

Abstract
Limited clinical data are available regarding the utility of multi-kinase inhibition in neuroblastoma. Repotrectinib (TPX-0005) is a multi-kinase inhibitor that targets ALK, TRK, JAK2/STAT and Src/FAK which have all been implicated in the pathogenesis of neuroblastoma. We evaluated the preclinical activity of repotrectinib monotherapy and in combination with chemotherapy as a potential therapeutic approach for relapsed/refractory neuroblastoma. In vitro sensitivity to repotrectinib, ensartinib, and cytotoxic chemotherapy was evaluated in neuroblastoma cell lines. In vivo anti-tumor effect of repotrectinib monotherapy, and in combination with chemotherapy, was evaluated using a genotypically diverse cohort of patient derived xenograft (PDX) models of neuroblastoma. Repotrectinib had comparable cytotoxic activity across cell lines irrespective of ALK mutational status. Combination with chemotherapy demonstrated increased antiproliferative activity across several cell lines. Repotrectinib monotherapy had notable anti-tumor activity and prolonged event-free survival compared to vehicle and ensartinib in PDX models (p<0.05). Repotrectinib plus chemotherapy was superior to chemotherapy alone in ALK-mutant and ALK wild-type PDX models. These results demonstrate that repotrectinib has anti-tumor activity in genotypically diverse neuroblastoma models, and that combination of a multi-kinase inhibitor with chemotherapy may be a promising treatment paradigm for translation to the clinic.
BACKGROUND:
Neuroblastoma is the most common pediatric extracranial solid tumor with more than 650 cases in North America per year. Despite aggressive medical and surgical therapy, patients with high-risk disease have 5-year overall survival rates of 40–50% (1,2), with even poorer outcomes in patients with relapsed/refractory disease. Therefore, development of novel treatment strategies is essential to improve survival rates among these patients. Preclinical work has demonstrated that aberrant activation and signaling through ALK, PI3K/AKT, MAPK, PLCγ, TrkB, Src/FAK, and JAK/STAT pathways contribute to deregulated proliferation and therapy resistance in neuroblastoma.(3–9) Thus far, therapeutic strategies that target a singular pathway have not resulted in significant clinical benefit. For example, the administration of ALK inhibitors in vitro and in vivo demonstrates potent inhibition of cell growth in ALK-mutant cell lines and tumor growth inhibition in xenograft models (3,10–14), but overall response rates in early phase clinical studies of single-agent ALK inhibitors in neuroblastoma patients range from 9–20%.(15–17) Therefore, the evaluation of rational combination therapies incorporating novel agents that inhibit multiple relevant signaling pathways represents a treatment paradigm that may lead to more effective and durable treatment responses in high-risk tumors.
Repotrectinib (TPX-0005) is a novel macrocyclic multi-kinase inhibitor that has the potential to exploit several therapeutic vulnerabilities of neuroblastoma and was specifically designed to overcome multiple common mechanisms of therapy resistance. In addition to bypassing solvent front and gatekeeper mutations of ALK, ROS1, and TRK (18,19), repotrectinib effectively inhibits multiple critical signaling pathways mediating proliferation, survival, angiogenesis, and drug resistance, including JAK2/STAT and Src/FAK, with low nanomolar kinase inhibitory profiles.(18–22) Taken together, these distinct features position repotrectinib as a promising targeting agent that seeks to address and overcome mechanisms of therapy resistance encountered by earlier generation multi-kinase inhibitors.
The activity of repotrectinib has recently been demonstrated in ALK-mutant neuroblastoma cell lines and in a cell line-derived xenograft model.(11) Based on the promising clinical activity in patients with ROS1-rearranged solid tumors and tyrosine kinase inhibitor resistant tumors (23), and preclinical work demonstrating anti-tumor activity in solid tumors without ALK, ROS1, or NTRK aberrations (24,25), we hypothesize that repotrectinib may have activity extending beyond ALK-mutant neuroblastoma. We further hypothesize that combining repotrectinib with cytotoxic chemotherapy may augment its activity. To evaluate the applicability of repotrectinib, singly and in combination with chemotherapy, we assembled and treated a clinically representative, diverse cohort of neuroblastoma PDX models representing the molecular spectrum of tumor phenotypes and provide preclinical rationale for further exploration within the context of a clinical trial.
METHODS:
Drugs
Repotrectinib was provided by Turning Point Therapeutics. Irinotecan and temozolomide were obtained commercially from SelleckChem. Ensartinib (E543721) was obtained commercially from LKT Laboratories.
Cell lines and culture
The neuroblastoma cells lines, SH-SY5Y (ATCC Cat#CRL-2266, RRID:CVCL_0019), SK-N-DZ (ATCC Cat#CRL-2149, RRID:CVCL_1701) and IMR-32 (ATCC Cat#CCL-127, RRID:CVCL_0346) were obtained from the American Type Culture Collection. Kelly (DSMZ Cat# ACC-355 RRID:CVCL 2092) and LA-N-5 (DSMZ CAT#ACC-673, RRID:CVCL_0389) were obtained from the German Collection of Microorganisms and Cell Cultures GmbH. NB-1 and GI-ME-N cell lines were provided by Dr. Frank Speleman. Cells were cultured in treated flasks in DMEM/F12 media (Corning™) supplemented with 10% fetal bovine serum (Corning™), L-glutamine (Gibco), non-essential amino acids (Gibco), and antibiotic-antimycotic (Gibco) at 37 °C and 5% CO2. Cells were passaged twice weekly. Cell lines were obtained between 2014 and 2021 and cell line identities were verified by STR profiling. Mycoplasma testing was performed prior to all in vitro drug screens using the MycoAlert-Plus™ kit (Lonza).
Dose response assays
The assay was conducted in a 384-well format (Corning), and cells were seeded at optimized seed densities empirically determined for each cell line to achieve exponential growth during the assay period. Dose-response studies were performed using serial dilutions ranging from 100 µM to 0.001µM with each concentration performed in triplicate. The high controls (HC) contained 1% DMSO (v/v) and the low controls (LC) contained 1µM of killer mix(26) (combination of six cytotoxic drugs) in 1% DMSO (v/v). Cells were incubated at 37ºC and 5% CO2 and viability assayed with Alamar Blue after 96 hours of drug treatment. Fluorescence intensity was read on the Cytation 5 Cell Imaging Multi-Mode Reader (Biotek, RRID:SCR_019732). For the screening analysis, the Z factor was used to determine the quality of the assay, and is defined as 1–3 x (standard deviation of the HC + standard deviation of the LC)/ HC average – LC average). Values between 0.5–1.0 represent an excellent performance of the assay, and a marginal assay shows values between 0–0.5.(27) Dose response curves were fitted with a logistic 4-parameter sigmoidal equation using SigmaPlot 13.0 (Systat Software, Inc., RRID:SCR_003210). Percent inhibition was defined as (HC average – obtained value)/(HC average – LC average) x 100.
