bound to the synthetic inhibitor ML-SI3, which binds to the same site as the synthetic agonist ML-SA1. Electrophysiology experiments show that ML-SI3 can affect ML-SA1 activation but not activation by the native lipid agonist PI(3,5)P2.
Introduction
TRP (Transient receptor potential) channels are widely regulated by various sensations and ligands revealing important physiological roles in cells (Ramsey et al., 2006; Venkatachalam and Montell, 2007). The technology revolution in cryogenic electron microscopy (cryo-EM) has considerably accelerated the determination of most TRP channels at near atomic resolution (Gao et al., 2016; Grieben et al., 2017; Huynh et al., 2016; Liu et al., 2021; Paulsen et al., 2015; Shen et al., 2016; Suo et al., 2020; Wilkes et al., 2017; Zubcevic et al., 2016). These structures have provided remarkable insights into the functional mechanisms and molecular dynamics of this important ion channel family. Mucolipin TRP channels include three distinct members: TRPML1, TRPML2 and TRPML3. TRPML1, which is ubiquitously expressed, regulates lysosomal calcium signaling, lipid trafficking and autophagy-related processes (Fine et al., 2020; Venkatachalam et al., 2015; Wang et al., 2014). More importantly, loss-of-function TRPML1 mutants lead to the lysosomal storage disease Mucolipidosis type IV (MLIV), a neurodegenerative disorder characterized by aberrant development, cognitive defects and vision loss (Altarescu et al., 2002; Eichelsdoerfer et al., 2010; Sun et al., 2000).
All TRPML channels have been shown to be positively modulated by various small-molecule synthetic compounds (e.g. ML-SA1 and MK6-83) (Chen et al., 2014; Shen et al., 2012). Furthermore, TRPML1 is activated by the endogenous lipid phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) (Dong et al., 2010; Zhang et al., 2012) and application of the mTOR inhibitor, rapamycin (Zhang et al., 2019). These channels can also be inhibited by a synthetic compound ML-SI3 (Samie et al., 2013), and the lipids sphingomyelin and PI(4,5)P2 (Zhang et al., 2012). Importantly, the structures of mammalian TRPML1 and TRPML3 (Chen et al., 2017; Hirschi et al., 2017; Li et al., 2017; Schmiege et al., 2017; Zhang et al., 2017; Zhou et al., 2017) reveal that TRPML1 and TRPML3 both form a classic tetrameric channel: each subunit contains six transmembrane helices (S1-S6), two pore helices (PH1 and PH2) and a ~30kD lumenal/extracellular domain (Figure 1A). We have shown that the cytosolic extensions of S1-S3 (labeled as IS1-IS3), which include several basic amino acids, form an unique feature called the Mucolipin Domain that binds to either PI(3,5)P2 or PI(4,5)P2, indirectly regulating the activity of TRPML1 (Fine et al., 2018). Mutations on these corresponding residues in either TRPML1 (Fine et al., 2018) or TRPML3 (Hirschi et al., 2017) and the deletion of IS1 and IS2 in mouse TRPML1 (Chen et al., 2017) has been shown to reduce the PI(3,5)P2 activation of TRPMLs.
Figure 1. Overall Structure of ML-SI3-bound TRPML1.
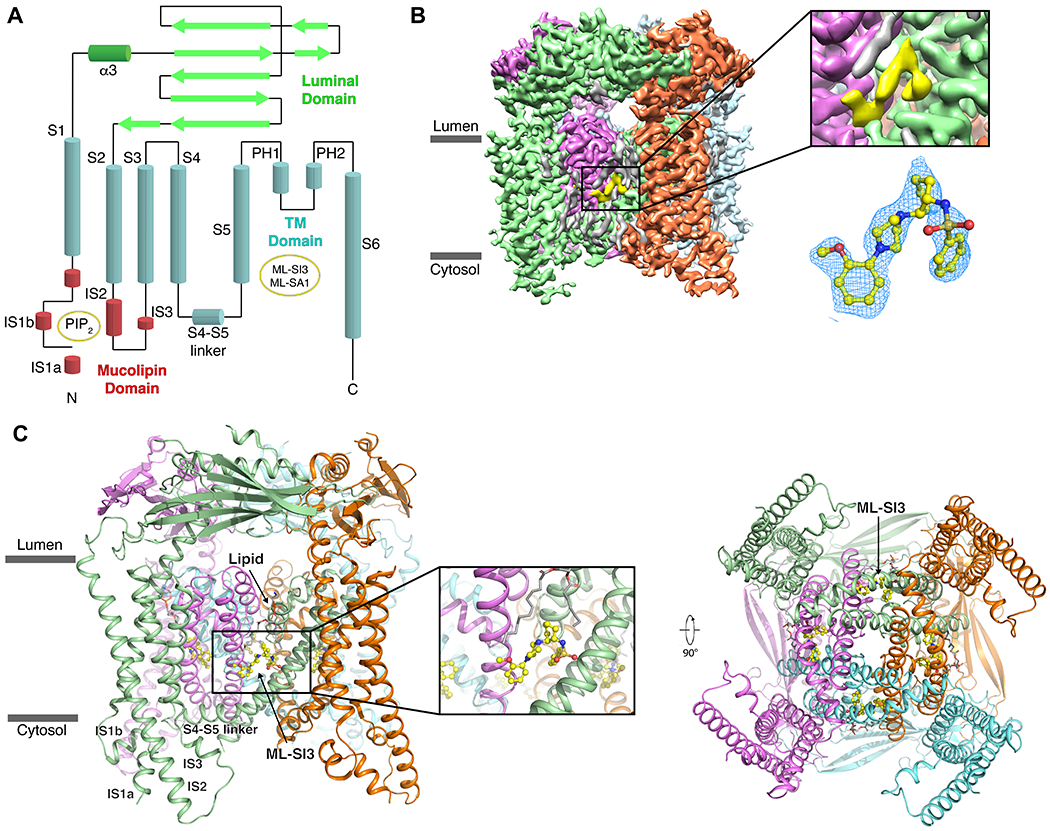
(A) A 2D schematic of TRPML1 with the various domains colored, and the binding sites indicated in yellow circles. (B) The cryo-EM map of TRPML1-ML-SI3 with the protomers colored individually and ML-SI3 in yellow. A zoom-in on the density that represents ML-SI3 with the EM-density in mesh (blue) fitted to the molecular structure (yellow sticks). (C) Structural model of TRPML1–ML-SI3 with a zoom-in on ML-SI3 and its binding pocket. Subunits are represented by the same colors as in B.