Combination drug screen
For the drug combination screen, the fixed ratio method(28) was used, where the concentrations of the drugs (drug 1: repotrectinib or ensartinib, drug 2: irinotecan, drug 3: temozolomide) were set at 1:1:0.1–1 the 50% growth inhibitory concentration (IC50). Ratios were determined based on drug solubility characteristics at the specified concentrations. The IC50 determined in the dose-response assays was used to establish the concentrations of each drug for the combination to assure that the IC50 was approximately at the middle of the serial dilutions. 1.25 fold dilutions were done for a total of 23 points for the combinations in triplicate. The effect of the combination was assessed via the combination index (CI) obtained with the Chou and Talalay Method(29) using CalcuSyn Software version 2 (BIOSOFT, RRID:SCR_020251). The CI values are defined as synergistic if CI<1, additive if CI=1, or antagonistic if CI>1. A semiquantitative CI annotation system (synergism, moderate synergism, slight synergism, nearly additive, slight antagonism, and moderate antagonism) has been defined previously.(30)
Immunoblotting and analysis
For in vitro analysis, approximately 5×106 cells were seeded in 10 cm culture dishes and FBS-starved for 24 hours prior to treatment. Cells were treated with either DMSO or repotrectinib (at concentrations of 10 nM, 100 nM, and 1 mM) for one hour and then harvested with RIPA buffer (#89901; ThermoFisher Scientific) supplemented with Halt Protease & Phosphatase Inhibitor Cocktail (#1861284; ThermoFisher Scientific). For in vivo pharmacodynamic analyses, tumors were harvested 3 hours post-dose on the 3rd day of treatment and tumors flash frozen in liquid nitrogen. Tumor lysates were obtained using RIPA buffer + protease/phosphatase inhibitor cocktail. Protein concentration was measured using the BCA assay (#23210; ThermoFisher Scientific) and 20 μg of total protein was analyzed on SDS-PAGE gels and transferred to nitrocellulose membrane using standard methods. Membranes were probed with the following primary antibodies: phospho-ALK (Y1278) (#6941; Cell Signaling Technologies, CST, RRID:AB_10860598), phospho-ALK (Y1586) (#3348; CST, RRID:AB_659803), Pan Trk (#92991; CST, RRID:AB_2800196), phospho-TrkA/TrkB (#4621; CST, RRID:916186), FAK (#13009; CST, RRID:AB_2798086), phospho-FAK (#3283; CST, RRID:AB_2173659), Src (#2109; CST RRID:AB_2106059), phospho-Src (#2101; CST, RRID:AB_331697), PI3K (#4257; CST, RRID: AB_659889), AKT (#75692; CST, RRID:AB_2716309), phospho-AKT (#4060; CST, RRID:AB_2315049), STAT3 (#9139; CST, RRID:AB_331757), phospho-STAT3 (#9131; CST, RRID:AB_331586), MEK1/2 (#4694; CST, RRID:AB_10695868), phospho-MEK1/2 (#9154; CST, RRID:AB_2138017), Erk1/2 (#9102; CST, RRID:AB_330744), phospho-Erk1/2 (9101; CST, RRID:AB_331646), RPS6 (#2217; CST, RRID:AB_331355), phospho-RPS6 (#4858; CST, RRID:AB_916156), phospho-ALK (Y1604) (#ab62185; Abcam, RRID:AB_955656), phospho-PI3K (#ab138364; Abcam, RRID:AB_2892208), ALK (#398791; Santa Cruz, RRID:AB_2889357), tubulin (#T9026; Sigma, RRID:AB_477593). Secondary antibodies were obtained from LI-COR: anti-mouse IgG (#926–68072, RRID:AB_10953628) and anti-rabbit (#926–32213, RRID:AB_621848). Signal detection and image capturing were performed using the LI-COR Odyssey scanner system.
Xenograft therapeutic studies
All mice were maintained under barrier conditions and experiments were conducted in accordance with and with the approval of Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Animal Care and Use Committee (IACUC, protocol #16–08-011). Patient derived tumor tissue to generate PDX models was obtained under the MSKCC Institutional Review Board (IRB)-approved protocols #17–387 and #06–107 with written informed consent provided by the subject or legally authorized representative. PDX mouse models were established by implanting tumor cells subcutaneously into non-obese diabetic/severe combined immunodeficiency interleukin-2R gamma null, HPRT null (NSGH) mice (Jackson Labs, IMSR Cat# JAX:012480, RRID: IMSR_JAX:012480). Two mice of each model were enrolled into each treatment arm as a preliminary screening single mouse trial (2 mice per model per treatment) adapted from Gao et al.(31) Once the tumor reached a volume of 150–200 mm3, mice were assigned to treatment groups using block randomization with two animals per group. Mice were treated in one of three groups for four weeks (treatment 5 days on, two days off for all arms): (1) repotrectinib 20 mg/kg via oral gavage (PO) twice daily, (2) ensartinib 25 mg/kg PO twice daily, and (3) vehicle (0.5% carboxymethylcellulose (CMC)/1% Tween-80) PO twice daily. Tumors were measured by caliper measurement twice weekly and tumor volume (TV) was calculated as follows: TV = width2 X ½ length. Aligned with clinical response criteria,(32,33) responses were categorized as complete response (CR, >95% reduction from baseline or no measurable tumor), partial response (PR, >50% reduction), stable disease (SD, <50% reduction but not more than 100% increase), or progressive disease (PD, >100% increase). Overall response rate was defined as the total of responses categorized as CR or PR. To account for variable tumor growth rates across different PDX models, the timepoint at which each individual model’s tumor volume change was analyzed corresponded to the day of treatment failure (>100% increase in tumor volume relative to baseline) in each respective model’s vehicle-treated animals.
For xenograft studies using combination therapy, a minimum of 3 mice were included in each of four treatment arms: (1) repotrectinib 15 mg/kg PO twice daily (5 days on, 2 days off) continuously throughout treatment cycle; (2) irinotecan 6 mg/kg/dose via intraperitoneal injection (IP) and temozolomide 25 mg/kg/dose PO daily days 1–5 and 15–29 of each 28 day cycle; (3) repotrectinib 15 mg/kg PO twice daily (5 days on, 2 days off) throughout 28 day treatment cycle + irinotecan 6 mg/kg/dose IP and temozolomide 25 mg/kg/dose PO daily on days 1–5 and 15–29 of each 28 day cycle; and (4) vehicle (0.5% CMC/1% Tween-80) PO twice daily. Mean relative tumor volumes (RTV) were calculated for vehicle control (C) and each treatment arm (T). The drug associated with mean RTV T/C ratio of < 15% are considered highly active, < 45% but > 15% are considered to have intermediate activity, and > 45% are considered to have low levels of activity.(34)
Statistical analysis
For in vivo statistical analysis, the Mann-Whitney-Wilcoxon method was used to evaluate differences in distribution of tumor volume between treatment groups. Vardi’s test was used to evaluate difference in area under the curve (AUC) between treatment groups. Event-free survival (EFS) was defined as the percentage of mice that survived at any given time point without an event. Accounting for inter-operator variability of caliper measurements(35) and consistent with clinical treatment response definitions (33,36), an event was defined as a 100% increase in tumor volume relative to baseline measurements or death. Kaplan-Meier survival curves were compared using the log-rank test. Statistical analysis was performed using R software (v3.5.0). Waterfall plots and tumor volume curves for in vivo analysis were generated with GraphPad Prism (v8.4.1 [RRID:SCR_002798]). Statistical significance was defined as p values < 0.05.