The structures of synthetic agonist ML-SA1 bound TRPML1 and TRPML3 have previously been determined (Schmiege et al., 2017; Zhou et al., 2017). In TRPML1, ML-SA1 binds to a hydrophobic pocket that is created by the S5, S6 and PH1 of one subunit and the S6 of the neighboring subunit. This binding triggers the opening of lower gate but not the selectivity filter (Schmiege et al., 2017). Interestingly, in ligand-free structures of other TRPML family members, including our apo and PIP2 bound structures of TRPML1, no endogenous ligand density is observed in this binding site (Fine et al., 2018; Schmiege et al., 2017). Indeed, this binding site is quite distinct from other agonist binding sites even among the larger TRP channel super family (Fine et al., 2020). Typically, the S4-S5 linker plays an important role in engaging the agonist or antagonist to regulate TRP channel activity (Cao et al., 2013; Liu et al., 2020). Although previous structural studies show the molecular basis of how ML-SA1 and phosphoinositides regulate the activity of TRPML1, the molecular mechanism of how ML-SI3 inhibits TRPML1 channel activation remain unclear. In this manuscript, we report a 2.9 Å cryo-EM structure of human TRPML1 bound to ML-SI3 that reveals the ligand-channel interaction details. The structure along with electrophysiological characterizations show that ML-SI3 can block ML-SA1-mediated activation of TRPML1 but not affect the PI(3,5)P2-mediated activation of the channel.
Results
To study the binding site of ML-SI3 in TRPML1, we expressed and purified TRPML1 according to our previously established protocol (Fine et al., 2018; Schmiege et al., 2017). ML-SI3 was supplemented to the protein at a final concentration of 0.5 mM for grid preparation. The structure of the TRPML1-ML-SI3 complex was solved at 2.9-Å resolution (Figures 1B–C, S1–S3 and Table 1), and most of the side chains and structural elements are well resolved in the cryo-EM map (Figure S2). The structures of ML-SI3 bound TRPML1 and apo-TRPML1 share a similar conformation with an R.M.S.D. of 1.4 Å, while the ML-SI3 structure is slightly different from the ML-SAl-bound TRPML1 structure with an R.M.S.D. of 2.4 Å. The main difference in the channel pore between the two structures is the open lower gate in the ML-SA1-bound structure (Figure 2A and B).
Table 1.
Cryo-EM data collection, refinement and validation statistics
| TRPML1–ML-SI3 (EMD-23828) (PDB 7MGL) |
|
|---|---|
| Data collection and processing | |
| Magnification | 75758 |
| Voltage (kV) | 300 |
| Electron exposure (e–/Å2) | 80 |
| Defocus range (μm) | 1.2 to 2.6 |
| Pixel size (Å) | 0.66 |
| Symmetry imposed | C4 |
| Initial particle images (no.) | 837,138 |
| Final particle images (no.) | 508,465 |
| Map resolution (Å) | 2.9 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 2.7-4.5 |
| Refinement | |
| Initial model used (PDB code) | 5WJ5 |
| Model resolution (Å) | 2.9 |
| FSC threshold | 0.5 |
| Map sharpening B factor (Å2) | −130 |
| Model composition | |
| Non-hydrogen atoms | 15,476 |
| Protein residues | 1,888 |
| Ligands | 8 |
| B factors (Å2) | |
| Protein | 47.15 |
| Ligand | 36.32 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.717 |
| Validation | |
| MolProbity score | 2.02 |
| Clashscore | 5.45 |
| Poor rotamers (%) | 3.10 |
| Ramachandran plot | |
| Favored (%) | 94.87 |
| Allowed (%) | 5.13 |
| Disallowed (%) | 0 |
Figure 2. Structural comparisons and Binding interfaces of the TRPML1–ML-SI3.