RESULTS:
Repotrectinib inhibits tumor growth and prolongs survival in patient derived neuroblastoma xenograft models
To determine the anti-tumor activity of repotrectinib in vivo, we evaluated its anti-tumor effect in several pediatric PDX models (Table 1), utilizing a modification of the Single Mouse Trial (SMT) design.(31) Eight discrete neuroblastoma models were included in the therapeutic study. For each model, 2 animals were evaluated on each treatment arm (n=16 per treatment group). When evaluating the change in tumor volume from baseline between treatment groups, there was a statistically significant difference between repotrectinib and vehicle treatment (median tumor volume change of 14% and 149%, respectively, p=0.005) and ensartinib versus vehicle (73% and 149%, respectively, p=0.005) (Figure 1a). In comparing the repotrectinib and ensartinib treatment cohorts, there was a trend toward reduced tumor volume in the repotrectinib group, although statistical significance was not reached at the p=0.05 threshold (14% and 73%, p=0.12, Figure 1a). When stratifying change in relative tumor volume by treatment arm based on ALK status, ALK-mutant models treated with ensartinib or repotrectinib had statistically significantly reduced change in relative tumor volume compared to the vehicle arm (p=0.005 and p=0.014, respectively). However, ALK WT models treated with repotrectinib showed greater relative tumor volume change from baseline compared to vehicle (p=0.041), in contrast to those treated with ensartinib compared to vehicle (p=0.10) (Figure S1). Furthermore, event-free survival was analyzed, and repotrectinib yielded a significant survival advantage when compared to the vehicle or ensartinib groups in the total neuroblastoma cohort (n=8 models) and the ALK-mutant neuroblastoma cohort (n=5 models) (p<0.05, via log-rank test in all treatment comparisons, Figures 1b–c). In the ALK WT cohort, the repotrectinib treatment arm also had improved event-free survival compared to vehicle (p=0.04, via log-rank) supporting activity of repotrectinib in both ALK mutant and WT models.
Table 1.
Neuroblastoma Patient Derived Xenograft Models Used in Repotrectinib Monotherapy Therapeutic Study
| PDX Model | Sample Type for PDX Generation | Genomic Profile | ||
|---|---|---|---|---|
| ALK Status | MYCN Status | Other relevant genomic aberrations1 (incl. chromosome 1p, 11q, 17q loss/gain) | ||
| MSKNBL-40352 | Primary Resection | Amplified and R1275Q | Amplified | N/A |
| MSKNBL-12717 | Metastatic Lesion at Primary Resection | ALK F1245I | WT | N/A |
| MSKNBL-30595 | Metastatic Relapse | ALK F1174V | Amplified | TERT promoter mutation, TP53 R249S |
| MSKNBL-93255 | Metastatic Relapse | ALK F1174L | Amplified | 1p loss, 17q gain |
| MSKNBL-80717 | Metastatic Relapse | ALK R1275Q | Amplified | 1p loss |
| MSKNBL-47976 | Metastatic Relapse | WT | Amplified | 1p loss, 17q gain |
| MSKNBL-41817 | Metastatic Relapse | WT | WT | 11q gain, 17q gain |
| MSKNBL-82180 | Metastatic Relapse | WT | WT | ATRX E553*, 17q gain |
no PDX models were characterized by mutations in ROS1, NTRK1–3, JAK, FAK, or SRC genes; WT= wild type
Figure 1: Repotrectinib Monotherapy In Vivo Therapeutic Drug Studies.
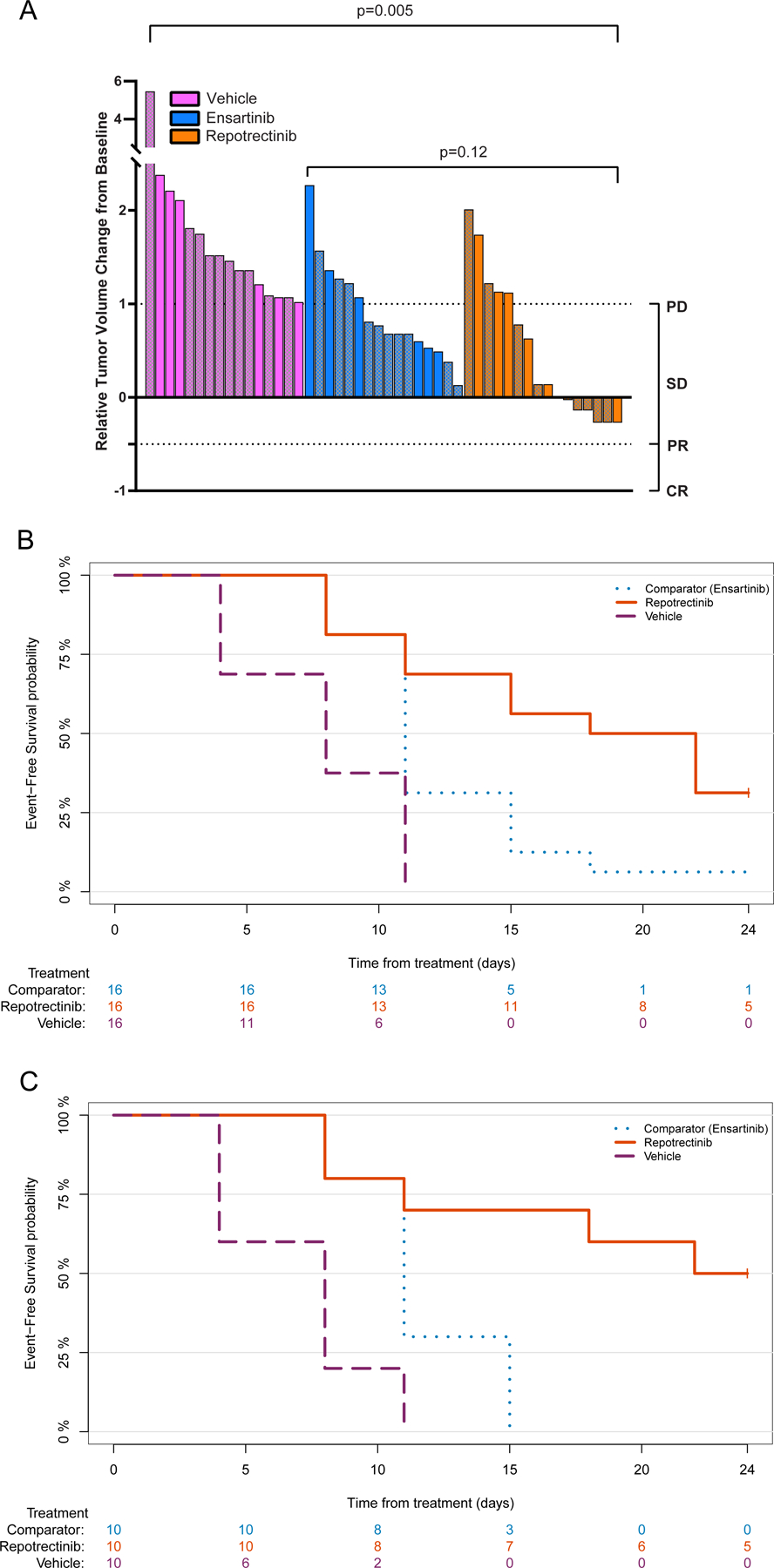
(A) Waterfall plot of repotrectinib monotherapy, ensartinib, and vehicle in PDX models; Solid bars represent ALK WT models and patterned bars represent ALK-mutant models (B) Kaplan-Meier estimates of event-free survival in repotrectinib monotherapy therapeutic studies in the overall cohort (repotrectinib vs vehicle p<0.0001, repotrectinib vs ensartinib =0.02, via log rank) and (C) the ALK-mutant subgroup (repotrectinib vs vehicle p<0.0001, repotrectinib vs ensartinib =0.007, via log rank).
Repotrectinib inhibits proliferation of neuroblastoma cell lines as monotherapy and shows additivity in combination with cytotoxic chemotherapy
We evaluated the anti-proliferative effect of repotrectinib and ensartinib in seven neuroblastoma cell lines (Table S1). Treatment of cells with repotrectinib or ensartinib demonstrated a dose-dependent anti-proliferative effect for both drugs. Repotrectinib and ensartinib effects were most pronounced in NB-1 (ALK amplified) and LAN-5 (ALK R1275Q) and least potent in SK-N-DZ (ALK WT, MYCN amplified, TP53 mutant) (Table 2; Figure S2). When stratifying the IC50 of repotrectinib by various genomic factors including ALK status (presence of mutation or amplification), MYCN status (presence of amplification) or TP53 status (presence of mutation), there was no clear segregation of sensitivity based on molecular alterations (Figure 2a). The IC50 of irinotecan and temozolomide used as single agents was also calculated in all cell lines in anticipation of combination therapy (Table 2; Figure S2). Temozolomide IC50 values were high (114–1500 μM) and reflect the requirement for autocatalysis of temozolomide in culture medium to its active metabolite, MTIC.(37) However, the observed IC50 values of temozolomide are similar to previously determined values in the literature.(38)
Table 2.
Drug Sensitivities in Neuroblastoma Cell Lines
| Compound Name | Description | IC50 Value by Cell Line (μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| SK-N-DZ | SH-SY5Y | Kelly | NB-1 | GI-ME-N | IMR-32 | LAN-5 | ||
| Repotrectinib | Multi-kinase inhibitor of ALK, TRK, JAK2/STAT, Src/FAK | 2.3 ± 0.1 | 1.3 ± 0.1 | 0.74 ± 0.04 | 0.15 ± 0.01 | 1.2 ± 0.1 | 0.65 ± 0.03 | 0.36 ± 0.03 |
| Ensartinib | ALK/MET inhibitor | 2.0 ± 0.1 | 1.5 ± 0.1 | 0.7 ± 0.1 | 0.11 ± 0.01 | 2.7 ± 0.2 | 1.1 ± 0.1 | 0.32 ± 0.03 |
| Temozolomide | Alkylating agent | 144 ± 14 | 130 ± 4 | 218 ± 10 | ~1500 | ~800 | 114 ± 4 | 173 ± 10 |
| Irinotecan | Topoisomerase I inhibitor | 2.9 ± 0.1 | 1.36 ± 0.03 | 2.2 ± 0.1 | 2.8 ± 0.1 | ~25 | 0.28 ± 0.01 | 0.74 ± 0.03 |
Figure 2: Repotrectinib Anti-Proliferative Activity in Neuroblastoma Cell Lines as Monotherapy and in Combination with Chemotherapy.
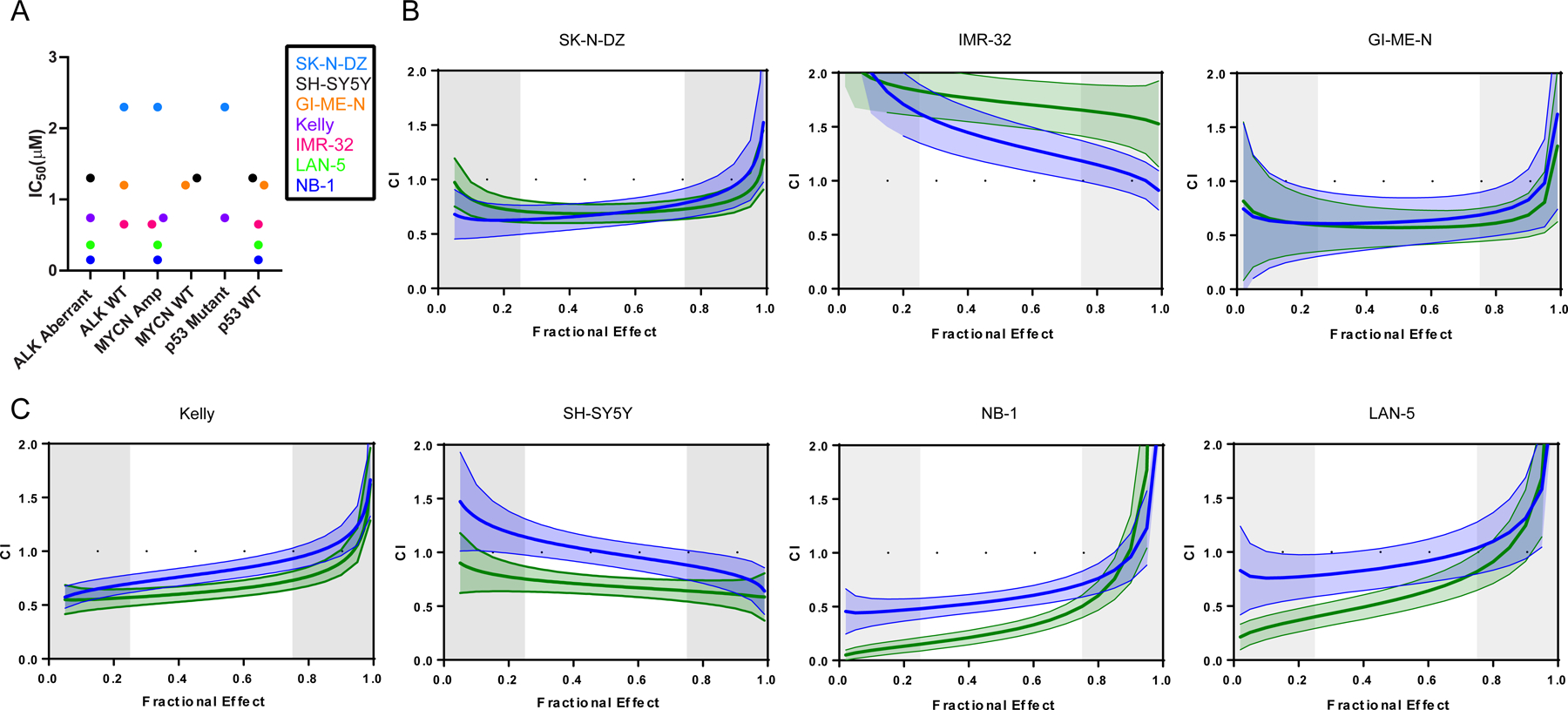
(A) Repotrectinib cytotoxicity (IC50) in neuroblastoma cell lines tested stratified by ALK status, MYCN status, and TP53 status; Combination index (CI) of IRI/TMZ/REPO (blue) or IRI/TMZ/ENS (green) in ALK WT (B) or ALK aberrant (C) neuroblastoma cell lines; solid line: median fractional effect; shaded regions: 1.96 x standard error; IRN=irinotecan; TMZ=temozolomide; REPO=repotrectinib, ENS=ensartinib
To assess for combinatorial activity of repotrectinib with chemotherapy, the fractional effect points (fraction of cells affected at each respective treatment dose) were plotted against the combination index (CI) for each unique cell line and treatment condition. The combination index defines the combination of two or more drugs as synergistic (CI<1), additive (CI=1), or antagonistic (CI>1). Targeted inhibition (with repotrectinib or ensartinib) in combination with irinotecan and temozolomide demonstrated a robust cytotoxic effect (additive to synergistic) in all ALK aberrant cell lines and two ALK WT cell lines. This is effectively demonstrated by evaluating the CI values generated in the interquartile range (25%−75%) of the fractional effect points for each cell line and treatment condition (Figure 2b–c, Table S2). For example, when evaluating Kelly, the CI of irinotecan and temozolomide treatment ranges from 0.868–1.194 (slight synergism to slight antagonism). With the addition of repotrectinib and ensartinib, CI values range from 0.699–0.930 and 0.570–0.73, respectively, suggesting a non-antagonistic effect range (all CI <1). The non-antagonistic effect of targeted inhibition plus chemotherapy holds true in nearly every cell line, treatment, and fractional effect combination evaluated in ALK aberrant cell lines as well as two ALK WT cell lines (SK-N-DZ and GI-ME-N). Overall, these findings suggest a cooperative effect when combining growth inhibitory agents with chemotherapy in vitro across a phenotypically diverse panel of neuroblastoma cell lines.