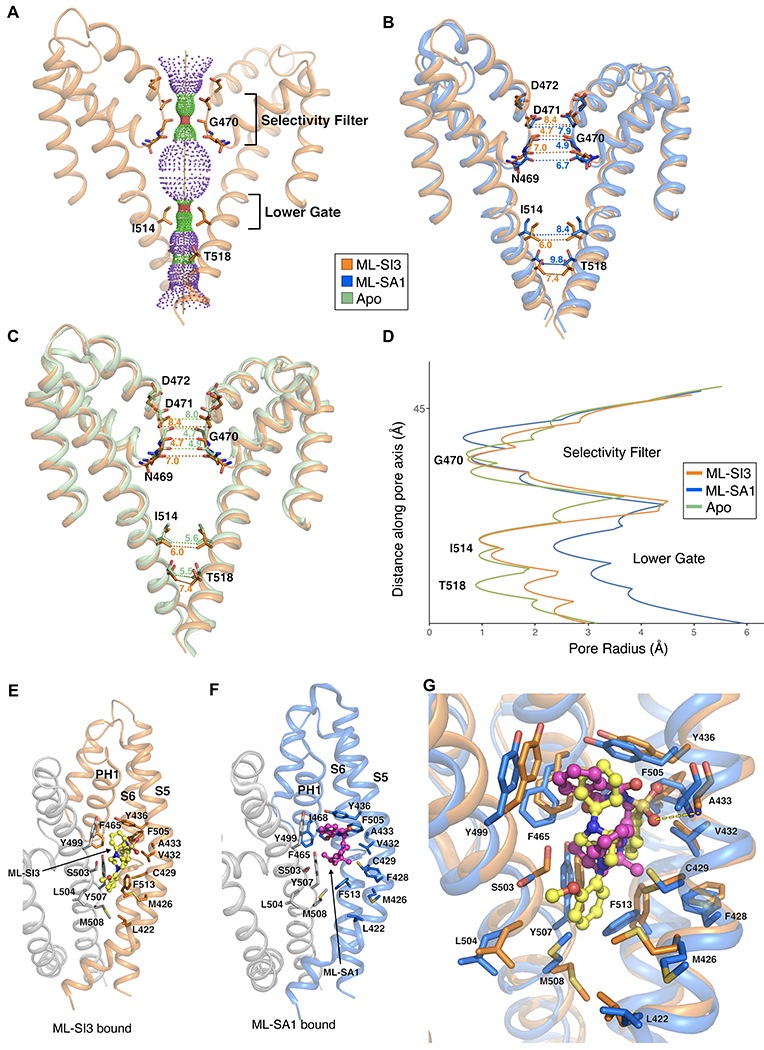
(A) Solvent accessible pathway along the ion permeation pore of ML-S13-bound TRPML1 generated by the program HOLE. (B) Superimposition of PH1, PH2 and S2 of ML-S13-bound TRPML1 (orange) and ML-SA1-bound TRPML1 (blue, PDB: 5WJ9) structures. Distances from the text are numbered and indicated by dotted lines. (C) Superimposition of PH1, PH2, and S2 of ML-S13-bound TRPML1 (orange) and apo TRPML1 (green, PDB: 5WJ5) structures. Distances from the text are numbered and indicated by dotted lines. Residues in the selectivity filter and lower gate are rendered as sticks. (D) Comparison of the pore radius (calculated by HOLE) for ML-SI3-bound (orange), ML-SA1-bound (blue) and Apo (green) TRPML1 structures. (E) The interaction details of TRPML1 bound to ML-SI3, with key residues labeled. (F) The interaction details of TRPML1 bound to ML-SA1, with key residues labeled. (G) Superimposition of ML-SI3 (yellow/orange) and ML-SA1 (pink/blue) binding interactions with key residues represented as sticks, and the polar interaction between A433 and ML-SI3 represented by a dashed yellow line.
The overall structure of ML-S13-bound TRPML1 presents a closed conformation (Figure 2B and D). The conformation of the selectivity filter and the lower gate is consistent with that in the apo-TRPML1 (Figure 2C and D). The cryo-EM map of ML-SI3 clearly indicates the position and orientation of the compound in the channel (Figure 1B). The residues Phe465 in PH1, Leu422, Met426, Cys429, Val432, Ala433 and Tyr436 in S5, Phe505 and Phe513 in S6 along with the residues Tyr499, Ser 503, Leu504, Tyr507 and Met508 in S6 of the neighboring subunit engage the ML-SI3 (Figure 2E). This binding site is consistent with the ML-SA1 binding site that we previously observed (Figure 2F). Notably, ML-SI3 is larger than ML-SA1 interacting with additional residues and the neighboring subunit not involved with ML-SA1 binding including Leu422, Met426 and Met508 (Figure 2E and F). We previously reported that ML-SA1 activation of the channel is dependent on the π-π interactions between the agonist and the aromatic rings of residues F505 and F513 (Schmiege et al., 2017). These interactions force S6 away from the central axis, opening the lower gate. However, in the ML-SI3 bound structure, the aromatic rings of F505 and F513 no longer form these π-π interactions, and as a result the S6 is not forced away from the central axis and the lower gate remains in a closed conformation (Figure 2G).
To validate our observation, we used a tetracycline inducible HEK-293 cell line expressing TRPML1 with four leucine to alanine mutations (L/A) (Vergarajauregui and Puertollano, 2006). The mutations disrupt the lysosomal signaling motif, increasing expression of TRPML1 on the cell surface for subsequent electrophysiological characterization via whole-cell patch clamp. Earlier reports show effective stimulation of TRPML1 inward rectifying cation currents at a working ML-SA1 concentration in the low micromolar range. However, the values are highly dependent on methodology (i.e. Ca2+ fluorescence, planar bilayer recordings, and lysosomal patch clamp) with limited demonstration of the concentration ranges required for whole-cell electrophysiology (Dong et al., 2008; Leser et al., 2021). Environmental factors (i.e. extracellular pH and cationic constituents) also contribute to the range of activities reported for TRPML agonists (Dong et al., 2008; Li et al., 2017).
To ascertain the effectiveness of the agonist on surface expressing TRPML1-L/A we determined the EC50 (relative mean effective concentration) of ML-SA1 in the presence of 1.5 mM extracellular Ca2+at both physiological lysosomal and extracellular pH (4.6 and 7.4, resp.). The EC50 of ML-SA1 at lysosomal pH is 9.7 μM with a diminished maximal response and an EC50 of 15.3 μM observed at pH 7.4 (Figure 3A). The reduction in activity at neutral pH is indicative of how both pH and extracellular Ca2+ down regulate TRPML1 activity as it moves from the lysosome to the cell surface (Li et al., 2017). Our observed EC50 values for ML-SA1 are comparable to earlier reports and represent a baseline for characterizing whole-cell electrophysiological activation of the surface expressing TRPML1-L/A by ML-SA1.
Figure 3. Functional analysis of TRPML1 ligands.