Repotrectinib inhibits phosphorylation of multiple pathways implicated in oncogenesis in an ALK aberrant cell line and xenograft model
In order to evaluate target engagement and downstream signaling inhibition by repotrectinib in the most sensitive (NB-1) and least sensitive (SK-N-DZ) neuroblastoma cell lines, we treated cells with increasing concentrations of repotrectinib and evaluated changes in signaling pathways that have been shown to be targeted by repotrectinib (ALK/p-ALK, Trk/p-Trk, FAK/p-FAK, Src/p-Src, PI3K/p-PI3K, AKT/p-AKT, STAT3/p-STAT3, MEK 1/2/p-MEK1/2, Erk1/2/p-Erk1/2, S6/p-S6) (Figure 3a–c). Both cell lines showed reductions in p-FAK and p-Src at the highest repotrectinib treatment concentration (1 μM). While SK-N-DZ showed a more marked reduction in p-Trk, NB-1 showed clear dose-dependent abrogation of p-PI3K, p-AKT, p-STAT3, pMEK1/2, pErk1/2, and p-S6 expression compared to SK-N-DZ. These findings suggest that more effective down-regulation of multiple pathways implicated in cell growth and proliferation may contribute to increased sensitivity to repotrectinib. As the single agent IC50 of repotrectinib in SK-N-DZ (2.3 μM) is higher than that of NB-1, treatment at the maximal treatment concentration (1μM) may have precluded demonstration of pathway inhibition.
Figure 3. Repotrectinib treatment of ALK aberrant cell line and xenograft model shows target engagement of downstream effector pathways.
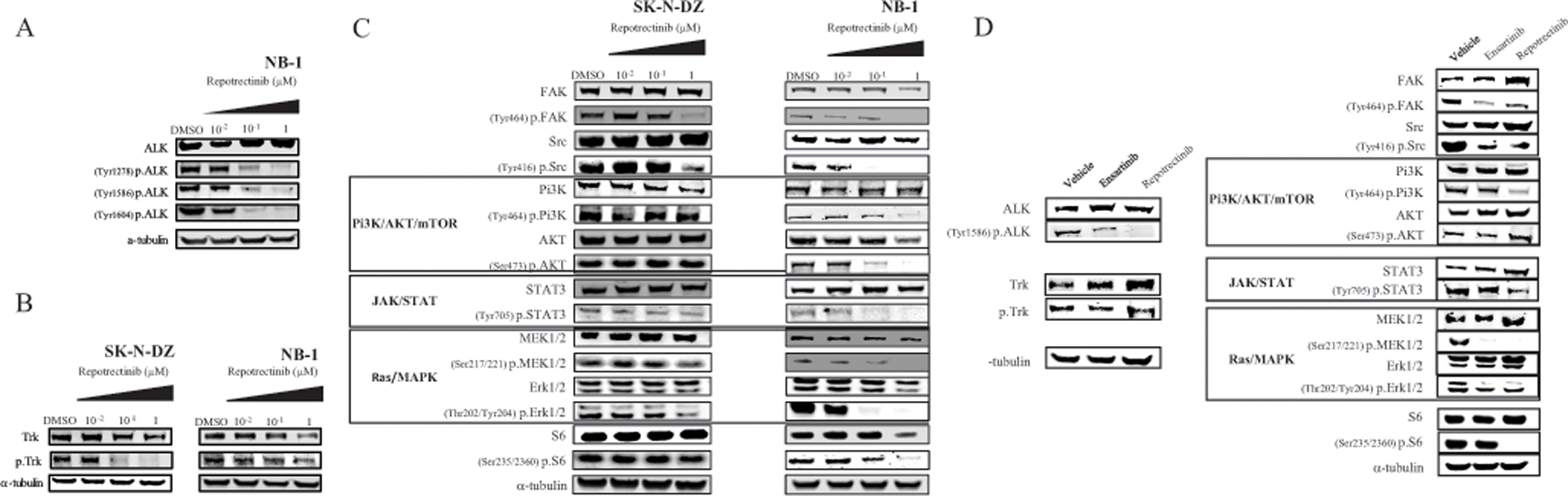
In vitro: protein levels were assessed by western blot analysis after treatment of cells with the indicated concentrations of repotrectinib: (A) Total ALK and p-ALK in NB-1; (B) Trk and p-Trk in SK-N-DZ and NB-1; (C) using the indicated antibodies to assess multiple downstream signaling pathways in SK-N-DZ and NB-1. (D) In vivo: MSKNBL-40352 (ALK 1275Q, ALK amplified, MYCN amplified) tumor lysates from the three treatment arms (vehicle, ensartinib, and repotrectinib (TPX-0005) were analyzed by western blot using the indicated antibodies.
To further assess pharmacodynamic efficacy of repotrectinib, we utilized an ALK amplified (ALK R1275Q, ALK amplification, MYCN amplification) neuroblastoma PDX model (MSKNBL-40352). Tumors were collected after 6 doses (3 days) of repotrectinib treatment, and immunoblot analysis of tumors revealed a similar pattern of ALK, PI3K/AKT and MAPK pathway inhibition (Figure S3, Figure 3d).