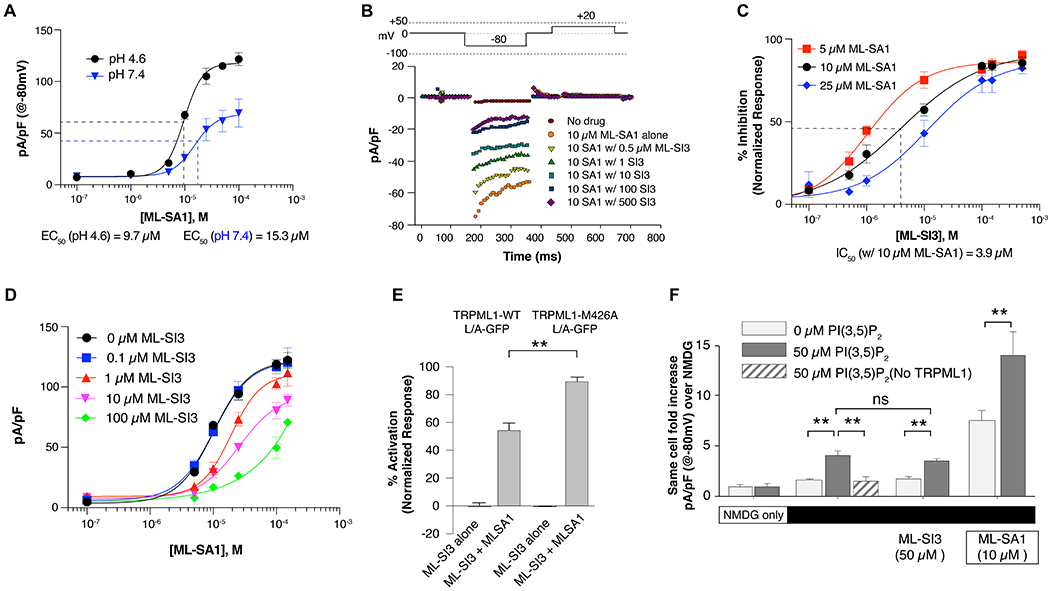
(A) EC50 determinations of ML-SA1 on induced TRPML1-L/A expressing HEK cells at pH 4.6 (black) and pH 7.4 (blue). Subsequent experiments are performed at pH 4.6 only. Current density is determined using real-time capacitive values determined prior to voltage pulses. All data are represented as mean ± SEM. (B) Representative inward rectifying TRPML1-L/A currents using the voltage pulse recording described (top) with increasing amounts of ML-SI3 applied prior to application of 10 μM ML-SA1. (C) IC50 determinations of normalized inhibition of ML-SA1 induced currents at 5, 10, and 25 μM ML-SA1 (red, black, blue, resp.). Normalized current densities are determined as the difference between maximal ML-SA1 activity with no inhibitor and baseline current density with no drug at −80 mV. (D) Rightward competitive shift in EC50 values of ML-SA1 with increasing amount of ML-SI3. (E) Composite results reveal a significant loss of inhibition in M426A cells during co-application of ML-SA1 and ML-SI3. Student t-test, p ** <0.001. (F) PI(3,5)P2 mediated currents in TRPML1-L/A expressing HEK cells show a 4-fold increase in same-cell Na-currents in TRPML1-L/A expressing cells when 50 μM PI(3,5)P2 is present in the cytoplasmic solution. This increase is not inhibited by application of ML-SI3 and is absent in cells lacking expression of the channel while also synergistically increased when ML-SA1 is applied.
Next, we examined whether ML-SI3 functions as a competitive inhibitor to ML-SA1 as our structural observations imply. Using a step pulse protocol, we activated TRPML1 inward rectifying currents at −80 mV at pH 4.6 with a working concentration of 10 μM ML-SA1. Upon addition of ML-SI3, current decreases in a dose-dependent manner (Figure 3B). However, not all the ML-SA1 current is blocked, even with fairly large concentrations of ML-SI3 (50:1 molar ratio). To clarify these findings, we determined the IC50 of ML-SI3 at three concentrations of ML-SA1. Pre-treatment with antagonist gave an IC50 of 3.9 μM when using a working agonist concentration of 10 μM, and a small right shift in IC50 values was observed ranging from 1-10 μM with increasing concentrations of agonist (Figure 3C). These results favor the competitive nature of agonist and antagonist, despite the maximal effective inhibition of TRPML1-induced currents at only 80%.
We examined if a classical right shift in the EC50 values of ML-SA1 could be observed at varying concentrations of ML-SI3. At low concentrations of ML-SI3, only a small change is observed in the ML-SA1 dose response curve (Figure 3D). As the concentration of ML-SI3 concentration is increased, the EC50 value for ML-SA1 doubles to nearly 20 μM indicating a clearly competitive rightward shift in the dose response curve. Taken together, these results support our structural observation that ML-SA1 shares a similar binding site with ML-SI3. To further validate the ML-SI3 binding site, we mutated Met426, which is responsible for engaging ML-SI3 but not ML-SA1 (Figure 2G). The electrophysiological experiment showed that the ML-SA1 was able to activate either wild type or M426A mutant (Figure S4); however, unlike wild type TRPML1, ML-SI3 fails to considerably inhibit the channel for M426A mutant expressing cells (Figure 3E), directly supporting our observations.
Based on our current structural observation, ML-SI3 does not seem to interfere with the binding of PI(3,5)P2, but rather ML-SI3 and PI(3,5)P2 may independently regulate the activity of TRPML1. Currently, it is not clear if ML-SI3 can inhibit lipid regulation of TRPML1 as initial in vitro characterizations of ML-SI3 in cells typically employed only co-treatment with synthetic agonists like ML-SA1 or MK-683. To validate our hypothesis that ML-SI3 and PI(3,5)P2 can independently regulate TRPML1 activity, we isolated the Na+-dependent current through TRPML1 and determined the whole-cell contribution of cytoplasmic dialysis of PI(3,5)P2 on TRPML1 activity. We then investigated if the same cells exhibited either an inhibition of the PI(3,5)P2-specific current during ML-SI3 application or a synergistic increase in activity during application of ML-SA1, which has been reported previously (Schmiege et al., 2017).
As shown in Figure 3E, in the absence of extracellular Na, utilizing the large non-conducting cation N-methyl-D-glucamine (NMDG) as the primary cation, no significant increase in current density is detected in either the presence or absence of cytoplasmic PI(3,5)P2 (50μM; −80 mV). When external NMDG is replaced with the conductive ion Na, there is a 4-fold increase in same-cell current density in cells internally dialyzed with 50μM PI(3,5)P2. This increase in current density is absent in cells lacking expression of TRPML1-L/A, demonstrating an isolation of the PI(3,5)P2-dependent Na conductance through TRPML1 while substantiating our earlier reports showing specific phosphoinositide stimulation (Fine et al., 2018) (Figure 3F). When ML-SI3 is supplemented at 50 μM, there is no decrease in the observed PI(3,5)P2-dependent Na current density supporting our hypothesis that ML-SI3 and PI(3,5)P2 independently regulate channel activity. Finally, when ML-SA1 is added to the extracellular bath, current density increases 7-fold over basal conditions, and 14-fold with both ML-SA1 and PI(3,5)P2 which is consistent with earlier reports that show ML-SA1 and PI(3,5)P2 cooperate to stimulate TRPML1. Our structural observations and analysis reveal that ML-SI3 works to competitively inhibit TRPML1 through interaction with the same hydrophobic binding pocket as ML-SA1 and is independent of TRPML1 regulation by phosphoinositides.
Discussion
The functional inhibitory properties of ML-SI3 raise several interesting conjectures. The classical shift in the EC50 of ML-SA1 points to a competitive inhibition by ML-SI3. However, there are some complexities in defining ML-SI3 as a purely competitive inhibitor. First, the inability of high concentrations of ML-SI3 to completely inhibit the ML-SA1-derived channel activity may result from the way each ligand interacts with the hydrophobic pocket: e.g. ML-SI3 has both a larger size and a shallower penetration into the pocket when compared to ML-SA1 (Figure 2E–G). Second, because reversibility or wash out of these compounds can take several minutes, potentially due to the differences in binding, it is possible that the order in which the ligands are applied may result in unique allosteric interactions that could contribute to the results. Recent reports involving ML-SI3 and a related agonist ML-SA5 further exacerbate these complexities by revealing that ML-SI3 may have enantiomer-specific activities. For TRPML1, the (+)-and (−)-enantiomers have varying levels of inhibition, while for TRPML2 and TRPML3, the (+)-enantiomer reverses its effect, displaying weak stimulatory properties (Leser et al., 2021). More physiologically relevant, however, is the way in which ML-SI3 does not impact the stimulatory effect of PI(3,5)P2 (Figure 3F), despite the synergistic increase in TRPML1 activity and open probability observed with both ML-SA1 and PI(3,5)P2 (Chen et al., 2017). This raises the question: what is the physiological role of ML-SI3 on TRPML1 signaling?
ML-SI3 has been reported to inhibit ML-SA1 or related agonist MK6-83 induced autophagy in cells (Scotto Rosato et al., 2019; Wang et al., 2015). However, in the absence of artificial TRPML1 agonists, previous work showed that classical starvation induced autophagy was also inhibited by treatment with ML-SI3 (Scotto Rosato et al., 2019). To date, the only known endogenous agonist identified for TRPML1 is PI(3,5)P2. It has been hypothesized that PI(3,5)P2 and the antagonist PI(4,5)P2 function mainly to regulate localized activity of TRPML1 activity through differential accumulation of the lipids within different compartments of the cell. This begs the question, if ML-SI3 can inhibit autophagic responses during cellular starvation, are there endogenous ligands that bind to the hydrophobic groove where ML-SI3 and ML-SA1 bind? Recently, a study showed that the bacterial macrolide rapamycin directly bound and activated TRPML1, independent of rapamycin’s classical inhibition of its mammalian target, mTOR (Zhang et al., 2019). However, rapamycin is not an endogenous ligand and the binding site for rapamycin on TRPML1 remains unclear. Together, these reports suggest that there is a complex association between TRPML1 and autophagic events, and that the hydrophobic groove created by S5 and S6 where ML-SA1 and ML-SI3 bind is a likely target for a currently unknown, autophagy inducing ligand.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaochun Li (xiaochun.li@utsouthwestern.edu).
Materials availability
Any unique reagents/materials used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The 3D cryo-EM density map of ML-S13-bound TRPML1 has been deposited in the Electron Microscopy Data Bank under accession numbers EMD-23828. Atomic coordinates for the atomic model of ML-SI3-bound TRPML1 have been deposited in the Protein Databank under the accession number 7MGL.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Spodoptera frugiperda Sf9 cells for baculovirus expression were cultured in Sf-900™ III SFM media (Thermo Fisher Scientific) at 27°C with shaking (135 rpm). HEK293S GnTI− cells used for protein expression were cultured in FreeStyle™ 293 media (Thermo Fisher Scientific) supplemented with 2% Fetal Bovine Serum (Corning) and 1% Pen Strep (Thermo Fisher Scientific) at 37°C with shaking (130 rpm). HEK cells for electrophysiology were cultured in DMEM media supplemented with 10% Fetal Bovine Serum (Coming) and 1% Pen Strep (Thermo Fisher Scientific).
METHOD DETAILS
Protein expression and purification
Human TRPML1 was purified according to our previous report (Schmiege et al., 2017). Briefly, the protein was cloned into pEG BacMam with an N-terminal Flag tag and subsequently expressed using baculovirus transfection of HEK293S GnTF cells grown at 37°C for 48 hours. Cells were then harvested and lysed by sonication in buffer A (20 mM HEPES pH7.5, 150 mM NaCl) with 1 mM PMSF and 5 μg ml−1 of leupeptin. The lysate was centrifuged at low speed and the resulting supernatant was incubated with 1% (w/v) lauryl maltose neopentyl glycol (MNG, Anatrace) for 1 hour at 4°C. After incubation, the lysate was centrifuged again, and the supernatant was incubated with anti-Flag M2 resin for 1 hour at 4°C. The protein was concentrated and purified by Superdex-200 (GE Healthcare) size-exclusion chromatography in a buffer containing 20 mM Sodium Acetate pH 5.0, 150 mM NaCl and 0.06% (w/v) digitonin (Sigma). The peak fractions were collected and concentrated to 5-7 mg/ml for grid preparation. TRPML1 mutants were generated using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies).