Repotrectinib in combination with chemotherapy shows superior anti-tumor activity compared to chemotherapy alone in ALK-mutant neuroblastoma xenograft models
To evaluate the comparative anti-tumor activity of chemotherapy alone versus in combination with repotrectinib in vivo, we evaluated an ALK-mutant neuroblastoma PDX model (MSKNBL-30595, MYCN amplified, ALK F1174V, TERT promoter mutation) and an ALK wild type neuroblastoma PDX model (MSKNBL-82180, MYCN wild type, ALK wild type, ATRX E553*). The two neuroblastoma PDX models were selected as they showed treatment resistance to repotrectinib in the monotherapy therapeutic studies (Figure S3). We enrolled mice onto one of four treatment arms: (1) repotrectinib, (2) irinotecan and temozolomide, (3) repotrectinib plus irinotecan and temozolomide, and (4) vehicle control. All treatments were tolerated well with no signs of systemic toxicity or weight loss (Figure S4). In the ALK-mutant model (n=10 per treatment arm), when comparing best response (% tumor reduction from baseline) after two cycles of chemotherapy, the repotrectinib plus chemotherapy cohort had significantly reduced tumor volume compared to all other treatment groups, including chemotherapy alone (p < 0.001, Mann-Whitney-Wilcoxon test, Figure 4a). Furthermore, when comparing average tumor volume throughout the treatment interval, repotrectinib plus chemotherapy was superior to treatment with chemotherapy alone (p=0.025, Vardi’s test, Figure 4b). When evaluating the role of repotrectinib plus chemotherapy in the ALK wild type model (n=3 per treatment arm with exception of irinotecan/temozolomide arm, n=4, due to rolling enrollment of study animals), both irinotecan/temozolomide and repotrectinib plus irinotecan/temozolomide induced rapid complete responses (Figure 4c). Median time to complete response in the repotrectinib plus chemotherapy arm was 28 days (range 21–38 days) compared to 44 days in the chemotherapy alone arm. Relative tumor volumes (RTV) were calculated for vehicle control (C) and each treatment arm (T) at day 18 (last day with measurable tumor in all mice). The mean RTVs were 74%, 4.8%, and 4.3% in the repotrectinib, chemotherapy, and repotrectinib plus chemotherapy arms respectively.
Figure 4: Repotrectinib Plus Chemotherapy In Vivo Therapeutic Drug Studies.
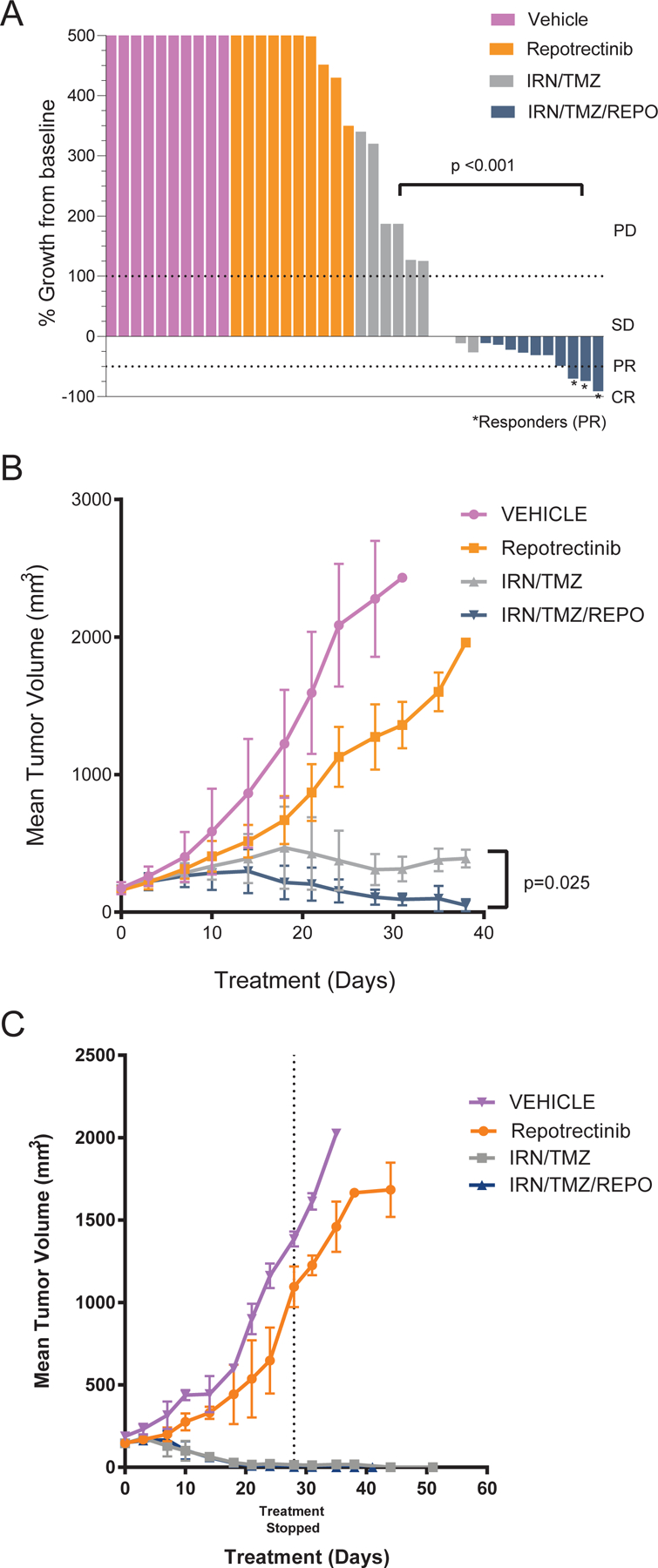
(A) Waterfall plot of best response after 2 cycles of therapy (MSKNBL-30595, ALK 1174V, MYCN amp, TERT promoter mutation); Tumor volume increase beyond 500% not shown here; (B) Mean tumor volume of MSKNBL-30595 in combination therapy study (n=10 mice per arm); Treatment ongoing throughout study interval. (C) Mean tumor volume of MSKNBL-82180 (ALK WT, MYCN WT, ATRX mutation n=3 mice per arm, exception: IRN/TMZ n=4); IRN=irinotecan; TMZ=temozolomide; REPO=repotrectinib.
DISCUSSION:
Due to its macrocyclic structure, small size, and broad kinase inhibitory profile, repotrectinib may provide additional benefit compared to earlier generation kinase inhibitors trialed in neuroblastoma and may augment treatment response when used in combination with chemotherapy. In preclinical studies published to date, repotrectinib has shown potent activity in models characterized by ALK-, ROS-, and NTRK-fusions,(19) with in vitro cytotoxicity observed in several neuroblastoma cell lines, and in vivo anti-tumor effect in one cell-line derived neuroblastoma xenograft model.(11) In our study, we extend these findings by confirming the anti-tumor effects of repotrectinib across a genomically heterogeneous cohort of PDX models that recapitulate the diverse genomic landscape of neuroblastoma seen in the clinic. We also explore the applicability of repotrectinib in ALK-mutant and ALK WT disease and demonstrate potential therapeutic benefit of repotrectinib in combination with a clinically relevant chemotherapy backbone for neuroblastoma.
Our in vivo studies evaluating repotrectinib monotherapy included PDX models with variable mutational status of the following clinically relevant genes in neuroblastoma: MYCN, ALK, TERT, and ATRX. This diverse genotypic representation of neuroblastoma models underscores the applicability and clinical translatability of our preclinical results based upon the prevalence of recurrent molecular alterations observed in neuroblastoma patients. With regard to tumor growth inhibition in pediatric neuroblastoma PDXs, repotrectinib monotherapy slowed tumor growth significantly compared to the vehicle control (p=0.005), with the tested cohort inclusive of ALK-WT and ALK-mutant models. Repotrectinib treated PDXs also had improved EFS compared to the other two treatment groups (p<0.05). However, despite promising preclinical findings with repotrectinib monotherapy, the overall observed clinical response rates are sub-optimal and suggests the need for combination therapy with additional drugs, such as cytotoxic agents, for improved anti-tumor control.