Whole-cell patch clamp electrophysiology
HEK 293 cells stably expressing a tetracycline-inducible expression of TRPML1-L/A were a gift from Casma Therapeutics. Cells were induced for 24-48 hours prior to experimentation with 4 μM tetracycline containing DMEM media supplemented with 10% Fetal Bovine Serum and 1% Pen Strep. Prior to recordings, cells were deplated with trypsin (0.05%, Sigma) and isolated cells resuspended in cell culture medium were diluted in recording buffer and transferred to a custom chamber on an inverted Nikon TE2000U microscope using a 60x oil immersion, 1.45-NA objective. Patch clamp recordings of cell electrical parameters were performed as described previously (Fine et al., 2011; Shen et al., 2012), using Matlab based Capmeter v7.2 (Wang and Hilgemann, 2008) with a National Instruments digital acquisition board and an Axopatch 200B patch clamp amplifier. Square-wave voltage perturbation (20 mV; 0.5 kHz) was employed for capacitance measurement, input resistances were 2-10 MΩ, and the apparent cell resistances were 0.5-2 GΩ. External solutions were adjusted to RT (23-25 °C) in gravity-fed parallel solution lines with an outlet flow velocities of 2 to 5 mm/s allowing extracellular solution changes within 2-3 seconds. Upon membrane rupture, input resistance was low (2-10 MΩ) to allow rapid diffusion of PI(3,5)P2 (Echelon Biosciences) during cytoplasmic dialysis. A minimum of 180 seconds after rupture was utilized prior to electrical recordings to ensure sufficient dialysis. Boroscilicate glass pipettes were fire polished and back-filled with the cytoplasmic solutions containing in mM: 120 cesium methanesulfonate, 4 NaCl, 10 EGTA, 2 MgCl2, 20 HEPES, 2 Tris-ATP, pH 7.2 with CsOH (~25 mM). Extracellular solutions were set to pH 7.4 (Figure 3A) and contained in mM: 140 NaCl, 5 KC1, 10 glucose, 20 HEPES, 1 MgCl2, 1.5 CaCl2 and 8 NaOH or were set to pH 4.6 (Figure 3) and contained in mM 140 sodium gluconate, 5 KCl, 10 glucose, 10 HEPES, 10 MES, 1 MgCl2, 1.5 CaCl2 and 8 HCl. For PI(3,5)P2 current determination sodium gluconate was replaced with 140 N-methyl-D-glucamine (NMDG) and 140 gluconic acid and a baseline current density at −80 mV was determined. NMDG was replaced with standard sodium gluconate based solution to determine the Na-current through TRPML1 with or without PI(3,5)P2. Uninduced cells lacking TRPML1-L/A expression were used to determine specificity. Stock concentrations of ML-SA1 or ML-SI3 in DMSO and were diluted and added to the appropriate bath solution immediately prior to recording. Inward-rectifying TRPML1 currents were detected using a voltage pulse protocol described in Figure 3B with a holding potential of 0 mV, TRPML1 mediated current at −80 mV for 200 msec and inactivated at +20 mV.
For mutant binding functional assessment in Figure 3E and Figure S4 transient expression of GFP tagged TRPML1-L/A was performed in HEK 293T cells using Fugene 6 HD (Promega) and selected for expression using standard epifluorescence microscopy as described above. To determine the level of ML-SI3 mediated inhibition, whole-cell patch clamp was performed as described above and a voltage ramp from −150 to + 50 mV was applied throughout the experiment and values at −100 mV were used for statistical analysis. A basal response was determined without any ligand perfusion. ML-SI3 alone (10 μM) was applied as a negative control followed by application of ML-SI3 and ML-SA1 (10 μM, each). Finally, maximal response was determined after washout and reapplication of ML-SA1 alone (10 μM). Percent activation was determined as the percentage of the difference between the maximal ML-SA1 alone response and ligand free conditions.
EM Sample Preparation and Imaging
A protein sample was added to Quantifoil R1.2/1.3 400 mesh Au holey carbon grids (Quantifoil), blotted with Vitrobot Mark IV (FEI), and frozen in liquid ethane. For ML-SI3 bound protein, the protein in a buffer containing 20 mM sodium acetate pH 5.0, 150mM NaCl and 0.06% digitonin was incubated with 0.5 mM ML-SI3 (gift from Casma Therapeutics, dissolved in DMSO as a 50 mM stock) on ice for 30 minutes before grid preparation and freezing. The grids were imaged with a 300keV Titan Krios (FEI) with a Gatan K2 Summit direct electron detector (Gatan). Data were collected at 0.66 Å per pixel with a dose rate of 23 electrons per physical pixel per second. Images were recorded for 1.5-second exposure in 30 subframes to give a total dose of 80 electrons per Å2 using Serial EM (Mastronarde, 2005).
Imaging Processing and 3D reconstruction
Dark subtracted images were first normalized by gain reference that resulted in a pixel size of 0.66 Å per pixel. Drift correction was performed using the program MotionCor2 (Zheng et al., 2017). The contrast transfer function (CTF) was estimated using CTFFIND4 (Rohou and Grigorieff, 2015). To generate templates for automatic picking, around 3,000 particles were manually picked and classified by 2D classification in RELION 3 (Zivanov et al., 2018). After automatic picking, the micrographs were manually inspected and low-quality images and false positive particles were removed. The subsequent particles were extracted and subjected to 2D classification. 3D classification was carried out using the map of human TRPML1 (EMD-8840) low-pass filtered to 60 Å as the initial model. The best class out of the four classes, containing ~500k particles, was selected and refined without a mask in RELION 3. This refinement yielded a 3.2 Å map, and was then refined two more times with masks. The particles were polished using the Bayesian Polishing in RELION 3, and finally, post-processing was performed with a mask yielding a final resolution map of 2.92 Å. Resolution was estimated using the FSC 0.143 criterion. The entire model was built in COOT (Emsley et al., 2010).