When evaluating repotrectinib monotherapy in vitro, comparable cytotoxic activity is seen with its comparator in class, ensartinib, and single-agent IC50’s for repotrectinib did not segregate based on ALK, MYCN, or TP53 mutational status of the cell lines tested. While the combination of growth inhibitory agents, such as repotrectinib, and cell cycle specific chemotherapy has the potential to be antagonistic, our findings do not demonstrate a significant antagonistic effect, and provide rationale for further exploring this multi-agent regimen. In combining repotrectinib with chemotherapy in vitro, additive to synergistic cytotoxicity was noted across almost all neuroblastoma cell lines tested. Interestingly, the repotrectinib single agent IC50 of each cell line did not directly correlate with the degree of additivity to synergy seen in combination with chemotherapy (irrespective of ALK mutational status). This suggests that there are likely complex signaling interactions contributing to sensitivity or resistance to combination therapy dependent on each cell line’s specific genotype that need to be further elucidated. Overall, our results are comparable to those of Krytska et al. who demonstrated that crizotinib synergizes with cyclophosphamide and topotecan in vitro and in vivo.(39) Similar to Krytska et. al., we evaluated a chemotherapy backbone with known anti-neuroblastoma activity. However, our findings show that the additive to synergistic effect of repotrectinib plus chemotherapy was not limited to ALK-aberrant, WT TP53 models. In addition to the ALK-aberrant models with functional p53, we show evidence of additivity to synergism in two TP53 mutant cells lines including an ALK WT cell line (SK-N-DZ). These findings suggest that non-ALK dependent signaling (JAK2/STAT, Src/FAK) may be contributing to the antiproliferative effect. Taken together, these results, along with the applicability of irinotecan and temozolomide as a rational salvage regimen in patients with neuroblastoma and several other solid tumors, support the clinical translation of this novel drug combination.(40,41)
The cooperative cytotoxicity noted in vitro with repotrectinib plus chemotherapy treatment was recapitulated in vivo. The MSKNBL-30595 PDX model (MYCN amplified, ALK F1174V, TERT promoter mutation) which demonstrated rapid progression when treated with repotrectinib monotherapy, was noted to have statistically significant reduced tumor volume in the repotrectinib plus irinotecan/temozolomide treatment arm compared to chemotherapy alone when evaluating the best response after two cycles of therapy (Figure 4a). Additionally, the repotrectinib plus chemotherapy arm had significantly lower average tumor volume throughout the therapeutic study (Figure 4b). While the sample size was small when evaluating the effect of repotrectinib in combination with chemotherapy in the MSKNBL-82180 model (MYCN WT, ALK WT, ATRX mutant), our results show rapid and durable complete responses with ongoing disease control beyond completion of scheduled drug treatments, as well as a trend toward faster time to response with the addition of multi-kinase inhibition to chemotherapy.
Limited published clinical data are available regarding the role of multi-kinase inhibitor therapy (monotherapy(42,43) or in combination with chemotherapy) in neuroblastoma. There has been a more robust experience with ALK inhibitor monotherapy in this population, but so far this has not yet been applied as a tractable therapeutic option likely due to the upregulation of compensatory signaling pathways seen in preclinical models. These mechanisms include AXL phosphorylation potentiating MAPK signaling and the development of an epithelial-to-mesenchymal transition (EMT) phenotype leading to treatment resistance, among other mechanisms.(44) Repotrectinib inhibits the JAK2/STAT and Src/FAK pathways (of which both can interact with downstream MAPK signaling),(18,19) and suppressed activation of Src, FAK, MEK1/2, and Erk1/2 were demonstrated in a repotrectinib treated ALK-aberrant cell line and xenograft model. Therefore, multi-kinase inhibition achieved with repotrectinib may contribute to broader efficacy across neuroblastoma models resistant to earlier generation inhibitors (e.g. ALK 1174L mutations with conferred crizotinib resistance). Other preclinical studies confirm that inhibition of the JAK/STAT and Src/FAK pathways can abrogate tumor growth either as single-agent therapy or in combination with other targeted inhibitors through re-establishing sensitivity in the setting of on-treatment resistance.(45) For example, in preclinical models of KRAS-mutant non-small cell lung cancer (i.e. without ALK, ROS1, or NTRK aberrations), repotrectinib treated models demonstrated tumor growth inhibition with notable downregulation of p-FAK, p-Src, and p-Erk by immunoblotting. Further, synergistic activity was seen when repotrectinib was administered in combination with trametinib, likely due to targeting of the upregulated compensatory signaling pathways that promote drug resistance.(24) When considering the role of the Src pathway in neuroblastoma specifically, the Src/ABL inhibitor, bosutinib, had potent anti-neuroblastoma activity in vitro and in vivo, and enhanced the effect of conventional cytotoxic chemotherapy.(46) Based on these preclinical findings, it is reasonable to hypothesize that broad, multi-kinase pathway inhibition by repotrectinib contributes to therapeutic efficacy in neuroblastoma xenograft models and warrants future investigation.
In summary, repotrectinib is a promising next generation multi-kinase inhibitor with activity directed toward a number of signaling pathways implicated in neuroblastoma biology. This novel agent has activity in a wide array of genotypically distinct neuroblastoma models irrespective of ALK, MYCN, or TP53 status. When repotrectinib is combined with chemotherapy in vitro and in vivo, superior anti-proliferative and anti-tumor effect is demonstrated most notably in ALK-aberrant tumors. There is an ongoing multi-center, international, first in pediatrics phase 1/2 trial evaluating repotrectinib monotherapy in pediatric patients with relapsed and refractory solid tumors (NCT04094610). Further work is needed to better define the mechanisms of treatment responses and resistance in order to identify populations that may enrich for response to repotrectinib, such as ALK-mutant neuroblastoma. Based on the promising preclinical data presented, a subsequent clinical trial has been developed to evaluate the clinical utility of repotrectinib in combination with cytotoxic chemotherapy in pediatric patients.
Supplementary Material
Acknowledgements:
This research was supported by the Gold Ribbon Riders and Cancer Center Support Grant P30 CA008748. Dr. O’Donohue is partially supported by the NIH/NCATS Grant # UL1-TR-002384, Swim Across America Young Investigator Award, Kristen Ann Carr Fund, Margaux’s Miracle, and TeamConnor. Dr. Dela Cruz is partially supported by The Paulie Strong Foundation, The Grayson Fund, and The Willens Family Fund. We would like to thank Turning Point Therapeutics for supplying repotrectinib for these preclinical studies, Dr. Frank Speleman for providing the NB-1 and GI-ME-N cell lines, and Joseph Olechnowicz (Memorial Sloan Kettering Department of Pediatrics, Senior Editor) for his editorial assistance.
Footnotes
COIs: Dr. O’Donohue reports non-financial support from Turning Point Therapeutics during the conduct of the study (supply of TPX-0005); non-financial support and other support from Turning Point Therapeutics outside the submitted work.
References:
- 1.Howlader NA, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations) Bethesda, MD. National Cancer Institute; 2012. [Google Scholar]
- 2.Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33(27):3008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer cell 2014;26(5):682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16(17):4353–62. [DOI] [PubMed] [Google Scholar]
- 5.Cole KA, Maris JM. New strategies in refractory and recurrent neuroblastoma: translational opportunities to impact patient outcome. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(9):2423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer research 2002;62(22):6462–6. [PubMed] [Google Scholar]
- 7.Radi M, Brullo C, Crespan E, Tintori C, Musumeci F, Biava M, et al. Identification of potent c-Src inhibitors strongly affecting the proliferation of human neuroblastoma cells. Bioorganic & Medicinal Chemistry Letters 2011;21(19):5928–33 [DOI] [PubMed] [Google Scholar]
- 8.Megison ML, Stewart JE, Nabers HC, Gillory LA, Beierle EA. FAK inhibition decreases cell invasion, migration and metastasis in MYCN amplified neuroblastoma. Clinical & Experimental Metastasis 2013;30(5):555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeNardo BD, Holloway MP, Ji Q, Nguyen KT, Cheng Y, Valentine MB, et al. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PloS one 2013;8(12):e82513-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;9(9):941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cervantes-Madrid D, Szydzik J, Lind DE, Borenas M, Bemark M, Cui J, et al. Repotrectinib (TPX-0005), effectively reduces growth of ALK driven neuroblastoma cells. Sci Rep 2019;9(1):19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455(7215):930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George RE, Sanda T, Hanna M, Fröhling S, Ii WL, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer cell 2012;22(1):117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. The Lancet Oncology 2013;14(6):472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulte JH, Moreno L, Ziegler DS, Marshall LV, Zwaan CM, Irwin MS, et al. Final analysis of phase I study of ceritinib in pediatric patients with malignancies harboring activated anaplastic lymphoma kinase (ALK). Journal of Clinical Oncology 2020;38:10505-. [Google Scholar]
- 17.Goldsmith KC, Kayser K, Groshen SG, Chioda M, Thurm HC, Chen J, et al. Phase I trial of lorlatinib in patients with ALK-driven refractory or relapsed neuroblastoma: A New Approaches to Neuroblastoma Consortium study. Journal of Clinical Oncology 2020;38(15_suppl):10504-. [Google Scholar]
- 18.Cui J, et al. TPX-0005 Investigator’s Brochure, TP Therapeutics 2016;1.
- 19.Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov 2018;8(10):1227–36. [DOI] [PubMed] [Google Scholar]
- 20.Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 2016;35(8):939–51. [DOI] [PubMed] [Google Scholar]
- 21.Thakur R, Trivedi R, Rastogi N, Singh M, Mishra DP. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci Rep 2015;5:10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan JP, et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014;5(17):7328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho BC, Drilon AE, Doebele RC, Kim D-W, Lin JJ, Lee J, et al. Safety and preliminary clinical activity of repotrectinib in patients with advanced ROS1 fusion-positive non-small cell lung cancer (TRIDENT-1 study). Journal of Clinical Oncology 2019;37(15_suppl):9011-. [Google Scholar]
- 24.Cui JJ, Zhai D, Deng W, Rodon L, Lee N, Murray B. Abstract 1958: Repotrectinib increases effectiveness of KRAS-G12C inhibitors in KRAS-G12C mutant cancer models via simultaneous SRC/FAK/JAK2 inhibition. Cancer research 2020;80(16 Supplement):1958-. [Google Scholar]
- 25.Murray B, Deng W, Zhai D, Rodon L, Lee N, Cui JJ. Abstract 1957: Repotrectinib increases effectiveness of MEK inhibitor trametinib in KRAS mutant cancer models via simultaneous SRC/FAK/JAK2 inhibition. Cancer research 2020;80(16 Supplement):1957-.32060148 [Google Scholar]
- 26.Mahida JP, Antczak C, Decarlo D, Champ KG, Francis JH, Marr B, et al. A synergetic screening approach with companion effector for combination therapy: application to retinoblastoma. PloS one 2013;8(3):e59156-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999;4(2):67–73. [DOI] [PubMed] [Google Scholar]
- 28.Tallarida RJ, Raffa RB. Testing for synergism over a range of fixed ratio drug combinations: replacing the isobologram. Life Sci 1996;58(2):Pl 23–8. [DOI] [PubMed] [Google Scholar]
- 29.Chou T-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer research 2010;70(2):440–6. [DOI] [PubMed] [Google Scholar]
- 30.Chou T-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacological Reviews 2006;58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 31.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21(11):1318–25. [DOI] [PubMed] [Google Scholar]
- 32.Oxnard GR, Morris MJ, Hodi FS, Baker LH, Kris MG, Venook AP, et al. When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst 2012;104(20):1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James K, Eisenhauer E, Christian M, Terenziani M, Vena D, Muldal A, et al. Measuring response in solid tumors: unidimensional versus bidimensional measurement. J Natl Cancer Inst 1999;91(6):523–8. [DOI] [PubMed] [Google Scholar]
- 34.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer 2007;49(7):928–40. [DOI] [PubMed] [Google Scholar]
- 35.Kersemans V, Cornelissen B, Allen PD, Beech JS, Smart SC. Subcutaneous tumor volume measurement in the awake, manually restrained mouse using MRI. J Magn Reson Imaging 2013;37(6):1499–504. [DOI] [PubMed] [Google Scholar]
- 36.Oxnard GR, Morris MJ, Hodi FS, Baker LH, Kris MG, Venook AP, et al. When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst 2012;104(20):1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. The oncologist 2000;5(2):144–51. [DOI] [PubMed] [Google Scholar]
- 38.Piskareva O, Harvey H, Nolan J, Conlon R, Alcock L, Buckley P, et al. The development of cisplatin resistance in neuroblastoma is accompanied by epithelial to mesenchymal transition in vitro. Cancer letters 2015;364(2):142–55. [DOI] [PubMed] [Google Scholar]
- 39.Krytska K, Ryles HT, Sano R, Raman P, Infarinato NR, Hansel TD, et al. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(4):948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bagatell R, London WB, Wagner LM, Voss SD, Stewart CF, Maris JM, et al. Phase II study of irinotecan and temozolomide in children with relapsed or refractory neuroblastoma: a Children’s Oncology Group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(2):208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyers PA, Ambati SR, Slotkin EK, Cruz FD, Wexler LH. The addition of cycles of irinotecan/temozolomide (i/T) to cycles of vincristine, doxorubicin, cyclophosphamide (VDC) and cycles of ifosfamide, etoposide (IE) for the treatment of Ewing sarcoma (ES). Journal of Clinical Oncology 2018;36(15_suppl):10533-. [Google Scholar]
- 42.Robinson GW, Gajjar AJ, Gauvain KM, Basu EM, Macy ME, Maese LD, et al. Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. Journal of Clinical Oncology 2019;37(15_suppl):10009-. [Google Scholar]
- 43.Perisa MP, Storey M, Streby KA, Ranalli MA, Skeens M, Shah N. Cabozantinib for relapsed neuroblastoma: Single institution case series. Pediatr Blood Cancer 2020;67(7):e28317. [DOI] [PubMed] [Google Scholar]
- 44.Debruyne DN, Bhatnagar N, Sharma B, Luther W, Moore NF, Cheung NK, et al. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016;35(28):3681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346(6216):1480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bieerkehazhi S, Chen Z, Zhao Y, Yu Y, Zhang H, Vasudevan SA, et al. Novel Src/Abl tyrosine kinase inhibitor bosutinib suppresses neuroblastoma growth via inhibiting Src/Abl signaling. Oncotarget 2017;8(1):1469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.