Model Refinement and Validation
The model was refined in real space using PHENIX (Adams et al., 2010) and also in reciprocal space using Refmac with secondary-structure restraints and stereochemical restraints (Brown et al., 2015; Murshudov et al., 1997). For cross validation, the final model was refined against one of the half maps from the final 3D refinement. The resulting model was used to calculate the model-versus-map FSC curves against the same half map and the other half map, respectively, using the Comprehensive validation module in PHENIX (Adams et al., 2010). MolProbity (Chen et al., 2010) was used to validate the geometries of the model. Local resolutions were estimated RELION 3. Structure figures were generated using PyMOL (http://www.pymol.org) and Chimera (Pettersen et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in Matlab and SigmaPlot (SigmaStat) while IC50 and EC50 values were calculated with a four-parameters analysis in Prism (GraphPad). Current-voltage relations represent the mean of at least 4 individual voltage pulses per cell, and agonist induced current density changes represent the mean and s.e.m. of at least 3 independent cells per condition, n = number of individual cells per data point. For Figure 3A, n= 3-6. For Figure 3C, n= 3-6. For Figure 3D, n= 4-5. For Figure 3E, n= 9. For Figure 3F, 0 μM PI(3,5)P2 n= 13; 50 μM PI(3,5)P2 n= 6; 50 μM PI(3,5)P2 without TRPML1 n= 9. Student t-test was performed in SigmaPlot (p ** <0.001; SigmaStat) and all data are represented as mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Digitonin | Acros Organics | Cat#407565000 |
| Lauryl maltose neopentyl glycol | Anatrace | Cat#NG310 |
| Phenylmethylsulfonyl fluoride | Goldbio | Cat#P-470-25 |
| Leupeptin | Peptides International | Cat#ILP-4041 |
| ML-SI3 | Casma Therapeutics | N/A |
| ML-SA1 | Tocris Bioscience | Cat#4746 |
| PI(3,5)P2 | Echelon Biosciences | Cat#P-3508 |
| Deposited data | ||
| Atomic coordinates of ML-SI3-bound TRPML1 | This paper | PDB: 7MGL |
| 3D cryo-EM map of ML-SI3-bound TRPML1 | This paper | EMD-23828 |
| Atomic coordinates of apo TRPML1 | Schmiege et al, 2017 | PDB: 5WJ5 |
| 3D cryo-EM map of apo TRPML1 | Schmiege et al, 2017 | EMD-8840 |
| Experimental models: Cell lines | ||
| HEK293S GnTI- | ATCC | CRL-3022 |
| Sf9 | ATCC | CRL-1711 |
| HEK293-tet-TRPML1-L/A | Casma Therapeutics | N/A |
| Recombinant DNA | ||
| pEG BacMam | This paper | N/A |
| pEG BacMam-TRPML1 | This paper | N/A |
| Software and algorithms | ||
| Serial EM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM |
| CTFFIND 4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| MotionCor 2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-software |
| RELION 3 | Zivanov et al., 2018 | http://www2.mrclmb.cam.ac.uk/relion |
| GraphPad Prism8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MatLab 2015 | Mathworks | https://www.mathworks.com/products/matlab.html |
| Sigma Plot 14 | Systat Software | https://systatsoftware.com/products/sigmaplot/ |
| Capmeter V7.2 | Wang and Hilgemann, 2008 | https://sites.google.com/site/capmeter |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| Refmac | Brown et al., 2015 Murshudov et al., 1997 | https://www.ucl.ac.uk/~rmhasek/refmac.html |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| PyMOL | PyMOL | http://www.pymol.org |
| Other | ||
| R1.2/1.3 400 mesh Au holey carbon grids | Quantifoil | Cat#1210627 |
| Superose 6, 10/300 GL | GE Healthcare | Cat#17-5172-01 |
| ANTI-FLAG M2 Affinity Gel | Millipore Sigma | Cat#A2220 |
Highlights:
A 2.9Å cryo-EM structure of ML-SI3-bound TRPML1 reveals a closed conformation.
The TRPML1 inhibitor ML-SI3 binds the same cavity as its agonist ML-SA1.
ML-SI3 competes with ML-SA1 to block the channel activity.
TRPML1 inactivation via ML-SI3 is independent of phosphoinositide regulation.
Acknowledgement
The data were collected at the UT Southwestern Medical Center; we thank D. Stoddard for assistance in data collection. We thank J. Fortanet and A. Jha (Casma Therapeutics) for providing the ML-SI3 and HEK293-tet-TRPML1-L/A cell line. This work was supported by the Endowed Scholars Program in Medical Science of UT Southwestern Medical Center (to X.L.). P.S. is supported by NIH T32GM131963. X.L. is a Damon Runyon-Rachleff Innovator supported by the Damon Runyon Cancer Research Foundation (DRR-53S-19) and a Rita C. and William P. Clements Jr. Scholar in Biomedical Research at UT Southwestern Medical Center.
Declaration of interests
M.F. was partially supported by the Casma Therapeutics and X.L receives consultant income and holds shares from Casma Therapeutics on the study of TRPML1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference:
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarescu G, Sun M, Moore DF, Smith JA, Wiggs EA, Solomon BI, Patronas NJ, Frei KP, Gupta S, Kaneski CR, et al. (2002). The neurogenetics of mucolipidosis type IV. Neurology 59, 306–313. [DOI] [PubMed] [Google Scholar]
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, and Murshudov G (2015). Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta crystallographica Section D, Biological crystallography 71, 136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao E, Liao M, Cheng Y, and Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, et al. (2014). A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nature communications 5, 4681. [DOI] [PubMed] [Google Scholar]
- Chen Q, She J, Zeng W, Guo J, Xu H, Bai XC, and Jiang Y (2017). Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nature 550, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, and Xu H (2008). The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, et al. (2010). PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nature communications 1, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelsdoerfer JL, Evans JA, Slaugenhaupt SA, and Cuajungco MP (2010). Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel. The Journal of biological chemistry 285, 34304–34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta crystallographica Section D, Biological crystallography 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine M, Li X, and Dang S (2020). Structural insights into group II TRP channels. Cell calcium 86, 102107. [DOI] [PubMed] [Google Scholar]
- Fine M, Llaguno MC, Lariccia V, Lin MJ, Yaradanakul A, and Hilgemann DW (2011). Massive endocytosis driven by lipidic forces originating in the outer plasmalemmal monolayer: a new approach to membrane recycling and lipid domains. The Journal of general physiology 137, 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine M, Schmiege P, and Li X (2018). Structural basis for PtdInsP2-mediated human TRPML1 regulation. Nat Commun 9, 4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D, and Cheng Y (2016). TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieben M, Pike AC, Shintre CA, Venturi E, El-Ajouz S, Tessitore A, Shrestha L, Mukhopadhyay S, Mahajan P, Chalk R, et al. (2017). Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nature structural & molecular biology 24, 114–122. [DOI] [PubMed] [Google Scholar]
- Hirschi M, Herzik MA Jr., Wie J, Suo Y, Borschel WF, Ren D, Lander GC, and Lee SY (2017). Cryo-electron microscopy structure of the lysosomal calcium-permeable channel TRPML3. Nature 550, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh KW, Cohen MR, Jiang J, Samanta A, Lodowski DT, Zhou ZH, and Moiseenkova-Bell VY (2016). Structure of the full-length TRPV2 channel by cryo-EM. Nature communications 7, 11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leser C, Keller M, Gerndt S, Urban N, Chen CC, Schaefer M, Grimm C, and Bracher F (2021). Chemical and pharmacological characterization of the TRPML calcium channel blockers ML-SI1 and ML-SI3. European journal of medicinal chemistry 210, 112966. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang WK, Benvin NM, Zhou X, Su D, Li H, Wang S, Michailidis IE, Tong L, Li X, et al. (2017). Structural basis of dual Ca(2+)/pH regulation of the endolysosomal TRPML1 channel. Nat Struct Mol Biol 24, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Reese R, Vu S, Rouge L, Shields SD, Kakiuchi-Kiyota S, Chen H, Johnson K, Shi YP, Chernov-Rogan T, et al. (2020). A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Reese R, Vu S, Rouge L, Shields SD, Kakiuchi-Kiyota S, Chen H, Johnson K, Shi YP, Chernov-Rogan T, et al. (2021). A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 109, 273–284 e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Armache JP, Gao Y, Cheng Y, and Julius D (2015). Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, and Clapham DE (2006). An introduction to TRP channels. Annu Rev Physiol 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, et al. (2013). A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Developmental cell 26, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiege P, Fine M, Blobel G, and Li X (2017). Human TRPML1 channel structures in open and closed conformations. Nature 550, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A, et al. (2019). TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat Commun 10, 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, et al. (2012). Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen PS, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE, and Cao E (2016). The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell 167, 763–773 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS Jr., Bove C, Kaneski CR, Nagle J, Bromley MC, et al. (2000). Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Human molecular genetics 9, 2471–2478. [DOI] [PubMed] [Google Scholar]
- Suo Y, Wang Z, Zubcevic L, Hsu AL, He Q, Borgnia MJ, Ji RR, and Lee SY (2020). Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 105, 882–894 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, and Montell C (2007). TRP channels. Annual review of biochemistry 76, 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Wong CO, and Zhu MX (2015). The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S, and Puertollano R (2006). Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7, 337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TM, and Hilgemann DW (2008). Ca-dependent nonsecretory vesicle fusion in a secretory cell. The Journal of general physiology 132, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, et al. (2015). Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America 112, E1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhang X, Gao Q, and Xu H (2014). TRPML1: an ion channel in the lysosome. Handbook of experimental pharmacology 222, 631–645. [DOI] [PubMed] [Google Scholar]
- Wilkes M, Madej MG, Kreuter L, Rhinow D, Heinz V, De Sanctis S, Ruppel S, Richter RM, Joos F, Grieben M, et al. (2017). Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nature structural & molecular biology 24, 123–130. [DOI] [PubMed] [Google Scholar]
- Zhang S, Li N, Zeng W, Gao N, and Yang M (2017). Cryo-EM structures of the mammalian endo-lysosomal TRPML1 channel elucidate the combined regulation mechanism. Protein & cell 8, 834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen W, Gao Q, Yang J, Yan X, Zhao H, Su L, Yang M, Gao C, Yao Y, et al. (2019). Rapamycin directly activates lysosomal mucolipin TRP channels independent of mTOR. PLoS biology 17, e3000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li X, and Xu H (2012). Phosphoinositide isoforms determine compartment-specific ion channel activity. Proceedings of the National Academy of Sciences of the United States of America 109, 11384–11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li M, Su D, Jia Q, Li H, Li X, and Yang J (2017). Cryo-EM structures of the human endolysosomal TRPML3 channel in three distinct states. Nature structural & molecular biology 27, 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubcevic L, Herzik MA Jr., Chung BC, Liu Z, Lander GC, and Lee SY (2016). Cryo-electron microscopy structure of the TRPV2 ion channel. Nature structural & molecular biology 23, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 3D cryo-EM density map of ML-S13-bound TRPML1 has been deposited in the Electron Microscopy Data Bank under accession numbers EMD-23828. Atomic coordinates for the atomic model of ML-SI3-bound TRPML1 have been deposited in the Protein Databank under the accession number 7MGL.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.